
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA syn outer-sphere oxidative addition: the reaction mechanism in Pd/Senphos-catalyzed carboboration of 1,3-enynes†
Ziyong
Wang
 a,
Walid
Lamine
b,
Karinne
Miqueu
*b and
Shih-Yuan
Liu
*ab
a,
Walid
Lamine
b,
Karinne
Miqueu
*b and
Shih-Yuan
Liu
*ab
aDepartment of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467-3860, USA. E-mail: shihyuan.liu@bc.edu
bUniversité de Pau et des Pays de l'Adour, E2S UPPA/CNRS, Institut des Sciences Analytiques et de Physico-Chimie pour l'Environnement et les Matériaux IPREM UMR 5254, Hélioparc, 2 Avenue P. Angot, 64053 Pau Cedex 09, France. E-mail: karinne.miqueu@univ-pau.fr
First published on 17th January 2023
Abstract
We report a combined experimental and computational study of Pd/Senphos-catalyzed carboboration of 1,3-enynes utilizing DFT calculations, 31P NMR study, kinetic study, Hammett analysis and Arrhenius/Eyring analysis. Our mechanistic study provides evidence against the conventional inner-sphere β-migratory insertion mechanism. Instead, a syn outer-sphere oxidative addition mechanism featuring a Pd-π-allyl intermediate followed by coordination-assisted rearrangements is consistent with all the experimental observations.
Introduction
Tetra-substituted alkenes are prevalent structural motifs among bioactive compounds and natural products.1 Representative examples include illudol, cassiabudanol A, brasilenol and tamoxifen. They also serve as synthetic intermediates for downstream functionalization.2 However, synthesis of tetra-substituted alkenes3 in a stereo-defined manner remains nontrivial. Among numerous methods developed to date, transition metal-catalyzed difunctionalization of alkynes4 has exhibited remarkable efficiency owing to the simultaneous introduction of two desired units across an alkyne, an inexpensive and readily available feedstock, and therefore has continuously drawn substantial attention. In this context, transition metal-catalyzed carboboration of internal alkynes5 allows expedient synthesis of tetra-substituted alkenyl boronates6 which are useful precursors to stereo-defined tetra-substituted alkenes. These alkenyl boronates have long been recognized as versatile building blocks7 for their participation in Suzuki–Miyaura cross coupling8 and in various derivatizations.9 The carboboration approach features a diverse combination of carbon and boron moieties and all involve syn-selective addition of the boron group and metal across a π-bond of an alkyne (Scheme 1a). Suginome et al. pioneered nickel- or palladium-catalyzed intramolecular carboboration of alkynes tethered with the chloroborane moiety.10 Similarly, extensions to intermolecular carboboration with various carbon nucleophile sources were achieved.11 More recently, in copper-catalyzed three-component coupling systems, carboboration has been accomplished by a copper-boryl migratory insertion process with an alkyne to give an alkenyl copper intermediate, which can then be quenched by various electrophilic carbon sources.12 Despite these breakthroughs, regio- and diastereoselectivity issues for internal unsymmetrical alkynes remain generally challenging in stereoselective carboborations to produce tetra-substituted alkenes.12a–e Furthermore, enolate nucleophiles have not been demonstrated as reagents in carboboration reactions. In 2021, our group reported a new cis-selective enolate carboboration reaction (Scheme 1b) of internal 1,3-enynes accompanying the discovery of a new family of carbon-bound boron enolates (C–boron enolates) generated by a kinetically controlled halogen exchange between chlorocatecholborane and silylketene acetals.13 These unquaternized C–boron enolates14 are demonstrated to activate 1,3-enyne substrates in the presence of a Pd0/Senphos ligand complex. A remarkable feature is that this transformation provides access to the highly substituted dienyl boron building blocks in high site-, regio-, and diastereoselectivity.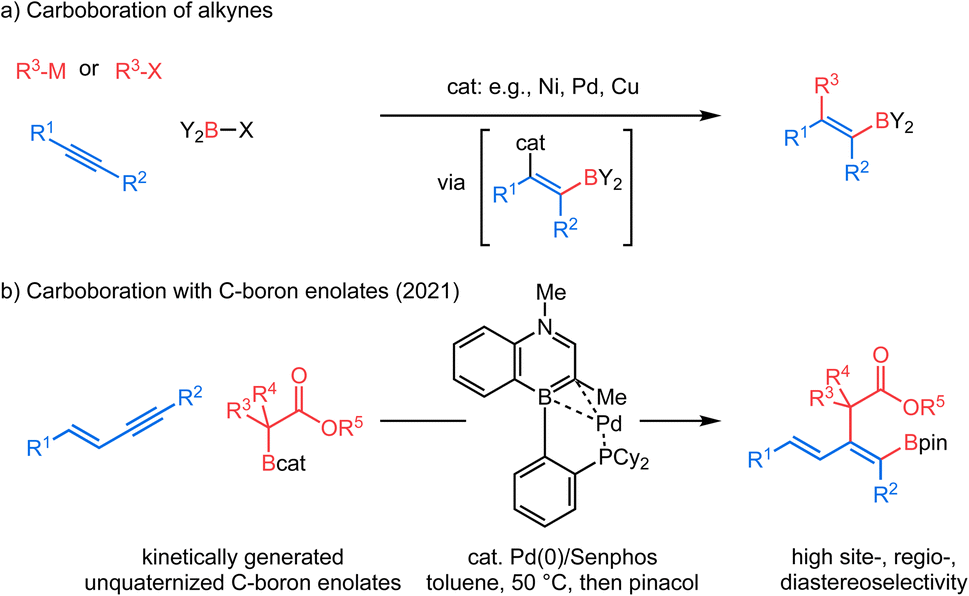 | ||
| Scheme 1 Tetrasubstituted alkenyl boronates via carboboration reactions. | ||
The consistently high selectivity of the reaction and the underexplored carbon-bound boron enolates inspired our interest in understanding the reaction mechanism. In our recently reported trans-hydroboration15 and trans-cyanoboration16 of 1,3-enynes catalyzed by a Pd0/Senphos ligand complex, an unusual “outer-sphere” oxidative addition mechanism featuring a Pd-π-allyl intermediate is proposed.17,18 Our initial mechanistic hypothesis was analogous (Scheme 2a): the presence of Senphos ligand L enables (COD)Pd(CH2TMS)2 to reductively eliminate 1,2- bis(trimethylsilyl)-ethane to form the active LPd0 species I,19 which then binds to the 1,3-enyne. The resulting LPd0–enyne complex II is then activated by the Lewis acidic C–boron enolate to furnish the outer-sphere oxidative adduct III. An enolate equivalent then attacks Pd-π-allyl to yield the product with concomitant regeneration of LPd0 species I.20 Our initial hypothesis provided a plausible explanation for the observed high site-, regio-, and diastereoselectivity. However, many mechanistic details of the C–B bond cleavage of the C–boron enolate and the C–C and C–B bond formation in the product are vaguely defined. For example, mechanistic divergence arises as the C–boron enolate could potentially approach the LPd0–enyne complex II either from the same side with respect to the Pd complex (“syn” outer-sphere oxidative addition) or from the opposite side (“anti” outer-sphere oxidative addition, Scheme 2b). In addition, the conventional “inner-sphere” oxidative addition21/β-migratory insertion/reductive elimination sequence (Scheme 2c) could not be ruled out completely, calling for new mechanistic evidence. We believe a detailed mechanistic investigation is therefore critical to clarify this mechanistic puzzle as well as to understand the origin of the selectivity and the behavior of carbon-bound boron enolates. In this article, we report our mechanistic study, starting with computational investigation (DFT calculations) which has been well correlated with 31P NMR study, kinetic study, Hammett analysis, Arrhenius analysis and Eyring analysis to obtain a more complete picture. Our mechanistic analysis does not support the inner-sphere mechanism and instead offers compelling evidence for Pd-π-allyl intermediacy as well as reveals full details for each elementary step of the new catalytic cycle, including a series of coordination-assisted rearrangements. Additionally, we also compare the ligand performance of Senphos and its carbonaceous version to probe the intrinsic effect of BN/CC isosterism. Collectively, the fundamental insights from this work should further expand the application of the Senphos ligand toward new reaction development.
