
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-safety and high-efficiency electrolyte design for 4.6 V-class lithium-ion batteries with a non-solvating flame-retardant†
Li
Chen
 ab,
Qingshun
Nian
a,
Digen
Ruan
a,
Jiajia
Fan
a,
Yecheng
Li
a,
Shunqiang
Chen
a,
Lijiang
Tan
a,
Xuan
Luo
a,
Zhuangzhuang
Cui
a,
Yifeng
Cheng
c,
Changhao
Li
c and
Xiaodi
Ren
*a
ab,
Qingshun
Nian
a,
Digen
Ruan
a,
Jiajia
Fan
a,
Yecheng
Li
a,
Shunqiang
Chen
a,
Lijiang
Tan
a,
Xuan
Luo
a,
Zhuangzhuang
Cui
a,
Yifeng
Cheng
c,
Changhao
Li
c and
Xiaodi
Ren
*a
aSchool of Chemistry and Materials Science, University of Science and Technology of China, Anhui 230026, China. E-mail: xdren@ustc.edu.cn
bKey Laboratory of Structure and Functional Regulation of Hybrid Materials, Anhui University, Ministry of Education, Hefei, 230601, China
cState Grid Anhui Electric Power Research Institute, China
First published on 28th December 2022
Abstract
Nonflammable electrolytes are critical for the safe operation of high-voltage lithium-ion batteries (LIBs). Although organic phosphates are effective flame retardants, their poor electrochemical stability with a graphite (Gr) anode and Ni-rich cathodes would lead to the deterioration of electrode materials and fast capacity decay. Herein, we develop a safe and high-performance electrolyte formulation for high-voltage (4.6 V-class) LIBs using flame-retarding ethoxy(pentafluoro) cyclotriphosphazene (PFPN) as a non-solvating diluent for the high-concentration carbonate–ether hybrid electrolyte. In contrast to conventional nonflammable additives with restricted dosage, the high level of PFPN (69% mass ratio in our electrolyte design) could significantly increase the electrolyte flash point and protect the favored anion-rich inner solvation sheath because of its non-solvating feature, thus preventing solvent co-intercalation and structural damage to the Gr anode. The nonflammable electrolyte could also form a stable LiF-rich cathode electrolyte interphase (CEI), which enables superior electrochemical performances of Gr‖LiNi0.8Mn0.1Co0.1O2 (NMC811) full cells at high voltages (∼82.0% capacity retention after 1000 cycles at 4.5 V; 89.8% after 300 cycles at 4.6 V) and high temperatures (50 °C). This work sheds light on the electrolyte design and interphase engineering for developing practical safe high-energy-density LIBs.
1. Introduction
To achieve the ambitious goal of carbon neutrality, the ever-increasing demand for electric vehicles and grid energy storage has greatly stimulated the development of lithium-ion batteries (LIBs).1–3 To further improve the energy density of LIBs, considerable efforts have been devoted to exploring high-voltage LiNixMnyCo1−x−yO2 (NMC) cathode materials with high Ni content (e.g., LiNi0.8Mn0.1Co0.1O2, NMC811).4 However, the increased energy density is usually accompanied by shortened cycle life and more prominent battery safety issues, thus hindering their large-scale applications. One of the primary failure mechanisms of Ni-rich NMC cathodes is their incompatibility with state-of-the-art organic carbonate-based electrolytes. The highly reactive Ni4+ on the surface of delithiated NMC cathodes could induce severe electrolyte side reactions, which are closely associated with phase transformation, interfacial impedance increase and capacity decay of the cathode.5 On the graphite (Gr) anode, the solid electrolyte interphase (SEI) derived from ethylene carbonate is not robust enough to withstand high temperature, short circuit or overcharging conditions. Moreover, conventional carbonate electrolytes are highly volatile and flammable (with low flash points, e.g., 16 °C for dimethyl carbonate). Therefore, catastrophic thermal runaway reactions in the LIBs could be triggered by the decomposition of the anode SEI and subsequent exothermic reactions between electrodes and electrolyte.4,5Momentous efforts have been focused on developing electrolytes for high-safety LIBs, for example, by adding flame-retarding additives or adjusting the solvation structure of the electrolyte.6 Organic phosphates (e.g., triethylphosphate, TEP, and trimethylphosphate, TMP) are among the most studied co-solvents for nonflammable electrolytes.1 Despite their ability to inhibit the combustion chain reactions as hydrogen radical scavengers, they suffer from poor electrochemical compatibility with the Gr anode. Previous studies indicate that the cycling stability of the LIBs will decline rapidly when the ratio of phosphate co-solvent added exceeds 10 wt%.1,6,7 Phosphate molecules could decompose on the Gr anode and deteriorate the anode SEI, which results in Gr exfoliation and continuous electrolyte decomposition.6,8 To inhibit the undesired reaction of phosphate solvent, high concentration electrolytes (HCE) with high salt/solvent ratios (e.g., 3.3 M LiFSI in TMP) were developed to enrich anions in the inner Li+ solvation sheath and promote their reactions on the electrode/electrolyte interface.1 With the formation of an inorganic-rich SEI, the electrochemical stability of the Gr anode could be greatly improved.1 To address the issues of high cost, high viscosity, and poor wettability for HCEs, non-solvating hydrofluoroethers (e.g., bis(2,2,2-trifluoroethyl) ether (BTFE) and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE)) were added as diluents without disrupting the featured anion-rich solvation structure, which is thus referred to as localized high concentration electrolytes (LHCEs).9 Recent studies indicate that phosphate-based LHCE with a unique solvation structure displays excellent electrochemical performance in LIBs and lithium metal batterie.9–11 Xu et al. reported TMP-based LHCE with additives to improve the long-term cycling stability of LIBs in the voltage range of 2.5–4.4 V.6 However, hydrofluoroether diluents typically have low flash points (e.g., 1 °C for bis(2,2,2-trifluoroethyl) ether), which might still be problematic for battery safety.12 Therefore, the development of functional electrolytes with excellent flame-retarding capability and electrochemical stability on reactive electrodes is desired.
In this work, we design an LHCE formulation using flame-retarding ethoxy(pentafluoro) cyclotriphosphazene (PFPN) as the non-solvating diluent for the high-concentration carbonate–ether hybrid electrolyte. Fluorophosphazenes have been typically used as electrolyte additives with <10% percentage to avoid deteriorating the electrode stability.13–16 Their usage at high levels has seldom been reported to maximize the safety benefits. Our electrolyte design contains an extremely high percentage of PFPN (69% in mass ratio) as the diluent to significantly increase the electrolyte flash point. In addition, the unique anion-rich inner solvation sheath with carbonate–ether hybrid solvent significantly improves the electrochemical stabilities of both electrodes. The co-intercalation of solvent molecules into Gr layers could be inhibited by the effective SEI on the Gr anode. The designed LHCE can also form a highly robust fluorine-rich CEI, thus achieving excellent electrochemical properties (4.5 V, ∼82.0% capacity retention after 1000 cycles), and high-voltage stability (4.6 V, 89.8% after 300 cycles) in Gr‖NMC811 cells. Moreover, the CEI and SEI with rich inorganic species can enable excellent high-temperature (50 °C) cycling stability of NMC811-based LIBs under high voltage conditions.
