DOI:
10.1039/D3RA07321A
(Paper)
RSC Adv., 2023,
13, 35369-35378
In situ synthesis of g-C3N4/Ti3C2Tx nano-heterostructures for enhanced photocatalytic H2 generation via water splitting†
Received
27th October 2023
, Accepted 15th November 2023
First published on 4th December 2023
Abstract
Herein, we demonstrated the in situ synthesis of g-C3N4/Ti3C2Tx nano-heterostructures for hydrogen generation under UV visible light irradiation. The formation of the g-C3N4/Ti3C2Tx nano-heterostructures was confirmed via powder X-ray diffraction and supported by XPS. The FE-SEM images indicated the formation of layered structures of MXene and g-C3N4. HR-TEM images and SAED patterns confirmed the presence of g-C3N4 together with Ti3C2Tx nanosheets, i.e., the formation of nano-heterostructures of g-C3N4/Ti3C2Tx. The absorption spectra clearly showed the distinct band gaps of g-C3N4 and Ti3C2Tx in the nano-heterostructure. The increase in PL intensity and broadening of the peak with an increase in g-C3N4 indicated the suppression of electron–hole recombination. Furthermore, the nano-heterostructure was used as a photocatalyst for H2 generation from water and methylene blue dye degradation. The highest H2 evolution (1912.25 μmol/0.1 g) with good apparent quantum yield (3.1%) and an efficient degradation of MB were obtained for gCT-0.75, which was much higher compared to that of the pristine materials. The gCT-0.75 nano-heterostructure possessed a high surface area and abundant vacancy defects, facilitating the separation of charge carriers, which was ultimately responsible for this high photocatalytic activity. Additionally, TRPL clearly showed a higher decay time, which supports the enhancement in the photocatalytic activity of the gCT-0.75 nano-heterostructure. The nano-heterostructure with the optimum concentration of g-C3N4 formed a hetero-junction with the linked catalytic system, which facilitated efficient charge carrier separation also responsible for the enhanced photocatalytic activity.
1. Introduction
The excessive use of fossil fuels satisfies the energy demand in society but simultaneously generates significant pollution in the ecosystem, and also results in the depletion of fossil fuel reserves. Consequently, this excessive usage will totally deplete fossil fuel reservoirs in the near future. Thus, to keep the eco-system pollution free together with the availability of an ample amount of useful energy, the search for alternative energy sources was initiated. To address the above-mentioned issues, research in the area of solar energy emerged and newer technologies have also been developed to harvest solar energy in useful form.1 The most important way to harvest solar energy is the use of photovoltaic technology, which directly converts solar energy into electricity. However, photovoltaic technology requires the development of highly efficient solar cells. Alternatively, a major way to harvest solar energy is the use of the photocatalysis technique where solar energy is also utilized to generate hydrogen via water splitting over semiconductor oxides or sulphides.2,3 Thus, to increase the efficiency of H2 generation via photocatalytic water splitting, many researchers have focused their research on tuning the band gap of semiconductors for the maximum absorption of solar light and also techniques for the separation of photo-generated charge carriers.4–6 However, due to the weak visible-light absorption and quick charge recombination of a single semiconductor, it shows low photocatalytic efficiency. Therefore, the creation of heterojunctions, metal/cationic doping, and coupling co-catalysts have been proposed to increase the photocatalytic activity of semiconductors. In this case, numerous substances, including graphene, carbon nanotubes, graphitic carbon nitride, carbon quantum dots, and noble metals, have been utilised as co-catalysts to improve the photocatalytic efficacy.7,8 However, it is essential to identify effective and affordable catalytic systems to improve their photocatalytic efficiency for commercial applications.9,10
Recently, many researchers have reported the properties of two-dimensional (2D) graphene, serving as inspiration for the development of graphitic carbon nitride (g-C3N4)-based photocatalysts. Due to the unique physicochemical characteristics of its 2D structure, 2D g-C3N4 shows benefits compared to conventional bulk g-C3N4 such as a large surface area, a short charge diffusion route, and a large number of active sites.11–14 Single or few-layer g-C3N4 nanosheets with high photocatalytic performance have been fabricated using liquid or thermal exfoliation techniques. Wu et al. reported that graphene-like g-C3N4 nanosheets have a large specific area and quick charge transport capabilities, resulting a greater hydrogen evolution rate than bulk g-C3N4.15 However, after the photocatalytic process, it is challenging to recycle the tiny g-C3N4 nanosheets from the suspension. Further, during the photocatalytic process, 2D g-C3N4 nanosheets have a tendency to stack together, resulting in a reduction in their photocatalytic performance.16–19 This problem can be solved by creating composite catalytic systems with other materials, which can also form heterostructures, resulting in efficient charge transport ability.20–23 Dong et al. reported the assembly of 2D g-C3N4 into a 3D interconnected porous network, which harvested abundant photons owing to the multireflection inside the 3D-linked structure.24
Recently, MXenes, a unique family of 2D layered transition metal carbides, carbonitrides and nitrides, have received attention for the development of coupled catalytic systems. The synthesis of Ti3C2 was first reported in 2011 by Gogotsi et al.,25 and soon after, many researchers reported the synthesis and properties of layered materials such as Ti3C2. For the synthesis of MXenes, the selective etching of the ‘A’ layers from the MAX phases (Mn+1XnTx) is frequently performed. The primary group of IIIA and IVA elements is represented by the letter ‘A’.26,27 Mn+1XnTx is the general formula for MXene, where M is a transition metal (Sc, Ti, Cr, Mo, Ta, Zr, Nb, Hf, V, and Nb) element, X denotes C and/or N, and Tx denotes surface terminations such as –F, –O, and –OH, and n is 1, 2, or 3.28,29 Due to the presence of hydroxy groups or terminal oxygen species on their surfaces, MXene materials show similar conductivity toward transition metal carbides.30–34 Furthermore, considering the conducting behaviour and layered structure of MXenes, they will be good co-catalysts for the development of coupled catalysts to separate photogenerated charge carriers efficiently.
In this regard, herein, we synthesized an MXene (Ti3C2Tx) from the MAX (Ti3AlC2) phase and prepared nano-heterostructures of a g-C3N4/Ti3C2Tx composite with different proportions. These nano-heterostructures were characterised thoroughly and utilized for photocatalytic H2 generation via water splitting. It is noteworthy that the nano-heterostructures demonstrated excellent H2 evolution under UV visible light.
2. Experimental
2.1 Materials
Titanium aluminium carbide powder MAX (Ti3AlC2) (99.8%) was supplied commercially by Aritech Chemazone Private Limited. Hydrofluoric acid (HF, 48% AR/ACS) and melamine were provided by Loba Chemicals Ltd. PVDF filter paper was procured from Microgram Ltd. All chemicals were utilized as received without further purification.
2.2 Synthesis of Ti3C2Tx
MXene was synthesised from MAX (Ti3AlC2) via HF etching approach.35,36 For this purpose, 20 mL of 48% aqueous HF solution was transferred to a 100 mL Teflon beaker. Subsequently, 1 g of Ti3AlC2 powder was slowly added to the beaker (up to 30 min) and stirred for 24 h at room temperature. The resulting solution was centrifuged at 3500 rpm to yield a Ti3C2Tx slurry. This slurry was washed several times with deionized (DI) water, and then filtered with a 0.22 μm pore size polyvinyl difluoride (PVDF) filter paper. Subsequently, the resulting powder (Ti3C2Tx) was dried in a vacuum oven at 80 °C for 24 h. The dried powder was obtained and identified as MXene (Ti3C2Tx).