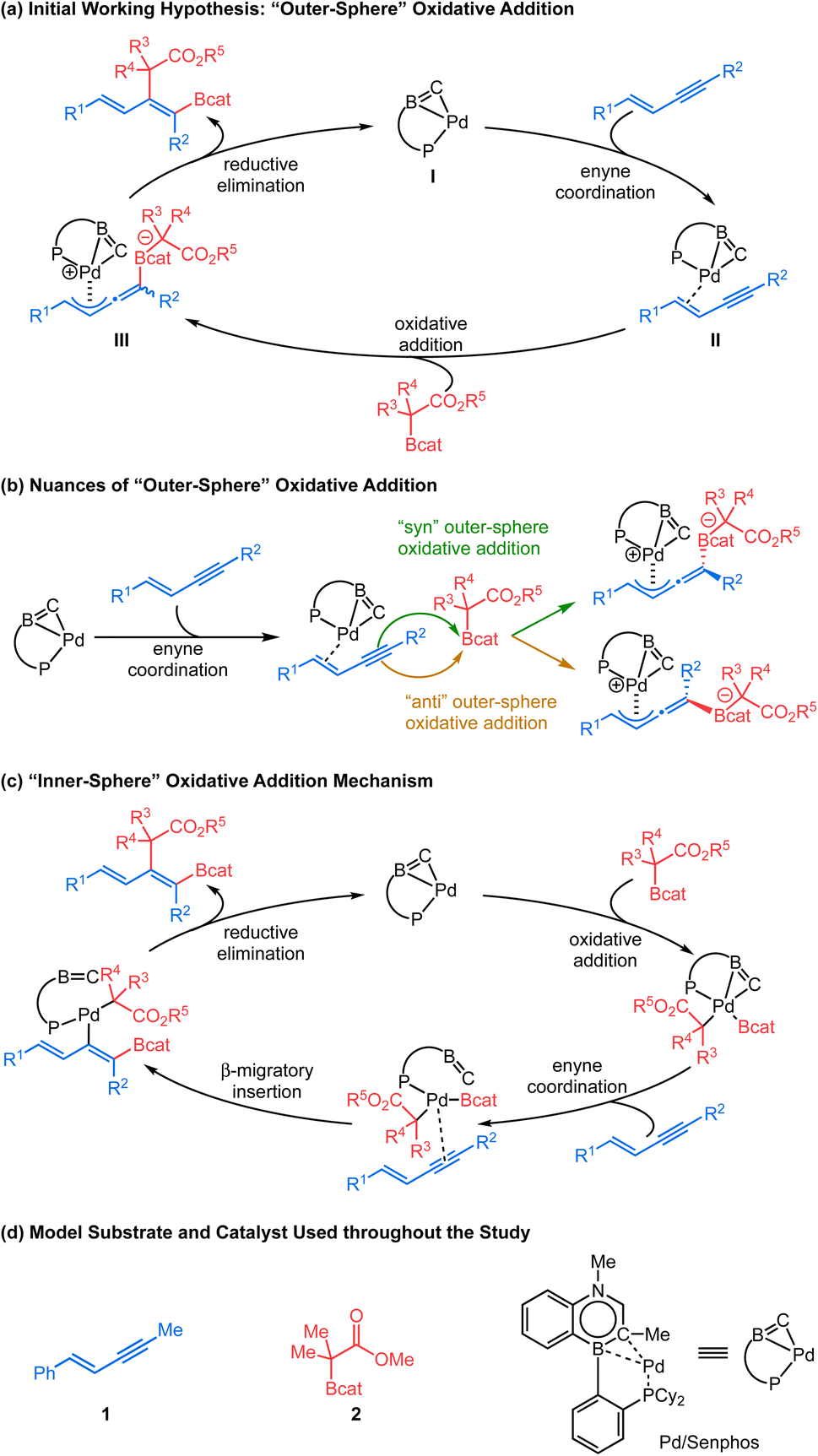 | ||
| Scheme 2 Mechanistic consideration. | ||
Results and discussion
We selected 1 and 2 as model substrates (Scheme 2d) and performed DFT calculations to explore the mechanism at the SMD(toluene)-TPSS-D3(BJ)/SDD+f(Pd), 6-31G**(other atoms) level of theory (see the ESI† for Computational details).22,23 Given the observed selectivity of the reaction, different pathways leading to the cis-addition product have been considered theoretically. We first studied the inner-sphere mechanism involving direct oxidative addition of the C–boron enolate to the Pd(0)/Senphos complex. Upon oxidative addition, two approaches of the C–boron enolate were considered leading to two products, with the Bcat moiety cis (path Ia, Fig. 1, Ia-Int1) or trans (see path Ib, Ib-Int1 Fig. S1 in ESI†) to the phosphorus atom of the Pd(II)/Senphos complex. Then, the 1,3-enyne coordinates to the Pd(II)/Senphos complex to form Ia-Int2. Compared to the initial reactants, this π-complex (Ia-Int2) is strongly uphill in energy (path Ia, Fig. 1, ΔG: 40.9 kcal mol−1; path Ib, Fig. S1, ΔG: 36.5 kcal mol−1 (see the ESI)†), which may be due in part to the loss of the η2-BC coordination of the Senphos ligand on Pd and to unfavorable steric interactions induced by the coordination of the enyne. Consequently, the β-migratory insertion, which is the rate-determining step, proceeds with an inaccessible activation barrier computed at 41.8 kcal mol−1 for path Ia (Fig. 1) and 38.2 kcal mol−1 for path Ib (see the ESI†). Lastly, the activation barrier for the reductive elimination is energetically less demanding (ΔG‡: 18.7 kcal mol−1 from starting materials, path Ia (Fig. 1); ΔG‡: 32.8 kcal mol−1, path Ib (see the ESI†)) than the β-migratory insertion step. Overall, the inner-sphere mechanism is predicted to be energetically unfeasible and is not consistent with the reported relatively mild catalytic cis-carboboration reaction conditions.13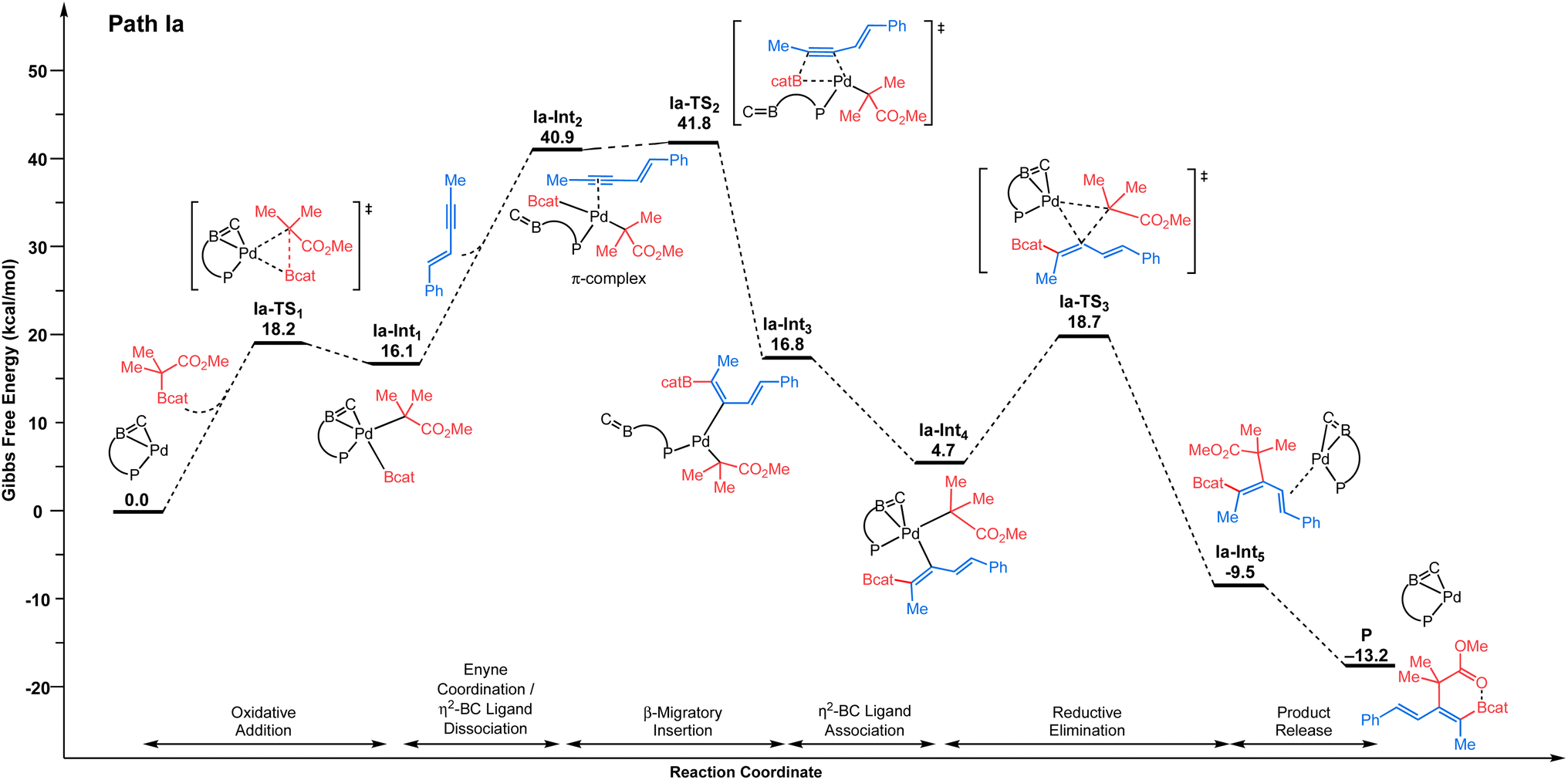 | ||
| Fig. 1 Energy profile (ΔG in kcal mol−1) computed at the SMD(toluene)-TPSS-D3(BJ)/SDD+f(Pd), 6-31G**(other atoms) level of theory for the inner-sphere mechanism involving direct oxidative addition pathway of the C–boron enolate to Pd. Path Ia is shown: Bcat moiety cis to phosphorus of the 1,4-azaborine-Senphos ligand. For Path Ib: Bcat moiety trans to phosphorus of the 1,4-azaborine-Senphos ligand, see the ESI.† | ||
We then examined the outer-sphere oxidative addition mechanism (Scheme 2a and b), where the palladium center is not directly involved in the cleavage of the C–B bond of the C–boron enolate. As illustrated in Fig. 2, this mechanism starts with the enyne coordinating to the palladium center. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the enyne is coordinated in a quasi-symmetric fashion to the metal center (Pd⋯C distances: 2.183–2.202 Å).24 The formation of the π-complex (Pd–enyne) is thermodynamically favorable by 19.1 kcal mol−1 compared to the initial reactants. Then, the C–boron enolate approaches this π-complex in a fashion syn to the Pd to engage in a syn outer-sphere oxidative addition (see the ESI† for details of the anti outer-sphere oxidative addition). Calculations predict that the activation barrier for the syn outer-sphere oxidative addition step is ΔG‡: 16.2 kcal mol−1 from the Pd–enyne π-complex. The “activated” quaternized C–boron enolate in Int1 then undergoes a coordination-assisted (Pd⋯O distance: 2.138 Å in the transition state TS2) rearrangement to form a Pd–O-enolate (Int2) with an activation barrier of 22.8 kcal mol−1 from the resting state. This coordination assistance in the