
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The α-alkylation of ketones in flow†
Ella
Cooper‡
,
Emma
Alcock‡
,
Mark
Power
and
Gerard
McGlacken
 *
*
School of Chemistry and Analytical and Biological Chemistry Research Facility, University College Cork, T12 YN60 Cork, Ireland. E-mail: g.mcglacken@ucc.ie
First published on 11th May 2023
Abstract
The α-deprotonation and alkylation of ketones is a fundamental transformation in organic chemistry. However, the apparent simplicity of this process belies its complexity. Oftentimes experimental conditions are non-ideal, and yields are low. Herein, we directly target these issues, and provide a continuous flow methodology which leads to excellent yields, reduces reaction time, avoids cryogenic temperatures, minimises exposure to alkyllithiums/alkylhalides, and can be scaled-out.
The α-deprotonation and alkylation of ketones is a fundamental transformation in organic chemistry, taught at undergraduate level.1 Ketone lithium enolates are utilised in particular,2 and are applied ‘pervasively’3 both in academia2 and industry.4 The abundance of commercially available ketones which are potentially enolisable (>10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000)5 bolsters the applicability of simple alkylation protocols.
000)5 bolsters the applicability of simple alkylation protocols.
Although a conceptually simple transformation, the practical α-alkylation of a ketone (Fig. 1) is plagued with issues. Firstly, the transformation can be low yielding.6–8 This is usually as a result of either: i) incomplete enolisation, ii) incomplete alkylation or iii) problematic side reactions. Aldol-type reactions/condensations, O-alkylation, dialkylation and persistent presence of starting ketone, are the most common issues. To alleviate these problems and effect some control over the reactivity of highly energetic lithium enolate intermediates, cryogenic reaction conditions (usually −78 °C) are employed. Efficient ketone alkylation remains a key goal and a highly rewarding endeavour for organic chemists.9
 | ||
| Fig. 1 The α-alkylation of ketones: issues alleviated in continuous flow. | ||
Seminal reports by Enders and Corey10 described an alternative approach for the synthesis of α-substituted ketones. In this work, N,N-dimethylhydrazones (DMHs) were used as ketone surrogates allowing access to a range of α-substituted ketones in good yields. Despite the widespread application of the DMH strategy,11 the use of dimethylhydrazine in stoichiometric quantities, and issues around the formation of toxic by-products formed upon ketone reinstatement, makes this alternative protocol a very unattractive choice. The classic Stork enamine protocol12 has advantages, but again is not a direct alkylation of ketones.
Overall, we envisaged that the direct alkylation of ketones could be improved upon, and a number of the issues (vide supra) could be obviated by using continuous flow technology. In addition to the challenges associated with the previously discussed issues, reactor clogging due to aggregation of organometallic speciation,13,14 was a concern of ours, but we anticipated that use of a peristaltic pump, at least during the process of alkylation and LiBr formation, would obviate these issues.
Listed as one of IUPACs top emerging technologies in chemistry in 2019,15 continuous flow chemistry has emerged from an enabling technology to a new platform for improved chemistry. The development of continuous flow chemistry can provide a plethora of benefits, including the discovery of new reactivity patterns, improved reaction efficiency, enhanced safety, and scalability.16–20 Pioneering work by Yoshida21 encapsulates these benefits via the use of organolithiums in flow.22
An excellent report by Kappe involving the continuous flow alkylation of esters has been reported.23 Specific issues “especially in the case of enolates derived from ketones” was noted by Kappe. The pointed divergent reactivity of ketones and esters (related to the comparatively lower pKa24 and higher electrophilicity25 of ketones) requires new methodology/engineering development, and a scale-out for both reactions in flow has yet to be reported to the best of our knowledge.
Herein, we document a successful continuous flow methodology for the synthesis of α-alkylated ketones in good to excellent yields. Numerous advantages over the corresponding batch reaction are detailed and include: i) reduced reaction times; ii) elimination of cryogenic conditions, iii) increased safety profile; and iv) facile scale-out.
