
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual ligand approach increases functional group tolerance in the Pd-catalysed C–H arylation of N-heterocyclic pharmaceuticals†
Igor
Beckers
a,
Aram
Bugaev
 b and
Dirk
De Vos
*a
b and
Dirk
De Vos
*a
aDepartment of Microbial and Molecular Systems, Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: dirk.devos@kuleuven.be
bThe Smart Materials Research Institute, Southern Federal University, Sladkova 174/28, 344090 Rostov-on-Don, Russia
First published on 10th January 2023
Abstract
The excellent functional group tolerance of the Suzuki–Miyaura cross-coupling reactions has been decisive for their success in the pharmaceutical industry. Highly diversified (hetero)aromatic scaffolds can be effectively coupled in the final step(s) of a convergent synthetic route. In contrast, electrophilic Pd catalysts for non-directed C–H activation are particularly sensitive to inhibition by coordinating groups in pharmaceutical precursors. While C–H arylation enables the direct conversion of (hetero)aromatics without preinstalled functional or directing groups, its functional group tolerance should be increased to be viable in late-stage cross-couplings. In this work, we report on a dual ligand approach that combines a strongly coordinating phosphine ligand with a chelating 2-hydroxypyridine for the highly robust C–H coupling of bicyclic N-heteroaromatics with aryl bromide scaffolds. The catalyst speciation was studied via in situ XAS measurements, confirming the coordination of both ligands under the reaction conditions. The C–H activation catalyst was shown to be tolerant to a wide range of pharmaceutically relevant scaffolds, including examples of late-stage functionalization of known drug molecules.
Introduction
Since their discovery, Pd-catalysed cross-coupling reactions have enabled both the lab-scale preparation of bi(hetero)aryl drug candidates1–4 and the industrial production of pharmaceuticals.5,6 In the recent surge in research related to protein kinase inhibitors7–9 and anti-viral small molecules,10 the coupling of bicyclic N-heteroaromatics has become increasingly important.11–14 By mimicking the purine nucleobases which are omnipresent in genetic material or signalling molecules (e.g. ATP or cGMP), these are often key structural features in potent inhibitors. Such scaffolds bear unique patterns of polar, often chelating substituents and N-heteroatoms that are indispensable for selective binding with the drug target. However, these coordinating functionalities often hinder the activity of metal-based catalysts.15–18 The development of ligands, in particular advanced phosphine ligands, gave access to the C–X/C–M cross-coupling of (hetero)aryl halides and -metals bearing a wide range of pharmaceutically relevant substituents (Scheme 1).19,20 In contrast, catalysts boasting the highest activity for non-directed C–H arylation, i.e. C–H/C–X coupling using low Pd loadings below 1 mol%, rely on phosphine-free conditions with acetate or carbonate bases21–34 or on the use of stoichiometric metal additives.27,35,36 While the minimal Pd loading is a highly attractive feature of such phosphine-free catalytic systems, the weakly coordinated electrophilic catalyst is particularly sensitive towards unwanted reactant or product inhibition by metal-coordinating moieties. Indeed, the scope of these methodologies was so far restricted towards simple scaffolds, with the absence of important pharmaceutical functionalities, for example chelating 2-aminopyridine motifs or metal-coordinating groups such as sulfones or amides.24 New catalysts should therefore be developed that combine a functional group tolerance similar to the traditional Suzuki–Miyaura couplings with the high activity in C–H activation as previously encountered in the reported phosphine-free systems. Recent examples have demonstrated that a dual ligand approach can be effective in promoting the performance of Pd catalysts in C–H activation reactions.37–39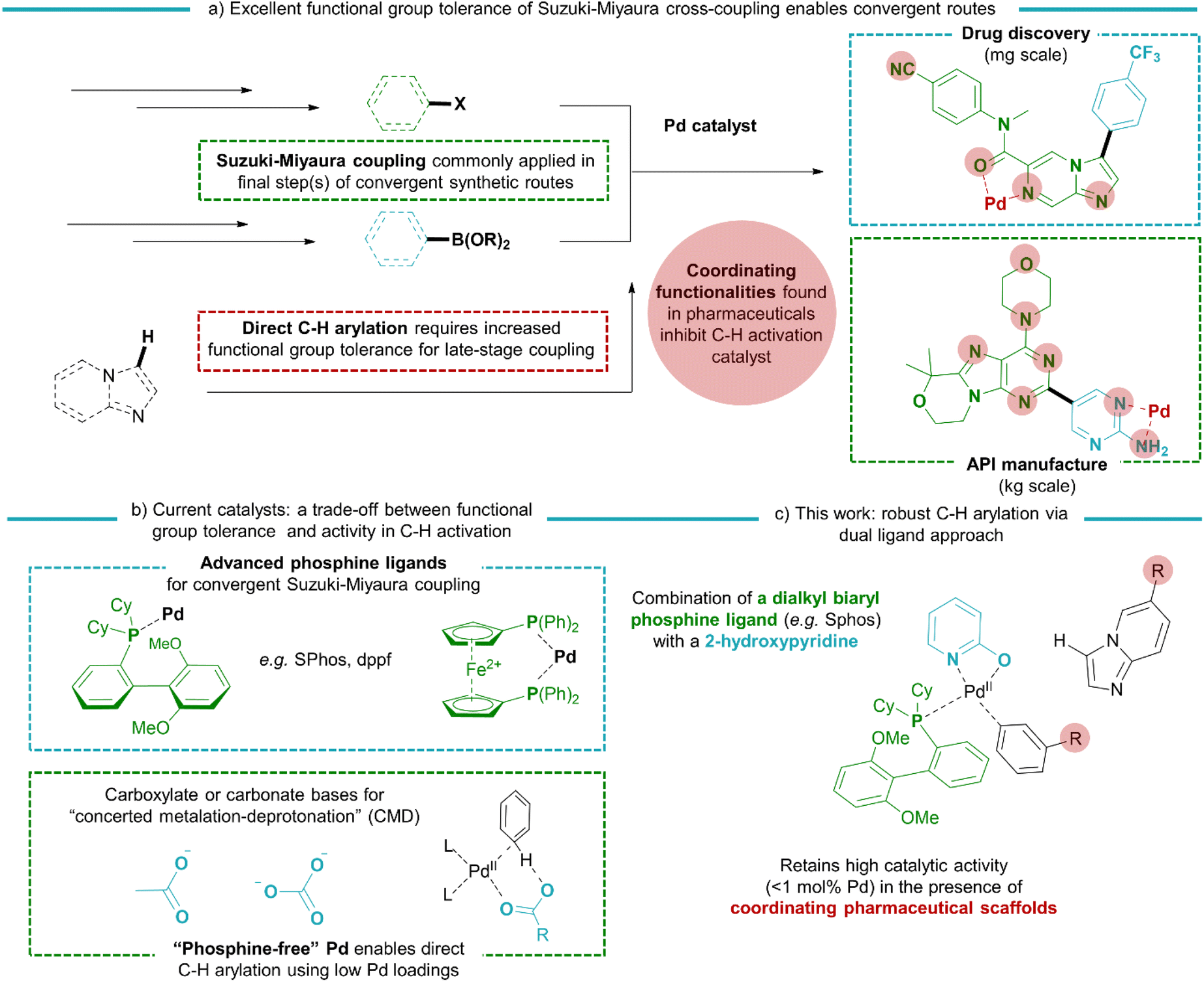 | ||
