
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chelating chloride using binuclear lanthanide complexes in water†‡
Carlson
Alexander
 a,
James A.
Thom
a,
Alan M.
Kenwright
b,
Kirsten E.
Christensen
a,
Thomas Just
Sørensen
ac and
Stephen
Faulkner
*a
a,
James A.
Thom
a,
Alan M.
Kenwright
b,
Kirsten E.
Christensen
a,
Thomas Just
Sørensen
ac and
Stephen
Faulkner
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stephen.faulkner@chem.ox.ac.uk
bDepartment of Chemistry, University of Durham, South Road, Durham DH1 3LE, UK
cNano-Science Centre and Department of Chemistry, University of Copenhagen, 2100 København Ø, Denmark
First published on 27th December 2022
Abstract
Halide recognition by supramolecular receptors and coordination complexes in water is a long-standing challenge. In this work, we report chloride binding in water and in competing media by pre-organised binuclear kinetically inert lanthanide complexes, bridged by flexible –(CH2)2– and –(CH2)3– spacers, forming [Ln2(DO3A)2C-2] and [Ln2(DO3A)2C-3], respectively. These hydrophilic, neutral lanthanide coordination complexes are shown to bind chloride with apparent association constants of up to 105 M−1 in water and in buffered systems. Hydroxide bridging was observed in these complexes at basic pH, which was proven to be overcome by chloride. Thus, these lanthanide complexes show promise towards chloride recognition in biology and beyond. The results described here have clearly identified a new area of anion coordination chemistry that is ripe for detailed exploration.
1 Introduction
Anions, including DNA, halides, carbonates, phosphates, sulfates, and nitrates are everywhere in the natural world, and play a key role in defining the biology and the environment.1,2 Despite this, the discussion of coordination chemistry was framed from the perspective of the metal ion for many years; it is only more recently that anions have been given the attention they deserve.3,4 Anion recognition relies on bonding of the guest anions to a host which exploits coulombic or supramolecular forces in general. In solution, solvent molecules can also bind to both guest and host, meaning that anion binding can be hard to achieve in water.1,5 While a host can almost invariably bind to a variety of guests, selectivity for a single type of anion is difficult to achieve.1,3–5 Selective anion recognition requires careful design to match host and guest, so some of these supramolecular systems can be challenging to synthesise and often involve intricate architectures of considerable structural complexity, yet minimal practical applicability.4aNature provides inspiration towards anion binding: protein-based interactions include bacterial sulfate binding protein and phosphatase enzymes, which selectively recognise sulfate and phosphate under physiological conditions via a network of hydrogen bonds from neutral donors. In these proteins, anion affinity is increased by a hydrophobic environment providing complementarity of charge and a shape that minimises the receptor desolvation energy.6 These interactions have inspired biomimetic receptors that integrate additional binding sites within the synthetic host, creating a structured binding pocket with high geometric complementarity.1,3–5 A range of interactions such as the macrocyclic effect,1,2 and conventional ion-pairing have been exploited in anion recognition;5a the latter based on electrostatics,1 and supported by hydrogen bonding,2 halogen bonding,7 anion–π interactions,8 and coordinate bond using Lewis acids such as metals.3,4
Here, we focus on chloride binding. Chloride is the most abundant physiological anion9 and plays important roles in neuronal growth (104–115 mM in extracellular fluid),10 and development of the central nervous system (115–130 mM in cerebrospinal fluid) through the functions of chloride channels. The intracellular chloride concentration (up to 70 mM in eukaryotic cell types) is crucial in moderating neuronal excitability and neurotransmission.11 Tracking chloride in living systems is vital if we are to understand diseases related to chloride transport defects such as cystic fibrosis and epilepsy.9,11,12 Apart from its importance as the counterion of life,9,12 sequestering salt from sea water (559 mM of chloride in surface sea water)13 for clean drinking water is an ongoing challenge.14 However, developing suitably selective receptors for chloride in water continues to be a challenge.1,4,5,15
The large enthalpy and free energy of hydration of halides (Table 1) make chloride recognition challenging in water.16d Halide anions exhibit different levels of hydration in different solvents as reflected by the Hofmeister series of anion hydrophobicity.17 Across the series,§ the decrease in charge density results in increasing lipophilicity and weaker hydration.7 However, the solvation of the host must also be considered in designing an effective receptor for anion recognition.1,3–5
| Anion | Radius (Å) | n hyd | −ΔG°hyd (kJ mol−1) | −ΔH°hyd (kJ mol−1) |
|---|---|---|---|---|
| HO− | 1.33 | 2.7 | 430 | 520 |
| F− | 1.33 | 2.7 | 465 | 510 |
| Cl− | 1.81 | 2 | 340 | 367 |
| Br− | 1.96 | 1.8 | 315 | 336 |
| I− | 2.2 | 1.6 | 275 | 291 |
