
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homoleptic and heteroleptic ketodiiminate zinc complexes for the ROP of cyclic L-lactide†
Eduard Glöcklera,
Leon Kappa,
Christoph Wölpera,
Marcel Schumacher b,
André H. Gröschelb and
Stephan Schulz*ac
b,
André H. Gröschelb and
Stephan Schulz*ac
aFaculty of Chemistry, University of Duisburg-Essen, Universitätsstraße 7, 45141 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bFaculty of Chemistry, University of Münster and Center for Soft Nanoscience (SoN), Busso-Peus-Strasse 10, 48149 Münster, Germany
cCenter for Nanointegration Duisburg-Essen (CENIDE), Carl-Benz-Straße 199, 47057 Duisburg, Germany
First published on 12th October 2023
Abstract
Homo- and heteroleptic ketodiiminate zinc complexes L12Zn2 (1, L1 = [Me2NC2H4NC(Me)CH]2CO), L2(ZnCp)2 (2, L2 = [Me2NC3H6NC(Me)CH]2CO, Cp = C5H5) and L2HZnCp* (3, Cp* = C5Me5) were synthesized and characterized by 1H and 13C NMR and IR spectroscopy as well as by elemental analysis and single crystal X-ray diffraction (sc-XRD, 2, 3). The catalytical activity of heteroleptic complexes 2 and 3 were tested in the ring-opening polymerization (ROP) of L-lactide. Homobimetallic complex 2 showed the highest activity and selectivity for the synthesis of cyclic polylactide (cPLLA; TOF = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460 h−1) at 100 °C in toluene solution, while linear polymers are formed with mononuclear complex 3.
460 h−1) at 100 °C in toluene solution, while linear polymers are formed with mononuclear complex 3.
Introduction
The use of polymers derived from renewable resources is considered an environmentally friendly and sustainable alternative to the use of petroleum-based plastics.1–3 In recent years, the physicochemical properties, biodegradability, and biocompatibility of aliphatic polyester have been extensively studied.4–9 Polylactide (PLA) is one of the most important polymers used in agriculture, packaging, pharmaceutical and biomedical fields. The physicochemical properties of such kind of polyesters can be modified by their molar mass, stereochemistry, typological structure and polymer architecture.4,10,11 Cyclic polyesters such as cyclic polylactide (cPLA) are also of interest since their properties differ from their linear counterparts in terms of melting temperature (Tm), glass transition temperature (Tg), viscosity, hydrodynamic volume, thermostability and morphology, respectively.12–18 However, unlike their linear and branched counterparts, the synthesis of cPLA with high molar mass continues to be a difficult scientific problem.19,20Ring-closure and ring-expansion reactions are typically used for the synthesis of cyclic polymers.21,22 Unfortunately, ring-closure reactions often gave impure cyclic polymers with relatively low molar masses resulting from side-formation of linear polymers.12 In contrast, ring extension reactions often created cyclic polymers with higher molecular weights.13 One of the most effective ring-expansion catalysts for the synthesis of cyclic polylactide are N-heterocyclic carbenes (NHCs).14–17 Mechanistic analyses of the synthesis of cPLA revealed that neutral carbenes start the polymerization process by entering a zwitterionic state. The monomer is cyclized through transesterification, known as zwitterionic ring-opening polymerization (zROP). Carbene-catalysed reactions typically have a limited capacity for chain growth and, consequently, the production of high molecular weight polymers.14–17 In contrast, the addition of alcohols as initiators produced linear polylactides with higher molecular masses.23–25 In 2021, Arnold et al. reported a highly active and highly selective homogeneous Ce(III)-NHC catalyst for the synthesis of high average molecular weight cyclic polylactides with turn-over-frequencies (TOFs) up to 864000 h−1. This catalyst efficiently produces cyclic PLA from rac/L-lactide and other aliphatic cyclic polyesters, i.e., ε-caprolactone or β-butyrolactone. The high activity results from synergistic effects between the Lewis acidic Ce(III) centre and the N-heterocyclic carbene.26
In the last decades, main group metals such as alkaline or earth alkaline metals27–29 as well as zinc have been intensively studied as catalysts for the ROP of lactides and lactones due to their high activity, low toxicity and biocompatibility.30–41 In marked contrast, these type of metal complexes are rarely known for the preparation of cyclic polylactides. Recently, our group reported the synthesis of binuclear magnesium complexes and their capability for the synthesis of cPLA and cPCL with high TOF values up to 712800 h−1 under mild reaction conditions.42 We herein report the synthesis of mono- and homodinuclear zinc ketodiiminate complexes and their catalytic activity for the synthesis of cPLA.