2. Results and discussion
Our electrolyte design takes full advantage of the high electrochemical stability feature of the high-concentration inner solvation structure and the high flame-retarding capability of the PFPN diluent in the outer solvation sheath. For the inner solvation complex, fluoroethylene carbonate (FEC) was first selected for its ability to form a fluorinated SEI on the Gr anode.12,17–19 Its fluorine-donating ability, along with the enhanced reactivity of the bis(fluorosulfonyl)imide (FSI−) anion in the concentrated solvation structure, is beneficial for forming an inorganic-rich highly-durable SEI.20 However, phase separation was induced when mixing PFPN with the FEC-based HCE (LiFSI-2FEC in molar ratio), which is likely due to the high polarity of the FEC solvent (dielectric constant of 110), as shown in Fig. 1a and b. The immiscibility issue was successfully addressed with the addition of an ether cosolvent with low polarity (1,2-dimethoxyethane or DME, dielectric constant of 7.2), which may have favored interactions with PFPN molecules. To understand the effect of solvents with different dielectric constants on the miscibility of electrolytes, various electrolytes were compared at the same molar ratio (Table S3†). The dielectric constant of sulfone and most carbonates have relatively large dielectric constants, which causes incompatibility of the electrolyte with the PFPN diluent. In comparison, ethers with a low dielectric constant, such as DME and THF, can achieve good electrolyte miscibility. Thus, a carbonate–ether hybrid electrolyte is designed to solve the immiscibility issue. More importantly, ether was introduced into the solvation structure because of its unique synergy with anions to induce the formation of a LiF-enriched cathode–electrolyte interphase (CEI) on high-voltage cathodes, as revealed by recent studies.21,22 The concentrated electrolyte (E-conc) contains LiFSI, DME and FEC with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:0.5. Using highly fluorinated PFPN as the diluent, the PFPN-based LHCE was prepared with an LiFSI:PFPN molar ratio of 1:3 (abbreviated as E-PFPN) (Fig. 1a and b). In contrast, two electrolytes with different contents of phosphate (TMP, with a LiFSI:TMP molar ratio of 1:1 or 1:3) were prepared to investigate the effect of phosphate on the electrochemical performance of the LIBs with E-conc (the abbreviations are E-TMP, and E-TMP-3). Besides, a conventional carbonate electrolyte, 1 M LiPF6 in EC/DEC/EMC (1:1:1 by vol%) (E-carbonate), was chosen as the baseline electrolyte. The detailed compositions of the electrolytes mentioned above are summarized in Table S1.†
:1.5:0.5. Using highly fluorinated PFPN as the diluent, the PFPN-based LHCE was prepared with an LiFSI:PFPN molar ratio of 1:3 (abbreviated as E-PFPN) (Fig. 1a and b). In contrast, two electrolytes with different contents of phosphate (TMP, with a LiFSI:TMP molar ratio of 1:1 or 1:3) were prepared to investigate the effect of phosphate on the electrochemical performance of the LIBs with E-conc (the abbreviations are E-TMP, and E-TMP-3). Besides, a conventional carbonate electrolyte, 1 M LiPF6 in EC/DEC/EMC (1:1:1 by vol%) (E-carbonate), was chosen as the baseline electrolyte. The detailed compositions of the electrolytes mentioned above are summarized in Table S1.†
 | ||
| Fig. 1 (a) Molecular structure of the solvents in the electrolyte of LiFSI/FEC/PFPN and E-PFPN. (b) Photos of different electrolytes without and with DME cosolvent. (c) and (d) Raman spectra of different solvents and electrolytes. (e) Molecular structures of TMP and PFPN, and the solubility test of LiFSI in PFPN. (f)–(h) Contact angle tests of various electrolytes on a Celgard separator. (i)–(k) Schematic illustrations of solvation structures in different electrolytes. (l)–(n) Photos of ignition tests of glass fibers soaked with different electrolytes. | ||
To reveal the electrolyte solvation structures, Raman spectra of different electrolytes and pure solvents are exhibited in Fig. 1c and d. The vibration signals of free DME (820 and 847 cm−1 signals for CH2 rocking and C–O stretching vibrations, respectively) and FEC (729 cm−1 for C–O stretching vibration) molecules become very weak in E-conc, suggesting that they mostly coordinate with Li+ due to the high salt/solvent ratio. The P–O–C stretching vibration of TMP shifts from 748 cm−1 in the free state to 764 cm−1 in E-TMP due to the formation of Li+-coordinated TMP.31 As 10 mg LiFSI was added to 825 mg PFPN, negligible dissolution was observed (Fig. 1f). The supernatant was removed by centrifugal filtration, and the remaining salt was dried in a vacuum oven at 120 °C for 24 hours. The weight of the undissolved LiFSI was 9.6 mg. The result implies the very limited Li+-solvating ability of the PFPN diluent. Therefore, the high salt/solvent ratio in the Li+ solvation sheath is maintained on replacing TMP with PFPN. The P–O–C stretching vibration of PFPN at 720 cm−1 does not shift in E-PFPN (Fig. 1d), confirming that the PFPN is not involved in the inner Li+ solvation sheath in the PFPN-diluted LHCE. Thus, the solvent structure of E-conc will not be destroyed by the PFPN diluent in the E-PFPN, and DME and FEC molecules are still coordinated with Li+.
The key physical properties of the chosen electrolytes, for instance, ionic conductivity, wettability, viscosity, flash point, and contact angle test, were conducted with the results shown in Fig. 1g–i, S1, and Table S2.† The E-conc and E-TMP displayed higher ionic conductivities than E-PFPN at 25 °C. The ionic conductivity of E-PFPN decreases to 2.22 mS cm−1 due to a much lower bulk ion concentration (0.83 m) and the intensified electrostatic interactions between Li+ and anions in the LHCE.21,32,33 The contact angle tests on Celgard 2500 separators were used to characterize the wettability of different electrolytes (Fig. 1f–h). E-PFPN has the best wettability with a contact angle of 21.4°, which is much smaller than that of the E-conc (71.9°) and E-TMP (64.5°). The wettability tests of various electrolytes on the Celgard 2500 separators also showed the better wettability of E-PFPN than that of E-carbonate, E-conc, E-TMP, and E-PFPN. Moreover, E-PFPN has smaller viscosity than E-conc and E-TMP (Table S2†) due to the presence of the PFPN diluent. The high wettability and smaller viscosity of the electrolyte design with the high level of PFPN demonstrate its excellent rate capability and high ionic conductivity.
In addition, the flame-retardant properties of electrolytes were tested by ignition experiments (Fig. 1l–n, S2, and Videos S1–S5†). Both the E-carbonate and E-conc are highly flammable due to the abundance of organic solvent molecules (Fig. 1l, S2, Videos S1 and S2†). Although E-TMP cannot be ignited in a short time, intensive burning was still observed after ignition (Fig. 1m and Video S3†). It indicates that the DME/FEC molecules are intensively involved in the burning process, and TMP cannot immediately quench the radical chain reactions (Fig. 1m). Moreover, the ignition test of E-TMP-3 is shown in Fig. S2 and Video S4.† E-TMP-3 requires to be ignited three times before burning on increasing the amount of TMP. Despite the high content of TMP existing in E-TMP-3, a large number of free DME/FEC molecules will cause the electrolyte to be ignited. In stark contrast, E-PFPN cannot be ignited after several attempts and essentially only generates smoke with the contact of torch flame (Fig. 1n and Video S5†), demonstrating its excellent flame-retarding capability. The PFPN molecules would produce large amounts of P- and F-containing radical scavenges during the pyrolysis process, thus effectively quenching H and O-based free radicals generated from the combustion of organic solvents.14,34 Moreover, the flash points of different electrolytes were tested (Table S2†). In contrast to the low flash point of DME (−2 °C), the flash point of E-conc increases significantly to 72 °C due to the concentrated solvation structure with low amounts of solvent molecules. As in previous reports, the drawbacks of high volatility and high flammability of ether and carbonate solvents can be overcome to a large degree owing to the much lower content of organic solvents in the high concentration electrolyte.35,36 However, the flash point of E-TMP decreases significantly. It is likely that TMP will participate in the solvation structure of E-TMP, which will eventually lead to an increase in free DME molecules (Fig. 1j). In sharp contrast, E-PFPN has no measurable flash point before ∼121 °C (the boiling point of PFPN). The significantly increased flash point and the ignition test result for E-PFPN suggest that the large percentage of PFPN (50% in molar ratio or 69% in mass ratio) in E-PFPN is critical for the superior flame-retarding ability of the electrolyte (Fig. 1k).
A linear sweep voltammetry (LSV) experiment was used to evaluate the electrochemical stability of electrolytes with a Super-P carbon/PVDF composite as the working electrode, and Li foil as the counter and reference electrodes.37 As shown in Fig. S3,† E-PFPN shows a higher oxidation potential higher than 4.6 V, indicating its feasibility for high voltage battery applications. Moreover, a constant voltage floating test was conducted to compare leakage currents on the Al electrode at 4.5 V (Fig. S4†). E-PFPN exhibits the lowest leakage current (∼1 μA cm−2) relative to other electrolytes during the 60 h test. From the SEM post-analysis of the Al electrodes (Fig. S5†), no noticeable Al corrosion could be observed in the E-PFPN. It indicates that a concentrated solvation structure is beneficial for inhibiting undesired electrolyte side reactions and protecting the Al current collector. The immediate current drop at the beginning of the leakage current test implies that the PFPN diluent also plays a vital role in the passivation process. Fig. S6† shows that the CV curves were also measured using Li‖Gr cells filled with different electrolytes. The intercalation/deintercalation peaks observed in these electrolytes indicate the formation of protective SEI layers on Gr anodes.
Gr‖NMC811 full cells were assembled to evaluate the electrochemical performance of different electrolytes. Fig. 2a–c, S7 and S8† show the cyclic performance of Gr‖NMC811 cells at a cut-off voltage of 4.5 V and 25 °C. In the first formation cycle at 1/10C, Gr‖NMC811 cells using E-carbonate, E-conc, E-TMP, and E-PFPN display discharge capacities and initial coulombic efficiencies (ICE) of 187.3 mA h g−1 (80.1%), 201 mA h g−1 (84.8%), 194.4 mA h g−1 (80.33%), and 199.8 mA h g−1 (85.5%), respectively (Fig. 2b and S8†). The higher ICEs in E-conc and E-PFPN indicate that they have less side reactions with Gr anodes. As shown in Fig. 2c, the discharge capacity of E-carbonate cells declines to 150.2 mA h g−1 after 500 cycles with a capacity retention of 83.8%. The critical role of EC in protecting the Gr anode and the anodic stability of carbonate contributes to the cell cycling performance at a high voltage. In contrast, the E-conc cell displays a slower capacity fading rate than the E-carbonate before 400 cycles. The high-concentration electrolytes can adjust the Li+ solvation structure to produce an anion-derived SEI layer with inorganic components to suppress the co-intercalation of the solvent molecules into the Gr anode.38,39 Thus, its high ICE and stable long-term cycling are likely from the robust inorganic-rich passivation film on the Gr anode in E-conc. However, after introducing TMP as a cosolvent into E-conc, E-TMP shows a dramatic capacity fading (Fig. 2a and c). After 130 cycles, the capacity of the cells in E-TMP was only 35.3 mA h g−1. Due to its high dielectric constant (ε = 21.3), TMP would participate in the Li+ solvation structure to form the Li+ solvation sheath in E-TMP. Thus, an effective SEI protective layer cannot be formed, so the co-intercalation of the solvent molecule into the Gr anode induces the structural exfoliation of Gr during repeated cycling.6,40 The capacity fading was intensified further in E-TMP-3 (3.3 mA h g−1 after 30 cycles) (Fig. S8†), which further proves the disadvantage of TMP cosolvent in the electrolyte. After replacing TMP with PFPN, E-PFPN shows an outstanding cycling performance at 4.5 V, with a capacity retention of 82.0% after 1000 cycles (154.3 mA h g−1) (Fig. 2a, c and S7c†). Although DME is acting as the main solvent in E-PFPN, the Gr‖NMC811 full cells can still maintain a long cycle stability. Previous work indicates that ether-based LHCE with finely tuned structures and composition of solvation sheaths can achieve a uniform and robust solid electrolyte interphase (SEI) on the Gr anode.41,42 To confirm this, the electrolyte (LiFSI/DME/PFPN, 1:2:3 by molar ratio) without adding FEC is used to assemble Gr‖NMC811 full cells. As shown in Fig. S9,† the electrolyte without FEC still maintains good cycle stability. Thus, the co-intercalation of ether solvent molecules into Gr can be also effectively suppressed in E-PFPN. Moreover, as PFPN will not influence the Li+ solvation sheath, it does not hinder the formation of an effective SEI on the Gr anode despite its high percentage in the electrolyte. In addition, 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE) is used as the diluent to configure E-TTE (LiFSI/DME/FEC/TTE, 1:1.5:0.5:3 by molar ratio). As shown in Fig. S10,† the high-concentration carbonate–ether hybrid electrolyte with the TTE diluent also displayed stable cycle performance in Gr‖NMC811 cells at 1/3C charge/discharge rate and 25 °C (∼85.0% after 300 cycles at 4.5 V). Thus, the design of carbonate–ether hybrid LHCE has great potential for improving the cycle stability of Gr‖NMC811 cells.
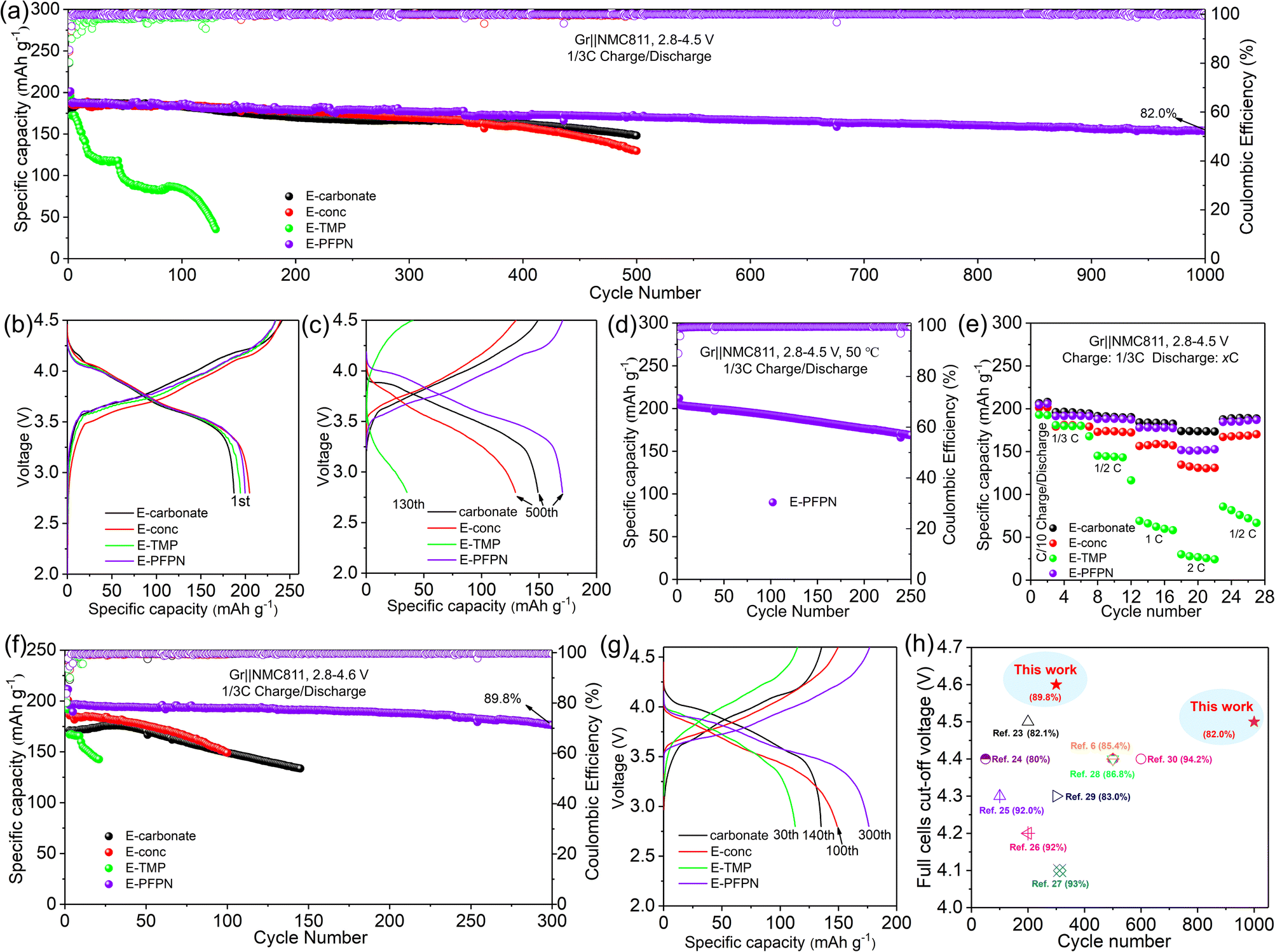 | ||
| Fig. 2 Electrochemical properties of various electrolytes. (a) Cycling stabilities at 1/3C rate and 25 °C at a cut-off voltage of 4.5 V; charge–discharge curves at (b) the first formation cycle at 1/10C rate and (c) the subsequent cycles at 1/3C rate. (d) Cycling stabilities with 1/3C rate at 50 °C at a cut-off voltage of 4.5 V. (e) Discharge rate capability of Gr‖NMC811 cells. (f) Cycling stabilities at a cut-off voltage of 4.6 V. (g) Charge–discharge curves at the subsequent cycles at 1/3C rate and 4.6 V. (h) The comparison of the capacity retention rate of the cells with that in previously reported work.6,23–30 | ||
Increasing temperature typically accelerates the parasitic reactions between the electrolytes and both the high-Ni NMC cathode and Gr anode. To further evaluate the passivation ability of E-PFPN, the cycling performance of Gr‖NMC811 cells at an elevated temperature (50 °C) was investigated. As seen in Fig. 2d and S11,† the Gr‖NMC811 cells with E-PFPN as the electrolyte reveal excellent high-temperature cycling performance after 250 cycles at a cut-off voltage of 4.5 V with a capacity retention of 82.6%. The Gr‖NMC811 cells with E-PFPN demonstrate outstanding cycling stability and less voltage polarization even at a higher temperature, suggesting that the formed SEI on Gr and CEI on NMC811 are highly effective. Moreover, the discharge rate capabilities of Gr‖NMC811 cells with various electrolytes at a cut-off voltage of 4.5 V were investigated, as shown in Fig. 2e. E-PFPN displays better discharge rate performance compared to E-Conc and E-TMP, which can be assigned to lower interfacial resistance (the R2 and R3) as evidenced by the electrochemical impedance spectroscopy (EIS) results in Fig. 3a. The charge–discharge curve of Gr‖NMC811 cells at a discharge rate of 1C shown in Fig. S12 and S13† indicated that E-PFPN has great potential to enable high energy density LIBs with superior safety capability.