The extent of solvation of the host, guest, and supramolecular assemblies depends on the solvent. Polar protic solvents such as water and methanol strongly solvate charged species, forming hydrogen bonds with both the anion and the host molecule.1,5 By contrast, in less polar aprotic solvents such as dichloromethane, the anion is weakly solvated and neutral receptors are able to function.5,17 Strong electrostatic or metal–ligand interactions offer the potential to overcome the anion hydration energy and allow binding in biological media.3,4
We previously reported a lanthanide tethered rotaxane that binds to chloride in apolar media (Fig. 1b). In this, the first binding of chloride involves chelation to the metal, a second binding event occurs by encapsulation within the rotaxane.18 Gale, Davis et al. have reported cholapods made of a steroidal framework containing squaramide groups in axial positions which bind to chloride and other anions of tetraethylammonium salts with ∼1014 M−1 affinity in wet chloroform, employing a variation of Cram's extraction procedure.19 Flood et al. have reported a triazolo cage which binds to chloride with ∼1017 M−1 affinity in dichloromethane, which is very effective in sequestering chloride from water and has been shown to prevent brine-accelerated corrosion when coated onto mild steel (Fig. 1a).20 Beer et al. have designed rotaxane hosts based on iodotriazole halogen bonding and amide hydrogen bonding with the halogens, but it recognises iodide (Ka = 2200 M−1) better than chloride (Ka = 55 M−1) and bromide in water.21 Other chloride binding supramolecular agents have been extensively reviewed elsewhere.1,5,15
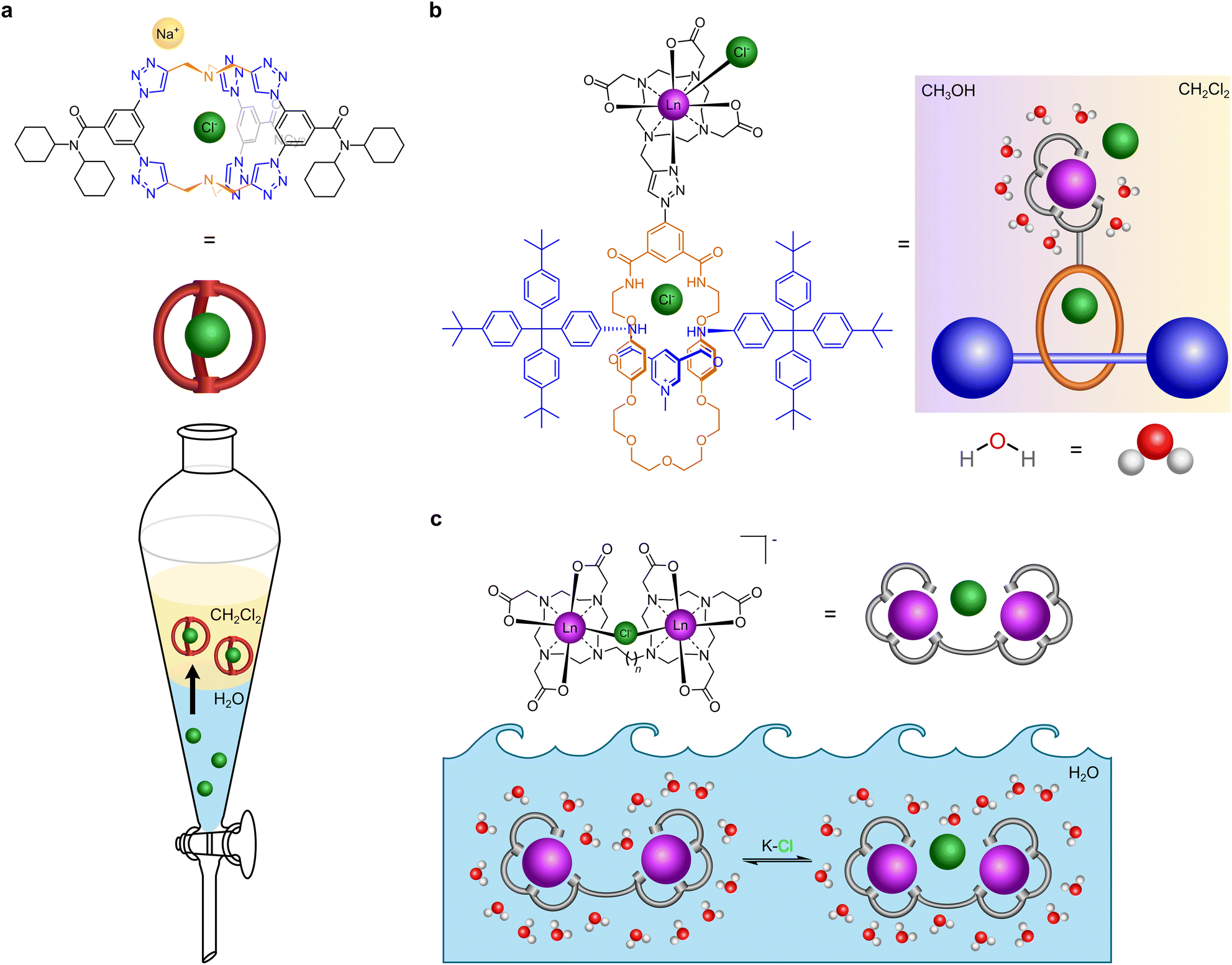 | ||
| Fig. 1 Cartoon illustration of receptors that recognise chloride. (a) Receptor that extracts chloride from water to dichloromethane (adapted from ref. 20). (b) Receptor that binds to chloride in mixed media (dichloromethane and methanol) (adapted from ref. 18). (c) Our receptor that binds to chloride in water. | ||
Lanthanide complexes are an attractive choice for developing anion receptors due to their ability to report anion binding by MRI, luminescence, and optical imaging.22 The luminescence applications use lanthanide complexes as luminescent tags, exploiting stable complexes in which their luminescence signal is enhanced via the antenna principle where chromophores sensitise lanthanide luminescence and overcome the inherently low molar absorption coefficients associated with f–f transitions.22,23 Furthermore, the necessity for kinetic stability in clinical use ensures that macrocyclic ligands related to DOTA are well understood in competitive media with high anion concentrations.4,22,23 In such systems, anion chelation is achievable on the metal site without the dissociation of the lanthanide complex.4
Anion recognition by lanthanide complexes can reflect either collisional quenching of excited states by anions,24 or anion binding.4 While luminescence can be modulated both by collision and anion binding, the latter generates a ‘turn-off’ event in MRI since the functioning of MRI contrast agents requires the presence of water molecules in the inner-coordination sphere which exchange with the bulk water.24 Thus, isostructural MRI and luminescent probes can be constructed with suitable lanthanide ions for targeted anion binding thereby allowing one design platform to exploit bio-imaging combining paramagnetism and luminescence, an exclusive feature offered by the lanthanides.4,22 Halides are strong Lewis bases which coordinate to metals that are hard Lewis acids.25 Using this HSAB approach, several mononuclear Ln(III) complexes comprising cryptands,26 and cyclen derived C2 (ref. 27) and C4 symmetric28,29 ligands have been reported to chelate fluoride at the metal centre, but none have been reported to bind chloride in water.
Binuclear lanthanide(III) complexes of two heptadentate DO3A-based binding domains tethered by aryl spacers were studied for binding organic dicarboxylate anions at physiological pH;30 sensing biologically relevant anions such as phosphate, methylphosphate, double-stranded DNA and a DNA hairpin loop;31 and as a pH sensor.32 We have explored this binuclear approach by incorporating kinetically inert charge neutral lanthanide DO3A centres bridged by different m-xylyl scaffolds to achieve reversible binding of dinicotinate and isophthalate guests with high affinity at physiological conditions, but this requires methanol to improve the solvation of these guests.33
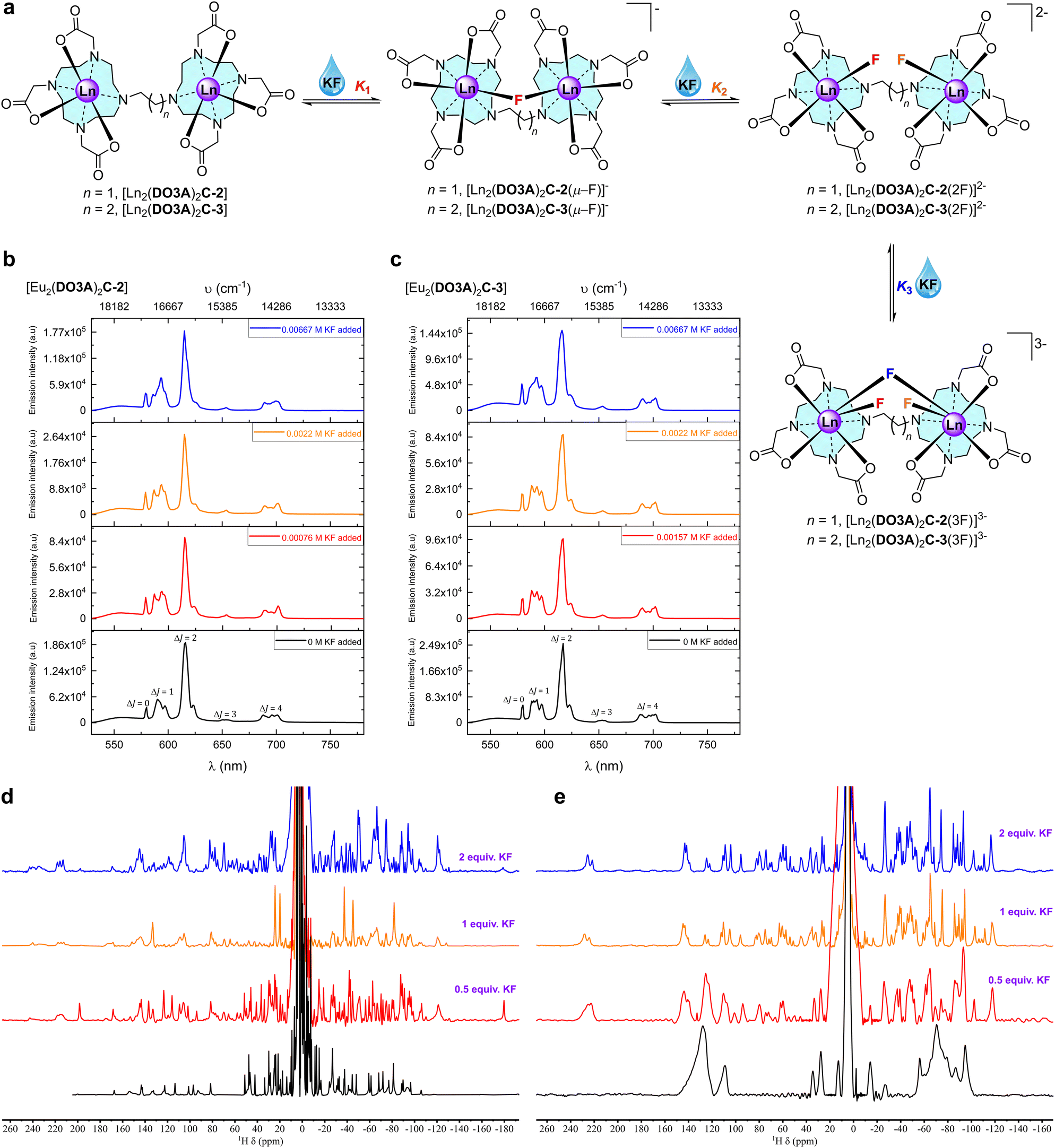
In this work, we report how binuclear complexes can be tailored to bind chloride ions in water under conditions where mononuclear complexes4,26–30 and free lanthanide ions34 do not interact with chloride. We hypothesised that coordinatively unsaturated neutral binuclear lanthanide(III) complexes of DO3A ligands bridged by ethane and propane linkers ([Ln2(DO3A)2C-2] and [Ln2(DO3A)2C-3]) would create a pre-organised pocket between the metal centres capable of hosting halides coordinated to the lanthanide metal centres. Furthermore, having a short and flexible spacer between the coordinatively unsaturated hard Lewis acids, which act as acceptors would increase the steric strain thereby limiting their conformational freedom,35 where subsequent halide binding would lead to a smaller entropy loss in the host. Both binuclear complexes respond to halides under physiological conditions and the binding can be followed by luminescence and NMR spectroscopy. A neutral mononuclear lanthanide propargyl DO3A complex [Ln(pDO3A)] was23 used to compare the relative effectiveness of mononuclear complexes.