| Scheme 1 General introduction of the research described in this work: (a) the functional group tolerance as a decisive feature in the success of Suzuki–Miyaura coupling reaction in pharmaceutical industry, (b) advances in phosphine ligands strongly control the coordination sphere of the Pd catalyst, in contrast to the weakly coordinated electrophilic catalysts that are highly active in direct arylation via C–H activation, and (c) the catalyst developed in this work combines a highly coordinated environment (via dual ligand approach) for functional group tolerance with high activity in C–H activation. | ||
With the regiospecific C–H activation of N-heterocyclic imidazopyridines as a starting point, this work describes a dual ligand approach that enables their C–H coupling with (hetero)aryl bromide scaffolds bearing a wide variety of medicinally relevant functional groups. By combining advanced phosphine ligands used in the conventional cross-coupling reactions with a 2-hydroxypyridine ligand to accelerate the C–H activation, a highly active and robust catalyst for direct arylation with superior functional group tolerance was obtained. Both ligands coordinate strongly to the homogeneous Pd catalyst, as shown by in situ XAS measurements. While strictly maintaining Pd catalyst loadings of 1 mol% or less, the scope of the direct C–H arylation featuring high activity was extended towards the convergent cross-coupling of pharmaceutically relevant imidazo[1,2-a]pyridines and related N-heterocyclic scaffolds. This was demonstrated in the preparation of 50 compounds, including examples of late-stage diversification of drug molecules.
Results and discussion
Ligand screening and optimisation of reaction conditions
Considering that the advanced phosphine ligands gave rise to excellent functional group tolerance in late-stage Suzuki–Miyaura coupling of pharmaceutically relevant motifs, we hypothesized that a robust catalyst could be obtained if such Pd-phosphine complexes could be reconciled with a fast C–H activation. However, it was previously shown that their electron-donating nature hinders the (hetero)aromatic C–H activation. Indeed, the highest turnover numbers in C–H arylation of N-heterocycles with aryl halides are previously obtained by relying on phosphine-free conditions to render the catalyst more electrophilic.23,24 We therefore searched for ligands that can accelerate the heteroaromatic C–H activation while being compatible with a (dialkylbiaryl) phosphine ligand. Our initial screening involved catalytic amounts of potential C–H activation ligands in combination with triphenylphosphine, 0.1 mol% of Pd2dba3 as the Pd precursor and KOH as the external base (Table 1). The reactions were ended early (after 3 h) to obtain yields that are representative for the initial reaction rate. A wide range of ligands were evaluated that were previously found to be effective in literature for various state-of-the-art (hetero)aromatic direct C–H arylations, as well as oxidative couplings and C–H alkenylations. The