 | ||
| Fig. 3 (a) EIS of Gr‖NMC811 cells using the studied electrolytes after two formation cycle and the inset is the equivalent circuit. (b) and (c) XRD patterns of the pristine Gr anode and the cycled Gr anodes in E-conc, E-TMP, and E-PFPN after 50 cycles at 4.5 V. Electrochemical performance of various electrolytes (E-conc, E-TMP, and E-PFPN) in Li‖Gr half cells: (d) first cycle charge/discharge voltage curves at C/10, (e) cycling capacity for Li+ de-intercalation and (f) CE of the Li‖Gr cells, where the Li‖Gr cells were cycled at 1/5C with the cut-off voltage range of 0.01–1.2 V and 1C = 372 mA h g−1 based on the weight of the Gr active material. | ||
When the cut-off voltage of Gr‖NMC811 cells was increased to 4.6 V, all the cells except E-TMP exhibited higher discharge capacity in the initial formation cycle at 1/10C (Fig. 2f and S14†). The capacity of the electrolyte with TMP as the cosolvent shows a large capacity fading after 30 cycles (Fig. 2g). Fig. 2f shows a clear contrast of the capacity retentions in different electrolytes during cycling, and E-PFPN offers obvious advantages over other electrolytes (89.8% capacity retention after 300 cycles). Besides TMP, triethyl phosphate (TEP) was also added to the electrolytes (E-TEP and E-TEP-3) to verify its effect on the long-term cycling performance of Gr‖NMC811 (Fig. S15 and S16†). Similar to E-TMP, TEP-based nonflammable electrolytes show poor cycling performance, further indicating that a large proportion of phosphates in the electrolyte as flame retardants is unfavorable to the cycling stability of LIBs. Very recently, fluorophosphazenes have demonstrated improved electrochemical performances for Gr and Li metal anodes as the diluent of LHCEs.11,43 However, this study of E-PFPN first proves the advantages of such nonflammable electrolyte design in terms of the long-term stability Gr‖NMC811 full cells at such high voltages and temperatures over other reported electrolytes (Fig. 2h).
For differential scanning calorimetry (DSC) experiments of safety performance, charged NMC811 cathodes (to 4.5 V) are tightly sealed in high-pressure crucibles with different electrolytes. As shown in Fig. S17,† despite the close onset exothermic temperatures, the amounts of heat release in different electrolytes are significantly different. Due to the solvation structure involving TMP, E-TMP showed a greatly increased heat release (905.5 J g−1) than E-conc (671 J g−1). More importantly, a minimum heat release is also observed in E-PFPN (499 J g−1). The improved thermal stability of E-PFPN is likely related to the existence of the high level of flame-retardant PFPN and the formation of a protective CEI.
To verify the stability of Gr anodes, Li‖Gr half cells were tested in different electrolytes (Fig. 3d–f and S18†). As for E-PFPN, an initial charge/discharge capacity of 341.5 and 359.3 mA h g−1 and an initial CE of 95.05% are gained in the Li‖Gr cell (Fig. 3d). Moreover, no additional plateau was observed on the discharge curve, indicating that the co-intercalation of an ion solvation sheath into Gr layers did not occur. From the 3rd cycle (Fig. S18†), the Gr anode shows a stable discharge capacity of 344.5 mA h g−1 and a CE of 99.47% in E-PFPN. Fig. 3e and f indicate that the best electrochemical stability was obtained in E-PFPN with the high cell CE approaching 100% during cycling. However, the low reversible capacity of Gr at 25 °C with the E-TMP electrolyte shown in Fig. 3e may come from two aspects. Firstly, the low capacity of Gr is probably caused by the high viscosity, and poor electrode wettability of E-TMP at room temperature;44 Secondly, the solvent co-intercalation destroys the structure of the layered Gr anode, and thereby the Gr/Li cell with E-TMP exhibits a low capacity and low initial coulombic efficiency. Thus, an effective protective layer on Gr was formed during the formation cycles in the E-PFPN electrolyte.
The electrochemical performance and the XRD patterns (Fig. 3b and c) of the Gr anodes cycled in E-conc and E-PFPN (higher intensity of (002) peaks) demonstrate the enhanced structural integrity of Gr layers in these electrolytes. However, after cycling in E-TMP, the Gr anode shows noticeable crystal structure changes. The SEM images were collected to investigate the effect of different electrolytes on the Gr anodes (Fig. S19†). The cycled Gr anodes in full cells using E-conc and E-PFPN have uniform and smooth surface morphologies. However, fragmented structures were observed for the cycled Gr anode in E-TMP, indicating the deterioration effect of TMP on the structure of the Gr anode. Under high-resolution transmission electron microscopy (HR-TEM), the SEI layer on the Gr anode after cycling in E-conc was relatively compact and thin with slight Gr exfoliation observed (marked by the red dotted line) (Fig. 4a). The interlayer spacing of the exfoliated Gr layer (0.382 nm) is larger than that of the Gr layer (0.355 nm) after cycling, indicating that the Gr layer is partially destroyed. However, after cycling in E-TMP, the Gr anode was covered by a non-uniform and thick SEI (Fig. 4b). Moreover, the Gr anode displays a large amorphous domain (marked by the green dotted line), which is caused by the deterioration of the SEI by the co-intercalation of TMP during cycling.6 In contrast, the SEI of the Gr anode formed in E-PFPN was uniform and thin (8–9 nm), and Gr exfoliation was effectively suppressed (Fig. 4c). The well-maintained Gr layered structure with a much thinner SEI corresponds to the high-capacity retention of the Gr‖NMC811 cells using E-PFPN.
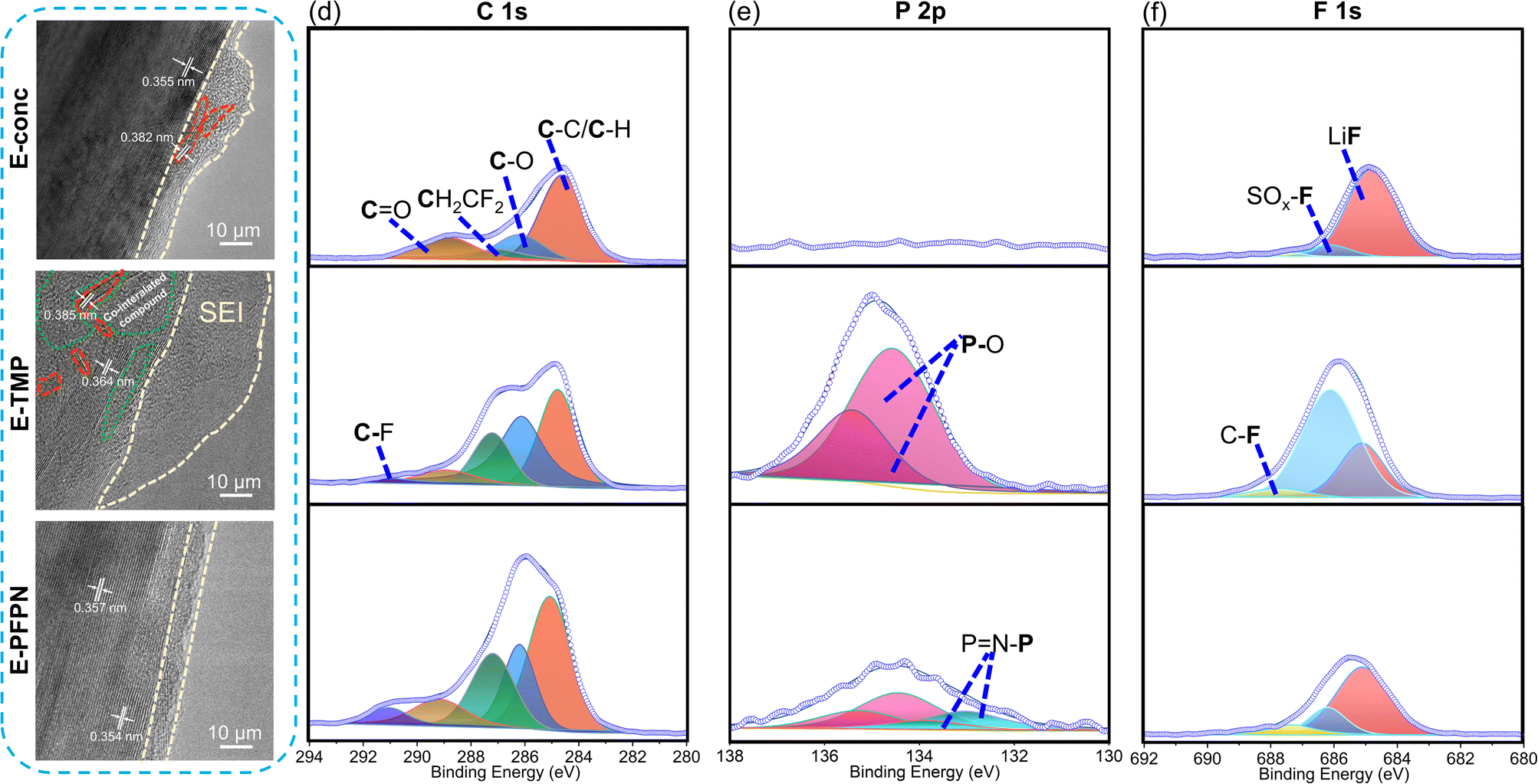 | ||
| Fig. 4 HR-TEM of Gr electrodes after 50 charge/discharge cycles in different electrolytes: (a) E-conc, (b) E-TMP, and (c) E-PFPN. XPS spectra of Gr electrodes after 50 charge/discharge cycles in different electrolytes: (d) C 1s, (e) P 2p, and (f) F 1s. | ||
XPS analyses were also performed to characterize the detailed compositions of the SEIs on the cycled Gr anodes after 50 charge/discharge cycles (Fig. 4d–f). As shown in Fig. 4d, all the electrolytes show roughly similar SEI compositions, including the C–C/C–H, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–F, and CF2–CH2 (PVDF) species on the surfaces of Gr anodes. The organic carbonaceous species (C–C, C–O, and CO) in the SEI layers are likely from the decompositions of solvent molecules (FEC and DME) on the Gr anode. As discussed above, FEC is effective in constructing a stable SEI on the Gr anode. The higher proportions of CO (Li2CO3, 289.2 eV) in the C 1s and O 1s (Fig. S20†) of E-conc and E-PFPN than those in E-TMP may indicate the higher reactivity of FEC in the concentrated solvation structures. As shown in Fig. 4e, the P 2p signals from the SEIs confirmed that TMP or PFPN was involved in the SEI formation process (Fig. 4e, f and S20†). The much stronger P–O signal intensity in E-TMP suggests more apparent TMP decompositions on the surface of Gr, which is undesirable for the anode stability. In the F 1s spectra (Fig. 4f), the peaks at 688.0 eV, 686.1 eV, and 684.9 eV correspond to the organic C–F, SO2–F, and LiF species, respectively.41 Both E-conc and E-PFPN induce the enrichments of LiF in the SEI layers, indicating that FEC and FSI− play essential roles in the protection of the Gr anode. However, the lower ratio of LiF and the higher ratio of SO2–F in E-TMP suggest the incomplete decomposition of FSI−, in agreement with the N 1s, and S 2p spectra (Fig. S20†). This is because TMP molecules enter the inner solvation sheath and change the reactivity of salt anions. Compared with LiF, with high physical and electrochemical stabilities, the SO2–F species are not as effective in protecting Gr anodes. In addition, the P–O signal from the P 2p XPS spectrum suggests the possible participation of PFPN in SEI formation. This difference with the recent result of hexafluorocyclotriphosphazene on the Gr anode may be due to the additional ethoxy functional group of PFPN and the reaction intermediates from decompositions of carbonate/ether solvents.43 Based on the above analysis, the thin, robust SEI formed in E-PFPN by the synergy of FEC and LiFSI can hinder solvent co-intercalation into the Gr and enable the long-term cycling performance of Gr anodes (Fig. 6). Moreover, to further explore the reasons for the quick fading of Gr‖NMC811 full cells of E-TMP, the cycling performance of Gr‖NMC811 full cells at a lower charge/discharge voltage was measured. The experimental results of the cycling performance are shown in Fig. S21.† Although the charge–discharge cut-off voltage has dropped to 4.3 V, the capacity of E-TMP still drops rapidly after 8 cycles. Thus, the quick fading of Gr‖NMC811 full cells with E-TMP as the electrolyte may be mainly due to the deterioration of the Gr anode, rather than the cathode side.