2 Results and discussion
2.1 Synthesis and characterisation
Synthesis of the proposed complexes is shown in Scheme 1. Detailed synthetic protocol, purification, and characterisation is reported in the ESI.‡ Cyclen was tri-N-alkylated using t-butyl bromoacetate to form the triester, DO3A(t-BuO)3. Subsequent alkylation with 1,2-dibromoethane and 1,3-dibromopropane yielded the ethane and propane bridged bis-macrocycles, (DO3A(t-BuO)3)2C-2 or (DO3A(t-BuO)3)2C-3. Following purification and deprotection with trifluoroacetic acid, the pro-ligands (DO3A)2C-2 and (DO3A)2C-3 were obtained. These were reacted with the appropriate lanthanide triflate salt in methanolic solution under reflux and worked-up to afford the complexes [Ln2(DO3A)2C-2] and [Ln2(DO3A)2C-3]. Single crystal X-ray structures of (DO3A(t-BuO)3)2C-2 (Fig. S122; Table S9‡), pDO3A(t-BuO)3 (Fig. S123; Table S10‡), [Ln2(DO3A)2C-3] and [Ln(pDO3A)] (Ln = Eu(III) and Yb(III)) were obtained. The binuclear complexes crystallised as a cluster containing 12 metal centres via μ-oxo bridges from the carbonyl oxygens, maintaining the connectivity and neutrality of the complexes (Fig. S124; Table S11‡). A crystal structure of [Lu(pDO3A)] has been reported as a dimer, linked by a carbonate.18 In our case, [Ln(pDO3A)] (Ln = Eu(III) and Yb(III)) crystallised without any anion bound to the metal centre (Fig. S126; Table S13‡).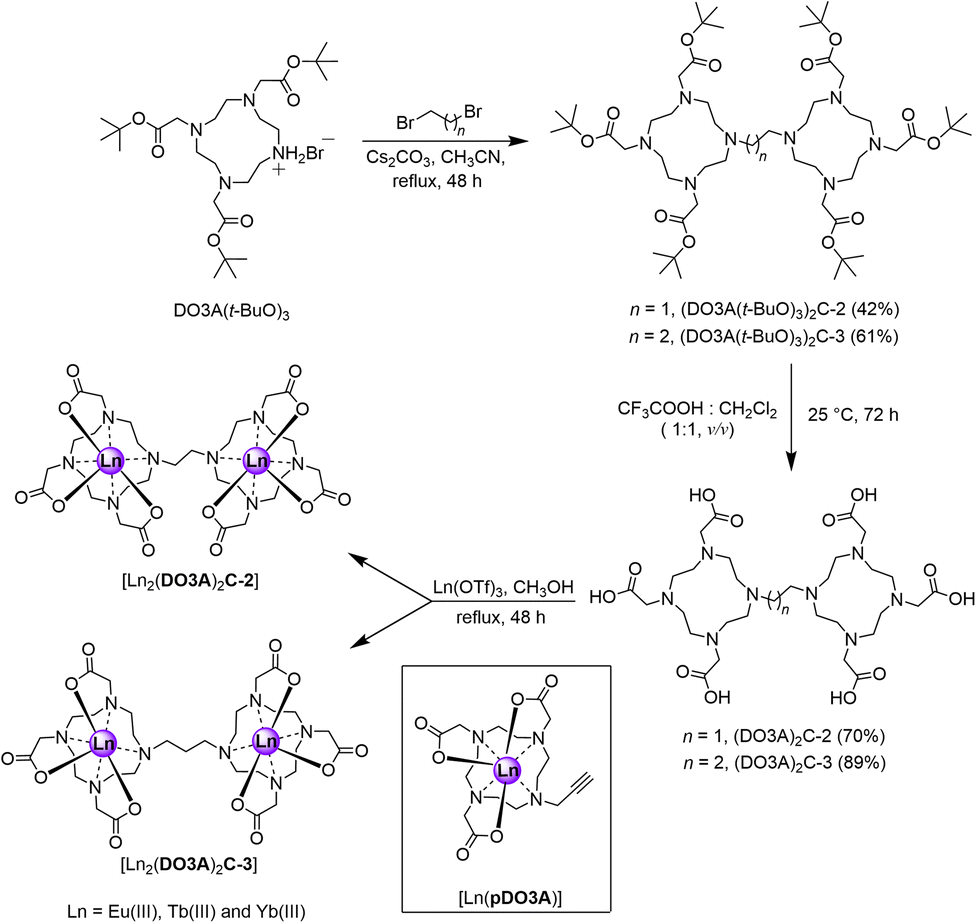 | ||
| Scheme 1 Synthetic scheme of proposed binuclear complexes. The mononuclear complex used as a model system is shown inside the black box. | ||
These kinetically robust binuclear complexes23 exhibit variations in behaviour with pH. At neutral pH, the 1H NMR spectra of the paramagnetic Eu(III) and Yb(III) complexes consist of broad lines; however, at pD 10.12, the lines are sharp and well defined (Fig. S28, S31, S34, and S36‡). This observation can be explained by the rapid inter-conversion between diastereomeric forms of the compound, in which the coordination environment at the lanthanide centre varies between Square Antiprism (SAP) and Twisted Square Antiprism (TSAP) geometries. At high pH, interchange between these species was found to be slow on the NMR timescale, resulting in sharp spectral lines. Furthermore, luminescence spectra of [Eu2(DO3A)2C-2] and [Eu2(DO3A)2C-3] at pH 4 and 7 showed no significant change (Fig. S62–S65‡), but at pH 9.9, the form and shape of the emission spectra was changed, which implies hydroxide binding to the metal centres. A decrease in the hydration number of the complexes can be inferred from luminescence lifetime measurements at increasing pH (Fig. S112 and S115; Tables S1 and S4‡). These results complement the NMR observations that chelation to hydroxide is observed at basic pH.
2.2 Effect of chloride on the mono- and binuclear Ln(III) complexes
No fluoride chelating Ln(III) complexes have been observed to bind chloride at the metal centre.26–29 Since the binuclear complexes in this work are neutral species, a neutral monometallic Ln(III) complex [Ln(pDO3A)] was used as a model system to study halide binding. Steady-state luminescence titration of [Eu(pDO3A)] with potassium chloride (KCl) salt in deionised water did not result in any binding event (Fig. S108 and S109‡).Chloride binding by the binuclear Eu(III) complexes were then explored. Steady-state luminescence titrations were performed in deionised water, phosphate buffer at pH 7.4, and in CHES buffer at pH 9.9. In the case of deionised water and in phosphate buffer, the emission intensity decreased with increasing KCl addition, but the opposite was the case in CHES buffer (Fig. 2, S74–S78 and S94–S99‡). It should be noted that chloride can act as a PeT quencher, reducing the observed lanthanide centred emission intensity.24 However, a single binding event (K1) was observed in all three cases and the binding strength increased as we move into buffered systems (Table 2). These results could be suggestive of competitive binding of phosphate: indeed, the changes to the form of the spectra in phosphate buffer (in comparison with the spectra in water) are strongly suggestive of phosphate binding in line with other mono and binuclear lanthanide complexes derived from DO3A.4,31,37
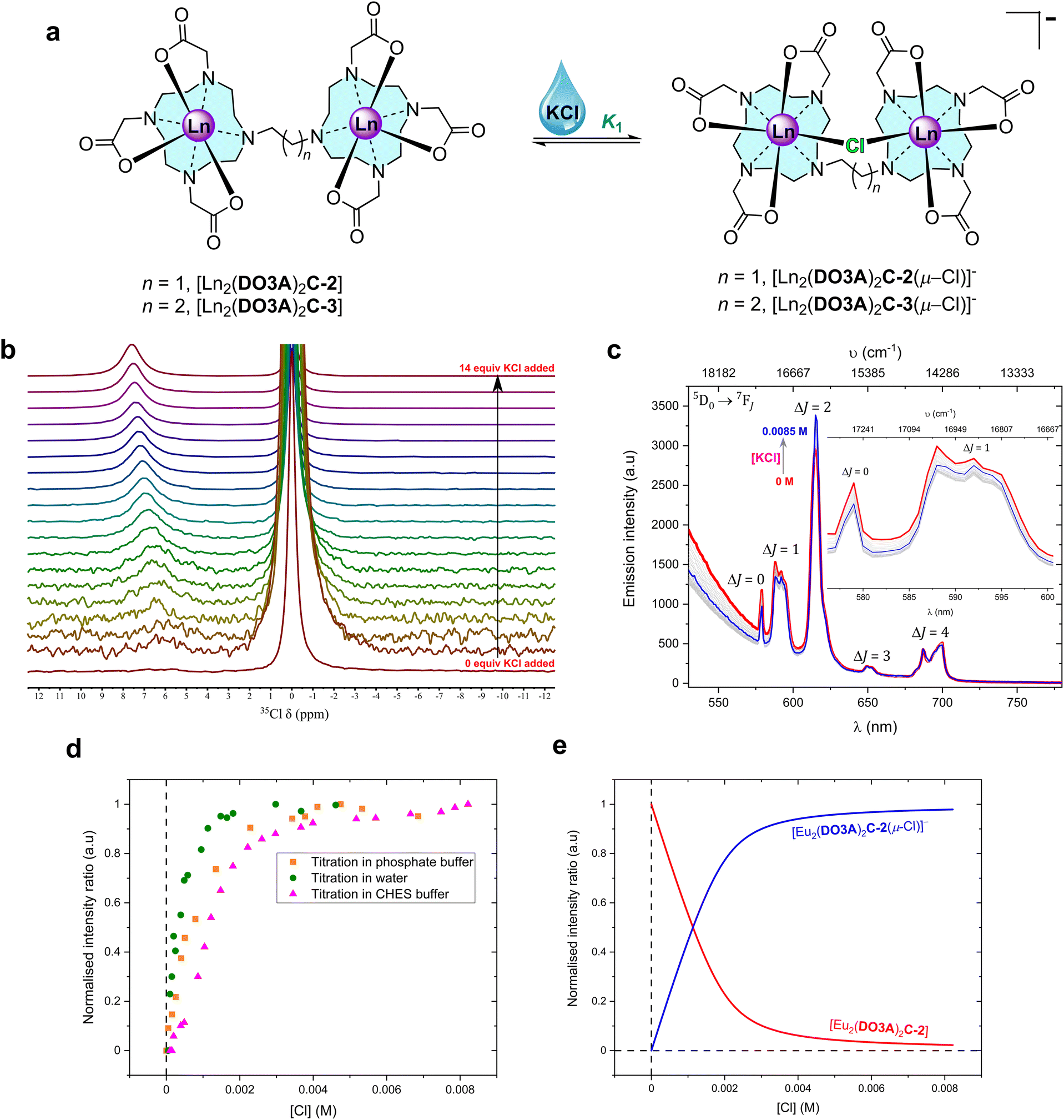 | ||
Fig. 2 Chloride chelation by the binuclear complexes in water. (a) Structural representation of the binding events in water. (b) 49 MHz 35Cl NMR titration spectra of 0.035 M [Tb2(DO3A)2C-3] with increasing additions of KCl (stock concentration, 1.4 M) in D2O at 298 K (Fig. S52 for binding isotherm‡). Each spectrum was recorded with a capillary tube insert containing saturated KCl in D2O (δ = 0 ppm) (non-dilution method used). (c) Steady-state luminescence titration spectra of 1 mM [Eu2(DO3A)2C-2] (λex = 393 nm) against KCl (stock concentration, 0.02 M) in 0.01 M CHES buffer (pH 9.98) at 22 °C; spectrum of neat complex solution ( ), spectra upon the additions of KCl (grey), spectrum upon the final addition of KCl ( ), spectra upon the additions of KCl (grey), spectrum upon the final addition of KCl ( ) (non-dilution method used). The inset expands the ΔJ = 0 and ΔJ = 1 emission bands (Fig. S78 for binding isotherm‡). (d) Normalised emission trend of ΔJ = 2/ΔJ = 1 obtained from steady-state emission titrations for 1 mM [Eu2(DO3A)2C-2] against KCl in deionised water, 0.01 M phosphate buffer (pH 7.4), and 0.01 M CHES buffer (pH 9.9) at 22 °C. (e) Normalised model of speciation obtained from ΔJ = 2/ΔJ = 1 bands from steady-state emission titration spectra of [Eu2(DO3A)2C-2] against KCl in 0.01 M CHES buffer (pH 9.9) at 22 °C. ) (non-dilution method used). The inset expands the ΔJ = 0 and ΔJ = 1 emission bands (Fig. S78 for binding isotherm‡). (d) Normalised emission trend of ΔJ = 2/ΔJ = 1 obtained from steady-state emission titrations for 1 mM [Eu2(DO3A)2C-2] against KCl in deionised water, 0.01 M phosphate buffer (pH 7.4), and 0.01 M CHES buffer (pH 9.9) at 22 °C. (e) Normalised model of speciation obtained from ΔJ = 2/ΔJ = 1 bands from steady-state emission titration spectra of [Eu2(DO3A)2C-2] against KCl in 0.01 M CHES buffer (pH 9.9) at 22 °C. | ||
| Halide | Mediab | Binding constant (K in M−1)c,d | |
|---|---|---|---|
| [Eu2(DO3A)2C-2] | [Eu2(DO3A)2C-2] | ||
| a The binding of halides with the complexes were studied by steady-state luminescence titrations with varying concentration of potassium halides (non-dilution method used) and binding isotherm generated using DYNAFIT®. b All buffers were maintained at 0.01 M in deionised water: PBS = phosphate buffer saline at pH 7.4; PB = phosphate buffer at pH 7.4; CHES = N-cyclohexyl-2-aminoethanesulfonic acid at pH 9.98; Tris–HCl = tris(hydroxymethyl)aminomethane hydrochloride at pH 7.4. c K 1 = first binding event; K2 = second binding event; K3 = third binding event – deduced from binding isotherm generated using DYNAFIT®. d ± is the coefficient of variation in percentage for each binding event obtained from binding isotherm plotted in DYNAFIT® by employing trust-region algorithm in confidence interval at 95% probability level. Confidence intervals for all binding constants are given in the ESI under each binding isotherm. | |||
| Cl− | Water | K 1 = 2800 (±7%) | K 1 = 4550 (±3.9%) |
| PB | K 1 = 9400 (±12.2%) | K 1 = 6640 (±7.3%) | |
| CHES | K 1 = 7460 (±11.2%) |
K
1 = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210 (±7%) 210 (±7%) |
|
|
|||
| F− | Water |
K
1 = 600000 (±39.7%) |
K
1 = 10480 (±5.9%) |
|
K
2 = 144000 (±33.5%) |
K
2 = 10950 (±4%) |
||
| K 3 = 394.7 (±2.3%) | K 3 = 6570 (±3.6%) | ||
|
|||
| Methanol |
K
1 = 54800 (±11.8%) |
K
1 = 179000 (±13.2%) |
|
|
K
2 = 20200 (±10.6%) |
K
2 = 32000 (±13%) |
||
| K 3 = 0.002843 (±0.6%) | K 3 = 728 (±7.5%) | ||
|
|||
| PBS | K 1 = 4030 (±2.9%) | K 1 = 5350 (±6.9%) | |
| K 2 = 18.57 (±1%) | |||
|
|||
| Tris–HCl |
K
1 = 106000 (±38.9%) |
K 1 = 1670 (±14.7%) | |
| K 2 = 219.1 (±0.8%) | K 2 = 116.4 (±1.7%) | ||
|
|||
| CHES |
K
1 = 13600 (±9.9%) |
K 1 = 3840 (±9.6%) | |
| K 2 = 3494 (±2.8%) | |||
NMR studies of chloride binding at lanthanide centres present difficulties due to the interference of the quadrupolar relaxation from the low symmetry 35Cl nucleus with the paramagnetic metal centre resulting in broad 1H and 35Cl NMR signals. However, dipolar 35Cl NMR chemical shifts induced by the addition of chloride to axially symmetric [Ln(DOTA)]− complexes give well-resolved 35Cl NMR spectra that can be interpreted as evidence of interaction with chloride.36 By contrast, when [Tb(pDO3A)] was added to a solution of KCl in deuterium oxide, no new peaks were observed in 35Cl NMR, and the chemical shift of the chloride resonance was unchanged, suggesting that chloride does not bind in our model system (Fig. S45‡).
The 35Cl NMR spectra of [Tb2(DO3A)2C-3] in deuterium oxide display chemical shift upon the addition of KCl (Fig. 2b). This shift can be ascribed to fast exchange between bound and free chloride; this is borne out by the fact that increasing addition of KCl increases the observed paramagnetic shift. Therefore, 35Cl NMR titrations were pursued with [Tb2(DO3A)2C-3] against KCl in D2O. The resulting chemical shifts were fitted to generate a binding isotherm. A strong first binding event followed by a very weak second binding event was observed (Fig. S47, S49, S51 and S52‡). Titrations were performed at 4 different temperatures in D2O in order to understand the thermodynamics of chloride binding to [Tb2(DO3A)2C-3] (Fig. S46–S52‡). The resulting van't Hoff plot suggested this chelation to be exothermic with high negative enthalpy (Fig. S53‡). By comparison with the results reported for [Tb(DOTA)]− where chloride binding is axial and a negative shift results (Fig. S45‡),36 this suggests that chloride ions bind at the equatorial position on the metal centres in [Tb2(DO3A)2C-3] (Fig. 2). Similarly, 35Cl NMR titrations were performed with [Tb2(DO3A)2C-2] against KCl in D2O at 4 different temperatures. Although chlorine bound chemical shifts were observed, the resulting binding isotherms were sigmoidal in shape which was difficult in deducing meaningful binding events. However, they suggest the likely competition between chloride and hydroxide in occupying the binding site between the metal centres in [Tb2(DO3A)2C-2].
After establishing chloride binding by the binuclear Eu(III) and Tb(III) complexes in water, the association was further evaluated by high resolution ESI-Mass spectrometry. The mass spectral peak of a chloride bound to the Eu(III) binuclear complexes in deionised water were found at m/z 1051.1486 and m/z 1065.1640 for C-2 and C-3 complexes, respectively, and confirmed by the calculated isotopic distribution pattern (Fig. S54 and S55‡), thus further supporting the formation of the ternary complexes [Ln2(DO3A)2C-2(μ-Cl)]− and [Ln2(DO3A)2C-3(μ-Cl)]−.
2.3 Exploring chloride binding mode using fluoride
To investigate the mechanism of halide binding in binuclear lanthanide complexes, fluoride binding was explored by titrating the complexes with solutions of potassium fluoride (KF) salt. Fluoride is known to bind strongly to a range of complexes,26–29 though it can be neglected as a potential interferent in biological applications as the concentration of free fluoride is very low (70 μM in surface sea water,13 and 20–210 μM for human consumption as recommended by WHO38). However, we can use the fluoride interaction to further investigate the mode of chloride binding.Steady-state emission titrations revealed that three fluoride ions are successively bound to the binuclear lanthanide complexes as the fluoride concentration was increased in deionised water. These three binding events were also observed in methanol, where the experimental observables at each event were better resolved, presumably due to better solvation of KF, and the lack of competing hydroxides.39 The binding isotherms were fitted to three binding events; all three were needed to generate an acceptable fit (Fig. 3).
 | ||
Fig. 3 (a) Steady-state emission titration spectra of 1 mM [Eu2(DO3A)2C-2] against KF (stock concentration, 0.02 M) in deionised water at 22 °C. Spectrum of neat complex solution ( in bold), spectra upon the additions of KF ( in bold), spectra upon the additions of KF ( to to  ), spectrum upon the final addition of KF ( ), spectrum upon the final addition of KF ( in bold) (non-dilution method used). The inset expands the ΔJ = 0 and ΔJ = 1 emission bands and the asterisks highlight the emission maxima used in quantifying the binding of fluoride to the complex (Fig. S79 for binding isotherm‡). (b) Normalised trend of selected emission wavelengths in ΔJ = 0 and ΔJ = 1 transitions obtained from the titration of [Eu2(DO3A)2C-2] against KF in deionised water at 22 °C used in studying and quantifying binding. (c) Normalised model of speciation obtained from 580 nm emission in the steady-state emission titration spectra of [Eu2(DO3A)2C-2] against KF in deionised water at 22 °C. in bold) (non-dilution method used). The inset expands the ΔJ = 0 and ΔJ = 1 emission bands and the asterisks highlight the emission maxima used in quantifying the binding of fluoride to the complex (Fig. S79 for binding isotherm‡). (b) Normalised trend of selected emission wavelengths in ΔJ = 0 and ΔJ = 1 transitions obtained from the titration of [Eu2(DO3A)2C-2] against KF in deionised water at 22 °C used in studying and quantifying binding. (c) Normalised model of speciation obtained from 580 nm emission in the steady-state emission titration spectra of [Eu2(DO3A)2C-2] against KF in deionised water at 22 °C. | ||
From the luminescence titrations (Fig. 3, S72, S73 and S79‡ for [Eu2(DO3A)2C-2]; Fig. S84, S85, S92, and S93‡ for [Eu2(DO3A)2C-3]), it can be hypothesised that the speciation involves binding of first fluoride (K1) by chelation between the metal centres. Excess addition of KF generates the second binding event (K2) which involves chelation of fluoride to each metal, while the third binding event (K3) forms a bridging fluoride in addition to the fluoride chelated to each metal (Fig. 4).
 | ||
| Fig. 4 Fluoride chelation by the binuclear complexes in aqueous media. (a) Structural representation of binding events in aqueous media. (b) Stacked steady-state luminescence of 1 mM [Eu2(DO3A)2C-2] with KF in methanol at 22 °C. The spectra are arranged as per the binding events deduced from the luminescence titration spectra (Fig. S72‡) and binding isotherm (Fig. S73‡). (c) Stacked steady-state luminescence of 1 mM [Eu2(DO3A)2C-3] with KF in methanol at 22 °C. The spectra are arranged as per the binding events deduced from the luminescence titration spectra (Fig. S92‡) and binding isotherm (Fig. S93‡). (d) 500 MHz stacked paramagnetic 1H NMR spectra of [Yb2(DO3A)2C-2] with increasing KF in deuterium oxide at 298 K. (e) 500 MHz stacked paramagnetic 1H NMR spectra of [Yb2(DO3A)2C-3] with increasing KF in deuterium oxide at 298 K. | ||
When titrations with the binuclear Eu(III) complexes and KF were performed in competing (phosphate buffered saline at pH 7.4), non-competing (Tris–HCl buffer at pH 7.4), and basic (CHES buffer at pH 9.98) media, the binding strength as well as the number of fluoride bound to the binuclear Eu(III) complexes were lowered and no more than two binding events were observed (Fig. S66–S71‡ for [Eu2(DO3A)2C-2] and Fig. S84–S91‡ for [Eu2(DO3A)2C-3]). A single crystal X-ray structure of [Yb2(DO3A)2C-3(2F)]2− was obtained (Fig. 5) which verifies our hypothesis on K2 mode of fluoride binding to the binuclear complexes. In luminescence titrations, the emission spectral change caused by fluoride is so high due to its strong influence on the crystal-filed of the metal centre as a result of its small-size with highly dense charge and strong electrostatic interaction in comparison with chloride.16d,29a–c It is noteworthy from the above observations that although fluoride chelation by the binuclear systems suffers from competing ions in buffers, chloride binding is enhanced amidst competition from other ions (Table 2).
