
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable continuous flow synthesis of β-aminocarbonyls via acid-catalyzed hydration of N-Boc-2-azetines†
Michael
Andresini‡
 a,
Marco
Colella‡
a,
Roberta Savina
Dibenedetto
a,
Elena
Graziano
a,
Giuseppe
Romanazzi
b,
Andrea
Aramini
c,
Leonardo
Degennaro
*a and
Renzo
Luisi
*a
a,
Marco
Colella‡
a,
Roberta Savina
Dibenedetto
a,
Elena
Graziano
a,
Giuseppe
Romanazzi
b,
Andrea
Aramini
c,
Leonardo
Degennaro
*a and
Renzo
Luisi
*a
aFLAME-Lab, Flow Chemistry and Microreactor Technology Laboratory Department of Pharmacy – Drug Sciences University of Bari “A. Moro”, Via E. Orabona 4 – 70125 Bari, Italy. E-mail: renzo.luisi@uniba.it
bDICATECh, Politecnico di Bari, Via E. Orabona, 4 – 70125 Bari, Italy
cDepartment of Discovery, Dompé Farmaceutici S.p.A, Via Campo di Pile, L'Aquila I-67100, Italy
First published on 13th September 2023
Abstract
In this work we report a waste-minimized continuous flow process for the synthesis of β-aminocarbonyls through hydration of N-Boc-2-azetines promoted under acid-catalysed conditions. The greenness of the process is harnessed by the use of flow technology for handling a biphasic segmented flow and the use of an inline liquid–liquid separator allowing the recovery of both the eco-friendly organic solvent (CPME) and the reusable acidic aqueous phase.
Introduction
β-Aminocarbonyl compounds serve as valuable building blocks in the synthesis of biologically and medicinally significant molecules.1–3 These compounds are also privileged scaffolds in the field of peptidomimetics and molecular recognition.4 In fact, β-aminocarbonyl compounds are valuable intermediates in organic synthesis, especially in the production of β-amino alcohols,5 β-amino acids,6 and lactams,7 which are crucial in various pharmaceutical and natural product synthesis.8 Due to the wide range of applications of β-aminocarbonyls, there is an ongoing effort to develop new synthetic methods for preparing these compounds. One of the most widely used method for their preparation is the popular Mannich reaction, involving the multi-component condensation of a nonenolizable aldehyde, an enolizable carbonyl compound and a primary or secondary amine.9 The original strategy has then been deeply studied and further adapted, leading to the development of diverse Mannich-type transformations,10 that allow the preparation of β-aminocarbonyls from different substrates including aldimines and α-imino esters (Scheme 1a).11,12 Otherwise, aza-Michael reactions of α,β-unsaturated carbonyls with various nitrogen nucleophiles provide access to the target products (Scheme 1b).13 Considering their wide application in synthesis, the preparation of β-aminocarbonyls bearing a cleavable function linked to the nitrogen is highly valuable. In this regard, Peelen and coworkers described the ZnCl2 catalyzed addition of carbon nucleophiles to Fmoc-protected N,O-acetals en route to Fmoc-protected β-aminocarbonyls (Scheme 1c).14 Very recently, Wang et coworkers reported the successful combination of Ni and HAT photocatalysis for the ring opening of N-tosyl styrenyl aziridines with aldehydes furnishing N-tosyl β-amino ketones (Scheme 1d).15 Thereof, the development of novel methods for the preparation of protected β-aminocarbonyls would be impactful. Moreover, it is desirable to establish synthetic protocols that not only guarantee high product yields but also meet sustainability requirements. In this work, we report an efficient approach for the preparation of N-Boc-protected β-aminocarbonyls through the acid-catalyzed hydration of strained 2-substituted azetines.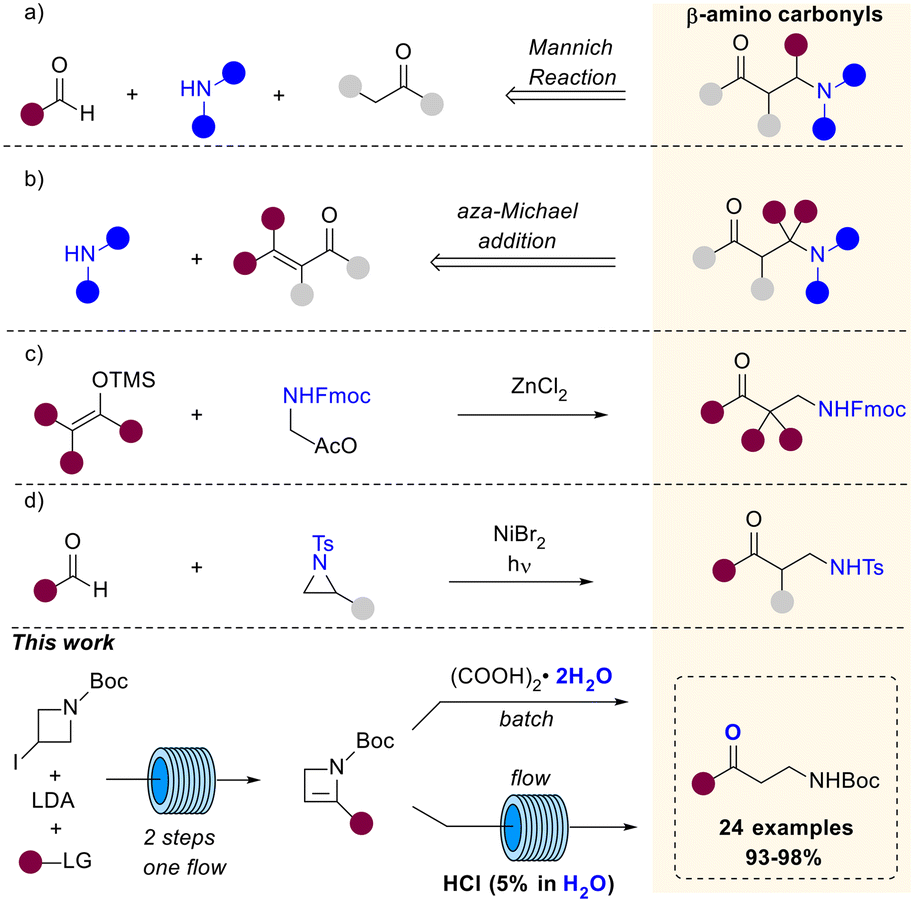 | ||