syn outer-sphere oxidation pathway is responsible for a 17.2 kcal mol−1 lower overall rate-limiting energy barrier in comparison to the anti outer-sphere oxidative addition (or 25.1 kcal mol−1 lower energy barrier when directly comparing the C–B bond breaking step, see the ESI† for the anti energy profile). The O→Pd bonding in TS2 is apparent in the NBO analysis (see the ESI†) as donor–acceptor interactions with a total stabilizing energy ΔE(2) of 43.9 kcal mol−1 (∑O→Pd interaction). The natural localized molecular orbital (NLMO) associated with the main nO→Pd interaction shows a major contribution of the oxygen lone pair (77%) mixed with contributions from Pd (5.3%).
C double bond of the enyne is coordinated in a quasi-symmetric fashion to the metal center (Pd⋯C distances: 2.183–2.202 Å).24 The formation of the π-complex (Pd–enyne) is thermodynamically favorable by 19.1 kcal mol−1 compared to the initial reactants. Then, the C–boron enolate approaches this π-complex in a fashion syn to the Pd to engage in a syn outer-sphere oxidative addition (see the ESI† for details of the anti outer-sphere oxidative addition). Calculations predict that the activation barrier for the syn outer-sphere oxidative addition step is ΔG‡: 16.2 kcal mol−1 from the Pd–enyne π-complex. The “activated” quaternized C–boron enolate in Int1 then undergoes a coordination-assisted (Pd⋯O distance: 2.138 Å in the transition state TS2) rearrangement to form a Pd–O-enolate (Int2) with an activation barrier of 22.8 kcal mol−1 from the resting state. This coordination assistance in the syn outer-sphere oxidation pathway is responsible for a 17.2 kcal mol−1 lower overall rate-limiting energy barrier in comparison to the anti outer-sphere oxidative addition (or 25.1 kcal mol−1 lower energy barrier when directly comparing the C–B bond breaking step, see the ESI† for the anti energy profile). The O→Pd bonding in TS2 is apparent in the NBO analysis (see the ESI†) as donor–acceptor interactions with a total stabilizing energy ΔE(2) of 43.9 kcal mol−1 (∑O→Pd interaction). The natural localized molecular orbital (NLMO) associated with the main nO→Pd interaction shows a major contribution of the oxygen lone pair (77%) mixed with contributions from Pd (5.3%).
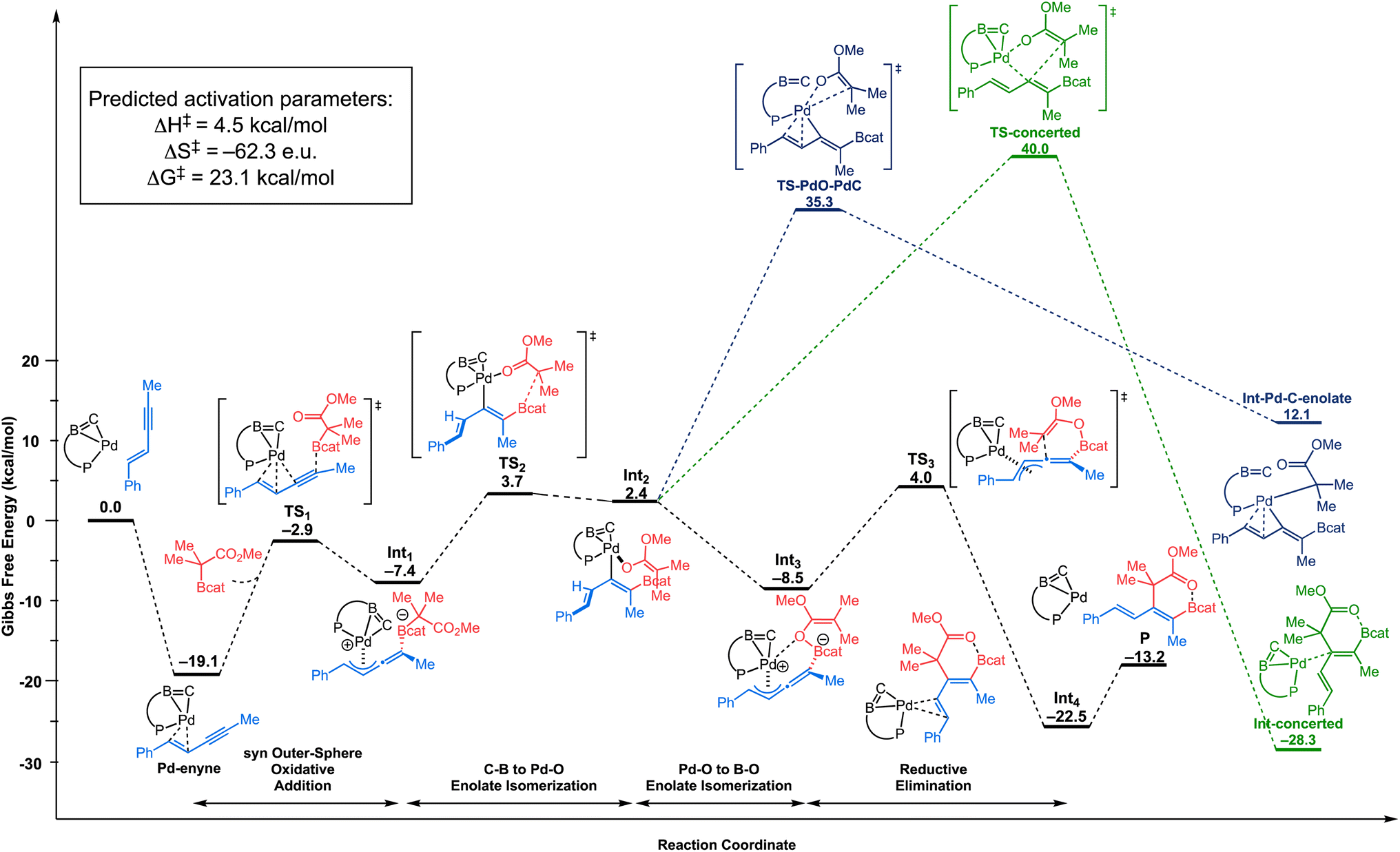 | ||
| Fig. 2 Energy profiles (ΔG in kcal mol−1) computed at the SMD(toluene)-TPSS-D3(BJ)/SDD+f(Pd), 6-31G** (other atoms) level of theory for the outer-sphere oxidative addition mechanism, involving B–C bond cleavage with assistance of O→Pd interaction, Pd–O/B–O isomerization and reductive elimination (black path). Concerted reductive elimination (green path), and Pd–O/Pd–C isomerization prior to reductive elimination (blue path) were also considered. | ||
From Int2, a direct reductive elimination transition state (TS-concerted, green path, Fig. 2) via a concerted 5-membered transition state (Pd–O distance: 2.373 Å and C⋯C distance: 2.477 Å) has been located with a barrier of ΔG‡: 59.1 kcal mol−1 from the resting state, suggesting that this C–C bond-forming step is unlikely to take place under the relatively mild reaction conditions. Thus, an isomerization was considered before the reductive elimination step. Two possibilities were examined: (1) Pd–O-enolate to Pd–C-enolate isomerization (blue path, Fig. 2), and (2) Pd–O-enolate to B–O-enolate isomerization (black path, Fig. 2). From the resting state, the barrier for the Pd–O-enolate to Pd–C-enolate isomerization to furnish Int–Pd–C-enolate (blue) was found to be ΔG‡: 54.4 kcal mol−1 whereas Pd–O-enolate to B–O-enolate isomerization (black) to form Int3 is predicted to be barrierless.
