
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved catalytic performance in gas-phase dimethyl ether carbonylation over facile NH4F etched ferrierite
Dexin Zhang abc,
Kang Li*b,
Junli Chena,
Changyu Sun*a,
Zhi Lib,
Jie Leib,
Qinlan Maa,
Pan Zhangc,
Yong Liuc and
Lin Yangc
abc,
Kang Li*b,
Junli Chena,
Changyu Sun*a,
Zhi Lib,
Jie Leib,
Qinlan Maa,
Pan Zhangc,
Yong Liuc and
Lin Yangc
aCollege of Chemical Engineering and Environment, China University of Petroleum, Beijing 102249, China. E-mail: 2565874326@qq.com; cysun@cup.edu.cn
bLuoyang R & D Center of Technologies of Sinopec Engineering (Group) Co., Ltd., Luoyang 471003, China. E-mail: likang.segr@sinopec.com
cSchool of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: 18980632893@163.com
First published on 5th December 2023
Abstract
Gas-phase dimethyl ether (DME) carbonylation to methyl acetate (MA) initiates a promising route for producing ethanol from syngas. Ferrierite (FER, ZSM-35) has received considerable attention as it displays excellent stability in the carbonylation reaction and its modification strategy is to improve its catalytic activity on the premise of maintaining its stability as much as possible. However, conventional post-treatment methods such as dealumination and desilication usually selectively remove framework Al or Si atoms, ultimately altering the intrinsic composition, crystallinity, and acidity of zeolites inevitably. In this study, we successfully prepared a series of hierarchical ZSM-35 materials through post-treatment with NH4F etching, which dissolved framework Al and Si at similar rates and preferentially attacked the defective sites. Interestingly, the produced pore systems effectively penetrated the [100] plane, offering elevated access to both the 8-membered ring (8-MR) and 10-membered ring (10-MR) channels. The physicochemical and acid properties of the pristine and NH4F etched ZSM-35 samples were comprehensively characterized using various techniques, including XRD, XRF, FESEM, HRTEM, Nitrogen adsorption–desorption, NH3-TPD, Py-IR, 27Al MAS NMR, and 29Si MAS NMR. Under moderate treatment conditions, the intrinsic microporous structure, acid properties, and crystallinity of zeolite were retained, leading to superior catalytic activity and stability with respect to the pristine sample. Nonetheless, severe NH4F etching disrupted the crystalline framework and created additional defective sites, bringing about faster deposition of coke precursors on the interior Brønsted acid sites (BAS) and decreased catalytic performance. This technique provides a novel and efficient method to slightly enhance the micropore and mesopore volume of industrially pertinent zeolites through a straightforward post-treatment, thus elevating the catalytic performance of these zeolites.
Introduction
Ethanol (EtOH) is considered as a potential alternative gasoline additive, green solvent, and a crucial raw material to produce numerous industrial chemicals and polymers.1,2 Currently, the main methods of EtOH production are biomass fermentation and ethylene hydration,3 both of which are limited by the high cost and scarcity of petroleum resources, respectively. On the other hand, Syngas (CO, CO2, and H2 majorly) has abundant feedstocks, including natural gas, biomass, coal, and industrial emissions.4–7 Therefore, the catalytic conversion of syngas to EtOH is an attractive and competitive route for EtOH production.8 However, direct synthesis of EtOH from syngas is restricted due to the usage of expensive Rh-based catalysts, tedious procedures, and low productivity of EtOH.1,2,9–11 Furthermore, indirect synthesis of EtOH from syngas via methanol (MeOH) or dimethyl oxalate (DMO) has been investigated, which is often hindered by the use of costly Rh or Ir organometallic complexes as catalysts and corrosive iodide compounds as promoters, low CO conversion, low EtOH selectivity and high energy consumption for product separation.5,12–18 Alternatively, indirect synthesis of EtOH from syngas via dimethyl ether (DME) has attracted widespread attention due to its moderate reaction temperature (423–513 K), high atom economy, high selectivity, and low-cost zeolites.19–23 In this route, methyl acetate (MA) is firstly synthesized by the DME carbonylation reaction, which is an important intermediate reaction, and then EtOH is prepared by MA hydrogenation. The zeolites used in the DME carbonylation reaction chiefly include mordenite (MOR),19,24–31 ferrierite (FER, ZSM-35),8,19–22,32–45 and some novel zeolites (EU-12,46 SSZ-13,47 HSUZ-4,48,49 Al-RUB-41,50 and T51). Among them, MOR has the highest DME conversion and MA selectivity, but it suffers from severe deactivation owing to the presence of larger 12-membered ring (12-MR) channels. In contrast, ZSM-35 exhibits good catalytic stability in the DME carbonylation reaction because its smaller 10-membered ring (10-MR) channels inhibit carbonaceous depositions, while its catalytic activity is relatively lower on account of the smaller 10-MR channel and the FER cage [82686458]. Unfortunately, these novel zeolites require long synthesis duration and are more prone to deactivation.As we all know, ZSM-35 is a two-dimensional microporous zeolite consisting of a 10-MR channel (0.54 × 0.42 nm) along the [001] direction and perpendicularly intersected an 8-member ring (8-MR) channel (0.48 × 0.35 nm) parallel to the crystallographic direction of [010]. The intersection of the 8-MR channel and the 6-membered ring (6-MR) channel parallel to the 10-MR channel results in an ellipsoidal cage (FER cage) with a diameter of 0.6–0.7 nm accessible through 8-MR windows.52,53 The DME carbonylation reaction selectively occurs at the Brønsted acid site (BAS) located in the 6-MR zone of the 8-MR channel.20,54,55 According to the previously reported reaction mechanism, DME adsorbed on BAS can generate surface methoxy species, which react with CO to form surface acetyl groups. These acetyl intermediates then react with additional dissociated DME to produce MA by simultaneously regenerating the surface methoxy species. CO effectively reacts with surface methoxy species to form surface acetyl intermediates, with the CO insertion step being the rate determining step (RDS) of the DME carbonylation reaction.19,56 ZSM-35 is more desirable because of its outstanding stability and its modification aims to improve its catalytic activity while maintaining excellent stability. Bottom-up measures mainly contain the synthesis of nano-sized ZSM-35 zeolite38,40,42 and FER/MOR composite zeolite.57 Top-down approaches primarily involve alkaline treatment33,41 and the modification of loaded metal ions.58 For example, Feng et al. successfully synthesized nanosheet and nano-sized HZSM-35 (NZ35) zeolites using trimethylcetylammonium hydroxide (TMCAH) and piperidine (Py) as organic structure-directing agents (OSDAs), separately. These zeolites exhibited superior catalytic activity compared to the commercial reference HZSM-35 (CZ35) zeolite due to the larger amount of active sites and shorter diffusion pathways of NZ35 zeolite.38,40 Nevertheless, the addition of special OSDAs often brings about higher production costs and health-safety-environment issues, which constrains their further industrialized applications. On the contrary, post-synthesis treatments are more readily implemented in practical applications. Generally, chemical etching selectively dissolves framework Al or Si atoms, changing the intrinsic composition, crystallinity, and acidity of zeolites unavoidably.59 Recently, it has been reported that NH4F etching can introduce hierarchical porosity in zeolites without altering the pristine composition and is also subject to the framework composition.30,60–72 Taking advantage of double hydrolysis equilibrium in the HF-NH4F or NH4F solution, the existence of etching species (e.g., HF, HF2−) guarantees controlled and unbiased extraction of Al and Si. To the best of our knowledge, ZSM-35 has not been treated with an NH4F solution under simultaneous ice bath and ultrasonication, and the NH4F etched ZSM-35 zeolite has not been applied in the DME carbonylation reaction.
Herein, miscellaneous hierarchical ZSM-35 zeolites were obtained by NH4F etching for different processing time under ice bath, ultrasonic, and stirring conditions. Making the best of NH4F etching, the moderate etched ZSM-35 possessed diverse mesoporosity but retained the interior microporous structure, acid properties, and crystallinity well, which was elucidated through systematical characterizations such as XRD, XRF, FESEM, HRTEM, nitrogen adsorption–desorption, NH3-TPD, Py-IR, 27Al MAS NMR, and 29Si MAS NMR. Compared with the pristine sample, the properly etched ZSM-35 displayed higher catalytic activity and stability in the DME carbonylation reaction. The relationship between catalytic performance and physicochemical properties of the zeolites was investigated in detail.