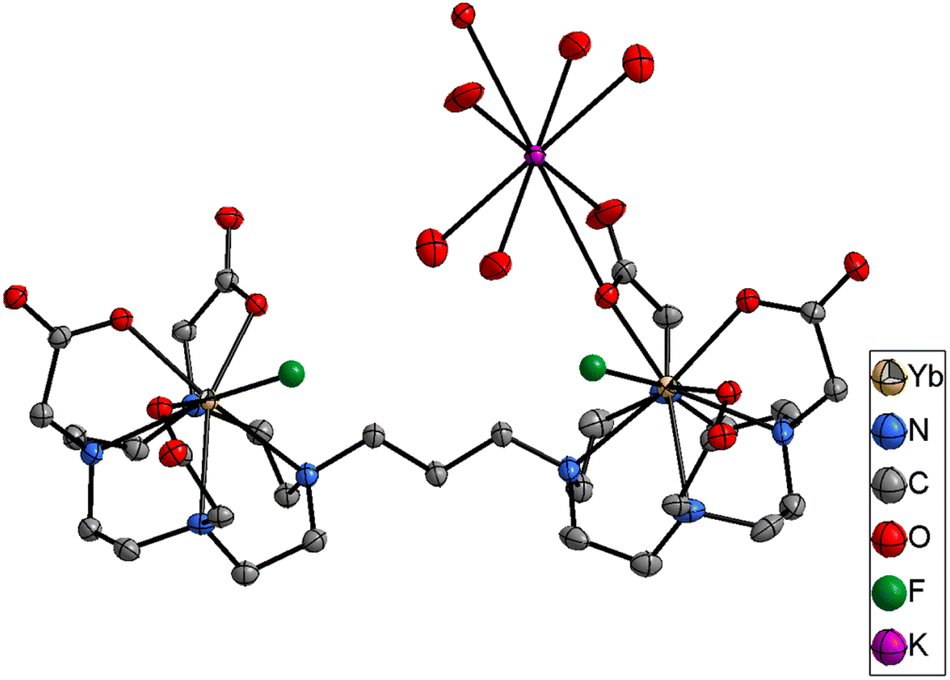 | ||
| Fig. 5 Single crystal X-ray of [Yb2(DO3A)2C-3(2F)]2−. H atoms and water omitted for clarity. Thermal ellipsoid drawn at 30% probability level. | ||
As expected, in luminescence lifetime measurements on the binuclear Eu(III) complexes, upon increasing the concentration of fluoride in deionised water, methanol and their deuterated counterparts, a decrease in the hydration number accounting for the displacement of inner-sphere water by the coordination of fluoride ions was observed (Fig. S110, S111, S113 and S114; Tables S2, S3, S5 and S6‡). Surprisingly, no change in the luminescence lifetime was observed during the addition of chloride with Eu(III) complexes which we hypothesise due to a counteracting interference between Photoinduced electron transfer (PeT) from chloride and intermetallic luminescence quenching.
The 1H NMR spectra of Eu(III) and Yb(III) binuclear complexes sharpen with increasing addition of fluoride due to its chelation involving both the metal centres which slows the rate of interconversion between the SAP and TSAP isomers (Fig. 4d, e, S27, S29, S30, S32, S33, S35 and S37‡). New signals are observed that extend the NMR spectral range in both the positive and negative chemical shift regions suggesting equatorial binding of fluoride with respect to the metal centre: axial binding of fluoride commonly results in a decrease or inversion of the local crystal field by destabilising axially oriented mj states, while equatorial binding destabilises other mj states.27–2919F NMR spectra of binuclear Eu(III) complexes in deuterium oxide contain a small peak at around −450 ppm for the fluoride bound to the complexes (Fig. S42 and S43‡) which is similar to the chemical shift reported for fluoride binding in mononuclear complexes.27d,28,29
2.4 Control experiments with mononuclear complexes
In [Eu(pDO3A)], no chloride chelation was observed (Fig. S45, S108, and S109‡). However, when this complex was titrated against KF in deionised water and methanol, a single binding event was observed (Table 2, Fig. S104–107‡). Results from time-resolved lifetime, paramagnetic 1H NMR, and 19F NMR are similar to the interaction of fluoride to the binuclear systems (Fig. S38–S40, S43, S116 and S117; Tables S7 and S8‡). Although fluoride chelation was observed in these binuclear systems, it does not need the binuclear cavity which chloride demands.26–292.5 Chloride selectivity over other halides on binuclear Ln(III) complexes
In contrast to fluoride, significant interference for a chloride receptor can be envisioned from the heavier halides. Titration of the binuclear Eu(III) complexes against bromide and iodide solutions in deuterium oxide and deionised water gave broadened NMR spectra and no observable changes to the fine structure in the luminescence spectra (Fig. S80–S83‡ for [Eu2(DO3A)2C-2]; Fig. S100–S103‡ for [Eu2(DO3A)2C-3]) indicating that these ions bind very weakly, if at all. Thus, we have a highly selective receptor for chloride.3 Conclusion
Binuclear lanthanide complexes can bind to chloride ions in aqueous solution provided that two lanthanide ions can bind to chloride – reducing intermetallic repulsions. This offers new scope for the development of chloride specific/selective molecular probes that can be exploited in competitive biological media. Much remains to be done, but the results described here clearly identify a new area of anion coordination chemistry that is ripe for detailed exploration. With the current need for chloride binding receptors in water for aquifers and in vivo, these hydrophilic binuclear complexes are a good beginning. This work is a possible solution to the decades old challenge of chloride recognition in water, which opens up new avenues to various applications from biology to sensors and beyond.Data availability
ESI is available which contains detailed methods and materials, detailed synthesis and characterization, titrations (NMR and luminescence), speciation models, luminescence lifetimes and single crystal X-ray data. Crystallographic information for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2201928 for (DO3A(t-BuO)3)2C-2, CCDC 2201929 for pDO3A(t-BuO)3, CCDC 2201930 for [Eu2(DO3A)2C-3], CCDC 2201931 for [Yb2(DO3A)2C-3], CCDC 2201932 for [Yb2(DO3A)2C-3(2F)]2−, CCDC 2201933 for [Eu(pDO3A)], and CCDC 2201934 for [Yb(pDO3A)].Author contributions
Conceptualisation, S. F.; supervision, S. F., A. M. K. and T. J. S.; methodology, C. A., S. F., A. M. K., T. J. S. and K. E. C.; investigation, C. A., K. E. C. and J. A. T.; validation, C. A.; formal analysis, C. A., T. J. S., K. E. C. and S. F.; data curation, C. A. and K. E. C.; visualisation, C. A.; writing – original draft, C. A., T. J. S. and S. F.; writing – review & editing, C. A., T. J. S., A. M. K., S. F., K. E. C. and J. A. T.; resources, S. F. and K. E. C.; funding acquisition, S. F. and C. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Oxford for support. C. A. acknowledges the RSC Researcher Mobility Grant (grant no. MI9-0393) for partial financial support towards this project and Research England GCRF QR fund for providing partial scholarship. C. A. thanks Dr Leila Hill for advice on using DYNAFIT® and thermodynamic study, Dr Nicholas H. Rees for helpful advice and support on 35Cl NMR, Faulkner group members (Dr Grace McMullon, Dr Daniel Kovacs, Cameron Gray, Dr Deborah Sneddon, and Clara von Randow), and Zongyao Zhang for suggestions incorporated in this work.Notes and references
- (a) P. D. Beer and P. A. Gale, Anion recognition and sensing: The state of the art and future perspectives, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS; (b) J. W. Steed and J. L. Atwood, Supramolecular chemistry, John Wiley and sons, New York, 3rd edn, 2022 Search PubMed; (c) Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. Kim and J. L. Sessler, Macrocycles as ion pair receptors, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- D. Dalkara, G. Zuber and J. P. Behr, Intracytoplasmic delivery of anionic proteins, Mol. Ther., 2004, 9, 964–969 CrossRef CAS PubMed.