From the isomerized B–O-enolate Int3, “reductive elimination” proceeds with an activation barrier of ΔG‡: 12.5 kcal mol−1, leading to Int4, which is the cis-carboboration product bound to the Pd catalyst. Finally, the cis-carboboration product is released from the Pd catalyst, and a new cycle can begin. The cis-carboboration product features an O→B interaction (B⋯O distance: 1.662 Å), in agreement with the observed 11B NMR signal of the product at 17 ppm.
Based on the computational study, the most favorable pathway to form the cis-carboboration product involves: (i) a syn outer-sphere oxidative addition, (ii) coordination-assisted B–C bond cleavage to form a Pd–O-enolate, (iii) Pd–O-enolate to B–O-enolate isomerization, and (iv) reductive elimination via an enolate attack to a Pd-π-allyl species. Overall, DFT calculations predict the Pd–enyne complex (Pd–enyne) to be the resting state and the reductive elimination (TS3) to be the rate-limiting step with an overall rate-limiting barrier of 23.1 kcal mol−1. It is worth noting that TS2 (B–C to Pd–O isomerization, overall barrier of 22.8 kcal mol−1 from the resting state) is very close in energy with TS3.25 The assistance of an O→Pd interaction between the CO2Me moiety and the Pd metal center in the syn outer-sphere oxidative addition pathway is critical in lowering the overall activation barrier to allow for sufficient reactivity under mild conditions.26
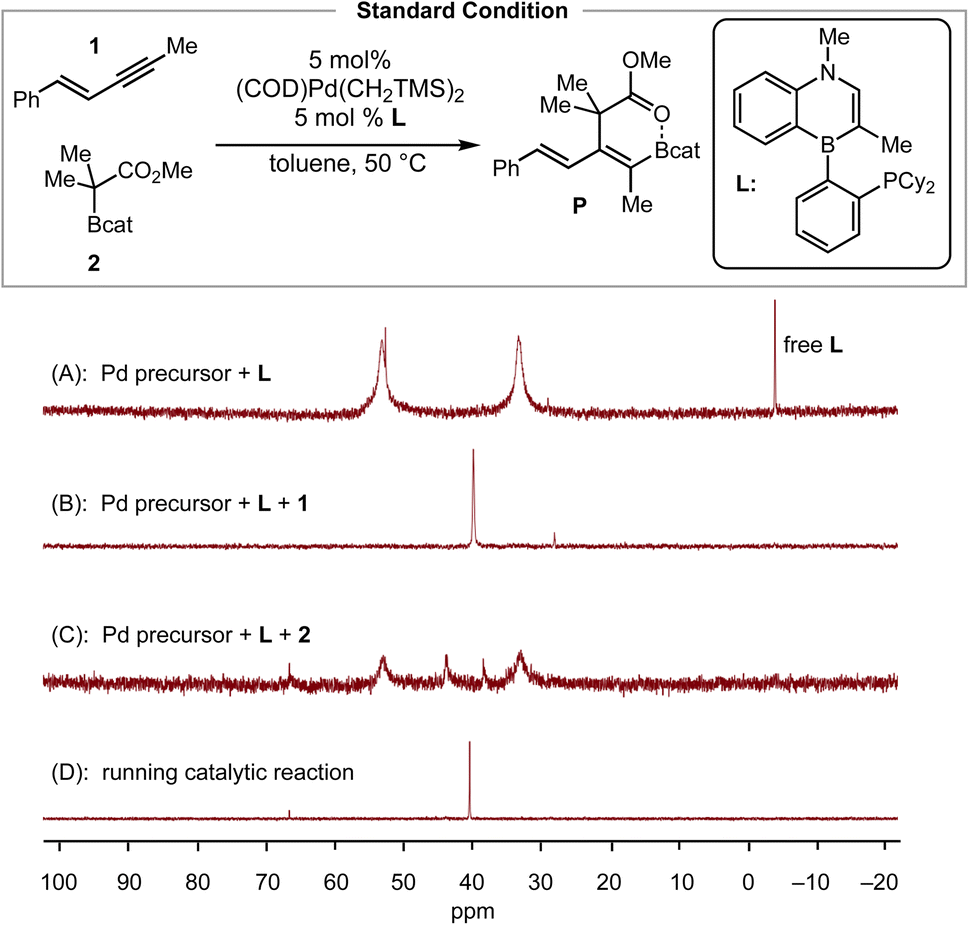
To validate the conclusion of the computational study, we performed the following experimental studies: (1) resting state determination via31P NMR, (2) initial-rate kinetics to obtain reaction orders of the reactants and the catalyst, (3) Hammett analysis, and (4) determination of activation parameters. We began with 31P NMR characterization of the reaction mixture to determine the likely resting state of the catalyst. The toluene-d8 solution of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (COD)Pd(CH2TMS)2 and L showed two broad signals (δP = 53.2 and δP = 33.3 ppm) and one sharp signal (δP = 52.7 ppm) (Scheme 3A). Next, the addition of 20 equiv. of enyne 1 to this solution, as a simulation of real catalytic reaction conditions, resulted in the observation of a sharp resonance (δP = 39.9 ppm, Scheme 3B), which we tentatively assign as the enyne-bound Pd complex (Pd–enyne). On the other hand, the addition of 40 equivalents of C–boron enolate 2 to a 1:1 mixture of the Pd precursor and L generated three additional signals (δP = 66.0, δP = 43.1, and δP = 38.5 ppm) with two broad signals remaining as major peaks (Scheme 3C). Under standard conditions for the catalytic reaction, the 31P NMR spectrum exhibited a major sharp peak (δP = 40.4 ppm) that matches nicely the signal observed for the enyne bound Pd π-complex (Scheme 3D). This peak persisted throughout the course of the reaction for ca. 4 hours and disappeared when the starting material was consumed. Thus, the observed 31P data are consistent with the Pd–enyne being the resting state of the catalytic cycle, which is in agreement with the DFT calculations (Fig. 2).