Experimental
Chemicals and reagents
Commercially available chemicals and reagents were purchased and utilized without any additional purification. Commercial Na-ZSM-35 zeolites were purchased from Nankai Catalyst Factory Co., Ltd., China. NH4NO3 (analytical grade) and NH4F (analytical grade) were both supplied by Luoyang Chemical Reagent Factory Co., Ltd., China. The feed gas (N2/DME/CO = 5/5/90 in molar ratio) and the pure N2 were obtained from Luoyang Huapu Gas Technology Co., Ltd., China.Treatments with NH4F solution
Prior to NH4F etching, the commercial Na-ZSM-35 zeolites underwent a three-fold ion exchanged process with a 1 mol L−1 NH4NO3 water solution at a liquid–solid ratio of 8 under mechanical stirring of 200 r/min at 80 °C for 3 h. The resulting solid products were then centrifuged, thoroughly washed with deionized water, dried at 120 °C for 12 h, and calcined under an air atmosphere at 550 °C for 6 h with a heating rate of 1 °C min−1 to obtain the H-ZSM-35 zeolites. The above H-ZSM-35 zeolites were subsequently treated with a 25 wt% NH4F aqueous solution using a liquid-to-solid ratio of 8. The treatment was carried out in an ice bath (277 K) for 5, 10, and 20 min under mechanical agitation of 200 rpm and ultrasonic irradiation of 60 kHz. The resultant precipitates were quickly centrifuged, thoroughly rinsed with 80 °C preheated deionized water several times, and dried at 120 °C for 12 h. The NH4F treated H-ZSM-35 zeolites were successively mixed with a certain proportion of citric acid, nitric acid, sesbania powder, SB powder (high-purity macroporous alumina), and deionized water to produce the catalyst precursors. After kept at room temperature for 8 h and dried at 120 °C for 12 h, the catalyst precursors were calcined in air at 550 °C for 6 h with a ramp of 1 °C min−1 and eventually sieved into 0.42–0.84 mm. The NH4F etched H-ZSM-35 samples for 5, 10, and 20 min were designated as F5, F10, and F20, respectively. For comparison, the pristine H-ZSM-35 sample was denoted as P.Characterization
Wide-angle powder X-ray diffraction (XRD) patterns were obtained using a PANalytical B.V. diffractometer equipped with a monochromatic Cu-Kα radiation source of 0.15406 nm. The instrument was operated at an accelerated voltage of 40 kV and a current of 40 mA in the 2θ region of 5–40° with a fixed scanning speed of 10° min−1.X-ray fluorescence (XRF) spectra were acquired by an S8 TIGER spectrometry conducted at 50 kV and 40 mA. Before analysis, the sample was melted into a sheet with lithium borate and ammonium iodide.
Field-emission scanning electron microscopy (FESEM) images were observed with a Thermo Scientific Apreo 2C microscope at a voltage of 30 kV, a resolution of 2 nm, and a magnification range of 20–200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times. Prior to the measurements, the sample was dried, uniformly dispersed in ethanol and deposited onto holey copper grids. Gold sputtering was performed on the sample to enhance surface conductivity.
000 times. Prior to the measurements, the sample was dried, uniformly dispersed in ethanol and deposited onto holey copper grids. Gold sputtering was performed on the sample to enhance surface conductivity.
High-resolution transmission electron microscopy (HRTEM) images were captured on a Talos F200S G2 appliance with an accelerating voltage of 200 kV. To prepare the sample, a small amount of thoroughly milled sample was scattered in an ethanol solution and subjected to ultrasonic oscillation for 20–30 minutes. The solution containing the dissolved sample was added dropwise onto a copper grid covered with a thin carbon film and then dried at 60 °C.
Nitrogen adsorption–desorption isotherms were recorded via an automatic ASAP2460 gas adsorption analyzer at a temperature of 77 K. Ahead of the adsorption experiments, the samples were outgassed and dehydrated under vacuum environment at 350 °C for 10 h. The total specific surface area (SBET) was determined using the Brunauer–Emmett–Teller (BET) theory. The total pore volume (Vtotal) was calculated from the amount of nitrogen adsorbed at a relative pressure (P/P0) of 0.99. The t-plot method was applied to estimate the micropore area (Smicro) and micropore volume (Vmicro). The pore size distribution of micropores and mesopores was determined by the Barrett–Joyner–Halenda (BJH) model and the Horvath–Kawazoe (HK) model, respectively.
Temperature-programmed desorption of ammonia (NH3-TPD) measurements were manipulated on a BELCAT-M analyzer installed with a thermal conductivity detector (TCD) to measure the surface acidity of the zeolites. Approximately 100 mg of sample was loaded in a U-shape quartz tube reactor and pretreated at 600 °C for 1 h under a He flow (40 ml min−1) to remove adsorbed water and contaminants. After cooling down to 150 °C, the sample was exposed to a 10 vol% of NH3/He mixture (40 ml min−1) for 2 h, followed by flushing the sample with a He flow (40 ml min−1) at the same temperature for 1 h again to eliminate physisorbed NH3 molecules. Once the baseline was stable, the desorption patterns of NH3 were monitored by the TCD in flowing He (40 ml min−1) from 150 °C to 700 °C at a heating rate of 10 °C min−1.
Fourier transform infrared spectra of pyridine adsorption (Py-IR) analysis were performed over a BRUKER VERTEX 80 V spectrometer. The sample was scanned by a mercury cadmium telluride (MCT) detector operating at 77 K with 4000–1000 cm−1 wavenumber range, 4 cm−1 spectral resolution, and 32 scans. The sample was pressed into a self-supporting wafer with a diameter of 13 mm and a weight of 15 mg, which was successively loaded into an in situ transmission cell decorated with CaF2 windows. Prior to pyridine adsorption, the sample was treated at 400 °C for 2 h under a vacuum condition to get rid of any adsorbed pollutants and moisture. When the temperature was reduced to 150 °C, the background spectrum was collected. Pyridine was introduced into the in situ cell until the sample was fully saturated at 573 K for 0.5 h, and then the physisorbed pyridine was released at 150 °C for 0.5 h. Thereafter, the Py-IR spectra were recorded at 150 °C.