| Scheme 1 Preparation of β-aminocarbonyl compounds. a) Mannich-type reaction; b) aza-Michael addition; c) ZnCl2 catalyzed addition of carbon nucleophiles to Fmoc-protected N,O-acetals; d) photocatalyzed ring opening of N-tosyl styrenyl aziridines with aldehydes; bottom: this work. | ||
First, we investigated the reactivity of 2-substituted azetines with water in the presence of oxalic acid as the proton donor. Subsequently, we targeted the development of a more sustainable method for such transformation, employing eco-friendly CPME (cyclopenthyl methyl ether) as the organic solvent and reusable solution of HCl 5% (w/w) in water for catalysis. The optimized protocol combines a continuous segmented flow process followed by the inline liquid–liquid separation that enables the recovery of both the acidic aqueous solution and the organic solvent. The sustainability of the method, which furnished the desired products in excellent yields, was evaluated through the calculation of various green metrics.
Results and discussion
Batch hydration of 2-azetines
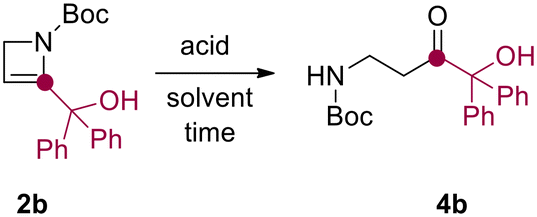
Relying on our experience in the field of flow technology,16–22 we have recently developed a sustainable continuous flow protocol to access either C3-functionalized azetidines or C2-functionalized 2-azetines by using 3-iodo N-Boc-2-azetidine as common starting material.23 This synthetic protocol was proved to be highly efficient and robust for the preparation of several C2-functionalized 2-azetines. It is worth pointing out that N-Boc-2-azetines are strained endocyclic ene-carbamates. Hence, we envisioned that these compounds could be hydrated en route to β-aminocarbonyls. In this context, Baumann and co-workers reported the preparation of γ-aminoketones from N-Boc-pyrrolidines via water addition to an N-acyliminium intermediate.24 Surprisingly, while the ring-opening of azetidine leading to α-aminoketones and diversely β-functionalized amines have been recently reported, the acid-promoted hydration of 2-azetines to the putative β-aminocarbonyls remains unexplored.25,26The initial stages of this synthetic progress were marked by the unexpected discovery that NMR analysis of purified compound 2b using commercially available CDCl3 yielded a changing ratio of 2b and 4b over time. However, employing CDCl3 aged on K2CO3 allowed for obtaining NMR spectra of pure 2b, indicating that the acidic content and residual water in commercial CDCl3 likely influenced the results. In order to confirm our intuition, we performed a NMR monitoring over time (i.e. 0–12 h) on azetine 2a using commercial CDCl3 (Scheme 2). As expected, the quantitative conversion of 2a to β-aminoaldehyde 4a was observed after 12 h. NMR monitoring revealed the formation of a reaction intermediate after 6 hours, and complete disappearance of the signals of 2a after 9 hours (Scheme 2). This intermediate was thoroughly analyzed (see ESI†) and ascribed to 2-hydroxy azetidine 3. This transient hemiaminal 3 completely disappeared in the next 3–6 hours leading to exclusive formation of β-aminoaldehyde 4a (Scheme 2). Similarly, the dissolution of azetine 2b in commercial CDCl3 resulted in a faster and quantitative conversion to β-aminoketone 4b, suggesting that this reaction could smoothly proceed also employing varied 2-substituted-2-azetines. With this evidence in hand, we decided to optimize the reaction and develop a general synthetic protocol for the easy formation of β-aminocarbonyl derivatives.
