
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo birds one stone: β-fluoropyrrolyl-cysteine SNAr chemistry enabling functional porphyrin bioconjugation†
Guo-Qing
Jin
 a,
Jing-Xiang
Wang
a,
Jianhua
Lu
a,
Hang
Zhang
a,
Yuhang
Yao
a,
Yingying
Ning
a,
Hua
Lu
a,
Song
Gao
abc and
Jun-Long
Zhang
*ab
a,
Jing-Xiang
Wang
a,
Jianhua
Lu
a,
Hang
Zhang
a,
Yuhang
Yao
a,
Yingying
Ning
a,
Hua
Lu
a,
Song
Gao
abc and
Jun-Long
Zhang
*ab
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhangjunlong@pku.edu.cn
bChemistry and Chemical Engineering Guangdong Laboratory, Shantou 515031, P. R. China
cSpin-X Institute, School of Chemistry and Chemical Engineering, State Key Laboratory of Luminescent Materials and Devices, Guangdong-Hong Kong-Macao Joint Laboratory of Optoelectronic and Magnetic Functional Materials, South China University of Technology, Guangzhou 510641, China
First published on 17th January 2023
Abstract
Bioconjugation, a synthetic tool that endows small molecules with biocompatibility and target specificity through covalent attachment of a biomolecule, holds promise for next-generation diagnosis or therapy. Besides the establishment of chemical bonding, such chemical modification concurrently allows alteration of the physicochemical properties of small molecules, but this has been paid less attention in designing novel bioconjugates. Here, we report a “two birds one stone” methodology for irreversible porphyrin bioconjugation based on β-fluoropyrrolyl-cysteine SNAr chemistry, in which the β-fluorine of porphyrin is selectively replaced by a cysteine in either peptides or proteins to generate novel β-peptidyl/proteic porphyrins. Notably, due to the distinct electronic nature between fluorine and sulfur, such replacement makes the Q band red-shift to the near-infrared region (NIR, >700 nm). This facilitates intersystem crossing (ISC) to enhance the triplet population and thus singlet oxygen production. This new methodology features water tolerance, a fast reaction time (15 min), good chemo-selectivity, and broad substrate scope, including various peptides and proteins under mild conditions. To demonstrate its potential, we applied porphyrin β-bioconjugates in several scenarios, including (1) cytosolic delivery of functional proteins, (2) metabolic glycan labeling, (3) caspase-3 detection, and (4) tumor-targeting phototheranostics.
Introduction
Bioconjugation represents an innovative synthetic strategy allowing the complementary usage of both chemical and biological approaches to construct functional molecules.1,2 Toward this goal, a number of efficient and selective bioconjugations have been discovered over the decades and have succeeded in attaching small molecules to biomolecules of interest (e.g., peptide, protein, nucleic acid, or carbohydrate) via a covalent bond.3–8 In most cases, such modification is assumed not to affect the intrinsic properties of the small molecules. However, as a matter of fact, any formation of a new bond more or less perturbs the electronic nature of the small molecule and even influences its physicochemical properties and functions. To date, it is still far from well recognized that a significant opportunity exists in implementing bioconjugation: that is, to tailor the structural and functional diversity of small molecules beyond establishing a robust connection with biomolecules.9–11 Such recognition is particularly relevant to fabricating porphyrin bioconjugates for diagnostic or therapeutic purposes, as described in this work.Porphyrin and its derivatives are time-honoured photosensitizers that share a similar tetrapyrrole core to naturally occurring light-harvesting antennas and redox cofactors, with extensive applications in functional materials, catalysis, and biomedicines.12–17 As artificial synthetic macrocycles, they are subject to chemical manipulation to create desirable “drug-like” entities featuring structural diversity. Biomolecules, such as oligonucleotides,18,19 peptides,20 proteins,21 and carbohydrates12,22 have been used to endow synthetic porphyrins with biocompatibility and tumour-specificity. Among them, meso-aryl groups have mainly been chosen as conjugation sites due to lower steric hindrance and the rich synthetic repertoire of aryl groups. However, meso-aryl is perpendicular to the tetrapyrrolic plane and hardly delivers electronic engagement in π-conjugation and the related excited states. In contrast, fine-tuning of the π-periphery enables facile, efficient modulation of photophysical properties, but an efficient and selective β-bioconjugation approach has not been achieved due to peripherial steric hindrance and relatively inert reactivity. To fill the knowledge gap, we aimed to develop a general approach toward functional porphyrin β-bioconjugates with better perceived photophysical properties.
Sulfur, often embodied in cysteine, plays an important role in post-translational and site-selective bioconjugation owing to its relatively low abundance in proteins and inherently high nucleophilicity.23–26 Through the rational design of electrophilic warheads such as maleimides,27 a new irreversible C–S bond typically formed with a cysteine residue has been extensively exploited in designing bio-probes,28 small molecule inhibitors,29 and antibody–drug conjugates.30 However, the β-pyrrolic ring in porphyrins is not electrophilic enough to react with cysteine under physiological conditions. To increase their electrophilicity, pioneering work has demonstrated that the fluorination of aromatics enhances their reactivity, namely fluorine-cysteine SNAr chemistry by Pentelute,31 Derda,32 Diness,33 and Harran34 (Fig. 1). This was ascribed to the highest electronegativity of fluorine among all elements in the periodic table, which increases the electrophilicity of the neighbouring carbon in perfluoroarenes31–33 and octafluorocyclopentenes34 so as to react with a cysteine residue.35 In the contest for porphyrin bioconjugates, fluorine-thiol replacement at the meso-pentafluorophenyl group with thiosugars22 or sulfhydryl-terminated polyethylene glycol36 has been explored to improve biocompatibility, but such conjugation seldom altered photophysical properties. Thus, exploiting a new synthetic modification at the π-periphery is of importance to simultaneously achieve biocompatibility and tailor photophysical properties.
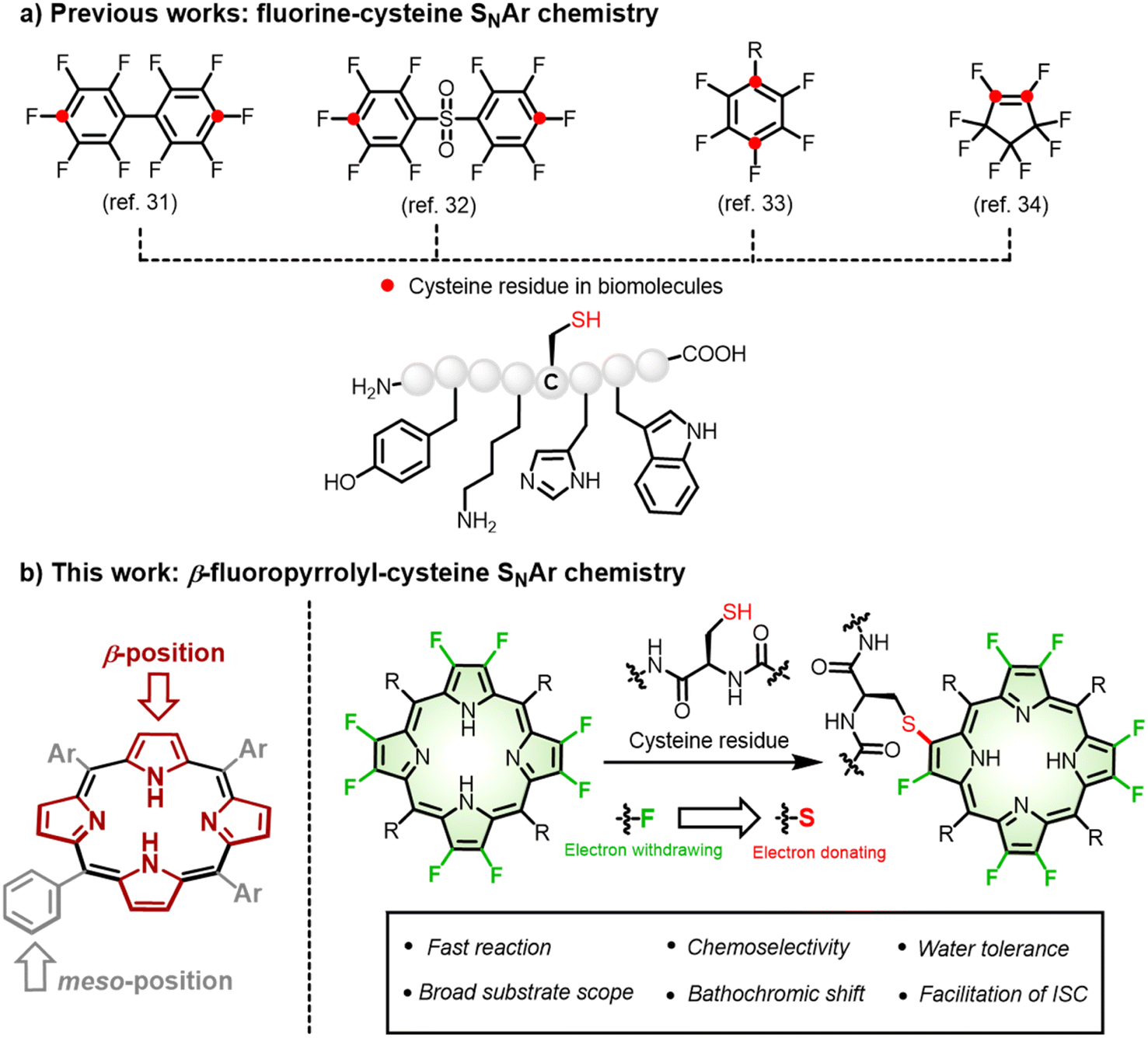 | ||
| Fig. 1 (a) Reported fluorine-cysteine replacement chemistry and corresponding reactive sites (red) of perfluoro-reagents. (b) β-fluoropyrrolyl-cysteine SNAr chemistry for irreversible porphyrin bioconjugation. | ||
An anticipated bonus for such fluorine-thiol exchange at the β-periphery is manipulation of the excited states and related photophysical properties arising from the distinct electronic nature between fluorine and sulfur.37–39 As a manifestation of β-fluoropyrrolyl-cysteine SNAr chemistry, we first performed computational calculations to predict the photophysical properties before and after β-fluorine-thiol replacement. We then prepared a series of water-soluble β-octafluoroporphyrins and investigated β-fluorine-thiol replacement with peptides/proteins under physiological conditions (Fig. 1). To our delight, β-peptidyl porphyrins were obtained quickly (15 min) and selectively (mono-peptidyl conjugates) with good yields (80–99%). As predicted by the computational calculations, the obtained β-peptidyl porphyrins displayed a bathochromic shift of the porphyrinic Q band to the NIR region and population of triplet states in favour of ROS generation that has been validated to develop tumour-targeted photosensitizers. Finally, we demonstrated several potential application scenarios, including (1) cytosolic delivery of functional proteins, (2) metabolic glycan labeling, (3) caspase-3 detection, and (4) tumour-targeting phototheranostics. Therefore, given that the importance of porphyrins and their derivatives in biological studies and biomedicines, the synthetic approach described here provided generally feasible access to endow biocompatibility, tumour specificity, and tunable photophysical properties.