Results and discussion
Synthesis and characterization
Reactions of ketodiimine L1H2 with two equivalents of either Cp2Zn or Cp*2Zn (Cp = C5H5, Cp* = C5Me5) in toluene at 25 °C gave the homoleptic complex 1 (Scheme 1). In situ 1H-NMR spectra of the reactions showed the disappearance of the NH-protons at 9.99 ppm and simultaneous formation of CpH (2.69, 6.08–6.50 ppm) and Cp*H (0.98–1.00, 1.74–1.80, 3.56–3.58 ppm), respectively (Fig. S7 and S8†). In contrast, reactions of ketodiimine L2H2 with two equivalents of Cp2Zn and Cp*2Zn in toluene at 25 °C gave the homobinuclear complex 2 and the mononuclear complex 3. Complex 3 was also obtained from the reaction of L2H2 with one equivalent of ZnCp*2, while reactions of L1H2 and L2H2 with one equivalent of ZnCp2 as well as of L1H2 with one equivalent of ZnCp*2 gave no pure compounds. | ||
| Scheme 1 Synthesis of dinuclear homoleptic and mononuclear heteroleptic ketodiiminate zinc complexes 1–3. | ||
Complexes 2 and 3 were purified by recrystallization from solutions in toluene at −30 °C. The 1H NMR spectra of 2 (Fig. S11†) shows resonance due to the doubly deprotonated ligand L2 (4.25 ppm) and the Cp groups (6.32 ppm). All resonances are shifted to higher field compared to those of L2H2. In addition, broad triplet and multiplet signals indicate hindered rotations. In contrast, the 1H NMR spectrum of complex 3 (Fig. S14†) is more complex due to the asymmetric nature of the mono-deprotonated ligand L2H. The Cp* unit shows only one signal (1.89 ppm) caused by the fluctuation of the Cp* ring.
Complexes 2 and 3 crystallize in the triclinic space group P1 (Fig. 1 and 2). The Zn atoms are tetrahedrally coordinated by the N, N, O heteroatoms of the ligand L1/2 and one η1-bonded Cp/Cp* group. The Zn–O bond lengths in complexes 2 (2.0266(8) Å) and 3 (1.9821(16) Å) as well as the Zn–N bond lengths in 2 (Zn(1)–N(1) 1.9403(12) Å, Zn(1)–N(3) 2.1311(13) Å) and 3 (Zn(1)–N(1) 1.985(2) Å, Zn(1)–N(3) 2.127(2) Å) are elongated compared to the sum of the covalent radii.43 In addition, the sum of the N(1)–Zn(1)–O/N(2)–Zn(2)–O (99.59(4)°/98.30(4)°), N(1)–Zn(1)–N(3)/N(2)–Zn(2)–N(4) (92.25(4)°/92.63(5)°) and O–Zn(1)–N(3)/O–Zn(2)–N(4) (110.57(4)°/111.71(4)°) bond angles of 2 are slightly larger than the sum of the N(1)–Zn(1)–O (96.37(8)°), N(1)–Zn(1)–N(3) (94.34(9)°), O–Zn(1)–N(3) (105.16(5)°) of compound 3, most likely resulting from the bulky Cp* substituents in compound 3.
 | ||
| Fig. 1 ORTEP representation of solid-state structure of complex 2. H atoms have been omitted for clarity and thermal ellipsoids are shown with 50% probability level. Selected bond lengths [Å] and angles [°]: Zn(1)–O 2.0266(8), Zn(1)–N(1) 1.9403(12), Zn(1)–N(3) 2.1311(13), Zn(1)–C(18) 2.0950(13), Zn(2)–O 2.0266(9), Zn(2)–N(2) 1.9437(11), Zn(2)–N(4) 2.1582(11), Zn(2)–C(23) 2.1051(12), N(1)–Zn(1)–O 99.59(4), N(1)–Zn(1)–C(18) 128.71(5), O–Zn(1)–C(18) 111.80(4), N(1)–Zn(1)–N(3) 92.25(4), O–Zn(1)–N(3) 110.57(4), C(18)–Zn(1)–N(3) 111.80(5), N(2)–Zn(2)–O 98.30(4), N(2)–Zn(2)–C(23) 126.99(5), O(1)–Zn(2)–C(23) 114.01(4), N(2)–Zn(2)–N(4) 92.63(5), O–Zn(2)–N(4) 111.71(4), C(23)–Zn(2)–N(4) 111.01(5). | ||
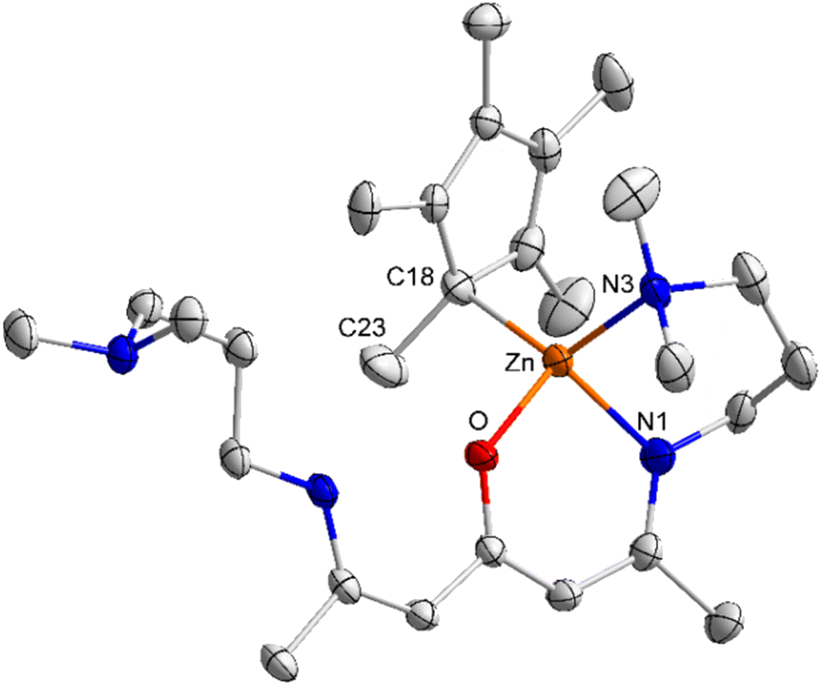 | ||
| Fig. 2 ORTEP representation of the solid-state structure of 3. H atoms and the second orientation of the side arm are omitted for clarity and thermal ellipsoids are shown with 50% probability level. Selected bond lengths [Å] and angles [°]: Zn(1)–O(1) 1.9821(16), Zn(1)–N(1) 1.985(2), Zn(1)–C(18) 2.087(2), Zn(1)–N(3) 2.127(2), O(1)–Zn(1)–N(1) 96.37(8), O(1)–Zn(1)–C(18) 107.63(8), N(1)–Zn(1)–C(18) 123.83(10), O(1)–Zn(1)–N(3) 105.16(8), N(1)–Zn(1)–N(3) 94.34(9), C(18)–Zn(1)–N(3) 124.98(8), C(23)–C(18)–Zn(1) 103.59(18). | ||
Catalytic studies
Complexes 1–3 were tested in the ROP of L-lactide (L-LA) at 100 °C in toluene in the absence of any co-initiator. The catalytic activity of each complex was tested with a monomer-to-metal ratio of 200:1 up to 1000:1 (Table 1). The resulting polymers were analysed by 1H NMR spectroscopy, while the Mn values were determined by gel permeation chromatography (GPC). Complex 1 turned out to be catalytically inactive, whereas complex 2 showed a high activity, and more than 90% of the monomer was converted within minutes (Table 1, entries 1–6). The conversion of 200 equivalent of L-LA occurred in 1 min, corresponding to a TOF value of 10800 h−1.
| Entry | Catalyst | [M]o/[Zn]o | Time [min] | Conversionb [%] | Mn(GPC)c [kg mol−1] | Mn(Theo)d [kg mol−1] | Đe | TOFf [h−1] |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: all polymerizations were performed 100 °C in toluene.b Determined by 1H NMR spectroscopy.c Measured by GPC at 40 °C in CHCl3, relative to poly(ethylene oxide) standards.d Mtheon at 100% conversion = [M]0/[Zn]0 × mol. wt of monomer.e Measured by GPC.f TOFs were calculated as (mol of monomer consumed)/(mol of catalyst × time of polymerization).g At ambient temperature (25 °C) in toluene or CH2Cl2. | ||||||||
| 1 | 2 | 200:1 |
1 | 90 | 22.3 | 25.9 | 1.52 | 10800 |
| 2 | 2 | 400:1 |
1.5 | 93 | 37.2 | 53.61 | 1.88 | 14880 |
| 3 | 2 | 600:1 |
2 | 97 | 55.3 | 83.9 | 1.59 | 17460 |
| 4 | 2 | 800:1 |
3 | 90 | 51.5 | 103.8 | 1.47 | 14400 |
| 5 | 2 | 1000:1 |
4 | 80 | 58.2 | 115.3 | 1.47 | 12000 |
| 6 | 2 | 200:1 + 200 |
1 + 1 | 98 | 56.0 | 56.5 | 1.58 | 10855 |
| 7 | 2 | 200:1 + 200 + 200 |
1 + 1 + 4 | 98 | 54.6 | 84.7 | 2.28 | 5880 |
| 8 | 3 | 200:1 |
6 | 99 | <10 | 28.5 | — | 1980 |
| 9 | 3 | 400:1 |
14 | 99 | <10 | 57.1 | — | 1697 |
| 10 | 3 | 600:1 |
300 | 85 | 11.7 | 73.5 | 1.02 | 102 |
| 11 (ref. 36) | Ig | 200:1 |
210 | 94 | 8.1 | 27.1 | 1.4 | 54 |
| 12 (ref. 37) | IIg | 200:1 |
35 | 93 | 27.2 | 26.8 | 1.2 | 318 |
| 13 (ref. 42) | IIIg | 200:1 |
15 | 97 | 31.9 | 27.9 | 1.58 | 776 |
The resulting Mn of 22.3 kg mol−1 is close to the theoretically expected value (25.9 kg mol−1), and a moderate dispersity Đ of 1.52 was obtained. The activity of complex 2 is almost maintained when the monomer-to-metal ratio is increased up to 1000:1. However, the gap between the expected and obtained Mn values of the polymers increased with increasing dilution of the catalyst. Thus, only polymers with a maximum Mn of 58.2 kg mol−1 and moderate Đs (1.47–1.88) were obtained. Moreover, complex 2 showed “living” polymerization character in the ROP of L-LA using a monomer-to-metal ratio of 200:1 in toluene at 100 °C. After full conversion of the initially added L-LA, a second fraction of 200 eq. of L-LA was added, whose turnover occurred after 70 seconds (Table 1, entry 6). The GPC data of the resulting polymer still showed a monomodal peak with a similar Đ compared to the 200:1 obtained polymer (Fig. S15†), while the Mn value of the resulting polymer was higher than that formed with only 200 eq. of L-LA. In marked contrast the addition of a third fraction of 200 eq. of L-LA, which was fully converted within 4 minutes, did not result in an increased Mn value of the resulting polymer (Table 1, entry 7). This indicates that the molecular weight of the resulting cyclic polymer is somehow restricted to >60 kg mol−1, which is supported by the broadened Đ value of 2.28.