- J. W. Steed, Coordination and organometallic compounds as anion receptors and sensors, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- (a) S. J. Butler and D. Parker, Anion binding in water at lanthanide centres: from structure and selectivity to signalling and sensing, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC; (b) S. E. Bodman and S. J. Butler, Advances in anion binding and sensing using luminescent lanthanide complexes, Chem. Sci., 2021, 12, 2716–2734 RSC.
- (a) C. H. Park and H. E. Simmons, Macrobicyclic amines. III. Encapsulation of halide ions by in,in-1, (k + 2)-diazabicyclo[k.l.m]alkaneammonium ions, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS; (b) K. Worm and F. P. Schmidtchen, Molecular recognition of anions by zwitterionic host molecules in water, Angew. Chem., Int. Ed., 1995, 34, 65–66 CrossRef CAS; (c) F. P. Schmidtchen, Hosting anions. The energetic perspective, Chem. Soc. Rev., 2010, 39, 3916–3935 RSC; (d) S. Kubik, Anion recognition in water, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC; (e) M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Discovery of a cyclic 6 + 6 hexamer of D-biotin and formaldehyde, Chem. Sci., 2014, 5, 2647–2650 RSC; (f) M. A. Yawer, V. Havel and V. Sindelar, A bambusuril macrocycle that binds anions in water with high affinity and selectivity, Angew. Chem., Int. Ed., 2015, 54, 276–279 CrossRef CAS PubMed; (g) M. Lisbjerg, B. E. Nielsen, B. O. Milhøj, S. P. A. Sauer and M. Pittelkow, Anion binding by biotin[6]uril in water, Org. Biomol. Chem., 2015, 13, 369–373 RSC; (h) M. J. Langton, C. J. Serpell and P. D. Beer, Anion recognition in water: Recent advances from a supramolecular and macromolecular perspective, Angew. Chem., Int. Ed., 2015, 55, 1974–1987 CrossRef PubMed; (i) P. S. Cremer, A. H. Flood, B. C. Gibb and D. L. Mobley, Collaborative routes to clarifying the murky waters of aqueous supramolecular chemistry, Nat. Chem., 2018, 10, 8–16 CrossRef CAS PubMed; (j) Y. Chen, G. Wu, L. Chen, L. Tong, Y. Lei, L. Shen, T. Jiao and H. Li, Selective recognition of chloride anion in water, Org. Lett., 2020, 22, 4878–4882 CrossRef CAS PubMed; (k) S. Kubik, When molecules meet in water-recent contributions of supramolecular chemistry to the understanding of molecular recognition processes in water, ChemistryOpen, 2022, 11, e202200028 CrossRef CAS PubMed.
- (a) J. W. Pflugrath and F. A. Quiocho, Sulphate sequestered in the sulphate-binding protein of Salmonella typhimurium is bound solely by hydrogen bonds, Nature, 1985, 314, 257–260 CrossRef CAS PubMed; (b) H. Luecke and F. A. Quiocho, High specificity of a phosphate transport protein determined by hydrogen bonds, Nature, 1990, 347, 402–406 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen bonding in supramolecular chemistry, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Anion–π interactions in supramolecular architectures, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- S. C. Dodani, in Building better chloride sensors, ed. E. G. Berg, Chem. Eng. News, 2018, (Dec 28) Search PubMed.
- D. E. C. Cole, J. Shafai and C. R. Scriver, Inorganic sulfate in cerebrospinal fluid from infants and children, Clin. Chim. Acta, 1982, 120, 153–159 CrossRef CAS PubMed.
- (a) D. Reuter, K. Zierold, W. H. Schröder and S. Frings, A depolarizing chloride current contributes to chemoelectrical transduction in olfactory sensory neurons in situ, J. Neurosci., 1998, 18, 6623–6630 CrossRef CAS PubMed; (b) P. Bregestovski, T. Waseem and M. Mukhtarov, Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity, Front. Mol. Neurosci., 2009, 2, 1–15 Search PubMed; (c) T. J. Jentsch, V. Stein, F. Weinreich and A. A. Zdebik, Molecular structure and physiological function of chloride channels, Physiol. Rev., 2002, 82, 503–568 CrossRef CAS PubMed; (d) D. Arosio and G. M. Ratto, Twenty years of fluorescence imaging of intracellular chloride, Front. Cell. Neurosci., 2014, 8, 258 Search PubMed.
- J. N. Tutol, W. Peng and S. C. Dodani, Discovery and characterization of a naturally occurring, turn-on yellow fluorescent protein sensor for chloride, Biochemistry, 2019, 58, 31–35 CrossRef CAS PubMed.
- M. E. Q. Pilson, An Introduction to the Chemistry of the Sea, Cambridge University Press, New York, 2nd edn, 2012 Search PubMed.
- Y. Guo and R. G. Compton, A bespoke chloride sensor for seawater: Simple and fast with a silver electrode, Talanta, 2021, 232, 122502 CrossRef CAS PubMed.
- (a) X. Wu, A. M. Gilchrist and P. A. Gale, Prospects and challenges in anion recognition and transport, Chem, 2020, 6, 1296–1309 CrossRef CAS; (b) L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Progress in anion receptor chemistry, Chem, 2022, 8, 46–118 CrossRef CAS.
- (a) R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef; (b) Y. Marcus, Thermodynamics of solvation of ions. Part 5. Gibbs free energy of hydration at 298.15 K, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC; (c) Y. Marcus, The thermodynamics of solvation of ions. Part 2. The enthalpy of hydration at 298.15 K, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 339–349 RSC; (d) Y. Marcus, A simple empirical model describing the thermodynamics of hydration of ions of widely varying charges, sizes, and shapes, Biophys. Chem., 1994, 51, 111–127 CrossRef CAS.
- F. Hofmeister, Zur Lehre von der Wirkung der Salze (About the Science of the Effect of Salts), Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 CrossRef.
- C. Allain, P. D. Beer, S. Faulkner, M. W. Jones, A. M. Kenwright, N. L. Kilah, R. C. Knighton, T. J. Sørensen and M. Tropiano, Lanthanide appended rotaxanes respond to changing chloride concentration, Chem. Sci., 2013, 4, 489–493 RSC.
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, High-affinity anion binding by steroidal squaramide receptors, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Chloride capture using a C–H hydrogen-bonding cage, Science, 2019, 365, 159–161 CAS.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Halogen bonding in water results in enhanced anion recognition in acyclic and rotaxane hosts, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- (a) M. C. Heffern, L. M. Matosziuk and T. J. Meade, Lanthanide probes for bioresponsive imaging, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed; (b) A. de Bettencourt-Dias. Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, John Wiley and Sons, New York, 2014 Search PubMed.
- T. J. Sørensen and S. Faulkner, Multimetallic lanthanide complexes: Using kinetic control to define complex multimetallic arrays, Acc. Chem. Res., 2018, 51, 2493–2501 CrossRef PubMed.