screening included N-protected amino acids,40–45 2-pyridones,46–51 thioether52–57 and carboxylate58,59 ligands (Scheme 2). The carboxylate ligands were tested via in situ formation of catalytic quantities of the corresponding carboxylic acid (L2–L4) with the stoichiometric KOH base. However, only low yields (<5%) of the resulting C–H arylated imidazo[1,2-a]pyridine was observed via GC analysis. The tested mono-protected amino acids (L5–L8) were also largely ineffective as ligands for this C–H arylation with Pd-phosphine catalysts, with the highest yield of 6.8% obtained for N-acetylphenylalanine (L8). No C–H arylation products were detected with the S-containing ligands ethyl phenyl sulfide and thiophenoxyacetic acid (L9 and L10). We then moved towards the 2-pyridone scaffold previously discovered by Yu and co-workers. The ligand 3,5-bis(trifluoromethyl)pyridin-2(1H)-one (L11), which was found to be highly effective in the C–H alkenylation of a wide range of aromatics, resulted in only 6.0% yield for this C–H arylation reaction in combination with a phosphine ligand. A similar result was found for the 5-nitro substituted 2-pyridone (L12). Gratifyingly, when we increased the basicity of the 2-pyridone ligand by omitting the electron-withdrawing groups (L13) or replacing them with an electron-donating group (L14), superior catalytic activity was observed with yields of 35.0 and 41.9% respectively. Two other 2-substituted pyridines, 2-mercaptopyridine (L15) and 2-pyridinesulfonic acid (L16) did not result in considerable yields.|
|
|||
|---|---|---|---|
| Entry | Phosphine ligand | C–H activation ligand | Yielda (%) |
| a Yields as determined via GC analysis. b Reaction performed for 18 h. | |||
| 1 | Triphenyl-phosphine (L1) | — | n.d. |
| 2 | Triphenyl-phosphine (L1) | Acetic acid (L2) | <5 |
| 3 | Triphenyl-phosphine (L1) | Benzoic acid (L3) | <5 |
| 4 | Triphenyl-phosphine (L1) | Pivalic acid (L4) | <5 |
| 5 | Triphenyl-phosphine (L1) | Ac-Gly-OH (L5) | n.d. |
| 6 | Triphenyl-phosphine (L1) | Ac-Val-OH (L6) | <5 |
| 7 | Triphenyl-phosphine (L1) | Ac-Met-OH (L7) | <5 |
| 8 | Triphenyl-phosphine (L1) | Ac-Phe-OH (L8) | 6.8 |
| 9 | Triphenyl-phosphine (L1) | Ethyl phenyl sulfide (L9) | n.d. |
| 10 | Triphenyl-phosphine (L1) | Thiophenoxyacetic acid (L10) | n.d. |
| 11 | Triphenyl-phosphine (L1) | 3,5-Bis(trifluoromethyl) pyridin-2(1H)-one (L11) | 6.0 |
| 12 | Triphenyl-phosphine (L1) | 5-Nitro-2-hydroxypyridine (L12) | 6.3 |
| 13 | Triphenyl-phosphine (L1) | 2-Hydroxypyridine (L13) | 35.0 |
| 14 | Triphenyl-phosphine (L1) | 5-Methyl-2-hydroxypyridine (L14) | 41.9 |
| 15 | Triphenyl-phosphine (L1) | 2-Mercaptopyridine (L15) | n.d. |
| 16 | Triphenyl-phosphine (L1) | 2-Pyridinesulfonic acid (L16) | 5.9 |
| 17 | — | 2-Hydroxypyridine (L13) | 9.8 |
| 18 | Tricyclohexyl-phosphine (L17) | 2-Hydroxypyridine (L13) | 42.8 |
| 19 | Tri(t-butyl) phosphine (L18) | 2-Hydroxypyridine (L13) | 19.0 |
| 20 | SPhos (L19) | 2-Hydroxypyridine (L13) | 43.5 |
| 21 | XPhos (L20) | 2-Hydroxypyridine (L13) | 40.7 |
| 22 | BrettPhos (L21) | 2-Hydroxypyridine (L13) | 45.5 |
| 23 | SPhos (L19) | 2-Hydroxypyridine (L13) | 97.8b |
| 24 | SPhos (L19) | — | n.d. |
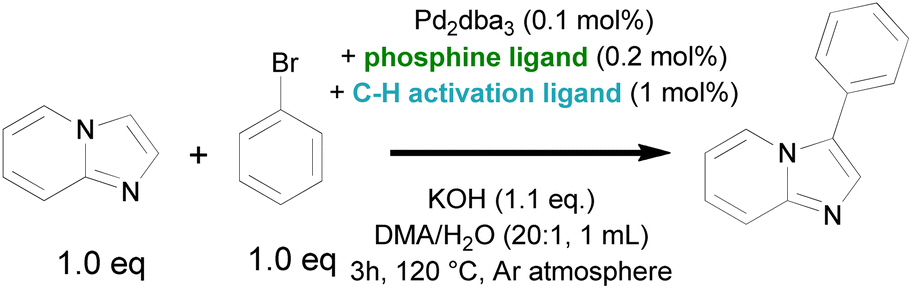
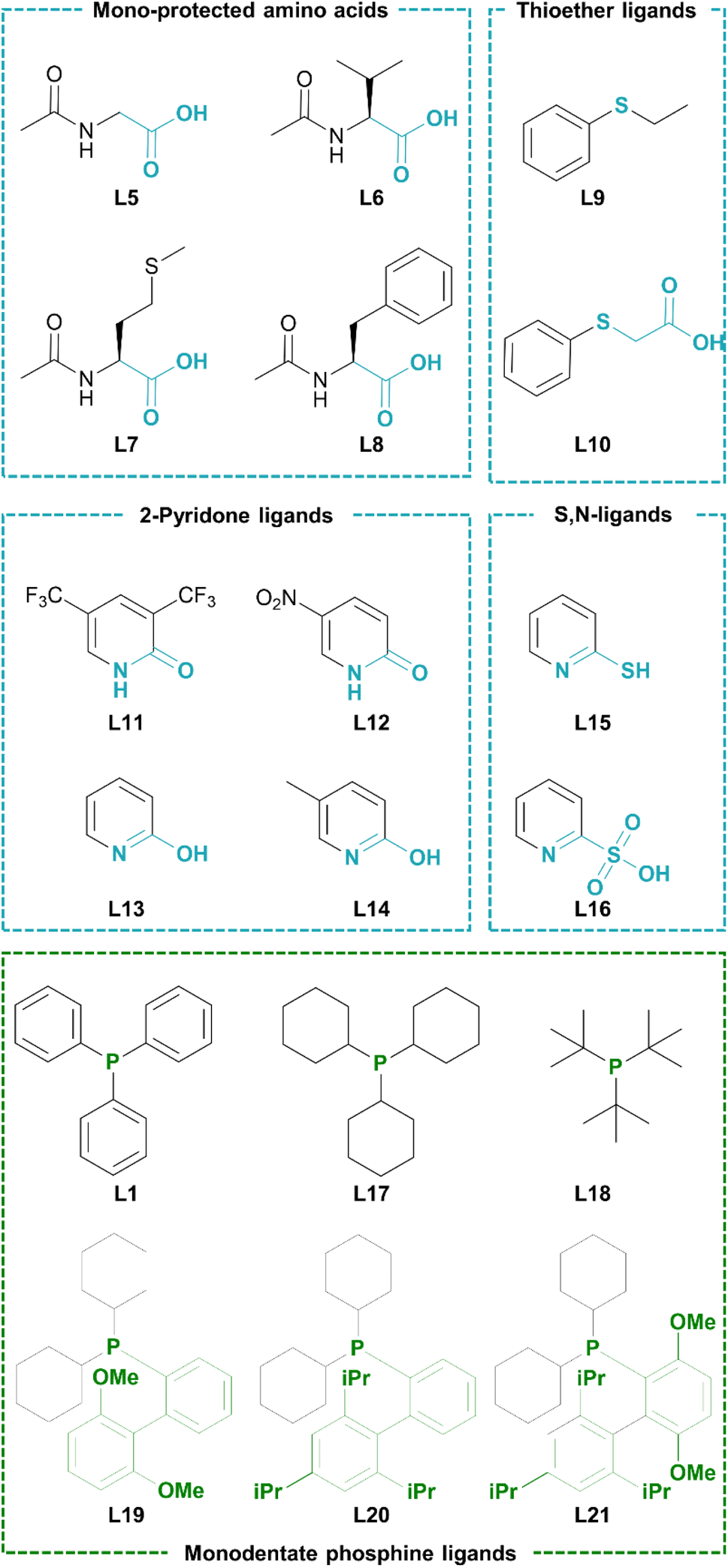 | ||
| Scheme 2 Screening of compatible ligand combinations of a C–H activation ligand (blue) and a conventional phosphine ligand (green) for the model C–H arylation of imidazo[1,2-a]pyridine. | ||
