
Insight into CO selective chemisorption from syngas mixtures through Li2MnO3; a new H2 enrichment material†
Carlos
Hernández-Fontes
 a,
Daniel G.
Araiza
a,
Gabriela
Díaz
b and
Heriberto
Pfeiffer
*a
a,
Daniel G.
Araiza
a,
Gabriela
Díaz
b and
Heriberto
Pfeiffer
*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito exterior s/n, Ciudad Universitaria, Del. Coyoacán, Ciudad de México, CP 04510, Mexico. E-mail: pfeiffer@materiales.unam.mx
bInstituto de Física, Departamento de Física Química, Universidad Nacional Autónoma de México, Ciudad de México, CP 04510, Mexico
First published on 17th October 2022
Abstract
Carbon oxides separation from a hydrogen stream is essential to achieve ideal energy systems. In this context, lithium manganate (Li2MnO3) is the first alkaline ceramic reported for the selective chemisorption of CO under non-oxidative conditions. The Li2MnO3 was synthesized, characterized, and dynamically and isothermally tested for selective CO chemisorption from syngas mixtures (H2 + CO) by thermogravimetry, GC and DRIFTS techniques, using different H2, CO, and CO2 flow compositions. Afterwards, the kinetic parameters were determined by fitting the isothermal data to the modified Jander–Zhang model to CO capture. All results showed that addition of H2 shifted the CO capture to lower temperatures and highly increased its kinetics and its efficiency. Moreover, it was elucidated that the presence of carbon oxides has a “shielding effect” over the surface of the Li2MnO3, minimizing or inhibiting the reaction between the Li2MnO3 and H2. Finally, cyclic CO capture and desorption, using syngas flows, presented good efficiencies and stability, although the CO capture efficiency slightly diminished over 10 cycles. Therefore, Li2MnO3 is a promising material for H2 purification systems.
Introduction
Due to environmental implications, eliminating carbon oxides (CO and CO2) has been a significant scientific challenge. Additionally, separating these acid gases from hydrogen is a key point for ideal clean energy systems. Within this context, 90% of hydrogen production is generated using reforming reaction processes, which spawn huge amounts of carbon oxides.1 Several of these reforming reactions mainly produce H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO syngas mixtures, where the carbon monoxide separation becomes harder than in the CO2 case, as an oxidation process is usually required.2 Thus, developing efficient systems for hydrogen separation from CO is an urgent topic to work with to reduce carbon oxide emissions and improve high purity hydrogen production. For example, in energy applications, the elimination of CO is significant since the H2 is intended to feed fuel cells, which are prone to be poisoned by CO.
:CO syngas mixtures, where the carbon monoxide separation becomes harder than in the CO2 case, as an oxidation process is usually required.2 Thus, developing efficient systems for hydrogen separation from CO is an urgent topic to work with to reduce carbon oxide emissions and improve high purity hydrogen production. For example, in energy applications, the elimination of CO is significant since the H2 is intended to feed fuel cells, which are prone to be poisoned by CO.
It is already known that H2 production is enhanced if CO2 capture materials are added to the process, which is regarded as sorption-enhanced reforming (SER) technologies.3,4 This technology performs the H2 and carbon oxides production simultaneously through different reactions, trapping the produced carbon oxides. These multiple processes not only reduce the carbon oxides concentrations of the final effluent gas, but shifts the thermodynamic reaction equilibrium towards H2 production. Within this context, calcium oxide-containing ceramics seem to possess excellent properties as carbon oxides captors within the sorption-enhanced processes,5–8 although other alkaline and earth-alkaline ceramics have also been analysed, such as MgO and Li4SiO4.9–11 All these works have evidenced that SER processes exhibit great potential in decarbonizing hydrogen production.
Although the CO2 capture capacities of lithium and sodium ceramics has been analysed and probed in the last two decades,12–15 some of these ceramics seem to possess similar capacities for bifunctionally working as CO oxidants and subsequently as CO2 captors.16–21 For example, it has been shown that Li2CuO2 and Li5FeO4 can chemically trap CO,20,21 in the presence and absence of oxygen, by forming lithium carbonate implying the copper and iron partial or total reductions. These materials are able to trap CO2 and/or CO according to reactions 1 to 5.
| Li2CuO2 + CO2 → Li2CO3 + CuO | (1) |
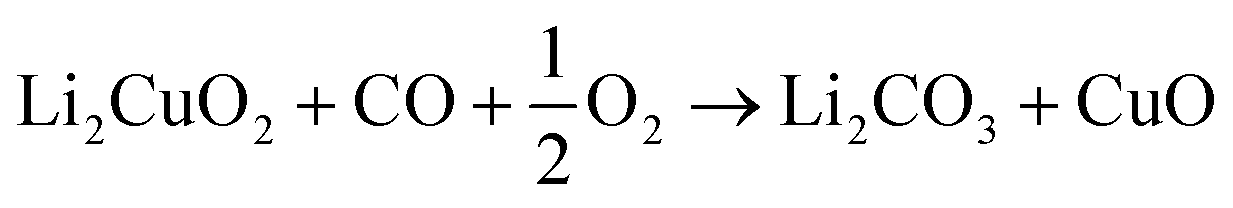 | (2) |
| Li2CuO2 + CO → Li2CO3 + Cu | (3) |
| Li5FeO4 + 2CO2 → 2Li2CO3 + LiFeO2 | (4) |
| Li5FeO4 + 2CO + O2 → 2Li2CO3 + LiFeO2 | (5) |
In a similar context, it was recently published that Li2MnO3 and LiMnO2 ceramics can perform the bifunctional work of CO oxidation and subsequent capture only in oxygen absence. Moreover, these ceramics do not trap CO2 if it is not coming from the CO oxidation previous step.22 Thus, these lithium manganates selectively trap CO in moderate and high-temperature ranges in non-oxidative atmospheres (reactions 6 and 7).
| 2Li2MnO3 + CO → Li2CO3 + 2LiMnO2 | (6) |
| 2LiMnO2 + CO → Li2CO3 + 2MnO | (7) |
Based on the above, this work aims to analyse further the CO oxidation–capture of the Li2MnO3 material, this time in the presence of hydrogen, employing a syngas flow mixture (CO + H2). This goal was achieved by exploring the bulk and surface reactivity through TGA, GC, and DRIFTS techniques, which helped to establish the selectivity towards the CO oxidation–capture over the H2 oxidation.
Experimental
The Li2MnO3 powder was synthesized by conventional solid-state reaction, employing lithium oxide (Li2O, Aldrich) and manganese(II) oxide (MnO, Meyer) as it has been reported previously.22,23 The precursors were mixed using a 5 wt% excess of lithium oxide to compensate for the lithium sublimation.24 Then, a pellet was prepared by uniaxially pressing the powders mixture (pressure of 40 MPa) and fired at 900 °C for 12 h in an air atmosphere.The crystal structure of the as-prepared sample was determined by X-ray diffraction (XRD); besides, it was microstructurally characterized by N2 adsorption–desorption (N2 ads–des) isotherms and scanning electron microscopy (SEM). The XRD analysis was carried out at room temperature (RT) using a Siemens D5000 diffractometer with Co Kα-radiation. The crystalline phase was identified by matching the corresponding Joint Committee Powder Diffraction Standards (JCPDS) files. Then, N2 ads–des isotherms were measured at 77 K using a Minisorp II instrument from Bel-Japan employing a multipoint technique. Before measurement, the sample was degassed under vacuum at room temperature for 12 hours. The BET model was used to determine the specific surface area (SBET). Finally, the microscopy analysis was carried out in a JEOL JMS-7600 F microscope, obtaining secondary electron images.
Thermogravimetric analyses (TGA) were performed on a thermogravimetric balance (Q500HR from TA Instruments) to determine the capability of the material to capture selectively CO from a syngas mixture. First, the sample was dynamically heated from 30 to 950 °C at 5 °C min−1, under different gas flow compositions with a total flow rate of 60 mL min−1. CO, H2, and a mixture of CO + H2 were used as reactant atmospheres, diluted in nitrogen (N2) to a 5 vol%. The mixture is also known as syngas (CO + H2).
Afterwards, the CO and H2 oxidation properties of Li2MnO3 were evaluated using a catalytic reactor system (CATLAB MicroReactor, Hiden Analytical). In these tests, 100 mg of Li2MnO3 were dynamically heated from 30 to 850 °C at 5 °C min−1 using 60 mL min−1 of an equimolar syngas mixture (CO + H2). The gas products were analysed using a mass spectrometer (QGA, Hiden Analytical). Additionally, some thermogravimetric isothermal experiments were carried out in the same syngas flow conditions, from 575 to 675 °C. Initially, the sample was heated to the isotherm's temperature at 20 °C min−1, under an N2 flow as an inert atmosphere. Once the specific temperature was reached, the flow was switched to a syngas mixture flow, and the isotherms were thermogravimetrically measured for 5 hours. Besides, all the solid products of the isotherms were characterized by XRD, using the same equipment and conditions as those used to characterize the as-synthesized sample, to elucidate the crystal phase evolution and the carbonated or hydroxylated products.
Furthermore, the analysis of the surface reactivity of the Li2MnO3 sample was achieved through DRIFTS analyses. Experiments were performed using an environmentally controlled PIKE DRIFTS cell with SeZn windows coupled to a Thermo-Scientific Nicolet iS50 spectrometer with a DTGS detector. Absorbance spectra were obtained by collecting 64 scans at 4 cm−1 of resolution. Tests were done in helium (He) as a reference, and the following gas mixtures: CO at 5 vol% (He balance), H2 at 5 vol% (He balance), and CO2 at 5 vol% (Ar balance), using a total flow of 60 mL min−1. Also, CO, H2, and CO2 were diluted with He flow to achieve a 2.5% volume concentration for each gaseous reactant. The heating rate was set to 10 °C min−1, and spectra were collected each 2 min (around 17 °C per spectrum). Complementarily, the H2 + CO2 reactivity of the material was thermogravimetrically studied from 30 to 950 °C using a flow composition of both gases diluted in N2 to 5 vol% with a total flow rate of 60 mL min−1. Then at 550 °C, an ATR-FTIR was carried on to unravel any carbonated or hydroxylated species formation.
Finally, Li2MnO3 was tested in ten cycles of CO capture under a syngas mixture flow. The chemisorption steps were carried on for 3 h at 650 °C using the same syngas described above. Then, the desorption and regeneration steps were performed for 3 h under synthetic air (N2 at 80 vol% and O2 at 20 vol%) at a higher temperature (800 °C), based on the fact that the CO2 desorption and regeneration of the material are improved at higher temperatures than the CO capture step.