 | ||
| Scheme 2 NMR evidence of the acid-promoted ring opening of 2-azetines in CDCl3. | ||
First, we engaged in the choice of a suitable solvent for this process. Due to the scarce solubility of azetine 2b in either alcoholic solvents (i.e. MeOH or EtOH) or ethereal solvents (i.e. THF or Et2O), and based on the NMR evidence, we considered exploring the hydration of 2-azetines using an halogenated solvent. Notably, the use of dichloromethane ensured a satisfying solubility. At this stage of our research, we excluded the use of aqueous acidic solutions in order to avoid working with a multiphasic liquid–liquid process which would have been problematic to scale-up using traditional batch reactors.27 Therefore, azetine 2b, selected as the model substrate, was transformed under different reaction conditions as reported in Table 1. The role of water was assessed and, as expected, the reaction did not proceed in absence of water even for prolonged reaction times (entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid | Equiv. | Solventb | Time | 4b yielda (%) |
| a Calculated by 1HNMR on the crude reaction mixture. b Treated with Na2SO4 or distilled over CaH2. c Unidentified side-products detected. | |||||
| 1 | — | DCM | 16 h | — | |
| 2 | Oxalic acid·2H2O | 0.5 | DCM | 1 h | 80 |
| 3 | Oxalic acid·2H2O | 0.5 | DCM | 1.5 h | 98 |
| 4 | Oxalic acid·2H2O | 0.5 | DCM/H2O | 1.5 h | 98 |
| 5 | Acetic acid | 1.0 | DCM | 1.5 h | 70c |
| 6 | TsOH | 0.1 | CDCl3 | 0.5 h | 50 |
| 7 | TsOH | 1.0 | CDCl3 | 2 h | 90 |
We reasoned that oxalic acid dihydrate (0.5 equiv.) could have been the best promoter for the reaction providing the needed proton and the water. In fact, complete conversion of 2b was observed after 1.5 hours (entries 3 and 4). The use of not-anhydrous acetic acid resulted in 70% yield of the desired product 4b (entry 5), and several unidentified side products that made difficult the purification step. The use of TsOH in sub-stoichiometric (0.1 equiv.) or stoichiometric (1.0 equiv.) amount was additionally tested in an NMR tube (see ESI†) providing evidence on the role of water and enabling the preparation of 4a in 50% and 90% yield after 0.5 and 2 h respectively (entries 6 and 7). The observed results from optimization experiments and NMR monitoring supported the hydration steps reported in Scheme 3.
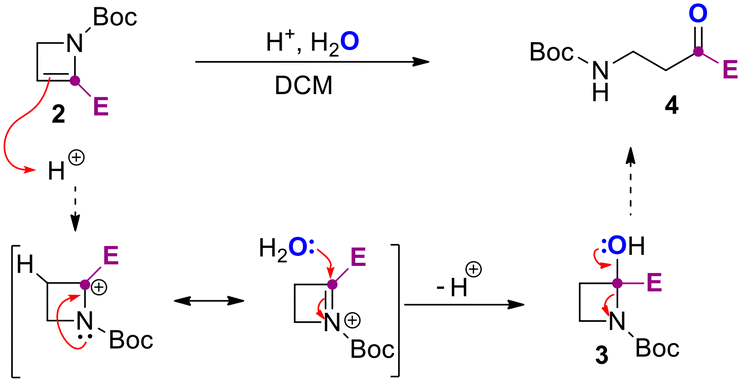 | ||
| Scheme 3 Acid-promoted ring opening of 2-azetines with water. | ||
In detail, azetine 2 is first protonated at C3 to provide a stabilized azetidinium ion that undergo nucleophilic addition of water leading to hemiaminal 3. Spontaneous ring-opening of 3 provide the β-aminocarbonyl derivative 4. Next, by using the optimal conditions reported in Table 1, entry 3, we decided to explore the scope of the reaction for several C2-functionalized azetines 2 prepared using our previously developed continuous flow protocol.22 As reported in Scheme 4, the process proceeds under mild conditions using oxalic acid in CH2Cl2 at 25 °C allowing for the preparation of β-aminocarbonyls 4a–z in almost quantitative yields. The protocol was found to be highly efficient regardless of the nature of the substituent installed at the C2 position of the N-Boc-2-azetines (2a–z) and occurred with high chemoselectivity and functional group tolerance. For example, in the case of 4d no potentially competitive hydration of the double bond was observed. The reaction proved effective in the presence of haloalkyl moieties (4q, r) as well as with aliphatic, aromatic and cyclic alcohols (4b–i, 4k–p), also bearing additional functional group as in the case of 4n, obtained from azetine 2n whose relative configuration has been previously determined.28 Heterosubstituted ketones 4s–u could also be obtained in very high yields (>95%) leaving untouched the heterocyclic core. Interestingly, an unusual and difficult to make acylsilane 4j could be easily prepared in good yield using this strategy. To further test the usefulness of this methodology, the β-aminopropanoyl group was installed on natural or biorelevant chiral scaffolds such as (−)-carvone and O-methyl-estrone. The corresponding products 4w and 4z formed quantitatively and with high chemoselectivity and preserving the stereochemistry. Remarkably, the method was successfully applied also to a 2,3 disubstituted azetine (2v) obtaining the corresponding β-aminoketone 4v.
 | ||
| Scheme 4 Reaction scope. | ||
Continuous flow synthesis of β-aminocarbonyls
After evaluating the feasibility of the method for synthesizing a wide range of β-aminocarbonyls from 2-azetines, we focused on optimizing a procedure that would fulfill sustainability and safety requirements. In fact, the use of dichloromethane and oxalic acid arises safety concerns mostly related to toxicity issues. To this end, we first considered the use of a more sustainable and safe solvent and identified CPME (cyclopentyl methyl ether) as a promising candidate also in terms of solubility capability.29 Unfortunately, the reaction of azetine 2b with oxalic acid in CPME was poorly effective with a conversion to 4b of 15% (Table 2, entry 1). At this point, we decided to consider a different catalytic system to promote the hydration of 2-azetines in more sustainable media. Pleasingly, azetine 2b could be quantitively transformed in compound 4b employing an acidic aqueous solution (HCl, 5% (w/w)) and performing the reaction in CPME under biphasic conditions (Table 2, entry 2). With this preliminary results in hand, we investigated the development of a continuous flow method which could ensure reduced reaction times while overcoming the technological issues associated with the scalability of multiphasic transformations, including the separation and recycle of immiscible solvents. In this regard, the efficient scaling-up of several Brønsted acid-catalyzed reactions has already been achieved through the development of continuous flow methods. These methods enable enhanced control over key reaction parameters, which often leads to improved time-productivities.30–32 Moreover, the microfluidic technology allows an efficient control over multiphasic reactions, as for immiscible liquid–liquid systems, by producing a reproducible segmented flow regime. Notably, the use of an inline liquid–liquid separator can ensure the efficient separation of two phases before collecting the product.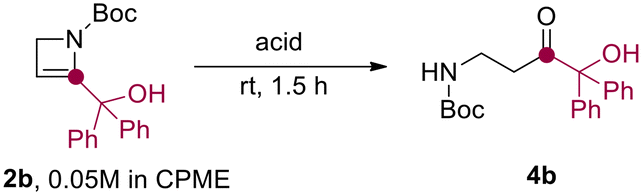
Bearing this in mind, we explored the possibility to separate, recover and reuse the acidic solution after the flow reaction, enhancing the sustainability of the method. To achieve our goal, we built a continuous flow set up including a final inline separation step performed by a Zaiput separator (SEP-10) equipped with a hydrophobic PTFE OB-900 membrane. The efficiency of the liquid–liquid separation was first assessed by fluxing CPME and HCl(aq) 5% (w/w) delivered by two