With the 2-hydroxypyridine as a simple and effective ligand for the C–H activation, we proceeded with the screening of various phosphine ligands. Without the phosphine counterpart, the catalytic system only resulted in poor yields. This confirms the synergy between both ligands. The tricyclohexylphosphine ligand (L17) performed better than triphenylphosphine, suggesting that the C–H activation can occur with a more bulky phosphine ligand attached to a less electrophilic Pd(II) center. In contrast, the use of tri(t-butyl)phosphine (L18) resulted in lower yields. The best yields were obtained when combining the 2-hydroxypyridine ligand with the dialkylbiaryl ligands such as SPhos (L19), XPhos (L20) and BrettPhos (L21). Performing the reaction with this ligand combination overnight resulted in complete conversions with close to quantitative yield.
Other reaction parameters were also screened, such as the solvent and the nature of the base (see ESI, Tables S1 and S2†). Tetramethylurea emerged as a performant alternative to the harmful DMA solvent,60 and weaker bases such as K3PO4 were successfully employed, which is important in the coupling of more base-sensitive reactants.
Spectroscopic study of the catalyst speciation via XAS
The catalytic system comprising of both a phosphine and 2-pyridone ligand was further investigated via extended X-ray absorption fine structure (EXAFS) spectroscopy (at the Pd K-edge). The spectroscopic study allowed in situ monitoring of the catalyst speciation in solution phase under conditions close to the optimal reaction conditions. The measured ligand combinations were either triphenylphosphine (L1) or SPhos (L19) and 2-hydroxypyridine (L13) with Pd2dba3 as the catalyst precursor in DMA. To obtain high-quality EXAFS spectra, the catalyst concentration was increased by a 100-fold and the addition of base to the reaction mixture was omitted. Analysis of the resulting EXAFS spectra confirmed that in both cases, the dual ligated species were formed at room temperature as revealed by the appearance of Pd–N (or Pd–O, which cannot be clearly distinguished in EXAFS) and Pd–P contributions at distances of approximately 2.1 and 2.3 Å respectively (Scheme 3). After addition of 4-bromothioanisole as a model aryl bromide bearing a metal-coordinating group, the sample with SPhos as the ligand was heated to 90 °C. The Pd–Br contribution is emerging in the resulting EXAFS spectrum, which shows that the oxidative addition is occurring. Both the Pd–P and Pd–N/O peaks remain present under these conditions, meaning that these ligands were not replaced by unwanted coordination of the reactant (see also ESI, Sections 4 and 5†).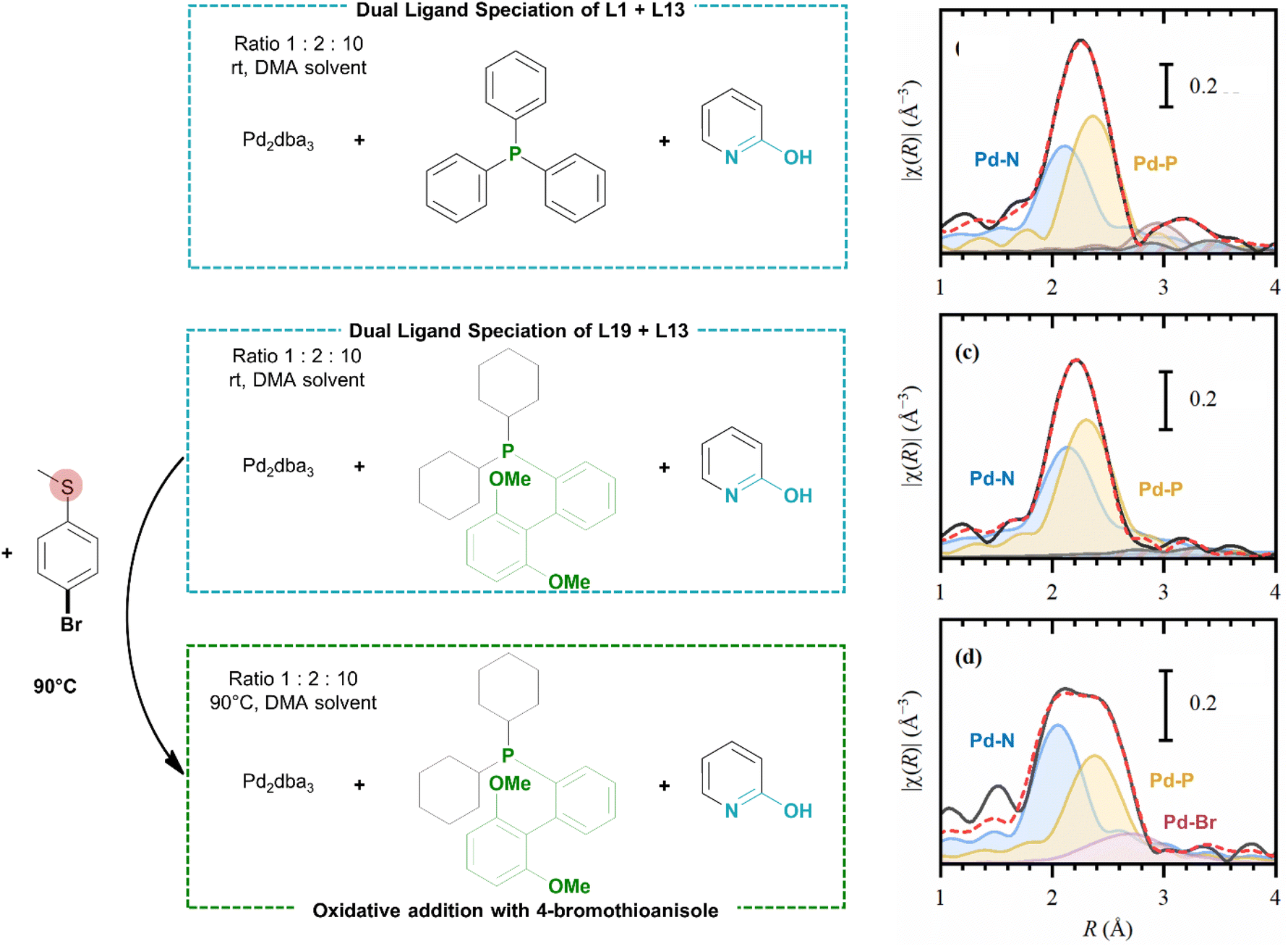 | ||
| Scheme 3 In situ measurement of Pd catalyst under the corresponding reaction conditions (left) and the analysed spectroscopic results (right). Experimental (solid black) and theoretical (dashed red) phase-uncorrected in situ EXAFS data (phase-corrected) for the Pd catalyst under the reaction conditions (see ESI† part 3 for details). Coloured lines with background correspond to different scattering pathways: Pd–N: blue, Pd–P: yellow, Pd–Br: purple. | ||
Functional group tolerance of the catalyst
To thoroughly evaluate the functional group tolerance of the dual ligated catalysts, we assessed a large series of coordinating substituents on the bromoarene or the N-heterocyclic reactant that are regularly encountered in medicinal chemistry. Functionalities such as free amides, chelating 2-arylpyridines or sulfoxides have so far been absent in the scope of state-of-the-art electrophilic C–H activation catalysts. In comparison, the current catalyst is not strongly hindered by unwanted coordination of the reactants. Especially the use of a dialkylbiaryl phosphine ligand such as SPhos limits the negative effect of (chelating) functional groups, rendering the catalyst more robust. In traditional cross-couplings, the reaction scope was also expanded to such coordinating (hetero)aromatic scaffolds by the use of advanced phosphine ligands.61 The compatibility of the 2-pyridone ligand with the same phosphine ligands is a strong asset in terms of reaction scope (Scheme 4). Next to the additional protection of the catalyst towards unwanted reactant or product coordination, the electron-donating nature of the phosphine ligand accelerates the oxidative addition step, enabling the facile coupling of bromoarenes with either electron-withdrawing or electron-donating substituents. This was confirmed in the evaluation of a large series of aryl bromide reactants in the reaction scope (1–19). Moreover, reactants with coordinating functionalities, including a variety of N-containing (nitrile, amines and amides) as well as S-containing (thioether, sulfoxide and sulfone) groups were also effectively coupled with low Pd loadings using the SPhos ligand (20–28, 36). Interestingly, a range of heteroaryl bromides are also tolerated (29–35), even when the product scaffolds bear the chelating 2-pyridinyl or 2-aminopyridinyl motifs. In the latter cases, the reaction exclusively proceeds with the choice of SPhos as a dialkylbiaryl phosphine ligand. The reaction also occurred smoothly with other substituents on the heteroaromatic ring, including electron-donating, -withdrawing as well as sterically hindering substituents on the C2 position (37–42). The C–H activation could also be expanded towards other bicyclic N-heteroaromatic scaffolds, such as imidazo[1,2-a]pyrazine, indazole, imidazo[1,5-a]pyridine and xanthine (43–46). The functional group tolerance of the catalyst was further demonstrated in the late-stage diversification of several marketed drug compounds, such as zolimidine, doxofylline and pentoxifylline (47–49). Finally, the catalyst was also effective in the convergent coupling of a potent anti-malarial kinase inhibitor in pre-clinical research (50).62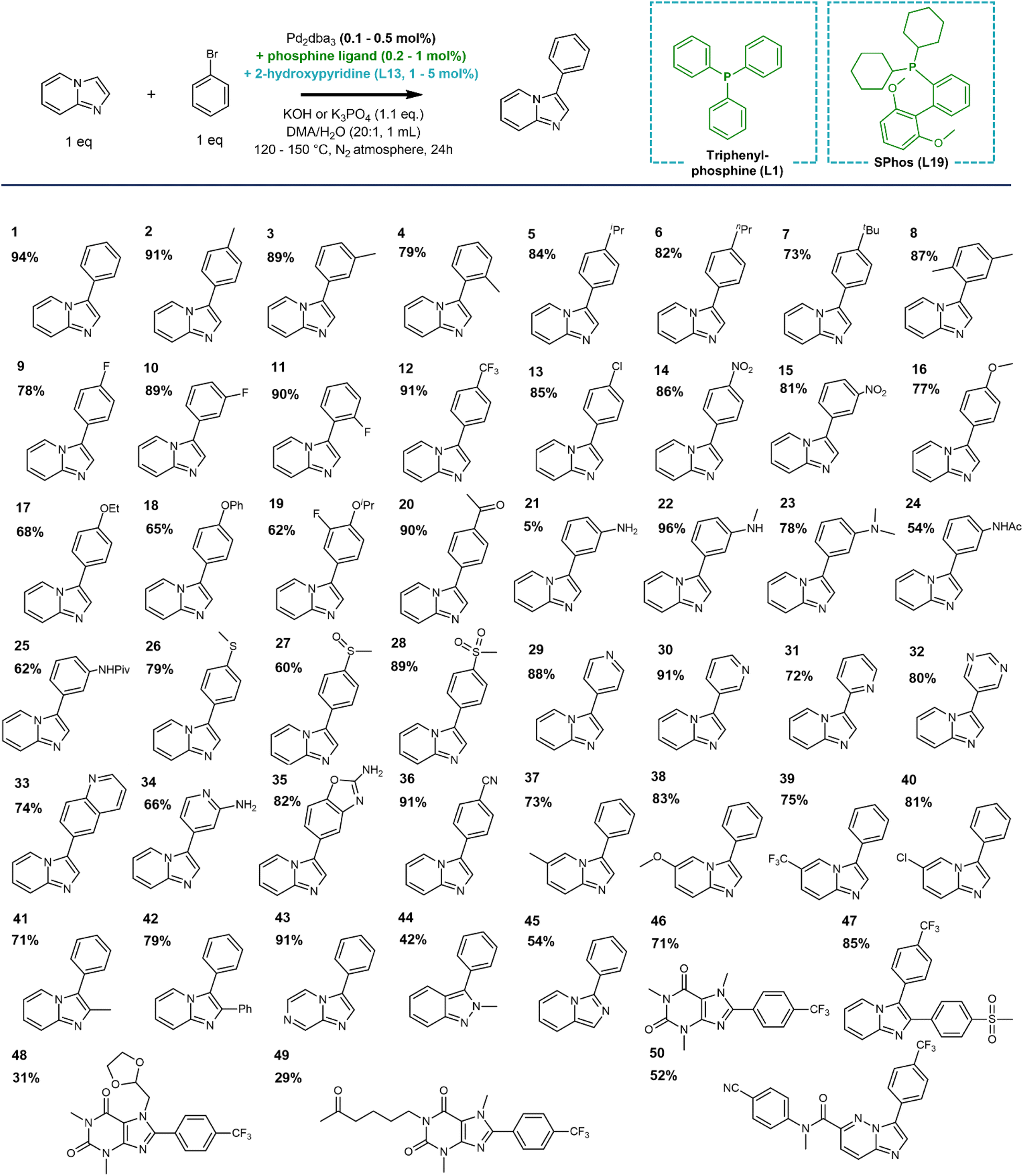 | ||
| Scheme 4 Substrate scope of the Pd catalyst includes a wide range of bromoarenes containing electron-withdrawing, electron-donating and coordinating substituents, as well as different bicyclic N-heteroaromatic scaffolds. Several examples of drug diversification and a convergent synthesis of a pre-clinical protein kinase inhibitor are also given. Triphenylphosphine was used as the ligand for entries 1–4, 9–15, 29, 30, 33, 36–44 and SPhos for 5–8, 16–28, 31, 32, 34, 35, 45–50. Yields (%) given correspond to isolated yields. | ||
Experimental
General information
All chemicals were obtained from commercial suppliers or prepared according to known synthetic procedures. An additional purification via column chromatography was performed for the heterocyclic reactants, and the other chemicals were used as received. N,N-Dimethylacetamide (>99.9%, HPLC grade) was used as the solvent without further drying steps. The Pd2dba3 precursor was stored under inert, moisture-free atmosphere. GC analysis was performed on a Shimadzu GC-2010 Pro equipped with a CP-Sil 5 CB column and flame ionization detector. The NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer. Column chromatography was performed on 70–230 mesh silica 60 (Merck) as the stationary phase. For TLC analysis, silica gel on TLC Al foils with fluorescent indicator (254 nm) was used. The standard MS spectra were obtained via GC-MS (Agilent 6890 equipped with HP-5MS column and a 5973 MSD mass spectrometer using electron impact ionization). The MS data of the products featuring a high boiling point or limited stability were acquired using a Water's Radian ASAP mass spectrometer.General reaction procedure
Potassium hydroxide pellets were crushed to a fine powder and weighed in a 10 mL glass vial (0.11 mmol, 6.17 mg) under ambient atmosphere. Alternatively, a 2.2 M KOH solution in Milli-Q water was prepared and added volumetrically to the reaction. N,N-Dimethylacetamide solvent (DMA, 1 mL) with 5 vol% of H2O (50 μL), imidazo[1,2-a]pyridine (0.1 mmol, 10.1 μL) and bromobenzene (0.1 mmol, 10.5 μL) were also added volumetrically by means of a micropipette or calibrated syringe. A Teflon-coated magnetic stirring bar was added. The resulting reaction mixture was sealed with a septum-covered crimp cap and flushed by exposing the vial to argon flow. A stock solution of Pd2dba3 (0.01 mmol, 9.16 mg) with triphenylphosphine (0.02 mmol, 5.25 mg) and 2-hydroxypyridine (0.1 mmol, 9.51 mg) was prepared by weighing the corresponding solids in a septum-covered glass vial, purging with argon gas and volumetric addition of purged DMA solvent (5 mL). The catalyst stock solution was stirred or sonicated until the Pd precursor was fully dissolved. A catalytic quantity of Pd2dba3, triphenylphosphine and 2-hydroxypyridine stock solution (50 μL) was transferred to the reaction mixture under inert conditions via a calibrated syringe. The reaction vial was then inserted in a heating block (120 °C) equipped with magnetic stirring (500 rpm). After reaction, the product mixtures were cooled down to room temperature, and a sample of the product mixture was analyzed via GC. The detailed procedures on the preparation and isolation of compounds 1–50, as well as their corresponding characterization, are given in ESI, Sections 4 and 5.†XAS procedure
Pd K-edge XAS measurements were performed at BM23 beamline of the ESRF. The samples were measured inside closed glass vials purged with inert gas. The chemicals were added volumetrically by a calibrated syringe. The vials were placed in the oil bath for temperature control. XAS spectra were collected in situ in transmission geometry by measuring the beam intensity before and after the sample using ionization chambers (Fig. S2–S4†). The energy was scanned from 24.1 to 25.2 keV by Si(311) double-crystal monochromator (ca. 40 s per scan). Palladium foil was measured simultaneously with the sample using the third ionization chamber for energy calibration. The data processing and analysis was performed in Demeter software.63 For EXAFS fitting, theoretical calculations were performed for the geometry models obtained from DFT calculations.Conclusions
To conclude, we have explored a dual ligand approach to overcome unwanted catalyst coordination to polar substituents and chelating motifs, which is often the bottleneck in the C–H coupling of pharmaceutically relevant compounds. A synergy between the conventional phosphine ligands and basic 2-hydroxypyridines was found, leading to increased functional group tolerance while maintaining the high activity in the coupling of aryl bromides with purine-like N-heterocycles. The synthetic utility of the increased functional group tolerance was demonstrated for a range of diversified scaffolds, including examples of late-stage, convergent C–H couplings. This flexible, generic catalyst will facilitate the preparation of such pharmaceuticals in the future.Data availability
This study did not generate large datasets.Author contributions
IB has conceived the research concept and performed the experiments. IB has also investigated the scope of the reaction and purified the chemical compounds. AB has performed the analysis via XAS and interpreted the resulting spectra. DDV has supervised the project and contributed to the research concept. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the NMBP-01-2016 Program of the European Union’s Horizon 2020 Framework Program H2020/2014-2020/under grant agreement no [720996]. We thank the European Synchrotron Radiation Facility and Kirill Lomachenko for the XAS measurements at BM23 beamline of ESRF. A.L.B. acknowledge the Strategic Academic Leadership Program of the Southern Federal University (“Priority 2030”). DDV is grateful to KU Leuven for support in the frame of the CASAS Metusalem project and to Research Foundation Flanders (FWO) for project funding (G0781118N, G0D0518N). We also thank Martijn Thonnon and Gerben Bernaerts for their assistance with the experimental work. Furthermore, we are grateful to Frederick Martens and Monica Oliva for their help with the acquisition of MS data.Notes and references
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed.
- M. J. Buskes and M.-J. Blanco, Molecules, 2020, 25, 3493 CrossRef CAS PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709 CrossRef PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- A. Stumpf, A. McClory, H. Yajima, N. Segraves, R. Angelaud and F. Gosselin, Org. Process Res. Dev., 2016, 20, 751 CrossRef CAS.
- P. Cohen, D. Cross and P. A. Jänne, Nat. Rev. Drug Discovery, 2021, 20, 551 CrossRef CAS PubMed.
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Nat. Rev. Drug Discovery, 2021, 20, 839 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2015, 100, 1 CrossRef CAS PubMed.
- C. S. Adamson, K. Chibale, R. J. M. Goss, M. Jaspars, D. J. Newman and R. A. Dorrington, Chem. Soc. Rev., 2021, 50, 3647 RSC.
- V. Asati, A. Anant, P. Patel, K. Kaur and G. D. Gupta, Eur. J. Med. Chem., 2021, 225, 113781 CrossRef CAS PubMed.
- D. J. Baillache and A. Unciti-Broceta, RSC Med. Chem., 2020, 11, 1112 RSC.
- P. Sharma, C. Larosa, J. Antwi, R. Govindarajan and K. A. Werbovetz, Molecules, 2021, 26, 4213 CrossRef CAS PubMed.
- P. Perlíková and M. Hocek, Med. Res. Rev., 2017, 37, 1429 CrossRef PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383 CrossRef CAS PubMed.
- Y. J. Liu, H. Xu, W. J. Kong, M. Shang, H. X. Dai and J. Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed.
- L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- S. El Kazzouli, J. Koubachi, N. El Brahmi and G. Guillaumet, RSC Adv., 2015, 5, 15292 RSC.
- J. Aziz and S. Piguel, Synthesis, 2017, 49, 4562 CrossRef CAS.
- T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296 CrossRef CAS PubMed.
- H. Y. Fu, L. Chen and H. Doucet, J. Org. Chem., 2012, 77, 4473 CrossRef CAS PubMed.
- B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050 CrossRef CAS PubMed.
- S. A. Ohnmacht, A. J. Culshaw and M. F. Greaney, Org. Lett., 2010, 12, 224 CrossRef CAS PubMed.
- N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926 CrossRef CAS PubMed.
- X. Wang, D. V. Gribkov and D. Sames, J. Org. Chem., 2007, 72, 1476 CrossRef CAS PubMed.
- A. Ben-Yahia, M. Naas, S. El Kazzouli, E. M. Essassi and G. Guillaumet, Eur. J. Org. Chem., 2012, 3, 7075 CrossRef.
- R. Bhatt, N. Bhuvanesh, K. N. Sharma and H. Joshi, Eur. J. Inorg. Chem., 2020, 532 CrossRef CAS.
- R. Bhaskar, A. K. Sharma and A. K. Singh, Organometallics, 2018, 37, 2669 CrossRef CAS.
- X.-X. He, Y.-F. Li, J. Huang, D.-S. Shen and F.-S. Liu, J. Organomet. Chem., 2016, 803, 58 CrossRef CAS.
- J.-S. Ouyang, Y.-F. Li, D.-S. Shen, Z. Ke and F.-S. Liu, Dalton Trans., 2016, 45, 14919 RSC.
- B.-T. Luo, H. Liu, Z.-J. Lin, J. Jiang, D. S. Shen, R. Z. Liu, Z. Ke and F. S. Liu, Organometallics, 2015, 34, 4881 CrossRef CAS.
- S. Islam and I. Larrosa, Chem.–Eur. J., 2013, 19, 15093 CrossRef CAS PubMed.
- Y. Liang and S. F. Wnuk, Molecules, 2015, 20, 4874 CrossRef CAS PubMed.
- H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497 CrossRef CAS PubMed.
- L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J. Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 13831 CrossRef CAS PubMed.
- L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Erwing and J. Q. Yu, J. Am. Chem. Soc., 2019, 141, 14870 CrossRef CAS PubMed.
- L. Hu, P. X. Shen, Q. Shao, K. Hong, J. X. Qiao and J. Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134 CrossRef CAS PubMed.
- H. T. Kim, H. Ha, G. Kang, O. S. Kim, H. Ryu, A. K. Biswas, S. M. Lim, M. H. Baik and J. M. Joo, Angew. Chem., Int. Ed., 2017, 56, 16262 CrossRef CAS PubMed.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746 RSC.
- C. A. Salazar, J. J. Gair, K. N. Flesch, I. A. Guzei, J. C. Lewis and S. S. Stahl, Angew. Chem., Int. Ed., 2020, 59, 10873 CrossRef CAS PubMed.
- B. F. Shi, N. Maugel, Y. H. Zhang and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed.
- Y. Lu, D. H. Wang, K. M. Engle and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed.
- L. Y. Liu, K. S. Yeung and J. Q. Yu, Chem.–Eur. J., 2019, 25, 2199 CrossRef CAS PubMed.
- N. Mandal and A. Datta, J. Org. Chem., 2020, 85, 13228 CrossRef CAS PubMed.
- S. Qian, Z. Q. Li, M. Li, S. R. Wisniewski, J. X. Qiao, J. M. Richter, W. R. Ewing, M. D. Eastgate, J. S. Chen and J. Q. Yu, Org. Lett., 2020, 22, 3960 CrossRef CAS PubMed.
- S. Liu, Z. Zhuang, J. X. Qiao, K. S. Yeung, S. Su, E. C. Cherney, Z. Ruan, W. R. Ewing, M. A. Poss and J. Q. Yu, J. Am. Chem. Soc., 2021, 143, 21657 CrossRef CAS PubMed.
- J. Cheng, L. Xiao, S. Qian, Z. Zhuang, A. Liu and J. Yu, Angew. Chem., Int. Ed., 2022, 61, 1 Search PubMed.
- K. Naksomboon, C. Valderas, M. Gómez-Martínez, Y. Álvarez-Casao and M. Á. Fernández-Ibáñez, ACS Catal., 2017, 7, 6342 CrossRef CAS PubMed.
- B. J. Gorsline, L. Wang, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 9605 CrossRef CAS PubMed.
- L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821 CrossRef CAS PubMed.
- Y. Álvarez-Casao and M. Á. Fernández-Ibáñez, Eur. J. Org. Chem., 2019, 8, 1842 CrossRef.
- K. Naksomboon, J. Poater, F. M. Bickelhaupt and M. Á. Fernández-Ibáñez, J. Am. Chem. Soc., 2019, 141, 6719 CrossRef CAS PubMed.
- O. Tamura, T. Yamada, Y. Hashimoto, K. Tanaka and N. Morita, J. Org. Chem., 2020, 85, 12315 CrossRef PubMed.
- D. Zhao, W. Wang, S. Lian, F. Yang, J. Lan and J. You, Chem.–Eur. J., 2009, 15, 1337 CrossRef CAS PubMed.
- I. Beckers, B. Krasniqi, P. Kumar, D. Escudero and D. De Vos, ACS Catal., 2021, 11, 2435 CrossRef CAS.
- A. Lüttringhaus and H. W. Dirksen, Angew. Chem., Int. Ed., 1964, 3, 260 CrossRef.
- K. L. Billingsley, K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 3484–3488 CrossRef CAS PubMed.
- C. W. McNamara, M. C. S. Lee, C. S. Lim, S. H. Lim, J. Roland, A. Nagle, O. Simon, B. K. S. Yeung, A. K. Chatterjee, S. L. McCormack, M. J. Manary, A.-M. Zeeman, K. J. Dechering, T. R. S. Kumar, P. P. Henrich, K. Gagaring, M. Ibanez, N. Kato, K. L. Kuhen, C. Fischli, M. Rottmann, D. M. Plouffe, B. Bursulaya, S. Meister, L. Rameh, J. Trappe, D. Haasen, M. Timmerman, R. W. Sauerwein, R. Suwanarusk, B. Russell, L. Renia, F. Nosten, D. C. Tully, C. H. M. Kocken, R. J. Glynne, C. Bodenreider, D. A. Fidock, T. T. Diagana and E. A. Winzeler, Nature, 2013, 504, 248 CrossRef CAS PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2021, 12, 537–541 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data, methods for XAS measurements, synthetic procedures and chemical details of isolated compounds. See DOI: https://doi.org/10.1039/d2sc04911b |
| This journal is © The Royal Society of Chemistry 2023 |