- (a) D. Parker, K. Senanyake and J. A. G. Williams, Luminescent sensors for pH, pO2, halide and hydroxide ions using phenanthridine as a photosensitiser in macrocyclic europium and terbium complexes, J. Chem. Soc., Perkin Trans. 2, 1998, 2129–2139 RSC; (b) D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Being excited by lanthanide coordination complexes: Aqua species, chirality, excited-state chemistry, and exchange dynamics, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS PubMed.
- R. G. Pearson, Hard and soft acids and bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) R. M. Scarborough Jr and A. B. Smith III, Synthesis and chemical properties of lanthanide cryptates, J. Am. Chem. Soc., 1977, 99, 7087–7089 CrossRef PubMed; (b) E. L. Yee, O. A. Gansow and M. J. Weaver, Electrochemical studies of europium and ytterbium cryptate formation in aqueous solution. Effects of varying the metal oxidation state upon cryptate thermodynamics and kinetics, J. Am. Chem. Soc., 1980, 102, 2278–2285 CrossRef CAS; (c) J. P. Cross, A. Dadabhoy and P. G. Sammes, The sensitivity of the lehn cryptand–europium and terbium(III) complexes to anions compared to a coordinatively saturated systems, J. Lumin., 2004, 110, 113–124 CrossRef CAS.
- (a) R. Tripier, C. Platas-Iglesias, A. Boos, J.-F. Morfin and L. J. Charbonnière, Towards fluoride sensing with positively charged lanthanide complexes, Eur. J. Inorg. Chem., 2010, 2735–2745 CrossRef CAS; (b) L. M. P. Lima, A. Lecointre, J.-F. Morfin, A. de Blas, D. Visvikis, L. J. Charbonniere, C. Platas-Iglesias and R. Tripier, Positively charged lanthanide complexes with cyclen-based ligands: Synthesis, solid-state and solution structure, and fluoride interaction, Inorg. Chem., 2011, 50, 12508–12521 CrossRef CAS PubMed; (c) T. Liu, A. Nonat, M. Beyler, M. Regueiro-Figueroa, K. N. Nono, O. Jeannin, F. Camerel, F. Debaene, S. Cianferani-Sanglier, R. Tripier, C. Platas-Iglesias and L. J. Charbonniere, Supramolecular luminescent lanthanide dimers for fluoride sequestering and sensing, Angew. Chem., Int. Ed., 2014, 53, 7259–7263 CrossRef CAS PubMed; (d) S. J. Butler, Quantitative determination of fluoride in pure water using luminescent europium complexes, Chem. Commun., 2015, 51, 10879–10882 RSC.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Conformational and coordination equilibria on DOTA complexes of lanthanide metal ions in aqueous solution studied by 1H-NMR spectroscopy, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- (a) O. A. Blackburn, N. F. Chilton, K. Keller, C. E. Tait, W. K. Myers, E. J. L. McInnes, A. M. Kenwright, P. D. Beer, C. R. Timmel and S. Faulkner, Spectroscopic and crystal field consequences of fluoride binding by [Yb·DTMA]3+ in aqueous solution, Angew. Chem., Int. Ed., 2015, 54, 10783–10786 CrossRef CAS PubMed; (b) O. A. Blackburn, A. M. Kenwright, P. D. Beer and S. Faulkner, Axial fluoride binding by lanthanide DTMA complexes alters the local crystal field, resulting in dramatic spectroscopic changes, Dalton Trans., 2015, 44, 19509–19517 RSC; (c) O. A. Blackburn, J. D. Routledge, L. B. Jennings, N. H. Rees, A. M. Kenwright, P. D. Beer and S. Faulkner, Substituent effects on fluoride binding by lanthanide complexes of DOTA-tetraamides, Dalton Trans., 2016, 45, 3070–3077 RSC; (d) A. Rodríguez-Rodríguez, Á. Arnosa-Priesto, I. Brandariz, D. Esteban-Gómez and C. Platas-Iglesias, Axial ligation in Ytterbium(III) DOTAM complexes rationalized with multireference and ligand-field ab initio calculations, J. Phys. Chem. A, 2020, 124, 1362–1371 CrossRef PubMed.
- A. J. Harte, P. Jensen, S. E. Plush, P. E. Kruger and T. Gunnlaugsson, A dinuclear lanthanide complex for the recognition of bis(carboxylates): formation of Terbium(III) luminescent self-assembly ternary complexes in aqueous solution, Inorg. Chem., 2006, 45, 9465–9474 CrossRef CAS PubMed.
- C. M. Andolina and J. R. Morrow, Luminescence resonance energy transfer in heterodinuclear LnIII complexes for sensing biologically relevant anions, Eur. J. Inorg. Chem., 2011, 154–164 CrossRef CAS.
- J. D. Moore, R. L. Lord, G. A. Cisneros and M. J. Allen, Concentration-independent pH detection with a luminescent dimetallic Eu(III)-based probe, J. Am. Chem. Soc., 2012, 134, 17372–17375 CrossRef CAS PubMed.
- (a) J. A. Tilney, T. J. Sørensen, B. P. Burton-Pye and S. Faulkner, Self-assembly between dicarboxylate ions and a binuclear europium complex: Formation of stable adducts and heterometallic lanthanide complexes, Dalton Trans., 2011, 40, 12063–12066 RSC; (b) L. R. Hill, T. J. Sørensen, O. A. Blackburn, A. Brown, P. D. Beer and S. Faulkner, Self-assembly between dicarboxylate ions and binuclear europium complexes: moving to water-pH dependence and effects of buffers, Dalton Trans., 2013, 42, 67–70 RSC; (c) T. J. Sørensen, L. R. Hill and S. Faulkner, Thermodynamics of self-assembly of dicarboxylate ions with binuclear lanthanide complexes, ChemistryOpen, 2015, 4, 509–515 CrossRef PubMed.
- N. Kofod, M. S. Thomsen, P. Nawrocki and T. J. Sørensen, Revisiting the assignment of innocent and non-innocent counterions in lanthanide(III) solution chemistry, Dalton Trans., 2022, 51, 7936–7949 RSC.
- M. Tropiano, O. A. Blackburn, J. A. Tilney, L. R. Hill, M. P. Placidi, R. J. Aarons, D. Sykes, M. W. Jones, A. M. Kenwright, J. S. Snaith, T. J. Sørensen and S. Faulkner, Using remote substituents to control solution structure and anion binding in lanthanide complexes, Chem. – Eur. J., 2013, 19, 16566–16571 CrossRef CAS PubMed.
- C. C. Bryden, C. N. Reilley and J. F. Desreux, Multinuclear nuclear magnetic resonance study of three aqueous lanthanide shift reagents: complexes with EDTA and axially symmetric macrocyclic polyamino polyacetate ligands, Anal. Chem., 1981, 53, 1418–1425 CrossRef CAS.
- L. R. Tear, M. L. Maguire, M. Tropiano, K. Yao, N. J. Farrer, S. Faulkner and J. E. Schneider, Enhancing 31P NMR relaxation rates with a kinetically inert gadolinium complex, Dalton Trans., 2020, 49, 2989–2993 RSC.
- J. Fawell, K. Bailey, E. Chilton, E. Dahi, L. Fewtrell and Y. Magara, Fluoride in drinking-water, World Health Organization, IWA Publishing, London, 2006 Search PubMed.
- G. T. Hefter, Solvation of fluoride ions. 3. A review of fluoride solvation thermodynamics in nonaqueous and mixed solvents, Rev. Inorg. Chem., 1989, 10, 185–224 CAS.
Footnotes |
| † This manuscript is dedicated to Prof. David Parker on his retirement from the University of Durham. |
| ‡ Electronic supplementary information (ESI) available: Detailed methods and materials, detailed synthesis and characterisation, titrations (NMR and luminescence), speciation models, luminescence lifetimes, and single crystal X-ray data. CCDC 2201928–2201934. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05417e |
| § Hofmeister series of anion hydrophobicity (taken from ref. 17): CO32− > SO42− > S2O32− > H2PO4− > HO− > F− > HCO2− > CH3CO2− > Cl− > Br− > NO3− > I− > CIO4− > SCN−. |
| This journal is © The Royal Society of Chemistry 2023 |