O, C–F, and CF2–CH2 (PVDF) species on the surfaces of Gr anodes. The organic carbonaceous species (C–C, C–O, and CO) in the SEI layers are likely from the decompositions of solvent molecules (FEC and DME) on the Gr anode. As discussed above, FEC is effective in constructing a stable SEI on the Gr anode. The higher proportions of CO (Li2CO3, 289.2 eV) in the C 1s and O 1s (Fig. S20†) of E-conc and E-PFPN than those in E-TMP may indicate the higher reactivity of FEC in the concentrated solvation structures. As shown in Fig. 4e, the P 2p signals from the SEIs confirmed that TMP or PFPN was involved in the SEI formation process (Fig. 4e, f and S20†). The much stronger P–O signal intensity in E-TMP suggests more apparent TMP decompositions on the surface of Gr, which is undesirable for the anode stability. In the F 1s spectra (Fig. 4f), the peaks at 688.0 eV, 686.1 eV, and 684.9 eV correspond to the organic C–F, SO2–F, and LiF species, respectively.41 Both E-conc and E-PFPN induce the enrichments of LiF in the SEI layers, indicating that FEC and FSI− play essential roles in the protection of the Gr anode. However, the lower ratio of LiF and the higher ratio of SO2–F in E-TMP suggest the incomplete decomposition of FSI−, in agreement with the N 1s, and S 2p spectra (Fig. S20†). This is because TMP molecules enter the inner solvation sheath and change the reactivity of salt anions. Compared with LiF, with high physical and electrochemical stabilities, the SO2–F species are not as effective in protecting Gr anodes. In addition, the P–O signal from the P 2p XPS spectrum suggests the possible participation of PFPN in SEI formation. This difference with the recent result of hexafluorocyclotriphosphazene on the Gr anode may be due to the additional ethoxy functional group of PFPN and the reaction intermediates from decompositions of carbonate/ether solvents.43 Based on the above analysis, the thin, robust SEI formed in E-PFPN by the synergy of FEC and LiFSI can hinder solvent co-intercalation into the Gr and enable the long-term cycling performance of Gr anodes (Fig. 6). Moreover, to further explore the reasons for the quick fading of Gr‖NMC811 full cells of E-TMP, the cycling performance of Gr‖NMC811 full cells at a lower charge/discharge voltage was measured. The experimental results of the cycling performance are shown in Fig. S21.† Although the charge–discharge cut-off voltage has dropped to 4.3 V, the capacity of E-TMP still drops rapidly after 8 cycles. Thus, the quick fading of Gr‖NMC811 full cells with E-TMP as the electrolyte may be mainly due to the deterioration of the Gr anode, rather than the cathode side.
Moreover, the evolutions of the CEI on the cathodes in different electrolytes were characterized by HR-TEM to understand the effect of electrolytes on cathode stability. After 50 cycles at 4.5 V, the thicknesses of CEIs on the NMC811 cathodes are 12–18 nm for E-conc and 16–29 nm for E-TMP, respectively (Fig. 5a and b). In contrast, the CEI on the NMC811 cathodes in E-PFPN is much thinner (2–5 nm) and more uniform (Fig. 5c). The XPS analysis of NMC811 cathodes was also conducted to reveal the reasons behind the differences in cycling stability. As shown in C 1s spectra (Fig. 5d), the intensities of C–O components in E-conc and E-PFPN are lower compared to that in E-TMP, implying less organic species from solvent decomposition. The concentrated solvation structures in E-conc and E-PFPN with low proportions of free solvent molecules suppressed the solvent decompositions on the NMC811 cathodes. Major differences between different electrolytes could be observed in the F 1s spectra. In E-conc and E-TMP, the main F 1s signal comes from C–F, which may come from the PVDF binder and the decomposition of FEC solvent. In addition, there is a high proportion of the SO2–F signal from the incomplete decomposition of FSI− in the CEI of E-TMP. The decomposition of TMP and the deposition of P–O species on the cathode were not desirable for the cathode protection at high voltages. In comparison, the CEI formed in E-PFPN is enriched with LiF (Fig. 5e). A large amount of LiF species in the CEI indicates that the PFPN diluent has a critical role in the CEI formation process. The P–F and PN–P species in E-PFPN (Fig. 5e, f and S22†) also suggested that PFPN was decomposed on the cathode surface. The electrolyte effect on the cathode stability could be further verified by the SEM images of the cycled cathodes shown in Fig. S23.† The NMC811 cathode in the E-TMP shows severe cracks in bulk particles after cycling (yellow rectangles in the SEM images, Fig. S24c†). Nevertheless, the cathode particles in E-PFPN remain intact (Fig. S24d†). The results indicate that the uniform protective layer enriched in LiF in E-PFPN would effectively passivate the highly active cathode surface at high voltages during long-term cycling (Fig. 6).
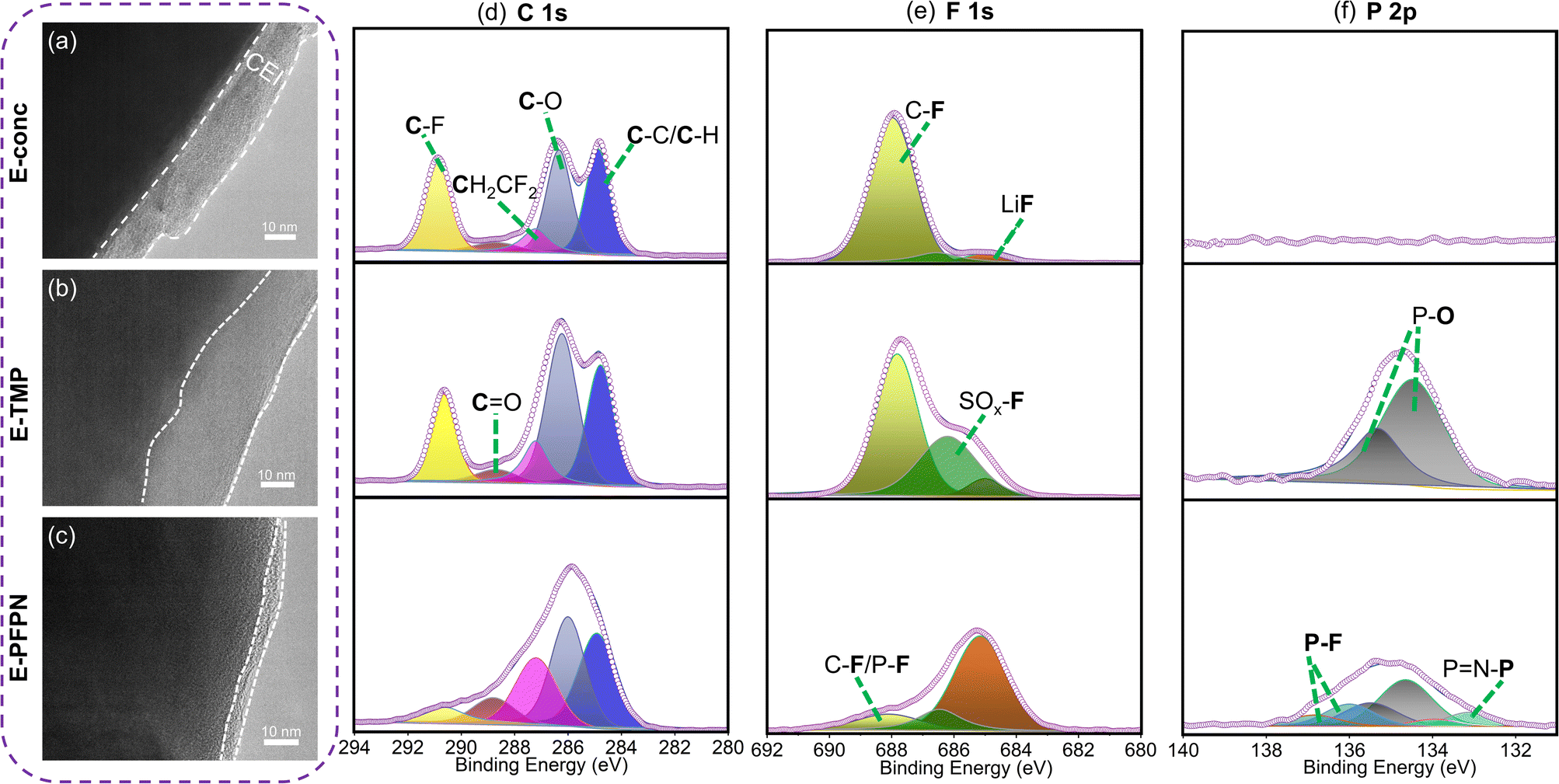 | ||
| Fig. 5 HR-TEM images of NMC811 cathodes after 50 cycles in Gr‖NMC811 cells using different electrolytes: (a) E-conc; (b) E-TMP; (c) E-PFPN. XPS spectra of NMC811 cathodes after 50 cycles in different electrolytes: (d) C 1s, (e) F 1s, and (f) P 2p. | ||
 | ||
| Fig. 6 Schematic of the relationship between the solvation structure, SEI, and CEI composition in E-PFPN electrolyte. | ||
3. Conclusions
In summary, with the combination of the carbonate–ether mixture solvent and the fluorophosphazene-based flame-retarding diluent, we successfully designed a nonflammable LHCE for LIBs with a Gr anode and high-voltage cathode. E-PFPN can solve the dilemma of flame retardants and their poor compatibility with Gr anodes and Ni-rich cathodes. Specifically, the Gr‖NMC811 cells employing E-PFPN exhibit outstanding cycling performance with a capacity retention of 82.0% after 1000 cycles at 4.5 V and 89.8% after 300 cycles at 4.6 V compared to those with E-carbonate, E-conc, and E-TMP. Moreover, it also can achieve superior cycling stability at a high temperature (50 °C) (82% capacity retention after 250 cycles at 4.5 V). E-PFPN enables the formation of thin, robust, and conductive interphases enriched with inorganic species on both electrodes, thus ensuring long-term cycle stability of high-voltage LIBs. Our work demonstrates the great potential of nonflammable functional electrolyte design for long-life, high-safety, and high-energy-density LIBs.4. Experimental
4.1 Electrolyte and electrode preparation
The LiFSI was kindly provided by Nippon Shokubai (Japan) and dried under vacuum at 100 °C for 24 hours before use. TMP, TEP, DME, and EC solvents were purchased from Aladdin Co., Ltd. PFPN was purchased from Shanghai Bide Pharmaceutical Technology Co., Ltd, and its purity was higher than 98%. All the salts are of battery grade and dried at 100 °C in a vacuum drying oven overnight. All the solvents were dried with molecular sieves for seven days before use and used without further purification. The carbonate of 1 M LiPF6 EC/DEC/EMC (1:1:1 by vol) was purchased from DodoChem Co. The electrolytes with different formulations shown in Table S1† were prepared in a glovebox filled with high purity argon (H2O < 0.1 ppm and O2 < 0.1 ppm). The Gr anode (2.10 mA h cm−2, active material loading: 5.80 mg cm−2) was received from Guangdong Canrd New Energy Technology Co., Ltd. Single crystal NMC811 powder was purchased from Shenzhen Kejing materials technology co., Ltd. The NMC811 cathode disks (a diameter of 12 mm) were prepared by coating a slurry of mixing the NMC811 powder, Super P (Aladdin), and 10 wt% polyvinylidene fluoride (PVDF, Aladdin) in N-methyl-2-pyrrolidone (NMP, Aladdin) (80:10:10 by wt%) on Al foil (NMC811, 1.77 mA h cm−2 at 4.5 V, active material loading: 8.35 mg cm−2). All the electrode disks were dried under vacuum at 110 °C for over 24 hours and kept in the glove box.
4.2 Characterization studies
The contact angles of various electrolytes on a Celgard 2500 separator were measured with a contact angle meter SL200B (Solon Tech. Co., Ltd). The viscosity of various electrolytes was measured with a DVNext rheometer (Brookfield. Co., Ltd) at 25 °C. After 50 charge/discharge cycles, the cells were disassembled in the glove box with a disassembly tool to obtain the Gr anode and NMC811 cathode. All the recycled Gr anodes and NMC811 cathodes were rinsed with pure DME 3 times to remove residue and then dried in small bins of the glove box for 2 hours. The recycled Gr anode and NMC811 cathode's surface chemistry were investigated by using an X-ray photoelectron spectrometer (XPS) on a physical electronic Quantera scanning X-ray microprobe (Al Kα X-ray source). A Bruker D8 Venture X-ray diffractometer (XRD) was used to perform the analysis of Gr anode and cathode materials. SEM images were obtained on a Gemini SEM 450 at an accelerating voltage of 5 kV and a current of 86 pA. The self-designed hermetically sealed container filled with argon was used in the glove box to transfer samples of the SEM and XPS instruments to eliminate the effects of ambient oxygen and moisture. The Gr anode and NMC811 cathode will be scraped off and ultrasonically dispersed in N,N-dimethylacetamide to obtain the transmission electron microscope (TEM) images. Then, the above dispersion is dropped on the copper TEM grids (200 mesh, Tedpella, Inc.) in the glove box. A 300 kV FEI Talos F200X transmission electron microscope was used to obtain the high-resolution TEM images. Raman spectroscopy (LABRAM, HR) with 532 nm laser excitation was used to estimate the solvation structure of different electrolytes. For the flammability test, glass microfiber filters soaked with the electrolytes were directly exposed to a torch flame. A specific interval close to the torch flame was taken several times to determine whether the electrolyte can be ignited. The flash points of electrolytes were determined with a flash point tester MINIFLASH FLP (Grabner Instruments) by using a modified continuously closed cup. The linear sweep voltammetry (LSV), cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS) experiments were conducted using a Biologic potentiostat (VMP-3). DSC was carried out by adding 5 mg of electrolyte and 3 mg of dry NMC811 electrode material into tightly sealed high-pressure crucibles at a rate of 5 °C min−1 from room temperature to 350 °C.4.3 Electrochemical measurements
Gr‖Li half-cells and full cells of NMC811‖Gr were assembled in a standard 2032-type coin cell in the glove box. All cells use the Celgard 2500 separator sandwiched between the Gr anode and the NMC811 cathode. 75 μL electrolyte was added in all coin cells to thoroughly wet the separator and electrodes. Gr‖Li half-cells were tested at 1/10C with a cutoff voltage range of 0.01–1.2 V, where the 1C rate was 372 mA h g−1 based on the active material weight of the Gr anode. NMC811‖Gr full-cells were tested at 1/3C with a cutoff voltage range of 2.8–4.5 V and 2.8–4.6 V, respectively, where the 1C rate was 200 mA h g−1 based on the active material weight of the NMC811 cathode. NMC811‖Gr full-cells were also tested at 1/3C with a cutoff voltage range of 2.8–4.5 V at 50 °C. The discharge test with different rates of NMC811‖Gr full cells was measured. To avoid corrosion of the stainless steel electrode shell by electrolyte at a high voltage, an Al-coated cathode shell was used. An additional Al foil with a diameter of 19 mm was added between the NMC811 cathode and Al-coated stainless steel shell. The slurry containing only Super P and PVDF (1:1 wt%) was coated on Cu foil for linear sweep voltammetry (LSV) of the studied electrolytes, with a potential range of 2.0–5.5 V (vs. Li/Li+) and a scan rate of 1.0 mV s−1. The cyclic voltammetry (CV) profiles of Gr‖Li half-cells with studied electrolytes were obtained at 0–2.0 V, and the scan rate was 0.1 mV s−1. The EIS of Gr‖NMC811 cells using the studied electrolytes after two formation cycles was measured with a frequency ranging from 1 M to 0.1 Hz, and the input signal amplitude is 5 mV.
Data availability
Data will be made available on request.Author contributions
X. D. Ren designed and guided this experiment. L. Chen did the electrochemical experiments and wrote the original draft. Q. S. Nian, Y. C. Li, and X. Luo conducted the material characterizations. D. G. Ruan, J. J. Fan, S. Q. Chen, and L. J. Tan helped analyze the XPS and Raman data. Z. Z. Cui, Y. F. Chen, and C. H. Li helped analyze the electrochemical results. X. D. Ren supervised the whole work and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 22179124 and 21905265), Anhui Provincial Natural Science Foundation (2021 Anhui Energy Internet Joint Fund Project, 2108085UD04) and the Fundamental Research Funds for the Central Universities (WK3430000007).Notes and references
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2018, 3, 22–29 CrossRef CAS.
- X.-G. Yang, T. Liu and C.-Y. Wang, Nat. Energy, 2021, 6, 176–185 CrossRef CAS.
- T. Liu, L. Lin, X. Bi, L. Tian, K. Yang, J. Liu, M. Li, Z. Chen, J. Lu, K. Amine, K. Xu and F. Pan, Nat. Nanotechnol., 2019, 14, 50–56 CrossRef CAS PubMed.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- X. Feng, D. Ren, X. He and M. Ouyang, Joule, 2020, 4, 743–770 CrossRef CAS.
- H. Jia, Y. Xu, X. Zhang, S. D. Burton, P. Gao, B. E. Matthews, M. H. Engelhard, K. S. Han, L. Zhong, C. Wang and W. Xu, Angew. Chem., Int. Ed., 2021, 60, 12999–13006 CrossRef CAS PubMed.
- K. Takada, Y. Yamada and A. Yamada, ACS Appl. Mater. Interfaces, 2019, 11, 35770–35776 CrossRef CAS PubMed.
- Z. Zeng, X. Jiang, B. Wu, L. Xiao, X. Ai, H. Yang and Y. Cao, Electrochim. Acta, 2014, 129, 300–304 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- X. Wang, W. He, H. Xue, D. Zhang, J. Wang, L. Wang and J. Li, Sustainable Energy Fuels, 2022, 6, 1281–1288 RSC.
- C. Zhang, S. Gu, D. Zhang, J. Ma, H. Zheng, M. Zheng, R. Lv, K. Yu, J. Wu, X. Wang, Q.-H. Yang, F. Kang and W. Lv, Energy Storage Mater., 2022, 52, 355–364 CrossRef.
- J. Xia, R. Petibon, A. Xiao, W. M. Lamanna and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A1637–A1645 CrossRef CAS.
- L. Xia, Y. Xia and Z. Liu, J. Power Sources, 2015, 278, 190–196 CrossRef CAS.
- X. Li, W. Li, L. Chen, Y. Lu, Y. Su, L. Bao, J. Wang, R. Chen, S. Chen and F. Wu, J. Power Sources, 2018, 378, 707–716 CrossRef CAS.
- H. Li, Z. Wen, D. Wu, W. Ji, Z. He, F. Wang, Y. Yang, P. Zhang and J. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 57142–57152 CrossRef CAS PubMed.
- L. Zhang, F. Min, Y. Luo, G. Dang, H. Gu, Q. Dong, M. Zhang, L. Sheng, Y. Shen, L. Chen and J. Xie, Nano Energy, 2022, 96, 107122 CrossRef CAS.
- N. von Aspern, G. V. Röschenthaler, M. Winter and I. Cekic-Laskovic, Angew. Chem., Int. Ed., 2019, 58, 15978–16000 CrossRef CAS PubMed.
- B. T. Young, D. R. Heskett, C. C. Nguyen, M. Nie, J. C. Woicik and B. L. Lucht, ACS Appl. Mater. Interfaces, 2015, 7, 20004–20011 CrossRef CAS PubMed.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- M. S. Kodaimati, S. Lian, G. C. Schatz and E. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8290–8295 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- X. Ren, P. Gao, L. Zou, S. Jiao, X. Cao, X. Zhang, H. Jia, M. H. Engelhard, B. E. Matthews, H. Wu, H. Lee, C. Niu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28603–28613 CrossRef CAS PubMed.
- Y. Wu, D. Ren, X. Liu, G.-L. Xu, X. Feng, Y. Zheng, Y. Li, M. Yang, Y. Peng, X. Han, L. Wang, Z. Chen, Y. Ren, L. Lu, X. He, J. Chen, K. Amine and M. Ouyang, Adv. Energy Mater., 2021, 11, 2102299 CrossRef CAS.
- H. Liu, A. J. Naylor, A. S. Menon, W. R. Brant, K. Edström and R. Younesi, Adv. Mater. Interfaces, 2020, 7, 2000277 CrossRef CAS.
- K. Beltrop, S. Klein, R. Nölle, A. Wilken, J. J. Lee, T. K. J. Köster, J. Reiter, L. Tao, C. Liang, M. Winter, X. Qi and T. Placke, Chem. Mater., 2018, 30, 2726–2741 CrossRef CAS.
- J. Vidal Laveda, J. E. Low, F. Pagani, E. Stilp, S. Dilger, V. Baran, M. Heere and C. Battaglia, ACS Appl. Energy Mater., 2019, 2, 7036–7044 CrossRef CAS.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- H. Jia, Y. Xu, S. D. Burton, P. Gao, X. Zhang, B. E. Matthews, M. H. Engelhard, L. Zhong, M. E. Bowden, B. Xiao, K. S. Han, C. Wang and W. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 54893–54903 CrossRef CAS PubMed.
- X. Cao, Y. Xu, L. Zhang, M. H. Engelhard, L. Zhong, X. Ren, H. Jia, B. Liu, C. Niu, B. E. Matthews, H. Wu, B. W. Arey, C. Wang, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 2529–2534 CrossRef CAS.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia, X. Ren, P. Gao, Z. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Wang, C. Wang, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2000368 CrossRef CAS.
- H. Nakagawa, M. Ochida, Y. Domi, T. Doi, S. Tsubouchi, T. Yamanaka, T. Abe and Z. Ogumi, J. Power Sources, 2012, 212, 148–153 CrossRef CAS.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia, X. Ren, P. Gao, Z. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Wang, C. Wang, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2070098 CrossRef CAS.
- N. Piao, X. Ji, H. Xu, X. Fan, L. Chen, S. Liu, M. N. Garaga, S. G. Greenbaum, L. Wang, C. Wang and X. He, Adv. Energy Mater., 2020, 10, 1903568 CrossRef CAS.
- H. Sun and Q. Wei, ECS Trans., 2013, 50, 349–354 CrossRef.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- M. He, X. Li, N. G. Holmes, R. Li, J. Wang, G. Yin, P. Zuo and X. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 38296–38304 CrossRef CAS PubMed.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- X. L. Yao, S. Xie, C. H. Chen, Q. S. Wang, J. H. Sun, Y. L. Li and S. X. Lu, J. Power Sources, 2005, 144, 170–175 CrossRef CAS.
- L.-L. Jiang, C. Yan, Y.-X. Yao, W. Cai, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3402–3406 CrossRef CAS PubMed.
- Y.-X. Yao, X. Chen, C. Yan, X.-Q. Zhang, W.-L. Cai, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed.
- S.-J. Tan, Y.-F. Tian, Y. Zhao, X.-X. Feng, J. Zhang, C.-H. Zhang, M. Fan, J.-C. Guo, Y.-X. Yin, F. Wang, S. Xin and Y.-G. Guo, J. Am. Chem. Soc., 2022, 144, 18240–18245 CrossRef CAS PubMed.
- L. O. Valøen and J. N. Reimers, J. Electrochem. Soc., 2005, 152, A882 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05723a |
| This journal is © The Royal Society of Chemistry 2023 |