Results and discussion
Computational studies
To predict the photophysical properties of porphyrins after β-fluorine-thiol replacement, we performed density functional theory (DFT) and time-dependent DFT (TDDFT) calculations based on 2,3,7,8,12,13,17,18-octafluoro-5,10,15,20-tetrakisphenyl porphyrin (P1) and β-sulfur substituted porphyrins (P2 and P3). The energy diagram and the corresponding nodal patterns of the frontier molecular orbitals (FMOs) are shown in Fig. 2. The first singlet excited states (S1 states) of P1–3 were mainly attributed to the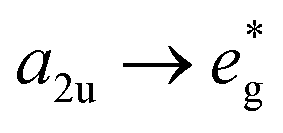 transition. β-Fluorine-thiol replacement introduced interference from the anti-bonding orbitals of the sulfur atoms, destabilizing the energy level of the HOMO (highest occupied molecular orbital). Hence, the energy gaps between HOMO and LUMO (lowest unoccupied molecular orbital) for P1–3 decreased in the order of P1 (2.8 eV) > P2 (2.64 eV) > P3 (2.58 eV) as replacement of the β-sulfur atom increased, indicating gradually red-shifted absorption. On the other hand, the anti-bonding orbitals of the sulfur atoms effectively stabilized the energy level of low-lying triplet states (Tn states). As shown in Table S5,† there were three triplet states (T1–T3) below the S1 state of P2 or P3, while only two nearly degenerate triplet states (T1–T2) existed below the S1 state of unsubstituted P1.
transition. β-Fluorine-thiol replacement introduced interference from the anti-bonding orbitals of the sulfur atoms, destabilizing the energy level of the HOMO (highest occupied molecular orbital). Hence, the energy gaps between HOMO and LUMO (lowest unoccupied molecular orbital) for P1–3 decreased in the order of P1 (2.8 eV) > P2 (2.64 eV) > P3 (2.58 eV) as replacement of the β-sulfur atom increased, indicating gradually red-shifted absorption. On the other hand, the anti-bonding orbitals of the sulfur atoms effectively stabilized the energy level of low-lying triplet states (Tn states). As shown in Table S5,† there were three triplet states (T1–T3) below the S1 state of P2 or P3, while only two nearly degenerate triplet states (T1–T2) existed below the S1 state of unsubstituted P1.
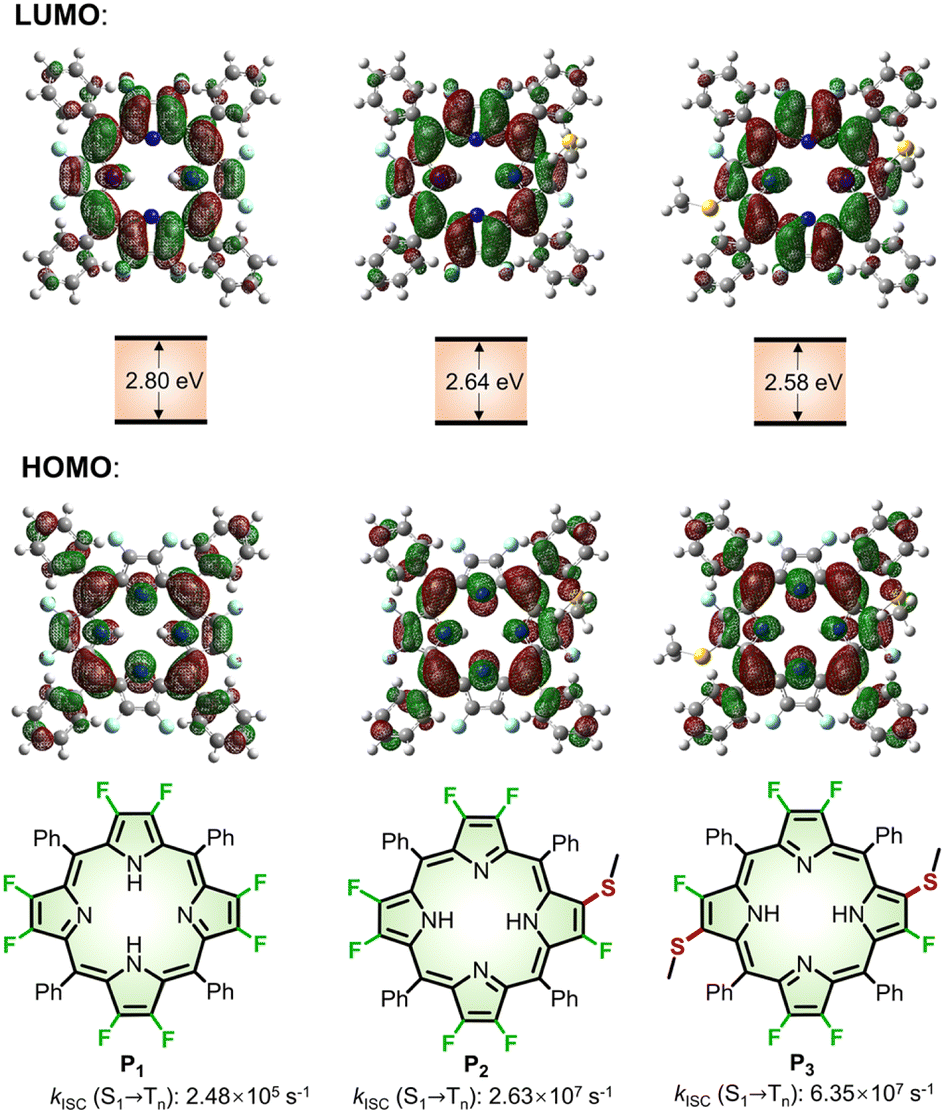 | ||
| Fig. 2 Nodal patterns of the frontier molecular orbitals (FMOs), HOMO–LOMO energy gaps, and kISC for (S1 → Tn) of P1–3 based on DFT and TDDFT calculations. | ||
We theoretically explored the dynamics of the excited states, including spin–orbit coupling (SOC) constants and intersystem crossing (ISC) rates before and after β-fluorine-thiol replacement. As a result, the SOC constants significantly increased from 0.0418 cm−1 (P1) to 0.604 cm−1 (P2) and 1.26 cm−1 (P3) and the singlet–triplet energy gap (ΔES-T) decreased from 1.24 eV (P1) to 0.0432 eV (P2) and 0.0405 eV (P3) after β-fluorine-thiol exchange, leading to a tremendous enhancement of the ISC rate constants (kISC) from the S1 to the Tn state. In particular, as the number of β-sulfur atoms increased, the kISC (S1 → Tn) of 2.63 × 107 s−1 (P2) and 6.35 × 107 s−1 (P3) were estimated to be 2 orders of magnitude higher than 2.48 × 105 s−1 (P1). Therefore, these computational results encouraged us to exploit β-fluoropyrrolyl-cysteine SNAr chemistry in pursuit of porphyrin bioconjugates with red-shifted absorption and accelerated ISC processes.
Optimizing the reaction condition
To optimize the reaction condition (Fig. 3a), we started with bioconjugation between P4 and glutathione (GSH, a), the most abundant tripeptide in living cells.40 As displayed in Table 1, entry 1, the reaction between P4 (1 mM) and a (2 mM) in Tris buffer (pH 8.0) for 15 min afforded β-peptidyl porphyrins with a yield of 68%. High-performance liquid chromatography (HPLC) analysis (Fig. S2–S11†) showed mono- (P4-a) and di-peptidyl porphyrins (P4-2a), as displayed in a ratio of 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 (Table 1, entry 1). Daylight had a trivial effect on β-bioconjugation (Table 1, entry 2). By decreasing GSH concentrations (Table 1, entries 3–6), mono-peptidyl porphyrins (P4-a) (>99% yield) were selectively obtained after β-fluorine-cysteine replacement (Table 1, entry 6). Inspired by a reported reaction-optimizing strategy by tuning the ratio of reactants,41,42 here largely excessive β-octafluoroporphyrin (4 equivalents) could effectively facilitate intermolecular SNAr replacement between porphyrin and GSH to achieve mono-substitution. A shortened reaction time (5 min) lowered the yield to 58% (Table 1, entry 7). Otherwise, when the reaction process was prolonged to 30 min, more di-substituted products were observed (13%, Table 1, entry 8). Increasing the pH to 8.8 led to a moderate yield of 59% (Table 1, entry 9). In contrast, lowering the pH to 7.4 gave a lower yield (46%) with P4-a as the dominant product (Table 1, entry 10). Next, we measured the reaction rates using the model reaction between P4 and a in Tris buffer (pH 8.0), which gave a rate constant of 1.44 M−1 s−1 according to the time-course study (Fig. S12, see details in the “Methods” part of ESI†).
:13 (Table 1, entry 1). Daylight had a trivial effect on β-bioconjugation (Table 1, entry 2). By decreasing GSH concentrations (Table 1, entries 3–6), mono-peptidyl porphyrins (P4-a) (>99% yield) were selectively obtained after β-fluorine-cysteine replacement (Table 1, entry 6). Inspired by a reported reaction-optimizing strategy by tuning the ratio of reactants,41,42 here largely excessive β-octafluoroporphyrin (4 equivalents) could effectively facilitate intermolecular SNAr replacement between porphyrin and GSH to achieve mono-substitution. A shortened reaction time (5 min) lowered the yield to 58% (Table 1, entry 7). Otherwise, when the reaction process was prolonged to 30 min, more di-substituted products were observed (13%, Table 1, entry 8). Increasing the pH to 8.8 led to a moderate yield of 59% (Table 1, entry 9). In contrast, lowering the pH to 7.4 gave a lower yield (46%) with P4-a as the dominant product (Table 1, entry 10). Next, we measured the reaction rates using the model reaction between P4 and a in Tris buffer (pH 8.0), which gave a rate constant of 1.44 M−1 s−1 according to the time-course study (Fig. S12, see details in the “Methods” part of ESI†).
