
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolarization-induced nanohelixes of organic cocrystals from asymmetric components with dopant-induced chirality inversion†
Jinqiu
Chen
,
Canglei
Yang
,
Shuang
Ma
,
Zhiqi
Liu
,
Wenxin
Xiang
and
Jing
Zhang
 *
*
State Key Laboratory of Organic Electronics and Information Displays, Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications, 9 Wenyuan Road, Nanjing 210023, China. E-mail: iamjingzhang@njupt.edu.cn
First published on 17th January 2023
Abstract
Supramolecular chirality is essential for the development of functional materials. In this study, we report the synthesis of twisted nanobelts based on charge-transfer (CT) complexes using self-assembly cocrystallization starting from asymmetric components. An asymmetric donor, DBCz, and a typical acceptor, tetracyanoquinodimethane, were used to construct a chiral crystal architecture. An asymmetric alignment of the donor molecules induced polar ±(102) facets that, accompanied with free-standing growth, resulted in a twisting along the b-axis due to the electrostatic repulsive interactions. Meanwhile, the alternately oriented ±(001) side-facets were responsible for the propensity of the helixes to be right-handed. Addition of a dopant significantly enhanced the twisting probability by reducing the surface tension and adhesion influence, even switching the chirality preference of the helixes. In addition, we could further extend the synthetic route to other CT systems for formation of other chiral micro/nanostructures. Our study offers a novel design approach for chiral organic micro/nanostructures for applications in optically active systems, micro/nano-mechanical systems and biosensing.
Introduction
Understanding the relationships between the molecular conformations of hierarchical chiral structures, ensembles, and the self-assembly process is crucial for the development of functional materials and biosimulations.1 Unique morphologies that show helical ordering undergo rhythmic twisting motions that can range from nanometers to centimeters. Studies on the loss of elastic energy during the chiral crystallization of substances, such as elemental substances, simple salts, minerals, and organic molecular crystals, and, in particular, polymers, show how deformation works and the periodic twisting affects the size and shape of the crystals.2 The strain computed from the approximated elastic energy of such deformations helps us to distinguish their predominant driving mechanism by considering the possible causes, such as screw dislocations, thermal/mechanic/compositional fields, and heteromeric stress.3 Organic chiral molecules can be assembled into twisted arrangements in one direction by symmetry breaking,4 and chiral additives can further enhance the chiral expression in the form of helixes and/or preferably handedness, even in achiral systems.5 The surface charge ultimately accumulated from electrostatics and piezoelectricity is, in a few specific instances, another way to create a twisted arrangement in a material.6 For example, piezoelectric ZnO grew into crystalline nanohelixes/nanosprings, and nanorings through a vapor–solid growth process, due to the presence of polar surfaces.7 However, the stiffness and brittleness of the inorganic nanohelixes confined them to a small size of up to several tens of nanometers.Charge-transfer complexes are a new and emerging type of organic functional semiconducting material that has attracted considerable attention in the fields of tunable and high-performance charge transport, light emitting materials, photoelectric conversion, optical waveguidance, and stimuli-responsive materials.8 This type of construction has a specific arrangement consisting of molecular-level p–n junctions, and, interestingly, charge-transfer, π–π interactions, and hydrogen bonds between various molecules that make this interpenetrated network very responsive to external stimuli.9 Recently, we prepared twisted achiral charge-transfer complex crystals of pyrene and 4,8-bis(dicyanomethylene)-4,8-dihydrobenzo[1,2-b:4,5-b′]-dithiophene (DTTCNQ) via a self-assembly method in a polar solvent that functions due to the internal stress and solvent–solute interactions.10 A phase transition from a dynamically stable phase to a thermodynamically stable phase induces the spontaneous stress relaxation of the untwisting. Later, Kahr et al. reported a series of charge-transfer complexes with twisted morphologies synthesized from melts with a larger propensity to form twisted structures than the monocomponent systems, where the slightly rotated layers helped to increase the charge mobilities. Recently, some charge-transfer complexes have been found to belong to the polar space group for the unique aligned packing mode and even to be ferroelectric.11 Twisted conformations in polar charge-transfer complex systems may be predicted considering certain design principles, while the sense of twist, called chirality, can be also changed by the asymmetry of the precursor molecules and surface charge disturbance.