Results and discussion
The XRD pattern of the Li2MnO3 sample is presented in Fig. 1A, corresponding to the monoclinic crystalline phase (01-08-1953 PDF file) without any secondary phase, at least at the XRD detection limit (∼3%). Additionally, the N2 adsorption–desorption characterization (data not shown) corresponded to an isotherm type II (IUPAC classification25,26) without any hysteresis, typically observed in these ceramics synthesized by solid-state reaction. The BET model was employed to determine the specific surface area (<1 m2 g−1). Complementarily to the microstructural characterization, the SEM images show that the Li2MnO3 material was composed of polyhedral faceted particles of around 10 μm (Fig. 1B and C), which are dense packed in larger agglomerates of 40 mm, in good agreement with previous works.23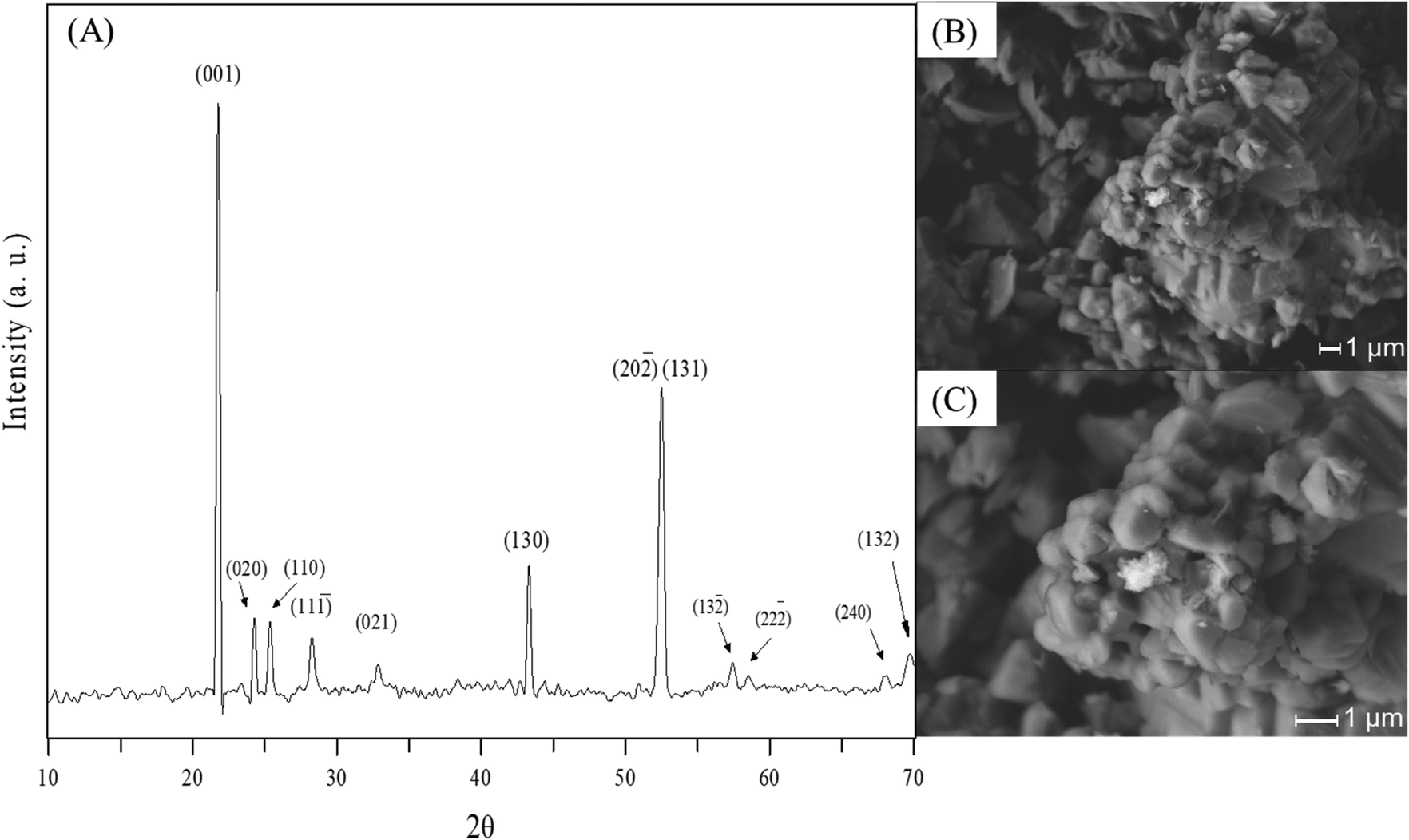 | ||
| Fig. 1 XRD pattern (A) and SEM images (B and C) of the pristine Li2MnO3 sample synthesized by solid-state reaction method. | ||
Afterwards, Li2MnO3 was evaluated for CO capture in the presence of hydrogen to unravel the viability of this material in other gas separation/purification processes. Fig. 2A shows the dynamic thermal mass evolution of Li2MnO3 in a CO + H2 flow (both at 5 vol%, N2 balance) and for comparison purposes in isolated CO and H2 flows (both at partial pressure equal to 0.05, N2 balance). As it can be seen, in the presence of CO, there is a weight increment starting at around 500 °C with a maximum near 700 °C, associated with the oxidation–capture processes of CO, in good agreement with previous reports.22,23 Furthermore, in a CO + H2 mixture, the mass loss evidenced in the pure hydrogen atmosphere was not observed under this syngas flow. At T > 370 °C in H2 flow (P = 0.05), a weight loss of 1.7 wt% occurs and increases with temperature up to 13.6 wt% at 950 °C, indicating probably the reaction between Li2MnO3 and H2, which reduces the Mn cations, from Mn4+ to Mn2+, and oxidizes H2 to produce water (H2O). The two compounds were later identified in the catalytic tests. Thus, all the processes observed in the H2 flow case imply the oxygen “loss” from the sample structure. Nevertheless, as there was no weight gain, there is no evidence of adsorbed hydroxylated species or water capture, although these species may have been produced and released through the gas flow. These results also suggests that Li2MnO3 does not present affinity to capture the produced hydrogen-containing compounds at high temperatures. In addition, it can be noticed that when CO was added to the reaction atmosphere (CO + H2 mixture), the initial weight loss of around 1.8 wt% shifted to lower temperatures (between 300 and 425 °C). The same behaviour was depicted in a CO flow (P = 0.05). The above has been attributed to a surface activation step, that leaded to the formation of the spinel structure Li1+xMn2−xO4, 0 < x < 0.33.22,23,27–30 Moreover, after the surface activation process in the syngas flow, almost the same weight increment as in a CO flow, up to 490 °C was observed. After this point, the process seems to slightly change for the superficial CO capture, which increased when H2 was added to the CO flow. However, the biggest change occurred at 580 °C, the temperature where the bulk CO capture seemed to start, whereas, in a CO flow, it started at a higher temperature (625 °C). It must be pointed out that the dynamic CO capture seems to be higher in H2 presence and shifts to lower temperatures, where the maximum weight gain was shifted from 700 °C to 670 °C. Finally, once the maximum dynamic CO capture was achieved, the decarbonation or decomposition took place. The temperature shifted to lower values when H2 was added to the reactive CO flow. However, regardless of the composition of the reductive atmosphere, all the dynamic experiments reached similar final weight values, which is associated with the maximum mass loss by the oxygen release from the Li2MnO3 structure in the temperature range of work.
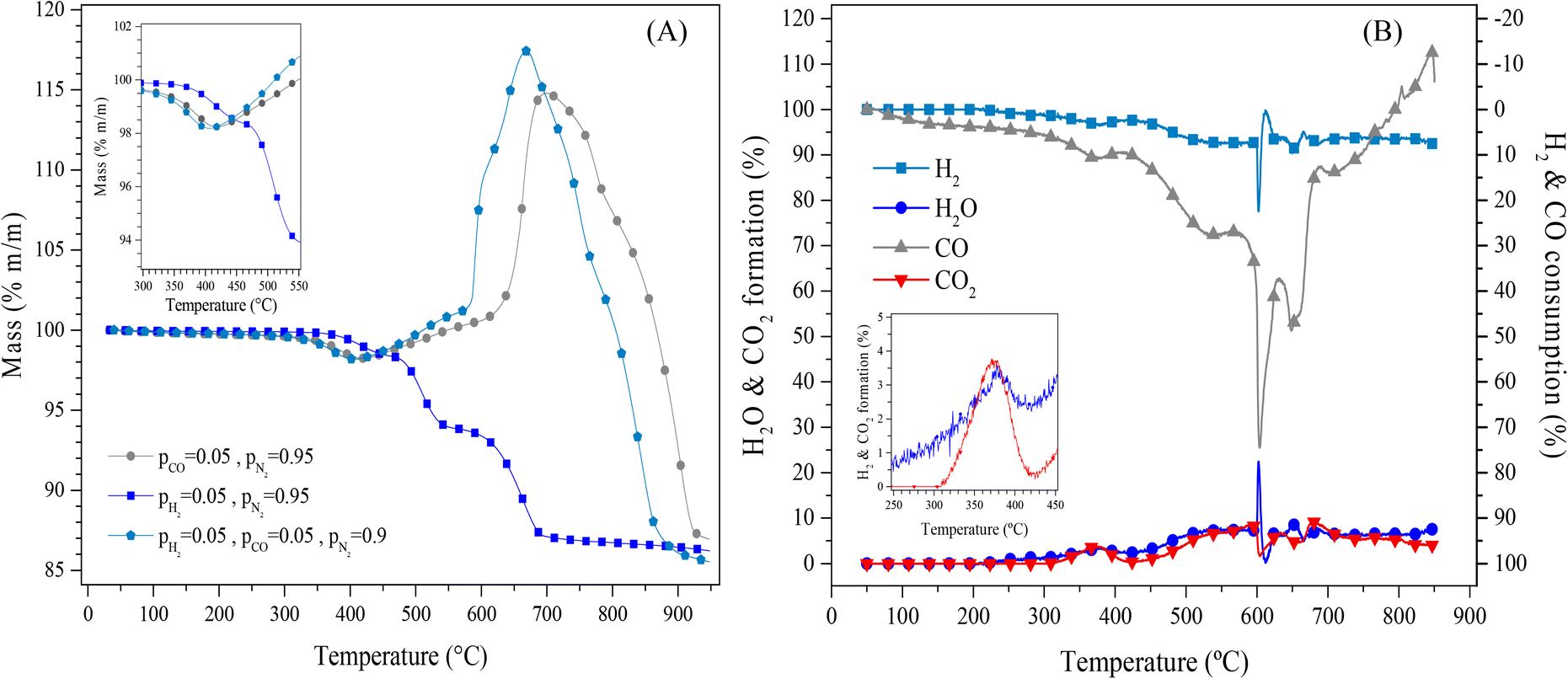 | ||
| Fig. 2 Dynamic experiments of the Li2MnO3 treated under different gas compositions between 30–950 °C (A). The square inset shows a zoom for the weight loss of the Li2MnO3 activation step. Dynamic evolutions in a catalytic reactor containing Li2MnO3 submitted to a syngas flow between 30 and 850 °C (B). The square inset shows a zoom for the obtained CO2 production between 300 and 450 °C. | ||
Perhaps, all the observed changes when both gases are mixed may be associated with a superficial reaction between Li2MnO3 and H2 to in situ produce water or hydroxyl groups at the surface, which may enhance the CO and CO2 capture as has been demonstrated in other works.31–33 However, as water or hydroxyl groups are not captured, these species should not be considered to contribute to the weight gain observed in the thermograms.
The evolved gas was analysed by mass spectrometry in a catalytic flow reactor (Fig. 2B) to unravel the mechanism of this process. As it can be seen, at temperatures below 300 °C, there was no H2 consumption, while a small CO conversion was observed. Then, between 300 and 425 °C, the CO consumption increased to produce CO2, in good agreement with the surface activation process of the Li2MnO3 in a reductive atmosphere. At higher temperatures, CO consumption increased to 27% at 568 °C and up to 75% at 604 °C, while H2 consumption slightly increased from 3% (T < 425°) to 7.5% and 18% at the same mentioned temperatures. It must be noticed that both the maximum CO consumption and H2 consumption (22.5%) were observed around the same temperature. Then, the CO consumption decreased to 50% at 650 °C and continued decreasing to 7.5% at 760 °C. Nonetheless, the H2 consumption remained almost the same (7.5%). Moreover, despite the high CO consumption, the CO2 production was below 10% for the whole temperature range, like the water production ascribed to the H2 consumption. The evidence of water production may be related to the high CO consumption and the lower CO2 production through the increase of the surface basicity of the material, which enhanced the CO and CO2 capture.33–35 Finally, over 760 °C, a negative consumption of CO is observed, suggesting that CO desorbs from the material.
Based on previous reports where the CO desorption from the Li2MnO3 material has not been evidenced,22 the generation of CO may be associated with the CO2 desorption by the thermal decomposition of Li2CO3, followed by the reaction of this carbon dioxide with hydrogen through the reverse water-gas shift reaction (eqn (8)).
The above correlates well with the observation that the water formation did not decrease. However, the dynamic thermogram in H2 (P = 0.05) indicated that over 720 °C, no more oxygen was released from the material (observed as weight loss), suggesting a different origin of the water oxygens, i.e., from the desorbed CO2. Also, the dynamic thermogram in syngas flow depicted the decarbonation or decomposition process at lower temperatures when H2 was absent in the reactive CO flow. Therefore, once the decarbonation occurs, different processes seem to occur: the CO desorption evidenced by this gas evolution measurements and the reverse water gas-shift reaction (RWGS) may occur between the desorbed CO2 and the H2 in the reactive flow. These reactions decrease the expected CO2 production. Based on all the thermogravimetric and gas evolution analyses (Fig. 2), these results provide evidence that the Li2MnO3 preferably reacts with CO instead of with H2, at least at temperatures below 700 °C.
| H2(g) + CO2(g) → H2O(V) + CO(g) | (8) |
Complementarily, the evolution of Li2MnO3 under a pure H2 flow (P = 0.05) by XRD (Fig. 3) was studied. In Fig. 3A, it can be observed three different decomposition processes. The first one corresponds to the evolution at 470 °C from Li2MnO3 to Li2MnO3−δ, related to the layered Li2MnO3 and the spinel Li1+xMn2−xO4, 0 < x < 0.33. This phase evolution is associated with the layered-to-spinel transition due to lattice oxygen release at the surface, allowing the ionic (Li1+, Mn4+/3+ and/or O2−) diffusion within the material. Then, these phases evolved to LiMnO2 at 560 °C, which implied the reduction of the manganese cations from Mn4+ to Mn3+ and the oxygen release from the crystal structure, leading to a rearrangement of the different species, through the mobility of Mn ions from octahedral sites to tetrahedral sites allowing the lithium diffusion from the transition metal (TM) layer to the lithium layer or corrugated lithium layer in the o-LiMnO2 compound.36–40 Finally, the LiMnO2 evolved to Li2O and MnO at 725 °C, due to reduction of the manganese cations Mn3+ to Mn2+ and the oxygen release that was evidenced by the dynamic thermogram (observed as a weight loss). Based on these results, eqn (9) describes the evolution of the Li2MnO3 crystal phase in a reductive atmosphere in well-agreement with previous works.22,23 However, as in this experiment, not any acid gas was present, the capture process was not evidenced by the dynamic thermogram (observed as a weight loss).
| Li2MnO3(s) → Li2MnO3−δ(s) → LiMnO2(s) → Li2O(s) + MnO(s) | (9) |
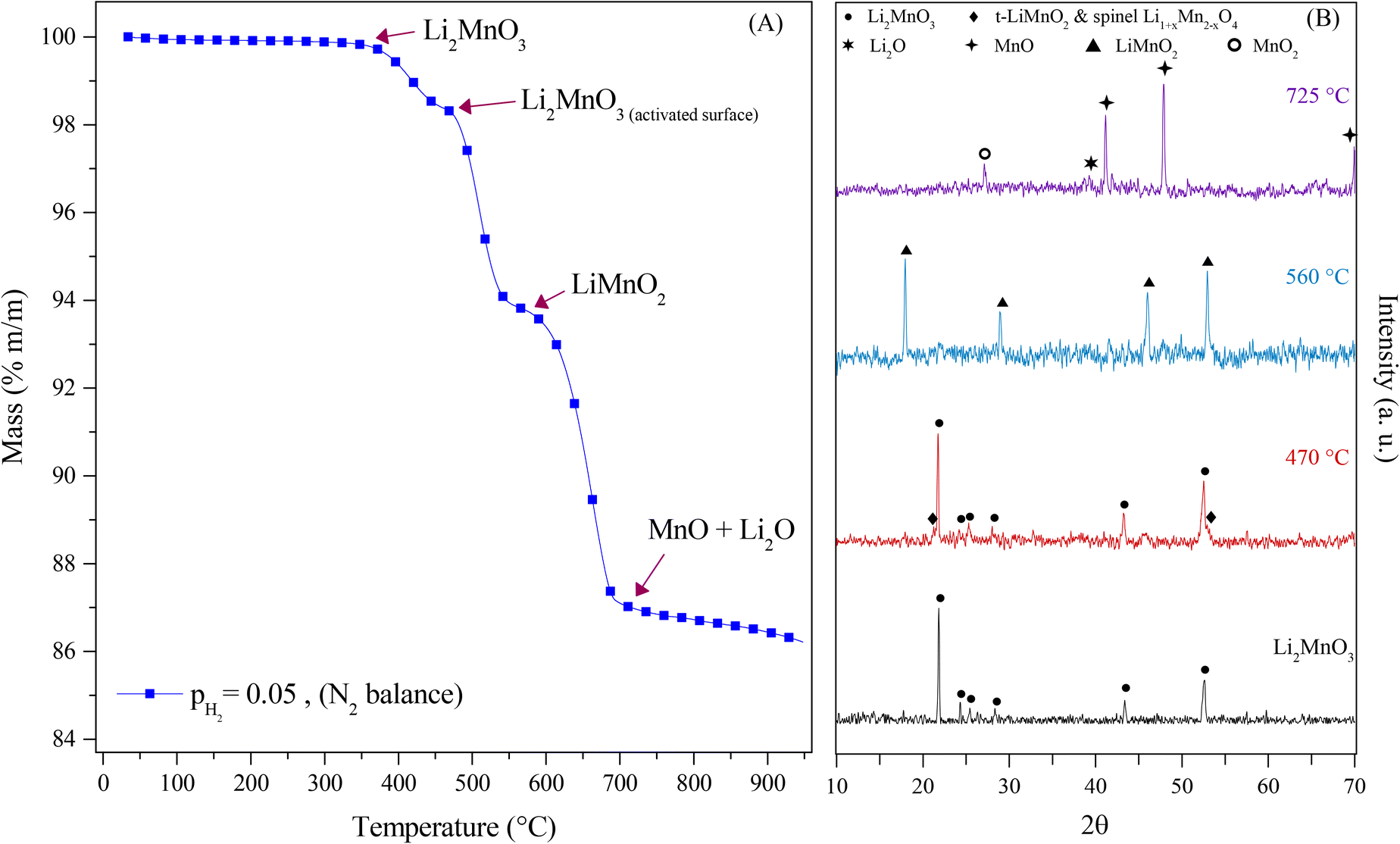 | ||
| Fig. 3 Dynamic thermogravimetric experiment of Li2MnO3 in H2 flow from 30 to 950 °C, showing the crystal phase evolution for each decomposition step (A). XRD patterns of the Li2MnO3, for comparison purposes, and the corresponding products of each step (B). | ||
To further analyse the preferential CO capture by Li2MnO3 in a syngas flow, different isothermal experiments were carried out between 575 and 675 °C (Fig. 4). All these isotherms presented an initial weight decrement of around 1.3 wt% during the first 7 minutes (square inset of Fig. 4), in good agreement with the dynamic thermograms presented above under CO and CO + H2 flows. Nevertheless, this process did not present dependence between time and temperature. In addition, the isotherms performed between 575 and 625 °C depicted two different increment behaviours. Initially, during the first 20 minutes, a weight increment of 8.77 wt% corresponded to an efficiency of 36.6% or 3.1 mmol of CO per gram of ceramic. It must be noticed that regardless of the temperature of these isotherms, this first capture was almost the same; however, this capture process seems to be faster when H2 is part of the gas flow in comparison to when there is only CO (P = 0.05, N2 balance).22 This behaviour may be associated with the enhanced superficial sorption process due to the in situ formation of some hydroxyl groups or water at the surface. Afterwards, a slight weight increment, no more than 5 wt%, was observed, which is related to the so-called diffusion process in the bulk chemisorption, which is favoured with the temperature increase, as shown at 625 °C by the highest weight increment for this process.
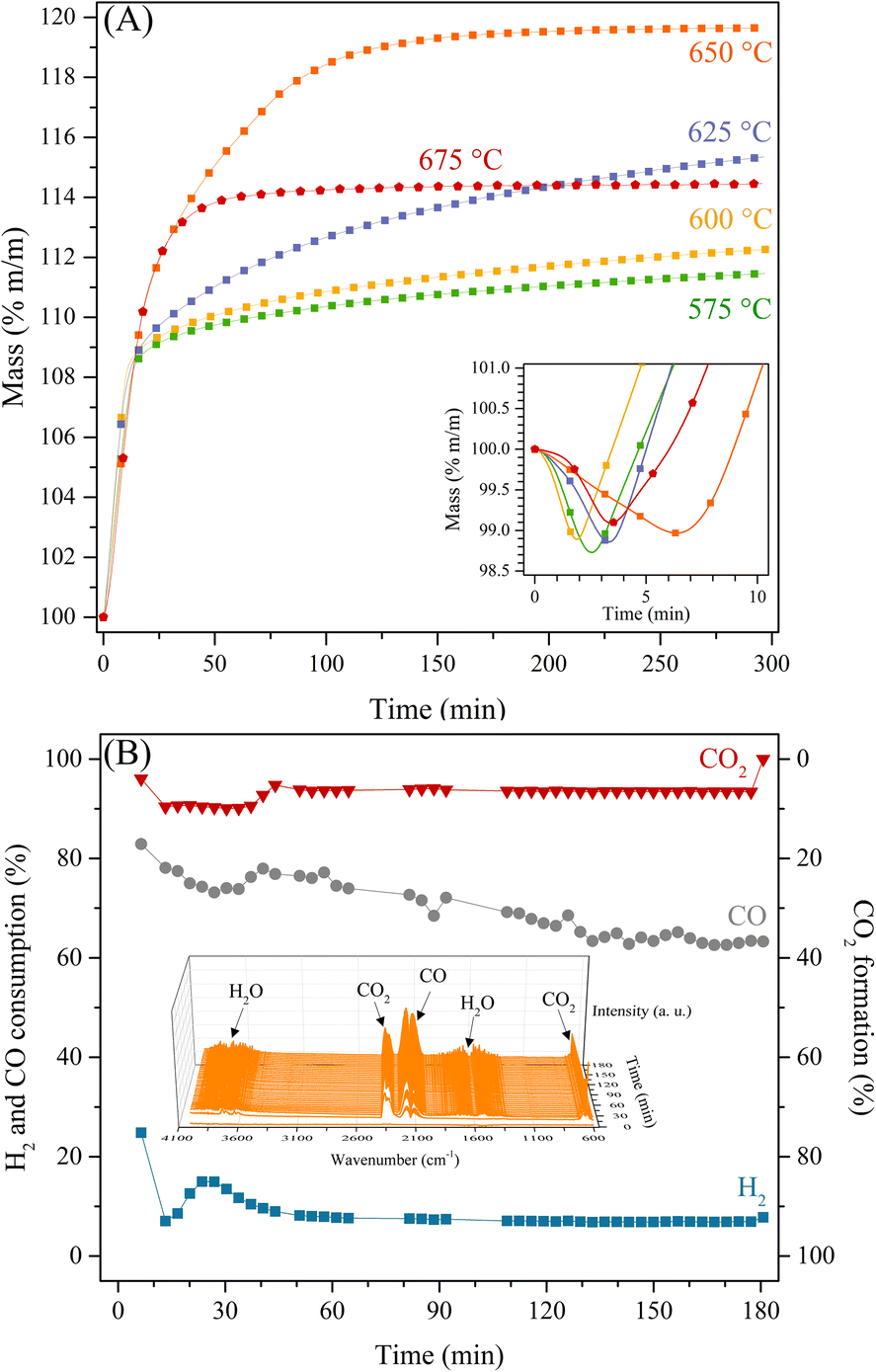 | ||
| Fig. 4 Thermogravimetric isothermal experiments of the Li2MnO3 sample using a syngas flow at different temperatures (575–675 °C) (A). In this inset, the initial mass was normalized in each experiment. Isothermal gas evolutions at 650 °C (B), where the square inset shows a representative FTIR obtained from the gas output. | ||
Then, at 650 °C the highest weight increment of 19.63 wt% (efficiency of 82%, or 7 mmol of CO per gram of ceramic) was observed, in which the superficial and bulk chemisorption processes became indistinguishable. Lastly, at higher temperatures, the CO capture decreases, as shown in the 675 °C isotherm, where the CO capture takes place during the first 60 minutes; then, the weight of the sample did not change for 4 hours. This behaviour may be associated with some equilibrium between the CO chemisorption, the decarbonation or decomposition process of the produced Li2CO3, and the oxygen release from the material, as it was observed in the dynamic thermograms. It seems, the H2 presence enhanced the CO capture at the surface and, once the surface was saturated, the diffusion of the Li1+ and O2− ions would be the limiting step of the CO capture, though, the oxidation process of CO to CO2 is also favoured with the increase of the temperature. Then, as the CO2 is produced at the surface, it can be captured as Li2CO3, or it reacts with the H2 through the reverse water-gas shift reaction to produce water and CO, which implies the oxygen release from the material. As CO2 is consumed, the capture of CO is also limited or inhibited. In addition, the CO, CO2 and H2 evolutions were determined at 650 °C (the best CO capture thermal condition), by using a catalytic reactor coupled to a gas chromatograph and FTIR cell gas system (Fig. 4B). These results show that CO selectivity (between 80–65% of sorption) is significantly higher than that of H2 (around 10% of consumption). Moreover, it must be pointed out that: i) CO2 release was consistently below 10%, ii) CH4 was not detected by GC or FTIR, and iii) H2O production was confirmed by the FTIR in the gas output (see square inset of Fig. 4B).
In addition, all these isothermal products were characterized by XRD (Fig. 5). It can be observed that the Li2MnO3 crystal phase completely evolved to o-LiMnO2, MnO and Li2CO3 at temperatures below 625 °C. Moreover, the amount of the secondary phase o-LiMnO2 tended to diminish with the increase of the temperature, as this phase was not identified at 650 °C or higher temperatures. However, the Li2CO3 and MnO amounts increased with temperature, confirming the CO capture in this temperature range. It is important to notice that this phase evolution is in good agreement with previous works.22,23 It should be mentioned that Li2CO3 relative peak intensities varied as a function of the isothermal temperature. This feature has been explained by microstructure changes or the formation of very diluted interstitial solid solutions.41 Furthermore, the formation of any hydroxylated specie, such as lithium hydroxide (LiOH) or manganese(II) hydroxide (Mn(OH)2) was not evidenced, at least by this technique, confirming that the hydrogen is not captured by the material. Moreover, the XRD results provide further information about the CO capture mechanism in a syngas mixture, as it was identified the o-LiMnO2 crystal phase in the isotherms where the observed capture was similar. In this sense, the reaction process must involve the evolution of Li2MnO3 to o-LiMnO2 and Li2CO3 (eqn (10)), which theoretical maximum capture is an increment of 12 wt%, a similar increase value to that observed in the isotherms. Nevertheless, the CO capture process seems to follow the reaction described in eqn (11), where the maximum capture corresponded to a weight increment of 18% and involves the evolution of the Li2MnO3 to MnO.
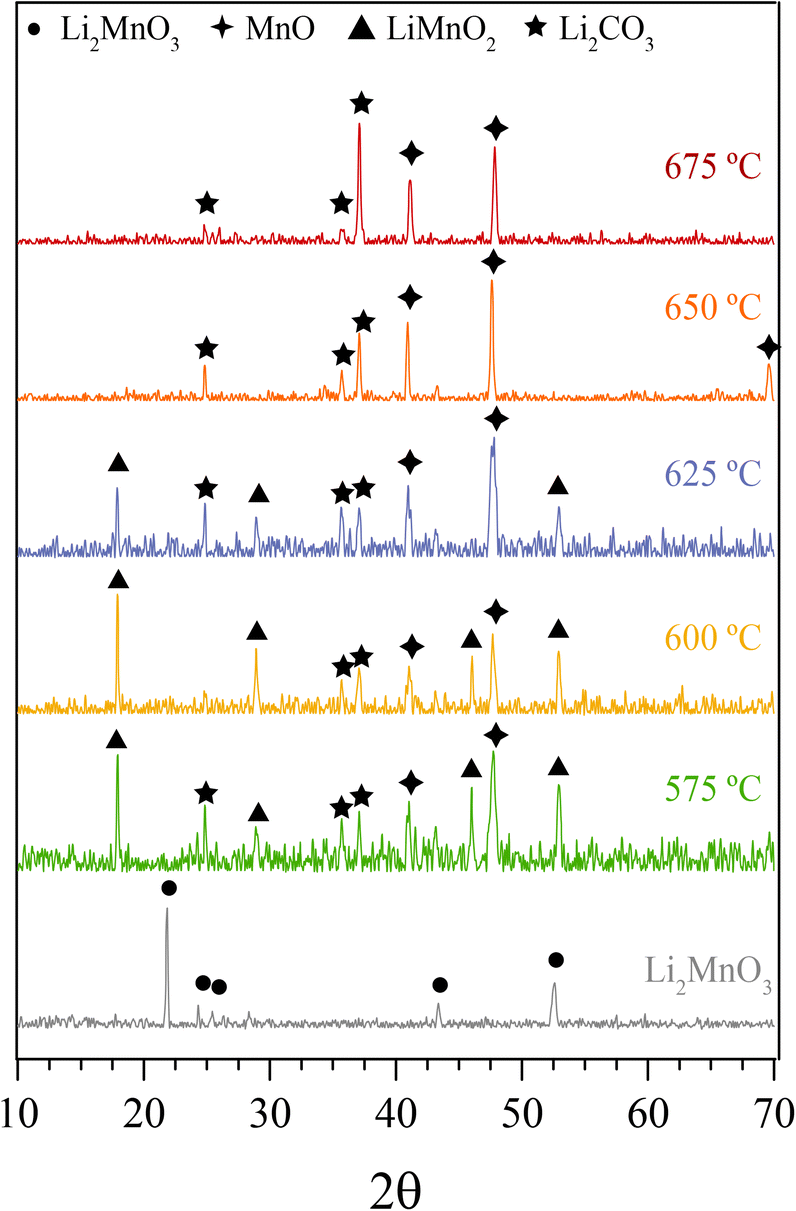 | ||
| Fig. 5 XRD patterns of isothermal solid products of Li2MnO3 submitted to a syngas mixture at different temperatures. The Li2MnO3 XRD pattern was added for comparison purposes. | ||
Although, the last is related to the reaction between CO and o-LiMnO2, which has lower kinetics and the process seemed to be kinetically limited by this reaction and the ion diffusions.22,42 Hence, the increase of the temperature favoured the Li1+ and O2− ion diffusions, and the reactivity of the LiMnO2 that is produced, leading to the total reaction of the Li2MnO3 to produce Li2CO3 and MnO, as it was observed at T ≥ 650 °C, and it is described on the eqn (12).
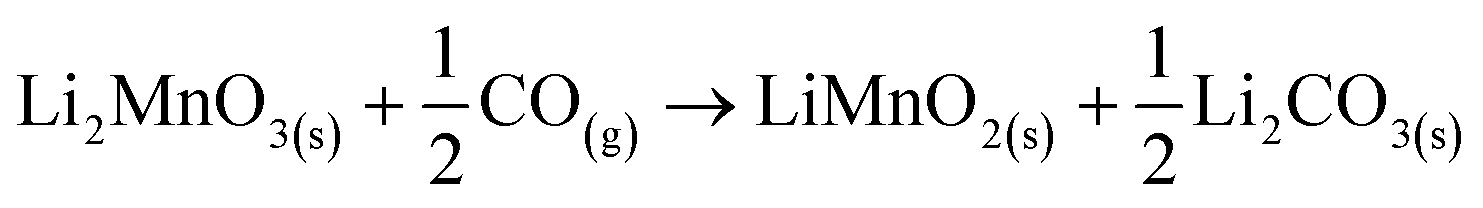 | (10) |
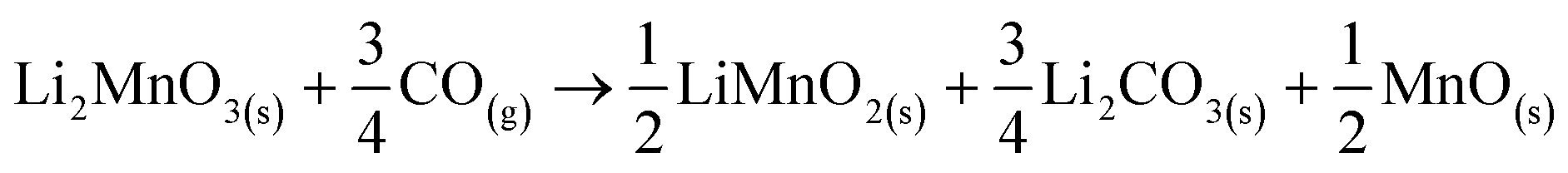 | (11) |
| Li2MnO3(s) + CO(g) → Li2CO3(s) + MnO(s) | (12) |
The surface characterization of the lithium manganate (Li2MnO3) was achieved through in situ DRIFT spectroscopy. Experiments were carried out by submitting the material under different gaseous atmospheres and recording the spectra as a function of the temperature between 30 and 600 °C. First, the Li2MnO3 surface was analysed under an inert He atmosphere (see ESI† Fig. S1). The first spectrum was recorded at room temperature (Fig. S1(a)†) evidencing the characteristic vibration bands of the material surface structure. An important vibration band at 753 cm−1 and two tiny contributions at 688 and 1043 cm−1 are linked to the structure of the manganate, specifically the metal–oxygen vibrations in octahedral coordination of the LiO6 and MnO6 units.43 Besides, the peak at 1432 cm−1 and the shoulder at 1473 cm−1 are associated with the asymmetric and symmetric stretching modes ν(CO3) of the Li2CO3 phase, respectively.44 Since the lithium precursor for the manganate synthesis was Li2O, the presence of the carbonate bands suggests the material reacts with CO2 from the environment at room temperature, forming the lithium carbonate, in agreement with Lux and Flood acid–base theory.45 Finally, the broad vibration band centred at 3424 cm−1 is related to the stretching mode ν(OH) of adsorbed hydroxyl species, which are also coming from the humidity of the ambient.
The evolution of the Li2MnO3 surface as a function of the temperature is presented in Fig. S1(b).† As temperature increases, hydroxyl species begin to disappear and above 150 °C, these are no longer visible, indicating their total desorption, as it would be expected. Besides, from around 200 °C, the intensity of the carbonate bands decreases with the temperature, although they remain visible up to the final spectrum. According to the literature, the Li2CO3 decomposition takes place over 730 °C.46 Thus, the presence of the carbonate bands in all the temperature range is expected and the decrease of their intensity is probably associated with some structural reorganization due to thermal effects or to a partial sample decarbonation. A similar behaviour (decrease in intensity) is observed in the vibrational bands coming from the structure of the Li2MnO3 material.
Afterwards, experiments were performed under various gaseous atmospheres such as CO2 (P = 0.025), CO (P = 0.025), H2 (P = 0.025) and an equimolar syngas mixture of CO + H2 (both at P = 0.025). Fig. 6 shows the absorbance DRIFT spectra recorded at three different temperatures (30, 300 and 600 °C) for all the gases used. At 30 °C, Fig. 6A, the Li2MnO3 material presents all the vibrational bands described above in the helium flow experiment (see Fig. S1†). Under hydrogen flow, no differences in the spectrum are noticed.
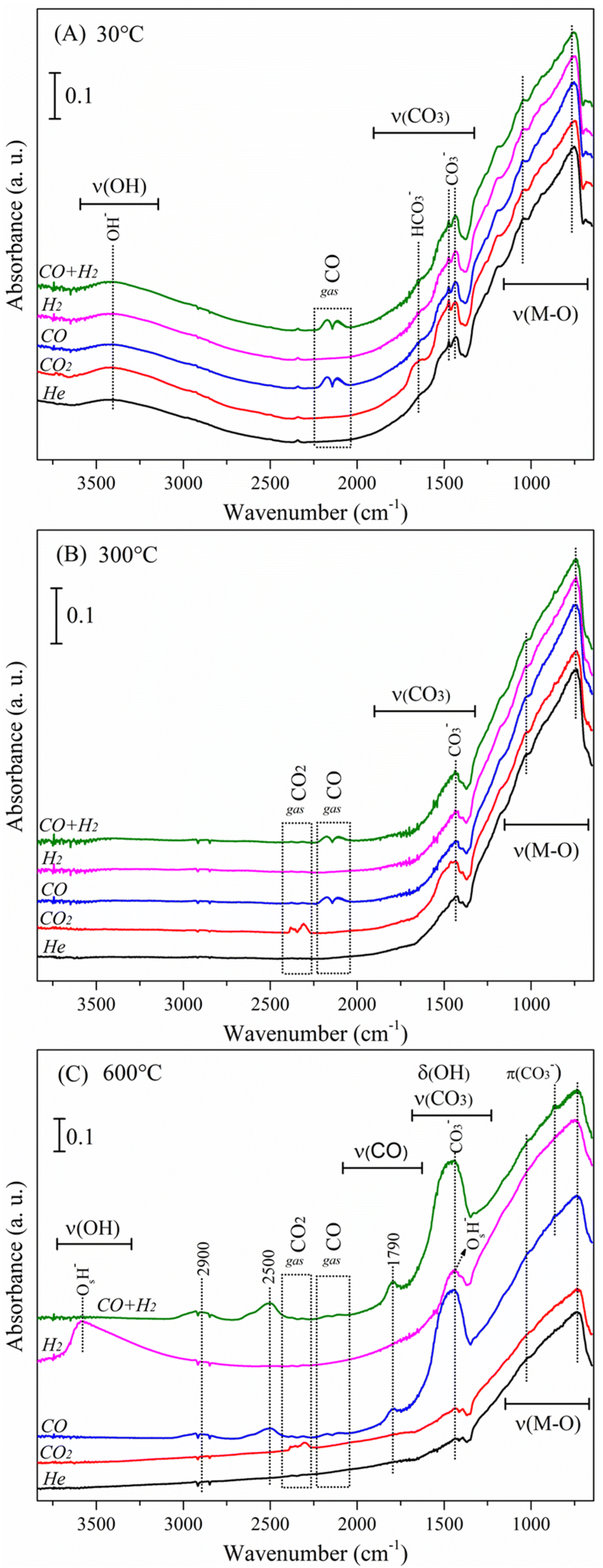 | ||
| Fig. 6 DRIFT spectra of the Li2MnO3 sample analysed under different reactive atmospheres: at 30 (A), 300 (B) and 600 °C (C). | ||
As expected, under CO and CO + H2 atmospheres, these spectra present two contributions at ca. 2111 and 2174 cm−1, linked to the R and P branches of gaseous CO.47 On the other hand, under a carbon dioxide atmosphere, the absence of CO2 vibrational bands indicates low concentration of this compound, as this was the first recorded spectrum (30 °C), and probably the steady state was not yet completely reached. Interestingly, a band at 1628 cm−1, related to adsorbed bicarbonate species (νs(CO3))48,49 develops importantly under this CO2 atmosphere. Bicarbonates typically appear at low temperatures, compared to other carbonates, due to their lower thermal stability.50 The mechanism proposed for the formation of adsorbed bicarbonate species is depicted in eqn (13). The adsorbed hydroxyl species (M–OH), identified in the pristine material, react with the flowing CO2 gas, producing adsorbed bicarbonate species (M–HCOOO). Identification of bicarbonates indirectly probes the presence of carbon dioxide.
| M–OH + CO2(g) → M–HCOOO | (13) |
DRIFT spectra recorded at 300 °C are shown in Fig. 6B. At this stage, hydrogen does not affect the Li2MnO3 surface structure or induce the formation of any adsorbed species. Experiments performed under CO and CO + H2 atmospheres present the characteristic vibrational bands related to carbon monoxide. However, compared to the spectra at 30 °C (Fig. 6A), the intensities of these signals are lower; this slight diminution suggests that part of the CO is reacting. Finally, vibrational bands located at ca. 2310 and 2380 cm−1 are associated with the ν3 asymmetric stretching vibrations of carbon dioxide,51 this time clearly observed in the flowing CO2 experiment. Besides, no bicarbonate bands were observed, in good agreement with the low thermal stability of this species. It is worth noting that, in all the reactive atmospheres, the intensity of adsorbed carbonate bands remained almost unaltered, indicating that no CO2 chemisorption has occurred at this stage, in well agreement with the thermogravimetric results.
Fig. 6C presents spectra recorded at 600 °C. The most important differences among the reactive atmospheres are observed at this final stage. Except for the bands related to the CO2, spectra recorded under helium and carbon dioxide atmospheres are almost the same. The above indicates that the presence of CO2 does not modify the surface structure of the Li2MnO3 material, indicating that this phase does not react with CO2 in non-reductive conditions, as it has been already reported.22,23 Besides, the adsorbed bicarbonates identified at low temperatures could be considered only as spectator species, as the band disappear above 100 °C, without affecting the chemisorption process. The complete evolution of the Li2MnO3 surface under a CO2 atmosphere, as a function of the temperature (30–600 °C), is presented in Fig. S2(a).†
In the spectrum recorded at 600 °C under flowing hydrogen, two important contributions are observed, one broad centred at 3589 cm−1 and other sharp at 1438 cm−1. These two vibrational bands are associated with the stretching (ν(OH)) and the bending (δ(OH)) modes of adsorbed hydroxyl species, respectively. According to the literature,52 two types of hydroxyl groups may be present in materials: i) those whose oxygen atoms do not belong to the ceramic lattice (e.g., adsorbed water), and ii) hydroxyl species that are formed from chemisorption of an H atom on a surface lattice oxygen atom. In this frame, these identified hydroxyl species arise after the H2 molecules react with the Li2MnO3 surface oxygen atoms. It is important to notice that both (δ(OH)) and ν(CO3) modes appear around the same wavenumber. However, under a hydrogen atmosphere, no source of carbon is fed into the system, therefore, the assignment of the vibrational band to hydroxyl species is solid. The complete evolution of the Li2MnO3 surface under a H2 atmosphere is presented in Fig. S2(b).† The temperature at which these new hydroxyl species appeared (T > 550 °C) matches with the decomposition of the Li2MnO3 to LiMnO2 through the reduction of the manganese ions from Mn4+ to Mn3+ (see Fig. 3).
Spectra recorded at 600 °C under CO-containing atmospheres are also shown in Fig. 6C. Since both CO and CO + H2 behave similarly, these are described together. The most important contribution is centred at 1438 cm−1, associated with the stretching mode ν(CO3) of the Li2CO3 phase. This indicates an important formation of the lithium carbonate at this stage, driven by the Li2MnO3 interaction with CO. The shape of this vibrational band, broader than the typically exhibited by the Li2CO3 material, suggests the presence of additional adsorbed carbonate species, while an incipient peak observed at ca. 860 cm−1, assigned to the out-of-plane vibration of the carbonate group π(CO3),53 corroborates the observation of these species. Contributions related to the CO gas almost vanished, indicating a high degree of conversion, which leads to the formation of the Li2CO3 phase and the presence of adsorbed carbonate species. Interestingly, the incipient band around 2320 cm−1 denotes the formation of CO2, produced from CO oxidation with surface oxygen atoms from the Li2MnO3 structure. The above observations are in well agreement with the temperatures range where these processes were depicted by the thermogravimetric analyses. Finally, three new contributions were identified at 1786, 2500 and 2900 cm−1 in these two CO-containing atmospheres (Fig. 6C). According to the literature, upon CO adsorption, carbonyl species appearing around 1800 cm−1 could be linked to bridged carbonyls.54 In that sense, the vibrational band located at 1786 cm−1 is proposed to arise from carbonyls (ν(CO)) adsorbed over partially reduced manganese sites CO–Mnδ+. The formation of this complex indirectly suggests some manganese diffusion up to the surface, linked to the Li2CO3 formation. Furthermore, since the appearance of this vibrational band matches with the intensity increase of carbonate bands and with those located at 2500 and 2900 cm−1, these probably arise from the same adsorbed species. On this regard, it has been reported that manganese carbonyl complexes present FTIR vibrational bands in the 1800–2500 cm−1 spectral region, explaining the presence of the 2500 cm−1 band.55 On the other hand, the bands located at 2500 and 2930 cm−1 could be related to overtone and combination bands of carbonates.56 In this sense, the band located at 2900 cm−1 is observed in all the temperature range, and its intensity correlates with the carbonate band at 1438 cm−1. Contrarily, the band at 2500 cm−1 appears only at high temperatures (T ≥ 535 °C) regardless of the presence or formation of carbonates at lower temperatures.
An alternative explanation of 1786, 2500, and 2930 cm−1 vibrational bands could be related to the presence of adsorbed formate species (HCOO).57–59 The first two bands would be associated with ν(CO) modes, whereas the last one with ν(H–C) modes. However, several considerations are worth mentioning to discard this hypothesis taking into account the fact that formate species could be produced through the hydrogenation of CO2: i) although CO is oxidized to CO2 and this could react with hydrogen to produce formate species, the three bands appear in both CO and CO + H2 experiment conditions, but in the former, there is no hydrogen source to produce the hydroxylated species, which is a necessary species during the formate formation mechanism;60 ii) in absence of hydrogen, hydroxylated species could arise from adsorbed hydroxyl species (coming from the humidity of the ambient), nonetheless, the thermal stability of those is limited, while the new bands appear at high temperatures (over 500 °C); iii) the bicarbonate species (eqn (13)) is an unavoidable intermediary during the formate formation, being its decomposition the limiting step of the formate production.61,62 However, bicarbonate presence was not evidenced in CO nor H2 + CO conditions, as well as its stability is limited at high temperatures; iv) methane could be produced during CO or CO2 hydrogenation and the band at 2900 cm−1 could be attributed to this species. Nevertheless, it was not observed the formation of methane (data not shown) during the analyses of evolved gas by mass spectrometry nor by GC in a H2 + CO flow (see Fig. 2B and 4B); v) some water must be produced at the surface due to the hydrogenation of CO or CO2, but in neither condition was observed the formation of water at the surface. Therefore, the first two bands (1786 and 2500 cm−1) are ascribed to CO–Mnδ+ species, while the last (2900 cm−1) is ascribed to overtone and combination bands of carbonates.
The complete evolution of the Li2MnO3 surface under CO and syngas (CO + H2) atmospheres is presented in Fig. 7. Interestingly, no evidence of hydroxyl species was observed along the CO + H2 experiment (Fig. 7B), confirming that the material reacts preferably with CO instead of with H2. The no identification of hydroxyl species agrees with the XRD characterization of the isothermal products and the dynamic thermogravimetric analyses. Some other differences are worth mentioning by taking a closer look at these experiments. First, the carbonate signal increases at around 500 °C under CO (Fig. 7A) and at 475 °C under the syngas mixture (Fig. 7B). In addition, the carbonyl CO–Mnδ+ species became visible near 550 °C under CO (Fig. 7A) and at around 535 °C in the syngas mixture (Fig. 7B), and the intensity of these bands was higher in the second case. In both cases, the CO2 signal starts decreasing when these carbonyls appear, as well as the carbonate signal increases. This observation suggests that, at this high-temperature regime, the CO oxidation to produce adsorbed carbon dioxide is less suitable, and instead the direct carbonate formation becomes favourable, which increase the CO adsorption as carbonyls over the manganese sites. In this scenario, the presence of hydrogen impacts the surface CO capture mechanism by reducing the temperature at which some events occur, although the path seems to be the same in both cases.
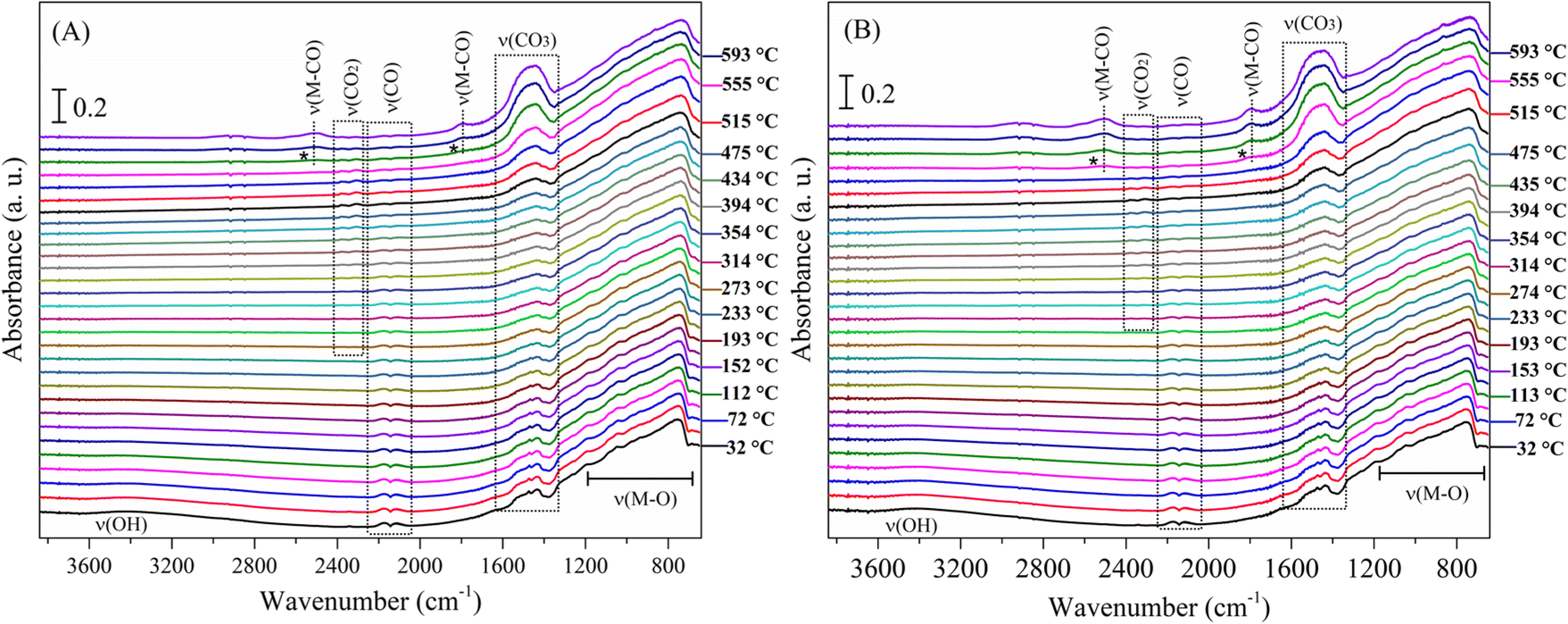 | ||
| Fig. 7 DRIFT spectra of Li2MnO3 as a function of temperature (30–600 °C), under CO (A) and CO + H2 (B) flows. | ||
To further understand the role of H2 during the CO capture in a syngas mixture, the Li2MnO3 was analysed thermogravimetrically in absence of carbon monoxide, i.e., under H2 + CO2 flow from 30 to 950 °C (both gases at P = 0.05 balanced in N2), as well as in a pure CO2 flow at same partial pressure, Fig. 8. For comparison purposes, the dynamic thermal behaviour of Li2MnO3 under a H2 flow (P = 0.05) was also included (Fig. 8). As it can be seen, in CO2 or H2 + CO2 flows, no mass change below 500 °C was depicted, though in H2 flow some partial reduction processes were taking place in the same range of temperature. Afterwards, it can be noticed that in a CO2 flow, the material does not suffer any mass loose up to 620 °C. In contrast, under H2 + CO2 flow, a mass loss no greater than 0.7 wt% was observed, while no carbonate formation was evidenced at this stage, followed by ATR-FTIR (data no shown). It must be pointed out that, in H2 flow in the same temperature range, the partial reduction of Li2MnO3 to LiMnO2 occurs. Based on these results, the surface-active sites of the material could be occupied by CO2, inhibiting or at least decreasing the interaction between Li2MnO3 and hydrogen, likely in H2 + CO condition. At higher temperatures (T > 620 °C), three phenomena must be emphasized: i) in H2, the Li2MnO3 is reduced from LiMnO2 to MnO and Li2O (see Fig. 3); ii) in CO2, a small weight loss of around 1.35 wt% was observed, and associated with the thermal modification of the manganese oxidation state at the surface;22,63 and iii) in H2 + CO2, a weight increment starting at 650 °C and with a maximum of 8.11 wt% at 830 °C is observed, associated with the CO2 capture as Li2CO3. These results are interesting because, as it has been reported, the Li2MnO3 is not capable of chemically capturing CO2 unless some structural modifications are carried out, such as the Mn partial reduction to form at the surface of the spinel-like structure Li1+xMn2−xO4, 0 < x < 0.33, which allows the Li1+ and O2− diffusion along the crystal structure.22,23 Moreover, the temperature range of the CO2 capture matches the temperature range where the manganese oxidation state is thermally modified, releasing molecular oxygen. Furthermore, in the H2 + CO DRIFT spectra, CO2 formation was observed but not the formation of hydroxylated species, so the hydrogen must not be interacting with the Li2MnO3 surface when some carbon oxides are part of the gas composition. Therefore, the hydrogen should be interacting with the CO2, through the RWGS reaction, producing some CO, which subtracts oxygen from the crystal structure, enabled by the high temperatures and enhanced by the thermal partial reduction of the manganese, allowing the CO2 capture. It must be pointed out that the RWGS reaction occurs at high temperatures, and CO2 partial pressure must shift the CO and or CO2 capture to higher temperatures as the Le Châtelier principle predicts. At last, in H2 + CO2 atmosphere, with the rise of the temperature, some kind of desorption or decomposition processes occur, which matches to half of the total of possible oxygen release from the material. Based on all these results, the presence of carbon oxides shields the surface of the Li2MnO3 from the reduction effect of H2. Then, these results suggest that the material does not react with H2 in CO or CO2 presence, as observed by DRIFTs, at least to its detection limit, as this is a surface-devoted technique.
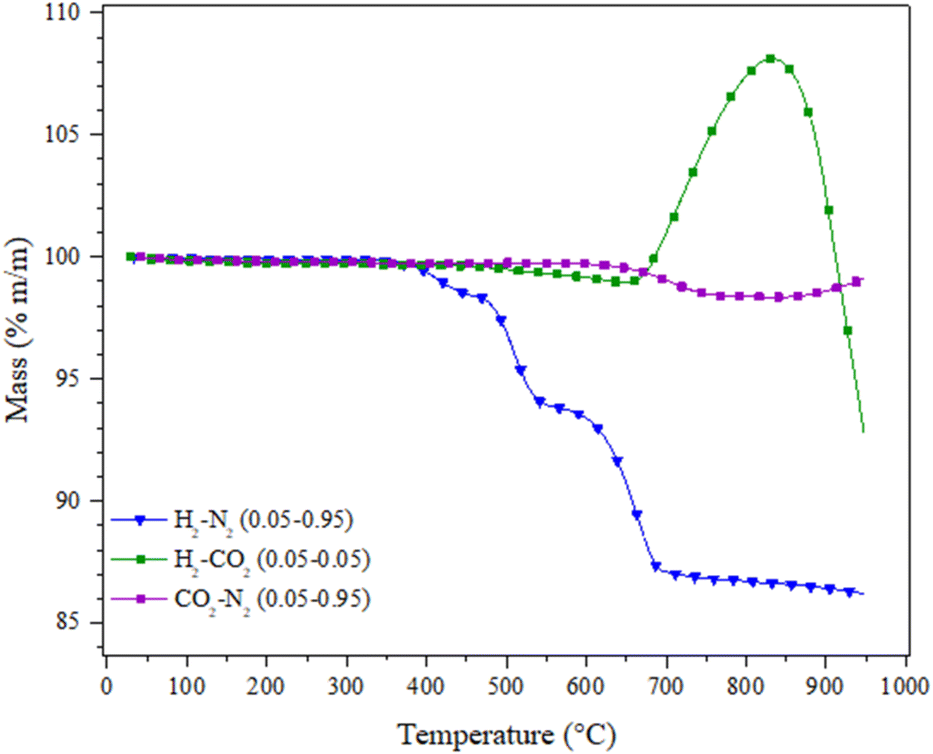 | ||
| Fig. 8 Dynamic experiments of the Li2MnO3 treated under different gas compositions between 30–950 °C. | ||
In the first case because the Li2MnO3 reacts preferably with CO and in the second case because of a shielding effect. All this is in good agreement with the increase of the intensity of the adsorbed carbonyl bands observed in the H2 + CO DRIFT spectra (see Fig. 7), as the produced CO2 reacts with H2 to produce CO and water (both identified by mass spectrometry, see Fig. 2B). The above process increases the adsorbed carbonyl CO–Mnδ+ species, enhancing the CO oxidation–capture mechanism.
An overview of the surface CO capture mechanism is as follows: i) CO interacts with the Li2MnO3 surface, ii) at elevated temperatures (∼300 °C) CO oxidizes into CO2, via the oxygens from the surface structure; iii) near 600 °C, the majority of the CO was converted into carbon dioxide, and chemisorbed by the material, forming the Li2CO3 phase, while some carbonates remained adsorbed over the surface; iv) the phase modification allows Mnδ+ species to become available over the surface, and part of the CO becomes adsorbed over these sites, where it would be either oxidized to CO2 or chemically captured as Li2CO3; v) in hydrogen presence the last two processes are enhanced due to the RWGS reaction by increasing the formation of adsorbed carbonyl CO–Mnδ+ species, a necessary step for the CO oxidation–capture mechanism.
After the whole characterization process, to further analyse the CO capture process in a syngas flow, the kinetics were determined. Thus, all these data were analysed through the kinetical modification of the Jander–Zhang model (eqn (14)),22,23,64 in which α represents the conversion value of the corresponding alkaline ceramic depending on time (t), Z is the proportion of Li2CO3 in the products based on the capture reaction and PCO is the partial pressure of CO.
| F(α) = (1 − (1 − Zα)1/3)3 = k(PCO)n1t | (14) |
In this work the CO concentration was not varied, therefore, the PCO and the kinetic parameter (n1) were considered as constants and incorporated into the rate constant (k). Then, all the isotherm data corresponding to the weight increments were fitted to the eqn (15). It must be pointed out that when the initial data (surface activation) and the bulk CO chemisorption–desorption equilibrium were incorporated to the mathematical model, these data did not correctly fit.
| F(α) = (1 − (1 − Zα)1/3)3 = kt | (15) |
Based on that, the obtained kinetic parameters for the CO capture in a syngas flow using Li2MnO3 were calculated from the corresponding isothermal mathematical fitting, as well as the Z parameters were calculated based on the chemistry reactions proposed after the analyses of the XRD results of the isothermal products (eqn (11) and (12)). All these calculated kinetic parameters are shown in Fig. 9A, presenting acceptable linearization values (R2 > 0.9). For comparison purposes in the same figure, the kinetic data of the CO capture of the Li2MnO3 was included, as these have been previously reported.22 It is evident through a direct comparison that the CO capture in the Li2MnO3 shows faster kinetics between 575 and 650 °C when hydrogen is part of the gas flow. Additionally, it must be noticed that in syngas flow conditions, the CO capture kinetics seemed to be similar between 575 and 625 °C, at least for the first and fastest capture process. However, these kinetic parameter values are at least one order of magnitude higher than those calculated for the CO capture in hydrogen absence. Furthermore, the kinetics in H2 presence increased from 2.45 × 10−7 to 6.21 × 10−7 s−1 with the temperature increment from 575 to 675 °C, which is associate with an improvement of the Li1+ and O2− ions diffusion, due to thermal effects. Therefore, the CO capture kinetics, in the Li2MnO3 sample, is significantly augmented in a syngas flow, probably due to the partial subtraction of oxygen from the crystal structure through the RWGS reaction between the H2 and the adsorbed CO2, increasing the adsorbed carbonyl species (CO–Mnδ+), boosting the CO capture. In such a case, the reaction process seems to fit the Eley–Rideal mechanism.65 To clarify, carbon dioxide is produced by the redox reaction between the Li2MnO3 and the CO, as proposed above, and not because of the water-gas shift reaction at this temperature range. On the other hand, at the beginning of this work it was proposed the in situ formation of some hydroxyl groups or water at the surface that immediately reacts with CO and/or CO2, enhancing the CO2 capture through the increase of the surface's basicity. Nevertheless, the lack of evidence of the formation of these species through the DRIFTS experiments, as well as the no detection of carbonates below 650 °C in H2 + CO2 flow, suggests that these hydroxylated species are not produced during the CO capture from a syngas flow, neither enhance the CO capture. Instead, the capture process is enhanced by the RWGS reaction, where water is produced at a low level based on the H2 consumption, around 7.5% (followed by mass spectrometry). Moreover, when H2 is not added to the flow mixture the CO capture kinetics is faster. In this case, this behaviour may be related to the oxygen availability from the material as there is no hydrogen oxidation from the RWGS reaction; also, as the diffusion process is activated, the Li2MnO3 can oxidize more CO and simultaneously capture it. Therefore, the best kinetic condition for the CO capture in a syngas flow using Li2MnO3 is at 650 °C due to the high selectivity for CO oxidation and the highest CO capture capability.
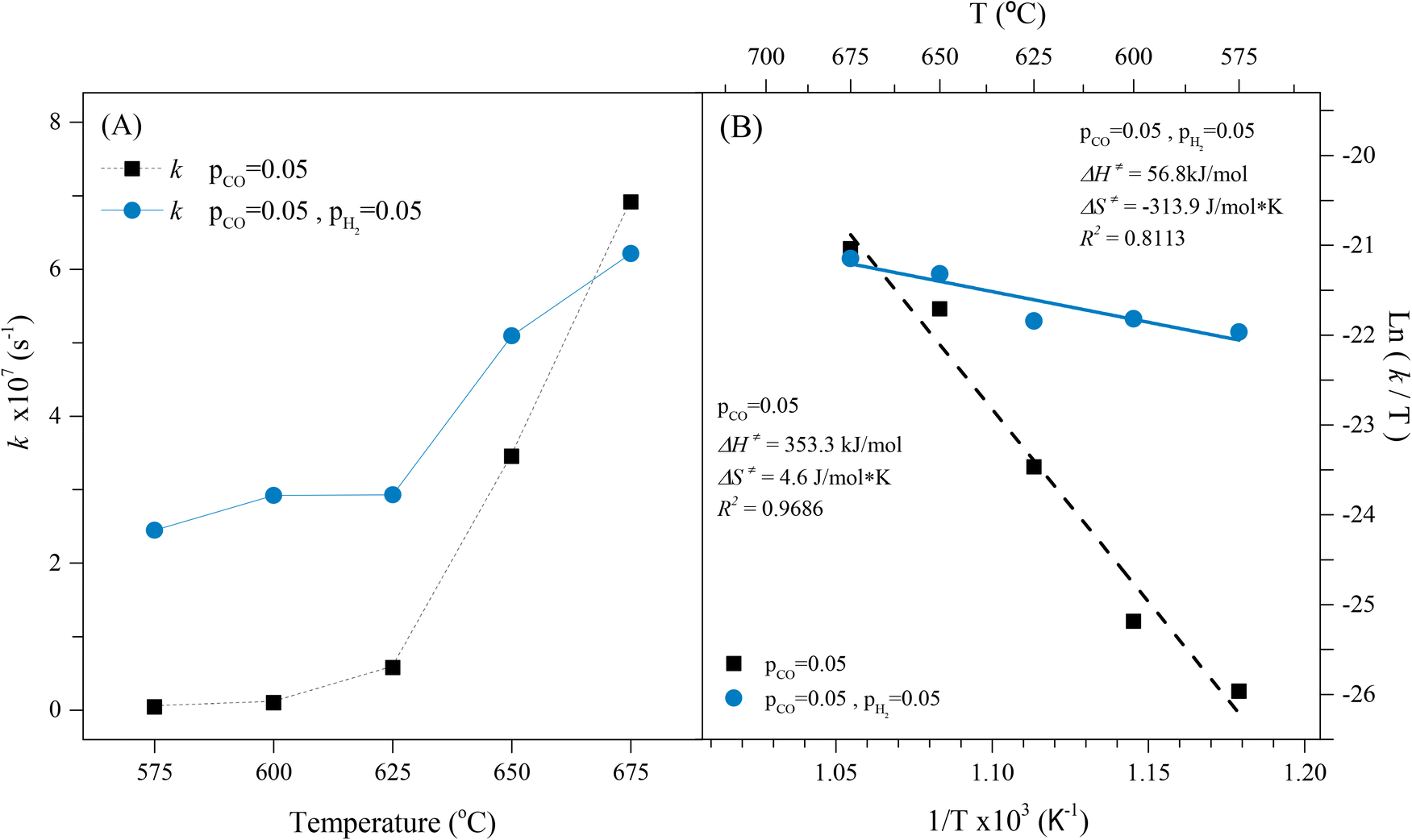 | ||
| Fig. 9 Rate constant values. k, of the isothermal CO capture from a syngas mixture on Li2MnO3 (A) and Eyring linear fitting of the apparent constant values (B). The kinetic data from the CO capture of Li2MnO3 was added for comparison purposes. | ||
Afterwards, the determinate k values were fitted to Eyring's model (Fig. 9B), which is typically used to describe solid–gas heterogeneous systems66,67 to calculate the activation enthalpy (ΔH≠) and entropy (ΔS≠). The calculated ΔH≠ (56.8 kJ mol−1) indicates that the CO capture by Li2MnO3 is less dependent on the temperature in hydrogen presence, in well agreement with the isothermal thermogravimetric results, where the first CO capture seemed to be similar regardless the temperature probably due to the formation of LiMnO2, which CO capture is thermally activated at higher temperatures. Hence, the thermal dependence in the CO capture by the Li2MnO3 may be closely related to the RWGS reaction and the formation of LiMnO2. On the other hand, the calculated ΔS≠ value (−313.9 J mol−1 K) is negative in comparison to the one reported in H2 absence, which is positive (4.6 J mol−1 K).22 This significant discrepancy must be related to a diminution of microstates in the activated complex compared to the probable microstates of the reagents (CO, H2 and Li2MnO3). This must be associated with the increase of the adsorbed carbonyls (CO–Mnδ+), reducing the microstates of the active complex. Furthermore, the increase of the CO adsorption enhances the CO2. In fact, it can be also related to a decrement of the molecularity of the whole Li2MnO3–CO–H2 system, as it has been defined as the molecules coming together to react in a single-step chemical reaction. Finally, the capability of the Li2MnO3 material to continuously capture CO in a syngas flow was studied through cyclical processes (Fig. 10A), in which the material could be regenerated after saturation. Two steps formed the cyclical process; the CO capture step and the decarbonation and regeneration step (exemplified in Fig. 10B). First, the CO capture process in a syngas flow was performed at 650 °C for 3 hours, as this was the best condition for selective CO oxidation with the highest CO capture and the best kinetics. Then, in the second step, the material was heated at 800 °C for 3 hours (heating rate 30 °C min−1) in synthetic air flow (PN2 = 0.80, PO2 = 0.20) to decompose the Li2CO3 and to recrystallize the Li2MnO3. This step must be done in oxygen presence because of the oxygen loss from the system through the desorption of CO2. In this sense, the oxygen deficiency in the system would limit or inhibit the Li2MnO3 regeneration. The results showed that the first CO capture presented an efficiency of 76% that decreased to 66% for the second cycle. This efficiency was similar over 6 more cycles (difference no larger than 4%), though, over the cycles the efficiency diminished to 55% after the tenth cycle. Besides, in all the regeneration steps, the weight loss was larger than the weight increment depicted in CO capture step. This feature is associated with the oxygen loss from the Li2MnO3 structure to form CO2. Nevertheless, during the last 40 minutes of the regeneration step, a weight increment can be observed, which may be related to the oxidation of the MnO to MnO2. It must be pointed out that in the regeneration step, three processes should take place: i) the CO2 desorption, ii) the oxidation of the manganese ions (Mn2+ → Mn4+), and iii) the insertion of lithium in the manganese(IV) oxide structure to recrystallize the Li2MnO3.
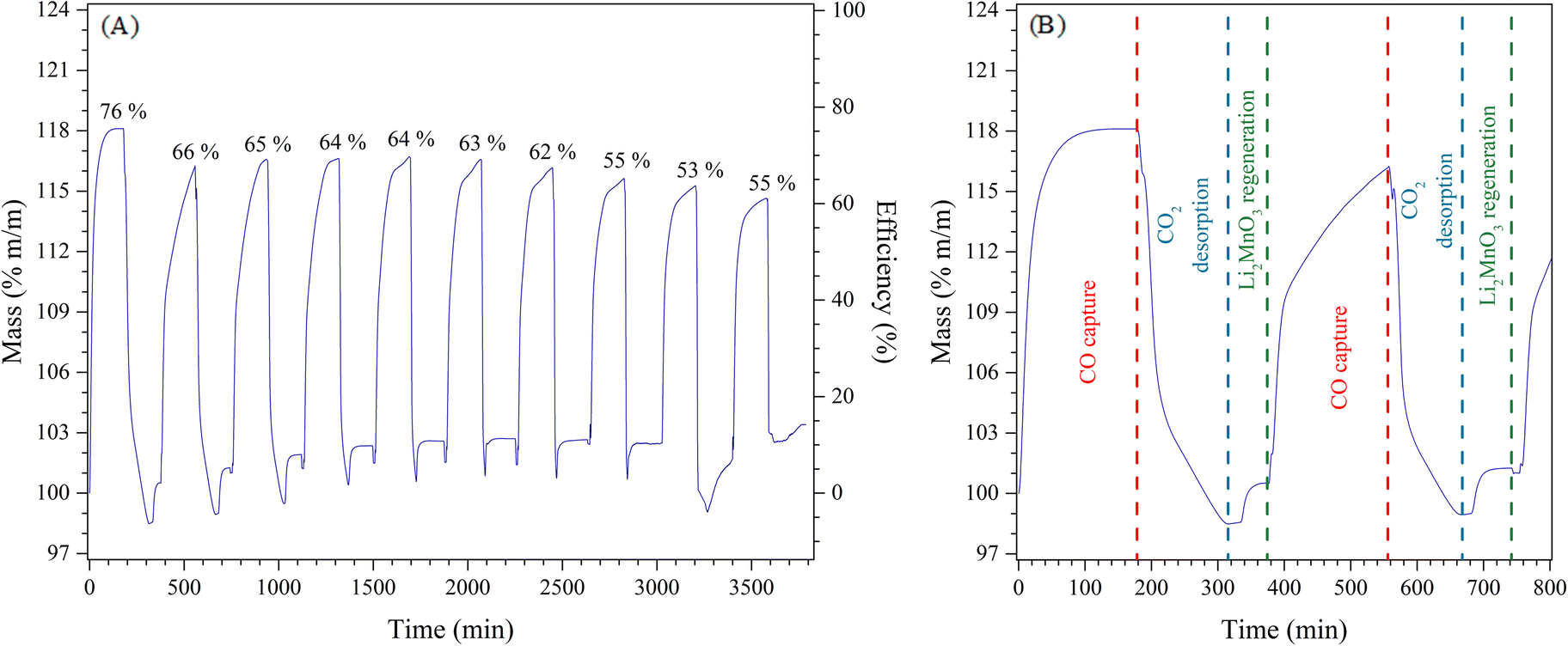 | ||
| Fig. 10 Cyclic behaviour of the CO capture step from a syngas mixture on Li2MnO3 and decarbonation–regeneration step using synthetic air (A). The first step (CO capture) was performed at 650 °C, while the second one at 800 °C, both were produced during 3 h periods (B). | ||
Then, the efficiency decrement over the cycles must be associated with the processes involved in these steps. Because the CO2 desorption was always larger than the CO capture, the Li2CO3 decomposition was not the limiting step. Conversely, the lithium diffusion in the manganese oxides structures to regenerate the Li2MnO3 has been widely treated as a limiting step in the cyclical process for battery applications.37,68–70 and CO capture processes.22 This behaviour is attributed to the energetic limitation of the lithium diffusion from the raw materials to the transition metal layer in the Li2MnO3 due to the stabilization of Mn3+ ions in octahedral sites, which is produced by a Jahn–Teller distortion effect in the manganese octahedrons (MnO6)2−.38,71,72 Moreover, in the regeneration step, some Li2O must be produced. Due to the high temperature, part of it must sublimate, implying incomplete recrystallization, which decreases the efficiency of the subsequent cycles. Hence, to further applications, the regeneration steps must be improved to avoid lithium losses and completely recrystallize the Li2MnO3.
Conclusions
In the present work, the fact that the Li2MnO3 is the first alkaline ceramic capable of selectively capturing CO from a syngas flow (CO + H2) in oxygen absence was evidenced. The Li2MnO3 was synthesized by solid-state reaction, characterized structurally and microstructurally, and then tested in H2, CO, and a syngas flow. In H2 flow, the Li2MnO3 is reduced to LiMnO2 at 560 °C and finally to MnO and Li2O at 725 °C. However, when there is CO in the reactive atmosphere, i.e., syngas flow, the Li2MnO3 reacts preferably with CO through an oxidation–capture process. Based on the results from in situ DRIFTS characterization, the surface CO capture mechanism was proposed to involve CO oxidation into CO2, carbon dioxide capture as Li2CO3, and adsorbed carbonates. At elevated temperatures, CO was adsorbed as carbonyls over manganese sites, subsequently oxidized to CO2 and or chemically captured. Moreover, under syngas flow, oxygen subtraction from the material surface is proposed to occur through the reverse water-gas shift reaction, following the Eley–Rideal mechanism between H2 and the produced CO2. This reaction increases the adsorbed carbonyls in the Li2MnO3 surface, which enhances the efficiency and the kinetics of the CO capture between 575 and 650 °C. Furthermore, the presence of carbon oxides seems to have a shielding effect over the material's surface, inhibiting the decomposition/reduction effect of H2 over the Li2MnO3 crystal phase, as neither hydroxyl groups nor water were identified in the final solid products and the DRIFTS analyses. Additionally, cyclic analysis revealed that the efficiency of the CO capture process decreased over the number of cycles from 76 to 55%; consequently, the regeneration step must be improved. Thus, Li2MnO3 may be of interest as a possible material capable of selectively capturing CO from syngas mixtures at high temperatures produced through reforming processes, avoiding the costly cold down processes.Author contributions
Carlos Hernández-Fontes: conceptualization, formal analysis, investigation, methodology and writing original draft. Daniel G. Araiza: investigation, methodology, writing review & editing. Gabriela Díaz: resources, writing review & editing. Heriberto Pfeiffer: conceptualization, resources, supervision, funding acquisition, writing Review & editing.Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
This work was financially supported by the 251801 SENER-CONACYT project. Authors thank to Adriana Tejeda, Omar Novelo, and Antonio Gómez-Cortés for technical support. Carlos Hernández-Fontes thanks to PNPC-CONACYT for his personal scholarship support, while D. G. Araiza acknowledges to SENER-CONACYT for the postdoctoral fellowship provided.References
- Y. Yan, V. Manovic, E. J. Anthony and P. T. Clough, Energy Convers. Manage., 2020, 226, 113530 CrossRef CAS.
- C. Wang, Y. Wang, M. Chen, D. Liang, Z. Yang, W. Cheng, Z. Tang, J. Wang and H. Zhang, Int. J. Hydrogen Energy, 2021, 46, 5852–5874 CrossRef CAS.
- J. Gandara-Loe, L. Pastor-Perez, L. F. Bobadilla, J. A. Odriozola and T. R. Reina, React. Chem. Eng., 2021, 6, 787–814 RSC.
- P. Cerqueira, M. A. Soria and L. M. Madeira, Energy Convers. Manage., 2021, 238, 114146 CrossRef CAS.
- M. S. Yancheshmeh, H. R. Radfarnia and M. C. Iliuta, ACS Sustainable Chem. Eng., 2017, 5, 9774–9786 CrossRef.
- M. Cortazar, S. Sun, C. Wu, L. Santamaria, L. Olazar, E. Fernandez, M. Artetxe, G. Lopez and M. Olazar, J. Environ. Chem. Eng., 2021, 9, 106725 CrossRef CAS.
- Y. Wang, M. Z. Memon, M. A. Seelro, W. Fu, Y. Gao, Y. Dong and G. Ji, Int. J. Hydrogen Energy, 2021, 46, 23358–23379 CrossRef CAS.
- Y. Xu, B. Lu, C. Luo, J. Chen, Z. Zhang and L. Zhang, Chem. Eng. J., 2021, 406, 126903 CrossRef CAS.
- N. Wang, Y. Feng, X. Guo and S. Ni, Int. J. Hydrogen Energy, 2021, 46, 10119–10130 CrossRef CAS.
- V. S. Sikarwar, N. R. Peela, A. K. Vuppaladadiyam, N. L. Ferreira, A. Mašláni, R. Tomar, M. Pohořelý, E. Meers and M. Jeremiáš, RSC Adv., 2022, 12, 6122–6132 RSC.
- S. Hernández-Castillo, H. Martínez-Hernández and J. A. Mendoza-Nieto, React. Chem. Eng., 2021, 6, 1412–1427 RSC.
- O. Akeeb, L. Wang, W. Xie, R. Davis, M. Alkasrawi and S. Toan, J. Environ. Manage., 2022, 313, 115026 CrossRef CAS.
- E. Victor and U. Great, J. Energy Res. Rev., 2021, 32, 46–64 CrossRef.
- C. Halliday and T. A. Hatton, Ind. Eng. Chem. Res., 2021, 60, 9313–9346 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- J. F. Gómez-García, J. A. Mendoza-Nieto, A. Yañez-Aulestia, F. Plascencia-Hernández and H. Pfeiffer, Fuel Process. Technol., 2020, 204, 106404 CrossRef.
- A. Yañez-Aulestia, M. A. Martínez-Cruz and H. Pfeiffer, J. Phys. Chem. C, 2020, 124, 16019–16031 CrossRef.
- A. Yañez-Aulestia, J. F. Gómez-García, J. A. Mendoza-Nieto, Y. Duan and H. Pfeiffer, Thermochim. Acta, 2018, 660, 144–151 CrossRef.
- B. Alcántar-Vázquez, Y. Duan and H. Pfeiffer, Ind. Eng. Chem. Res., 2016, 55, 9880–9886 CrossRef.
- H. Lara-Garcia, B. Alcántar-Vázquez, Y. Duan and H. Pfeiffer, J. Phys. Chem. C, 2016, 120, 3798–3806 CrossRef CAS.
- H. A. Lara-García, E. Vera, J. A. Mendoza-Nieto, J. F. Gómez-García, Y. Duan and H. Pfeiffer, Chem. Eng. J., 2017, 327, 783–791 CrossRef.
- C. Hernández-Fontes and H. Pfeiffer, Chem. Eng. J., 2022, 428, 131998 CrossRef.
- C. Hernández-Fontes and H. Pfeiffer, React. Chem. Eng., 2022, 7, 1573–1588 RSC.
- T. Takahashi and H. Watanabe, Fusion Eng. Des., 1989, 8, 399–405 CrossRef CAS.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids, Academic Press, San Diego, 1999 Search PubMed.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- X. Dong, Y. Xu, S. Yan, S. Mao, L. Xiong and X. Sun, J. Mater. Chem. A, 2015, 3, 670–679 RSC.
- M. Oishi, K. Shimoda, K. Ohara, D. Kabutan, T. Kawaguchi and Y. Uchimoto, J. Phys. Chem. C, 2020, 124, 24081–24089 CrossRef CAS.
- S. F. Amalraj, L. Burlaka, C. M. Julien, A. Mauger, D. Kovacheva, M. Talianker, B. Markovsky and D. Aurbach, Electrochim. Acta, 2014, 123, 395–404 CrossRef CAS.
- D. Y. W. Yu and K. Yanagida, J. Electrochem. Soc., 2011, 158, A1015 CrossRef CAS.
- O. Hernández-Rivas, A. Martínez, H. Pfeiffer and J. A. Mendoza-Nieto, Chem. Eng. J., 2020, 392, 123740 CrossRef.
- J. A. Mendoza-Nieto and H. Pfeiffer, RSC Adv., 2016, 6, 66579–66588 RSC.
- I. Yanase and T. Takano, Inorg. Chem. Commun., 2019, 104, 212–218 CrossRef CAS.
- I. Yanase, S. Onozawa, Y. Ohashi and T. Takeuchi, Powder Technol., 2019, 348, 43–50 CrossRef CAS.
- Y. Seo, S. H. Jo, C. K. Ryu and C. K. Yi, Chemosphere, 2007, 69, 712–718 CrossRef CAS PubMed.
- Y. Kan, Y. Hu, C.-K. Lin, Y. Ren, Y.-K. Sun, K. Amine and Z. Chen, Phys. Chem. Chem. Phys., 2014, 16, 20697–20702 RSC.
- K. Shimoda, M. Oishi, T. Matsunaga, M. Murakami, K. Yamanaka, H. Arai, Y. Ukyo, Y. Uchimoto, T. Ohta, E. Matsubara and Z. Ogumi, J. Mater. Chem. A, 2017, 5, 6695–6707 RSC.
- F. Kong, R. C. Longo, M. S. Park, J. Yoon, D. H. Yeon, J. H. Park, W. H. Wang, S. Kc, S. G. Doo and K. Cho, J. Mater. Chem. A, 2015, 3, 8489–8500 RSC.
- Z. Jadidi, T. Chen, P. Xiao, A. Urban and G. Ceder, J. Mater. Chem. A, 2020, 8, 19965–19974 RSC.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- P. Pasierb, R. Gajerski, S. Komornicki and M. Rekas, J. Therm. Anal. Calorim., 2001, 65, 457–466 CrossRef CAS.
- K. Kubota, Electrochemistry, 2020, 20, 00092 Search PubMed.
- A. D. Robertson and P. G. Bruce, Chem. Mater., 2003, 15, 1984–1992 CrossRef CAS.
- H. Zhang, B. Li, J. Wang, B. Wu, T. Fu and J. Zhao, RSC Adv., 2016, 6, 22625–22632 RSC.
- S. Frangini, Open Phys. Chem. J., 2009, 3, 1–7 CrossRef CAS.
- L. Shi, T. Qu, D. Liu, Y. Deng, B. Yang and Y. Dai, Process of thermal decomposition of lithium carbonate, in Miner. Met. Mater. Ser., Springer, 2020 Search PubMed.
- J. Bak and S. Clausen, Appl. Spectrosc., 1999, 53, 697–700 CrossRef CAS.
- Y. Wang, J. Zhao, T. Wang, Y. Li, X. Li, J. Yin and C. Wang, J. Catal., 2016, 337, 293–302 CrossRef CAS.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- C. Slostowski, S. Marre, P. Dagault, O. Babot, T. Toupance and C. Aymonier, J. CO2 Util., 2017, 20, 52–58 CrossRef CAS.
- M. Elavarasan, W. Yang, S. Velmurugan, J. N. Chen, T. C. K. Yang and T. Yokoi, Nanomater., 2022, 12, 332 CrossRef CAS.
- P. G. Lustemberg, M. V. Bosco, A. Bonivardi, H. F. Busnengo and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2015, 119, 21452–21464 CrossRef CAS.
- B. H. Solis, Y. Cui, X. Weng, J. Seifert, S. Schauermann, J. Sauer, S. Shaikhutdinov and H. J. Freund, Phys. Chem. Chem. Phys., 2017, 19, 4231–4242 RSC.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- H. M. Berends and P. Kurz, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- J. L. Bishop, S. J. King, M. D. Lane, A. J. Brown, B. Lafuente, T. Hiroi, R. Roberts, G. A. Swayze, J.-F. Lin and M. Sánchez Román, Earth Space Sci., 2021, 8, EA001844 Search PubMed.
- L. Jia, D. A. Bulushev and J. R. H. Ross, Catal. Today, 2016, 259, 453–459 CrossRef CAS.
- L.-Y. Wu, S.-R. Tong, S.-Q. Hou and M.-F. Ge, J. Phys. Chem. A, 2012, 116, 10390–10396 CrossRef CAS.
- H. A. Rojas, V. P. López, M. H. Brijaldo, S. Mancipe, J. J. Martínez, A. Gómez-Cortés, D. G. Araiza and G. Díaz, Mol. Catal., 2020, 498, 111262 CrossRef CAS.
- J. Schweicher, A. Bundhoo, A. Frennet, N. Kruse, H. Daly and F. C. Meunier, J. Phys. Chem. C, 2010, 114, 2248–2255 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
- X. Xiao, J. Gao, S. Xi, S. H. Lim, A. K. W. Png, A. Borgna, W. Chu and Y. Liu, Appl. Catal., B, 2022, 309, 121239 CrossRef CAS.
- C. Klingsberg and R. Roy, J. Am. Ceram. Soc., 1960, 43, 620–626 CrossRef CAS.
- Q. Zhang, D. Peng, S. Zhang, Q. Ye, Y. Wu and Y. Ni, AIChE J., 2017, 63, 2153–2164 CrossRef CAS.
- H. Shi, M. Huang, Y. Huang, L. Cui, L. Zheng, M. Cui, L. Jiang, H. Ibrahim and P. Tontiwachwuthikul, R. Soc. Open Sci., 2019, 6, 190311 CrossRef CAS PubMed.
- G. Lente, I. Fábián and A. J. Poë, New J. Chem., 2005, 29, 759–760 RSC.
- T. Engel and P. J. Reid, Thermodynamics, Statistical Thermodynamics, and Kinetics, Pearson Education, 3rd edn, 2006 Search PubMed.
- R. Kataoka, N. Taguchi, T. Kojima, N. Takeichi and T. Kiyobayashi, J. Mater. Chem. A, 2019, 7, 5381–5390 RSC.
- E. Lee and K. A. Persson, Adv. Energy Mater., 2014, 4, 1400498 CrossRef.
- C. James, Y. Wu, B. W. Sheldon and Y. Qi, Solid State Ionics, 2016, 289, 87–94 CrossRef CAS.
- A. R. Armstrong, N. Dupre, A. J. Paterson, C. P. Grey and P. G. Bruce, Chem. Mater., 2004, 16, 3106–3118 CrossRef CAS.
- Y. Il Jang, B. Huang, H. Wang, D. R. Sadoway and Y. M. Chiang, J. Electrochem. Soc., 1999, 146, 3217–3223 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00382a |
| This journal is © The Royal Society of Chemistry 2023 |