syringe pumps, producing a segmented flow after mixing through a T-shape micromixer (inner volume = 500 μL), into the system (see ESI† for further details). With our delight, no substantial cross-contamination between the organic and the aqueous phases was observed by NMR analysis of the recovered solutions. Therefore, we envisioned that this flow set-up would have been useful for the efficient transformation of 2-azetines into the corresponding β-aminocarbonyls with subsequent recovery of both the organic and aqueous phases. Hence, the flow process was optimized using azetine 2b (0.05 M in CPME) as model substrate under different conditions in terms of residence time and reaction temperatures. As reported in Table 3, the adopted conditions allowed to build a 3D contour map that allowed the identification of the most suitable flow conditions for this transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | Residence time (Rt, min) | Total flow rate (mL min−1) | R 1 volume (mL) | 4b yielda (%) |
| a Calculated by 1HNMR on the crude reaction mixture. b Isolated yield. | |||||
| 1 | 25 | 2.5 | 0.8 | 2 | 0 |
| 2 | 25 | 5.0 | 0.4 | 2 | 21 |
| 3 | 25 | 8.0 | 0.25 | 2 | 49 |
| 4 | 25 | 16.0 | 0.25 | 4 | 65 |
| 5 | 40 | 2.5 | 0.8 | 2 | 22 |
| 6 | 40 | 5.0 | 0.4 | 2 | 51 |
| 7 | 40 | 8.0 | 0.25 | 2 | 87 |
| 8 | 40 | 16.0 | 0.25 | 4 | 98b |
| 9 | 60 | 2.5 | 0.8 | 2 | 98b |
| 10 | 60 | 5.0 | 0.4 | 2 | 98b |
| 11 | 60 | 8.0 | 0.25 | 2 | 98b |
| 12 | 60 | 16.0 | 0.25 | 4 | 98b |
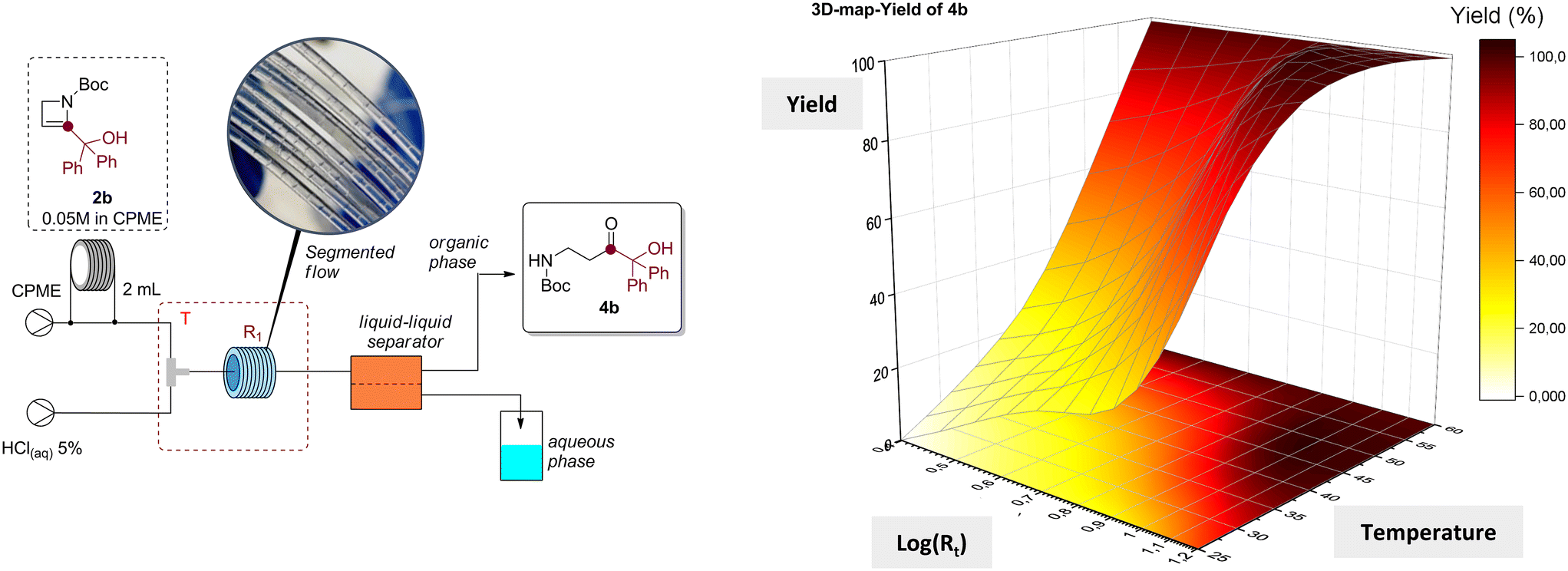
The target product 4b was obtained after evaporation of the organic phase in almost quantitative yield operating at 60 °C, reducing the residence time to 2.5 minutes (Table 3, entries 9–12). Full conversion was likewise obtained with 16 minutes of residence time and a temperature of 40 °C (Table 3, entry 8), while the quantitative formation of 4b could not be achieved within 16 minutes of residence time, and operating at room temperature (Table 3, entries 1–4). To guarantee the wider applicability of the protocol and avoid the potential formation of undesired products at higher temperature, we decided to explore the scope of the reaction at 40 °C and with a residence time of 16 minutes (Table 3, entry 8). To assess the efficiency of the method, selected azetines 2 were transformed under continuous flow conditions. Therefore, β-aminocarbonyls 4g, 4h, 4m, 4o, 4t, 4u, 4w and 4z were obtained in almost quantitative yields after distillation and recovery of CPME (Scheme 5).
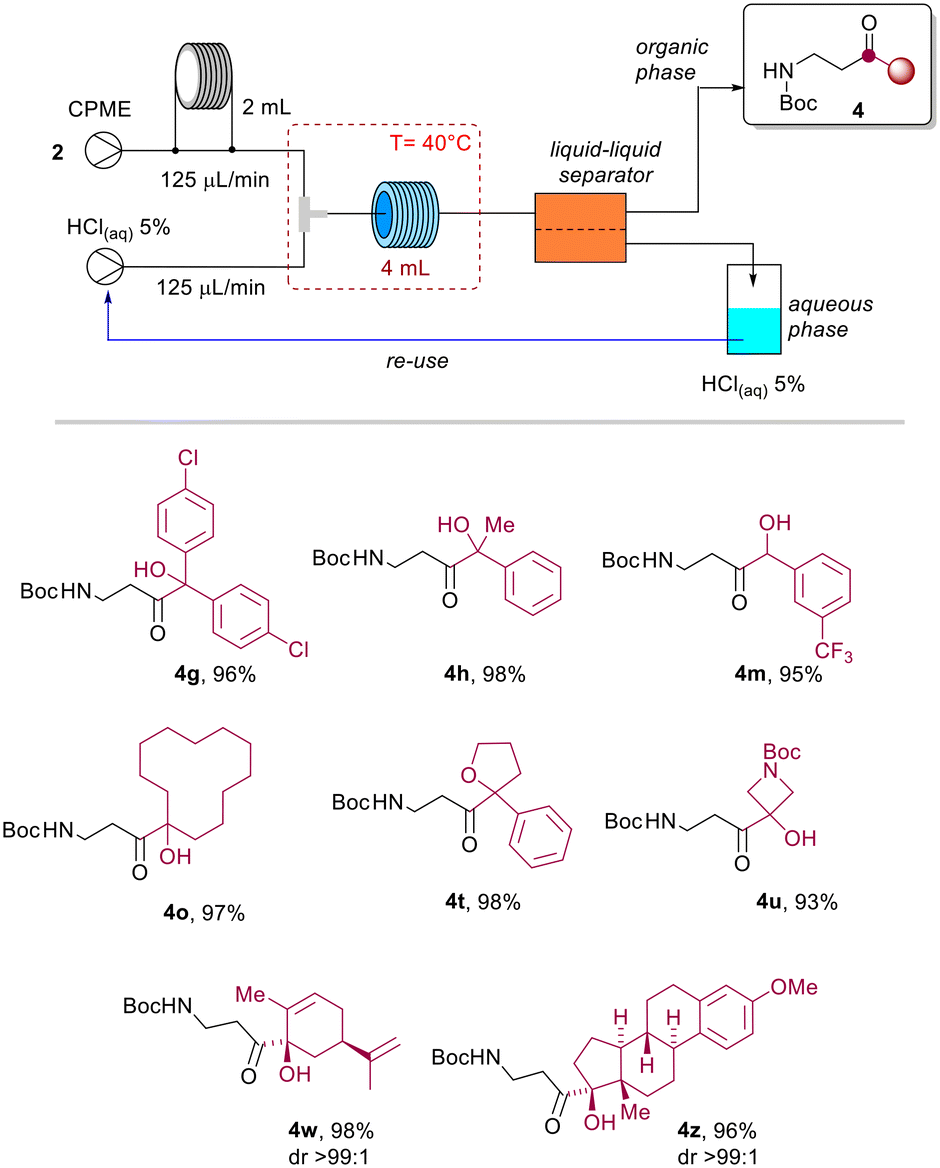 | ||
| Scheme 5 Scope of the continuous flow protocol (T = 40 °C, Rt = 16 min, R1 = 4 mL), isolated yields after CPME distillation. | ||
Interestingly, the aqueous acidic solution could be directly reused after separation in a closed-loop microfluidic setup. A 2.0 mmol scale reaction was conducted using azetine 2b (Scheme 6), where the aqueous acidic solution at the outlet of the liquid–liquid separator was directly recycled in the system by using a peristaltic pump (see ESI†). The method allowed the preparation of β-aminoketone 4b in 98% yield after CPME distillation (99.3% CPME recovered by distillation). It is worth noting that this process effectively combines microfluidic and separation technologies to fulfill the sustainability requirements that are imperative for large-scale chemical manufacturing.
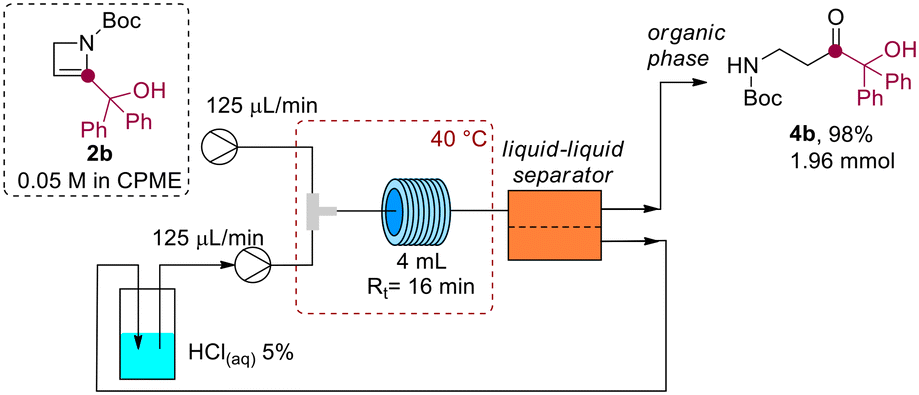 | ||
| Scheme 6 2.0 mmol synthesis of β-aminocarbonyl 4b with inline recycle of acidic solution. | ||
Green metric calculation for the synthesis of 4b
To evaluate the quantity of waste resulting from the optimized hydration of 2b, we computed the E-factor, a metric that expresses the amount of waste produced per unit of the desired product (kg/kg). Pleasingly, an excellent E-factor of 0.346 (being the ideal E-factor = 0) could be calculated for the continuous flow protocol involving the closed-loop liquid–liquid separation and solutions' recovery. In more details, the aqueous HCl 5% (w/w) solution was quantitively recovered after the reaction, while 99.3% of CPME was recovered by distillation. In striking contrast, a higher E-factor of 2.058, indicating a less environmentally friendly approach, was calculated for the batch process involving the use of oxalic acid as the catalyst and DCM as the solvent that was also recovered by distillation (95%). It is worth pointing out that the use DCM poses several concerns in terms of sustainability and hazard that are much better addressed and controlled with the replacement with CPME. To further assess the greenness of the flow method with respect to the batch processing, other metrics were compared, including atom efficiency (AE), material recovery parameter (MRP), reaction mass efficiency (RME), reaction yield (Rxn yield) and the space–time yield (STY), as reported in Fig. 1.33,34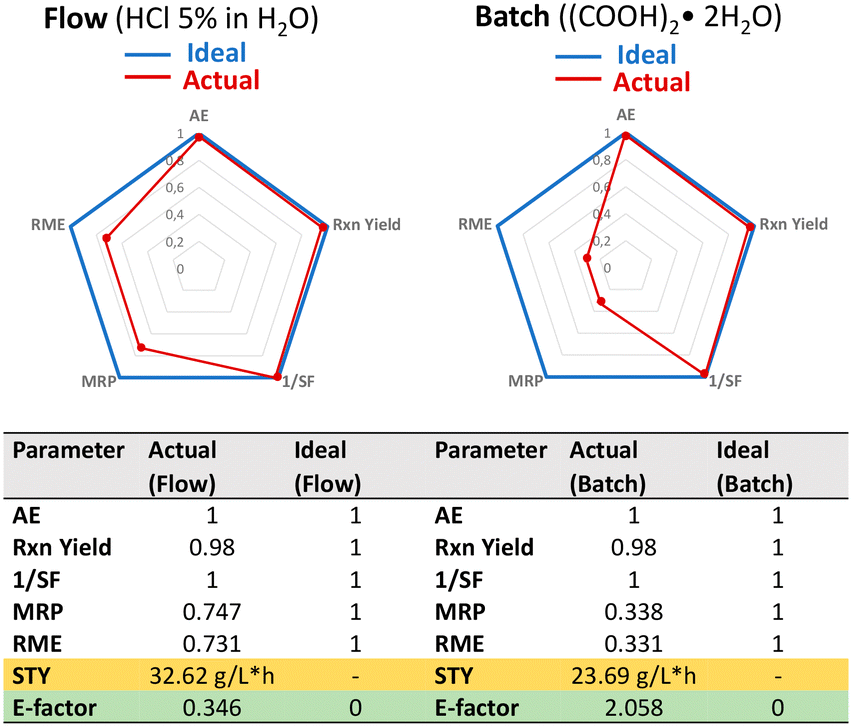 | ||
| Fig. 1 Comparison of green metrics for the batch protocol (oxalic acid catalyzed reaction) and continuous flow protocol (catalyzed by aqueous HCl with inline liquid–liquid extraction) for the preparation of 4b. | ||