Results and discussion
Initially, deprotonation of propiophenone using LDA26 and α-alkylation using benzyl bromide (BnBr) was chosen as the model system for our optimisation (Scheme 1). In batch, this transformation uses temperatures as low as −78 °C, and reaction times of up to 20 hours. This is a problematic reaction and, in our hands, we achieve variable yields of 25–45% in batch.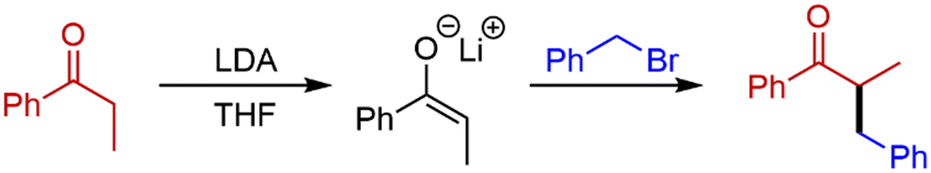 | ||
| Scheme 1 The α-alkylation of propiophenone using BnBr. | ||
Our initial continuous flow set-up consisted of two reaction zones: i) enolate formation and ii) electrophilic addition. The ketone substrate was delivered to the system via a commercially available HPLC/piston pump (Vapourtec R-series). With the aid of a second HPLC/piston pump, a solution of commercially available LDA was added to the system via a sample loop. The alkylating agent was introduced via a peristaltic pump (Vapourtec R-series). A 10 mL dual-core cooled reactor coil and a 10 mL heated reactor coil were used for all reactions. PFA reactor tubing was utilised, and the reagents were sequentially mixed using Teflon T-mixers (Table 1). An anhydrous, inert atmosphere and a pressure of 7 bar was maintained throughout.
|
|
||||
|---|---|---|---|---|
| Entry | RC 1 temp (°C) | RC 2 temp (°C) | Residence time (min) | Steady stateb (yield%) |
| a Unless otherwise noted, conditions are as follows: 1 equiv. of propiophenone, 1.5 equiv. of LDA, 1.2 equiv. of BnBr. b Determined by use of 1H NMR with 1,3,5-trimethoxybenzene as internal standard. | ||||
| 1 | −15 | rt | 5 | 0 |
| 2 | −70 | 75 | 5 | 15 |
| 3 | −45 | 75 | 5 | 39 |
| 4 | −30 | 75 | 5 | 53 |
| 5 | −15 | 75 | 5 | 64 |
| 6 | 0 | 75 | 5 | 70 |
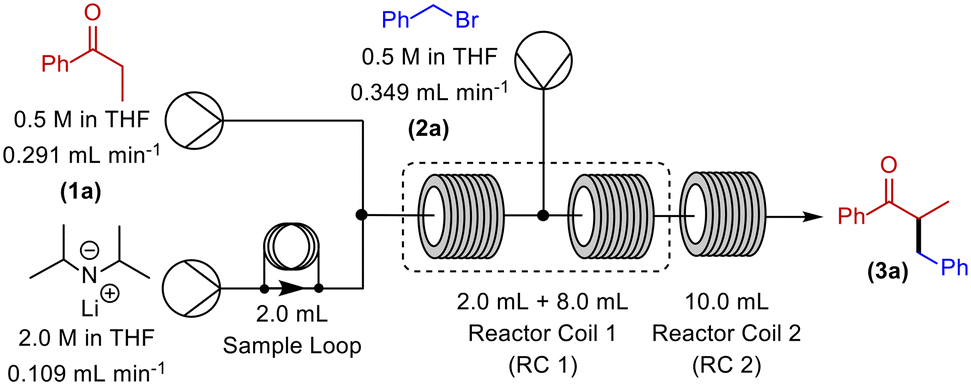
Initially, RC 1 was set at a temperature of −15 °C and RC 2 was set to room temperature (rt) (Table 1, entry 1). Disappointingly however, these conditions resulted in no observed α-benzylated product. Subsequently, we decreased the temperature of RC 1, and increased the temperature of RC 2 (Table 1, entry 2), which gave a steady state yield of 15%. Gratifyingly, a continual increase of the temperature of RC 1 to 0 °C (Table 1, entries 2–6) culminated in a yield of 70% of α-benzylated product, at steady state.
With these partially optimised reaction conditions in hand, we made two further alterations to our reaction conditions. Firstly, due to decreased volatility, we changed the alkylating agent employed to p-tert-butylbenzyl bromide (for comparison, in our hands, we achieved a 44% yield in batch using this electrophile). Secondly, we altered the configuration of the dual-core cooled reactor (RC 1) from (2 mL + 8 mL) configuration to (8 mL + 2 mL) configuration. This configurational swap allowed for a greater residence time for the enolate formation step, which we hoped would result in an increased yield of α-benzylated product. Using our previously optimised conditions, we initiated these reactions with the temperature of RC 1 set to 0 °C, which resulted in a 60% yield of α-benzylated product at steady state (Table 2, entry 1). Further increasing the temperature of RC 1 to room temperature resulted in a slight increase in the yield (Table 2, entry 2). In addition, the yield was further increased by lowering the equivalents of LDA (Table 2, entry 3). Extending the residence time from 5 min to 7.5 min resulted in a significant increase in the yield, to 80%, of α-benzylated product (Table 2, entry 4). Prolonging the residence time further to 10 min, gave a 90% steady state yield of α-benzylated ketone (Table 2, entry 5). However, any further increase in the residence time did not result in any improvement in yield (Table 2, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | LDA (equiv.) | Electrophile (equiv.) | RC 1 temp (°C) | Residence time (min) | Steady stateb (yield%) |
| a Unless otherwise noted, conditions are as follows: 1 equiv. of propiophenone, RC 2 temperature set to 75 °C. b Determined by use of 1H NMR with 1,3,5-trimethoxybenzene as internal standard. | |||||
| 1 | 1.5 | 1.2 | 0 | 5 | 60 |
| 2 | 1.5 | 1.2 | rt | 5 | 63 |
| 3 | 1.2 | 1.5 | rt | 5 | 70 |
| 4 | 1.2 | 1.5 | rt | 7.5 | 80 |
| 5 | 1.2 | 1.5 | rt | 10 | 90 |
| 6 | 1.2 | 1.5 | rt | 15 | 60 |

With the optimised reaction conditions in hand, we next sought to examine a small range of ketones and electrophiles tolerated within this methodology (Table 3). Initially, the reactions of a range of electrophiles, including benzyl and allylhalides were examined in the α-alkylation reaction of propiophenone (Table 3, entries 1–6). Pleasingly, in all cases, good to excellent yields (75–92%) of α-alkylated products were obtained at steady state. Altering the ketone substrate proved equally successful, with excellent yields obtained at steady state (92% & 95%) for the reaction of 2-phenyl acetophenone with both benzyl and allylhalides (Table 3, entries 7 & 8). Finally, we examined the reactivity of 3-pentanone in this methodology. This is a particularly difficult substrate to undergo α-alkylation and can produce very low yields.27 Gratifyingly, a good yield of 55% of the α-benzylated 3-pentanone was achieved at steady state (Table 3, entry 9). No evidence for the formation of additional side-products, for example, dialkylated product, was observed for this substrate. Finally, further purification is often not needed, but steady state yields translated very well to isolated yields (Table 3). In the case of cyclohexanone, the yield using flow chemistry is lower (34%) than that in batch (49%) (see ESI†), and investigations are ongoing to improve yields using symmetric ketones.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ketone | Electrophile | Product | Steady statea (yield%) | Isolatedb (yield%) |
| a Determined by use of 1H NMR with 1,3,5-trimethoxybenzene as internal standard. b Isolated yield after purification by column chromatography on SiO2. | |||||
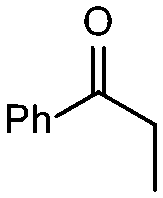
| 1 |
 1a
1a
|
 2b
2b
|
 3b
3b
|
90 | 81 |
| 2 |
 1a
1a
|
 2c
2c
|
 3c
3c
|
75 | 70 |
| 3 |
 1a
1a
|
 2d
2d
|
 3d
3d
|
85 | 72 |
| 4 |
 1a
1a
|
 2e
2e
|
 3e
3e
|
87 | 71 |
| 5 |
 1a
1a
|
 2f
2f
|
 3f
3f
|
92 | 75 |
| 6 |
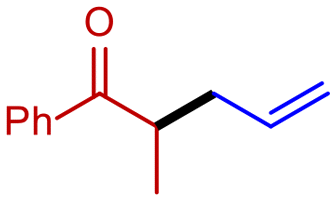
 1a
1a
|
 2g
2g
|
 3f
3f
|
80 | 71 |
| 7 |
 1b
1b
|
 2b
2b
|
 3g
3g
|
92 | 85 |
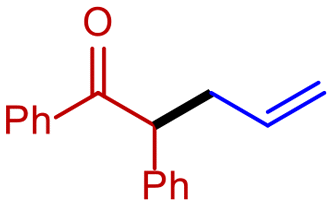
| 8 |
 1b
1b
|
 2f
2f
|
 3h
3h
|
95 | 80 |
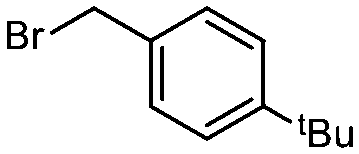
| 9 |
 1c
1c
|
 2b
2b
|
 3i
3i
|
55 | 53 |

Having demonstrated the versatility of this protocol, our next challenge was to scale-out our reaction to generate an appreciable quantity of α-alkylated ketone using this methodology. The α-allylation of 2-phenyl acetophenone was chosen as the reaction to conduct these investigations (Scheme 2). Our previously optimised reactor configuration was altered to include a 10 mL sample loop, allowing for a significantly increased loading of LDA to our system. All other reaction parameters remained unchanged. Pleasingly, a yield of 96% of α-alkylated ketone at steady state28 was achieved for this reaction, over a 45 min collection time.
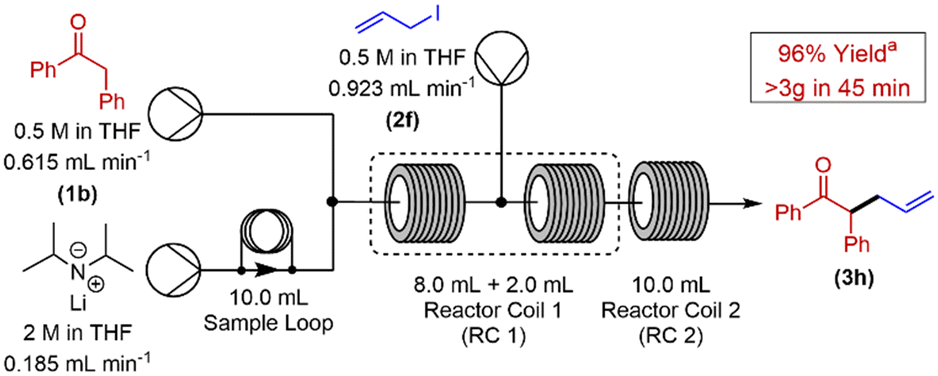 | ||
| Scheme 2 Continuous flow set-up for the scale out of 2-phenyl acetophenone α-allylation. aDetermined by use of 1H NMR with 1,3,5-trimethoxybenzene as internal standard. | ||
Conclusion
In conclusion, we provide an efficient methodology for the alkylation of ketones in continuous flow. Conveniently, commercial LDA can be used as a base in the deprotonation of several exemplar ketones along with a good variety of alkylhalides. The continuous flow protocol with good to excellent yields, provides numerous advantages over the corresponding batch reaction, including the avoidance of cryogenic temperatures, reduction in reaction time, lessening of side product formation and a convenient scale-out (>3 g of material in 45 min).Conflicts of interest
Vapourtec flow company helped in the early development of these reactions.Acknowledgements
Funding was provided through the IRC Enterprise Partnership Scheme (IRC EPSPG/2021/130) co-funded by Pfizer, Ringaskiddy, Ireland as the Industry Partner. We thank the Science Foundation Ireland (SFI/21/FFP-A/8784), and the Synthesis and Solid State Pharmaceutical Centre (SSPC) (SFI/12/RC/2275 and SFI/12/RC2275_P2). We thank D. Guthrie, C. Guthrie and R. C. Moses at Vapourtec for advice, and time on their instruments. We thank Dr Kishore Kumar for proof-reading this manuscript.Notes and references
- J. E. McMurry, Organic Chemistry, Cengage Learning, Boston, 2015 Search PubMed.
- (a) B. M. Trost and I. Flemming, Comprehensive Organic Synthesis, Paragamon, Amsterdam, 1991 Search PubMed; (b) J. Plaquevent, D. Cahard, F. Guillen and J. Green, Science of Synthesis, Georg Thieme Verlag, 2005, vol. 26, pp. 463–511 Search PubMed; (c) A. R. Katritzky and R. J. K. Taylor, Comprehensive Organic Functional Group Transformations II, Elsevier, Amsterdam, 2005 Search PubMed.
- L. R. Liou, A. J. McNeil, A. Ramirez, G. E. S. Toombes, J. M. Gruver and D. B. Collum, J. Am. Chem. Soc., 2008, 130, 4859–4868 CrossRef CAS PubMed.
- (a) R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253–258 CrossRef CAS; (b) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS PubMed; (c) G. Wu and M. Huang, Chem. Rev., 2006, 106, 2596–2616 CrossRef CAS PubMed.
- T. J. Donohoe, B. S. Pilgrim, G. R. Jones and J. A. Bassuto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11605–11608 CrossRef CAS PubMed.
- B. M. Trost and E. Keinan, Tetrahedron Lett., 1980, 21, 2591–2594 CrossRef CAS.
- B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 2846–2847 CrossRef CAS PubMed.
- B. M. Stoltz, N. B. Bennett, D. C. Duquette, A. F. G. Goldberg, Y. Liu, M. B. Loewinger and C. M. Reeves, in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2014, pp. 1–55 Search PubMed.
- F. Mo and G. Dong, Science, 2014, 345, 68–72 CrossRef CAS PubMed.
- (a) E. J. Corey and D. Enders, Tetrahedron Lett., 1976, 17, 11–14 CrossRef; (b) E. J. Corey and D. Enders, Tetrahedron Lett., 1976, 17, 3–6 CrossRef; (c) E. J. Corey and S. Knapp, Tetrahedron Lett., 1976, 17, 4687–4690 CrossRef.
- R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed.
- G. Stork, R. Terrell and J. Szmuszkovicz, J. Am. Chem. Soc., 1954, 76, 2029–2030 CrossRef CAS.
- T. Noël, J. R. Naber, R. L. Hartman, J. P. McMullen, K. F. Jensen and S. L. Buchwald, Chem. Sci., 2011, 2, 287–290 RSC.
- S. L. Poe, M. A. Cummings, M. P. Haaf and D. T. McQuade, Angew. Chem., Int. Ed., 2006, 45, 1544–1548 CrossRef CAS PubMed.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- M. Colella, A. Nagaki and R. Luisi, Chem. – Eur. J., 2020, 26, 19–32 CrossRef CAS PubMed.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- A. Hafner, P. Filipponi, L. Piccioni, M. Meisenbach, B. Schenkel, F. Venturoni and J. Sedelmeier, Org. Process Res. Dev., 2016, 20, 1833–1837 CrossRef CAS.
- J.-I. Yoshida, Basics of Flow Microeactors Synthesis, Springer, Tokyo, 2015 Search PubMed.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 CrossRef CAS.
- T. von Keutz, F. J. Strauss, D. Cantillo and C. O. Kappe, Tetrahedron, 2018, 74, 3113–3117 CrossRef CAS.
- F. G. Bordwell and J. A. Harrelson Jr, Can. J. Chem., 1990, 68, 1714–1718 CrossRef CAS.
- H. Neuvonen, K. Neuvonen, A. Koch, E. Kleinpeter and P. Pasanen, J. Org. Chem., 2002, 67, 6995–7003 CrossRef CAS PubMed.
- We also generated LDA in situ in flow from n-BuLi and diisopropylamine and used it in the formation of α-benzylated propriophenone from propiophenone and BnBr. However the yield was lowered to 42%. Overall, it proved advantageous and more convenient to use titrated commercial LDA, as we observed no reliability issues.
- G. A. Molander and C. del Pozo Losada, J. Org. Chem., 1997, 62, 2935–2943 CrossRef CAS PubMed.
- The product was sufficiently pure at this stage (see ESI† for details and spectra).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00229b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |