
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient neural network models of chemical kinetics using a latent asinh rate transformation†
Felix A.
Döppel
 a and
Martin
Votsmeier
*ab
a and
Martin
Votsmeier
*ab
aTechnical University of Darmstadt, Peter-Grünberg-Straße 8, 64287 Darmstadt, Germany. E-mail: martin.votsmeier@tu-darmstadt.de
bUmicore AG & Co. KG, Rodenbacher Chaussee 4, 63457 Hanau, Germany
First published on 11th July 2023
Abstract
We propose a new modeling strategy to build efficient neural network representations of chemical kinetics. Instead of fitting the logarithm of rates, we embed the hyperbolic sine function into neural networks and fit the actual rates. We demonstrate this approach on two detailed surface mechanisms: the preferential oxidation of CO in the presence of H2 and the ammonia oxidation under industrially relevant conditions of the Ostwald process. Implementing the surrogate models into reactor simulations shows accurate results with a speed-up of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Overall, the approach proposed in this work will significantly facilitate the application of detailed mechanistic knowledge to the simulation-based design of realistic catalytic systems. We foresee that combining this approach with neural ordinary differential equations will allow building machine learning representations of chemical kinetics directly from experimental data.
000. Overall, the approach proposed in this work will significantly facilitate the application of detailed mechanistic knowledge to the simulation-based design of realistic catalytic systems. We foresee that combining this approach with neural ordinary differential equations will allow building machine learning representations of chemical kinetics directly from experimental data.
1. Introduction
Detailed multi-scale modeling provides valuable insights into the complex phenomena of catalytic systems that typically occur in a wide range of time and length scales.1,2 While such highly complex models would allow for rational catalyst and reactor design3,4 they will be computationally infeasible for the foreseeable future.2,5 The computationally most demanding part of those simulations is the solution of the chemical kinetics that often takes 70% to 90% of the computation time for both gas-phase5,6 and surface-reactive1,7 systems. Therefore, there is huge interest in accelerating the kinetic calculations.1,2,8,9 This can be done by tabulating the kinetics or even a time integration step.9,10 Latter is often done for gas-phase reactive systems11–13 because the integration of the stiff ODE system resulting from the gas-phase kinetics is very time-consuming. For heterogeneous catalysis, timescales of surface reactions and the gas phase are usually separable via the steady state approximation.2,14–17 For each simulation time step, the surface kinetics can be solved for steady state separately to avoid unnecessarily small time steps in the computational fluid dynamics simulation. Even with that simplification, evaluating the surface kinetics still poses a severe computational bottleneck.1,7,18–201.1. State of the art
Several works are mapping steady-state solutions of surface kinetics in a tabulation approach. Those maps can be built before a simulation using pre-computed solutions or during a simulation with so-called on-the-fly techniques. Some of the most used on-the-fly techniques exploit prior solutions to estimate new queries like the in situ adaptive tabulation (ISAT)7,8,18,20–23 and piecewise reusable implementation of solution mapping (PRISM)24 technique. In contrast, agglomeration algorithms exploit similarities of open queries to reduce the number of calls to the kinetic solver.8,20,25 Surrogate models like splines have been extensively used to map pre-computed steady-state solutions of chemical kinetics for accelerating reactor simulations14,15,19,26–29 or even subsystems of the reactor.30,31 The (error-based modified) Shepard interpolation approach has been used to replace very demanding but detailed kinetic Monte-Carlo calculations in reactor simulations.17,32,33 Recently, machine learning techniques gained growing attention for modeling kinetic data because they can overcome the so-called curse of dimensionality.34 State of the art methods are random forests35,36 and neural networks,29 both of which have been used for accurate predictions of steady-state surface kinetics.1.2. Data transformation
Not only the model type but also the way data are presented to the model strongly determine its accuracy. Besides scaling also transforming data is known to make models of wide-range data such as chemical kinetics more efficient. Logarithmic transformations have been used for gas-phase mass fractions6,11,37 while preprocessing data as log(r), log(pi) and 1/T is well known to facilitate modeling of surface kinetics.14,15,17,19,27,29 This can be accounted to the fact that it makes the target function more linear over a wide range of reaction conditions17,29 as demonstrated in Eqn. S1–S3 in the ESI.† However, because these transformations rely on the logarithm a problem arises for source terms that are not strictly positive or strictly negative over the entire range of interest. This presents a substantial limitation as most systems of practical interest contain species that are both, consumed and produced depending on the reaction conditions. In our previous work we showed that this limitation can be overcome by modeling the rates of the rate-determining steps.29 Since elementary rates are always positive, they can be modeled accurately using the logarithm. Source terms can then be constructed as a linear combination of the modeled elementary rates. Choosing the rates of the rate-determining steps instead of e.g., the adsorption/desorption reaction avoids subtracting two very similar numbers, which would lead to unfavorable error propagation. However, this approach requires insights into the mechanism that are not always available for example when modeling experimental data. This leaves us with the challenge to accurately model source terms changing sign without prior insights into the reaction mechanism.In contrast to the logarithm, logarithm-like functions like the inverse hyperbolic sine asinh(x) are not limited to positive inputs but can process negative and zero values in a meaningful way. As shown in Fig. 1, asinh(x) behaves logarithmically for values |x| ≫ 1 while it is linear in the interval −1 < x < 1. This function is commonly used to analyze financial data when zero or negative values occur.38–40 Like economic data, the net rates of chemical kinetics usually span many orders of magnitude and assume both, positive and negative values or zero.
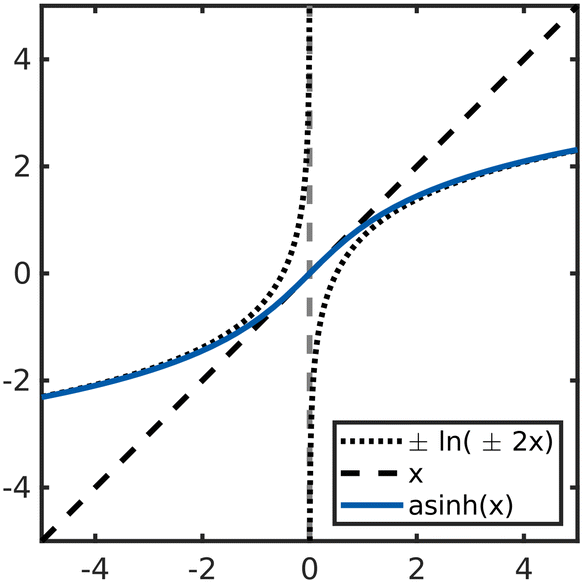 | ||
| Fig. 1 Plot of the asinh(x) function which approximates the logarithm of 2x for large positive and negative inputs while being linear near the origin. | ||
1.3. Scope of this work
In this work, we propose a neural network architecture specialized to efficiently model steady-state solutions of detailed surface kinetics thus removing the computational bottleneck from reactor simulations. It consists of two major points:1. We transform the rates using the logarithm-like function asinh(x) that can be applied to negative values and zero, which is crucial for modeling systems of practical interest e.g., when they include intermediate species.
2. We work with latent (hidden) representations of the transformed data. This means we embed data transformation directly into the model instead of the conventional preprocessing of data, see Fig. 2. This allows minimizing meaningful error metrics like the relative error of reaction rates while preserving the advantage of data transformation. In other words, we avoid spending model capacities to regions that are not important for its application in reactor simulations.
 | ||
| Fig. 2 Comparing conventional and latent training strategy. They differ in how data transformation is applied and which error metric is optimized during training. In both cases, a machine learning model predicts chemical rates using reaction conditions as inputs. The loss is computed to evaluate the prediction accuracy and the model parameters are updated accordingly. Conventionally, the transformation is applied to the data in a preprocessing step. The transformed values asinh(r) are then learned by a standard neural network. The disadvantage of this approach is that during training a loss function with respect to the transformed values has to be used instead of the actual error measure of interest. We propose to work with latent (hidden) representations of the transformed data. This means that a model with standard fully connected layers learns a latent representation of the transformed rates. Afterwards, the inverse of the transformation function is applied as a custom output activation in the final layer so that outputs represent the original rates. Hence, the error metric of interest can be optimized during training. If all parameters of the transformation function are fixed before the optimization, the inverse transformation can alternatively be implemented in a customized loss function. | ||
With this setup, neural networks can accurately model wide-range data changing sign such as chemical kinetics. No prior knowledge about the reaction mechanism is required, paving the way for learning kinetics directly from experimental data or highly detailed first principles simulations. The approach is validated by reactor simulations. The preferential oxidation of CO in the presence of H2 is simulated in a plug-flow reactor showing a speed-up of 100000 when using neural networks instead of solving the full mechanism. Further, we model the ammonia oxidation under conditions of the Ostwald process.
2. Methodology
2.1. Preferential oxidation of CO
Reaction rates rj (s−1) are calculated as
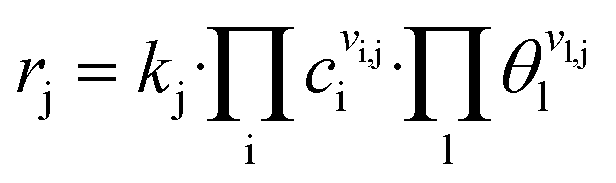 | (1) |
 | (2) |
 | (3) |
For each reaction condition given by a temperature and the partial pressures of CO, CO2, H2, H2O and O2, steady state surface coverages are calculated. This is done by integrating eqn (4) in time until dθl/dt = 0. Gas composition and temperature are assumed to be constant during this process. The obtained surface coverages are used in eqn (5) to calculate steady state source terms ṡi.29,42 Numerically, integration is performed using the DASPK solver43 with an integration time of 107 s, a relative tolerance of 10−6 and an absolute tolerance of 10−50.
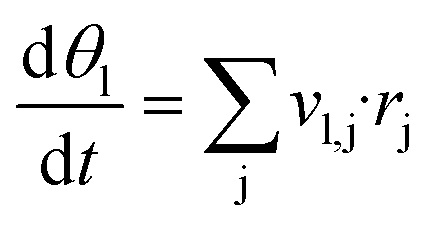 | (4) |
 | (5) |
| Quantity | Unit | Minimum | Maximum | Scaling |
|---|---|---|---|---|
| T | K | 280 | 600 | Reciprocal |
| p(H2) | atm | 8 × 10−2 | 8 × 10−1 | Logarithmic |
| p(O2) | atm | 1 × 10−7 | 4 × 10−2 | Logarithmic |
| p(H2O) | atm | 4 × 10−2 | 4 × 10−1 | Logarithmic |
| p(CO) | atm | 1 × 10−7 | 4 × 10−2 | Logarithmic |
| p(CO2) | atm | 4 × 10−2 | 4 × 10−1 | Logarithmic |
 | (6) |
A total pressure of 1 atm, a site concentration cPt of 26.3 mol m−3, a reactor length of 1 m divided into 200 cells and a gas velocity of 1 ms−1 are used. The resulting residence time τ is 1 s. The feed consists of 40% H2, 1% O2, 10% H2O, 1% CO and 10% CO2 with N2 as the balance species.
If conditions outside the input range defined in Table 1 occur, they are set to the corresponding minimum or maximum values to avoid extrapolation of the neural network models.
2.2. Ammonia oxidation in the Ostwald process
Reaction rates rj (s−1) are calculated as
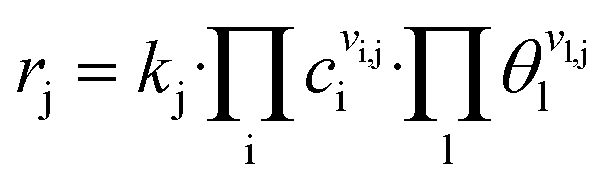 | (7) |
For each reaction condition given by a temperature and the partial pressures of NH3, O2, H2O, NO, N2O and N2, steady state surface coverages are calculated. This is done by integrating eqn (8) in time until dθl/dt = 0. Gas composition and temperature are assumed to be constant during this process. The obtained surface coverages are used in eqn (9) to calculate steady state source terms ṡi (mol m−2 s−1) using the site density Γ which is assumed to be 2.3 × 10−5 mol m−2.
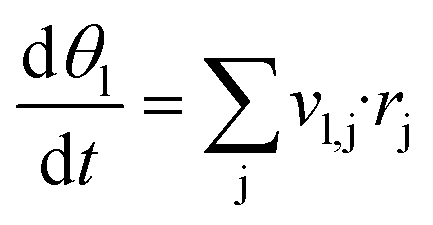 | (8) |
 | (9) |
Numerically, integration is performed using MATLAB's ode15s solver45 with an integration time of 1015 s, a relative tolerance of 10−8 and an absolute tolerance of 10−50.
The rate constants for surface reactions ksurfj are calculated as
 | (10) |
 | (11) |
 | (12) |
 | (13) |
 | (14) |
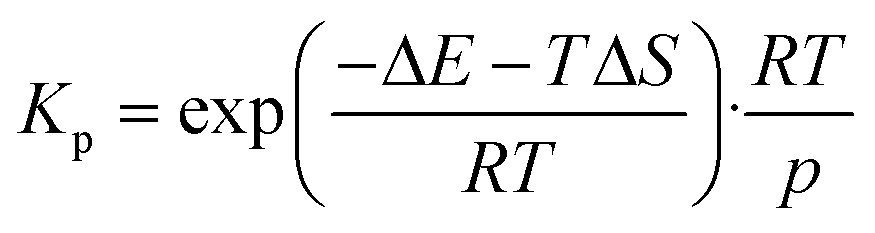 | (15) |
 | (16) |
We chose this mechanism because in contrast to simpler ammonia oxidation mechanisms considered in our earlier works15,26,28,30 it is more detailed and does not neglect the consumption of several gas species. In consequence, all species except NH3, H2O and N2 show source terms changing sign in the range of reaction conditions considered. Therefore, it is not possible to rely on modeling only strictly positive source terms and compute all other species source terms from the atom balance. Rather, at least one species with sign changing source terms has to be modeled for use in a reactor simulation. We focus on predicting the source terms of NH3, N2 and N2O.
| Quantity | Unit | Minimum | Maximum | Scaling |
|---|---|---|---|---|
| T | K | 103 | 1.3 × 103 | Reciprocal |
| p(NH3) | Pa | 0.5 | 6 × 104 | Logarithmic |
| p(O2) | Pa | 1.25 × 104 | 1 × 105 | Logarithmic |
| p(H2O) | Pa | 0.5 | 9 × 104 | Logarithmic |
| p(NO) | Pa | 0.5 | 6 × 104 | Logarithmic |
| p(N2O) | Pa | 0.5 | 3 × 103 | Logarithmic |
2.3. Neural networks
Neural networks are implemented using PyTorch.48 All neural networks are fully connected, use tanh activation and have an equal number of nodes in all hidden layers. The number of nodes per hidden layer is chosen to meet a total number of adjustable parameters up to 5000. Hidden layer weights are initialized using PyTorch's kaiming uniform function.| xT = (T/1 K)−1 | (17) |
| xp,i = log(pi/1 atm) | (18) |
 | (19) |
 | ||
| Fig. 3 Scheme of the recommended architecture. The neural network takes reaction conditions in form of temperature and partial pressures as input. Those values are transformed and linearly scaled before being fed into conventional hidden layers with tanh activation. The last layer holds a single node per gas phase species and contains y, a latent representation of the transformed target values. A hyperbolic sine activation is applied to obtain outputs in the form of source terms. | ||
The preprocessed thermo-chemical state is fed to the hidden layer(s) which are fully connected and use tanh activation. Per key species (CO and O2 for test case 1 and NH3, N2O and N2 for test case 2) we train a separate neural network with a single output node which contains a latent representation of the transformed source terms y = asinh (ṡ/z), see eqn (20). This node uses the inverse of this function z sinh(y) as activation to restore outputs in the form of the original source term target values ṡ. The only parameters to be learned are the weights in and out of the hidden layer(s) and optionally the parameter z of the sinh activation. To mimic the behavior of the well-known logarithmic transformation, we will choose the parameter z in a way that all modeled rates lie within the logarithmic part of the function, i.e. by assigning it the smallest absolute source term occurring in the training data.
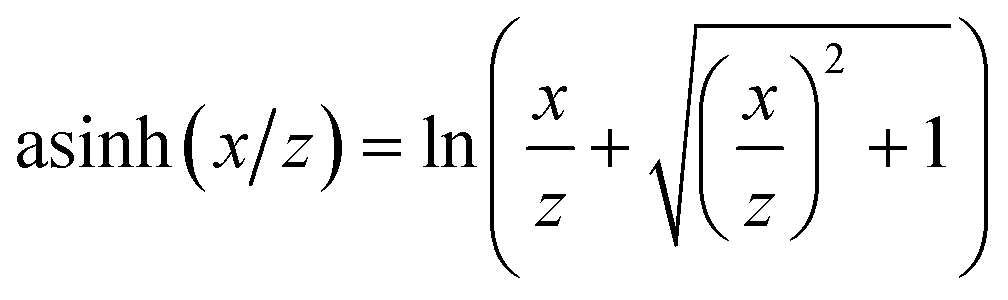 | (20) |
When alternatives to the hyperbolic sine function are discussed, the output activation is replaced by either z·nl−1 (y), gpow−1 (y, n) or exp(y).
In contrast to the latent transformation approach, the conventional approach computes transformed target values in a preprocessing step (asinh (ṡ/z) in our case) and uses a standard fully connected neural network to learn the transformed target values. Consequently, during training the differences between exact and estimated transformed values, e.g. measured by the root-mean-square error of transformed values, are minimized instead of a typical error metric of interest like the relative error of the source terms.
Direct modeling means dropping both, the input transformation in eqn (17) and (18) as well as the output activation. However, the original steady state source terms are used as targets.
For test case 1 we model CO and O2 source terms. The other species are calculated as follows:
 | (21) |
 | (22) |
 | (23) |
For test case 2 we model NH3, N2 and N2O source terms. The other species are calculated as follows:
 | (24) |
 | (25) |
 | (26) |
000 input–output pairs of reaction conditions and resulting steady state source terms for both test cases. The training set contains 25000, the validation set contains 5000 and the test set contains another 5000 input–output pairs. Every input–output pair is contained in only one of the three data sets. The data for the preferential oxidation test case are identical to the ones used in our previous work.29
We do not perform excessive hyper-parameter tuning as the focus of this work lies on the general modeling strategy for steady state source terms.
 | (27) |
![[script L]](https://www.rsc.org/images/entities/i_char_e144.gif) rel (eqn (28)) is minimized when source terms are used as target data.
rel (eqn (28)) is minimized when source terms are used as target data. | (28) |
The root mean squared absolute loss abs (eqn (29)) of transformed values is minimized when using transformed source terms as targets.
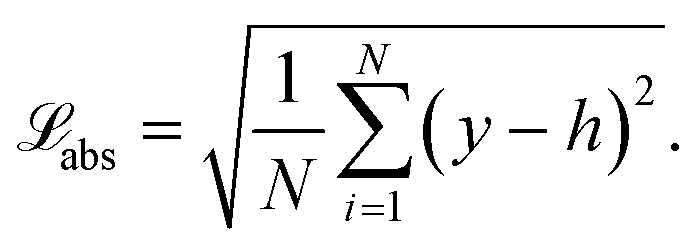 | (29) |
2.4. Hard- and software
Datasets for this work were produced using MATLAB Version R2021a and a faster fortran-based in-house code. Neural network training and inference were performed using PyTorch Version 1.10. Prediction times were measured using a Ryzen 7 5800X CPU @3800 MHz and a NVIDIA GFORCE RTX 3070 GPU running Linux Mint 20.3 as an operating system and averaged over 1000 identical calculations.3. Results and discussion
We discuss the proposed latent hyperbolic sine transformation in detail using the preferential oxidation of CO as a showcase mechanism. The obtained models are validated in a plug-flow reactor simulation and compared to our previous work based on approximating the rates of the rate-determining steps.29 A DFT-based mechanism for the ammonia oxidation under conditions of the industrial Ostwald process is used as a second test case. Finally, we discuss alternatives to the hyperbolic sine function.3.1. Test case 1: preferential oxidation of CO
The latent hyperbolic sine transformation will be presented in detail for the preferential oxidation of CO in the presence of H2 with a platinum catalyst. This system is important in H2 production for fuel cell applications42 and has been the first detailed surface mechanism modeled with neural networks in literature.29 It can be described by three global reactions:| 2CO + O2 → 2CO2 (CO oxidation) |
| 2H2 + O2 → 2H2O (H2 oxidation) |
| CO + H2O ⇄ CO2 + H2 (water-gas shift) |
As the mechanism contains five gas phase species and three elements, at least 5 − 3 = 2 species source terms must be modeled to fully describe the reaction progress in the system. Analogous to the procedure in our previous work29 we train a separate neural network to model the source terms of both key species O2 and CO each. The net production rates of all other species are derived via the atom balance. Therefore, the mass balance is always exactly closed.
The equilibrium of the CO and H2 oxidation reactions is fully on the right side, so that the source term of O2 is negative under all relevant reaction conditions. Consequently, a logarithmic transformation can be applied to model the O2 source terms.14,15,17,19,27,29 As typical for systems of practical relevance, other species in the mechanism (including CO) change the sign of their source term depending on the reaction conditions. For those, logarithmic transformation cannot be applied in a meaningful way. We propose the latent hyperbolic sine transformation, overcoming this limitation. Fig. 4a shows a histogram of the distribution of CO source term values while Fig. 4b shows the same data on a logarithmic scale.
 | ||
| Fig. 4 Histogram of the CO source term distribution: (a) in linear scale, and (b) in logarithmic scale. | ||
 | ||
| Fig. 5 Comparing the CO source term prediction accuracy of different neural network training strategies on 5000 unseen test data randomly sampled from the input range of Table 1. Using a standard feed-forward neural network without data transformation (“direct” modeling) does not yield accurate results. Using the asinh transformation conventionally, i.e. in a preprocessing step, reduces the prediction errors considerably. When the asinh transformation is implemented in a latent fashion, the models are even more accurate and yield application ready predictions with relative errors below 1%. All models contain about 5000 parameters distributed over one (625 nodes), three (48 nodes each) or five hidden layers (34 nodes each) and were trained with 25000 data points. | ||
Fig. 6 shows how the prediction error of the latent transformation models scales with the number of parameters. For example, application ready models with relative prediction errors of 1% can be obtained with 15 nodes each in five hidden layers (≈1000 parameters) and less than 15 minutes of training time (see Fig. S1 in the ESI†). As neural networks are usually deployed with orders of magnitude more parameters and layers, all models used in this work can be considered small. Wan et al. for example used about 180000 parameters for modeling chemical kinetics.51
 | ||
| Fig. 6 Relative prediction error of CO source terms dependent of the total number of learnable parameters in a neural network using the latent hyperbolic sine transformation. | ||
In our previous work we proposed another method dealing with chemical source terms changing sign by approximating the rates of the rate-determining step.29 As shown in Fig. S4 in the ESI,† it yields more accurate models than the latent hyperbolic sine transformation proposed in this work. That is achieved by exploiting detailed insights from a reaction path analysis. An analysis, however, is not feasible when dealing with experimental data or highly complex computational models. In contrast, the latent hyperbolic sine transformation is designed to work without any previous knowledge about the mechanism and therefore poses the first method to obtain accurate and lightweight surrogate models for detailed surface kinetics when dealing with experimental data or highly complex computational models.
In summary, the latent hyperbolic sine transformation works well because of two major points: 1. The inverse hyperbolic sine transformation brings target data into a similar order of magnitude and leads to a more linear input–output relation. 2. Using the transformation in a latent fashion gives full control over the training objective while maintaining the advantages of data transformation.
Fig. 7 shows that the concentration profiles obtained from the neural network models (dotted line) cannot be visually separated from the exact solution (full lines). The lower part of Fig. 7 shows, that the relative difference between both solutions is about 1% or lower. Note however, that calculating the neural network estimation of the source terms is approximately 50000 times faster than evaluating the exact steady state kinetics, see Table S5 in the ESI.† Using a consumer grade graphics card for inference increases the speed-up to 100000. As the obtained accuracy is well above that of the kinetic parameters, these results suggest that our method can be applied to much larger and more complex reaction mechanisms.
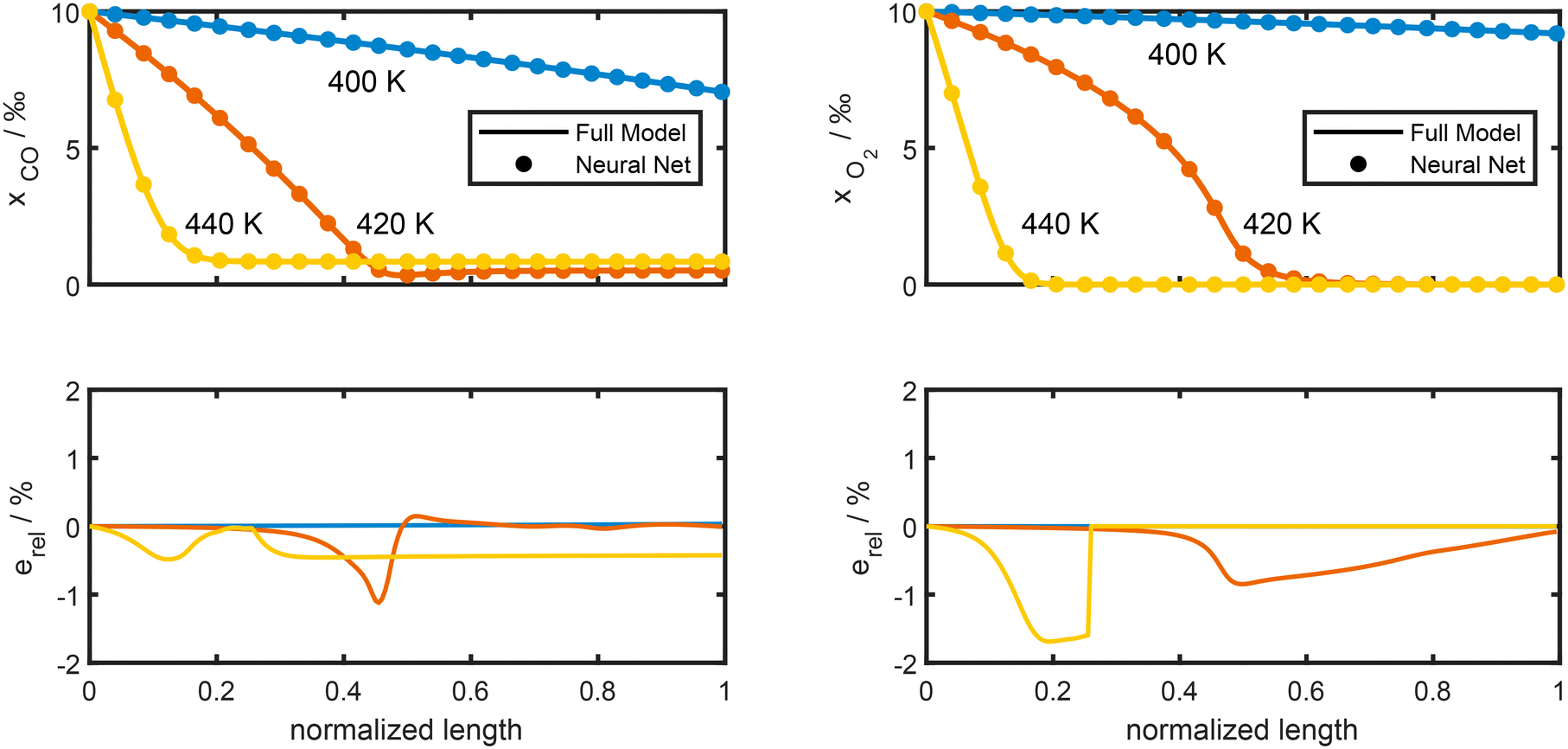 | ||
| Fig. 7 Plug-flow reactor model of the preferential oxidation of CO in H2 rich environments at three different temperatures. The upper part shows CO and O2 molar fractions along the reactor length. The neural network solution (dotted lines) cannot be visually separated from the exact solution (full lines). The lower part shows the relative difference between both solutions. None of the conditions shown were part of the 25000 training data which were randomly sampled from the input range of Table 1. Feed composition and other details are described in section 2.1.3. | ||
In summary, the neural network models obtained with latent hyperbolic sine transformation are well suited for replacing the computationally expensive steady state source term calculations associated with heterogeneous catalysis. They yield accurate solutions and speed-up the calculations by factor 100000.
3.2. Test case 2: ammonia oxidation in the Ostwald process
To test the generality of the latent hyperbolic sine transformation approach we apply it to a second detailed surface mechanism. We consider the DFT-based ammonia oxidation mechanism from Ma and Schneider44 for the Ostwald process under industrially relevant conditions.Neural networks are used to predict the steady state source terms as a function of temperature and gas composition. The training data set covers all industrially relevant reaction conditions of a medium pressure ammonia oxidation reactor and contains 25000 samples. As the mechanism contains six gas phase species and three elements, at least 6 − 3 = 3 species source terms (e.g. NH3, N2 and N2O) must be modeled to fully describe the reaction progress in the system.
Since ammonia is burned at high temperatures, NH3 source terms are negative for all training conditions. N2 shows only positive source terms because it is the thermodynamically favored product. Consequently, NH3 and N2 source terms can be modeled using the well-known logarithmic transformation. For both species a separate lightweight neural network with 63 nodes in a single hidden layer (≈500 parameters) is trained, resulting in relative prediction errors around 0.1%.
N2O source terms, however, do change sign and the logarithmic transformation cannot be applied. Again, using a standard feed-forward neural network without data transformation (“direct” modeling) does not yield usable models as it leads to relative prediction errors near 100%. Using the asinh transformation in the conventional way increases accuracy to about 15%. The latent variant of the asinh transformation performs even better leading to errors near 1%, see Fig. 8a. See section 2.3.1 for a detailed comparison between the three modeling approaches.
 | ||
| Fig. 8 Comparing different neural network training strategies. All models contain 63 nodes in a single hidden layer (≈500 parameters) and were trained with 25000 data points. (a) Comparing the N2O source term prediction accuracy of different neural network training strategies on 5000 unseen test data randomly sampled from the input range of Table 2. Using a standard feed-forward neural network without data transformation (“direct” modeling) does not yield accurate results. Using the asinh transformation conventionally, i.e. in a preprocessing step, reduces the prediction errors to approximately 15%. When the asinh transformation is implemented in a latent fashion, the models yield application ready predictions with relative errors near 1%. (b) Models are validated by comparing product selectivities at unseen Ostwald process conditions and zero ammonia conversion (10% NH3 in air at 5 bar). The model using asinh data transformation in the conventional way covers the general trend of selectivities but fails at lower temperatures. In contrast, the model using the latent asinh transformation is in excellent agreement with the full model over the whole temperature range. Besides N2O, neural network models of NH3 and N2 are used to fully describe the reaction progress in the system and use the well-known logarithmic transformation. | ||
The neural network models are validated by computing the product selectivities at Ostwald process conditions and zero ammonia conversion. None of these conditions are part of the training data set. Analogous to the original publication of the mechanism44 mass transfer was not considered. The model based on the newly proposed latent transformation is in excellent alignment with the results from the full kinetic model, see Fig. 8b. In contrast, the model based on the conventional data transformation shows significant deviations. Most notably it overestimates the N2O selectivity by an order of magnitude at lower temperatures.
Overall, the latent asinh transformation allows lightweight and therefore computationally cheap neural network models ready for use in reactor simulations.
3.3. Alternatives to the hyperbolic sine
While in this work, we focused on the inverse hyperbolic sine, there are other functions able to flatten unfavorable data distributions such as wide-range data changing sign.The Bi-Symmetric log transformation nl(x) was introduced by Webber to depict data that cover a wide range of scales and have both positive and negative components.52–54 It is defined as
 | (30) |
Power transformations pose another way to normalize the skewed distribution of wide-range data that assume both, positive and negative values. To this end, a generalized n-th root of x can be defined as
| gpow(x,n) = sgn(x)·|x|1/n | (31) |
The three functions asinh(x), nl(x) and gpow(x,n) perform similarly for CO source term predictions when applied with the latent approach, see Fig. S5a in the ESI.† However, for O2 source term predictions gpow(x,n) performs significantly worse than asinh(x), nl(x) and log(x), see Fig. S5b in the ESI.† This might be attributed to the fact, that the logarithmic rate transformation that is commonly used can be motivated by the Arrhenius equation and the power law expressions the rate calculations are based on and might therefore be ideal for transforming source term data without sign changes. Consequently, deviating from logarithm-like behavior can be expected to have a negative effect on accuracy. Since the adjustable parameter variant did not lead to higher accuracy, we conclude that the lowest target value occurring in the training data is a good initial guess for z. However, there seems to be no obvious initial guess for the parameter n of the generalized power function. The data shown use n = 12 for CO and n = 18 for O2 source terms as these values provided the most accurate results in an initial testing phase.
In summary, all three functions studied in this work are suitable for latent transformation of steady state source terms changing sign and perform about similar. We suggest using the inverse hyperbolic sine function to get started as nearly all numerical libraries provide an efficient implementation.
4. Conclusions and future work
This work proposes the latent hyperbolic sine transformation for efficient neural network models of detailed surface kinetics. As the standard logarithmic transformation is not applicable to source terms changing sign we introduced the asinh function that behaves similar to the logarithm but can deal with negative numbers and zero. Further, we work with latent (hidden) representations of the transformed data. This means we embed data transformation directly into the model instead of the conventional preprocessing of data. This allows to decouple the error metric optimized during training from the data transformation and therefore increases the model accuracy significantly.The development of the new approach is demonstrated using two test cases. The first test case is a detailed surface reaction mechanism describing the oxidation of CO in presence of H2 as well as the water-gas shift reaction. It includes 5 gas species, 9 surface species, and 36 reactions. Models are validated by implementing them in plug-flow reactor simulations. While the neural network-assisted solution is visually not separable from the exact solution, it is computed 100000 times faster. Neural network training used 25000 data points and takes less than an hour on a consumer grade PC.
The second test case is a detailed surface mechanism based on density functional theory calculations of the ammonia oxidation on platinum. The latent hyperbolic sine transformation increases model accuracy significantly and allows using very small and thus computationally efficient neural networks in detailed reactor simulations.
In our previous work, we reached similarly good results by performing a reaction path analysis to exploit the detailed insights into the reaction mechanism available.29 The present work, however, can produce accurate models of detailed surface kinetics without any previous knowledge about the underlying mechanism.
Currently, there is huge interest in determining kinetic models directly from experimental data. Especially neural ODEs56 are promising for generating a representation of the reaction kinetic ODEs from experimental data directly.57–59
In accordance with our findings, it is reported that the parameter z of the inverse hyperbolic sine transformation (eqn (20)) can substantially affect regression results.39 While several works developed strategies for finding the best value others even argue not to use this transformation at all, emphasizing that the optimal parameter value is not given by theory.40 In this work we embedded the transformation function into a neural network. This allows optimizing all transformation parameters automatically during training and could potentially be used to identify the optimal parameter value for related problems like economic analyses.
The concept of latent data transformation is not limited to neural networks and can be used in all machine learning methods that allow customizing the loss function. For this purpose we define the custom loss function * that applies the inverse of the desired data transformation f−1(x) to the model outputs h before comparing them to the target values y in the conventional loss function , see eqn (32). This approach yields the same results as embedding f−1(x) in the output layer of a neural network but does not allow optimizing transformation parameters like z from eqn (20) during training.
 | (32) |
Overall, the approach proposed in this work will not only significantly facilitate the application of detailed mechanistic knowledge in the simulation-based design of realistic catalytic systems, but it also presents a first step towards learning detailed surface kinetics directly from experimental data.
Author contributions
Felix A. Döppel: conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing – original draft, writing – review and editing and visualization. Martin Votsmeier: supervision, writing – review and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by BMBF in the framework of the project ML-MORE (contract 05M20RDA). The authors gratefully acknowledge the computing time provided to them on the high-performance computer Lichtenberg at the NHR Centers NHR4CES at TU Darmstadt. This is funded by the Federal Ministry of Education and Research, and the state governments participating on the basis of the resolutions of the GWK for national high performance computing at universities.References
- D. Micale, C. Ferroni, R. Uglietti, M. Bracconi and M. Maestri, Chem. Ing. Tech., 2022, 94, 634–651 CrossRef CAS.
- G. D. Wehinger, M. Ambrosetti, R. Cheula, Z.-B. Ding, M. Isoz, B. Kreitz, K. Kuhlmann, M. Kutscherauer, K. Niyogi, J. Poissonnier, R. Réocreux, D. Rudolf, J. Wagner, R. Zimmermann, M. Bracconi, H. Freund, U. Krewer and M. Maestri, Chem. Eng. Res. Des., 2022, 184, 39–58 CrossRef CAS.
- M. Bracconi, Chem. Eng. Process., 2022, 181, 109148 CrossRef CAS.
- G. D. Wehinger, Chem. Ing. Tech., 2022, 94, 1215–1216 CrossRef.
- T. S. Brown, H. Antil, R. Löhner, F. Togashi and D. Verma, Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), Springer International Publishing, 2021, vol. 12761, LNCS, pp. 23–39 Search PubMed.
- V. F. Nikitin, I. M. Karandashev, M. Y. Malsagov and E. V. Mikhalchenko, Acta Astronaut., 2022, 194, 376–382 CrossRef CAS.
- M. Bracconi, M. Maestri and A. Cuoci, AIChE J., 2017, 63, 95–104 CrossRef CAS.
- E. A. Daymo, M. Hettel, O. Deutschmann and G. D. Wehinger, Chem. Eng. Sci., 2022, 250, 117408 CrossRef CAS.
- S. Barwey and V. Raman, Energies, 2021, 14, 2710 CrossRef.
- X. Han, M. Jia, Y. Chang and Y. Li, Combust. Flame, 2022, 238, 111934 CrossRef CAS.
- A. J. Sharma, R. F. Johnson, D. A. Kessler and A. Moses, AIAA Scitech 2020 Forum, Reston, Virginia, 2020 Search PubMed.
- M. Haghshenas, P. Mitra, N. Dal Santo and D. P. Schmidt, Energies, 2021, 14, 1–15 CrossRef.
- J. Blasco, N. Fueyo, C. Dopazo and J. Ballester, Combust. Flame, 1998, 113, 38–52 CrossRef CAS.
- M. Votsmeier, Chem. Eng. Sci., 2009, 64, 1384–1389 CrossRef CAS.
- M. Votsmeier, A. Scheuer, A. Drochner, H. Vogel and J. Gieshoff, Catal. Today, 2010, 151, 271–277 CrossRef CAS.
- O. Deutschmann, Handbook of Heterogeneous Catalysis, Wiley-VCH, 2nd edn, 2008, ch. Comutatio, pp. 1811–1821 Search PubMed.
- S. Matera, M. Maestri, A. Cuoci and K. Reuter, ACS Catal., 2014, 4, 4081–4092 CrossRef CAS.
- J. M. Blasi and R. J. Kee, Comput. Chem. Eng., 2016, 84, 36–42 CrossRef CAS.
- B. Partopour and A. G. Dixon, Comput. Chem. Eng., 2016, 88, 126–134 CrossRef CAS.
- R. Uglietti, M. Bracconi and M. Maestri, React. Chem. Eng., 2020, 5, 278–288 RSC.
- S. Mazumder, Comput. Chem. Eng., 2005, 30, 115–124 CrossRef CAS.
- S. Pope, Combust. Theory Modell., 1997, 1, 41–63 CrossRef CAS.
- R. Uglietti, M. Bracconi and M. Maestri, React. Chem. Eng., 2018, 3, 527–539 RSC.
- S. R. Tonse, N. W. Moriarty, N. J. Brown and M. Frenklach, Isr. J. Chem., 1999, 39, 97–106 CrossRef CAS.
- S. Rebughini, A. Cuoci, A. G. Dixon and M. Maestri, Comput. Chem. Eng., 2017, 97, 175–182 CrossRef CAS.
- A. Scheuer, W. Hauptmann, A. Drochner, J. Gieshoff, H. Vogel and M. Votsmeier, Appl. Catal., A, 2012, 111–112, 445–455 CrossRef CAS.
- B. Partopour and A. G. Dixon, AIChE J., 2017, 63, 87–94 CrossRef CAS.
- M. Klingenberger, O. Hirsch and M. Votsmeier, Comput. Chem. Eng., 2017, 98, 21–30 CrossRef CAS.
- F. A. Döppel and M. Votsmeier, Chem. Eng. Sci., 2022, 262, 117964 CrossRef.
- A. Scheuer, O. Hirsch, R. Hayes, H. Vogel and M. Votsmeier, Catal. Today, 2011, 175, 141–146 CrossRef CAS.
- T. Nien, J. Mmbaga, R. Hayes and M. Votsmeier, Chem. Eng. Sci., 2013, 93, 362–375 CrossRef CAS.
- J. M. Lorenzi, T. Stecher, K. Reuter and S. Matera, J. Chem. Phys., 2017, 147, 164106 CrossRef PubMed.
- J. E. Sutton, J. M. Lorenzi, J. T. Krogel, Q. Xiong, S. Pannala, S. Matera and A. Savara, ACS Catal., 2018, 8, 5002–5016 CrossRef CAS.
- S. Matera, W. F. Schneider, A. Heyden and A. Savara, ACS Catal., 2019, 9, 6624–6647 CrossRef CAS.
- B. Partopour, R. C. Paffenroth and A. G. Dixon, Comput. Chem. Eng., 2018, 115, 286–294 CrossRef CAS.
- M. Bracconi and M. Maestri, Chem. Eng. J., 2020, 400, 125469 CrossRef CAS.
- W. Ji and S. Deng, arXiv, 2021, preprint, pp. 1–23, DOI:10.48550/arXiv.2108.00455.
- M. F. Bellemare and C. J. Wichman, Oxf. Bull. Econ. Stat., 2020, 82, 50–61 CrossRef.
- G. B. D. Aihounton and A. Henningsen, J. Econom., 2021, 24, 334–351 CrossRef.
- J. Mullahy and E. Norton, Why Transform Y? A Critical Assessment of Dependent-Variable Transformations in Regression Models for Skewed and Sometimes-Zero Outcomes, National bureau of economic research technical report, 2022 Search PubMed.
- A. B. Mhadeshwar and D. G. Vlachos, J. Phys. Chem. B, 2004, 108, 15246–15258 CrossRef CAS.
- W. Hauptmann, M. Votsmeier, H. Vogel and D. G. Vlachos, Appl. Catal., A, 2011, 397, 174–182 CrossRef CAS.
- P. E. Van Keken, D. A. Yuen and L. R. Petzold, Geophys. Astrophys. Fluid Dyn., 1995, 80, 57–74 CrossRef.
- H. Ma and W. F. Schneider, ACS Catal., 2019, 9, 2407–2414 CrossRef CAS.
- L. F. Shampine and M. W. Reichelt, SIAM J. Sci. Comput., 1997, 18, 1–22 CrossRef.
- NIST Chemistry WebBook, https://webbook.nist.gov/cgi/cbook.cgi?Source=1998CHA1-1951, Last accessed at 20.04.2020.
- M. Chase, C. Davies, J. Downey and D. Frurip, J. Phys. Chem. Ref. Data, 1998, 14, 927–1856 Search PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, Advances in Neural Information Processing Systems 32, Curran Associates, Inc., 2019, pp. 8024–8035 Search PubMed.
- T. Zhang, Y. Yi, Y. Xu, Z. X. Chen, Y. Zhang, W. E and Z.-Q. J. Xu, Combust. Flame, 2022, 245, 112319 CrossRef CAS.
- X. Huang, X. Li, W. Xiao and Z. Wei, AIChE J., 2023, 69, e17945 CAS.
- K. Wan, C. Barnaud, L. Vervisch and P. Domingo, Combust. Flame, 2020, 220, 119–129 CrossRef CAS.
- J. Whittaker, C. Whitehead and M. Somers, J. R. Stat. Soc., C: Appl. Stat., 2005, 54, 863–878 CrossRef.
- J. B. W. Webber, Meas. Sci. Technol., 2013, 24, 027001 CrossRef CAS.
- J. A. John and N. R. Draper, Appl. Stat., 1980, 29, 190 Search PubMed.
- B. Klumpers, T. Luijten, S. Gerritse, E. Hensen and I. Filot, SSRN Journal, 2023, 1–20 Search PubMed.
- R. T. Chen, Y. Rubanova, J. Bettencourt and D. K. Duvenaud, Adv. Neural. Inf. Process. Syst., 2018, 31, 1–13 Search PubMed.
- J. Yin, J. Li, I. A. Karimi and X. Wang, Chem. Eng. J., 2023, 452, 139487 CrossRef CAS.
- W. Ji and S. Deng, J. Phys. Chem. A, 2021, 125, 1082–1092 CrossRef CAS PubMed.
- G. S. Gusmão, A. P. Retnanto, S. C. da Cunha and A. J. Medford, Catal. Today, 2022, 417, 113701 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00212h |
| This journal is © The Royal Society of Chemistry 2023 |