The almost quantitative recovery of both the aqueous solution and organic solvent using the flow protocol enhanced the sustainability profile of the method, that approaches the ideal parameters. Due to the full recovery of the catalyst and the more efficient distillation of CPME, both the reaction mass efficiency (RME) and mass recover parameter (MRP) metrics for the flow protocol were improved if compared to the acid-catalyzed batch reaction (Fig. 1). Moreover, an enhanced space–time yield (32.62 g L−1 h−1vs. 23.69 g L−1 h−1) could also be noticed when using the continuous flow process.
Conclusions
In conclusion, our study reports an unprecedented preparation of β-aminocarbonyls via hydration of N-Boc-2-azetines promoted by acid catalysis. The reaction scope has been explored employing more than 20 diversely functionalized 2-azetines, including biorelevant chiral scaffolds, collecting the desired products in excellent yields. Moreover, a waste-minimized and time-saving continuous flow process has been developed, allowing the recovery of both the eco-friendly organic solvent (CPME) and the reusable acidic aqueous phase through inline liquid–liquid separation. The microfluidic technology combined with in-line separation additionally enabled the direct and continuous recycling of the aqueous catalytic phase employing a closed-loop set-up. The sustainability of the flow method has been assessed through the calculation of various green metrics that approached ideal reference values. Further studies concerning the development of catalytic multiphasic methods in microfluidic reactors involving the inline liquid–liquid separation are ongoing in our laboratory and will be presented in due course.Author contributions
R. L. conceptualization and methodology; R. L. and A. A. funding acquisition; R. L., L. D. writing – original draft; M. C., M. A., G. R. data curation, writing – review and editing; E. G., R. S. D. investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Debora Cannillo and Tiziana Cuocci for their support at the beginning of this investigation. This work has been funded by the European Union – Health and Digital Executive Agency (HADEA), project SusPharma grant agreement No 101057430. Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the Health and Digital Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.Notes and references
- M. Garbacz and S. Stecko, Org. Biomol. Chem., 2023, 21, 115–126 RSC.
- M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044 CrossRef PubMed.