2.3 Synthesis of g-C3N4
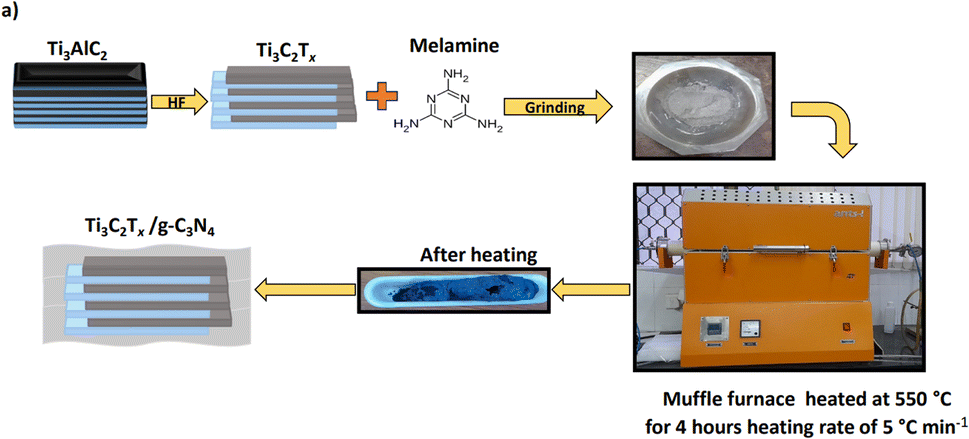
For the synthesis of g-C3N4, melamine was kept in a crucible with a lid and heated at 550 °C for 4 h in a muffle furnace at the heating rate of 5 °C min−1. After natural cooling, the obtained material was finely ground using an agate mortar and pestle. For 1 g melamine, 0.3 g g-C3N4 was obtained, i.e., almost 30% yield. Accordingly for the synthesis of g-C3N4/Ti3C2Tx, we employed an excess amount of melamine so to get the desired wt% composite material.
2.4 Synthesis of g-C3N4/Ti3C2Tx composites
For the synthesis of g-C3N4/Ti3C2Tx composites, different amounts of melamine (100, 75, 50, 25, and 5 wt%) were mixed with 0.5 g of Ti3C2Tx using a mortar and pestle, and then heated at 550 °C for 4 h in a muffle furnace at a heating rate of 5 °C min−1 under an N2 atmosphere. The in situ-formed composites were denoted as gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 for g-C3N4/Ti3C2Tx with ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.75:1, 0.5:1, 0.25:1 and 0.05:1, respectively. This process is schematically shown in Fig. 1.
:1, 0.75:1, 0.5:1, 0.25:1 and 0.05:1, respectively. This process is schematically shown in Fig. 1.
 |
| | Fig. 1 Schematic process for the synthesis of MXene (Ti3C2Tx) from MAX (Ti3AlC2) phase and g-C3N4/Ti3C2Tx nano-heterostructure. | |
2.5 Characterization
The prepared g-C3N4/Ti3C2Tx composites were analysed using various techniques. X-ray diffraction was conducted using a Bruker D-8 Advance equipped with an Ni-filtered Cu target, having Cu-Kα radiation with a wavelength of 1.5418 Å, in the 2θ range of 5–80° at a scan rate of 1° min−1. Field emission scanning electron microscopy (FE-SEM, HITACHI 4800) was used to examine the surface morphology. Field-emission TEM (FE-TEM) and selected area electron diffraction (SAED) were also recorded using a JEOL JEM-2200 FS at an operating voltage of 200 kV. To analyse the chemical states of the atoms and the composition of the surface elements, X-ray photoelectron spectroscopy (XPS) measurement was performed using an ESCALAB 250 (Thermo Electron, Al Kα) spectrometer. A Shimadzu UV-Vis-NIR spectrophotometer (Model UV-3600) was used to record at the room temperature diffuse reflectance UV-visible spectra in the wavelength range of 200 to 800 nm. Photoluminescence (PL) spectra were recorded using a Shimadzu RF-5301pc spectroflurophotometer. Further, time-resolved photoluminescence (TRPL) experiments were performed using a photoluminescence Horiba model spectrometer with an xenon arc lamp. Fourier transform infrared (FTIR) spectroscopy was recorded in transmittance mode using a Shimadzu FTIR spectrometer in the range of 500–4000 cm−1. The specific Brunauer–Emmett–Teller (BET) surface areas of the samples were determined using (Quanta chrome NOVA touch LX1) N2 adsorption isotherms at liquid N2 temperature. Prior to the BET analysis, the samples were degassed under vacuum for two hours at 150 °C.
2.6 Photocatalytic activity measurement
2.6.1 Photocatalytic H2 generation. To check the photocatalytic activity of the prepared nanostructures for H2 generation via water splitting, we used a 100 mL cylindrical quartz vessel and 400 W mercury vapor lamp. To measure the photocatalytic activity, 25 mL of distilled water was added to a quartz reactor containing 20 mg of prepared catalyst having 1 wt% preloaded Pt as a co-catalyst. To this mixture, 5 mL of methanol was added as a sacrificial reagent. To remove the dissolved gases, UHP grade N2 was bubbled for 10 min in the reaction mixture, which was later sealed with a septum. The sealed quartz reactor with septum was stirred under a 400 W mercury vapour lamp. After a certain duration, 500 μL of the generated gas was removed from the reactor using a gas-tight syringe, and the amount of H2 gas was measured using a gas chromatograph (Shimadzu: Model GC 2014) equipped with a 5 Å molecular sieve column. The standard fitted curve was used to calculate the amount of hydrogen gas produced. The same experimental procedure was also used to conduct catalyst recycling investigations.
2.7 Photocatalytic MB dye degradation
We utilised our previously described technique for the photocatalytic dye degradation studies.83,84 In a 150 mL conical flask, 50 mg of the prepared catalyst (Ti3C2Tx) was dispersed in 100 mL of 10 ppm methylene blue (MB) solution (10 ppm MB, pH = 7) and stirred for 15 min at room temperature in the dark to achieve adsorption–desorption equilibrium. Next, the conical flask containing catalyst and MB solution was stirred under a 400 W mercury vapour lamp kept vertically in a quartz condenser having a water circulation arrangement to minimize the heating effect. The whole reaction set-up was fixed in a closed wooden box. Aliquots of the aqueous solution were removed from the flask at predetermined intervals, centrifuged, and the UV-visible absorption spectra of the clear solutions recorded using a double-beam UV-visible spectrophotometer. The decrease in absorbance at the wavelength of 664 nm was employed to determine the extent of MB degradation as a function of time. To calculate the % of MB degradation, the maximum absorbance value at the wavelength of 664 nm was employed. The % MB degradation was quantified using eqn (1), as follows| | |
% MB degrades = [(C0 − Ct)/C0] × 100
| (1) |
where C0 is the initial concentration of MB and Ct is the concentration of MB after irradiation time ‘t’.
3 Results and discussion
3.1 XRD analysis
The crystal structure and phase of the synthesized nano-heterostructures were examined by XRD (Fig. 2). The XRD patterns of MAX (Ti3AlC2) show prominent peaks, similar to the previous report.37 After the chemical exfoliation of MAX with HF, a decrease in the intensity of the peak at the 2θ of 38.8° was observed, indicating the complete removal of Al from MAX and formation of MXene (shown as Ti3C2Tx).38–40 Further, the (002) and (004) planes corresponding to the peaks at 2θ = 8.7° and 18.1° of Ti3C2Tx shifted to a lower angle due to the increase in the interlayer spacing of Ti3C2Tx, indicating that Ti3AlC2 was successfully converted into Ti3C2Tx.41
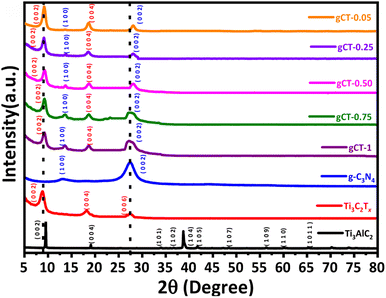 |
| | Fig. 2 XRD patterns of Ti3AlC2, Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 nano-heterostructures. | |
The XRD pattern of the g-C3N4 nanosheets shows two typical diffraction peaks at 12.2° and 27.8°, corresponding to the (100) plane with in-plane structural packing motif and (002) plane of interplanar stacking with conjugated aromatic segments, respectively.42 Furthermore, the formation of composites between MXene and g-C3N4 was confirmed by XRD. It was observed that the peak intensity of g-C3N4 decreased with a decrease in the concentration of g-C3N4. The broad peak at 27.5° validates the presence of g-C3N4 together with Ti3C2Tx (28.2°), confirming the formation of nano-heterostructures of g-C3N4/Ti3C2Tx with different weight ratios.
3.2 Morphological studies
Field-emission scanning electron microscopy was used to examine the surface morphology of Ti3C2Tx, g-C3N4 and its composite gCT-0.75 (Fig. 3). After the selective removal of Al from MAX (Ti3AlC2) by etching with HF, the formation of MXene (Ti3C2Tx) was observed, having a perfect accordion-like morphology. It showed layers with a thickness of around 90–140 nm (Fig. 3a and b),43,44 which were stacked periodically. In contrast, we observed wavy layers in the image of the pristine g-C3N4 (Fig. 3c and d), which were quite difficult to quantify.45,46 Further, the presence of g-C3N4 together with Ti3C2Tx was observed with a uniform distribution in the nano-heterostructure, gCT-0.75 (Fig. 3e and f). This validates the in situ formation of a nano-heterostructure between g-C3N4 and Ti3C2Tx with a weight ratio 0.75:1 (gCT-0.75). The FESEM images of gCT-1 (Fig. 3g and h), gCT-0.50 (Fig. 3i and j), gCT-0.25 (Fig. 3k and l) and gCT-0.05 (Fig. 3m and n) are shown in the ESI (Fig. S1†), also confirming the formation of heterostructures between g-C3N4 and Ti3C2Tx.
 |
| | Fig. 3 FE-SEM of Ti3C2Tx (a and b), g-C3N4 (c and d) and gCT-0.75 (e and f). | |
3.3 FE-TEM
Fig. 4 shows the field-emission and high resolution transmission electron microscopy images of gCT-0.75. The FE-TEM images (Fig. 4a–d) of the gCT-0.75 nano-heterostructure depicts the formation of a 2D layered morphology of nanosheets, confirming the formation of a nano-heterojunction between g-C3N4 and Ti3C2Tx.47–49 The HRTEM images indicate that the interplanar spacing in the gCT-0.75 sample is around 0.24 nm and 0.32 nm, corresponding to the (006) and (002) planes of Ti3C2Tx and g-C3N4, respectively (Fig. 4e).50–52 This image authenticates the formation of a nanocomposite between g-C3N4 and MXene. The SAED (Fig. 4f) pattern also confirms the existence Ti3C2Tx together with g-C3N4 and supports the XRD results. Overall, the HRTEM images indicate the presence of g-C3N4 and Ti3C2Tx nanosheets.
 |
| | Fig. 4 FE-TEM (a–c) and HRTEM (d and e) images and selected area electron diffraction pattern (SAED) (f) of gCT-0.75. | |
3.4 XPS
The chemical composition and oxidation state of the Ti3C2Tx, gCT-0.75 and g-C3N4 samples were determined using XPS. As shown in Fig. 5 and the ESI (Fig. S2†), Ti, C, O, F and N elements were detected in the XPS survey spectra of the Ti3C2Tx, gCT-0.75 (Fig. S2a†) and g-C3N4 (Fig. S2c†) samples. The Ti, C, O, F and N elements were further investigated by XPS (Fig. 5 and S2†). The Ti 2P spectrum (Fig. 5a) was fitted with four doublets of Ti 2p3/2 and Ti 2p1/2 with an area ratio of 2:1 and doublet separation of 5.8 eV. The Ti 2p3/2 spectrum exhibits primary deconvoluted peaks centred at 454.6, 455.4, 456.3, 459.1, 461.5 and 464.9 eV, corresponding to Ti–C, Ti2+, Ti3+, Ti–O, C–Ti–Tx and TiO2−xFx, respectively, which indicate the presence of Ti3C2Tx in the gC3N4-0.75 sample, in agreement with the previous report.53 The C 1s spectrum of Ti3C2Tx (Fig. 5b) exhibited five distinct peaks at 281.6, 284.4, 285.0, 286.1 and 288.4 eV, which are assigned to the C–Ti, C–Ti–Tx, C–C, CHx/–COO and C–F/–COO bonds, respectively.54 These peaks can be ascribed to the adventitious carbon (284.6 eV). In addition, the C 1s spectrum of gCT-0.75 (Fig. 5b) exhibited four distinct peaks at 281.0, 284.5, 286.8 and 287.8 eV, corresponding to C–Ti, C–C, C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and N–CN, which clearly indicate the presence of g-C3N4 in the gCT-0.75 sample.41,55 The N 1s spectrum of gCT-0.75 (Fig. S2b†) exhibited three distinct peaks at 397.9, 398.8 and 400.2 eV, corresponding to the C–NC, N–(C)3 and C–N–H bonds, respectively.56 The O 1s spectrum (Fig. 5c) exhibited four peaks at 529.9, 530.7, 531.8 and 533.2 eV, which are assigned to TiO2, Ti–Ox, Ti–OH and Ti–H2O, respectively.57 The peak at 531.4 eV shows the presence of –OH groups on the surface (formation of Ti–OH bond) of the gCT-0.75 sample. In the F 1s spectrum (Fig. 5d), the dominant peak located at 685.7 eV corresponds to the C–F species, while the peak at the lower binding energy of 684.7 eV corresponds to C–Ti-Tx from Ti3C2Tx.58 The C 1s spectra of the g-C3N4 sample shown Fig. S2d† exhibit three carbon species at 284.2, 286.8 and 287.8 eV, which are assigned to the adsorbed carbon species (C–C) on the g-C3N4 surface, sp2-hybridized carbon in the triazine rings (C–NC) and N-containing aromatic ring (N–CN), respectively. The carbon in N–CN is considered as the major carbon species in the g-C3N4 nanosheets.56 Three peaks were observed in the N 1s spectrum (Fig. S2e†), and the peaks located at binding energies of 398.2, 399.1, and 400.5 eV can be assigned to the sp2-hybridized nitrogen involved in the triazine rings (C–NC), tertiary nitrogen N–(C)3 groups, and free amino groups (C–N–H), respectively41,58 (XPS, ESI Table S1†). Overall, the presence of peaks at 286.1 eV and 286.8 eV corresponds to CHx/C–O and C–NC, respectively. Also, the increase in the intensity of the peak at 287.8 eV corresponding to N–CN indicates the formation of the g-C3N4/Ti3C2Tx nano-heterostructure (gCT-0.75).
C and N–CN, which clearly indicate the presence of g-C3N4 in the gCT-0.75 sample.41,55 The N 1s spectrum of gCT-0.75 (Fig. S2b†) exhibited three distinct peaks at 397.9, 398.8 and 400.2 eV, corresponding to the C–NC, N–(C)3 and C–N–H bonds, respectively.56 The O 1s spectrum (Fig. 5c) exhibited four peaks at 529.9, 530.7, 531.8 and 533.2 eV, which are assigned to TiO2, Ti–Ox, Ti–OH and Ti–H2O, respectively.57 The peak at 531.4 eV shows the presence of –OH groups on the surface (formation of Ti–OH bond) of the gCT-0.75 sample. In the F 1s spectrum (Fig. 5d), the dominant peak located at 685.7 eV corresponds to the C–F species, while the peak at the lower binding energy of 684.7 eV corresponds to C–Ti-Tx from Ti3C2Tx.58 The C 1s spectra of the g-C3N4 sample shown Fig. S2d† exhibit three carbon species at 284.2, 286.8 and 287.8 eV, which are assigned to the adsorbed carbon species (C–C) on the g-C3N4 surface, sp2-hybridized carbon in the triazine rings (C–NC) and N-containing aromatic ring (N–CN), respectively. The carbon in N–CN is considered as the major carbon species in the g-C3N4 nanosheets.56 Three peaks were observed in the N 1s spectrum (Fig. S2e†), and the peaks located at binding energies of 398.2, 399.1, and 400.5 eV can be assigned to the sp2-hybridized nitrogen involved in the triazine rings (C–NC), tertiary nitrogen N–(C)3 groups, and free amino groups (C–N–H), respectively41,58 (XPS, ESI Table S1†). Overall, the presence of peaks at 286.1 eV and 286.8 eV corresponds to CHx/C–O and C–NC, respectively. Also, the increase in the intensity of the peak at 287.8 eV corresponding to N–CN indicates the formation of the g-C3N4/Ti3C2Tx nano-heterostructure (gCT-0.75).
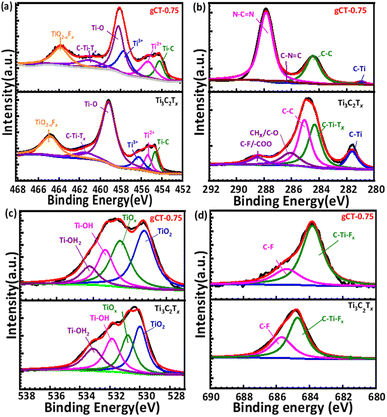 |
| | Fig. 5 XPS peak deconvolution of Ti 2p (a) C 1s (b) O 1s (c) and F 1s (d) for Ti3C2Tx and gCT-0.75. | |
3.5 FTIR
Fig. S3† shows the FTIR spectra of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05. MXene (Ti3C2Tx) does not show any peaks corresponding to C–C or C–Ti bonds.59 Further, g-C3N4 shows a vibration peak at 810 cm−1, corresponding to the symmetric stretching of the triazine unit, and peaks in the range of 1200 cm−1 to 1650 cm−1, which are ascribed to heterocyclic C–N and CN, respectively.60,61 The g-C3N4/Ti3C2Tx nano-heterostructure shows a peak at 810 cm−1 with a slight shift in the range of 790–810 cm−1, which indicates the formation of a nano-heterostructure between g-C3N4 and Ti3C2Tx.
3.6 Optical characterization
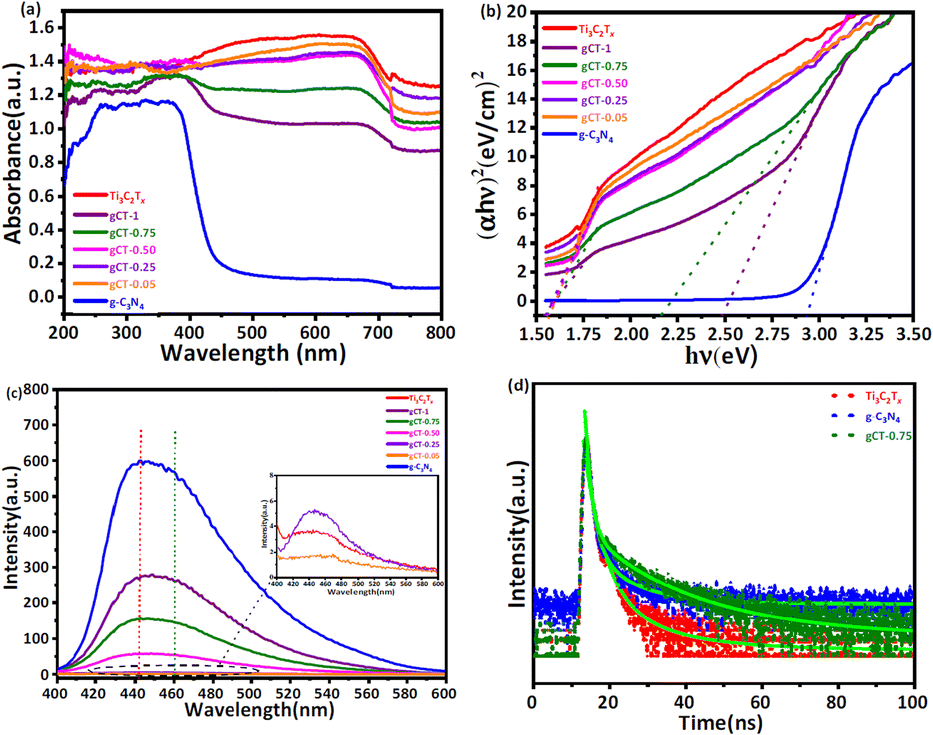
3.6.1 UV-visible analysis. The UV-Visible diffuse reflectance spectra (UV-vis DRS) of the Ti3C2Tx MXene and nano-heterostructure are shown in Fig. 6a. Ti3C2Tx exhibits an absorbance with a wide surface plasmon resonance (SPR) peak extending from 450 to 700 nm and centred at 670 nm. This SPR peak centred at 670 nm corresponds to Ti3C2Tx. The pristine g-C3N4 has an absorption band at about 460 nm. All the composites of g-C3N4/Ti3C2Tx showed an extended absorption compared to the pure g-C3N4 nanosheets. The absorbance of the g-C3N4/Ti3C2Tx nano-heterostructure gradually increased between 360 and 720 nm as the content of g-C3N4 decreased.41,62 All the prepared nano-heterostructures exhibited a greater absorbance compared to the pristine g-C3N4. This enhanced absorbance will help to capture a greater number of photons, resulting in the formation of more electron–hole pairs. Further, the formed heterojunction will also facilitate effective charge separation, resulting in a higher photocatalytic performance.63–65 The optical band gap energies of the as-prepared Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 were calculated using the Tauc plot, as shown in Fig. 6b, and the band gap values are listed in Table 1. The Tauc plot made it evident that two separate band gap values for gCT-1 and gCT-0.75 were formed, indicating the formation of a nano-heterostructure between g-C3N4 and Ti3C2Tx.
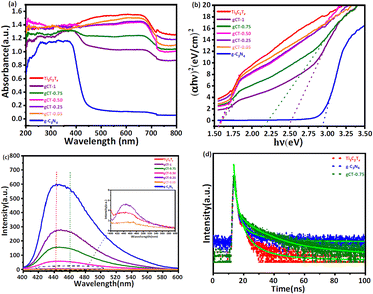 |
| | Fig. 6 (a) Diffuse reflectance UV-visible absorbance spectra of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05. (b) Tauc plots of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05. (c) Photoluminescence spectra of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05. (d) Time-resolved photoluminescence spectra of Ti3C2Tx, gCT-0.75 and g-C3N4. | |
Table 1 The band gap, specific surface area, pore volume (BJH), and average pore radius of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 catalyst
| S. No |
Catalyst name |
Band gap (eV) |
Specific surface area (m2 g−1) |
Pore volume BJH (cm3 g−1) |
Average pore radius (nm) |
| 1 |
Ti3C2Tx |
1.57 |
9.1 |
0.0346 |
1.69 |
| 2 |
g-C3N4 |
2.93 |
55.7 |
0.3028 |
10.69 |
| 3 |
gCT-1 |
1.54, 2.45 |
10.4 |
0.0403 |
7.07 |
| 4 |
gCT-0.75 |
1.56, 2.16 |
16.7 |
0.0635 |
7.75 |
| 5 |
gCT-0.50 |
1.56 |
8.8 |
0.0198 |
6.43 |
| 6 |
gCT-0.25 |
1.55 |
5.7 |
0.0179 |
4.81 |
| 7 |
gCT-0.05 |
1.58 |
3.8 |
0.0185 |
2.13 |
3.6.2 Photoluminescence (PL) spectroscopy. Photoluminescence spectra were recorded by dispersing 1 mg of prepared composite in 10 mL of DI water and sonicating it for 15 min. The pristine g-C3N4 showed the highest emission peak centred at 442 nm, corresponding to the band edge emission.66,67 As the concentration of g-C3N4 increased, the PL intensity centred at 442 nm also increased. Fig. 6c depicts the PL spectra of Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05, showing a decline in intensity with a decrease in the amount of g-C3N4. The higher PL intensity of g-C3N4 indicates the formation of a large number of charge carriers, which further reduced the recombination rate by the addition of MXene. The broadening of the emission peaks may be due to the radiative emission of the electrons in Ti3C2Tx.68,69 The Ti3C2Tx energy level is −0.635 V lower than that of the conduction band level of g-C3N4 (−1.165 V), which can facilitate the easy transfer of photogenerated electrons from the conduction band of g-C3N4 to Ti3C2Tx. This resulted in a red shift in the emission peak (∼460 nm), and subsequently broadening of the emission peak of the prepared nano-heterostructure.70,71 This is beneficial to separate photoelectrons and holes, creating electron transfer channels.
3.6.3 TRPL study. The lifetime of the charge carriers was also determined by time-resolved PL spectroscopy, as shown in Fig. 6d, and the biexponential model function was employed (eqn (2) and (3)), as follows| | |
R(t) = A1exp{−t/τ1} + A2exp{−t/τ2}
| (2) |
| |
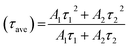 | (3) |
where A1 and A2 are the PL amplitudes and τ1 and τ2 are the decay times for the faster and slower components, respectively. The fast component (τ1) corresponds to the direct generation of free electrons and holes, whereas the slow component (τ2) results from the indirect formation of self-trapped excitations.72–74 The kinetic parameters are presented in Table S2, ESI.† The time constants for Ti3C2Tx were evaluated to be τ1 = 0.6801 ns and τ2 = 3.9214 ns, representing the electron trapping and formation time of electron–hole pairs. The time constants for g-C3N4 was evaluated to be τ1 = 0.7682 ns and τ2 = 4.0123 ns, respectively. Comparatively, gCT-0.75 showed longer time constants with τ1 = 0.9723 ns and τ2 = 4.5923 ns, respectively. The average lifetime (τave) of the entire fluorescence decay process was computed using eqn (3). According to the calculations, the average lifetime (τave) of Ti3C2Tx, gCT-0.75 and g-C3N4 is 1.8290, 2.7656 and 1.9390 ns, respectively. It was found that the τave of gCT-0.75 was almost 1.5-times that of Ti3C2Tx. The longer decay time shows the trapping of electrons, i.e., enhanced electron–hole separation.
3.7 Surface area analysis (using BET equation)
The Brunauer–Emmett–Teller (BET) specific surface area (SBET), pore size distribution, pore volume distribution and average pore radius of Ti3C2Tx, g-C3N4 and their nano-heterostructure are listed in Table 1. The specific surface area of Ti3C2Tx and g-C3N4 powder was measured to be 9.1 m2 g−1 and 55.7 m2 g−1, respectively. The measured specific surface areas of the synthesised nano-heterostructures gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 were 10.4, 16.7, 8.8, 5.7 and 3.8 m2 g−1, respectively.75 The specific surface area (SSA) was estimated using BET equation for the gCT-0.75 composite and found to be 16.7 m2 g−1, which is a factor of 1.84-greater than that of the as-synthesized Ti3C2Tx (9.1 m2 g−1). The increase in specific surface area (SSA) can be explained by the formation of a nano-heterostructure between Ti3C2Tx and g-C3N4 nanosheets, enlarging the interlayer spacing between the MXene multilayers after increasing the content of g-C3N4 in the composite.76 All the prepared nano-heterostructures, as shown in Fig. 7a, showed a clear hysteresis loop between the adsorption and desorption branches at relative pressures in the range of 0.4–1.0, indicating the existence of mesopores. At a relative pressure near to 1.0, the practically vertical tails indicated the existence of macropores. According to this, Ti3C2Tx mostly consists of macropores, while the gCT-0.75 sample typically has mesopores and macropores. The BJH (cm3 g−1) pore volume and average pore radius (nm) of the prepared nano-heterostructures, i.e., gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05, are shown in Table 1. The pore size distributions calculated from the nitrogen desorption branches for all the samples are depicted in Fig. 7b. The results show the presence of more mesopores in gCT-0.75 than Ti3C2Tx (almost 4.5-times more), as well as a very close pore size distribution with a peak centred at ca. 7.5 nm. This indicates the presence of more active sites and surface area available for photocatalytic reactions.
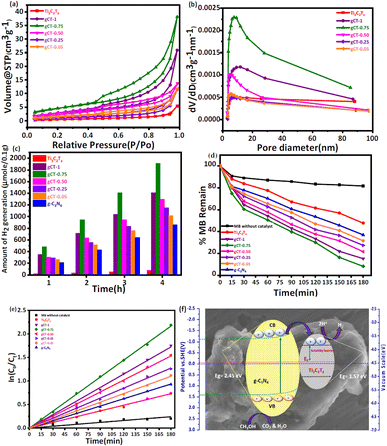 |
| | Fig. 7 (a) BET surface area using N2 adsorption–desorption isotherms of Ti3C2Tx, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05. (b) Pore size distributions calculated from N2 desorption isotherms for Ti3C2Tx, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05. (c) Photocatalytic H2 generation by Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 as a function of time. (d) Amount of MB remaining with irradiation time using Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 catalyst prepared at different temperatures. (e) Plots of lnC0/Ct versus irradiation time for photocatalytic MB degradation using prepared Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 catalysts. (f) Proposed mechanism of photocatalytic hydrogen evolution by Ti3C2Tx/g-C3N4. | |
3.8 Photocatalytic activity
Under a 400 W mercury vapour lamp, the photocatalytic performance of the prepared g-C3N4/Ti3C2Tx nanostructures was evaluated by monitoring H2 generation via water splitting and methylene blue (MB) dye degradation.
3.8.1 Photocatalytic H2 generation. The photocatalytic performance of Ti3C2Tx, g-C3N4 and their nano-heterostructures gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 was measured using photocatalytic hydrogen production from water under UV-visible light irradiation.59,77 As shown in Fig. 7c, the Ti3C2Tx sample exhibited the lowest H2 generation of around 74.90 μmol/0.1 g of H2 upon 4 h of irradiation. However, the prepared nano-heterostructures with different g-C3N4 to Ti3C2Tx ratios showed enhanced photocatalytic H2 generation. The cumulative amounts of H2 evolved in 4 h were 1410.21, 1912.25, 1296.23, 1150.32 and 1021.23 μmol/0.1 g for gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05, respectively (ESI, Tables S3 and S4†). The pristine g-C3N4 nanosheets showed H2 generation of around 860.98 μmol/0.1 g in 4 h. Among them, gCT-0.75 exhibited the highest hydrogen evolution (1912.25 μmol/0.1 g), which was almost 25.5-times higher than that of Ti3C2Tx (74.90 μmol/0.1 g). This higher photocatalytic H2 generation by gCT-0.75 is due to the formation of a nano-heterostructure between g-C3N4 and Ti3C2Tx.78 This formation of a nano-heterostructure led to the effective separation of photogenerated charge carriers due to existence of the coupled catalytic system. Yuliang et al. reported the H2 generation of around 88 μmol h−1 gcat−1 using g-C3N4/Ti3C2Tx.76 In the present case, gCT-0.75 showed the highest H2 generation of 1912.25 μmol/0.1 g among the prepared compositions. Further, the apparent quantum yield (AQY) was also calculated and found to be 0.2%, 1.4%, 2.3%, 3.1%, 2.1%, 1.9% and 1.7% for Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05, respectively79 (ESI, Table S5†). In the PL study, the PL intensity decreased with a decrease in the content of g-C3N4 in the case of gCT-0.75. This clearly shows the enhancement in electron–hole separation, which is responsible for its higher photocatalytic activity. Further, the higher decay time observed in the TRPL of gCT-0.75 exhibits higher charge carrier separation, which also supports its higher photocatalytic H2 evolution. Additionally, gCT-0.75 has a larger specific surface area (SSA), which provides a greater surface area and active sites for photocatalytic H2 generation. The optimum concentration of g-C3N4 facilitated fast electrons transport to the surface, which ultimately suppressed the charge carrier recombination. The optimised nano-heterostructures also facilitated higher electron hole separation due to self-trapping at the interface.
3.8.2 Dye degradation. The amount of MB dye degraded with time using the prepared catalysts is depicted in Fig. 7d. Upon 3 h of irradiation, the amount of MB degradation without the use of catalyst was found to be around 18.4%. The prepared Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 showed 52.3%, 63.1%, 84.7%, 91.7%, 78.3%, 72.6% and 67.4% MB degradation within 3 h of irradiation, respectively. In the case of Ti3C2Tx, g-C3N4 and their composite, the UV-visible absorbance spectra of MB dye with irradiation time are shown in ESI, Fig. S4.† Among, the prepared catalysts, gCT-0.75 showed the highest photocatalytic activity towards MB degradation. This is because Ti3C2Tx and g-C3N4 formed a hetero-junction, which facilitated the easy separation of photo-generated electron–hole pairs, making them available for further degradation reaction. Consequently, when Ti3C2Tx and the g-C3N4 nanosheets form a nanostructure, a linked catalytic system was created, resulting in increased photoactivity compared to the pristine material (ESI, Tables S6 and S7,† respectively). The rate constant for the photocatalytic MB degradation process was computed for the purpose of comparing the photocatalytic activity by plotting the graph of ln(C0/Ct) against irradiation time, as shown in Fig. 7e. The apparent rate constant is shown by the slope of the graph of ln(C0/Ct) vs. irradiation time, where C0 represents the starting concentration of MB and Ct the concentration at time ‘t’ (In Table 2), respectively. The observed rate constant values for the prepared Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 catalysts are 4.06 × 10−3 min−1, 5.2 × 10−3 min−1, 9.58 × 10−3 min−1, 12.05 × 10−3 min−1, 8.52 × 10−3 min−1, 7.08 × 10−3 min−1 and 6.12 × 10−3 min−1, respectively. In contrast, the MB degradation without catalyst showed a rate constant value of 1.35 × 10−3 min−1. This clearly shows that the gCT-0.75 nano-heterostructure has the highest overall photocatalytic activity among the nano-heterostructures and pristine materials. The reason for the enhancement in photocatalytic activity was also briefed in the previous photocatalytic H2 generation section. In summary, all the nano-heterostructures showed an enhancement in photocatalytic activity with an optimum concentration of g-C3N4.
Table 2 The MB degradation rate and H2 generation using Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25, and gCT-0.05 catalysts
| S. no. |
Catalyst |
Rate constant (min−1) with standard fitting error |
H2 generation μmol/0.1 g |
| 1 |
Ti3C2Tx |
4.06 × 10−3 ± 0.00010709 |
74.90 |
| 2 |
g-C3N4 |
5.2 × 10−3 ± 0.00017425 |
860.98 |
| 3 |
gCT-1 |
9.58 × 10−3 ± 0.00012244 |
1410.21 |
| 4 |
gCT-0.75 |
12.05 × 10−3 ± 0.00006253 |
1912.25 |
| 5 |
gCT-0.50 |
8.52 × 10−3 ± 0.00000981 |
1296.23 |
| 6 |
gCT-0.25 |
7.08 × 10−3 ± 0.00000798 |
1150.32 |
| 7 |
gCT-0.05 |
6.12 × 10−3 ± 0.00000730 |
1021.23 |
| 8 |
Without catalyst |
1.35 × 10−3 ± 0.00012001 |
— |
We also performed I–V photoconductivity measurements (ESI, Fig. S5†) on all the Ti3C2Tx, g-C3N4, gCT-1, gCT-0.75, gCT-0.50, gCT-0.25 and gCT-0.05 nano-heterostructures. The highest photoconductivity was obtained for gCT-0.75, which also supports the highest photocatalytic ability of this nano-heterostructure.
3.8.3 Reaction mechanism. Among the prepared g-C3N4/Ti3C2Tx catalysts, gCN-0.75 showed the highest photocatalytic H2 generation and MB dye degradation. A schematic of the band gap of the catalyst is shown in Fig. 7f. g-C3N4 showed a band gap of 2.93 eV and its conduction band (−1.165 V) level is higher than the H2 evaluation potential (1.765 V).81 The Fermi level (−0.53 V) of Ti3C2Tx just below the CB of g-C3N4 creates an easy path for the transfer of photogenerated CB electrons to the Ti3C2Tx level. This easy transfer minimizes the electron–hole recombination reaction, resulting in higher photocatalytic activity towards H2 generation and MB degradation. Specifically, upon exposure to light, the electrons in the valence band (VB) of g-C3N4 can be excited to the conduction band (CB), generating photo-induced electrons and holes. Further, the migration rate of the photoinduced electrons may be significantly increased due to higher electronic conductivity of Ti3C2Tx (hopping of the electrons from the CB of g-C3N4 to the CB of Ti3C2Tx), and also the intimate contact between the Ti3C2Tx and g-C3N4 nano-heterostructure.41,80 Moreover, the contact between Ti3C2Tx and g-C3N4 results in the formation of a hetero-junction (at the interface).
3.8.4 Recyclability of the hydrogen generation behaviour. Ti3C2Tx functions as an electron acceptor and promotes the transfer of electrons from g-C3N4 to Ti3C2Tx.82 Consequently, the photogenerated electrons collected on Ti3C2Tx can reduce H+ to produce H2 on its surface; meanwhile, methanol is oxidised into CO2 and H2O via the holes in the VB of g-C3N4. Hence, the nano-heterostructure of Ti3C2Tx and g-C3N4 can efficiently separate and transport the photo-induced electrons and holes, significantly improving the performance of the photocatalytic system.85,86 This enhanced activity was observed for gCN-0.75, given that this was the optimal combination of g-C3N4 and Ti3C2Tx for the formation of the coupled catalyst system among the prepared compositions, as discussed in the previous section.76 Furthermore, the stability and reusability of the catalyst were assessed using a recycling study of the most active sample for 3 cycles, i.e., for gCT-0.75. For the hydrogen generation process, the identical catalyst was recycled three times (Fig. 8a). It was observed that the catalytic activity of gCT-0.75 did not exhibit any significant loss after the second and third cycles. After the third cycle, the amount of H2 generated was approximately 7610.25 μmol of H2 per 0.1 g of recycled catalyst (i.e., 1912.25 μmol h−1 0.1 g−1), indicating the high stability of the catalyst towards hydrogen generation.41,59 We did not find any structural changes in the recycled sample via examination by XRD (Fig. 8b), clearly indicating the good stability of the catalyst.
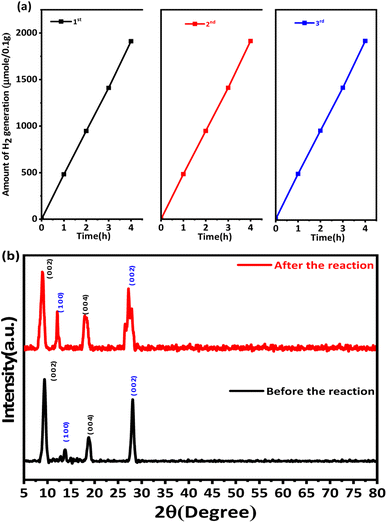 |
| | Fig. 8 (a) Recyclability of the hydrogen generation behaviour of gCT-0.75. (b) XRD patterns of gCT-0.75 before and after the photocatalytic reaction. | |
4. Conclusion
In summary, we demonstrated the process for the in situ synthesis of g-C3N4/Ti3C2Tx nanosheets, i.e., nano-heterojunction. The formation of g-C3N4 nanolayers on the surface of MXene was confirmed by FE-SEM and HR-TEM. In comparison to the pristine Ti3C2Tx (74.90 μmol/0.1 g), the optimised gCT-0.75 showed a higher photocatalytic hydrogen evolution rate, which was almost 25-times higher (1912.25 μmol/0.1 g). The effective charge separation at the interface between Ti3C2Tx and g-C3N4 is the main reason for the enhanced photocatalytic performance compared to the pristine MXene. Overall, the gCT-0.75 nano-heterostructure exhibited improved photocatalytic activity due to the enhanced average lifetime of photogenerated electron–hole pairs, creation of an appropriate nano-heterostructure between Ti3C2Tx and g-C3N4, optimal surface, and suitable energy band modification. This technique shows potential for the in situ fabrication of other metal oxide/sulfide hetero-structure photocatalytic systems.
Author contributions
ABT performed the experimental work, material characterisation, photocatalytic performance measurements and wrote the initial draft of the manuscript. SSA given the research scheme and assisted in designing the experiment, analysed the results and modified the manuscript. GGU assisted in photoconductivity measurements. SKK revised the manuscript and analysed the XPS data. BBK supervised the whole research work and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors are grateful to Dr Rahul Salunkhe, Assistant Professor, IIT Jammu and Dr Vinayak Kamble, Scientist, IISER, Trivandrum for XPS and TEM analysis respectively. One of the author SKK is thankful to INSA for senior scientist fellowship.
References
- J. Uribe-Toril, J. Ruiz-Real, J. Milan-Garcia and V. de Pablo, Energies, 2019, 12(6), 1120 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488(7411), 294–303 CrossRef CAS PubMed.
- M. N. Chong, B. Jin, W. K. Christopher and C. Saint, Water, 2010, 44, 2997–3027 CAS.
- X. Meng, N. Yun and Z. Zhang, Can. J. Chem. Eng., 2019, 97(7), 1982–1998 CrossRef CAS.
- X. Wang, G. Zhang, L. Yang, E. Sharman and J. Jiang, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8(5), 1369 CrossRef.
- Y. Zhao, S. Zhang, R. Shi, G. I. N. Waterhouse, J. Tang and T. Zhang, Adv. Mater. Today, 2020, 34, 78–91 CrossRef CAS.
- X. Li, J. Yu, S. Wageh, A. A. Al-Ghamdi and J. Xie, Small, 2016, 12(48), 6640–6696 CrossRef CAS PubMed.
- R. Wang, K. Lu, Z. Tang and Y. Xu, J. Mater. Chem. A, 2017, 5(8), 3717–3734 RSC.
- L. Wu, Q. Li, C. Yang, X. Ma, Z. Zhang and X. Cui, J. Mater. Chem. A, 2018, 6(42), 20947–20955 RSC.
- Z. Li and X. Meng, J. Alloys Compd., 2020, 830, 154699 CrossRef.
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed.
- M. Chen, Y. Zhang, L. Cui, Z. Cao, Y. Wang, W. Zhang, Y. Zheng, D. Sun, L. Zheng, S. Kang and D. Zhang, Chem. Eng. J., 2021, 422, 130089 CrossRef CAS.
- X. Zhang and P. Yang, Carbon, 2024, 216, 118584 CrossRef CAS.
- X. Zhang and P. Yang, Chem. Nano. Mat., 2023, 9, 6 Search PubMed.
- X. Lu, K. Xu, P. Chen, K. Jia, S. i. Liu and C. Wu, J. Mater. Chem. A, 2014, 2(44), 18924–18928 RSC.
- Y. Yu, D. Chen, W. Xu, J. Fang, J. Sun, Z. Liu, Y. Chen, Y. Liang and Z. Fang, J. Hazard. Mater., 2021, 416, 126183 CrossRef CAS PubMed.
- X. Zhang, K. Matras-Postolek and P. Yang, Mater. Today Adv., 2023, 17, 100355 CrossRef CAS.
- X. Zhang, X. Zhang, P. Yang and S. P. Jiang, J. Nanostruct. Chem., 2022, 12, 669–691 CrossRef CAS.
- X. Zhang, K. Matras-Postolek, P. Yang and S. Ping Jiang, J. Colloid Interface Sci., 2023, 636, 646–656 CrossRef CAS PubMed.
- X. Zhang and S. P. Jiang, Mater. Today Energy, 2022, 23, 100904 CrossRef CAS.
- X. Zhang, K. Matras-Postolek, P. Y. San and P. Jiang, Carbon, 2023, 214, 118337 CrossRef CAS.
- T. Song, X. Zhang, C. Xie and P. Yang, Carbon, 2023, 210, 118052 CrossRef CAS.
- T. Song, X. Zhang, K. Matras-Postolek and P. Yang, Carbon, 2023, 202, 378–388 CrossRef CAS.
- D. Liu, C. Li, J. Ge, C. Zhao, Q. Zhao, F. Zhang, T. Ni and W. Wu, Appl. Surf. Sci., 2022, 579, 152180 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Dahlqvist, B. Alling and J. Rosen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 220102 CrossRef.
- H. Yang, Y. Ma, X. Lv, B. Huang and Y. Dai, J. Catal., 2020, 387, 12–16 CrossRef CAS.
- H. Chang, X. Li, L. Shi, Y. Zhu and T. Yi, Chem. Eng., 2021, 421, 129944 CrossRef CAS.
- R. Tang, S. Xiong, D. Gong, Y. Deng, Y. Wang, L. Su, C. Ding, L. Yang and C. Liao, ACS Appl. Mater. Interfaces, 2020, 12(51), 56663–56680 CrossRef CAS PubMed.
- S. Tasleem and M. Tahir, Energy Fuels, 2023, 37(2), 1421–1440 CrossRef CAS.
- F. Xu, D. Zhang, Y. Liao, G. Wang, X. Shi, H. Zhang and Q. Xiang, Am. Ceram. Soc., 2020, 103(2), 849–858 CrossRef CAS.
- W. Yi, X. Du, M. Zhang, S. Yi, R. Xia, C. Li, Y. Liu, Z. Liu, W. Zhang and X. Yue, Nano Res., 2023, 16, 6652–6660 CrossRef CAS.
- M. Xiao, Y. Jiao, B. Luo, S. Wang, P. Chen, M. Lyu, A. Du and L. Wang, Nano Res., 2023, 16, 4539–4545 CrossRef CAS.
- Y. Zhang, N. Cao, X. Liu, F. He, B. Zheng, C. Zhao and Y. Wang, Sci. China Mater., 2023, 66, 2274–2282 CrossRef CAS.
- M. Naguib and Y. Gogotsi, Acc. Chem. Res., 2015, 48, 128–135 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29(18), 7633–7644 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- Y. Liu, X. Zhang, S. Dong, Z. Ye and Y. Wei, J. Mater. Sci., 2017, 52, 2200–2209 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6(2), 1322–1331 CrossRef CAS PubMed.
- A. Feng, Y. Yu, Y. Wang, F. Jiang, Y. Yu, L. Mi and L. Song, Mater. Des., 2017, 114, 161–166 CrossRef CAS.
- T. Su, Z. D. Hood, M. Naguib, L. Bai, S. Luo, C. M. Rouleau, I. N. Ivanov, H. Ji, Z. Qin and Z. Wu, Nanoscale, 2019, 11, 8138–8181 RSC.
- X. Yu, W. Yin, T. Wang and Y. Zhang, Langmuir, 2019, 35(8), 2909–2916 CrossRef CAS PubMed.
- S. Liu, M. Wang, G. Liu, N. Wan, C. Ge, S. Hussain, H. Meng, M. Wang and G. Qiao, Appl. Surf. Sci., 2021, 567, 150747 CrossRef CAS.
- S. Yang, P. P. Zhang, F. X. Wang, A. G. Ricciardulli, M. R. Lohe, P. W. Blom and X. L. Feng, Angew. Chem., Int. Ed., 2018, 57, 15491–15495 CrossRef CAS PubMed.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- M. Houa, J. Gao, L. Yanga, S. Guoa, T. Hub and Y. Lid, Appl. Surf. Sci., 2021, 535, 147666 CrossRef.
- C. Xu, C. Fan, X. Zhang, H. Chen, X. Liu, Z. Fu, R. Wang, T. Hong and J. Cheng, ACS Appl. Mater. Interfaces, 2020, 12(17), 19539–19546 CrossRef CAS PubMed.
- Y. Li, F. Zhang, Y. Chen, J. Lia and Y. Xu, Green Chem., 2020, 22, 163 RSC.
- X. Ding, Y. Li, C. Li, W. Wang, L. Wang, L. Feng and D. Han, J. Mater. Sci., 2019, 54, 9385–9396 CrossRef CAS.
- Z. Yan, Z. Sun, X. Liu, H. Jia and P. Du, Nanoscale, 2016, 8, 4748–4756 RSC.
- X. Guo, J. Duan, C. Li, Z. Zhang and W. Wang, J. Mater. Sci., 2020, 55, 2018–2203 CrossRef CAS.
- S. B. Kokane, R. Sasikala, D. M. Phase and S. D. Sartale, J. Mater. Sci., 2017, 52, 7077–7090 CrossRef CAS.
- Y. T. Liu, P. Zhang, N. Sun, B. Anasori, Q. Z. Zhu, H. Liu, Y. Gogotsi and B. Xu, Adv. Mater., 2018, 30, 1707334 CrossRef PubMed.
- S. Y. Cho, Y. Lee, H. J. Koh, H. Jung, J. S. Kim, H. W. Yoo, J. Kim and H. T. Jung, Adv. Mater., 2016, 28, 7020–7028 CrossRef CAS PubMed.
- Q. Y. Lin, L. Li, S. J. Liang, M. H. Liu, J. H. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–143 CrossRef CAS.
- S. Guo, Y. Tang, Y. Xie, C. Tian, Q. Feng, W. Zhou and B. Jiang, Appl. Catal., B, 2017, 218, 664–671 CrossRef CAS.
- C. F. Du, K. N. Dinh, Q. L. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127 CrossRef.
- S. A. Shah, T. Habib, H. Gao, P. Gao, W. Sun, M. J. Green and M. Radovic, Chem. Commun., 2016, 53, 400–403 RSC.
- Y. Sun, D. Jin, Y. Sun, X. Meng, Y. Gao, Y. Dall'Agnese, G. Chen and X. Wang, J. Mater. Chem. A, 2018, 6, 9124–9131 RSC.
- F. Zhao, P. Zhai, Y. Wei, Z. Yang, Q. Chen, J. Zuo, X. Gu and Y. Gong, Adv. Sci., 2022, 9(6), 2103930 CrossRef CAS PubMed.
- Q. Zhou, K. Qian, J. Fang, M. Miao, S. Cao and X. Feng, Composites, Part A, 2020, 134, 105899 CrossRef CAS.
- K. Maleski, C. E. Shuck, A. T. Fafarman and Y. Gogotsi, Adv. Opt. Mater., 2020, 9, 2001563 CrossRef.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, Nano Lett., 2017, 17, 384–391 CrossRef CAS PubMed.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11(4), 3752–3759 CrossRef CAS PubMed.
- C. Shaowen, S. Baojia, T. Tong, F. Junwei and Y. Jiaguo, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- T. Su, H. Tian, Z. Qin and H. Ji, Appl. Catal., B, 2017, 202, 364–373 CrossRef CAS.
- Y. Huang, H. Xu, H. Yang, Y. Lin, H. Liu and Y. Tong, ACS Sustainable Chem. Eng., 2018, 6, 2751–2757 CrossRef CAS.
- P. Wu, J. Wang, J. Zhao, L. Guo and F. E. Osterloh, J. Mater. Chem. A, 2014, 2, 20338–20344 RSC.
- G. Dong, D. L. Jacobs, L. Zang and C. Wang, Appl. Catal., B, 2017, 218, 515–524 CrossRef CAS.
- J. Kang, S. Byun, S. Kim, J. Lee, M. Jung, H. Hwang, T. Kim, S. Ho Song and D. Lee, ACS Appl. Energy Mater., 2020, 3(9), 9226–9233 CrossRef CAS.
- J. S. Kim, J. W. Oh and S. I. Woo, Catal. Today, 2017, 293–294, 8–14 CrossRef CAS.
- S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 RSC.
- Y. Yang, D. Zhang and Q. Xiang, Nanoscale, 2019, 11, 18797–18805 RSC.
- X. Li, Y. Bai, X. Shi, J. Huang, K. Zhang, R. Wang and L. Ye, Appl. Surf. Sci., 2021, 546, 149111 CrossRef CAS.
- J. Yang, X. Zhang, C. Xie, J. Long, Y. Wang, L. Wei and X. Yang, J. Electron. Mater., 2021, 50, 1067–1074 CrossRef CAS.
- Y. Sun, D. Jin, Y. Sun, X. Meng, Y. Gao, Y. Dall'Agnese, G. Chen and X. Wang, J. Mater. Chem. A, 2018, 6, 9124–9131 RSC.
- X. An, W. Wang, J. Wang, H. Duan, J. Shi and X. Yu, Phys. Chem. Chem. Phys., 2018, 20, 11405–11411 RSC.
- Y. Li, L. Ding, Y. Guo, Z. Liang, H. Cui and J. Tian, ACS Appl. Mater. Interfaces, 2019, 11(44), 41440–41447 CrossRef CAS PubMed.
- B. Lin, H. Li, H. An, W. Hao, J. Wei, Y. Dai, C. Ma and G. Yang, Appl. Catal., B, 2018, 220, 542–552 CrossRef CAS.
- P. Lin, J. Shen, X. Yu, Q. Liu, D. Li and H. Tang, Ceram. Int., 2019, 45(18), 24656–24663 CrossRef CAS.
- M. Nasri, M. F. Samsudin, A. Tahir and S. Sufian, Energies, 2022, 15(3), 955 CrossRef CAS.
- M. Zhang, J. Qin, S. Rajendran, X. Zhang and R. Liu, ChemSusChem, 2018, 11(24), 4226–4236 CrossRef CAS PubMed.
- N. Jawale, S. Arbuj, G. Umarji, M. Shinde, B. Kale and S. B. Rane, RSC Adv., 2023, 13, 2418–2426 RSC.
- S. R. Damkale, S. S. Arbuj, G. G. Umarji, S. B. Rane and B. B. Kale, RSC Adv., 2021, 11, 7587–7599 RSC.
- H. Yu, H. Jiang, X. Cao, S. Yao and Z. You, RSC Adv., 2023, 13, 16602 RSC.
- Y. Liao, X. Li, J. Fan and Q. Xiang, Nanoscale, 2021, 13, 9463–9504 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Govind G. Umarjia,
Sulbha K. Kulkarnia and
Bharat B. Kale
*a,
Govind G. Umarjia,
Sulbha K. Kulkarnia and
Bharat B. Kale