In this study, we report nanohelixes made from charge-transfer complexes, which grow along the b-axis via a solution state growth process, and have top and bottom surfaces with polar ±(102) faces. The twisting mechanism is attributed to electrostatic repulsion from an unbalanced alignment of highly asymmetric 7H-dibenzo[c,g]carbazole (DBCz) molecules. Unlike inorganic nanostructures, the dimensions of organic twisted structures can even reach the micrometer range. Upon doping of a second donor, the production of the nanohelixes was highly improved and the chirality was transformed from right-hand dominated to left-hand dominated.
Results and discussion
DBCz is a helical and noncentrosymmetric molecule with a dipole moment of 1.6 D, that is introduced as the crucial element to build a symmetry breaking system. The new DBCz–TCNQ complex made from the DBCz donor (D) (Fig. S1†) and the 7,7,8,8-tetracyanoquinodimethane (TCNQ) acceptor (A) is synthesized by blending their powders in chloroform. As the solvent evaporates, dark needle-like cocrystals are self-assembled at the bottom of the reaction vessel, and the crystal structure was confirmed using X-ray single crystal diffraction spectroscopy (Table S1†). The DBCz–TCNQ single crystal belongs to the monoclinic Pc space group with lattice constants a = 11.1150(6) Å, b = 7.4032(5) Å, c = 41.998(3) Å, and β = 93.750(5)°. The p–n molecular pairs pack along the b-axis to form D–A–D–A mixed stacking columns through π–π interactions with π–π distances of 3.22–3.29 Å (Fig. S2†). We consider the helical DBCz molecule to be divided into two parts (the upper segment that bends up and the lower segment that bends down) from the middle nitrogen atom. The sandwiched TCNQ molecule interacts with the upper segment of the adjacent DBCz molecule and lower part of the other DBCz. For the other half of the DBCz molecule, C–H⋯π interactions with another DBCz molecule and short C–H⋯N contacts with a TCNQ molecule from the adjoining column exist along the [−201] direction, forming a herringbone structure (Fig. 1a). The slipping angle is around 120°. These stacking layers connect by short C–H⋯N contacts, resulting in a stabilized three-dimensional framework of the supramolecular complex. Asymmetrical and helical DBCz molecules play a crucial role in the polarization of the conformation in the polar phase of chiral crystallization. The DBCz molecules adopt two helical configurations, P and M, and are evenly distributed in different π-stacking columns (Fig. S3†). The dihedral angles of the two naphthyl groups (upper and lower segment) in each configuration are 16.21°, 16.64°, and 9.69°, respectively (Fig. 1b). The dipole moments of the DBCz molecules, resulting from nitrogen atom substitution, have a 1/3 head-up alignment and a 2/3 head-down mode (Fig. 1c). The ±(102) facets are considered to be the polar surfaces and are the source of the presumed electrostatic interaction-induced twisting.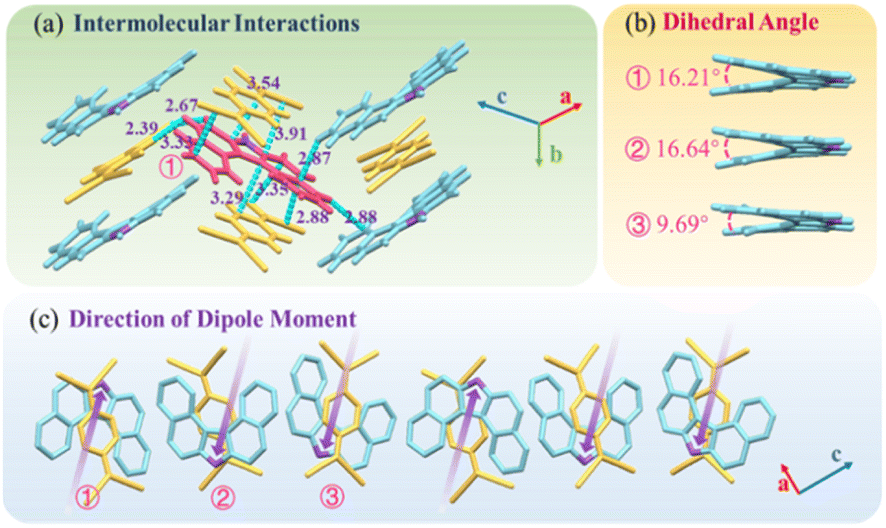 | ||
| Fig. 1 (a) Crystal structure of the DBCz–TCNQ complex, (b) dihedral angles of helical DBCz molecules in the cocrystals, and (c) dipole moment along the (102) face. | ||
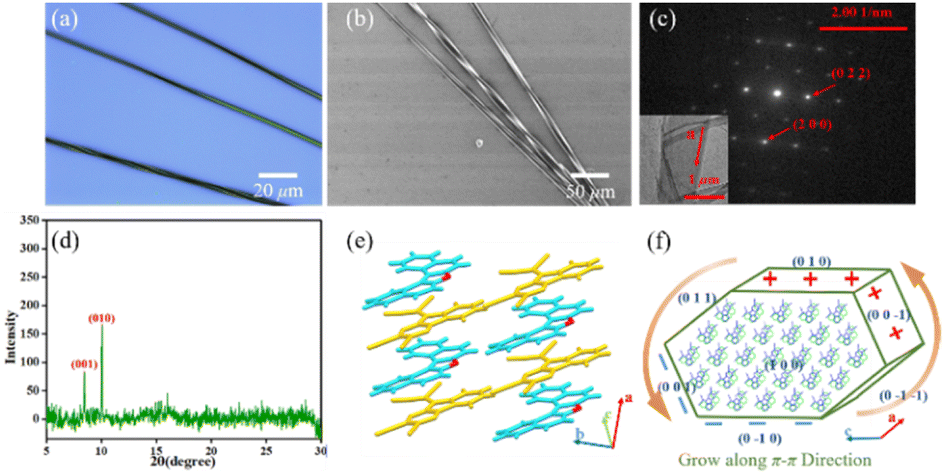
Next, binary DBCz–TCNQ nanohelixes were synthesized using a solution drop-casting approach (Fig. 2a). First, at a total concentration of ∼1.8 mg mL−1, DBCz and TCNQ powders with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were blended in acetonitrile. The mixed solution was drop-casted onto a silicon substrate after heating and sonication of the solution for full dissolution. Finally, the DBCz–TCNQ nanostructures were formed on the surface after evaporating for about 30 min. Straight nanobelts were formed at the edges and bunches of twisted nanocrystals formed around the center location (∼30% in area). The as-prepared nanobelts have a controlled morphology, and thick or thin belts were found to be straight. To examine the structures and chirality of the twisted DBCz–TCNQ nanobelts, optical microscopy (OM), scanning electron microscopy (SEM) at ambient conditions with the solvent, X-ray diffraction (XRD), and transmission electron microscopy (TEM) were performed (Fig. 2b–e and S4†). OM and SEM images reveal the belt-like architecture with widths of 100–2300 nm, a thickness of 50–800 nm, and lengths of up to several millimeters. Depending on the belt width, the pitch ranges from 500 nm to 10 μm. Fig. S5† shows the relationship between the width, thickness, and pitch, the twisting pitch is linearly proportional to the width and thickness. Being free-standing in solution, the thickening of the twisted belts lead to compensatory stress, where the relaxation of such stress causes a subsequent increase in the twist period.
:1 were blended in acetonitrile. The mixed solution was drop-casted onto a silicon substrate after heating and sonication of the solution for full dissolution. Finally, the DBCz–TCNQ nanostructures were formed on the surface after evaporating for about 30 min. Straight nanobelts were formed at the edges and bunches of twisted nanocrystals formed around the center location (∼30% in area). The as-prepared nanobelts have a controlled morphology, and thick or thin belts were found to be straight. To examine the structures and chirality of the twisted DBCz–TCNQ nanobelts, optical microscopy (OM), scanning electron microscopy (SEM) at ambient conditions with the solvent, X-ray diffraction (XRD), and transmission electron microscopy (TEM) were performed (Fig. 2b–e and S4†). OM and SEM images reveal the belt-like architecture with widths of 100–2300 nm, a thickness of 50–800 nm, and lengths of up to several millimeters. Depending on the belt width, the pitch ranges from 500 nm to 10 μm. Fig. S5† shows the relationship between the width, thickness, and pitch, the twisting pitch is linearly proportional to the width and thickness. Being free-standing in solution, the thickening of the twisted belts lead to compensatory stress, where the relaxation of such stress causes a subsequent increase in the twist period.
 | ||
| Fig. 2 (a) Self-assembly in solution of twisted and straight crystals of the DBCz–TCNQ complex. (b) OM image, (c) SEM image, and (d) XRD pattern of the helical DBCz–TCNQ nanobelts on the substrate. (e) TEM image and its corresponding selected area diffraction pattern of the twisted nanobelt. (f) Molecular arrangement and crystal plane distribution in the DBCz–TCNQ cocrystal. (g) Schematic diagram of the effect of different kinetic factors on the crystal morphology during drop-casting. | ||
Using SEM to examine hundreds of nanobelts prepared in situ on the substrate, it was found that more than 82% of the nanobelts were right-handed, showing preferable asymmetric twisting occurs using this growth procedure. The XRD pattern shows the crystallographic structure of the upper polar phase of the helix sample. The sharp peak at 9.1° indicated that the top surfaces of the nanobelts are the polar (102) faces, while the expected peak at 12.5° is attributed to the (006) faces and can be assigned as the lateral facets. The small surface energy of the crystal faces (Table S2:†E{102}s (55.9 kcal mol−1) < E{100}s (80.0 kcal mol−1) < E{002}s (101.4 kcal mol−1) < E{010}s (173.8 kcal mol−1)) in the equilibrium morphology of the DBCz–TCNQ cocrystal suggested the preferred orientation of the (102) faces reduces the total surface energy of the thermodynamically favored state. The comparable attachment energies of the (102) and (002) faces (E{002}s (−33.4 kcal mol−1) ≈ E{102}s (−40.4 kcal mol−1)) also predicted the exceptional exposed crystal area of the (102) and (002) faces (Table S3†). Combining TEM with selected area electron diffraction analysis (Fig. 2e), the self-assembled twisted nanobelts were demonstrated to grow along the [010] direction induced by charge-transfer (CT) and π–π interactions between the donor and acceptor molecules. The short-axis growth direction of the cocrystal was determined to be [−104] (Fig. 2f and S6†).
The electrostatic energy of the crystal originating from the ±(102) faces was computed as follows:

To test the theory that substrate surface induces the morphology, we made polymer solutions of the DBCz–TCNQ supramolecules, where the DBCz–TCNQ molecules were a minority, with a mass ratio of 5:95 for drop-casting self-assembly with the aim of overcoming the adhesion energy barrier. The macromolecules aggregated close to the surface, and a small amount of these supramolecules tend to crystallize into thin twisted nanostructures with a longitudinal dimension of 100 micrometers and a diameter of ∼200 nanometers (Fig. S7†). The dispersed and “protected” DBCz and TCNQ molecules immersed in viscous surroundings ensure that the polar facet-dominated nanohelix forms on a regular scale.
We treated the DBCz–TCNQ mixture solution with 5%, 10%, 15%, 20%, and 25% equiv. of a BCz additive (mass ratio of BCz:DBCz) to tune the assemblies by influencing the cocrystallization environment. Subsequent drop-casting of these doped solutions resulted in substantial morphological changes in both the twisting proportion and preferred chirality (Fig. 3). According to OM and SEM analysis, with 10% BCz doping, the total helix ratio across the assembly area was increased from the original 30% to a maximum of 85%, and the preferred handedness became left-handed with a proportion of 78% (Fig. 4a). This is further supported by the circular dichroism (CD) spectra of the different twisted nanobelts, obtained from a pristine binary blend and a 10% doped blend (Fig. 4b and S8†).
 | ||
| Fig. 3 (a) Right-handed and (b) left-handed helical nanobelts; (c) nuclei with multiple helical nanobelts; (d–i) helical nanobelts of DBCz–TCNQ system doped with different percentages of BCz (the doping percentage refers to the molar ratio of BCz molecules and DBCz molecules). | ||
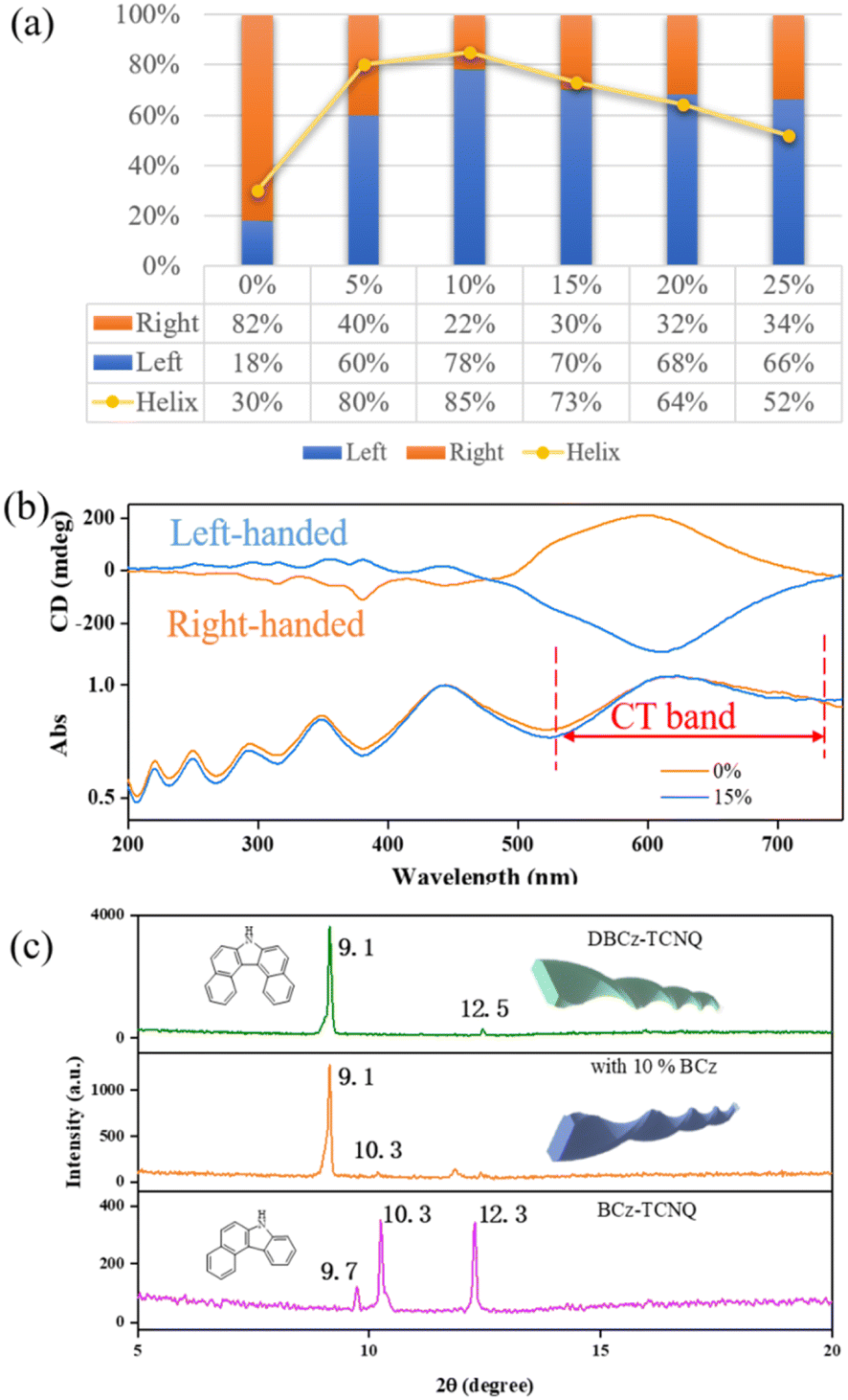 | ||
| Fig. 4 (a) Detail of the morphological changes of the supramolecular construct obtained from a blend solution with addition of varying equivalents of BCz, (b) CD spectra of DBCz–TCNQ nanohelixes assembled from a binary solution and 10% doped mixed solution at 25 °C. (c) XRD patterns of helical DBCz–TCNQ nanobelts, nanobelts doped with 10% BCz, and a pristine BCz–TCNQ complex. | ||
These optically active architectures have mirror-image spectra with positive and negative Cotton effects over the absorption range, and a highly improved Cotton effect in the red-shifted region of the CT band, indicating that the assemblies are effectively exciton coupled. As shown in Fig. S9,† the band that bathochromically shifted from about 500 nm to 900 nm in the binary cocrystal can be assigned to the CT band. This band is attributed to the charge-transfer interactions between the donor and acceptor molecules, hence the strong CD signal at about 500–700 nm arising from the CT influence. There exists a helical arrangement between the naphthalene plane of the DBCz molecule and the plane of the TCNQ molecule. The inverted left-handed helical arrangement in the π-conjugated region produces a strong negative CD signal due to the BCz doping.
XRD patterns of original helical belts are almost the same as that of 10% BCz doped nanobelts, except for several weak peaks arising from the BCz–TCNQ cocrystal (Fig. 4c). The crystal lattice is well maintained and nearly no visible destruction occurs, meanwhile, the large full width at half maxima value shows the existence of screw dislocations in the thin belt and explains the observed twisting ratio enhancement. Based on our previous results, BCz molecules have low solubility and are expected to nucleate with TCNQ before the DBCz units. The flow of molecules into the clusters of a nucleus would primarily be BCz molecules instead of DBCz, also explaining the high twisting ratio.
The surface charges on the DBCz–TCNQ belt are affected by doping with the BCz molecules and this changes the primary twist mode in the varied deformation orientation mechanism. To further confirm the generality of our design principle based on structural asymmetry considerations in chiral nanoarchitectures, we extended our strategy to other charge-transfer complexes, such as the dinaphtho[1,2-b:1′,2′-d]furan and 9-(dicyanomethylene)-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile (DNF–TCAF), 7H-dibenzo[c,g]carbazole and 2,2′-(benzo[1,2-b:4,5-b′]dithiophene-4,8-diylidene) dimalononitrile (DBCz–DTTCNQ), dinaphtho[1,2-b:1′,2′-d]furan and 7,7,8,8-tetracyanoquinodimethane (DNF–TCNQ) systems. Fig. 5a and b shows the optical and SEM images of the twisted form of the DNF–TCAF nanobelts after drop-casting of mixed solutions onto the substrate. It can be observed clearly that the helixes have lengths ranging from 100 μm to 1200 μm, widths of 1–40 μm, and thicknesses of about 100 nm–7 μm, with a high propensity for right-chirality of 43%. It is notable that in this system micrometer-sized crystal deformation is observed, which is attributed to the bi-asymmetric component construct. Judging from the TEM and XRD patterns (Fig. 5c and d), the supramolecular nanostructure grows along the a-axis (π–π stacking manner), with lateral facets of the (001) and (011) crystal planes. According to its crystal structure, the cocrystal belongs to the chiral P1 triclinic space group, with cell parameters of a = 7.023(10) Å, b = 9.909(14) Å, c = 11.109(14) Å, and β = 74.66(5)°. The donor and acceptor molecules alternately stack along the a-axis due to the π–π interactions (Fig. 5e). The main π-backbone of TCAF overlaps with the half of the DNF unit that bends down, while the DNF segment that bends up interacts with the malononitrile group of the TCAF molecule which bends about 7.8° to facilitate the donor–acceptor interactions. With the extension of the π-backbone, stronger π–π interactions and short C–H⋯N contacts confine the asymmetrical donor in one typical orientation. Therefore, the micro/nanostructures also have significant twisting polarization along the crystal surfaces and twisting behaviour occurs (Fig. 5f). The effective surface charge density of the {001} planes is about 0.0166 e Å−2 and that of the {010} facets is about 0.0231 e Å−2, from which the defined alignment results in a large electrostatic potential for the {001} and {010} crystal faces. The specific preference for right-chiral orientated helixes here originates from the stronger polarization of the {010} crystal faces. The DNF–TCNQ and DBCz–DTTCNQ morphologies arising from this design principle are shown in Fig. S10.† Moreover, an improvement in the probability of twisting and chirality inversion due to surface tension reduction and surface charge disturbance were found in these three supramolecular systems by adding the BCz dopant (Fig. S11†).
 | ||
| Fig. 5 (a) OM image, (b) SEM image, and (c) TEM image and its corresponding selected area diffraction pattern of one twisted nanobelt, (d) XRD pattern of helical DNF–TCAF micro/nanobelts on the substrate, (e) crystal structure, and (f) molecular arrangement and crystal plane distribution in DNF–TCAF cocrystal. | ||
Conclusions
In conclusion, we have shown the self-assembly of nanohelixes composed of a helical donor DBCz and an achiral acceptor TCNQ. Synergistic π–π and C–H⋯π interactions and C–H⋯N hydrogen bonds were used to build the supramolecular framework. The surface charge potentials were confirmed to be generated from an asymmetrical alignment of DBCz molecules of the ±(102) faces, which further induced a twist along the b-axis. The ±(001) side face with a constantly varied orientation facilitates the formation of a right-handed helix. Consequently, the addition of a BCz dopant can considerably enhance the twisting probability and even switch the chiral preference of the helixes due to adhesion, surface charge disturbance, and overcoming of surface tension. Furthermore, a series of crystalline mixed cocrystals comprising organic asymmetric components were developed to demonstrate that similar twisted nanostructures (and even microstructures) could be formed and that they demonstrate the same chiral properties upon addition of a dopant. This study represents a new design method for size and chirality controllable organic nanohelixes with potential applications in optically active, micro/nano-mechanical systems, and biosensing.Author contributions
Jinqiu Chen: data curation, formal analysis, investigation, validation, writing – original draft. Canglei Yang: formal analysis, investigation, supervision. Shuang Ma: data curation. Zhiqi Liu: data curation. Wenxin Xiang: data curation. Jing Zhang: conceptualization, formal analysis, investigation, methodology, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (61774087), 1311 Research Foundation of Nanjing University of Posts & Telecommunications and Jiangsu Specially Appointed Professor Foundation.Notes and references
- (a) L. L. Yang, X. X. Tan, Z. Q. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196 CrossRef CAS PubMed; (b) R. J. Dong, Y. F. Zhou, X. H. Huang, X. Y. Zhu, Y. F. Lu and J. Shen, Adv. Mater., 2015, 27, 498 CrossRef CAS PubMed; (c) P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 57, 8498 CrossRef CAS PubMed; (d) M. C. di Gregorio, L. J. W. Shimon, V. Brumfeld, L. Houben, M. Lahav and M. E. van der Boom, Nat. Commun., 2020, 11, 9 CrossRef PubMed.
- (a) A. G. Shtukenberg, J. Freudenthal and B. Kahr, J. Am. Chem. Soc., 2010, 132, 9341 CrossRef CAS PubMed; (b) A. G. Shtukenberg, Y. O. Punin, A. Gujral and B. Kahr, Angew. Chem., Int. Ed., 2014, 53, 672 CrossRef CAS PubMed; (c) Y. Yang, Y. J. Zhang and Z. X. Wei, Adv. Mater., 2013, 25, 6039–6049 CrossRef CAS PubMed; (d) M. H. Liu, L. Zhang and T. Y. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed.
- (a) Q. C. Lu, B. L. Huang, Q. H. Zhang, S. M. Chen, L. Gu, L. Song, Y. Yang and X. Wang, J. Am. Chem. Soc., 2021, 143, 9858 CrossRef CAS PubMed; (b) C. Q. Zhou, Y. Y. Ren, J. Han, X. X. Gong, Z. X. Wei, J. Xie and R. Guo, J. Am. Chem. Soc., 2018, 140, 9417 CrossRef CAS PubMed; (c) B. Biswas, D. Mitra, F. Kp, S. Bhat, A. Chatterji and G. Kumaraswamy, ACS Nano, 2021, 15, 19702 CrossRef CAS PubMed.
- (a) B. Yang, G. Zou, S. L. Zhang, H. L. Ni, H. F. Wang, W. Xu, C. Yang, H. Zhang, W. H. Yu and K. J. Luo, Angew. Chem., Int. Ed., 2021, 60, 10531 CrossRef CAS PubMed; (b) Y. Yang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 6039 CrossRef CAS PubMed.
- (a) B. Bosnich, J. Am. Chem. Soc., 2002, 89, 6143 CrossRef; (b) B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni and L. Bouteiller, Chem.–Eur. J., 2010, 16, 173 CrossRef CAS PubMed; (c) T. Yuan, Z. Sun, A. U. Mu, M. Zeng, A. J. Kalin, Z. Cheng, M. A. Olson and L. Fang, Chem.–Eur. J., 2018, 24, 16553 CrossRef CAS PubMed; (d) S. Goskulwad, D. D. La, M. A. Kobaisi, S. V. Bhosale, V. Bansal, A. Vinu, K. Ariga and S. V. Bhosale, Sci. Rep., 2018, 8, 11220 CrossRef PubMed; (e) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
- (a) W. L. Hughes and Z. L. Wang, J. Am. Chem. Soc., 2004, 126, 6703 CrossRef CAS PubMed; (b) R. S. Yang, Y. Ding and Z. L. Wang, Nano Lett., 2004, 4, 1309 CrossRef CAS; (c) J. K. Jian, Z. H. Zhang, Y. P. Sun, M. Lei, X. L. Chen, T. M. Wang and C. Wang, J. Cryst. Growth, 2007, 303, 427 CrossRef CAS; (d) R. S. Yang and Z. L. Wang, J. Am. Chem. Soc., 2006, 128, 1466 CrossRef CAS PubMed; (e) G. Z. Shen and D. Chen, J. Am. Chem. Soc., 2006, 128, 11762 CrossRef CAS PubMed; (f) M. J. Bierman, Y. K. A. Lau, A. V. Kvit, A. L. Schmitt and S. Jin, Science, 2008, 320, 1060 CrossRef CAS PubMed.
- (a) X. Y. Kong and Z. L. Wang, Nano Lett., 2003, 3, 1625 CrossRef CAS; (b) X. Y. Kong and Z. L. Wang, Appl. Phys. Lett., 2004, 84, 975 CrossRef CAS; (c) Z. L. Wang, ACS Nano, 2008, 2, 1987 CrossRef CAS PubMed.
- (a) W. Wang, L. X. Luo, P. Sheng, J. Zhang and Q. C. Zhang, Chem.–Eur. J., 2021, 27, 464 CrossRef CAS PubMed; (b) J. Zhang, J. Q. Jin, H. X. Xu, Q. C. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485 RSC; (c) J. F. Guo, Y. G. Zhen, H. L. Dong and W. P. Hu, J. Mater. Chem. C, 2021, 9, 16843 RSC; (d) S. Z. Li, Y. J. Lin and D. P. Yan, J. Mater. Chem. C, 2016, 4, 2527 RSC; (e) J. L. Han, D. Yang, X. Jin, Y. Q. Jiang, M. H. Liu and P. F. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013 CrossRef CAS PubMed; (f) Y. Q. Sun, Y. L. Le, H. L. Dong, Y. G. Zhen and W. P. Hu, J. Am. Chem. Soc., 2018, 140, 6186 CrossRef CAS PubMed; (g) S. J. Kan, S. Ahn, J. B. Kim, C. Schenck, A. M. Hiszpanski, S. Oh, T. Schiros, Y. L. Loo and C. Nuckolls, J. Am. Chem. Soc., 2013, 135, 10579 CrossRef.
- (a) I. V. Fedyanin, CrystEngComm, 2022, 24, 2591 RSC; (b) M. Boterashvili, M. Lahav, S. Shankar, A. Facchetti and M. E. van der Boom, J. Am. Chem. Soc., 2014, 136, 11926 CrossRef CAS PubMed; (c) L. J. Sun, W. G. Zhu, X. T. Zhang, L. Q. Li, H. L. Dong and W. P. Hu, J. Am. Chem. Soc., 2021, 143, 19243 CrossRef CAS PubMed; (d) W. G. Zhu, R. H. Zheng, Y. G. Zhen, Z. Y. Yu, H. L. Dong, H. B. Fu, Q. Shi and W. P. Hu, J. Am. Chem. Soc., 2015, 137, 11038 CrossRef CAS PubMed; (e) C. H. Liu, M. R. Niazi and D. F. Perepichka, Angew. Chem., Int. Ed., 2019, 58, 17312 CrossRef CAS PubMed.
- C. L. Yang, L. X. Luo, J. Q. Chen, B. Yang, W. Wang, H. B. Wang, G. K. Long, G. F. Liu, J. Zhang and W. Huang, Chem. Commun., 2021, 57, 10031 RSC.
- (a) T. Matsuura and H. Koshima, J. Photochem. Photobiol., C, 2005, 6, 7 CrossRef CAS; (b) A. S. Tayi, A. K. Shveyd, A. C. H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, the preparation of the DBCz/TCNQ single crystal, the intermolecular distances in the cocrystals, short-contact interactions, infrared absorption spectra, solid-state UV-vis-NIR absorption spectra, and CD spectra, AFM, and crystal data. CCDC 2169110 and 2214872. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05942h |
| This journal is © The Royal Society of Chemistry 2023 |