:1 mixture of (COD)Pd(CH2TMS)2 and L showed two broad signals (δP = 53.2 and δP = 33.3 ppm) and one sharp signal (δP = 52.7 ppm) (Scheme 3A). Next, the addition of 20 equiv. of enyne 1 to this solution, as a simulation of real catalytic reaction conditions, resulted in the observation of a sharp resonance (δP = 39.9 ppm, Scheme 3B), which we tentatively assign as the enyne-bound Pd complex (Pd–enyne). On the other hand, the addition of 40 equivalents of C–boron enolate 2 to a 1:1 mixture of the Pd precursor and L generated three additional signals (δP = 66.0, δP = 43.1, and δP = 38.5 ppm) with two broad signals remaining as major peaks (Scheme 3C). Under standard conditions for the catalytic reaction, the 31P NMR spectrum exhibited a major sharp peak (δP = 40.4 ppm) that matches nicely the signal observed for the enyne bound Pd π-complex (Scheme 3D). This peak persisted throughout the course of the reaction for ca. 4 hours and disappeared when the starting material was consumed. Thus, the observed 31P data are consistent with the Pd–enyne being the resting state of the catalytic cycle, which is in agreement with the DFT calculations (Fig. 2).
 | ||
| Scheme 3 31P NMR experiments for the detection of the resting state of the catalyst. | ||
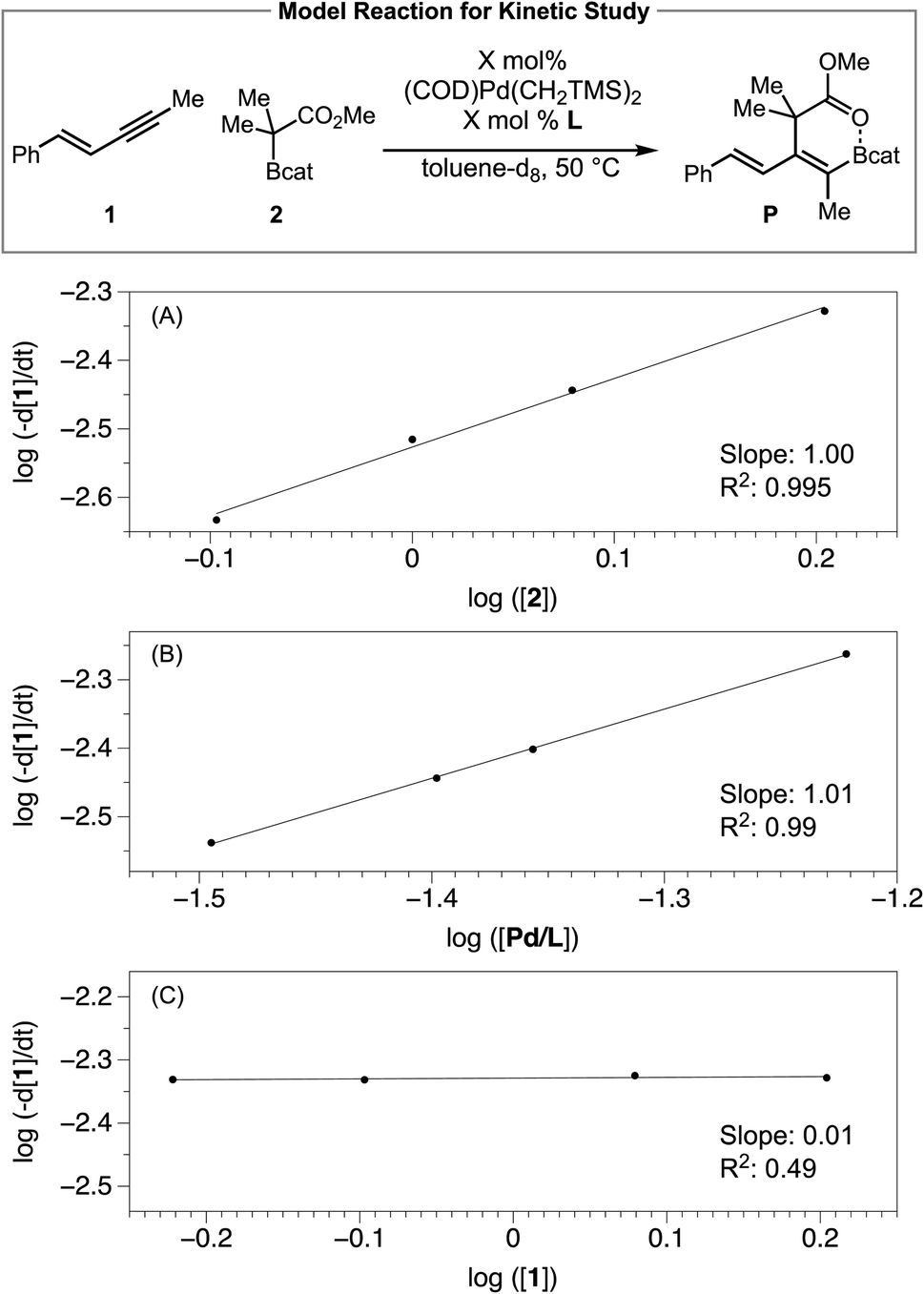
To probe the effect of each reaction component on the catalytic reaction, we then determined the kinetic orders in 1,3-enyne [1], C–boron-enolate [2] and [Pd/Ltotal], by measuring the initial rate over time in toluene-d8 solutions via1H NMR spectroscopy. Plotting the log(−d[1]/dt) vs. log([2]) (Scheme 4A), log(−d[1]/dt) vs. log([Pd/Ltotal]) (Scheme 4B) fitted two lines with slope 1.00 and 1.01, respectively, implying first order dependence on both C–boron-enolate [2] and [Pd/Ltotal]. Varying the concentration of enyne [1] did not influence the reaction rate (Scheme 4C), suggesting saturation kinetics (zero order dependence) with respect to [1]. Together with the conclusions from the 31P NMR study, the reaction order determination is consistent with the proposed syn outer-sphere oxidative addition pathway in Fig. 2 where the Pd–enyne complex is the resting state and the B–C bond cleavage (TS2) and/or reductive elimination (TS3) is the rate-limiting transition state.
 | ||
| Scheme 4 Initial-rate kinetic analysis. | ||
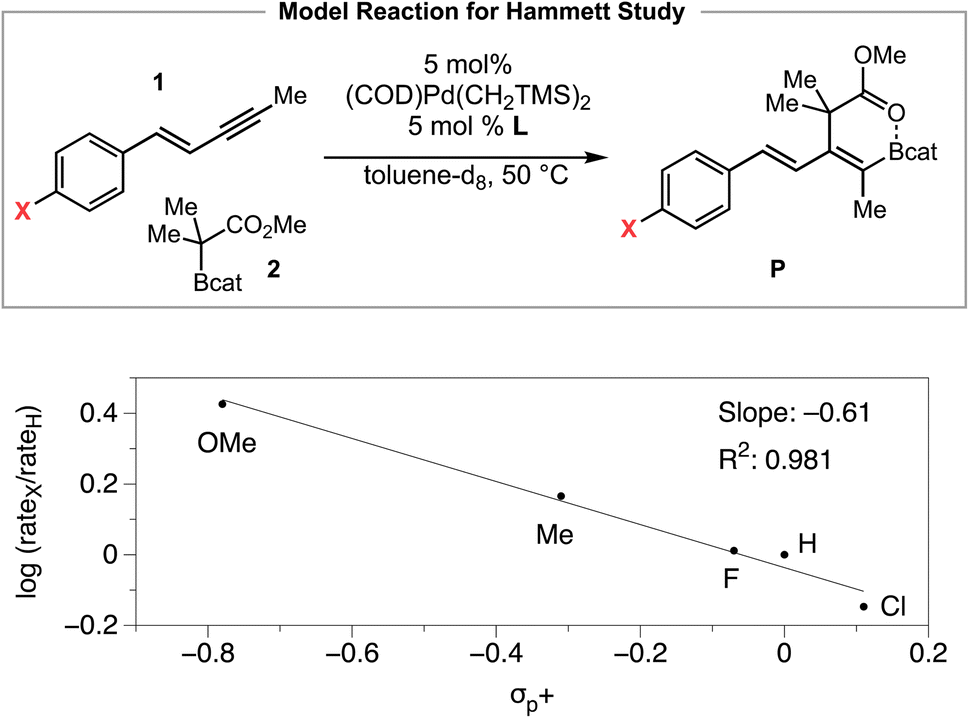
We then investigated the electronic effect of the aryl group in the 1,3-enyne on the reaction rate of the carboboration (Scheme 5). The Hammett analysis reveals that a 1,3-enyne bearing an electron-donating substituent reacts faster than an unsubstituted 1,3-enyne. A linear fit with reported σp+ constants is observed whereas less linear fit was obtained with σp or σp− constants, respectively.27 The ρ value determined from the Hammett plot (from σp+ values) was −0.61, implying the development of some positive charge in the 1,3-enyne substrate during the transition from the resting state to the rate-limiting transition state. Considering the multiple elementary processes involved (i.e., see Fig. 2, (i) syn outer-sphere oxidative addition (developing positive charge), (ii) coordination-assisted B–C bond cleavage to form a Pd–O-enolate (reduction of positive charge), (iii) Pd–O-enolate to B–O-enolate isomerization (development of positive charge)), and (iv) reductive elimination (reduction of positive charge), the relatively small magnitude of the ρ value could be interpreted as a net result of these opposing charge-generating processes.
 | ||
| Scheme 5 Hammett analysis. | ||
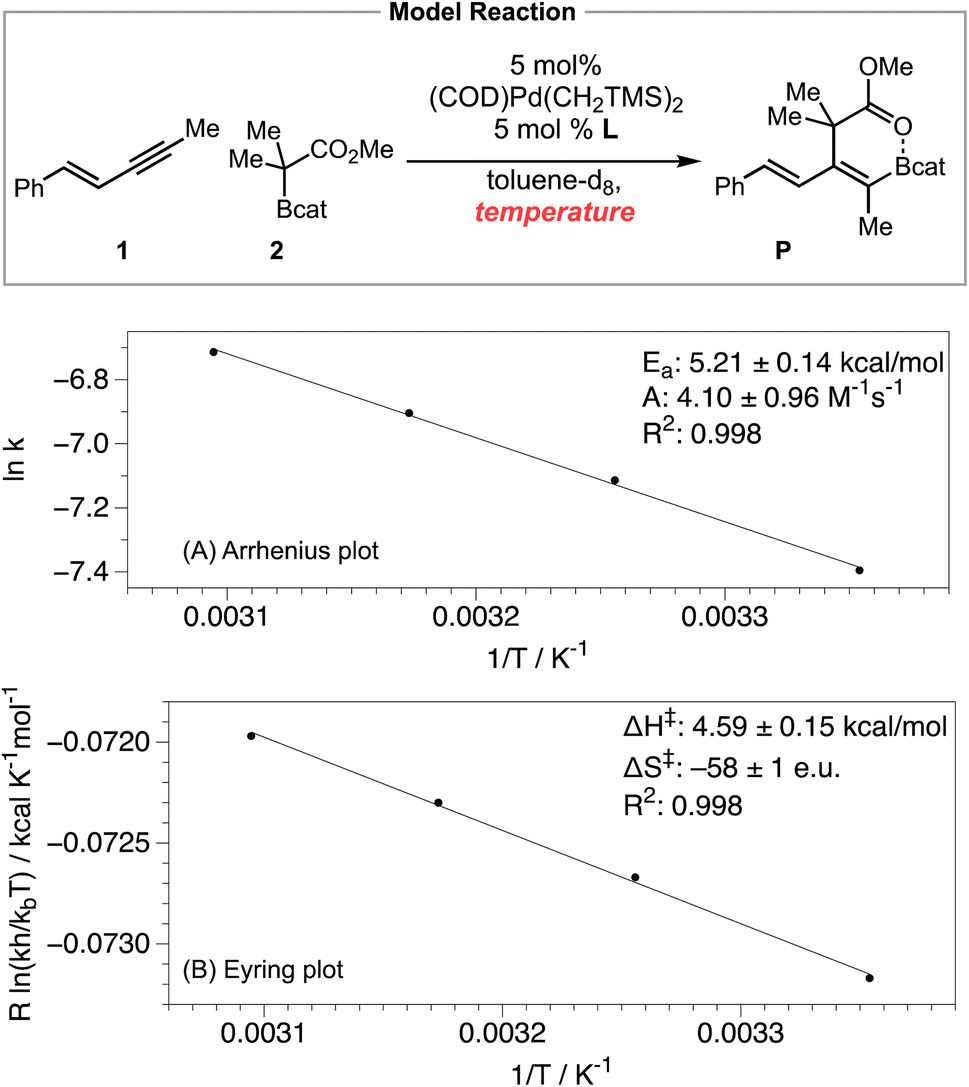
We also obtained the activation parameters of the carboboration by measuring the reaction rate in the temperature range of 25–50 °C. The Arrhenius plot (Scheme 6A) exhibited excellent linearity, with activation energy Ea = 5.21 ± 0.14 kcal mol−1, and preexponential factor A = 4.10 ± 0.96 M−1 s−1. Such a relatively small A value is consistent with a quite negative activation entropy for this reaction.28 We also performed an Eyring analysis (Scheme 6B) while acknowledging that the Eyring equation is theoretically only applicable to a single-step elementary reaction.29 The activation parameters determined via Eyring analysis are ΔH‡ = 4.59 ± 0.15 kcal mol−1, ΔS‡ = −58 ± 1 e.u., and ΔG‡ = 23.25 ± 0.22 kcal mol−1. While the exact values of the activation parameters should be interpreted with caution, the magnitude of the activation parameters is consistent with the prediction from the computational study (see Fig. 2, predicted ΔH‡ = 4.5 kcal mol−1, ΔS‡ = −62.3 e.u., and ΔG‡ = 23.1 kcal mol−1).
 | ||
| Scheme 6 Arrhenius and Eyring analysis. | ||
Achieving the rate-limiting transition state TS3 from the Pd–enyne resting state involves a bimolecular outer-sphere oxidative addition step and a highly conformationally organizing reductive elimination step. The very limited degrees of freedom in the rate-limiting TS3 (or in the energetically similar TS2) relative to the resting state are consistent with the predicted and experimentally observed Arrhenius preexponential factor A and activation entropy ΔS‡ values.
Finally, we compared the performance of Senphos (L) and its carbonaceous version (LCC ligand) under otherwise identical conditions by following the disappearance of the 1,3-enyne substrate over time via1H NMR analysis. As can be seen from Scheme 7, the initial rate of the reaction is similar when either the L (BN) or LCC ligand is used. However, the reaction stopped at ∼50% conversion after ca. 1.5 hours when the LCC ligand was employed while the Senphos L ligand enabled the reaction to go to completion over a period of 4 hours.
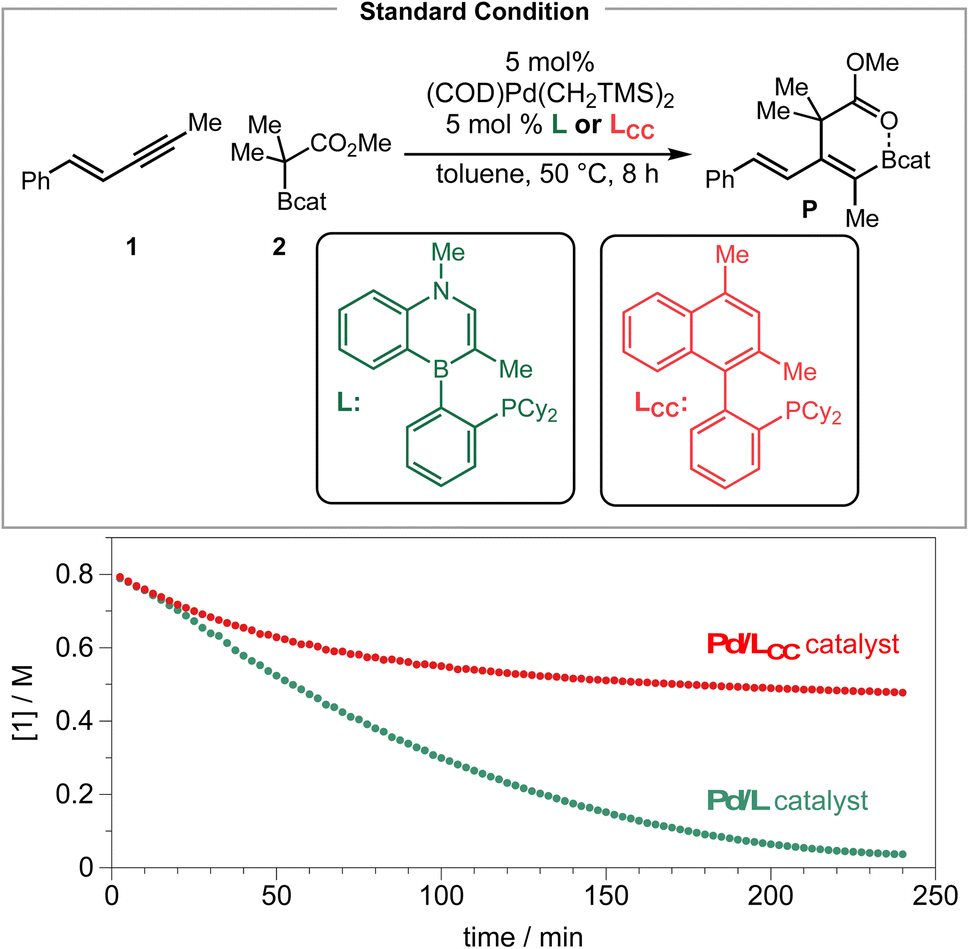 | ||
| Scheme 7 BN vs. CC: ligand performance comparison. | ||
The 4 hours reaction time correlates with the duration of where the corresponding resting-state Pd–enyne signal is observed by 31P NMR (see the ESI†). These results suggest that the Senphos ligand L generates a more long-lived Pd/L catalyst than the LCC ligand does and is consistent with the proposed unique κ2, η2-BC coordination mode of the ligand and the electron-donating borataalkene electronic structure of the BN-heterocyclic portion of the Senphos ligand.30,31
Conclusion
We have conducted a combined experimental and computational mechanistic investigation of the Pd/Senphos-catalyzed stereoselective carboboration reaction of internal 1,3-enynes with C–boron enolates. DFT calculations, spectroscopic characterization of reaction intermediates, initial-rate kinetics, linear free energy relationship analysis, and Arrhenius/Eyring analysis are inconsistent with an inner-sphere mechanism that is initiated by an oxidative addition of the C–boron enolates to the Pd(0)/L catalyst followed by alkyne β-migratory insertion and reductive elimination. Instead, our experimental and computational results are consistent with an outer-sphere oxidative addition mechanism where a Pd(0)/L–enyne complex is cooperatively activated by the Lewis acidic C–boron-enolate to furnish a zwitterionic Pd(II)/L π-allyl species. In contrast to the previously reported outer-sphere oxidative addition mechanism for the trans-selective hydroboration reaction where the Lewis acidic H-Bcat activator approaches the enyne anti to the Pd complex, the C–boron enolate activator approaches the enyne syn to the Pd complex. This syn outer-sphere oxidative addition pathway enables a critical coordination assisted rearrangement to form a Pd–O-enolate which then further rearranges to a B–O-enolate before the product-forming reductive elimination. Due to the lack of coordination-assisted Pd–O-enolate formation, the anti outer-sphere oxidative addition pathway is predicted to be more unfavorable than the syn outer-sphere oxidative addition mechanism by 17.2 kcal mol−1. Our experimental and computational data are consistent with the Pd–enyne complex being the resting state and the reductive elimination being the rate-determining step of the catalytic cycle. Furthermore, we establish that the BN heterocyclic Senphos ligand generates a significantly longer-lived active Pd catalyst species than its carbonaceous ligand does. Overall, this work highlights the mechanistic nuances associated with the emerging outer-sphere oxidative addition mechanism to leverage the versatile Pd-π-allyl intermediate for new catalytic bond-forming reactions. We also hope to take advantage of the stabilizing ability of the Senphos ligand family to develop new efficient catalytic processes.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