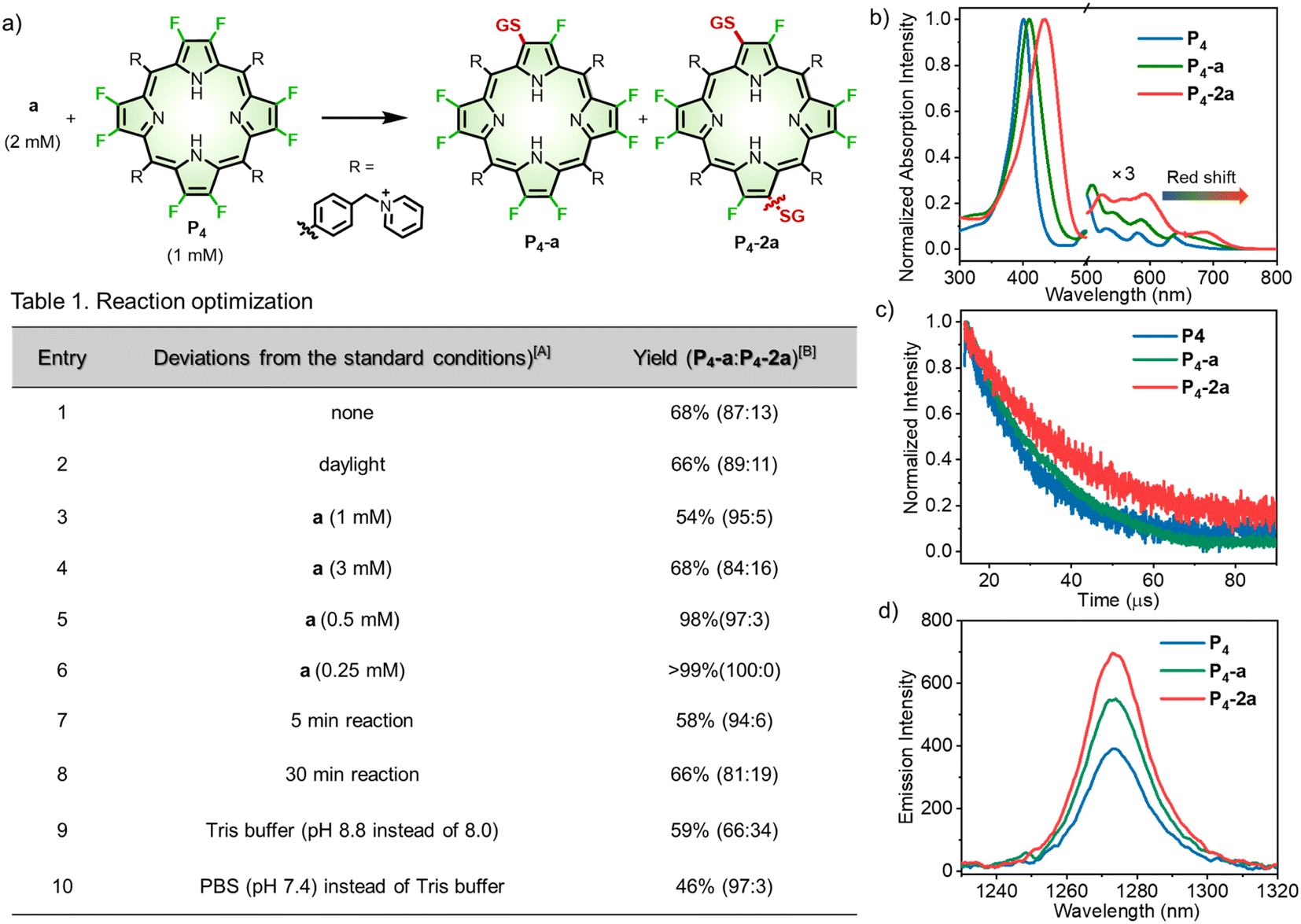 | ||
| Fig. 3 (a) Reaction optimization. A Standard conditions: a (2 mM) and P4 (1 mM) reacted in Tris buffer (pH 8.0) at room temperature in the dark for 15 min. B The yields were determined according to calibrated HPLC integral areas. (b) Normalized UV-visible absorption spectra of P4, P4-a and P4-2a in methanol. (c) The excited state lifetime decay curves of P4, P4-a and P4-2a in degassed methanol. (d) NIR emission of 1O2 at 1270 nm excited at 410 nm in toluene (0.1% DMSO). | ||
As the intracellular GSH concentration was 2–10 mM,43 we evaluated the stability of P4-a in GSH-abundant media (10 mM, pH 7.4, 37 °C) within 24 h (Fig. S13†). 67% of P4-a remained intact under such conditions. According to HPLC analysis, multi-substituted conjugates were formed, suggesting that P4-a partially reacts with GSH. However, such reactivity was inhibited when the pH was lowered to 6.8. After 24 h, P4-a was totally recovered, indicating the reaction was pH-sensitive, and that P4-a may remain intact in an acidic microenvironment, for example, tumors.43,44 To demonstrate the cell stability of P4-a, we treated HeLa cells with P4-a (10 μM) for different time periods. After the treatment, the HeLa cells were lysed using RIPA (Radio-Immunoprecipitation Assay) lysis buffer, then analysed by HPLC and sulfate polyacrylamide gel electrophoresis (SDS-PAGE). HPLC analysis for each cell lysate displayed the peak of P4-a alone, indicating P4-a did not react with cysteine-containing biomolecules in HeLa cells (Fig. S14†). Furthermore, SDS-PAGE analysis of the cell lysate showed no fluorescence from P4-a and Coomassie brilliant blue (CBB)-stained gel showed no molecular weight increases (Fig. S15†). These results also demonstrated no reaction between P4-a and cysteine-containing proteins; thus P4-a displayed good chemical stability in cells.
Photophysical properties
We then investigated the photophysical properties of P4, P4-a, and P4-2a. After the β-fluorine-sulfur replacement, the Soret band (401 nm) of P4 red-shifted to 409 nm and 433 nm for P4-a and P4-2a, respectively (Fig. 3b). Importantly, the Q bands of P4-a and P4-2a extended to 716 and 736 nm with remarkable red-shifts of 41 and 61 nm compared to P4 (675 nm). The bathochromic absorption after β-fluorine-sulfur replacement was consistent with that obtained in the computational studies. To the best of our knowledge, any known bioconjugation at meso-positions has not been reported to induce such large red-shifted Q-bands.21 By performing nanosecond transient absorption, the triplet decays for P4, P4-a, and P4-2a were determined to be 14, 19, and 24 μs in degassed methanol, respectively (Fig. 3c). The remarkably prolonged triplet lifetimes of P4-a and P4-2a compared to P4 revealed that β-fluoropyrrolyl-cysteine SNAr chemistry facilitated the intersystem crossing (ISC) process and enhanced the population of triplet state as well as the efficiency of oxygen sensitization, arising from the heavy atom effect of sulfur.45 Accordingly, the NIR emission of singlet oxygen (1O2) at 1270 nm upon irradiation of P4, P4-a, and P4-2a increased gradually with the extent of sulfur replacement (Fig. 3d). The quantum yields (ΦΔs) of singlet oxygen were determined in toluene using the comparative method referenced to TPP, as 75%, 84%, and 92% for P4, P4-a, and P4-2a (Fig. S16†). Furthermore, we examined the cell cytotoxicity of P4, P4-a, and P4-2a in the absence or presence of light (Fig. S17 and S18†). The photocytotoxicity, represented by the half-maximal inhibitory concentrations (IC50s) for P4 (1.97 ± 0.06 μM), P4-a (0.95 ± 0.03 μM), and P4-2a (0.51 ± 0.01 μM), was obtained under irradiation (LED laser (400–700 nm), 10 mW cm−2, 10 min). The results showed that β-sulfur substitution enhanced the 1O2-induced photocytotoxicity but slightly affected the dark cytotoxicity.Peptide scopes
Several amino acid residues possessed intrinsic nucleophilic reactivity toward C–F bonds; we investigated the cysteine-selectivity among the nucleophilic residues using P4 as a porphyrin substrate (Scheme 1). Peptide b containing several nucleophilic amino-acids (Cys, Ser, Lys, His, Arg, and Trp) was designed and used. In the context, we conducted the β-fluoropyrrolyl-cysteine SNAr reaction according to the reaction conditions (Table 1, entry 6). The reaction of P4 with b in Tris buffer (pH 8.0) for 15 min afforded conjugate P4-b in a yield of 90% (Fig. S19†). As a control, when iodoacetamide (IAA) was used to specifically block the cysteine of b, the subsequent addition of P4 could not give the P4-b product even after 24 h, indicating that conjugation of P4-b occurred at the cysteine residue. Encouraged by these preliminary results, we examined various peptides with cysteine at different positions (Fig. S20–S24†), including N- (c)/C-terminus (d), the middle of peptide (e), and cyclic peptide (f). Peptides c–f with hydrophobic and steric phenyl or isopropyl groups could efficiently conjugate with P4via β-fluorine-cysteine substitution and gave mono-peptidyl products in 68–99% yields. Of note, the biotinylated peptide (g) could react with P4 to afford P4-g in 99% yield.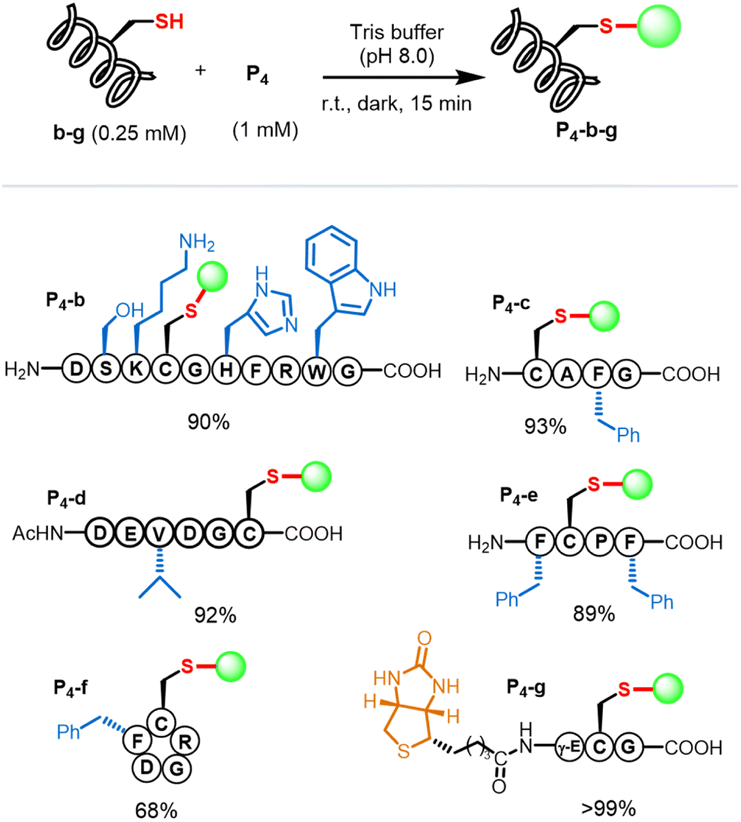 | ||
| Scheme 1 Scope of peptides. Reaction conditions: peptides b-g (0.25 mM) and P4 (1 mM) reacted in Tris buffer (pH 8.0) at room temperature in the dark for 15 min. The yield was determined according to calibrated HPLC integral areas of P4-b-g. | ||
Porphyrin scopes
On the other hand, to investigate the scope of porphyrins, we employed GSH as a model peptide under the optimized reaction conditions (Fig. S25–S32†). As shown in Scheme 2a, we used three types of β-octafluoroporphyrins containing cationic, anionic, and neutral meso-aryl groups, respectively. We found that, with the modification of pyridinium (P4), quaternary ammonium (P5–9), phosphonium (P10), or sulfonium (P11) cations, β-octafluoroporphyrins showed high reactivity toward GSH with yields of 83–99% (mono-peptidyl conjugates). Due to poor water solubility or steric hindrance, the PEGylated porphyrin (P12) could not conjugate with GSH under such reaction conditions. By adding dimethyl sulfoxide (DMSO) to facilitate the solubility of P12 (see details in the Methods part of ESI†), β-fluoropyrrolyl-cysteine replacement progressed smoothly with 80% yield of P12-a (Fig. S33†). The negatively charged sulfonate porphyrin (P13) also reacted with GSH in a complete conversion (>99%) monitored by UV-visible absorption (Fig. S34†), but the products could not be isolated under the HPLC conditions described above (see details in the “Methods” part of ESI†). High-resolution mass spectrometry (HRMS) confirmed the corresponding molecular weights of the formed conjugate P13-a (Fig. S35†).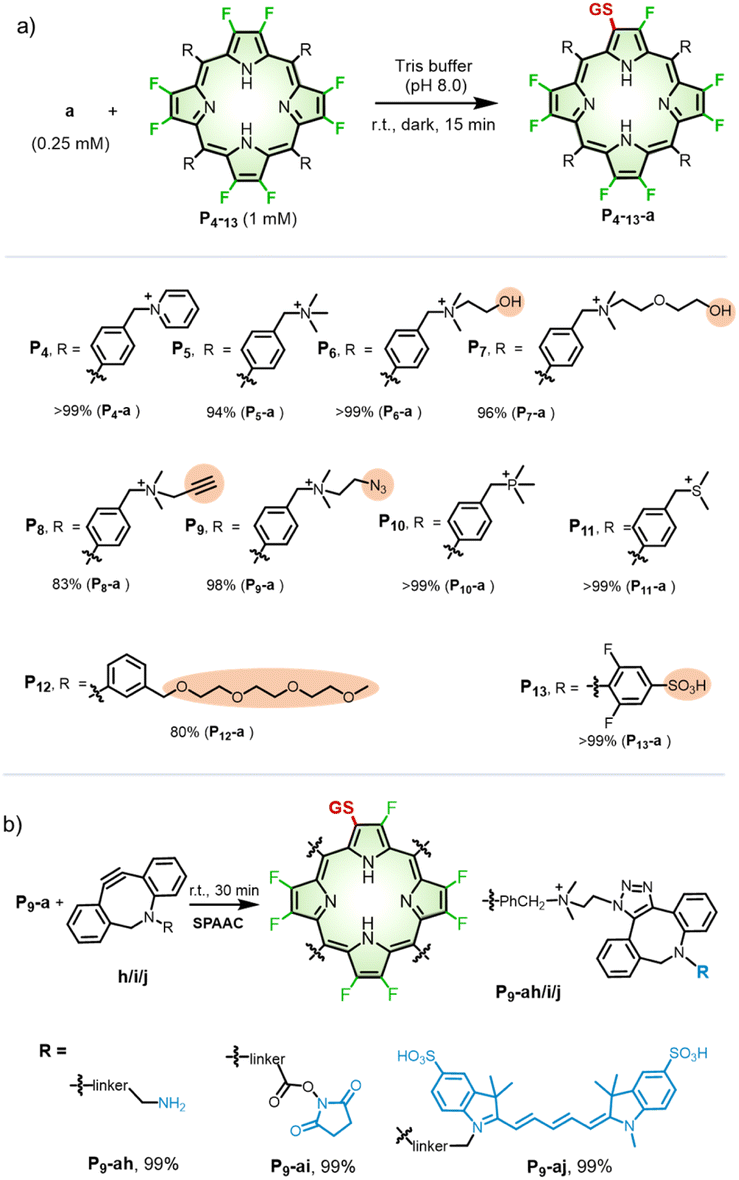 | ||
| Scheme 2 (a) Scope of β-octafluoroporphyrins. Reaction conditions: a (0.25 mM) and P4–13 (1 mM) in Tris buffer (pH 8.0) at room temperature in the dark for 15 min. For P12–13, the reaction conditions were partially changed, as described in the “Methods” part of ESI.† The yield was determined according to calibrated HPLC integral areas of P4–12-a. (b) SPAAC reaction of P9-a with DBCO derivatives (h–j). | ||
Note that, to test whether the β-fluoropyrrolyl-cysteine SNAr reaction was compatible with other bioconjugations such as Click chemistry,46 we used alkynyl (P8) or azido (P9) β-octafluoroporphyrin as the substrate to conjugate with GSH, which gave P8-a (83%) and P9-a (98%), respectively, as shown in Scheme 2a. To reveal the potential of multi-functionalization, we carried out a bioorthogonal strain-promoted alkyne–azide cycloaddition (SPAAC)47 between azido β-peptidyl porphyrin (P9-a) and dibenzocyclooctyne derivatives (DBCO, h–j), as shown in Scheme 2b. Specifically, P9-a was mixed and reacted with sulfo-DBCO-amine (h), DBCO-PEG4-NHS (i), or disulfo-Cy5-DBCO (j) in water for 30 min. These reactions afforded P9-ah, P9-ai, or P9-aj, respectively, whose molecular weights were confirmed by HRMS, in quantitative yields, as determined by HPLCs (Fig. S36–S38†). Therefore, we demonstrated the compatibility of the β-fluoropyrrolyl-cysteine SNAr reaction with Click chemistry that helps to establish porphyrin as a molecular platform with multiple functions.
Protein conjugation
We next extended the reaction to protein conjugation. Here, an enhanced green fluorescent protein (EGFP) with a genetically introduced cysteine48 was chosen as the model protein. We conducted the bioconjugation following the synthetic procedure shown in Fig. 4a (see details in the “Methods” part of ESI†). Firstly, EGFP (50 μM) was mixed with P4 (1 mM, 20 equiv.) in Tris buffer (pH 8.0) at room temperature for 8 hours. As shown in Fig. 4b, the mass peak (Obs. 30436 Da) of P4-EGFP, consistent with the calculated molecular weight (Cal. 30437 Da), was obtained with a mass spectrometer. As a control, when the cysteine residue in EGFP was blocked by excess IAA before reacting with P4, no P4-EGFP was observed but IAA-EGFP (Fig. S39†) alone. Similarly, conjugation of EGFP with P8 or P13 gave P8-EGFP or P13-EGFP. As shown in Fig. 4c, SDS-PAGE analysis showed that EGFP converted into the corresponding conjugates in >95% yield. Thus, either the positively or negatively charged β-octafluoroporphyrin could effectively conjugate with EGFP.