- E. F. Kleinmann in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and C. H. Heathcock, Pergamon, Oxford, 1991, vol. 2, p. 89 Search PubMed.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS PubMed.
- C. Wu, B. Ma, G.-Q. Chen and X. Zhang, Chem. Commun., 2022, 58, 12696 RSC.
- Y. Liang, F. Strieth-Kalthoff, P. Bellotti and F. Glorius, Chem Catal., 2021, 1, 1427 CrossRef CAS.
- S. Deketelaere, T. Van Nguyen, C. V. Stevens and M. D'hooghe, ChemistryOpen, 2017, 6, 301 CrossRef CAS PubMed.
- G. Lelais and D. Seebach, Biopolymers, 2004, 76, 206 CrossRef CAS PubMed.
- J. F. A. Filho, B. C. Lemos, A. S. de Souza, S. Pinheiro and S. J. Greco, Tetrahedron, 2017, 73, 6977 CrossRef.
- R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940 RSC.
- J. Jiang, H.-D. Xu, J.-B. Xi, B.-Y. Ren, F.-P. Lv, X. Guo, L.-Q. Jiang, Z.-Y. Zhang and W.-H. Hu, J. Am. Chem. Soc., 2011, 133, 8428 CrossRef CAS PubMed.
- J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2013, 135, 11799 CrossRef CAS PubMed.
- A. Y. Rulev, Eur. J. Org. Chem., 2023, 26(26) DOI:10.1002/ejoc.202300451 , in press.
- A. E. Hartman, C. L. Brophy, J. A. Cupp, D. K. Hodge and T. J. Peelen, J. Org. Chem., 2009, 74, 3952 CrossRef CAS PubMed.
- P. Fan, Y. Jin, J. Liu, R. Wang and C. Wang, Org. Lett., 2021, 23, 7364 CrossRef CAS PubMed.
- P. Musci, M. Colella, A. Sivo, G. Romanazzi, R. Luisi and L. Degennaro, Org. Lett., 2020, 22, 3623 CrossRef CAS PubMed.
- M. Colella, A. Tota, Y. Takahashi, R. Higuma, S. Ishikawa, L. Degennaro, R. Luisi and A. Nagaki, Angew. Chem., 2020, 59, 10924 CrossRef CAS PubMed.
- M. Andresini, S. Carret, L. Degennaro, F. Ciriaco, J. Poisson and R. Luisi, Chem. – Eur. J., 2022, 28(59) DOI:10.1002/chem.202202066 , in press.
- P. Musci, M. Colella, M. Andresini, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2022, 58, 6356–6359 RSC.
- M. Spennacchio, M. Colella, M. Andresini, R. S. Dibenedetto, E. Graziano, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2023, 59, 1373–1376 RSC.
- M. Andresini, L. Degannaro and R. Luisi, Beilstein J. Org. Chem., 2021, 17, 203–209 CrossRef CAS PubMed.
- P. Musci, T. Keutz, F. Belaj, L. Degennaro, D. Cantillo, C. O. Kappe and R. Luisi, Angew. Chem., 2021, 133, 6465–6469 CrossRef.
- M. Colella, P. Musci, D. Cannillo, M. Spennacchio, A. Aramini, L. Degennaro and R. Luisi, J. Org. Chem., 2021, 86, 13943–13954 CrossRef CAS PubMed.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, J. Org. Chem., 2021, 86, 14199–14206 CrossRef CAS PubMed.
- R.-C. Raclea, P. Natho, L. A. T. Allen, A. J. P. White and P. J. Parsons, J. Org. Chem., 2020, 85, 9375–9385 CrossRef CAS PubMed.
- M. Raulin, B. Drouillat, J. Marrot, F. Couty and K. Wright, Tetrahedron Lett., 2022, 94, 153710 CrossRef CAS.
- T. von Keutz, D. Cantillo and C. O. Kappe, J. Flow Chem., 2019, 9, 27–34 CrossRef CAS.
- E. Graziano, D. Cannillo, M. Spennacchio, P. Musci, L. Pisano, M. Andresini and M. Colella, Tetrahedron Green Chem, 2023, 1, 100003 CrossRef.
- U. Azzena, M. Carraro, L. Pisano, S. Monticelli, R. Bartolotta and V. Pace, ChemSusChem, 2018, 12, 40–70 CrossRef PubMed.
- A. S. Jadhav and R. V. Anand, Eur. J. Org. Chem., 2017, 2017, 3716–3721 CrossRef CAS.
- S. Miñoza, W.-C. Ke, Y.-Y. Yu, P. K. Keerthipati, K.-C. Chang, W.-C. Kao, Z.-N. Tsai and H.-H. Liao, Green Chem., 2022, 24, 9157–9167 RSC.
- D. Rigo, G. Fiorani, A. Perosa and M. Selva, ACS Sustainable Chem. Eng., 2019, 7, 18810–18818 CrossRef CAS.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
- J. Andraos and M. Sayed, J. Chem. Educ., 2007, 84, 1004 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00342f |
| ‡ M. A. and M. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |