
One-pot tandem synthesis of 5-ethoxymethylfurfural as a potential biofuel†
Gabriel Abranches Dias
Castro
 and
Sergio Antonio
Fernandes
*
and
Sergio Antonio
Fernandes
*
Grupo de Química Supramolecular e Biomimética (GQSB), Departamento de Química, CCE, Universidade Federal de Viçosa, Viçosa, MG 36570-900, Brazil. E-mail: santonio@ufv.br; sefernandes@gmail.com; Tel: (+55) 31 3612 6647
First published on 11th October 2022
Abstract
5-Ethoxymethylfurfural (EMF) is a potential biofuel, fuel additive and raw material in the chemical and beverage industry. It can be obtained from the etherification of 5-hydroxymethylfurfural (HMF) with ethanol catalyzed by acid. In this work, a green methodology was developed to produce this product through a one-pot tandem reaction, starting directly from fructose, eliminating the unnecessary process steps and the isolation of intermediates. For this, heating by microwave irradiation and p-sulfonic acid calix[4]arene (CX4SO3H) as an organocatalyst were used. EMF was obtained in 88% yield by heating a mixture of ethanol, fructose and CX4SO3H (1.0 mol%) at 140 °C for 20 min through the tandem one-pot microwave-assisted reaction.
Introduction
Due to the increasing environmental issues caused by the use of fossil fuels and the non-renewable nature of these resources, it has become a global concern to find alternative energy sources by the end of this century.1–4 In this context, biomass is the target of numerous studies, as it is an abundant, renewable and a sustainable source of energy and organic carbon.2,5,6Among the various substances that can be obtained from carbohydrates in biomass, we highlight 5-ethoxymethylfurfural (EMF), which is the product from the etherification reaction of 5-hydroxymethylfurfural (HMF) with ethanol.7,8 HMF can be obtained from the dehydration reaction of six-carbon (C6) carbohydrates, such as fructose.9
EMF has gained increasing attention as a potential biofuel and fuel additive.10,11 However, it has also been reported to have valuable applications in other areas,12 such as in the food industry as a flavor and aroma additive in beverages such as wine and beer13 and as a reaction substrate for the synthesis of various industrially interesting chemicals, such as ethyl levulinate, 2,5-diethoxymethylfuran, 5-ethoxymethylfurfuryl alcohol and cyclopentenone.14
As already mentioned, EMF is considered to possess adequate properties and great potential to be used as a biofuel or additive for gasoline and diesel in the fuel industry.12,14,15 This is mainly because it has a high energy density, which was previously estimated to be 30.3 MJ L−1. This value is higher than that of ethanol (23.5 MJ L−1) and similar to gasoline (31.1 MJ L−1) and diesel (33.6 MJ L−1).10,11 In addition, EMF also has other attractive features, such as a high boiling point (235 °C), being insoluble in water, high oxidation stability, low toxicity and adequate flow properties, and compared to fossil fuels such as diesel, it generates less soot, NOx and SOx emissions.10,16–18
Thus, considering the many potential applications and qualities of EMF, it is important to develop more sustainable and viable routes for its convenient production and commercial use. In this case, it is interesting and advantageous to synthesize EMF directly from C6 carbohydrates through one-pot tandem reactions.19–24 In this process, the carbohydrate is solubilized in ethanol and dehydrated in the presence of an acid catalyst and heating, leading to the formation of HMF. Subsequently, in a second cascade reaction step, HMF reacts with ethanol, leading to the formation of EMF. Accordingly, it is possible to reduce the unnecessary steps for the synthesis and purification of EMF, which require the use of more reagents, solvents, longer time and generation of more residues.8,18,25
In this case, catalysis plays a fundamental role in ensuring that these processes occur efficiently with the use of less energy, time and lower generation of chemical waste. The use of catalysts has been reported for the direct conversion of carbohydrates to EMF, which could convert fructose to EMF in good yields.26–30 In this context, organocatalysts have great potential for application in this chemical transformation, as they have been reported in important and diverse biorefinery processes.31,32 Nonetheless, organocatalysts are still rarely explored for the conversion of carbohydrates to EMF, and especially compared to metallic catalysts, they have many advantages, such as high selectivity, low cost, easy manipulation and the avoidance of metal contamination in the final product.31,33,34
Among the currently known organocatalysts, calix[n]arenes have stood out due to their wide applications in different chemical transformations,35–44 including biorefinery processes45–49 such as in the development of reaction methodologies for the dehydration of C6 carbohydrates.50,51 Herein, we focus on the one-pot tandem conversion of fructose to EMF by microwave (MW) assistance, employing p-sulfonic acid calix[4]arene (CX4SO3H) as an organocatalyst. To achieve the best results, different conditions such as temperature, reaction time, catalytic load and use of some other organocatalysts and H2SO4 were studied.
Experimental
Materials and instruments
All reagents and solvents used in this work were used as purchased and recommended for organic synthesis and analysis by the manufacturers (Sigma-Aldrich and Vetec).The experiments were carried out in a CEM Discover microwave irradiation reactor (MWI), which has an internal probe for monitoring and controlling the reaction temperature.
Chromatograms and mass spectra were obtained using a gas chromatograph coupled to a mass spectrometer, i.e., SHIMADZU GCMS-QP2010C Ultra mass spectrometer. Infrared spectra were recorded for neat samples using a FT-IR Varian 660 Fourier transform infrared spectrometer. Values are expressed in wavenumber (cm−1) and recorded in the range of 4000–400 cm−1. 1H NMR spectra were recorded at 25 °C in DMSO-d6 or CDCl3 on a Varian Mercury 300 spectrometer operating at 300 MHz for 1H and 75 MHz for 13C.
Synthesis of CX4SO3Si(n)
The CX4SO3H catalyst was synthesized according to a previously published methodology (ESI,† Fig. S1–S9).46Conversion of fructose to EMF
Specifically, 0.25 mmol of fructose (45.04 mg), 1.0 mL of ethanol (17 mmol) and 1.0 mol% of CX4SO3H (2.27 mg) were added to a sealed 10 mL Pyrex glass tube. Then, this mixture was placed in the MWI reactor, where it was heated at 140 °C for 20 min using a maximum power of 100 W. After the end of the reaction, the excess ethanol was removed using a rotary evaporator under reduced pressure. Subsequently, 4.0 mL of ethyl acetate was added to the tube to precipitate CX4SO3H. This mixture was centrifuged and the supernatant was transferred to a 5.0 mL volumetric flask, which had its volume made up with ethyl acetate. Finally, quantification was performed by GC-MS as described by Castro and Fernandes, in 2022, with slight modifications.51Analysis of EMF, ethyl levulinate and HMF
EMF, ethyl levulinate (EL) and HMF were quantified based on the external standard technique, with the addition of 1,3,5-trimethoxybenzene (TMB) as the internal standard. Standard solutions were prepared in ethyl acetate, with the EMF, EL and HMF concentrations in the range of 0.25–2.00 mg mL−1 and TMB at a fixed concentration of 1.00 mg mL−1.51 The calibration curves were obtained in relation to the ratio of the area of each of the analytes, i.e., EMF, EL and HMF, to the area of TMB, for each injected standard, as presented in the ESI† (Fig. S10). The yield of EMF, EL and HMF (X%) was calculated based on the calibration curve and eqn (1), as follows: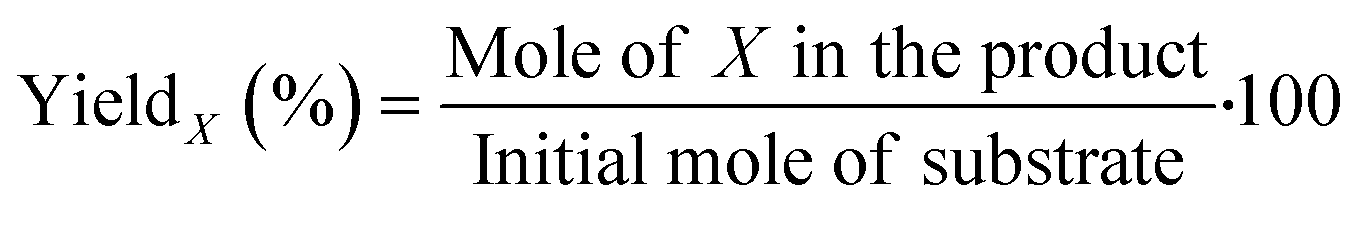 | (1) |
Spectroscopy data
The spectra of EMF, EL and HMF, as well as the materials obtained, are presented in the ESI† (Fig. S11–S21).EMF: yellow liquid, 88% yield. IR (cm−1) ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) max: 2972, 2866, 1673, 1520, 1186, 1095, 1019, 803. GC-MS (m/z) (abundancy %): 154 (20, M+), 125 (100), 109 (90), 97 (95), 81 (40), 69 (30), 53 (35), 41 (30). 1H NMR (CDCl3, 300 MHz) δ 1.23 (t, J = 7.0 Hz, 3H,), 3.79 (q, J = 7.0 Hz, 2H), 4.52 (s, 2H), 6.51 (d, J = 3.5 Hz, 1H), 7.20 (d, J = 3.5 Hz, 1H), 9.61 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 15.0, 64.8, 66.6, 111.0, 122.0, 153.0, 158.8, 177.7.
max: 2972, 2866, 1673, 1520, 1186, 1095, 1019, 803. GC-MS (m/z) (abundancy %): 154 (20, M+), 125 (100), 109 (90), 97 (95), 81 (40), 69 (30), 53 (35), 41 (30). 1H NMR (CDCl3, 300 MHz) δ 1.23 (t, J = 7.0 Hz, 3H,), 3.79 (q, J = 7.0 Hz, 2H), 4.52 (s, 2H), 6.51 (d, J = 3.5 Hz, 1H), 7.20 (d, J = 3.5 Hz, 1H), 9.61 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 15.0, 64.8, 66.6, 111.0, 122.0, 153.0, 158.8, 177.7.
EL: colorless liquid. IR (cm−1) max: 2984, 1721, 1367, 1160, 1025, 753. GC-MS (m/z) (abundancy %): 144 (4, M+), 129 (21), 99 (71), 43 (100). 1H NMR (CDCl3, 300 MHz) δ 1.24 (t, J = 7.1 Hz, 3H), 2.18 (s, 3H), 2.56 (t, J = 7.0 Hz, 2H), 2.74 (t, J = 7.0 Hz, 2H), 4.12 (q, J = 7.1 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 14.1, 28.0, 29.9, 37.9, 60.6, 172.7, 206.7.
HMF: yellow liquid. IR (cm−1) max: 3336, 2837, 1659, 1513, 1395, 1186, 1111, 764. GC-MS (m/z) (abundancy %): 126 (55, M+), 97 (100), 69 (35), 41 (65). 1H NMR (CDCl3, 300 MHz) δ 9.53 (s, 1H), 7.20 (d, J = 3.6 Hz, 1H), 6.49 (d, J = 3.6 Hz, 1H), 4.68 (s, 2H). 13C NMR (CDCl3, 75 MHz) δ 177.7, 160.8, 152.3, 123.0, 109.9, 57.5.
Reuse of CX4SO3H
The catalyst recycling experiment was conducted using a reaction model of 0.25 mmol fructose (45.04 mg), 1.00 mL of ethanol and 1.0 mol% CX4SO3H (2.27 mg), at a temperature of 140 °C for 20 min. At the end of this period, the system was cooled to room temperature and the ethanol evaporated under reduced pressure. Then, 4.0 mL of ethyl acetate was added to the reaction tube for the precipitation of CX4SO3H. Then, this mixture was centrifuged, and the supernatant removed for the quantification of EMF. The organocatalyst left in the tube was dried at 85 °C. subsequently, a new load of fructose (0.25 mmol) and 1.0 mL of ethanol were added to it for a new reaction cycle.Synthesis, identification, and quantification of different HMF ethers by 1H NMR
To evaluate the scope of the reaction, nine other alcohols besides ethanol were evaluated. After finishing each fructose conversion experiment in the respective ethers, an aliquot of the reaction mixture (200 μL) was transferred to a 5 mm NMR tube. Subsequently, 400 μL of 0.045 mol L−1 TMB solution in DMSO-d6 was added and the 1H NMR spectrum was acquired. To perform the yield calculations, the following equation (eqn (2)) was used, where N = number of nuclei that absorb in the frequency, n = amount of matter (mol); and A = integration area.40,47,50,52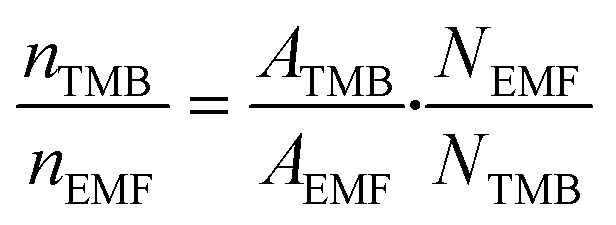 | (2) |
Results and discussion
Evaluation of the effect of time, temperature and CX4SO3H amount
We started our study by evaluating how the reaction proceeded with time (Fig. 1), keeping all the other parameters constant, as follows: 140 °C, 1.00 mol% CX4SO3H, 0.25 mmol fructose and 1.00 mL ethanol. Fig. 1 shows the reaction behavior over time. It can be observed that the yield of HMF and EMF increased up to 5 min. Subsequently, the HMF yield started to decrease due to the etherification reaction, and the EMF yield increased up to the time of 20 min, when it reached the maximum value of 88%. After 15 min, the formation of ethyl levulinate (EL) was observed, which increased gradually. This phenomenon was also observed in previous works and occurs because EL is formed from the EMF rehydration reaction, which is favored at longer reaction times.53–55 It is worth mentioning that this product is also of great interest in the biofuel industry.46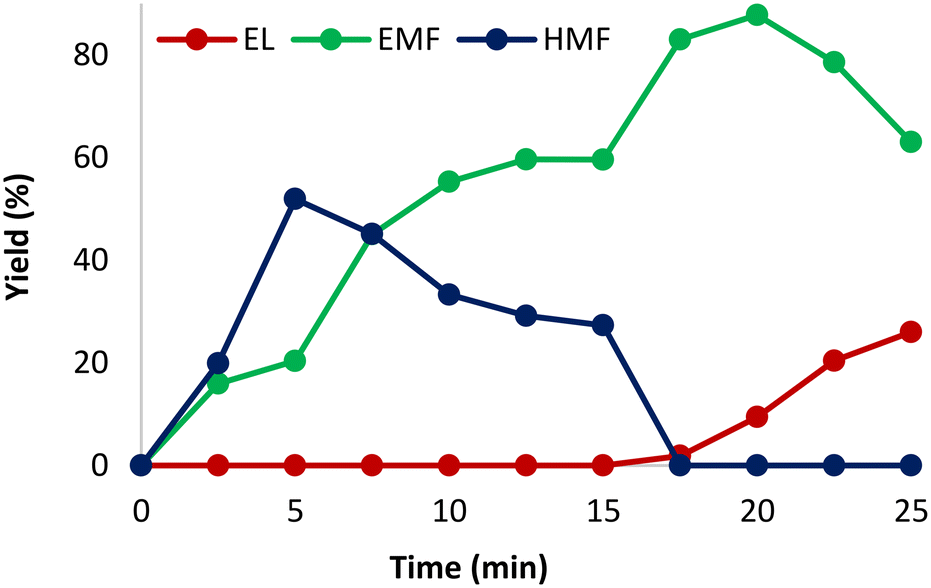 | ||
| Fig. 1 Variation in the reaction yield with time. Reagents and conditions: fructose (0.25 mmol), 1.0 mol% CX4SO3H (2.3 mg), MWI, 140 °C, and 1.0 mL of ethanol. | ||
Considering that at 20 min, the yield for EMF reached the maximum value, we fixed this experiment time, and then we continued our study, evaluating the effect of temperature on the reaction. It was observed that at temperatures lower than 140 °C, the EMF yield decreased (Table 1, entries 1–3). At 120 °C, the formation of EMF was not observed, and there was no by-product formed in the reaction (Table 1, entry 1). In contrast, when the temperature increased to 130 °C, EMF was obtained in 31% yield, HMF in 8% yield and EL was not detected (Table 1, entry 2). By increasing the reaction temperature to 150 °C, the yield of EMF dropped to 61% and that of EL increased to 38% (Table 1, entry 4). Therefore, the temperature of 140 °C was chosen to perform further experiments. The formation of a greater amount of EL was observed in both experiments where higher temperatures were used and that carried out for longer times, which has been reported in other studies. This is due to the instability of EMF at high temperatures in an acidic environment, which undergoes rehydration of the furan ring and leads to the formation of EL.8,16,17,25,56
|
|
|||||
|---|---|---|---|---|---|
| Entry | Temperature (°C) | CX4SO3H (mol%) | Yieldb (%) | ||
| EMF | HMF | EL | |||
| a Reagents and conditions: fructose (0.25 mmol), MWI, 1.0 mL of ethanol. b Yields were determined by GC-MS analysis using calibration curves. c Experiments performed in a sealed tube with CH in an oil bath. d Experiments performed in a sealed tube with CH in an oil bath for 120 min. | |||||
| 1 | 120 | 1 | 0 | 0 | 0 |
| 2 | 130 | 1 | 31 | 8 | 0 |
| 3 | 140 | 1 | 88 | 0 | 9 |
| 4 | 150 | 1 | 61 | 0 | 38 |
| 5 | 140 | 1.5 | 67 | 0 | 32 |
| 6 | 140 | 2.0 | 67 | 0 | 32 |
| 7 | 140 | 0.5 | 26 | 18 | 0 |
| 8 | 140 | 0 | 0 | 0 | 0 |
| 9c | 140 | 1 | 0 | 9 | 0 |
| 10d | 140 | 1 | 0 | 9 | 0 |

Finally, the effect of the amount of CX4SO3H on the reaction was evaluated. It was observed that when the amount of CX4SO3H was 1.5 and 2 mol%, the yield of EMF decreased to 67% and that of EL to 32% in both cases (Table 1, entries 5 and 6). By reducing the CX4SO3H load in half, the EMF yield dropped to 26% and formation of HMF was observed with 18% yield (Table 1, entry 7). Finally, when the experiment was carried out in the absence of the catalyst, no formation of EMF or any by-product was detected (Table 1, entry 8). This shows the fundamental importance of using the catalyst in this reaction.
An experiment was carried out via conventional heating (CH) using the optimal reaction conditions to verify the importance of the heating method. Consequently, only the formation of HMF was observed, with a yield of 9% (Table 1, entry 9). The result was the same when the experiment time was increased to 120 min (Table 1, entry 10). This difference observed in the experiments when using different heating sources can be explained based on the way energy is transferred to the system. CH consists of a combination of heat transfer in the form of conduction and convection, which causes a slow heating rate. Alternatively, MWI heats the system by applying MW energy directly to the molecules, thus generating a high rate of temperature rise due to ionic conduction and dipole rotation effects.57,58
Comparison with other Brønsted acids as catalysts
The catalytic effects of other Brønsted acids were evaluated (Table 2). For comparison with CX4SO3H, values corresponding to the same number of ionizable hydrogens were used. A decrease in EMF yield was observed for all the acids used (Table 2, entries 2–5), even for p-sulfonic acid calix[6]arene (CX6SO3H) (Table 2, entry 2). p-Hydroxybenzene sulfonic acid (PHSA) (Table 2, entry 3) and sulfuric acid (Table 2, entry 4) led to the formation of EMF in 82% and 62% yield, and an increase in EL yield of 18% and 37%, respectively. p-Toluene sulfonic acid (PTSA) provided 71% and 8% EMF and EL, respectively (Table 2, entry 5).|
|
||||
|---|---|---|---|---|
| Entry | Catalystc (mol%) | Yieldsb (%) | ||
| EMF | HMF | EL | ||
| a Reagents and conditions: fructose (0.25 mmol), MWI, 140 °C, 20 min, and 1.0 mL of ethanol. b Yields were determined by GC-MS analysis using calibration curves. c The concentration of H+ was kept constant. | ||||
| 1 | CX4SO3H (1.00) | 88 | 0 | 9 |
| 2 | CX6SO3H (0.67) | 72 | 6 | 7 |
| 3 | PHSA (4.00) | 82 | 0 | 18 |
| 4 | H2SO4 (2.00) | 62 | 0 | 37 |
| 5 | PTSA (4.00) | 71 | 0 | 8 |

The CX4SO3H catalyst presented the best result even taking care to keep the proton concentration constant for all the evaluated catalysts. It was previously reported that CX4SO3H has superior catalytic activity to other sulfonic acids, and this was justified based on its structure and conformation, which make it possible for its functional groups and cavity to undergo specific interactions with substrates. An example of the type of interaction that CX4SO3H can undergo is π–π hydrophobic interactions.51
Evaluation of other carbohydrates
To explore the substrate scope of the system developed with CX4SO3H, other carbohydrates were used as raw materials for the synthesis of EMF, under the same optimal conditions as that using fructose, and the results are summarized in Table 3. However, all the substrates used (sucrose and raffinose) had lower yields than that with fructose (Table 3, entries 1–3). This difference can be explained by the nature of the carbohydrate used. Fructose is a monosaccharide and a furanose and is more reactive than other hexoses, given that it is a ketose. Thus, considering that sucrose is a disaccharide composed of a fructose and a glucose molecule, it is expected that the yield of product will be lower (Table 3, entry 2), given that glucose is a pyranose and an aldose, and it needs to isomerize to fructose before undergoing the dehydration reaction. This is the same for raffinose, but it is a trisaccharide made up of a fructose, glucose, and galactose molecule. Thus, given that it consists of two pyranoses and one furanose, the yield when using it as a substrate was lower (Table 3, entry 3).25,59,60|
|
||||
|---|---|---|---|---|
| Entry | Carbohydrates | Yieldb (%) | ||
| EMF | HMF | EL | ||
| a Reagents and conditions: MWI, 140 °C, 20 min, 1.0 mL of ethanol and 1.0 mol% CX4SO3H. b Yield was determined by standard EMF calibration curve GC-MS analysis. c 0.125 mmol of sucrose. d 0.083 mmol of raffinose. | ||||
| 1 | Fructose | 88 | 0 | 9 |
| 2 | Sucrosec | 37 | 0 | 0 |
| 3 | Raffinosed | 17 | 0 | 0 |

Evaluation of other alcohols
Then, using the optimized conditions 20 min, 140 °C (MWI) and 1.00 mol% CX4SO3H, we examined the scope of the etherification reactions using different alcohols. We attempted the etherification based on the size of the carbon chain of primary alcohols and sterically hindered secondary and tertiary alcohols. In general, it was observed (Scheme 1) that the yields decreased with an increase in the size of the carbon chain of the alcohol, as in the examples of ET1 (88%), ET2 (54%), ET3 (34%) and ET4 (15%), as previously reported.6,62 It was experimentally observed that for these higher-chain alcohols, the formation of humins occurred. Another trend observed experimentally was the decrease in the yield of ether with sterically hindered alcohols. This effect can be seen by comparing ET2 (54%), ET6 (50%) and ET9 (7%). The yields were lower for sterically hindered nucleophiles (alcohols), as already reported in the literature.61–66 | ||
| Scheme 1 Scope of the reaction for different alcohols, employing CX4SO3H as an organocatalyst. Yield determined by 1H NMR using TMB with the standard internal. | ||
Comparison with other methodologies
The yield of EMF prepared in this work (Table 4, entry 1) is comparable in many cases and even higher to that in works reported in the literature where fructose was used as the substrate in a one-pot tandem reaction (Table 4). For example, Yang et al., in 2012,67 achieved 76% yield when phosphotungstic acid was used as the catalyst (0.8 mol%) at a temperature of 130 °C for 30 min, in MWI (Table 4, entry 2). Their experiments were performed using a mixture of ethanol and tetrahydrofuran (THF). Besides our superior yield of EMF (88%), these authors employed a longer reaction time and a non-recommended solvent (THF) in a sustainable context.68| Entry | Solvent | Catalyst | Conditions | EMF yield (%) | Ref. |
|---|---|---|---|---|---|
| Fe3O4@C–SO3H = sulfonated magnetic solid. MCC–SO3H = magnetic cellulose-derived carbonaceous solid acid. | |||||
| 1 | Ethanol | CX4SO3H (1.0 mol% = 5 wt%) | MWI, 140 °C, 20 min | 88.0 | This work |
| 2 | Ethanol and THF (62.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :37.5) :37.5) |
H3PW12O40 (0.8 mol% = 12.8 wt%) | MWI, 130 °C, 30 min | 76.0 | 67 |
| 3 | Ethanol | AlCl3 (10 mol% = 7.4 wt%) | CH, 100 °C, 11 h | 71.2 | 17 |
| 4 | Ethanol | Fe3O4@C–SO3H (55.5 wt%) | CH, 140 °C, 24 h | 67.8 | 56 |
| 5 | Ethanol | MCC–SO3H (2.8 wt%) | CH, 130 °C, 8 h | 63.2 | 16 |
| 6 | Ethanol | WS2 (30 mol%) | MWI, 160 °C, 15 min | 62.0 | 55 |
| 7 | Ethanol | MoS2 (30 mol%) | MWI, 160 °C, 30 min | 93.3 | 69 |
In 2013, Liu et al. used 10 mol% of AlCl3 to produce EMF, achieving 71.2% yield by heating fructose at 100 °C for 11 h in ethanol. In addition to using a large amount of catalyst (10-times higher), the authors took a longer time (33-times higher) to achieve a lower yield than the present work (Table 4, entry 3).17 As early as 2015, Yuan et al. achieved a yield of 67.8% employing a heterogeneous magnetic sulfonated catalyst (Fe3O4@C–SO3H) at a very high concentration (55.5 wt%), and CH at 140 °C for a long reaction time of 24 h.56 Yuan reported lower yields than that obtained in this work (88%), in addition to the catalyst loading being approximately 10-times greater and the reaction time 72-times longer (Table 4, entries 1 and 4).
In 2018, Chen et al. used magnetic cellulose-derived carbonaceous solid acid as a catalyst, at a concentration of 2.8 wt% and achieved a yield of 63.2% by heating fructose in the presence of ethanol at 130 °C for 8 h.16 Despite using approximately half of the catalyst load and a lower temperature (130 °C) than that evaluated in this work, the yield is approximately 25% lower, and the reaction time is 24-times longer (Table 4, entries 1 and 5).
In 2020, Quereshi et al. produced two works using different metallic sulfides by MWI heating. When tungsten disulfide was used55 (Table 4, entry 6), EMF was obtained in 62% yield, but under the extreme conditions of 30 mol% catalyst and 160 °C for 15 min.
When Quereshi et al. used molybdenum disulfide69 (Table 4, entry 7), they achieved a yield greater than 93%; however, they employed a very high catalyst dosage (30 mol%), at 140 °C for 30 min. In both studies, the authors used a much higher amount of catalyst (30 mol%) than that used in this work (Table 4, entries 1, 6 and 7).
Catalytic cycle
In the catalyst recycling study, it was observed that the yield dropped from 88% to 66% the first time the catalyst was reused, and from 66% to 47% in the third reuse cycle (Fig. 2). To verify if there was any change in the structure of the catalyst, 1H NMR experiments were performed with the catalyst recovered at the end of the third cycle. According to Fig. S22,† the presence of a triplet and a doublet at δ 6.63 and δ 7.06 ppm was observed, confirming the partial loss of the sulfonic groups, which explains the partial loss in catalytic activity.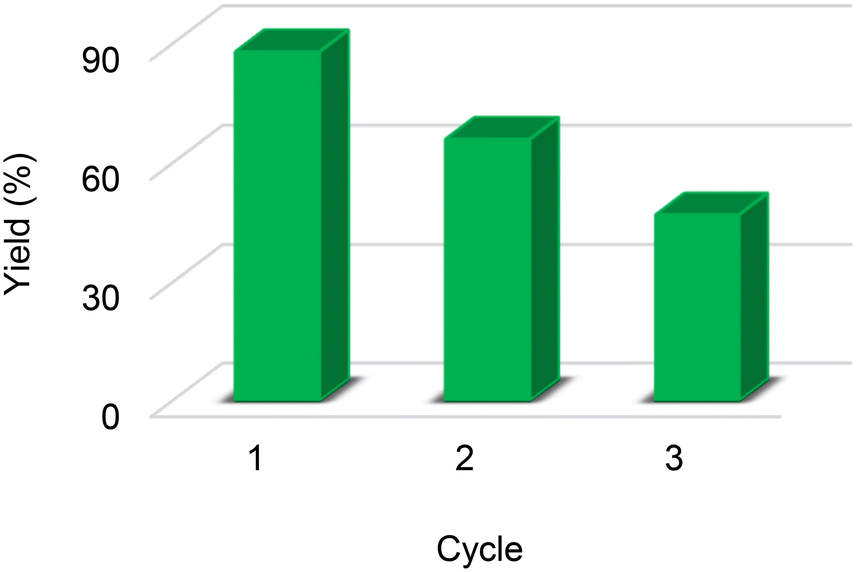 | ||
| Fig. 2 Reuse of CX4SO3H for the conversion of fructose to EMF. | ||
Mechanistic proposal
Based on the studies reported in the literature,51,70 the mechanism for the conversion of fructose to EMF was proposed, as presented in Scheme 2. Initially, the dehydration of fructose occurs in HMF, where in the first step, the protonation of the hydroxyl bound to the anomeric carbon of fructose occurs, followed by dehydration and formation of the oxonium ion (intermediate I, Scheme 2). Then, the β hydrogen is removed from the oxonium ion, forming an enol (intermediate II, Scheme 2), which is in tautomeric equilibrium with the keto form (intermediate III, Scheme 2). In the aldehyde, a second hydroxyl is protonated, followed by another dehydration, leading to the formation of an α,β-unsaturated aldehyde (intermediate IV, Scheme 2). Subsequently, secondary hydroxyl protonation followed by the loss of a water molecule leads to the formation of HMF (intermediate V, Scheme 2).51,70 Then, EMF is formed from HMF. For this chemical transformation, we propose a mechanism based on the works of Sacia et al., Elsayed et al., Balakrishnan et al. and Allen et al.61,71–74 Initially, the HMF hydroxyl is protonated, and then HMF loses a molecule of water, leading to the formation of an intermediate stabilized by electronic delocalization (intermediate VI, Scheme 2).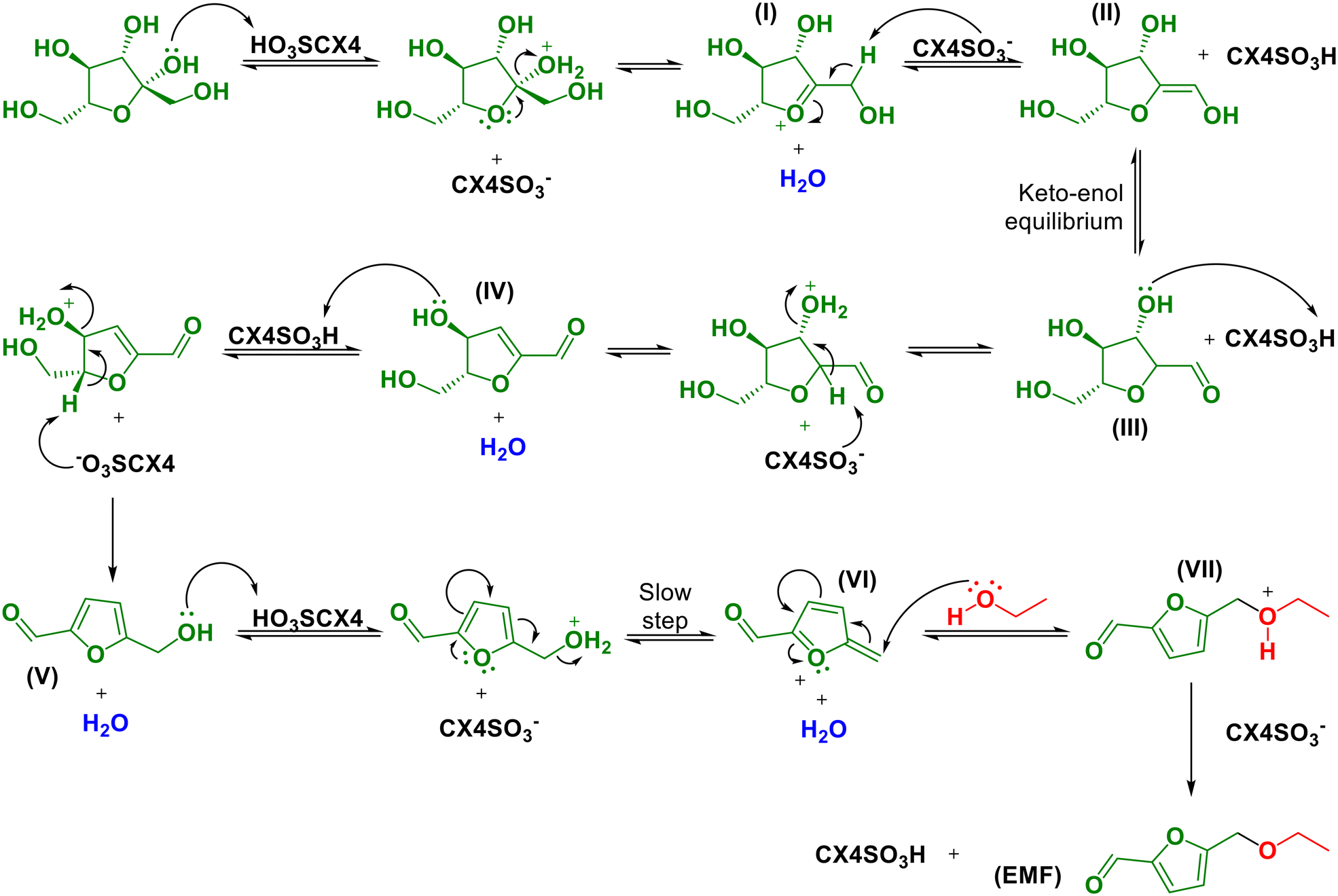 | ||
| Scheme 2 Mechanistic proposal for the conversion fructose for EMF one-pot tandem reaction. | ||
Sacia et al.72 discussed in their work that the elimination of the water molecule is the slowest step, and therefore is a limiting step in the formation of EMF. However, it is important to emphasize that this dehydration reaction of HMF forming intermediate VI is dominant in all experimental conditions, as demonstrated by DFT calculations in the work by Allen et al.61 After this step, ethanol undergoes nucleophilic attack of intermediate VI, leading to the formation of intermediate VII. After the removal of a proton, EMF is obtained (Scheme 2).
Conclusions
As a potential biofuel and fuel additive, EMF was produced in this work directly from fructose with a yield of 88% using an efficient and clean one-pot methodology, where the reaction occurs in tandem, without the addition of new reagents. In addition, we highlighted the other advantages of the developed methodology, as follows: (i) use of non-toxic catalyst, (ii) short reaction time, (iii) efficient heating system, (iv) metal-free process, (v) formation of two C–C π bonds and one C–O in sequence and (vi) process according to green chemistry. CX4SO3H showed good catalytic activity for the tandem one-pot conversion of fructose to EMF and brings new possibilities in the field of biomass valorization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support provided by Fundação de Amparo à Pesquisa do Estado de Minas Gerais – Brazil (FAPEMIG), Conselho Nacional de Desenvolvimento Científico e Tecnológico – Brazil (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES – Finance Code 001). SAF is supported by Research Fellowships from CNPq.Notes and references
- D. Kweku, O. Bismark, A. Maxwell, K. Desmond, K. Danso, E. Oti-Mensah, A. Quachie and B. Adormaa, Journal of Scientific Research and Reports, 2018, 17, 1–9 CrossRef.
- D. Y. Goswami and F. Kreith, Energy efficiency and renewable energy handbook, CRC Press, 2nd edn, 2016 Search PubMed.
- S. Sorrell, J. Speirs, R. Bentley, A. Brandt and R. Miller, Energy Policy, 2010, 38, 5290–5295 CrossRef.
- R. Draeger, B. S. L. Cunha, E. Müller-Casseres, P. R. R. Rochedo, A. Szklo and R. Schaeffer, Energy, 2022, 255, 124451 CrossRef CAS.
- P. Fornasiero and M. Graziani, Renewable resources and renewable energy - A global challenge, CRC Press, 2nd edn, 2012 Search PubMed.
- S. K. Brar, G. S. Dhillon and C. R. Soccol, Biotransformation of waste biomass into high value biochemicals, Springer, 1st edn, 2014 Search PubMed.
- M. M. Antunes, P. A. Russo, P. V. Wiper, J. M. Veiga, M. Pillinger, L. Mafra, D. V. Evtuguin, N. Pinna and A. A. Valente, ChemSusChem, 2014, 7, 804–812 CrossRef CAS PubMed.
- X. Jia, J. Ma, P. Che, F. Lu, H. Miao, J. Gao and J. Xu, J. Energy Chem., 2013, 22, 93–97 CrossRef CAS.
- M. A. A. Bin Abdul Rani, N. A. Karim and S. K. Kamarudin, Int. J. Energy Res., 2022, 1–55 Search PubMed.
- G. J. M. Gruter and F. Dautzenberg, Method for the synthesis of 5-hydroxymethylfurfural ethers and their use, U.S. Pat. Appl., 2011/0082304A1, 2011, vol. 1, pp. 1–8 Search PubMed.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS.
- H. Guo, T. Dowaki, F. Shen, X. Qi and R. L. Smith, ACS Sustainable Chem. Eng., 2022, 10, 9002–9021 CrossRef CAS.
- V. Pereira, M. Santos, J. Cacho and J. C. Marques, LWT--Food Sci. Technol., 2017, 75, 719–726 CrossRef CAS.
- X. Liu, D. Yu, H. Luo and C. Li, Front. Chem., 2022, 9, 1–7 RSC.
- D. Yu, X. Liu, J. Jiang, Y. Liu, J. Tan and H. Li, J. Chem., 2021, 2021, 16 Search PubMed.
- T. Chen, L. Peng, X. Yu and L. He, Fuel, 2018, 219, 344–352 CrossRef CAS.
- B. Liu, Z. Zhang, K. Huang and Z. Fang, Fuel, 2013, 113, 625–631 CrossRef CAS.
- X. Liu and R. Wang, Int. J. Chem. Eng., 2018, 2018, 10 CrossRef.
- H. Liu, X. Tang, W. Hao, X. Zeng, Y. Sun, T. Lei and L. Lin, J. Energy Chem., 2018, 27, 375–380 CrossRef.
- B. Srinivasa Rao, D. Dhana Lakshmi, P. Krishna Kumari, P. Rajitha and N. Lingaiah, Sustainable Energy Fuels, 2020, 4, 3428–3437 RSC.
- Y. He, L. Zhang, Y. Liu, S. Yi, H. Yu, Y. Zhu and R. Sun, Chem. Eng. J., 2022, 429, 132279 CrossRef CAS.
- Y. Chen, L. Peng, J. Zhang and L. He, Catal. Commun., 2020, 137, 105947 CrossRef CAS.
- G. Bharath, K. Rambabu, A. Hai, P. P. Morajkar, A. V. Salkar, S. W. Hasan, P. L. Show and F. Banat, Int. J. Energy Res., 2021, 45, 14680–14691 CrossRef CAS.
- G. Xu, S. Zhang, Z. Zheng, C. Wang, S. Wang and H. Tao, BioResources, 2020, 15, 3621–3635 CAS.
- X. Liu, H. Li, H. Pan, H. Zhang, S. Huang, K. Yang, W. Xue and S. Yang, J. Energy Chem., 2016, 25, 523–530 CrossRef.
- P. K. Kumari, B. S. Rao, D. Padmakar, N. Pasha and N. Lingaiah, Mol. Catal., 2018, 448, 108–115 CrossRef CAS.
- D. Gupta and B. Saha, Catal. Commun., 2018, 110, 46–50 CrossRef CAS.
- Z. Zhang, Y. Wang, Z. Fang and B. Liu, ChemPlusChem, 2014, 79, 233–240 CrossRef CAS.
- K. Zhao, Y. Xiang, X. Sun, L. Chen, J. Xiao and X. Liu, Front. Chem., 2022, 10, 1–8 Search PubMed.
- Y. Xiang, K. Zhao, S. Zhou, W. Zhao, Z. Zeng, X. Zhu and X. Liu, Sustainable Energy Fuels, 2022, 6, 2368–2376 RSC.
- S. Meninno, ChemSusChem, 2020, 13, 439–468 CrossRef CAS.
- D. Liu and E. Chen, Green Chem., 2014, 16, 964–981 RSC.
- K. Maruoka, B. List, H. Yamamoto and L. Zhu Gong, Chem. Commun., 2012, 48, 10703–10703 RSC.
- V. da G. Oliveira, M. F. D. C. Cardoso and L. da S. M. Forezi, Catalysts, 2018, 8, 605 CrossRef.
- W. F. de Paiva, I. B. Braga, J. V. de Assis, S. M. B. Castañeda, Á. G. Sathicq, V. Palermo, G. P. Romanelli, R. Natalino, M. J. da Silva, F. T. Martins, G. S. G. de Carvalho, G. W. Amarante and S. A. Fernandes, Tetrahedron, 2019, 75, 3740–3750 CrossRef CAS.
- P. A. D. S. Abranches, W. F. de Paiva, Â. de Fátima, F. T. Martins and S. A. Fernandes, J. Org. Chem., 2018, 83, 1761–1771 CrossRef CAS PubMed.
- T. R. M. Rezende, J. O. S. Varejão, A. L. L. de A. Sousa, S. M. B. Castañeda and S. A. Fernandes, Org. Biomol. Chem., 2019, 17, 2913–2922 RSC.
- D. L. Da Silva, S. A. Fernandes, A. A. Sabino and Â. De Fá, Tetrahedron Lett., 2011, 52, 6328–6330 CrossRef CAS.
- I. B. Braga, S. M. B. Castañeda, J. Vitor De Assis, A. O. Barros, G. W. Amarante, A. K. S. M. Valdo, F. T. Martins, A. F. D. P. Rosolen, E. Pilau and S. A. Fernandes, J. Org. Chem., 2020, 85, 15622–15630 CrossRef CAS PubMed.
- R. Natalino, E. V. V. Varejão, M. J. Da Silva, A. L. Cardoso and S. A. Fernandes, Catal. Sci. Technol., 2014, 4, 1369–1375 RSC.
- L. S. Santos, S. A. Fernandes, R. A. Pilli and A. J. Marsaioli, Tetrahedron: Asymmetry, 2003, 14, 2515–2519 CrossRef CAS.
- J. V. De Assis, P. A. S. Abranches, I. B. Braga, O. M. P. Zuñiga, A. G. Sathicq, G. P. Romanelli, A. G. Sato and S. A. Fernandes, RSC Adv., 2016, 6, 24285–24289 RSC.
- J. B. Simoes, D. L. da Silva, A. de Fatima and S. A. Fernandes, Curr. Org. Chem., 2012, 16, 949–971 CrossRef CAS.
- S. Shimizu, N. Shimada and Y. Sasaki, Green Chem., 2006, 8, 608–614 RSC.
- G. F. David, A. M. Ríos-Ríos, Â. de Fátima, V. H. Perez and S. A. Fernandes, Ind. Crops Prod., 2019, 134, 382–387 CrossRef CAS.
- G. A. D. Castro and S. A. Fernandes, Sustainable Energy Fuels, 2021, 5, 108–111 RSC.
- G. A. D. Castro, A. Santos, A. G. Sathicq, V. Palermo, G. Romanelli and S. Fernandes, React. Chem. Eng., 2022, 7, 2132 RSC.
- J. V. de Assis, P. A. S. Abranches, I. B. Braga, O. M. P. Zuñiga, A. G. Sathicq, G. P. Romanelli, A. G. Sato and S. A. Fernandes, RSC Adv., 2016, 6, 24285–24289 RSC.
- C. G. Almeida, I. F. Souza, N. A. Liberto, M. J. Da Silva, S. A. Fernandes and M. Le Hyaric, Monatsh. Chem., 2015, 146, 1927–1934 CrossRef CAS.
- S. de Paiva Silva Pereira, J. Oliveira Santana Varejão, Â. de Fátima and S. A. Fernandes, Ind. Crops Prod., 2019, 138, 111492 CrossRef.
- G. A. D. Castro and S. A. Fernandes, Catal. Lett. DOI:10.1007/s10562-022-04043-x.
- S. A. Fernandes, R. Natalino, M. J. Da Silva and C. F. Lima, Catal. Commun., 2012, 26, 127–131 CrossRef CAS.
- G. Xu, C. Chang, S. Fang and X. Ma, Renewable Energy, 2015, 78, 583–589 CrossRef CAS.
- G. Xu, B. Chen, Z. Zheng, K. Li and H. Tao, Asia-Pac. J. Chem. Eng., 2017, 12, 527–535 CrossRef CAS.
- S. Quereshi, E. Ahmad, K. K. K. Pant and S. Dutta, ACS Sustainable Chem. Eng., 2020, 8, 1721–1729 CrossRef CAS.
- Z. Yuan, Z. Zhang, J. Zheng and J. Lin, Fuel, 2015, 150, 236–242 CrossRef CAS.
- Z. Fang, R. L. S. Jr and X. Qi, Production of Biofuels and Chemicals with Microwave, 1st edn, 2015, vol. 3 Search PubMed.
- N. Sweygers, N. Alewaters, R. Dewil and L. Appels, Sci. Rep., 2018, 8, 1–11 CAS.
- H. Wang, T. Deng, Y. Wang, X. Cui, Y. Qi, X. Mu, X. Hou and Y. Zhu, Green Chem., 2013, 15, 2379–2383 RSC.
- Y. Yang, M. M. Abu-Omar and C. Hu, Appl. Energy, 2012, 99, 80–84 CrossRef CAS.
- M. C. Allen, A. J. Hoffman, T. W. Liu, M. S. Webber, D. Hibbitts and T. J. Schwartz, ACS Catal., 2020, 10, 6771–6785 CrossRef CAS.
- O. M. Portilla-Zuñiga, J. J. Martínez, M. Casella, D. I. Lick, Á. G. Sathicq, R. Luque and G. P. Romanelli, Mol. Catal., 2020, 494, 111125 CrossRef.
- M. J. da Silva, N. P. G. Lopes, S. O. Ferreira, R. C. da Silva, R. Natalino, D. M. Chaves and M. G. Texeira, Chem. Pap., 2021, 75, 153–168 CrossRef CAS.
- M. J. da Silva, L. A. S. Viana and M. G. Teixeira, C. R. Chim., 2020, 23, 93–103 CrossRef CAS.
- P. K. Sahoo, S. S. Gawali and C. Gunanathan, ACS Omega, 2018, 3, 124–136 CrossRef CAS PubMed.
- B. N. Moghadam, B. Akhlaghinia and S. Rezazadeh, Res. Chem. Intermed., 2016, 42, 1487–1501 CrossRef CAS.
- Y. Yang, M. M. Abu-Omar and C. Hu, Appl. Energy, 2012, 99, 80–84 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- S. Quereshi, K. K. Pant, S. Dutta and T. K. Naiya, Biomass Convers. Biorefin., 2020, 12, 641–3654 Search PubMed.
- R.-J. van Putten, J. G. de Vries, J. C. van der Waal, E. de Jong, C. B. Rasrendra and H. J. Heeres, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- I. Elsayed, M. A. Jackson and E. B. Hassan, Fuel Process. Technol., 2021, 213, 106672 CrossRef CAS.
- E. R. Sacia, M. Balakrishnan and A. T. Bell, J. Catal., 2014, 313, 70–79 CrossRef CAS.
- M. Balakrishnan, E. R. Sacia and A. T. Bell, Green Chem., 2012, 14, 1626–1634 RSC.
- S. Shinde and C. Rode, Catal. Commun., 2017, 88, 77–80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00348a |
| This journal is © The Royal Society of Chemistry 2023 |