The 27Al and 29Si solid-state magic angle spinning nuclear magnetic resonance (solid-state MAS NMR) spectra were both collected on a Bruker 400 M spectrometer using 4 mm probe head of zirconia rotors. The 27Al MAS NMR spectra were recorded at a resonance frequency of 104.3 MHz with a π/12 pulse width of 0.2 μs, a spinning rate of 14 kHz, and a recycle delay time of 1 s. The 29Si MAS NMR spectra were obtained at a resonance frequency of 119.2 MHz with a π/4 pulse width of 2.6 μs, a spinning rate of 8 kHz, and a recycle delay of 20 s. The chemical shifts of 27Al and 29Si were referenced to 1 mol L−1 Al(NO3)3 aqueous solution and Si(CH3)4 (TMS) solution, separately.
Catalytic performance evaluation
As presented in Fig. 1, the selective gas-phase DME carbonylation reaction was proceeded on a stainless tubular fixed-bed microreactor with a length of 800 mm and an inner diameter of 12 mm. The fixed reaction conditions were as follows: T = 220 °C, P = 1 Mpa, and weight hourly space velocity (WHSV) = 2000 ml g−1 h−1 for 70 h on stream using a feed gas composition of N2/DME/CO (mol%) = 5/5/90. In a typical run, 0.4 g of catalyst (0.42–0.84 mm) was loaded in the middle section of the reactor. The upper and lower ends of the reactor were filled with inert carrier quartz sand. Before the reaction, the catalyst was activated at 450 °C for 4 h under N2 environment with a flow rate of 30 ml min−1 to rule out any physisorbed contaminants and water. When the temperature was cooled down to 220 °C, the feedstock gas was injected into the reactor through a mass flow controller (MFC). The effluent gas from the reactor exit was kept at 160 °C and analyzed by an online Aglient 7890A gas chromatograph (GC) furnished with a thermal conductivity detector (TCD) and a flame ionization detector (FID) having a 100 m DBWAX capillary column to detect methanol, hydrocarbons, DME, MA, and other byproducts. DME conversion and MA selectivity were calculated based on the total carbon balance, assuming no significant coke formation on the zeolites. The turnover of frequency (TOF), DME conversion, and MA selectivity were represented at the maximum DME conversion (Max) and averaged value at a steady-state after 70 h on stream (SS). The specific TOF value (h−1) was calculated by using the equation of [(converted DME (mol))/(BAS (mol) in the 8-MR channels) × (h)]. Moreover, the deactivation rate (RD, %/h) was measured by comparing the maximum and steady-state DME conversion for 70 h on stream, where the RD (maximum) was designated as (Maximum DME conversion – DME conversion at 70 h on stream)/(required duration between maximum and final DME conversion).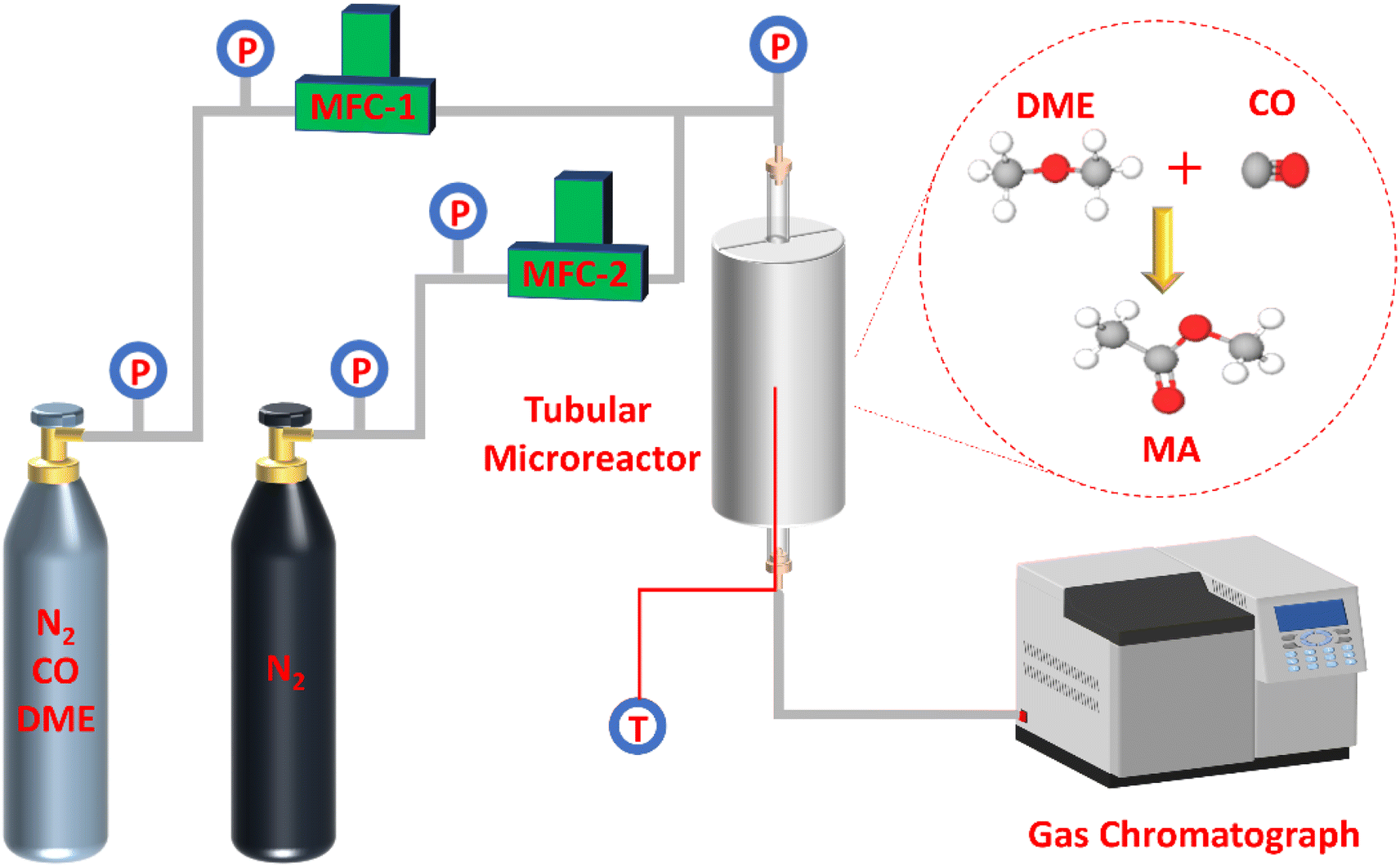 | ||
| Fig. 1 The schematic diagram of reaction apparatus system. | ||
Results and discussion
Physicochemical properties
The XRD patterns in Fig. 2(a) show that both the pristine and NH4F etched ZSM-35 derivatives exhibited a typical FER topology and the positions of the characteristic diffraction peaks at 2θ = 9.2, 25.2, and 25.7° (ref. 33) remained unchanged, revealing that the ZSM-35 zeolite structure was maintained after NH4F treatment without the formation of other impurities. The relative crystallinity (RC) of the zeolites was calculated by comparing the integrated areas of the three strongest characteristic diffraction peaks of FER, presuming that P possessed 100% crystallinity. As listed in Table 1, the RC of both F5 and F10 augmented compared to P, with F10 achieving the highest value of 139%. However, the diffraction pattern of F20 displayed a certain decline in peak intensity and the RC decreased to 96%. This may be attributed to short-term NH4F etching preferentially removed low-crystalline substances and amorphous regions,68,72 while long-term NH4F etching resulted in the dissolution of partial crystalline phases. Table 1 summarizes the bulk and framework Si/Al molar ratios of all samples from XRF and 29Si MAS NMR, respectively. The results show that NH4F treated ZSM-35 samples had similar Si/Al molar ratios to that of the parent P, indicating that the double hydrolysis of NH4F treatment effectively extracted Al and Si unbiasedly. This finding is consistent with previous studies.64,65,69 Namely, moderate NH4F etching is able to preserve the original crystallinity and Si/Al molar ratio of ZSM-35.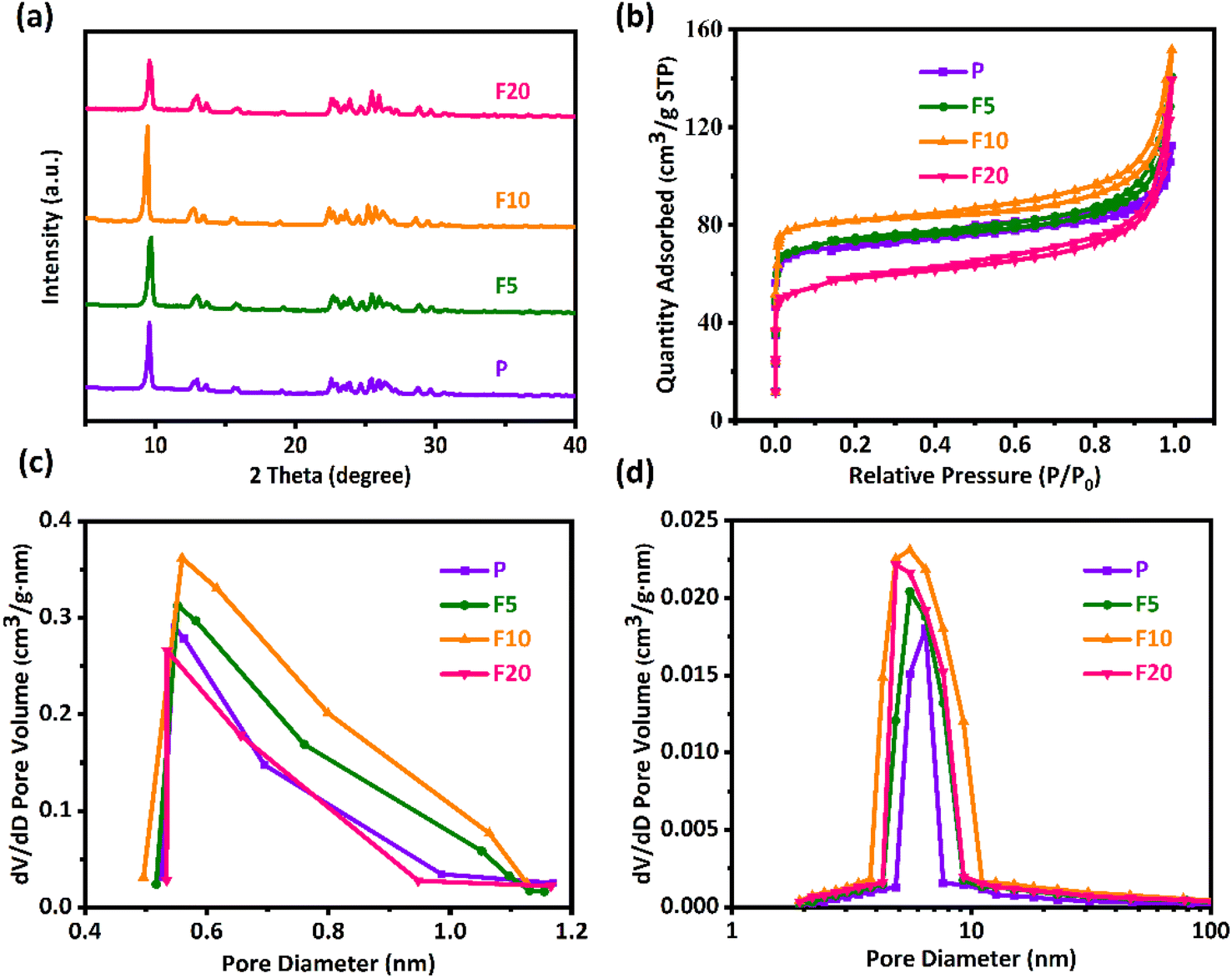 | ||
| Fig. 2 (a) XRD patterns. (b) Nitrogen adsorption–desorption isotherms. (c) Micropore size distribution profiles using HK model. (d) Mesopore size distribution profiles with BJH model of the pristine and NH4F etched ZSM-35 samples. | ||
| Sample | RCa (%) | Si/Al bulkb | Si/Al frameworkc | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Dp (nm)h | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| SBETd | Smicroe | Sextf | Vtotale | Vmicroe | Vmesog | |||||
a The relative crystallinity (RC) calculated by XRD.b Obtained by XRF.c Determined with 29Si MAS NMR data in Fig. 7 by the formula: 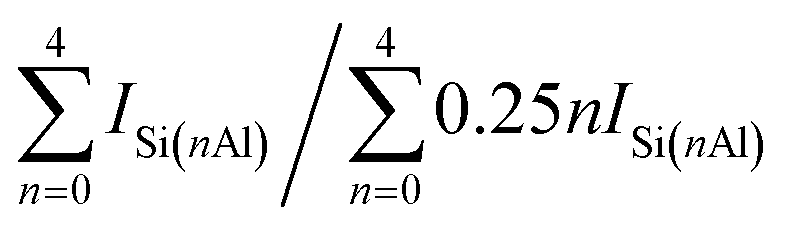 .d Calculated by the BET method.e Computed by the t-plot method.f Sext = SBET − Smicro.g Vmeso = Vtotal − Vmicro.h The average pore diameter (Dp) = 4 × Vtotal/SBET. .d Calculated by the BET method.e Computed by the t-plot method.f Sext = SBET − Smicro.g Vmeso = Vtotal − Vmicro.h The average pore diameter (Dp) = 4 × Vtotal/SBET. |
||||||||||
| P | 100 | 3.24 | 3.27 | 222.56 | 181.13 | 41.43 | 0.17 | 0.09 | 0.08 | 3.06 |
| F5 | 104 | 3.37 | 3.43 | 233.79 | 185.08 | 48.71 | 0.19 | 0.10 | 0.09 | 3.25 |
| F10 | 139 | 3.46 | 3.51 | 267.76 | 192.12 | 75.64 | 0.22 | 0.11 | 0.11 | 3.29 |
| F20 | 96 | 3.59 | 3.61 | 236.21 | 173.56 | 62.65 | 0.19 | 0.09 | 0.10 | 3.22 |
As displayed in Fig. 2(b), the nitrogen adsorption–desorption isotherms were utilized to determine the textural properties of the parent and NH4F etched ZSM-35 derivatives. It was discovered that all samples exhibited typical type IV isotherms, with a sharp increase at P/P0 less than 0.05, reflecting the presence of micropores, and an H4 hysteresis loop was observed in the P/P0 range of 0.4–1.0, which was related to the filling and emptying of mesopores through capillary condensation. The original sample P had some mesopores, likely due to the thinness of platelet crystals and the existence of hierarchical cumulative pores. Fig. 2(c) presents the micropore size distribution profiles, which were mainly concentrated on 0.5–1.2 nm. Compared with P, the micropore size distribution profiles of F5 and F10 were wider and higher, which may be ascribed to the preferential removal of defect sites by NH4F treatment, confirming the preservation of micropores (Table 1). Analogous results were observed in other researches.65,68 Nevertheless, the micropore size distribution profile of F20 was distinctly narrower and lower, suggesting that severe treatment probably disrupted the pore structure, in conformity with the lower RC of F20. It could be clearly seen from Fig. 2(d) that the mesopore size distribution profiles of all samples were primarily focused on 4–11 nm. With the elevated etching time, the Vmeso of all etched samples was higher than that of the original sample P (Table 1), which may be resulted from the formation of mesopores and the connection of small adjacent mesopores into larger mesopores at positions where Si and Al were nonselectively removed by NH4F treatment. As expected, the retained micropores and continuously increased mesopores contributed to the steady enhancement of average pore diameter (Dp) in F5 and F10. It should be pointed out, however, that the increase in SBET and Vmeso was not particularly remarkable, which is a distinctive feature of fluoride media dissolution.61 As mentioned earlier, the bi-fluoride anions lacked selectivity towards Al and Si, resulting in limited surface dissolution. The dissolution process was governed by the characteristics of crystal growth as defective zones were initially attacked.72 Under the current circumstances, the rectangular pores generated on the [100] plane manifest a helical growth of zeolite crystals. On the other hand, the Vmicro of F20 significantly reduced by 8% with respect to P, and the Sext of F20 decreased by 11% in comparison with F10. This can be attributable to the brutal treatment, capable of destroying the crystal structure, in agreement with observations from SEM.
SEM and TEM images were both employed to study the morphological changes after NH4F treatment. SEM images in Fig. 3 exhibit that P possessed a typical plate-like morphology of zeolites with 2D topology, consisting of stacked small flakes. The crystal surface appeared rough owing to the presence of low-crystallinity defective sites. After treatment with NH4F for 5 minutes, the surface defective sites were cleaned with some shallow etching marks observed and the edges melted, manifesting that the defective zones and peripheries were more susceptible to NH4F attack.65 With a treatment time of 10 minutes, some fissures and holes started to emerge on the surface, perforating into the interior of F10. This authenticates previous observations that defect zones are dissolved by such an indiscriminate chemical etching resulting ultimately in the formation of extended mesopores.71 After 20 minutes of treatment, the crystal structure was severely damaged, with large holes running through the crystals and crystals breaking down into many irregular debris. This indicates that the dissolution process occurred from the defective zones and rims on the outer surface to the interior of the crystals. It is worth noting that the formed pore systems penetrated the [100] plane, providing enhanced access to the 8-MR and 10-MR channels. TEM images of all samples in Fig. 4 show that the pristine sample had clear edges with dispersed amorphous materials on the surface, and did not exhibit conspicuous intracrystalline mesopores. Etched samples displayed intracrystalline mesopores, with larger and deeper cavities appearing as treatment time increased. This TEM study is in good accordance with the physisorption measurements.
 | ||
| Fig. 3 FESEM images of the pristine and NH4F etched ZSM-35 samples. | ||
 | ||
| Fig. 4 HRTEM images of the pristine and NH4F etched ZSM-35 samples. | ||
Acid properties
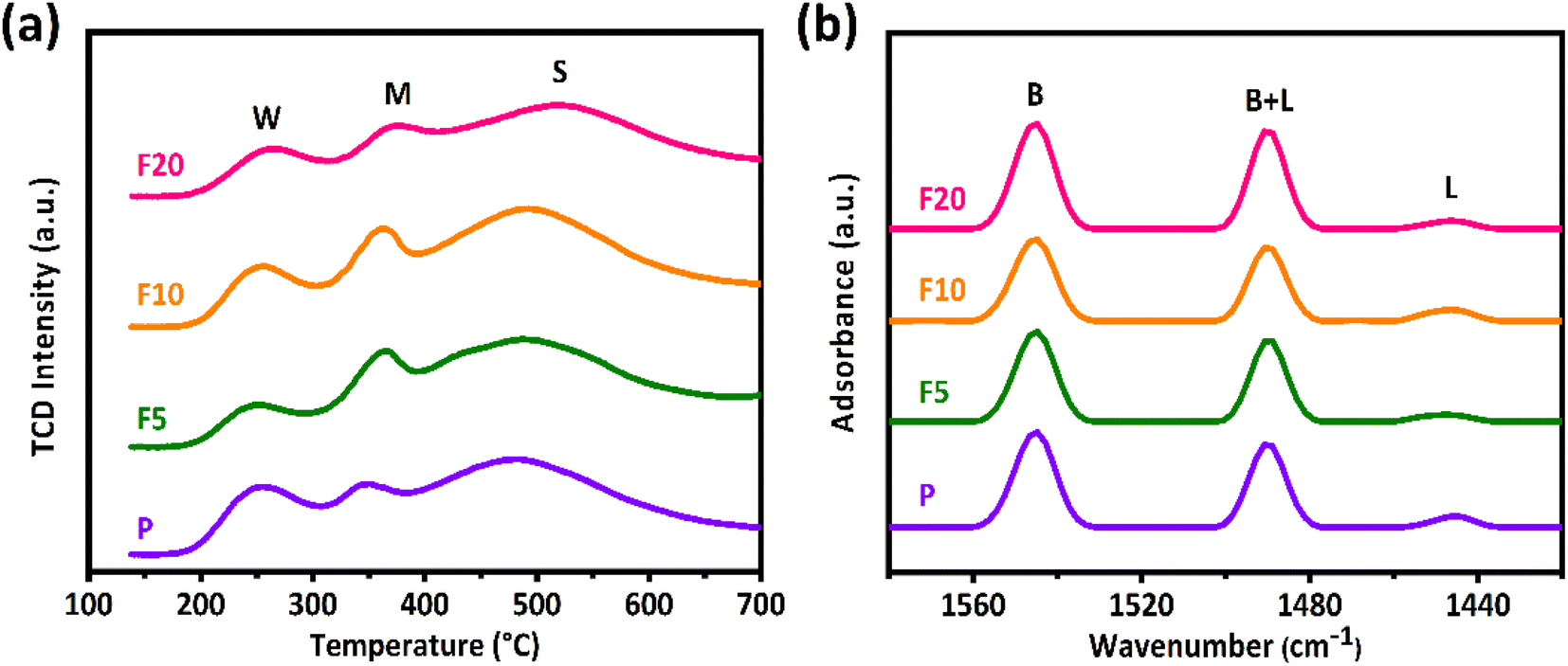
Fig. 6(a) displays the NH3-TPD profiles of the initial and NH4F etched ZSM-35 samples. NH3 is commonly applied to measure total acid sites as it can enter both the 10-MR and 8-MR channels of the zeolite. The NH3-TPD profiles were deconvoluted into three distinct peaks corresponding to three diverse types of acid sites: weak acidic sites (W) appeared at a desorption temperature of about 250 °C, medium acidic sites (M) at approximately 350 °C attributed to the Lewis acid sites (LAS) or defects, and strong acidic sites (S) at around 520 °C ascribed to the BAS.8,39 Their central positions of the desorption peaks did not alter notably. F5 and F10 exhibited a slight enhancement in strong acid sites (Table 2) compared with the initial sample P, which may be contributed to the retention of micropores and the formation of mesopores during the simultaneous removal of Al and Si upon NH4F etching. Notwithstanding, F20 showed a drop in strong acid sites owing to the partial collapse of the zeolite structure, as evidenced by XRD, XRF, and SEM observations.| Sample | NH3-TPDa (mmol g−1) | Py-IRb (mmol g−1) | BAS in 8 MRc (mmol g−1) | |||