We also investigated the catalytic activity of the mono-substituted zinc complex 3 in the ROP of L-LA. In situ 1H NMR spectroscopy showed full conversion of the monomers after 6 minutes for a monomer-to-metal ratio of 200:1 and after 14 minutes for a monomer-to-metal ratio of 400:1. GPC analyses showed that the obtained polymers had a lower molar mass than 10 kg mol−1, which made it difficult to determine the molecular weight distribution qualitatively (Table 1, entries 8 and 9). Also, a tremendous decrease in the catalytic activity of complex 3 was observed at a monomer-to-metal ratio of 600:1, converting 85% of the monomer after 5 hours, yielding a Mn of 11.74 kg mol−1 with a narrow Đ of 1.02 (Table 1, entry 10).
We already noted the enhanced catalytic activity of homobimetallic complexes compared to similar monometallic β-ketoiminate zinc and ketodiiminate zinc complexes.36,37,42 The higher catalytic activity of complex 2 compared to complex 3 most likely results from the presence of the second metal centre, allowing to proceed the polymerization process at two active centres at the same time. Homo- and heterobimetallic catalysts are widely investigated in recent years due to the potential beneficial effect of two metals, which can affect the mechanism, and thus the activity, of the catalyst.44,45 Comparing complex 2 with complex II (Scheme 2), significant higher polymerization rates were observed with complex 2 (TOF = 10800 h−1 (2); 318 h−1 (II)), while the formation of a polymer with higher molecular weight and lower molecular weight distribution was observed with catalyst II, indicating a less controlled polymerization process due to the bulkier Cp moieties. When comparing complex 2 with the similar Mg complex III, it is noticeable that complex III already showed a high catalytic activity (TOF = 776; Table 1, entry 13) under milder reaction conditions (25 °C). The resulting PLLA showed similar Mn and Đ values, but higher molecular weight PLLA was obtained with catalyst III and increasing monomer-to-metal ratio. These findings show that the catalytic activity is not only largely influenced by the metal centre (Zn vs. Mg) but also by the Cp* substituent. The introduction of a Cp*Zn moiety to ketodiiminate complexes yields lower molecular weight polymers.42,46
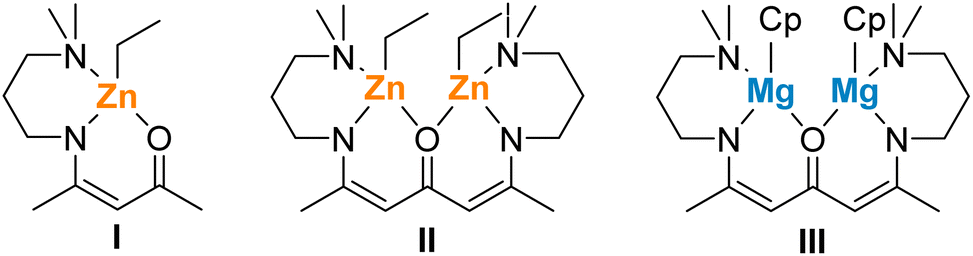 | ||
| Scheme 2 Catalytically active mono- and bimetalic catalysts I, II and III.36,37,42 | ||
Kinetic studies
Kinetic studies with complexes 2 and 3 were performed in toluene at 100 °C with monomer-to-metal ratios [LA]0/[Zn]0 of 200:1 in a J-Young NMR tube. The ln{[M]0/[M]t} ratio was calculated by integration of the Me proton peaks in the 1H NMR spectra from the polymer and the monomer (Fig. 3). We did not observe any induction period in the catalytic studies, and the plots of ln{[M]0/[M]t} versus time were for both complexes linear, showing a first-order dependency of the polymerization rates with respect to the monomer concentration. The apparent rate constants (kapp) were calculated from the slope of the observed plots (3.61 × 10−1 2, 1.47 × 10−1 3), and the homobimetallic complex 2 was found to be more active than the mononuclear complex 3.
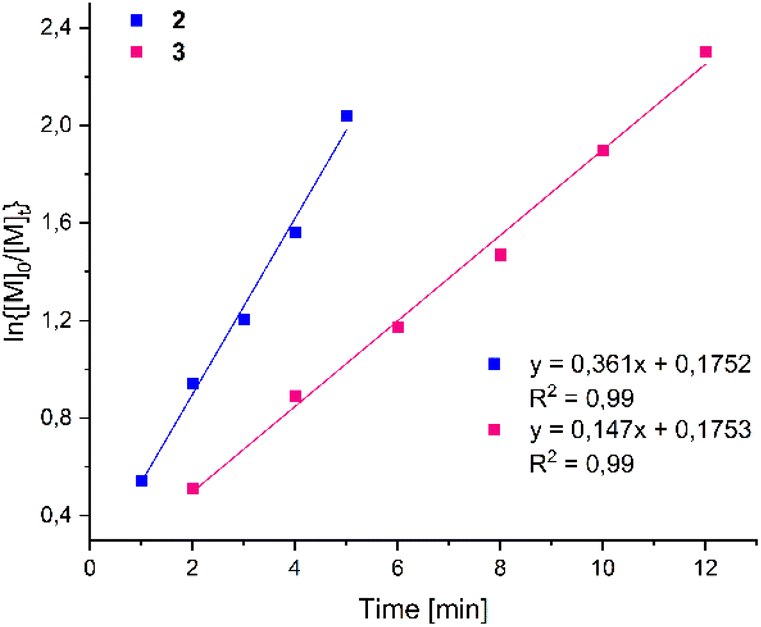 | ||
| Fig. 3 Semi-logarithmic plots of L-LA conversion at 100 °C initiated by complexes 2 and 3 versus time: [L-LA]0/[Zn]0 = 200 in toluene. | ||
In addition to the GPC analysis, the resulting PLLA was also analysed by 1H NMR and IR spectroscopy as well as by matrix-assisted laser desorption ionization time-of-flight (MALDI-ToF) mass spectroscopy. Linear PLLA was formed with the mononuclear zinc complex 3 as was proven by the detection of a hydroxyl (OH) chain-end group using both MALDI-ToF mass and IR spectroscopy (Fig. S22 and S23†).
In marked contrast, the MALDI-ToF spectra of the low-molecular-weight PLLA ([L-LA]:[Cat] = 50:1) obtained with complex 2 showed a peak series, which are equally separated by m/z = 72 au, which indicates the formation of cyclic polymer (72n + 23 is assigned to n(C3H4O2) + Na+, Fig. S20†). The absence of the polymer chain end was proven by 1H NMR and IR spectroscopy (Fig. S19 and S21†), confirming the selective formation of cyclic polylactide (cPLLA). In marked contrast, the formation of linear PLLA was observed with the similar binuclear ketodiiminate zinc complex II, while the ketodiiminate Mg complex III also gave cPLA. These findings indicate a rather strong influence of the Cp ligand on the polymerization mechanism, since the heteroleptic binuclear zinc complexes 2 and II only differ by one substituent (Cp in 2 vs. Et in II).
Since cyclic polymers were only formed with the bimetallic zinc complex 2 and previously reported magnesium complex III as well as its Cp*-substituted analogue, we assume the same polymerization mechanism as reported for the bimetallic ketodiiminate magnesium complexes.42 Both Zn atoms in complex 2 are initially coordinated by one lactide molecule. Nucleophilic attack of the Cp ligand at the electrophilic carbonyl unit of the coordinated lactide causes the ring opening followed by the formation of the catalytic active alkoxide species. The propagation process of the polymer performs through stepwise coordination of a lactide molecule to a zinc centre followed by insertion of an alkoxy group by breaking the Zn–O bond. The formation of the cyclic polymer occurs most likely by an intramolecular transesterification reaction, yielding a limited molecular weight polymer >60 kg mol−1 with moderate molecular weight distributions (Table 1, entries 1–7).42
Experimental
General experimental details
The reactions were performed under standard Schlenk and glovebox techniques under argon atmosphere, which was dried by passing through preheated Cu2O pellets and molecular sieves columns. Toluene and n-hexane were dried using a mBraun Solvent Purification System (SPS) and stored over molecular sieves (3 Å) under argon atmosphere. Deuterated solvents were dried over molecular sieves (3 Å). Karl Fischer titration of the dry solvents showed values below 2 ppm. Amines were commercially available (Sigma-Aldrich) and used as received. L-LA was sublimed three times and stored in a glovebox. ZnCp2 and ZnCp*2 were prepared according to literature methods.47 2,4,6-heptatrione was prepared according to a slightly modified literature procedure.48Materials and methods
1H and 13C NMR spectra were measured at 297 K in CD2Cl2 using a Bruker Avance 300 spectrometer with a QNP probe head (1H: 300 MHz, 13C: 75 MHz) or Bruker Avance 500 (1H: 400 MHz, 13C: 125 MHz) and referenced to the solvent shifts (CD2Cl2: 1H = 5.32 ppm, 13C = 53.84 ppm). IR spectra were determined using an ALPHA-T FT-IR spectrometer equipped with a single reflection ATR sampling module in a glovebox. MALDI-ToF mass spectra were recorded with a Bruker UltrafleXtreme MALDI-ToF mass spectrometer (Bruker Daltonik). The polymers were dissolved in THF (10 mg mL−1) using dihydroxy benzoic acid as the matrix in THF (20 mg mL−1). The mass spectra of the polymer samples were received using the reflective positive ion mode. An externally calibration was done using poly(methylmethacrylate) standards. Microanalyses were observed with a PerkinElmer Series 11 analyzer by the Elementaranalyse Labor of the University of Duisburg-Essen. Melting points were determined in glass capillaries sealed with grease and are not corrected.000 g mol−1 and evaluated with the WinGPC UniChrom software. For preparation, polymers were dissolved in CHCl3 (2 mg mL−1) and passed through a syringe filter with a pore size of 0.2 μm.[L1H2]. Yield: 8.44 g (85%). Mp: 64 °C. Anal. calc. for C15H30N4O: C, 63.81; H, 10.71; N, 19.83; Found: C, 63.8; H, 10.8; N 20.3; 1H NMR (300 MHz, CD2Cl2, 300 K): δ = 1.86 (s, 6H, NCCH3), 2.24 (s, 12H, N(CH3)2), 2.41 (t, 3JHH = 6.6 Hz, 4H, CH2N(CH3)2), 3.23–3.29 (m, 4H, CNCH2), 4.60 (s, 2H, OCCH), 9.99 (s, 2H, NH). 13C NMR (75.5 MHz, CD2Cl2, 300 K): δ = 19.6 (NCCH3), 41.5 (CH2N(CH3)2), 45.9 (CH2N(CH3)2), 60.2 (NCH2CH2), 95.8 (OCCH), 158.6 (CN), 190.2 (CO). ATR IR: ν = 2963, 2928, 2848, 2802, 2751, 1616, 1559, 1430, 1378, 1267, 1156, 1088, 1059, 1010, 857, 832, 786, 681, 620, 540 cm−1.
[L2H2]. Yield: 9.82 g (90%). Mp: 70 °C. Anal. calc. for C17H34N4O: C, 65.76; H, 11.04; N, 18.05. Found: C, 65.90; H, 11.26; N 18.35. 1H NMR (300 MHz, CD2Cl2, 300 K): δ = 1.62–1.71 (m, 4H, NCH2CH2), 1.86 (s, 6H, (NCCH3)2), 2.17 (s, 12H, N(CH3)2), 2.28 (t, 3JHH = 6.9 Hz, 4H, CH2N(CH3)2), 3.18–3.24 (m, 4H, CNCH2), 4.59 (s, 2H, OCCH), 10.08 (s, 2H, NH). 13C NMR (75.5 MHz, CD2Cl2, 300 K): δ = 19.4 (NCCH3), 29.4 (NCH2CH2), 41.3 (CH2N(CH3)2), 45.8 (CH2N(CH3)2), 57.3 (NCH2CH2), 95.5 (OCCH), 159.2 (CN), 190.3 (CO). ATR IR: ν 2963, 2928, 2848, 2802, 2751, 1616, 1559, 1430, 1378, 1267, 1156, 1088, 1059, 1010, 857, 832, 786, 681, 620, 540 cm−1.
L12Zn2 (1). Either ZnCp2 (277 mg, 1.42 mmol) or ZnCp*2(476 mg, 1.42 mmol), L1H2 (200 mg, 0.71 mmol). Yield: 155 mg (43%). Mp 210 °C (dec.). Anal. calc. for C30H56N8O2Zn2: C, 52.10; H, 8.16; N, 16.20. Found: C, 53.01; H, 8.02; N, 16.18. 1H NMR (400 MHz, C6D6, 300 K): δ = 1.89 (s, 12H, (NCCH3)2), 2.19 (s, 24H, N(CH3)2), 2.27–2.30 (br, 8H, CH2N(CH3)2), 3.09–3.15 (m, 3JHH = 6 Hz, 4H, NCH2CH2), 3.28–3.37 (m, 3JHH = 7 Hz, 4H, NCH2CH2), 4.82 (s, 4H, OCCH). 13C NMR (150 MHz, C6D6, 300 K): δ = 22.8 (NCCH3), 47.5 (CH2N(CH3)2), 52.1 (CH2N(CH3)2), 61.1 (NCH2CH2), 93.5 (OCCH), 166.8 (CN), 180.8 (CO). ATR IR: ν = 2950, 2908, 2855, 2844, 2716, 1626, 1533, 1464, 1411, 1398, 1326, 1287, 1268, 1253, 1241, 1204, 1179, 1096, 1080, 1020, 1003, 937, 860, 780, 728, 709, 644, 617, 556, 505, 463, 455, 443, 414 cm−1.
L2Zn2Cp2 (2). ZnCp2 (200 mg, 1.02 mmol), L2H2 (160 mg, 0.51 mmol). Yield: 275 mg (95%). Mp. 140 °C (dec.). Anal. calc. for C27H42Zn2N4O: C, 56.95; H, 7.42; N, 9.86. Found: C, 54.93; H, 7.41; N, 9.89. 1H NMR (300 MHz, CD2Cl2, 300 K): δ = 1.39–1.43 (m, 3JHH = 5.4 Hz, 4H, NCH2CH2), 1.74 (s, 6H, (NCCH3)2), 2.46 (s, 12H, N(CH3)2), 2.49 (br, 4H, CH2N(CH3)2), 3.05 (br, 2H, CNCH2), 4.23 (s, 2H, OCCH), 6.31 (s, 10H, C5H5). 13C NMR (75.5 MHz, CD2Cl2, 300 K): δ = 21.4 (NCCH3), 28.3 (NCH2CH2), 47.4 (CH2N(CH3)2), 49.8 (CH2N(CH3)2), 62.8 (NCH2CH2), 93.9 (OCCH), 109.7 (C5H5), 165.1 (CN), 176.2 (CO). ATR IR: ν = 3061, 2992, 2905, 2864, 2834, 1558, 1479, 1444, 1408, 1398, 1378, 1328, 1302, 1251, 1224, 1171, 1148, 1101, 1084, 1055, 1034, 1020, 998, 980, 906, 864, 837, 812, 781, 749, 720, 656, 617, 583, 556, 475, 443, 411 cm−1.
L2HZnCp* (3). ZnCp*2 (200 mg, 0.6 mmol), L2H2 (93 mg, 0.3 mmol). Yield: 75 mg (49%). Mp 210 °C. Anal. calc. for C27H48ZnN4O: C, 63.58; H, 9.49; N 10.98. Found: C, 64.22; H, 9.86; N, 10.11. 1H NMR (300 MHz, CD2Cl2, 300 K): δ = 1.42–1.49 (m, 3JHH = 5.4 Hz, 2H, NCH2CH2), 1.65–1.73 (m, 3JHH = 6 Hz, 2H, NCH2CH2), 1.82 (s, 3H, CNCH3), 1.87 (s, 3H, CNCH3), 1.89 (s, 15H, C5(CH3)5), 2.07 (s, 6H, N(CH3)2), 2.18 (s, 6H, N(CH3)2), 2.22–2.26 (t, 3JHH = 6 Hz, 2H, CH2N(CH3)2), 2.29–2.34 (t, 3JHH = 7.5 Hz, 2H, CH2N(CH3)2), 3.18–3.25 (q, 2H, 3JHH = 7 Hz, CNCH2), 3.28–3.31 (q, 2H, 3JHH = 4.5 Hz, CNCH2), 4.34 (s, 1H, OCCH), 4.44 (s, 1H, OCCH). 13C NMR (75.5 MHz, CD2Cl2, 300 K): δ = 12.3 (C5(CH3)5), 19.9 (NCCH3), 20.9 (NCCH3), 27.9 (NCH2CH2), 30.0 (NCH2CH2), 41.8 (NCH2CH2), 45.9 (CH2N(CH3)2), 47.6 (CH2N(CH3)2), 49.3 (NCH2CH2), 57.7 (CH2N(CH3)2), 62.0 (CH2N(CH3)2), 95.1 (OCCH), 96.0 (OCCH), 113.7 (C5(CH3)5), 155.1 (CN), 168.7 (CN), 182.2 (CO). ATR IR: ν = 3286, 2944, 2929, 2903, 2843, 2810, 2785, 2758, 1613, 1546, 1498, 1456, 1436, 1400, 1329, 1306, 1282, 1259, 1228, 1195, 1151, 1098, 1060, 1007, 971, 942, 896, 861, 837, 783, 762, 720, 645, 577, 477, 442 cm−1.
Conclusions
Ketodiiminate Zn complexes 1–3 were synthesized and the catalytic activity of the dinuclear (2) and mononuclear (3) heteroleptic complexes in the ROP of L-lactide studied. The highest catalytic activity was observed with complex 2, which selectively gave cyclic polylactide (cPLA) with low molecular weight >60 kg mol−1 and moderate Đ (1.47–1.88). In contrast, linear PLLA was obtained with complex 3. The polymerization reactions with complexes 2 and 3 is expected to proceed via coordination insertion mechanism (CIM), whereas cPLA is formed by cyclization of the polymer chains through intramolecular transesterification in case of complex 2.Author contributions
E. G. and L. K. performed the experiments including catalytic studies, C. W. the single crystal X-ray diffraction, and M. S. the GPC experiments. The work was supervised by A. H. G. and St. S. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, INST 20876/282-1 FUGG) and the University of Duisburg-Essen. D. C. and A. H. G. are also grateful for financial support from Evonik Industries and from the DFG through the Emmy Noether Programm (No. 376920678).Notes and references
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS PubMed.
- A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836 RSC.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523 CrossRef CAS PubMed.
- Synthesis Structure and Properties of Poly(lactid acid), ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, 2018, p. 303 Search PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC.
- J. Ahmed and S. K. Varshney, Int. J. Food Prop., 2011, 14, 37 CrossRef CAS.
- K. Van de Velde and P. Kiekens, Polym. Test., 2002, 21, 433 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217 CrossRef CAS.
- P. B. Yang, M. G. Davidson, K. J. Edler and S. Brown, Biomacromolecules, 2021, 22, 3649 CrossRef CAS PubMed.
- N. Halonen, P. S. Palvöglyi, A. Bassani, C. Fiorentini, R. Nair, G. Spigno and K. Kordas, Front. Mater., 2020, 7, 82 CrossRef.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251 CrossRef CAS.
- H. W. Beckham, Ring Polymers: Effective Isolation and Unique Properties, in Complex Macromolecular Architectures: Synthesis, Characterization, and Self-Assembly, ed. N. Hirao, A. Tezuka and F. Du Prez, Wiley, 2011, p. 791 Search PubMed.
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892 CrossRef CAS.
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585 CrossRef CAS PubMed.
- E. J. Shin, A. E. Jones and R. M. Waymouth, Macromolecules, 2012, 45, 595 CrossRef CAS.
- A. D. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627 CrossRef PubMed.
- N. Zaldua, R. Lienard, T. Josse, M. Zubitur, A. Mugica, A. Iturrospe, A. Arbe, J. De Winter, O. Coulembier and A. J. Müller, Macromolecules, 2018, 51, 1718 CrossRef CAS.
- N. Mase, Y. S. Moniruzzaman, Y. Nakaya, K. Sato and T. Narumi, Polymers, 2018, 10, 713 CrossRef PubMed.
- A. Meyer, S. M. Weidner and H. R. Kricheldorf, Polym. Chem., 2019, 10, 6191 RSC.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202 RSC.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 749 CrossRef CAS.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Möck, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046 CrossRef CAS PubMed.
- T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2004, 2504 RSC.
- A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881 RSC.
- R. W. F. Kerr, P. M. D. A. Ewing, S. K. Raman, A. D. Smith, C. K. Williams and P. L. Arnold, ACS Catal., 2021, 11, 1563 CrossRef CAS.
- S. Ghosh, D. Chakraborty and B. Varghese, Eur. Polym. J., 2015, 62, 51 CrossRef CAS.
- K. Devaine-Pressing, F. J. Oldenburg, J. P. Menzel, M. Springer, L. N. Dawe and C. M. Kozak, Dalton Trans., 2020, 49, 1531 RSC.
- X. Li, Z. Jia, X. Pan and J. Wu, Chem.–Asian J., 2019, 14, 662 CrossRef CAS PubMed.
- Y. Wang, W. Zhao, X. Lui, D. Cui and E. Y.-X. Chen, Macromolecules, 2012, 45, 6987 Search PubMed.
- M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276 RSC.
- Y. Sarazin, B. Lui, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069 CrossRef CAS PubMed.
- T. J. J. Whitehorne, B. Vabre and F. Schaper, Dalton Trans., 2014, 43, 6339 RSC.
- F. Drouin, P. O. Oguadinma, T. J. J. Whitehorne, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139 CrossRef CAS.
- S. Ghosh, P. M. Schäfer, D. Dittrich, C. Scheiper, P. Steiniger, G. Fink, A. N. Ksiazkiewicz, A. Tjaberings, C. Wölper, A. H. Gröschel and S. Schulz, ChemistryOpen, 2019, 8, 951 CrossRef CAS.
- C. Scheiper, D. Dittrich, C. Wölper, D. Bläser, J. Roll and S. Schulz, Eur. J. Inorg. Chem., 2014, 13, 2230 CrossRef.
- S. Ghosh, Y. Schulte, C. Wölper, A. Tjaberings, A. H. Gröschel, G. Haberhauer and S. Schulz, Organometallics, 2022, 41, 2698 CrossRef CAS.
- B. Theron, V. Vaillant-Coindrad, C. Balan, Y. Rousselin, J. Bayardon, R. Malacea-Kabbara and P. Le Gendre, Dalton Trans., 2023, 52, 7854 RSC.
- M. Fuchs, S. Schmitz, P. M. Schäfer, T. Secker, A. Metz, A. N. Ksiazkiewicz, A. Pich, P. Kögerler, K. Yu. Monakhov and S. Herres-Pawlis, Eur. Polym. J., 2020, 122, 109302 CrossRef CAS.
- I. D'Auria, V. Ferrara, C. Tedesco, W. Kretschmer, R. Kempe and C. Pellecchia, Appl. Polym. Mater., 2021, 8, 4035 CrossRef.
- Z. Peng, G. Xu, R. Yang, X. Guo, H. Sun and Q. Wang, Eur. Polym. J., 2022, 180, 111571 CrossRef CAS.
- E. Glöckler, S. Ghosh, C. Wölper, D. Coban, A. H. Gröschel and S. Schulz, Polyhedron, 2022, 222, 115918 CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186 CrossRef.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35 CrossRef CAS.
- L.-J. Wu, W. Lee, P. K. Ganta, Y.-L. Chang, Y.-C. Chang and H.-Y. Chen, Coord. Chem. Rev., 2023, 475, 214847 CrossRef CAS.
- P. Steiniger, P. M. Schäfer, C. Wölper, J. Henkel, A. N. Ksiazkiewicz, A. Pich, S. Herres-Pawlis and S. Schulz, Eur. J. Inorg. Chem., 2018, 4014 CrossRef CAS.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693 CrossRef CAS PubMed.
- M. W. Stoddart, J. H. Brownie, M. C. Baird and H. L. Schmider, J. Organomet. Chem., 2005, 690, 3440 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 1990, A46, 467 CrossRef CAS.
- G. M. Sheldrick, SHELXL-2017, Program for the Refinement of Crystal Structures. University of Göttingen, Göttingen, Germany, 2017, See also: CrossRef PubMed; G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. shelXle, A Qt GUI for SHELXL, Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A CIF file giving X-ray crystallographic data of complexes 2 and 3; 1H and 13C NMR spectra, IR spectra of complexes 2 and 3 as well as MALDI-TOF mass spectra and the crystallographic details of complexes 2 and 3 including structural plots (thermal ellipsoid style). CCDC 2269252 (2) and 2269253 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06529d |
| This journal is © The Royal Society of Chemistry 2023 |