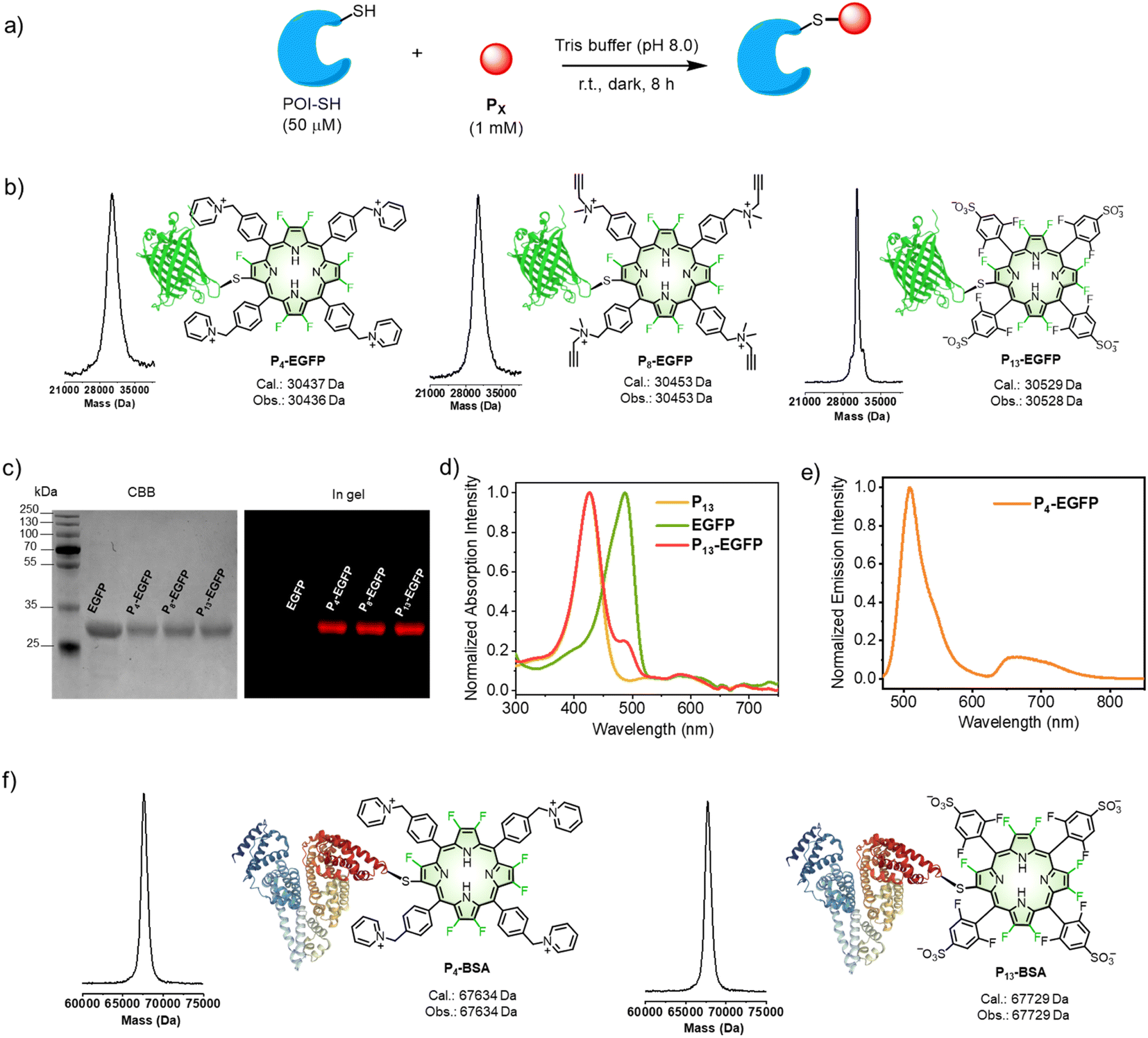 | ||
| Fig. 4 (a) β-Fluoropyrrolyl-cysteine SNAr chemistry for cysteine-selective bioconjugation of a protein of interest (POI). (b) Mass spectra of synthetic porphyrin–EGFP conjugates. Cal. (calculated mass); Obs. (observed mass). (c) SDS-PAGE of β-fluoropyrrolyl-cysteine SNAr reaction mixtures between EGFP and P4/P8/P13. The Coomassie brilliant blue (CBB)-stained gel showed increased protein weights. In-gel fluorescence scanning showed porphyrin emission (red). (d) UV-visible absorption spectra of P4, EGFP, and P4-EGFP in 1× PBS (pH = 7.4). (e) Fluorescence emission spectrum of P4-EGFP in 1× PBS (pH = 7.4). λex = 450 nm. (f) Mass spectra of synthetic porphyrin–BSA conjugates. | ||
We conducted fluorescence scanning for the same SDS-PAGE gel before CBB-staining (Fig. 4c). All porphyrin conjugates showed the red fluorescence attributed to porphyrin. In PBS (pH = 7.4) solution, P4-EGFP showed the characteristic UV-visible absorption of EGFP and P4 (Fig. 4d). Upon 450 nm excitation, it displayed two emission bands at 480–600 and 620–800 nm, assigned to the fluorescence of EGFP and P4, respectively (Fig. 4e).
Serum albumin is abundant in blood plasma, containing a free cysteine residue (cysteine 34),49 and it plays important roles in biomedical applications.43,50 When bovine serum albumin (BSA) was used, the observed mass peaks (67634 Da for P4-BSA and 67729 Da for P13-BSA) were obtained, consistent with the calculated values. This indicated BSA was modified with a single porphyrin (Fig. 4f). SDS-PAGE analysis showed increased molecular weights (CBB staining) compared with BSA alone, and showed that the gel fluorescence also arose from the porphyrin moiety (Fig. S40†).
Application scenarios
We intended to demonstrate the biocompatibility and biospecificity of porphyrin bioconjugates constructed via β-fluoropyrrolyl-cysteine SNAr chemistry. In most cases, the biocompatibility and biological targetability of porphyrin was realized by nano-modification13,51 or meso-bioconjugation.20,21 We demonstrated the feasibility of porphyrin β-bioconjugates for the intracellular delivery of functional proteins, fluorescent imaging of sialoglycoproteins, caspase-3 detection, and cancer-selective photodynamic therapy, demonstrating the very attractive application potential of this methodology.In this work, we prepared protein–PDS conjugates with the cysteine-initiated cryo-ring-opening polymerization (cryo-ROP) of 1,2-dithiolanes (Fig. 5a, see details in the “Methods” part of ESI†).48 After EGFP was mixed with lipoic acid (LA, 100 mM) at −30 °C, excess P13 or IAA was then added to cap the terminal free thiol, generating the protein–PDS conjugate denoted P13-EGFP-PDS or IAA-EGFP-PDS, respectively. CBB-stained non-reducing SDS-PAGE analysis (Fig. 5b) indicated that 76% and 54% of EGFP were converted into P13-EGFP-PDS and IAA-EGFP-PDS, respectively. Subsequently, we tested the cell-penetrating ability of P13-EGFP-PDS by incubating living HeLa cells with EGFP(i), P13-EGFP (ii), or P13-EGFP-PDS (iii) at pH 7.4. We observed pervasive and intense intracellular fluorescence in P13-EGFP-PDS-treated HeLa cells (Fig. 5c, iii), using confocal laser scanning microscopy. In contrast, no fluorescence was observed after treatment with free EGFP (Fig. 5c, i) or P13-EGFP (Fig. 5c, ii), revealing the good cell-penetrating capability of P13-EGFP-PDS.48
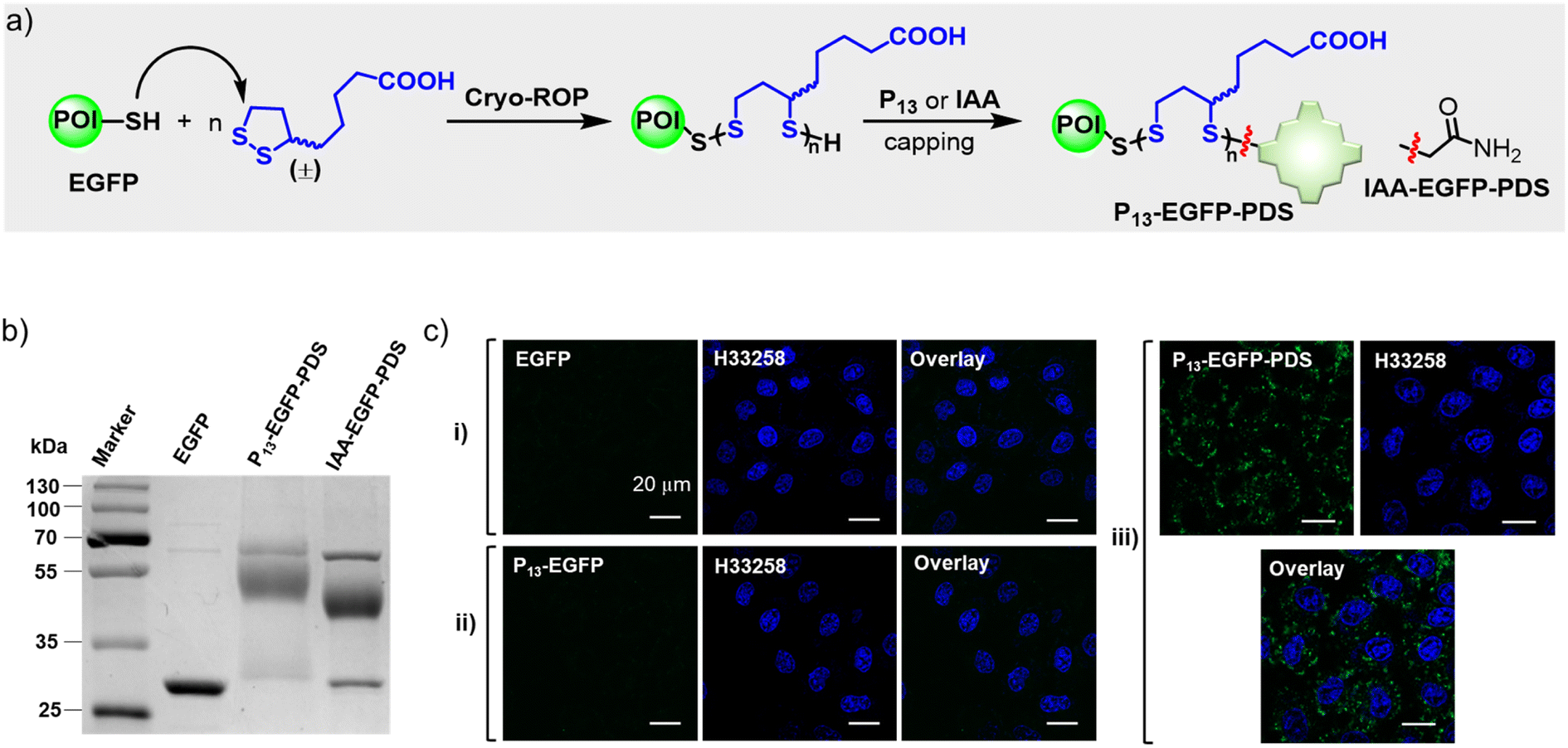 | ||
| Fig. 5 (a) Schematic illustration of the synthesis of protein–polydisulfide (PDS) conjugates via cryo-ring-opening polymerization (cryo-ROP) of lipoic acid (LA). (b) CBB-stained non-reducing SDS-PAGE of cryo-ROP reaction mixture capped by P13 or IAA. Conditions: pH = 7.0, EGFP concentration = 2.0 mg mL−1, [LA]0 = 100 mM, −30 °C, 2 h. (c) Fluorescence images of living HeLa cells treated with EGFP (250 nM, i), P13-EGFP (250 nM, ii), or P13-EGFP-PDS (250 nM, iii) at pH 7.4 for 1 h. λex = 450 nm; λem = 525/50 nm bandpass filter. | ||
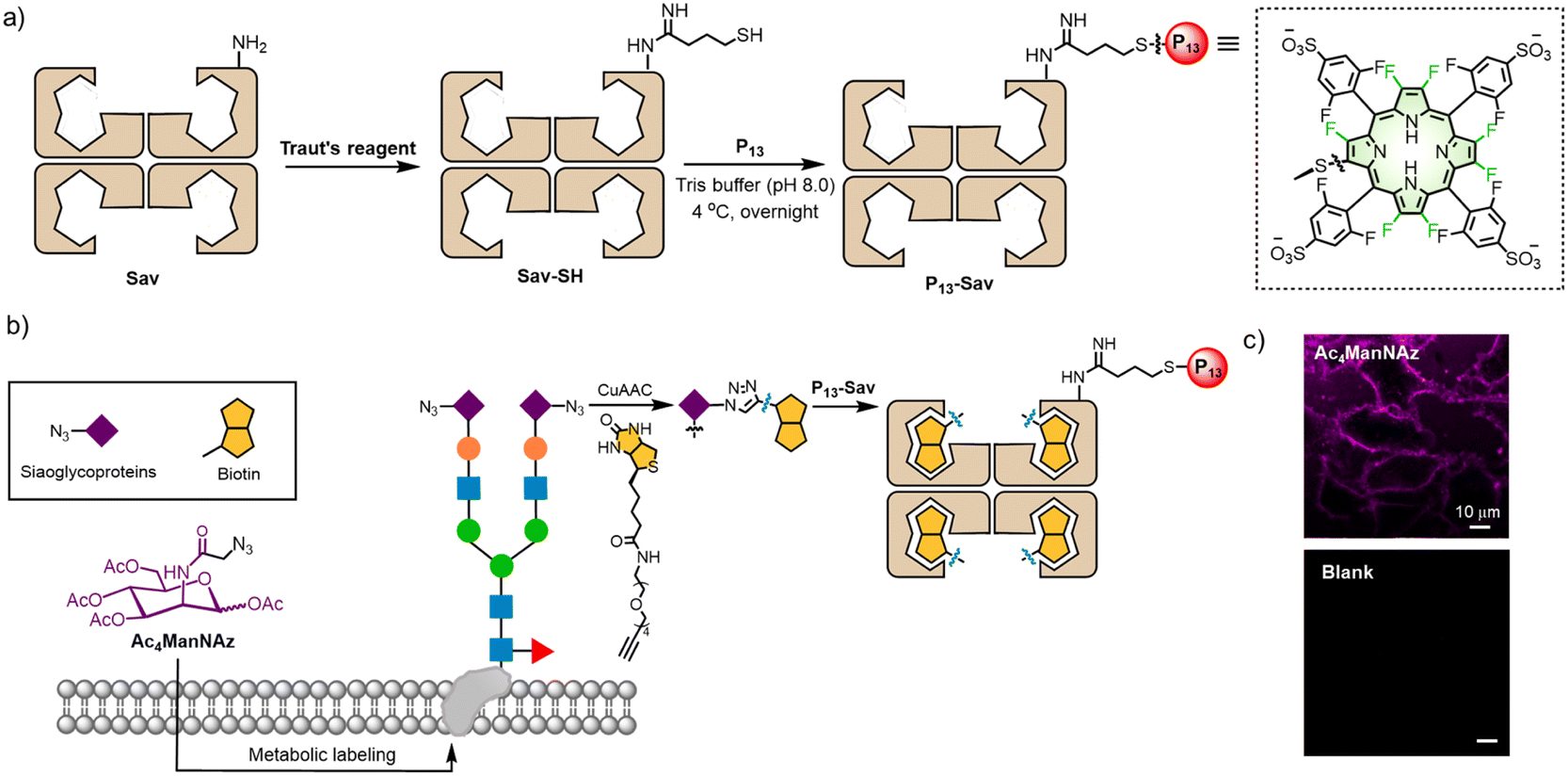 | ||
| Fig. 6 Schematic illustration of (a) the synthesis of P13-Sav. (b) Ac4ManNAz metabolic labeling. (c) Fluorescence images of sialoglycoproteins using P13-Sav. λex = 405 nm; λem = 647 nm longpass filter. Scale bar: 10 μm. | ||
To visualize sialic acids in vitro, we used MGL technology to endow the nascent sialoglycans with azido groups58 after incubation of HeLa cells with Ac4ManNAz (50 μM) for 2 days. Next, to selectively modify sialic acids with biotin warheads, we applied copper-catalysed azide–alkyne cycloaddition (CuAAC)59 between the biotin-PEG4-alkynyl group and the azido group on the cell surface after the cells were fixed (Fig. 6b). After washing and rinsing free biotin-PEG4-alkyne, P13-Sav was incubated with the HeLa cells, allowing the sialic acids to specifically recognize Sav-biotin. As shown in Fig. 6c, we observed intensive red fluorescence on the cell membranes, similar to previous studies using commercial Sav dyes.57 As a control, a blank group without Ac4ManNAz treatment showed no fluorescent signal. These results verified the feasibility of sialoglycoprotein labeling using P13-Sav, which is promising for MGL-assisted cancer phototheranostics.58
For this purpose, we designed a FRET (Förster resonance energy transfer) mechanism-based peptide probe P4-k (FITC-DEVDGC-P4),63 which consisted of fluorescein (FITC) as the fluorescence reporter, a reactive polypeptide (DEVDGC) linker whose C-terminal amide bond can be specifically hydrolyzed by caspase-3,64,65 and a porphyrin (P4) as the quencher of FITC (Fig. 7a). Treating P4-k (10 μM) with caspase-3 in PBS buffer gave strongly enhanced fluorescence, as shown in Fig. 7b (green line), while inhibiting the reactivity of caspase-3 using 5-[(S)-(+)-2(methoxymethyl)pyrrolidino]sulfonylisatin (MPS) showed no “turn-on” FITC fluorescence (blue line). Notably, the catalytic hydrolysis of P4-k by caspase-3 was also confirmed by HRMS, where the molecular ion peak with m/z = 979.3045 was detected, consistent with the calculated mass value ([M + H]+) of FITC-DEVD.
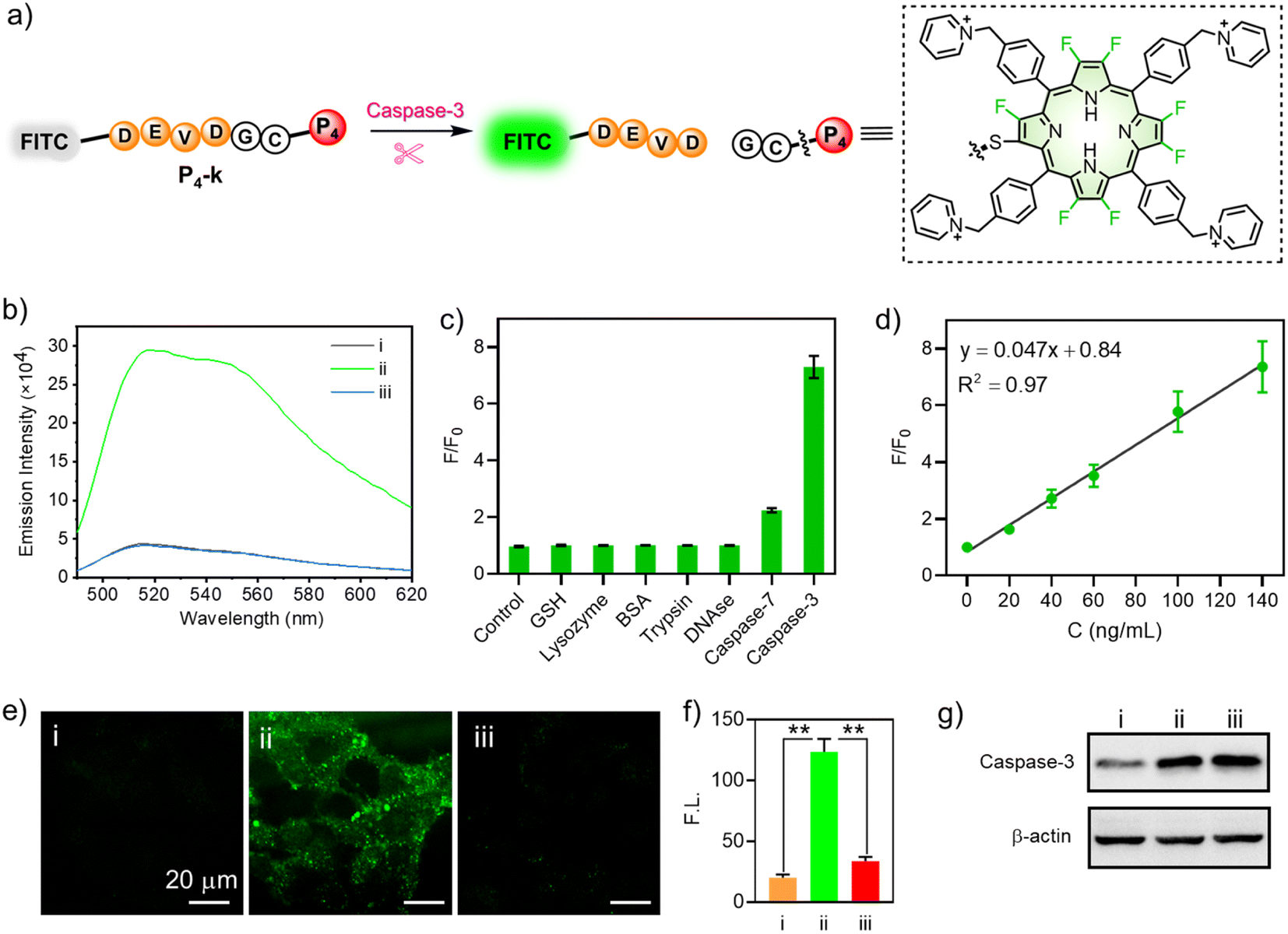 | ||
| Fig. 7 (a) Schematic illustration of P4-k as a fluorescent sensor for caspase-3. (b) Fluorescence spectra of P4-k (10 μM) in PBS (pH 7.4) with selected treatments. P4-k alone for i; caspase-3 + P4-k for ii; inhibitor + caspase-3 + P4-k for iii at 37 °C for 1 h. λex = 470 nm. (c) Fluorescence ratio (F/F0) response of P4-k to various species before (F0) and after (F) incubation with various species for 1 h in PBS (1×, pH 7.4). The concentration of GSH is 10 mM. The concentrations of lysozyme, BSA, trypsin, and DNAse are all 1 μg mL−1. The concentrations of caspase-7 and caspase-3 are 200 ng mL−1. (d) Linear fitting curve of F/F0 to the concentration of caspase-3 (0–140 ng mL−1). λex = 470 nm. The reaction was conducted in PBS (1×, pH 7.4) at 37 °C for 1 h. (e) Fluorescence microscope images. λex = 488 nm; λem = 525/50 nm bandpass filter. Scale bar: 20 μm. (f) Quantitative fluorescence intensity analysis of cell images in (e). (g) Expression level of caspase-3 of HeLa cells measured by Western blot assay. (i): HeLa cells were treated with P4-k (10 μM) for 1 h. (ii): HeLa cells underwent induced apoptosis by STS (3 μM) for 2 h, and were then cultured with P4-k (10 μM) for 1 h. (iii): HeLa cells were successively cultured with caspase-3 (10 μM) inhibitor and STS (3 μM) for 2 h, then with P4-k (3 μM) for 1 h. (*P < 0.05; **P < 0.01). | ||
To further demonstrate the specificity to caspase-3, we compared the fluorescence of P4-k in the presence of caspase-3, GSH, lysozyme, BSA, trypsin, DNAse, or caspase-7, respectively. As shown in Fig. 7c, only in the presence of caspase-3, did the fluorescence of P4-k show a 7-fold enhancement compared to the control. Note that the ratio of F/F0 increased linearly along with the concentration of caspase-3 (0–140 ng mL−1, Fig. 7d), demonstrating a correlation between the fluorescence intensity and caspase-3. We subsequently performed an enzymatic assay in living HeLa cells to demonstrate the potential of P4-k in detecting caspase-3 in vitro. As shown in Fig. 7e, when P4-k was incubated, the weak green fluorescence of FITC was observed, while the fluorescence was remarkably enhanced (ca. 6-fold) upon the addition of staurosporine (STS), a widely used drug to induce apoptosis (Fig. 7e(ii), and f). Moreover, when MPS (5-[(S)-(+)-2-(methoxymethyl)pyrrolidino]sulfonylisatin) was adopted to inhibit caspase-3 activity, the fluorescence barely increased (Fig. 7e(iii) and f). This is consistent with Western blotting analysis for the expression of caspase-3 in HeLa cells with or without STS and MPS (Fig. 7g). Together, we demonstrated the capability of β-peptidyl porphyrins for detecting the activity of caspase-3 in living cells.
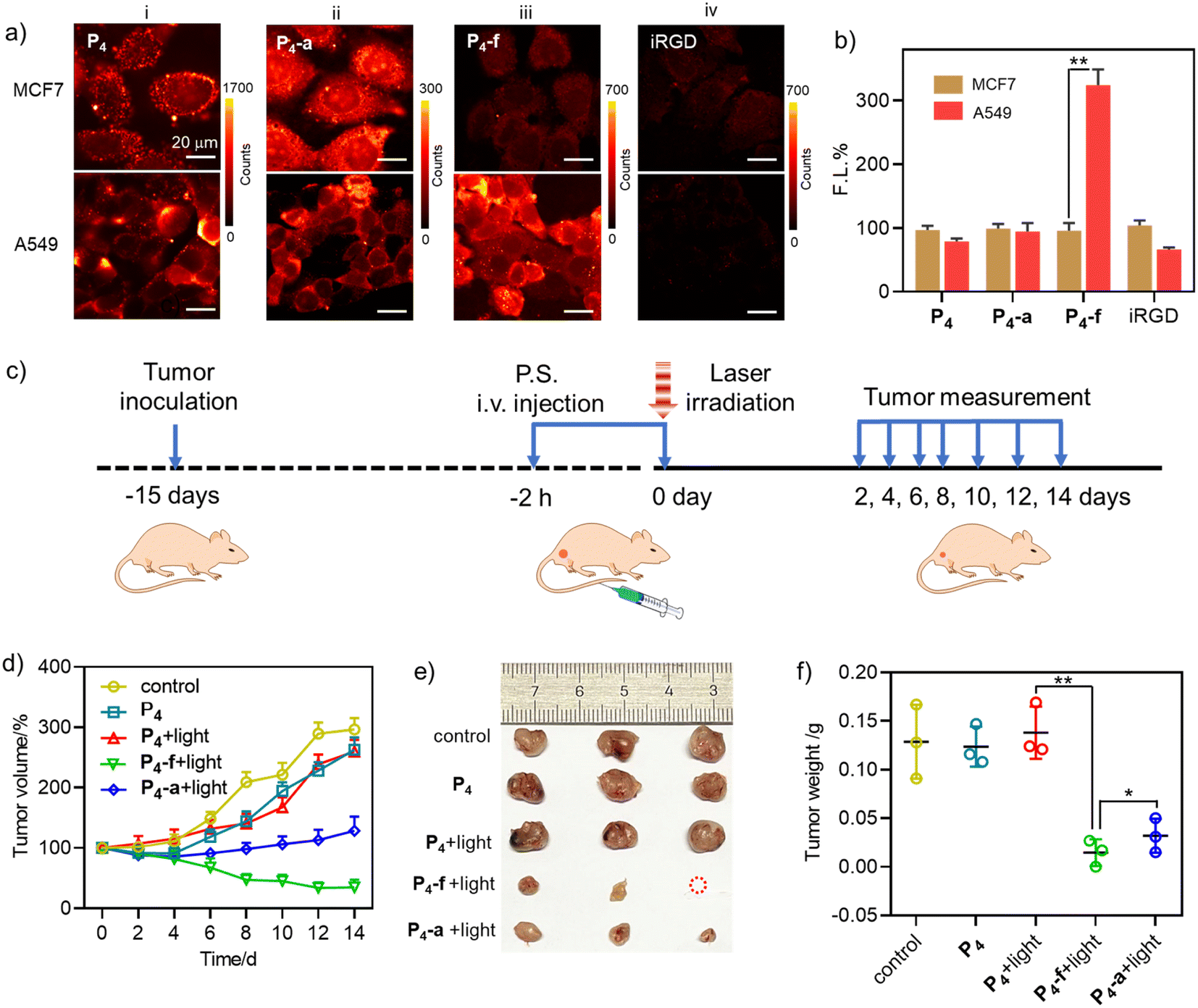 | ||
| Fig. 8 (a) Fluorescence images of A549 and MCF7 cells after incubation with P4 (8 μM, group i), P4-a (8 μM, group ii), or P4-f (8 μM, group iii) for 30 min. For group iv, the cells were incubated with iRGD (20 μM) for 1 h, then incubated with P4-f (8 μM) for 30 min. λex = 405 nm; λem = 647 nm longpass filter. (b) Quantitative fluorescence intensity analysis of fluorescence images of i–iv in (a). Red: A549 brown: MCF7. (*P < 0.05; **P < 0.01). The fluorescence of each compound in groups i–iv in MCF7 was calculated as 100%, and the fluorescence intensity in A549 was expressed as a percentage of that in MCF7. (c) Schematic illustration of the treatment regimen. (d) Tumor volume variation curves for the mice in the “control (PBS)”, “P4”, “P4 + light”, “P4-f + light”, and “P4-a + light” groups during the treatment period (n = 3, mean ± SD). (e) Photographs of the tumours dissected from the mice in different groups. (f) Tumor weights of the mice in different groups at the end of treatment (n = 3, mean ± SD). | ||
Then we evaluated the feasibility of P4, P4-a, and P4-f for in vivo PDT in A549 tumour mice by tail intravenous injection (Fig. 8c). The tumours were then subjected to irradiation (2 h post-injection, LED laser (400–700 nm), 100 mW cm−2, 10 min). During 14 days of tumour growth monitoring (Fig. 8d), we observed significant inhibition of tumour growth in the “P4-f + light” and “P4-a + light” groups. In comparison, other groups (“P4” or “P4 + light” groups) showed a minimal treatment effect. Note that tumour suppression was more effective in the “P4-f + light” group than that in the “P4-a + light” group due to the proven targeting ability of P4-f toward A549 cells. At the end of the treatment, the tumours were dissected and photographed. As shown in Fig. 8e and f, the tumour volumes and weights in the “P4-f + light” group were smaller than in the other groups. In particular, the tumour weights (0.0147 ± 0.0137 g) in the “P4-f + light” group were much lower than those in the control (0.129 ± 0.0380 g) and the “P4-a + light” groups (0.0320 ± 0.0175 g) due to the proven targeting ability of P4-f toward A549 cells. The mice body weights showed no abnormal change in any treatment group compared to the control group (PBS), indicating that the three photosensitizers had no obvious systemic toxicity (Fig. S42†). This biosafety was further supported by hematoxylin and eosin (H & E) staining, where no damage was found in major organ tissues such as the heart, liver, spleen, or kidney (Fig. S43†). Therefore, these results revealed the perspective of novel β-peptidyl porphyrins as tumour-targeting phototheranostics combining PDT and imaging.
Conclusions
We have developed an efficient and modular β-fluoropyrrolyl-cysteine SNAr preparation of irreversible peptide/protein–porphyrin bioconjugates. Due to the distinct electronic nature between fluorine and sulfur atoms, β-fluoropyrrolyl-cysteine SNAr chemistry represents a “two birds one stone” methodology that can not only covalently conjugate porphyrins with functional peptides/proteins but also induce intriguing photophysical properties, including facilitated ISC, red-shifted absorption, and increased singlet oxygen sensitization, which was highly favorable for phototheranostics. Moreover, these β-porphyrin bioconjugates have been applied in the intracellular delivery of EGFP, caspase-3 sensing in vitro, MGL, as well as in vivo tumor-targeting phototheranostics. β-Fluoropyrrolyl-cysteine SNAr chemistry provides unprecedented access to porphyrinic bioconjugates that enriches the repertoire of bioactive porphyrin. The present work highlights the promise of porphyrin bioconjugates as potential phototheranostics and the benefits that can accrue from manipulating the excited-state features of porphyrinoids. The fine-tuning of the structure and function achieved by the installation of β-sulfur atoms in porphyrin would also inspire research efforts to discover novel bioconjugation approaches based on other macrocycles and expand the functional range of small molecules for advanced materials and biomedical research.Author contributions
G.-Q. Jin and J.-L. Zhang planned and conducted the whole project experiments. J.-X. Wang and Y. Ning participated in chemical synthesis of β-octafluoroporphyrins. H. Zhang and J. Lu conducted the biological analysis and animal experiments. Y. Yao performed the theoretical calculations. J.-L. Zhang, S. Gao and H. Lu supervised the overall project. All authors contributed to the data analysis and the writing of the manuscript.Data availability
All data (NMR, HRMS, SDS-PAGE raw images, and Western blot raw images, etc.) have been provided in the main text and electronic ESI.†Conflicts of interest
The authors declare no competing financial interests. A Chinese patent (202111556065.4) has been applied that covers part of this work.Acknowledgements
We acknowledge financial support from the National Scientific Foundation of China (NSFC) (22131003, 21571007, 21621061, 21778002, and 21861162008). This work was supported by the High-performance Computing Platform of Peking University. This work was also supported by the Analytical Instrumentation Center of Peking University/the Center for Physicochemical Analysis and Measurements in ICCAS. All animal procedures were approved by the Institutional Animal Care and Use Committee of Sinoresearch (Beijing) Biotechnology Co., Ltd. (protocol number: 20211205).References
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, M. Weeks Amy, M. Hornsby, S. Lee Peter, V. Nichiporuk Rita, T. Iavarone Anthony, A. Wells James, F. D. Toste and J. Chang Christopher, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- Q. Luo, Y. Tao, W. Sheng, J. Lu and H. Wang, Nat. Commun., 2019, 10, 142 CrossRef PubMed.
- B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Nature, 2020, 585, 530–537 CrossRef CAS PubMed.
- Y. D. Gefei Li and J. Mo, Suwei Dong, Shin-ichiro Shoda and Xin-Shan Ye, CCS Chem., 2021, 4, 1930–1937 Search PubMed.
- R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, Chem, 2019, 5, 2243–2263 CAS.
- A. M.-H. Yip, C. K.-H. Lai, K. S.-M. Yiu and K. K.-W. Lo, Angew. Chem., Int. Ed., 2022, 61, e202116078 CrossRef CAS PubMed.
- S. Liu, C. Lin, Y. Xu, H. Luo, L. Peng, X. Zeng, H. Zheng, P. R. Chen and P. Zou, Nat. Chem., 2021, 13, 472–479 CrossRef CAS PubMed.
- H. Iwashita, E. Castillo, S. Messina Marco, A. Swanson Raymond and J. Chang Christopher, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018513118 CrossRef CAS PubMed.
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- M. A. Rajora, J. W. H. Lou and G. Zheng, Chem. Soc. Rev., 2017, 46, 6433–6469 RSC.
- Y. Ning, Y.-W. Liu, Z.-S. Yang, Y. Yao, L. Kang, J. L. Sessler and J.-L. Zhang, J. Am. Chem. Soc., 2020, 142, 6761–6768 CrossRef CAS PubMed.
- J. F. Arambula and J. L. Sessler, Chem, 2020, 6, 1634–1651 CAS.
- J.-Y. Liu, E. A. Ermilov, B. Röder and D. K. P. Ng, J. Porphyrins Phthalocyanines, 2013, 17, 831–835 CrossRef CAS.
- N. M. Casellas, G. Dai, E. Y. Xue, M. Jesús Vicente-Arana, D. K. P. Ng, T. Torres and M. García-Iglesias, Biomater. Sci., 2022, 10, 3259–3267 RSC.
- E. Stulz, Acc. Chem. Res., 2017, 50, 823–831 CrossRef CAS PubMed.
- P. Pathak, W. Yao, K. D. Hook, R. Vik, F. R. Winnerdy, J. Q. Brown, B. C. Gibb, Z. F. Pursell, A. T. Phan and J. Jayawickramarajah, J. Am. Chem. Soc., 2019, 141, 12582–12591 CrossRef CAS PubMed.
- F. Biscaglia and M. Gobbo, Pept. Sci., 2018, 110, e24038 CrossRef.
- J. Sandland and R. W. Boyle, Bioconjugate Chem., 2019, 30, 975–993 CrossRef CAS PubMed.
- P. M. R. Pereira, S. Silva, M. Bispo, M. Zuzarte, C. Gomes, H. Girão, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, Bioconjugate Chem., 2016, 27, 2762–2769 CrossRef CAS PubMed.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- H. Seki, S. J. Walsh, J. D. Bargh, J. S. Parker, J. Carroll and D. R. Spring, Chem. Sci., 2021, 12, 9060–9068 RSC.
- L. Xu, M. J. S. A. Silva, P. M. P. Gois, S. L. Kuan and T. Weil, Chem. Sci., 2021, 12, 13321–13330 RSC.
- Y. Zhang, C. Zang, G. An, M. Shang, Z. Cui, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2020, 11, 1015 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- H. Liu, W. Song, S. Zhang, K. S. Chan, Z. Guo and Z. Shen, Chem. Sci., 2020, 11, 8495–8501 RSC.
- C. Ma, Z. Xia, M. D. Sacco, Y. Hu, J. A. Townsend, X. Meng, J. Choza, H. Tan, J. Jang, M. V. Gongora, X. Zhang, F. Zhang, Y. Xiang, M. T. Marty, Y. Chen and J. Wang, J. Am. Chem. Soc., 2021, 143, 20697–20709 CrossRef CAS PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Compañón, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jiménez-Osés, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss and R. Derda, Chem. Sci., 2016, 7, 3785–3790 RSC.
- A. M. Embaby, S. Schoffelen, C. Kofoed, M. Meldal and F. Diness, Angew. Chem., Int. Ed., 2018, 57, 8022–8026 CrossRef CAS PubMed.
- T. Tsunemi, S. J. Bernardino, A. Mendoza, C. G. Jones and P. G. Harran, Angew. Chem., Int. Ed., 2020, 59, 674–678 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- J. Gonzales, N. V. S. D. K. Bhupathiraju, W. Perea, H. Chu, N. Berisha, V. Bueno, N. Dodic, J. Rozenberg, N. L. Greenbaum and C. M. Drain, Chem. Commun., 2017, 53, 3773–3776 RSC.
- J. Tang, M. A. Robichaux, K.-L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14706 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- N. V. S. D. K. Bhupathiraju, W. Rizvi, J. D. Batteas and C. M. Drain, Org. Biomol. Chem., 2016, 14, 389–408 RSC.
- J. Kvíčala, M. Beneš, O. Paleta and V. Král, J. Fluorine Chem., 2010, 131, 1327–1337 CrossRef.
- K. Ding, L. Wang, J. Zhu, D. He, Y. Huang, W. Zhang, Z. Wang, A. Qin, J. Hou and B. Z. Tang, ACS Nano, 2022, 16, 7535–7546 CrossRef CAS PubMed.
- X. Meng, Y. Yi, Y. Meng, G. Lv, X. Jiang, Y. Wu, W. Yang, Y. Yao, H. Xu and W. Bu, ACS Nano, 2022, 16, 4217–4227 CrossRef CAS PubMed.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. Lu, H. Wang, Z. Tian, Y. Hou and H. Lu, J. Am. Chem. Soc., 2020, 142, 1217–1221 CrossRef CAS PubMed.
- H. Choi, M. Kim, J. Jang and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22514–22522 CrossRef CAS PubMed.
- Q. Chen and Z. Liu, Adv. Mater., 2016, 28, 10557–10566 CrossRef CAS PubMed.
- J. Mao, Z. Xu and W. Lin, Curr. Opin. Chem. Eng., 2022, 38, 100871 CrossRef.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- H. Wang, Y. Hu, Y. Wang, J. Lu and H. Lu, Giant, 2021, 5, 100040 CrossRef CAS.
- Q. Laurent, R. Martinent, B. Lim, A.-T. Pham, T. Kato, J. López-Andarias, N. Sakai and S. Matile, JACS Au, 2021, 1, 710–728 CrossRef CAS PubMed.
- J. W. Trzciński, L. Morillas-Becerril, S. Scarpa, M. Tannorella, F. Muraca, F. Rastrelli, C. Castellani, M. Fedrigo, A. Angelini, R. Tavano, E. Papini and F. Mancin, Biomacromolecules, 2021, 22, 467–480 CrossRef PubMed.
- D.-e. Sun, X. Fan, Y. Shi, H. Zhang, Z. Huang, B. Cheng, Q. Tang, W. Li, Y. Zhu, J. Bai, W. Liu, Y. Li, X. Wang, X. Lei and X. Chen, Nat. Methods, 2021, 18, 107–113 CrossRef CAS PubMed.
- B. Cheng, Q. Tang, C. Zhang and X. Chen, Annu. Rev. Anal. Chem., 2021, 14, 363–387 CrossRef CAS PubMed.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed.
- A. G. Porter and R. U. Jänicke, Cell Death Differ., 1999, 6, 99–104 CrossRef CAS PubMed.
- J. F. Lovell, M. W. Chan, Q. Qi, J. Chen and G. Zheng, J. Am. Chem. Soc., 2011, 133, 18580–18582 CrossRef CAS PubMed.
- L. Zhang, J. Lei, F. Ma, P. Ling, J. Liu and H. Ju, Chem. Commun., 2015, 51, 10831–10834 RSC.
- P. Pyykkö, Angew. Chem., Int. Ed., 2006, 45, 28–29 CrossRef.
- D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed.
- Y. Jin, K. Xu, Y. Huang, H. Zhong and R. Zhao, Anal. Chem., 2021, 93, 2045–2052 CrossRef CAS PubMed.
- G. I. Vargas-Zúñiga, H. S. Kim, M. Li, J. L. Sessler and J. S. Kim, J. Porphyrins Phthalocyanines, 2021, 25, 773–793 CrossRef.
- S. Y. Y. Ha, R. C. H. Wong, C. T. T. Wong and D. K. P. Ng, Eur. J. Med. Chem., 2019, 174, 56–65 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06209g |
| This journal is © The Royal Society of Chemistry 2023 |