Z. W. performed the experimental mechanistic work guided by S.-Y. L. W. L. performed the computational work guided by K. M. All authors discussed the experiments and contributed to the writing of the manuscript and the ESI.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM136920, the Excellence Initiative of Université de Pau et des Pays de l'Adour I-Site E2S UPPA, and by Boston College start-up funds. The “Direction du Numérique” of the Université de Pau et des Pays de l’Adour and Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for supporting computational facilities. This work was also performed using HPC resources from GENCI-IDRIS (Grant 2023-[project AD010800045R1]). Z. W. was supported as a LaMattina Graduate Fellow in Chemical Synthesis. W. L. was funded as a postdoctoral fellow by I-Site E2S-UPPA. We also acknowledge the NIH-S10 (award: 1S10OD026910-01A1) and the NSF-MRI (award: CHE-2117246) for the support of Boston College's NMR facilities. We thank Prof. Anna Chrostowska and Dr Jean-Marc Sotiropoulos at UPPA for helpful discussions.References
- (a) A. S. Levenson and V. C. Jordan, Eur. J. Cancer, 1999, 35, 1628–1639 CrossRef CAS PubMed; (b) P. Prasit, Z. Wang, C. Brideau, C. C. Chan, S. Charleson, W. Cromlish, D. Ethier, J. F. Evans, A. W. Ford-Hutchinon, J. Y. Gauthier, R. Gordon, J. Guay, M. Gresser, S. Kargman, B. Kennedy, Y. Leblanc, S. Léger, J. Mancini, G. P. O'Neill, M. Ouellet, M. D. Percival, H. Perrier, D. Riendeau, I. Rodger, P. Tagari, M. Thérien, P. Vickers, E. Wong, L. J. Xu, R. N. Young, R. Zamboni, S. Boyce, N. Rupniak, M. Forrest, D. Visco and D. Patrick, Bioorg. Med. Chem. Lett., 1999, 9, 1773–1778 CrossRef CAS PubMed; (c) V. C. Jordan, J. Med. Chem., 2003, 46, 1081–1111 CrossRef CAS PubMed; (d) V. C. Jordan, J. Med. Chem., 2003, 46, 883–908 CrossRef CAS PubMed; (e) B. M. Trost, J. P. N. Papillon and T. Nussbaumer, J. Am. Chem. Soc., 2005, 127, 17921–17937 CrossRef CAS PubMed; (f) R. B. Williams, A. Norris, C. Slebodnick, J. Merola, J. S. Miller, R. Andriantsiferana, V. E. Rasamison and D. G. I. Kingston, J. Nat. Prod., 2005, 68, 1371–1374 CrossRef CAS PubMed; (g) K. Itami and J.-i. Yoshida, Bull. Chem. Soc. Jpn., 2006, 79, 811–824 CrossRef CAS; (h) N. F. McKinley and D. F. O'Shea, J. Org. Chem., 2006, 71, 9552–9555 CrossRef CAS PubMed; (i) V. C. Jordan, Eur. J. Cancer, 2008, 44, 30–38 CrossRef CAS PubMed; (j) D. Ganapathy and G. Sekar, Org. Lett., 2014, 16, 3856–3859 CrossRef CAS PubMed; (k) A. Lai, M. Kahraman, S. Govek, J. Nagasawa, C. Bonnefous, J. Julien, K. Douglas, J. Sensintaffar, N. Lu, K.-j. Lee, A. Aparicio, J. Kaufman, J. Qian, G. Shao, R. Prudente, M. J. Moon, J. D. Joseph, B. Darimont, D. Brigham, K. Grillot, R. Heyman, P. J. Rix, J. H. Hager and N. D. Smith, J. Med. Chem., 2015, 58, 4888–4904 CrossRef CAS PubMed; (l) S. Savage, A. McClory, H. Zhang, T. Cravillion, N.-K. Lim, C. Masui, S. J. Robinson, C. Han, C. Ochs, P. D. Rege and F. Gosselin, J. Org. Chem., 2018, 83, 11571–11576 CrossRef CAS PubMed; (m) W. Yin, W. Liang, L. Guo, J. Lei and F. G. Qiu, ACS Omega, 2018, 3, 4551–4556 CrossRef CAS PubMed.
- (a) H. K. Hall Jr, Angew. Chem., Int. Ed., 1983, 22, 440–455 CrossRef; (b) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS; (c) C. Dobler, G. M. Mehltretter, U. Sundermeier and M. Beller, J. Am. Chem. Soc., 2000, 122, 10289–10297 CrossRef; (d) Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed; (e) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed; (f) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed; (g) S. Kraft, K. Ryan and R. B. Kargbo, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS PubMed.
- (a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS PubMed; (b) D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552–4566 RSC; (c) P. Polák, H. Váňová, D. Dvořák and T. Tobrman, Tetrahedron Lett., 2016, 57, 3684–3693 CrossRef; (d) N. Mukherjee, S. Planer and K. Grela, Org. Chem. Front., 2018, 5, 494–516 RSC.
- (a) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727–3737 CrossRef CAS; (b) T. Besset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2015, 2015, 2765–2789 CrossRef CAS; (c) D. B. Huple, S. Ghorpade and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1348–1367 CrossRef CAS; (d) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327–1339 CrossRef CAS PubMed; (e) S. E. Bottcher, L. E. Hutchinson and D. J. Wilger, Synthesis, 2020, 52, 2807–2820 CrossRef CAS; (f) W. Liu and W. Kong, Org. Chem. Front., 2020, 7, 3941–3955 RSC; (g) B. Gao, D. Deng, D. Huang and X. Sun, Synthesis, 2021, 53, 3522–3534 CrossRef CAS; (h) S. Ghosh, R. Chakrabortty and V. Ganesh, ChemCatChem, 2021, 13, 4262–4298 CrossRef CAS; (i) J. Li, D. He, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2021, 8, 3502–3524 RSC.
- (a) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS; (b) J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2021, 11, 7513–7551 CrossRef CAS.
- (a) K. Itami, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670–14671 CrossRef CAS PubMed; (b) M. Shimizu, C. Nakamaki, K. Shimono, M. Schelper, T. Kurahashi and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 12506–12507 CrossRef CAS PubMed; (c) S. Mannathan, M. Jeganmohan and C. H. Cheng, Angew. Chem., Int. Ed., 2009, 48, 2192–2195 CrossRef CAS PubMed; (d) K. Endo, M. Hirokami and T. Shibata, J. Org. Chem., 2010, 75, 3469–3472 CrossRef CAS PubMed; (e) K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 1304–1307 CrossRef CAS PubMed; (f) S. Roscales and A. G. Csaky, Org. Lett., 2015, 17, 1605–1608 CrossRef CAS PubMed; (g) R. Fritzemeier and W. L. Santos, Chem.–Eur. J., 2017, 23, 15534–15537 CrossRef CAS PubMed; (h) M. Nogami, K. Hirano, M. Kanai, C. Wang, T. Saito, K. Miyamoto, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 12358–12361 CrossRef CAS PubMed; (i) Z. J. Kuang, H. H. Chen, J. X. Yan, K. Yang, Y. Lan and Q. L. Song, Org. Lett., 2018, 20, 5153–5157 CrossRef CAS PubMed; (j) N. Cabrera-Lobera, M. T. Quiros, W. W. Brennessel, M. L. Neidig, E. Bunuel and D. J. Cardenas, Org. Lett., 2019, 21, 6552–6556 CrossRef CAS PubMed; (k) W. H. Guo, H. Y. Zhao, Z. J. Luo, S. Zhang and X. G. Zhang, ACS Catal., 2019, 9, 38–43 CrossRef CAS; (l) M. Nogami, K. Hirano, K. Morimoto, M. Tanioka, K. Miyamoto, A. Muranaka and M. Uchiyama, Org. Lett., 2019, 21, 3392–3395 CrossRef CAS PubMed.
- (a) D. Hall, Boronic Acids Preparation and Applications in Organic Synthesis and Medicine Preface, 2005 CrossRef; (b) W. B. Reid, J. J. Spillane, S. B. Krause and D. A. Watson, J. Am. Chem. Soc., 2016, 138, 5539–5542 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CrossRef CAS PubMed.
- (a) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358–6359 CrossRef CAS PubMed; (b) A. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2005, 127, 15706–15707 CrossRef CAS PubMed; (c) M. Daini, A. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2008, 130, 2918–2919 CrossRef CAS PubMed.
- (a) M. Suginome, A. Yamamoto and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 2380–2382 CrossRef CAS PubMed; (b) M. Suginome, M. Shirakura and A. Yamamoto, J. Am. Chem. Soc., 2006, 128, 14438–14439 CrossRef CAS PubMed; (c) M. Daini and M. Suginome, Chem. Commun., 2008, 5224–5226 RSC.
- (a) R. Alfaro, A. Parra, J. Aleman, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2012, 134, 15165–15168 CrossRef CAS PubMed; (b) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2013, 15, 952–955 CrossRef CAS PubMed; (c) Y. Q. Zhou, W. You, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef CAS PubMed; (d) H. Y. Bin, X. Wei, J. Zi, Y. J. Zuo, T. C. Wang and C. M. Zhong, ACS Catal., 2015, 5, 6670–6679 CrossRef CAS; (e) T. Itoh, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 7528–7531 CrossRef CAS PubMed; (f) W. Su, T. J. Gong, Q. Zhang, Q. Zhang, B. Xiao and Y. Fu, ACS Catal., 2016, 6, 6417–6421 CrossRef CAS; (g) J. Zhao and K. J. Szabo, Angew. Chem., Int. Ed., 2016, 55, 1502–1506 CrossRef CAS PubMed; (h) A. Boreux, K. Indukuri, F. Gagosz and O. Riant, ACS Catal., 2017, 7, 8200–8204 CrossRef CAS; (i) T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS PubMed; (j) J. Mateos, E. Rivera-Chao and M. Fananas-Mastral, ACS Catal., 2017, 7, 5340–5344 CrossRef CAS; (k) B. Mun, S. Kim, H. Yoon, K. H. Kim and Y. Lee, J. Org. Chem., 2017, 82, 6349–6357 CrossRef CAS PubMed; (l) E. Rivera-Chao and M. Fananas-Mastral, Angew. Chem., Int. Ed., 2018, 57, 9945–9949 CrossRef CAS PubMed; (m) E. Rivera-Chao, M. Mitxelena, J. A. Varela and M. Fananas-Mastral, Angew. Chem., Int. Ed., 2019, 58, 18230–18234 CrossRef CAS PubMed; (n) J. del Pozo, S. C. Zhang, F. Romiti, S. B. Xu, R. P. Conger and A. H. Hoveyda, J. Am. Chem. Soc., 2020, 142, 18200–18212 CrossRef PubMed; (o) Z. H. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. M. Hou, ACS Catal., 2020, 10, 11685–11692 CrossRef CAS.
- Z. Wang, J. Wu, W. Lamine, B. Li, J.-M. Sotiropoulos, A. Chrostowska, K. Miqueu and S.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 21231–21236 CrossRef CAS PubMed.
- (a) A. Abiko, T. Inoue and S. Masamune, J. Am. Chem. Soc., 2002, 124, 10759–10764 CrossRef CAS PubMed; (b) N. J. Bell, A. J. Cox, N. R. Cameron, J. S. O. Evans, T. B. Marder, M. A. Duin, C. J. Elsevier, X. Baucherel, A. A. D. Tulloch and R. P. Tooze, Chem. Commun., 2004, 1854–1855 RSC; (c) Z. He, A. Zajdlik and A. K. Yudin, Dalton Trans., 2014, 43, 11434–11451 RSC; (d) E. W. H. Ng, K. H. Low and P. Chiu, J. Am. Chem. Soc., 2018, 140, 3537–3541 CrossRef CAS PubMed.
- S. M. Xu, Y. Z. Zhang, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 14566–14569 CrossRef CAS PubMed.
- Y. Z. Zhang, B. Li and S. Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 15928–15932 CrossRef CAS PubMed.
- (a) Y. N. Yang, J. L. Jiang, H. Z. Yu and J. Shi, Chem.–Eur. J., 2018, 24, 178–186 CrossRef CAS PubMed; (b) Y. Zhang, Z. Wang, W. Lamine, S. Xu, A. Chrostowska, K. Miqueu and S.-Y. Liu, J. Org. Chem., 2023 Search PubMed , jo-2022-02841d, preprint.
- For recent examples of Pd-catalyzed functionalization of 1,3-enynes with non-boron derived electrophiles, see: (a) H. Tsukamoto, T. Konno, K. Ito and T. Doi, Org. Lett., 2019, 21, 6811–6814 CrossRef CAS PubMed; (b) Q. He, L. Zhu, Z.-H. Yang, B. Zhu, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 17989–17994 CrossRef CAS PubMed; (c) B.-X. Xiao, B. Jiang, R.-J. Yan, J.-X. Zhu, K. Xie, X.-Y. Gao, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 4809–4816 CrossRef CAS PubMed; (d) Z.-L. He, Y. Zhang, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2022, 24, 6326–6330 CrossRef CAS PubMed.
- R. Takahashi, K. Kubota and H. Ito, Chem. Commun., 2020, 56, 407–410 RSC.
- (a) B. M. Trost and D. L. VanVranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- (a) C. S. Cho, T. Ohe and S. Uemura, J. Organomet. Chem., 1995, 496, 221–226 CrossRef CAS; (b) C. S. Cho and S. Uemura, J. Organomet. Chem., 1994, 465, 85–92 CrossRef CAS.
- (a) D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (b) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS; (c) J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed; (d) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (e) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (f) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Star-overov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghava-chari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Fores-man and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- DFT calculations show that the coordination of the alkene component of the 1,3-enyne to the Pd is energetically favored by 9.1 kcal mol−1 than the coordination of the alkyne unit, see the ESI† for details.
- For an example of a transition metal-catalyzed reaction featuring multiple rate-limiting steps, see: S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- The carboboration reaction when conducted at room temperature under otherwise identical conditions has a half-life of ca. 4 hours.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. E. Nicholas, Chemical kinetics: a modern survey of gas reactions, Halsted Press, 1976 Search PubMed.
- For an example of the application of the Eyring analysis to a multistep transition metal-catalyzed reaction, see: O. P. Schmidt and D. G. Blackmond, ACS Catal., 2020, 10, 8926–8932 CrossRef CAS.
- We believe that the strong κ2, η2-BC coordination mode of the Senphos ligand helps maintain the structure of the active Pd complex and avoids ligand dissociation and Pd black formation.
- S. M. Xu, F. Haeffner, B. Li, L. N. Zakharov and S. Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 6795–6799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, synthetic procedures, analytical data, NMR spectra and kinetic studies (PDF). Optimized Cartesian coordinates (XYZ). See DOI: https://doi.org/10.1039/d2sc05828f |
| This journal is © The Royal Society of Chemistry 2023 |