|---|---|---|---|---|---|---|
| W | M | S | BAS | LAS | ||
| a The acidic sites were tested by NH3-TPD, and the number of weak acid sites (designated as W), moderate acid sites (M), and strong acid sites (S) was calculated based on the integrated area at the maximum desorption temperatures of approximately 250, 350, and 520 °C, respectively.b The quantity of Brønsted acid sites (BAS) and Lewis acid sites (LAS) was measured by Py-IR, and their concentrations of BAS and LAS were determined using the equation of BAS = 1.88ABR2/W, LAS = 1.42ALR2/W, where AB and AL separately represent the integrated absorbance park area of BAS and LAS appeared at 1550 and 1450 cm−1, R is the radius of catalyst wafer, and W is the weight of wafer.c The amount of BAS in the 8-MR channels was computed by using the formula of [overall number of BAS (strong acid sites obtained by NH3-TPD analysis) – the quantity of BAS in the 10-MR channels harvested by Py-IR tests]. | ||||||
| P | 0.18 | 0.16 | 0.67 | 0.31 | 0.03 | 0.36 |
| F5 | 0.08 | 0.22 | 0.74 | 0.29 | 0.02 | 0.45 |
| F10 | 0.15 | 0.21 | 0.77 | 0.28 | 0.02 | 0.49 |
| F20 | 0.12 | 0.18 | 0.65 | 0.34 | 0.04 | 0.31 |
The Py-IR spectra of the parent and NH4F treated ZSM-35 samples are illustrated in Fig. 6(b). Pyridine, with a diameter of 0.58 nm, cannot access the 8-MR channels due to steric restrictions, making it suitable for selectively testing acidic sites in the 10-MR channels. The three characteristic absorption peaks correspond to BAS appeared at the wavenumber of 1550 cm−1, LAS at that of 1450 cm−1, and combined acid sites (BAS + LAS) at that of 1490 cm−1. The concentrations of BAS and LAS were quantified using the formula of BAS = 1.88ABR2/W, LAS = 1.42ALR2/W, where AB and AL separately represent the integrated absorbance park area of BAS and LAS, R is the radius of catalyst wafer, and W is the weight of wafer.41,42 It is widely accepted that the BAS in the 10-MR channels of ZSM-35 is responsible for the coke formation.39 As summarized in Table 2, the number of BAS in the 10-MR channels of F5 and F10 decreased mildly compared with P, which is in good accordance with the decline of their RD. On the other hand, the content of BAS in the 10-MR channels of F20 elevated with respect to P, coinciding with the enhancement of their RD. Furthermore, the number of BAS in the 8-MR channels can be calculated by subtracting the amount of BAS in the 10-MR channels identified by Py-IR from the total BAS corresponding to strong acidic sites measured by the NH3-TPD analysis.8,21,39,41,42 It is observed that the amount of BAS in the 8-MR channels of F5 and F10 were reserved, further verifying that appropriate NH4F etching extracted Al and Si at similar rates. As expected, the number of BAS in the 8-MR channels of F20 fell owing to severe NH4F etching. The quantity of BAS in the 8-MR channels was found to be maximized in F10 with a concentration of 0.49 mmol g−1, and minimized in F20 with that of 0.31 mmol g−1, which were well related with changes in catalytic activity.
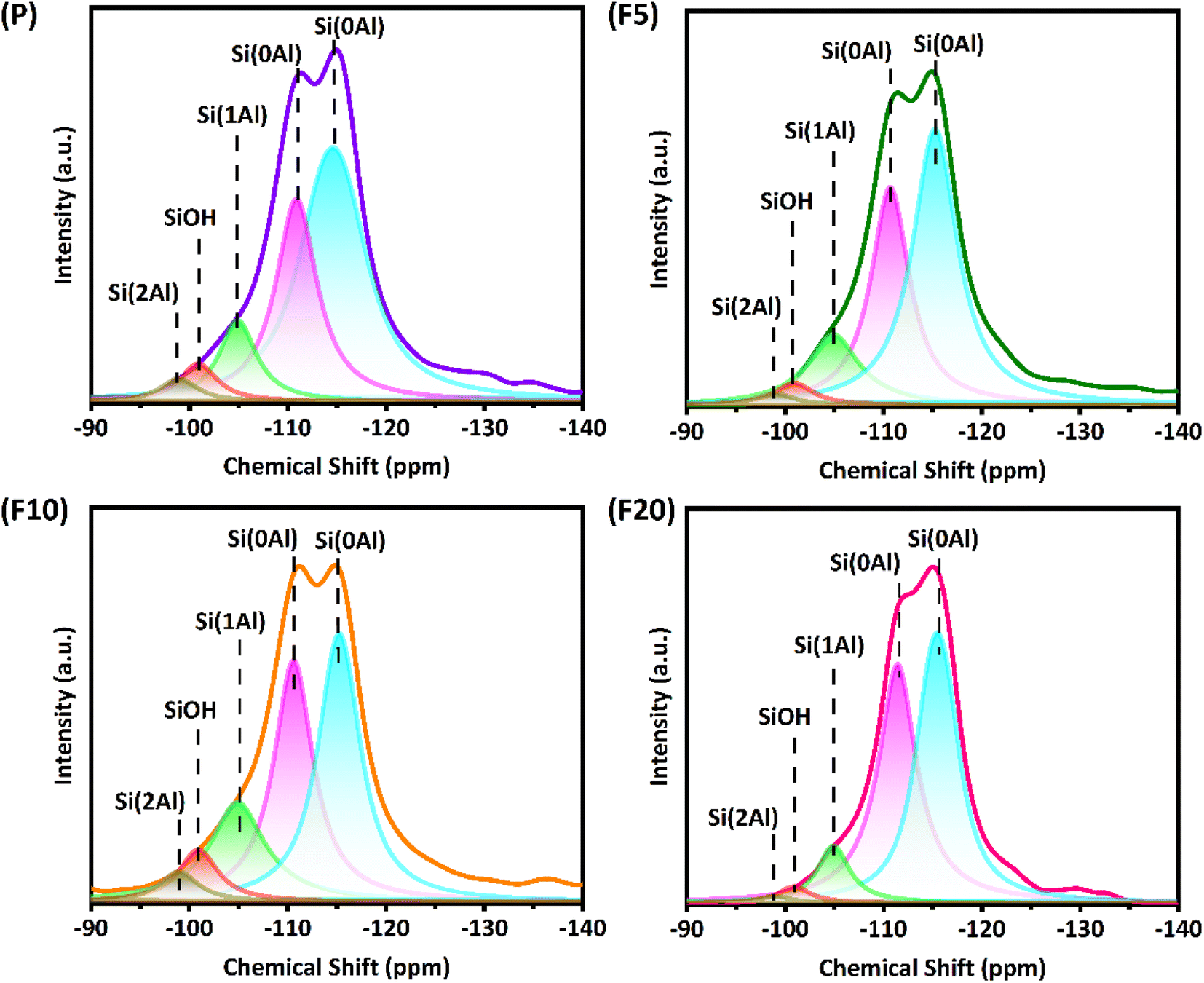
Generally, the gas-phase DME carbonylation activity is well correlated with the amount of BAS in the 8-MR channels and deactivation is also associated with the number of defect sites by coke formation.8,73,74 Accordingly, the highest number of BAS in the 8-MR channels on the F10 was responsible for the highest DME conversion with 37.6%, and the lower DME conversion on the F20 with 29.7% was ascribed to its lower crystallinity and a large quantity of defect sites. Those defect acidic sites on the pristine and NH4F modified ZSM-35 samples were substantiated by 27Al and 29Si solid-state MAS NMR spectra and the summarized results are illustrated in Fig. 5, 7, and Table 3. It can be found from the 27Al NMR spectra that all samples showed two peaks at around 55 and 0 ppm, corresponding to tetrahedral coordinated aluminum (framework aluminum) and octahedral coordinated aluminum (non-framework aluminum),19,20 respectively. All samples exhibited a spectacular tetrahedral aluminum peak and a relatively low octahedral aluminum peak, reflecting a limited content of non-framework aluminum. The defect ratio was designated as [(area of octahedral Al peak area)/(sum of tetrahedral and octahedral Al peak areas)] × 100%. As summed up in Table 3, the defect ratio can be expressed as the area of the octahedral aluminum peak divided by the total areas of the tetrahedral and octahedral aluminum peak.8 It is clear that the defect ratio gradually diminished by 15.6% and 22.2% in F5 and F10 compared with P, further confirming that NH4F treatment preferentially dissolved defective sites. On the contrary, the defect ratio of F20 increased by 8.2% with respect to P, possibly due to the rigorous treatment generating more non-framework aluminum. It can be discovered from the 29Si NMR spectra that all samples presented five peaks at approximately −99, −101, −105, −111, and −115 ppm, which were assigned to Si(2Al), SiOH, Si(1Al), and two Si(0Al) species,39 respectively. It was reported that the presence of Si(0Al) species may be associated with deactivation caused by selective coke formation.8 Compared with the pristine sample P, it can be apparently seen that Si(0Al) species dropped in F5 and F10, whereas enhanced in F20. This observation is in good accordance with the variation tendency of the defect ratio analyzed by 27Al MAS NMR. Additionally, the increased existence of Si(1Al) is likely correlated with a higher concentration of total BAS.44 For instance, F10 exhibited a Si(1Al) proportion of 19.23%, well in line with the higher content of strong BAS of 0.77 mmol g−1 (Table 2).
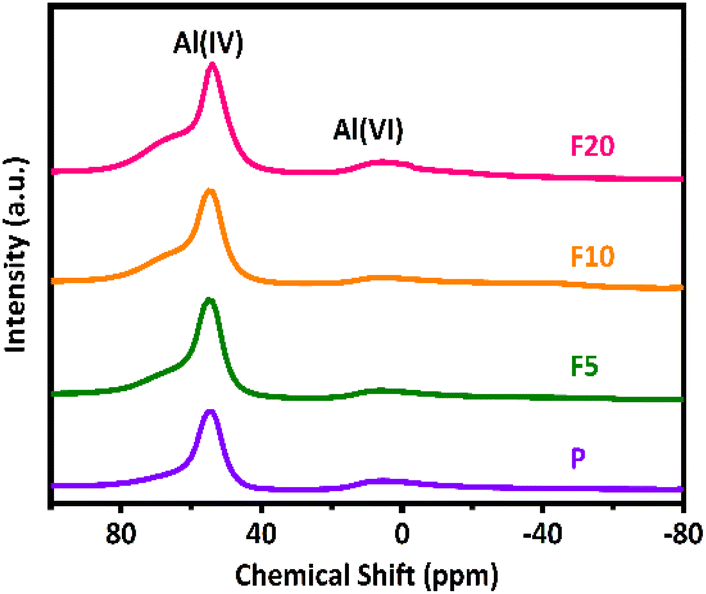 | ||
| Fig. 5 27Al MAS NMR spectra of the pristine and NH4F etched ZSM-35 samples. | ||
 | ||
| Fig. 6 (a) NH3-TPD profiles. (b) Py-IR spectra of the pristine and NH4F etched ZSM-35 sample. | ||
 | ||
| Fig. 7 29Si MAS NMR spectra of the pristine and NH4F etched ZSM-35 samples. | ||
| Sample | 27Al MAS NMRa | 29Si MAS NMRb | |||||
|---|---|---|---|---|---|---|---|
| Defect ratio (%) | Si(2Al) (%) | SiOH (%) | Si(1Al) (%) | Si(0Al) (%) | Si(0Al) (%) | Sum of Si(0Al) (%) | |
| a Defect ratio was calculated from 27Al MAS NMR peak after its separate deconvolution of tetrahedral peak at 55 ppm and octahedral peak at 0 ppm, and the defect ratio was defined as [(area of octahedral Al peak area)/(sum of tetrahedral and octahedral Al peak areas)] × 100%.b The deconvolution of 29Si MAS NMR peaks was operated with five characteristic peaks at −99, −101, −105, −111, and −115 ppm, corresponding to Si(2Al), SiOH, Si(1Al), and two Si(0Al) species, separately. | |||||||
| P | 22.75 | 3.04 | 4.96 | 10.51 | 28.83 | 52.66 | 81.49 |
| F5 | 19.19 | 2.95 | 2.29 | 14.66 | 32.61 | 47.49 | 80.10 |
| F10 | 13.62 | 3.12 | 8.01 | 19.23 | 32.39 | 37.25 | 69.64 |
| F20 | 27.62 | 1.01 | 2.41 | 8.36 | 42.54 | 45.68 | 88.22 |
Catalytic performance for DME carbonylation
The DME conversion and MA selectivity profiles of the pristine and NH4F treated ZSM-35 samples with time on stream are depicted in Fig. 8(a) and (b), separately. It was discovered that the DME conversion of all samples went through three stages: induction period, steady-state period, and deactivation period. During the induction period, the DME molecule reacted with the acidic proton presented in the zeolite, leading to the formation of a methoxy group and a methanol molecule. Owing to the secondary reaction generating additional methanol and trace hydrocarbons, the selectivity of the desired product, MA, was slightly lower during this induction period. Once the acid sites were occupied by methoxy species, the DME carbonylation reaction entered a steady-state period and CO insertion became the RDS. As the reaction progressed, the reaction eventually kicked off a deactivation period when the carbonaceous depositions reached a certain level, primarily due to the excessive carbonaceous depositions blocking the zeolite channels and covering the acidic centers.56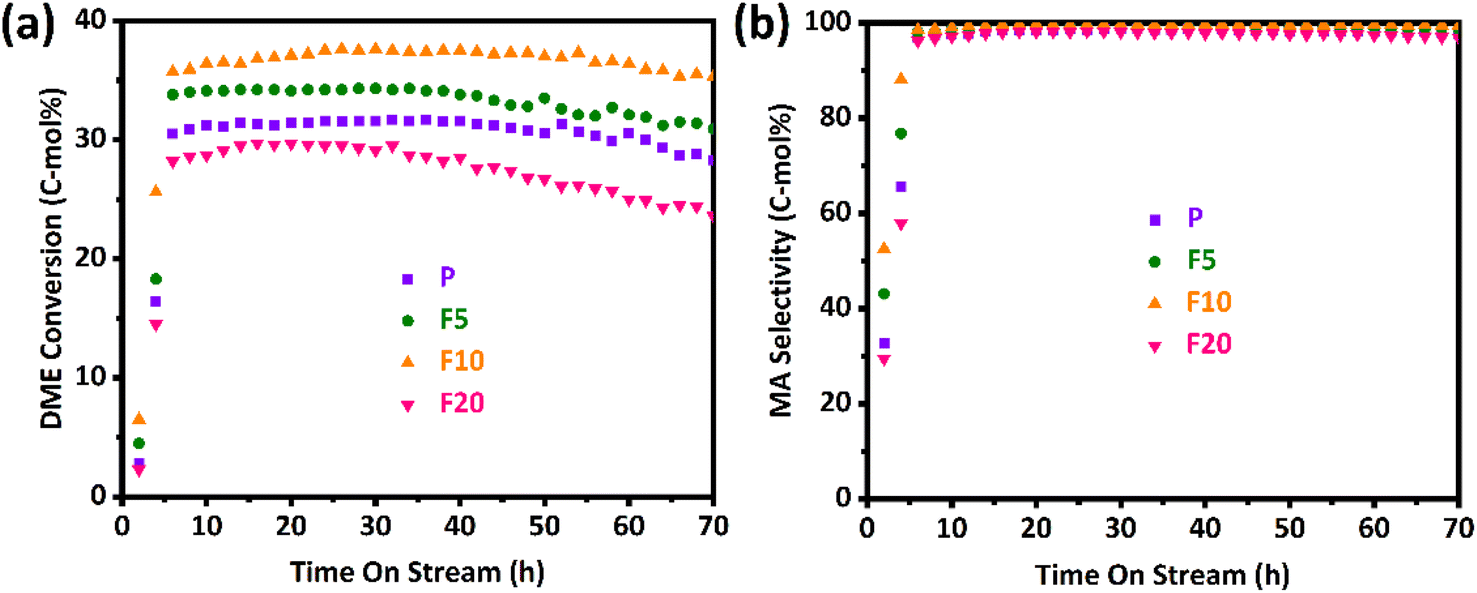 | ||
| Fig. 8 (a) DME conversion. (b) MA selectivity of the pristine and NH4F etched ZSM-35 samples with 0.4 g of catalyst for 70 h on stream (reaction conditions: T = 220 °C, P = 1 MPa, weight hourly space velocity (WHSV) = 2000 ml g−1 h−1, N2/DME/CO (mol%) = 5/5/90). | ||
As described in Fig. 8 and Table 4, the pristine sample P displayed an induction period of approximately 5 h, during which the DME conversion increased from 2.8% to about 30.5% and the MA selectivity elevated from 32.8% to around 97.1%. Apparently, P reached its maximum DME conversion of 31.7% at 32 h, and it retained at 28.3% even after 70 h on stream, indicating its eminent catalytic stability. The by-products mainly include methanol and trace hydrocarbons (principally CH4), and their contents gradually decrease to zero after the induction period. For ZSM-35, its 8-MR channel is connected with a 10-MR channel. The small size of 10-MR may inhibit the formation of carbonaceous species resulting in superior catalytic stability with respect to MOR zeolite whose 8-MR channel is interconnected with a 12-MR channel. The 10-MR channel in ZSM-35 also impedes the diffusion of the MA product molecule leading to reduced carbonylation activity. Removal of defective sites and formation of mesopores facilitate the transport of reactant DME and product MA molecules. With the increasing treatment time, the DME conversion, MA selectivity, and stability of F5 and F10 gradually improved, which might derive from a larger number of BAS in the 8-MR, higher crystallinity, and a smaller amount of defective sites.39,42 Specifically, the highest DME conversion of 37.6% and MA selectivity of 99.7% with the smallest RD of 0.05%/h were clearly observed on the F10, which had a larger amount of BAS in the 8-MR of 0.49 mmol g−1 and a lower defect ratio of 13.62%. Nevertheless, when the treatment time was prolonged to 20 minutes, the highest DME conversion and MA selectivity of F20 reduced to 29.7% and 98.2%, separately, with an enhancement in RD to 0.12%/h. This could be attributed to a lower relative crystallinity of 96% and a higher defect ratio of 27.62%. It is a common agreement that defective sites are responsible for preferential coke depositions. As plotted in Table 4, DME conversions between maximum value (max) and steady-state one (ss) were sharply altered with time on stream (from 37.6 to 35.3% even on the stable F10 with the smallest RD of 0.05%/h), however, MA selectivity with time on stream on all the FERs was less influenced by the deactivation behaviors. This observation reveals that the primary deactivation mechanisms of the FERs appear to be closely associated with the unavoidable blockage of BAS due to being covered with coke precursors.8,39,56,73,74 For instance, even on the least stable F20, DME conversion decreased from 29.7 to 23.6% (RD of 0.12%/h), however, MA selectivity reduced from 98.2 to 96.9%. Interestingly, specific TOF values calculated from the maximum DME conversion, defined as [(converted DME (mol))/(BAS (mol) in the 8-MR channels) × (h)], were found to be much larger on the P and F20 with the values of 3.93–4.28 h−1 and larger changes at a steady-state with 3.40–3.51 h−1 compared to the F5 and F10 with that of 3.40–3.43 h−1 and smaller changes at a steady-state with 3.07–3.22 h−1 at the present reaction condition. The smaller changes of TOFs on the F5 and F10 were primarily attributed to the stable reservation of active acidic sites even under coke depositions during a gas-phase DME carbonylation with the lower RD of 0.05–0.08%/h. Accordingly, it is advisable to choose an appropriate NH4F treatment time under the current experimental conditions, preferably between 5–10 minutes.
| Sample | Catalytic activitya | Deactivation rateb (RD, %/h) | |||||
|---|---|---|---|---|---|---|---|
| TOF (h−1) | DME conversion (%) | MA selectivity (%) | |||||
| Max | SS | Max | SS | Max | SS | ||
| a Catalytic activity was measured at the reaction conditions of T = 220 °C, P = 1 MPa, weight hourly space velocity (WHSV) = 2000 ml (g−1 h−1), N2/DME/CO (mol%) = 5/5/90 with 0.4 g of catalyst for 70 h on stream. The turnover of frequency (TOF), DME conversion and MA selectivity were represented at the maximum DME conversion (Max) and averaged value at a steady-state after 70 h on stream (SS). The specific TOF value (h−1) was calculated by using the equation of [(converted DME (mol))/(BAS (mol) in the 8-MR channels) × (h)].b The maximum deactivation rate (RD, %/h) was defined with the formula of [(maximum DME conversion – DME conversion at 70 h on stream)/(required duration from maximum DME to 70 h)]. | |||||||
| P | 3.93 | 3.51 | 31.7 | 28.3 | 98.7 | 97.8 | 0.09 |
| F5 | 3.40 | 3.07 | 34.3 | 30.9 | 99.1 | 98.7 | 0.08 |
| F10 | 3.43 | 3.22 | 37.6 | 35.3 | 99.7 | 99.5 | 0.05 |
| F20 | 4.28 | 3.40 | 29.7 | 23.6 | 98.2 | 96.9 | 0.12 |
Conclusions
Diverse hierarchical ZSM-35 zeolites were successfully synthesized through post-treatment using NH4F etching. This etching process preferentially removed defective sites and low-crystallinity regions, resulting in the formation of mesopores in their original positions. It is noteworthy that the created pore systems extended through the [100] plane, providing improved access to the 8-MR and 10-MR channels. The unique advantages of NH4F etching (e.g., indiscriminate, easy to implement and control) guaranteed that the intrinsic crystallinity, micropore volume, and amount of BAS in 8-MR channels were preserved under moderate conditions, leading to the higher DME conversion, higher MA selectivity, and smaller RD in the DME carbonylation reaction. Nevertheless, severe NH4F etching caused an increase in the defect ratio, which facilitated the formation of carbonaceous depositions. They could block the micropores and destroy the ZSM-35 platelet as well, generating a deactivation and instability of the NH4F etched zeolites. Hence, it is recommended to carefully opt an optimal duration of NH4F etching under the present experimental conditions, with a preference for a treatment period in the range of 5–10 minutes. This strategy offers a novel approach to slightly augment the micropore and mesopore volume of the most commercially relevant zeolites with a facile post-treatment. It enables the adsorption of larger molecules and further enhances the catalytic performance of these zeolites. Notwithstanding, the mass transfer effect and catalyst regeneration will be further studied in the future.Author contributions
Dexin Zhang: writing – original draft, investigation, project administration, supervision. Kang Li: resources, methodology, conceptualization. Junli Chen: software, writing – review & editing. Changyu Sun: resources, supervision, writing – review & editing. Zhi Li: resources, data curation. Jie Lei: validation, formal analysis. Qinlan Ma: investigation, writing – review & editing. Pan Zhang: software, writing – review & editing. Yong Liu: data curation, writing – review & editing. Lin Yang: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Sinopec Engineering (Group) Co., Ltd. Support Project (SER118020, China).Notes and references
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514 RSC
.
- H. Yue, X. Ma and J. Gong, Acc. Chem. Res., 2014, 47, 1483–1492 CrossRef CAS PubMed
- G. Liu, G. Yang, X. Peng, J. Wu and N. Tsubaki, Chem. Soc. Rev., 2022, 51, 5606–5659 RSC
- K. Göransson, U. Söderlind, J. He and W. Zhang, Renewable Sustainable Energy Rev., 2011, 15, 482–492 CrossRef
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed
- G. Prieto, S. Beijer, M. L. Smith, M. He, Y. Au, Z. Wang, D. A. Bruce, K. P. de Jong, J. J. Spivey and P. E. de Jongh, Angew. Chem., Int. Ed., 2014, 53, 6397–6401 CrossRef CAS PubMed
- W. Zhou, J. Kang, K. Cheng, S. He, J. Shi, C. Zhou, Q. Zhang, J. Chen, L. Peng, M. Chen and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12012–12016 CrossRef CAS PubMed
- H. S. Jung, H. Ham and J. W. Bae, Catal. Today, 2020, 339, 79–85 CrossRef CAS
- M. Gupta, M. L. Smith and J. J. Spivey, ACS Catal., 2011, 1, 641–656 CrossRef CAS
- D. Mei, R. Rousseau, S. M. Kathmann, V.-A. Glezakou, M. H. Engelhard, W. Jiang, C. Wang, M. A. Gerber, J. F. White and D. J. Stevens, J. Catal., 2010, 271, 325–342 CrossRef CAS
- Y. Choi and P. Liu, J. Am. Chem. Soc., 2009, 131, 13054–13061 CrossRef CAS PubMed
- Y. Zhu, X. Kong, X. Li, G. Ding, Y. Zhu and Y.-W. Li, ACS Catal., 2014, 4, 3612–3620 CrossRef CAS
- A. W. Budiman, J. S. Nam, J. H. Park, R. I. Mukti, T. S. Chang, J. W. Bae and M. J. Choi, Catal. Surv. Asia, 2016, 20, 173–193 CrossRef CAS
- Y. Ni, L. Shi, H. Liu, W. Zhang, Y. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2017, 7, 4818–4822 RSC
- J. S. Nam, A. Rong Kim, D. M. Kim, T. S. Chang, B. S. Kim and J. W. Bae, Catal. Commun., 2017, 99, 141–145 CrossRef CAS
- P. Ai, M. Tan, N. Yamane, G. Liu, R. Fan, G. Yang, Y. Yoneyama, R. Yang and N. Tsubaki, Chem. - Eur. J., 2017, 23, 8252–8261 CrossRef CAS PubMed
- P. Ai, M. Tan, P. Reubroycharoen, Y. Wang, X. Feng, G. Liu, G. Yang and N. Tsubaki, Catal. Sci. Technol., 2018, 8, 6441–6451 RSC
- Y. Chen and D. Liu, Fuel Process. Technol., 2018, 171, 301–307 CrossRef CAS
- T. Xu, H. Liu, Q. Zhao, S. Cen, L. Du and Q. Tang, Catal. Commun., 2019, 119, 96–100 CrossRef CAS
- J. Kim, H. Ham, H. S. Jung, Y. Wang, Y. He, N. Tsubaki, S. J. Cho, G. Y. Han and J. W. Bae, Catal. Sci. Technol., 2018, 8, 3060–3072 RSC
- H. Ham, H. S. Jung, H. S. Kim, J. Kim, S. J. Cho, W. B. Lee, M.-J. Park and J. W. Bae, ACS Catal., 2020, 10, 5135–5146 CrossRef CAS
- X. Gao, B. Xu, G. Yang, X. Feng, Y. Yoneyama, U. Taka and N. Tsubaki, Catal. Sci. Technol., 2018, 8, 2087–2097 RSC
- E. Zhan, Z. Xiong and W. Shen, J. Energy Chem., 2019, 36, 51–63 CrossRef
- K. Cao, D. Fan, L. Li, B. Fan, L. Wang, D. Zhu, Q. Wang, P. Tian and Z. Liu, ACS Catal., 2020, 10, 3372–3380 CrossRef CAS
- P. He, J. Wang, Y. Li, S. Huang, M.-Y. Wang, Y. Wang, J. Lv and X. Ma, Ind. Eng. Chem. Res., 2021, 60, 8085–8093 CrossRef CAS
- Z. Cheng, S. Huang, Y. Li, K. Cai, Y. Wang, M.-y. Wang, J. Lv and X. Ma, ACS Catal., 2021, 11, 5647–5657 CrossRef CAS
- F. Xu, J. Lv, C. Chen, Z. Hong, G. Zhao, L. Miao, W. Yang and Z. Zhu, Ind. Eng. Chem. Res., 2022, 61, 1258–1266 CrossRef CAS
- R. Liu, S. Zeng, T. Sun, S. Xu, Z. Yu, Y. Wei and Z. Liu, ACS Catal., 2022, 12, 4491–4500 CrossRef CAS
- L.-Y. Zhang, X.-B. Feng, Z.-M. He, F. Chen, C. Su, X.-Y. Zhao, J.-P. Cao and Y.-R. He, Chem. Eng. Sci., 2022, 256, 117671 CrossRef CAS
- S. Liu, Z. Cheng, Y. Li, J. Sun, K. Cai, S. Huang, J. Lv, S. Wang and X. Ma, Ind. Eng. Chem. Res., 2020, 59, 13861–13869 CrossRef CAS
- M. Boronat, C. Martinez-Sanchez, D. Law and A. Corma, J. Am. Chem. Soc., 2008, 130, 16316–16323 CrossRef CAS PubMed
- J. Liu, H. Xue, X. Huang, Y. Li and W. Shen, Catal. Lett., 2010, 139, 33–37 CrossRef CAS
- X. Li, X. Liu, S. Liu, S. Xie, X. Zhu, F. Chen and L. Xu, RSC Adv., 2013, 3, 16549 RSC
- L. Wang, P. Tian, Y. Yuan, M. Yang, D. Fan, H. Zhou, W. Zhu, S. Xu and Z. Liu, Microporous Mesoporous Mater., 2014, 196, 89–96 CrossRef CAS
- S. Y. Park, C.-H. Shin and J. W. Bae, Catal. Commun., 2016, 75, 28–31 CrossRef CAS
- Q. Wei, G. Yang, X. Gao, L. Tan, P. Ai, P. Zhang, P. Lu, Y. Yoneyama and N. Tsubaki, Chem. Eng. J., 2017, 316, 832–841 CrossRef CAS
- H. Ham, J. Kim, J. H. Lim, W. C. Sung, D. H. Lee and J. W. Bae, Catal. Today, 2018, 303, 93–99 CrossRef CAS
- X. Feng, P. Zhang, Y. Fang, W. Charusiri, J. Yao, X. Gao, Q. Wei, P. Reubroycharoen, T. Vitidsant, Y. Yoneyama, G. Yang and N. Tsubaki, Catal. Today, 2020, 343, 206–214 CrossRef CAS
- H. S. Jung, N. T. Xuan and J. W. Bae, Microporous Mesoporous Mater., 2021, 310, 110669 CrossRef CAS
- X. Feng, J. Yao, Y. Zeng, Y. Cui, S. Kazumi, R. Prasert, G. Liu, J. Wu, G. Yang and N. Tsubaki, Catal. Today, 2021, 369, 88–94 CrossRef CAS
- T. X. Nguyen, J. W. Moon, H. S. Jung, G. Y. Han and J. W. Bae, Korean J. Chem. Eng., 2021, 38, 1231–1239 CrossRef CAS
- E. J. Kim, X. Gao, J. Tian and J. W. Bae, Catal. Today, 2023, 411, 113822 CrossRef
- Z. Xiong, G. Qi, L. Bai, E. Zhan, Y. Chu, J. Xu, N. Ta, A. Hao, F. Deng and W. Shen, Catal. Sci. Technol., 2022, 12, 4993–4997 RSC
- Y. Guo, S. Wang, R. Geng, P. Wang, S. Li, M. Dong, Z. Qin, J. Wang and W. Fan, iScience, 2023, 26, 107748 CrossRef CAS PubMed
- W. C. Sung, H. S. Jung, J. W. Bae, J. Y. Kim and D. H. Lee, J. CO2 Util., 2023, 69, 102411 CrossRef CAS
- X. Feng, J. Yao, H. Li, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, Chem. Commun., 2019, 55, 1048–1051 RSC
- M. Lusardi, T. T. Chen, M. Kale, J. H. Kang, M. Neurock and M. E. Davis, ACS Catal., 2019, 10, 842–851 CrossRef
- Z. Xiong, E. Zhan, M. Li and W. Shen, Chem. Commun., 2020, 56, 3401–3404 RSC
- X.-B. Feng, J.-P. Cao, C. Su, Z.-M. He and X.-Y. Zhao, Fuel, 2022, 315, 123267 CrossRef CAS
- J. Yao, Q. Wu, J. Fan, S. Komiyama, X. Yong, W. Zhang, T. Zhao, Z. Guo, G. Yang and N. Tsubaki, ACS Nano, 2021, 15, 13568–13578 CrossRef CAS PubMed
- X.-B. Feng, F. Chen, Z.-M. He, X.-Y. Zhao and J.-P. Cao, Energy Fuels, 2022, 36, 14341–14348 CrossRef CAS
- P. Feng, X.-F. Chen, X.-J. Li, D. Zhao, S.-J. Xie, L.-Y. Xu and G.-Z. He, Microporous Mesoporous Mater., 2017, 239, 354–362 CrossRef CAS
- P. A. Vaughan, Acta Crystallogr., 1966, 21 Search PubMed
- P. Feng, G. Zhang, K. Zang, X. Li, L. Xu and X. Chen, Chem. Phys. Lett., 2017, 684, 279–284 CrossRef CAS
- P. Feng, G. Zhang, X. Chen, K. Zang, X. Li and L. Xu, Appl. Catal., A, 2018, 557, 119–124 CrossRef CAS
- P. Cheung, A. Bhan, G. J. Sunley, D. J. Law and E. Iglesia, J. Catal., 2007, 245, 110–123 CrossRef CAS
- X. Li, X. Chen, Z. Yang, X. Zhu, S. Xu, S. Xie, S. Liu, X. Liu and L. Xu, Microporous Mesoporous Mater., 2018, 257, 79–84 CrossRef CAS
- H. Ham, J. Kim, S. Park and J. W. Bae, Adv. Porous Mater., 2016, 4, 200–205 CrossRef
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS
- Z. Qin, L. Lakiss, J. P. Gilson, K. Thomas, J. M. Goupil, C. Fernandez and V. Valtchev, Chem. Mater., 2013, 25, 2759–2766 CrossRef CAS
- X. Chen, T. Todorova, A. Vimont, V. Ruaux, Z. Qin, J.-P. Gilson and V. Valtchev, Microporous Mesoporous Mater., 2014, 200, 334–342 CrossRef CAS
- Z. Qin, J.-P. Gilson and V. Valtchev, Curr. Opin. Chem. Eng., 2015, 8, 1–6 CrossRef
- F. Meng, X. Wang, S. Wang and Y. Wang, Catal. Today, 2017, 298, 226–233 CrossRef CAS
- Z. Qin, K. A. Cychosz, G. Melinte, H. El Siblani, J.-P. Gilson, M. Thommes, C. Fernandez, S. Mintova, O. Ersen and V. Valtchev, J. Am. Chem. Soc., 2017, 139, 17273–17276 CrossRef CAS PubMed
- Z. Qin, L. Pinard, M. A. Benghalem, T. J. Daou, G. Melinte, O. Ersen, S. Asahina, J.-P. Gilson and V. Valtchev, Chem. Mater., 2019, 31, 4639–4648 CrossRef CAS
- V. Babić, L. Tang, Z. Qin, L. Hafiz, J. P. Gilson and V. Valtchev, Adv. Mater. Interfaces, 2020, 8, 2000348 CrossRef
- G. Yang, Z. Qiu, J. Han, X. Chen and J. Yu, Mater. Chem. Front., 2020, 4, 2982–2989 RSC
- V. Babić, S. Koneti, S. Moldovan, N. Nesterenko, J.-P. Gilson and V. Valtchev, Microporous Mesoporous Mater., 2021, 314, 110863 CrossRef
- Z. Qin, S. Zeng, G. Melinte, T. Bučko, M. Badawi, Y. Shen, J. P. Gilson, O. Ersen, Y. Wei, Z. Liu, X. Liu, Z. Yan, S. Xu, V. Valtchev and S. Mintova, Advanced Science, 2021, 8, 2100001 CrossRef CAS PubMed
- Z. Qin, B. Wang, N. Asano, L. Wang, Y. Zhou, X. Liu, B. Shen, S. Mintova, S. Asahina and V. Valtchev, Inorg. Chem. Front., 2022, 9, 2365–2373 RSC
- Z. X. Qin, G. Melinte, J. P. Gilson, M. Jaber, K. Bozhilov, P. Boullay, S. Mintova, O. Ersen and V. Valtchev, Angew. Chem., Int. Ed., 2016, 55, 15049–15052 CrossRef CAS PubMed
- Z. X. Qin, B. Wang, N. Asano, L. J. Wang, Y. Zhou, X. M. Liu, B. J. Shen, S. Mintova, S. Asahina and V. Valtchev, Inorg. Chem. Front., 2022, 9, 2365–2373 RSC
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS PubMed
- H. Ham, H. S. Jung, H. S. Kim, J. Kim, S. J. Cho, W. B. Lee, M. J. Park and J. W. Bae, ACS Catal., 2020, 10, 5135–5146 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |