DOI:
10.1039/D3RA05867K
(Paper)
RSC Adv., 2023,
13, 29866-29878
N-(2-(Diphenylphosphino)ethyl)-2-alkyl-5,6,7,8-tetrahydro-quinolin-8-amines iron(II) complexes: structural diversity and the ring opening polymerization of ε-caprolactone†
Received
28th August 2023
, Accepted 27th September 2023
First published on 12th October 2023
Abstract
A series of N-(2-(diphenylphosphino)ethyl)-2-alkyl-5,6,7,8-tetrahydroquinolin-8-amines was prepared and used in individually reacting with iron chloride under nitrogen atmosphere to form their iron(II) complexes Fe1–Fe6. All compounds were characterized using FT-IR spectroscopy and elemental analyses, the organic compounds were confirmed with NMR measurements, and the iron complexes were submitted to single-crystal X-ray diffraction, revealing Fe1, Fe2, Fe4, Fe5, and Fe6 as either mono- or di-nuclear forms. Forming a binary system in situ with two equivalents of LiCH2SiMe3, all iron complexes Fe1–Fe6 efficiently initiated the ring opening polymerization of ε-caprolactone, achieving the TOF up to 8.8 × 103 h−1. More importantly, the resultant polycaprolactone (PCL) possessed high molecular weights with the Mn range of 9.21–24.3 × 104 g mol−1, being a rare case of the iron(II) catalyst in producing PCL with such high molecular weight. The 1H NMR and MALDI-TOF investigations demonstrated that the PCLs were linear features capped with a methoxy group or CH2SiMe3 or cyclic structure that varied with the molar ratio of [ε-CL]/Fe.
1. Introduction
Targeting environment-friendly materials with good biocompatibility and biodegradability, aliphatic polyesters, such as polylactide (PLA) and polycaprolactone (PCL), have attracted considerable attention in the past decades.1,2 In that, organometallic compounds have been extensively explored as conveniently effective catalysts to the ring opening polymerization (ROP) of their cyclic esters;3–7 meanwhile, its industrial catalyst commonly employs tin(II) octanoate, operated at a relatively higher temperature and producing the broader dispersive polyesters along with the residue of tin, which remains in the polymer, making it potentially harmful to mammals and/or ocean species.8 To develop non-toxic catalysts, the most abundant transition-metal iron-based catalysts have been found to be potential catalysts for the ROP of cyclic esters9,10 and are biocompatible.11–13 Interestingly, valence-variable iron catalysts have been found in redox-controlled ROP,14 iron salts, such as FeCl3, Fe2O3, and FeS, which displayed low efficiency toward ROP of lactides or ε-caprolactone (ε-CL) in bulk polymerization;15–17 in contrast, the FeCl3 hydrate salts are efficient catalysts toward the bulk polymerization of ε-CL, δ-valerolactone (δ-VL), and β-butyrolactone (β-BL).18 Employing initiators (water, isopropyl alcohol, benzyl alcohol, and 2-allyl phenol), commercial iron(III) salts, such as FeCl3, FeBr3, and perchlorate, were found to be efficient for the ROP of ε-CL.19 Organic iron salts, such as carboxylates, acetate, or porphyrins, showed very sluggish bulk polymerization of L-LA even at high temperature (120–210 °C, hours or days).20 Its ferric alkoxides, Fe5-(μ5-O)(OEt)13 and Fe2(OCMe2Ph)6, efficiently promoted the ROP of LA in a controllable manner, such as [LA]/[Fe] of 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 97% conversion in 21 min along with obtaining PLA with PDI as 1.17;21 moreover, other ferric alkoxides, Fe2(OCHPh2)6 and L2FeOCHPh2 (L as N,N′-bis(trimethylsilyl)benzamidinate), also efficiently achieved the ROP of ε-CL and rac-LA.22 However, the calixarene heteronuclear ferrous complexes bearing Fe–OAr bonds showed low efficiency toward ROP of ε-CL (requiring 41 h for 99% conversion at molar ratio of [ε-CL]/[Fe] = 700:1).23
:1 with 97% conversion in 21 min along with obtaining PLA with PDI as 1.17;21 moreover, other ferric alkoxides, Fe2(OCHPh2)6 and L2FeOCHPh2 (L as N,N′-bis(trimethylsilyl)benzamidinate), also efficiently achieved the ROP of ε-CL and rac-LA.22 However, the calixarene heteronuclear ferrous complexes bearing Fe–OAr bonds showed low efficiency toward ROP of ε-CL (requiring 41 h for 99% conversion at molar ratio of [ε-CL]/[Fe] = 700:1).23
Additionally, there are more iron complexes bearing various ligands explored for the ROP of cyclic esters,24–46 based on the chelating models of ligands used, which have been clarified with as monodentate (N-heterocyclic carbene, NHC),24 bidentate (N^N25–30 or N^O31,32), tridentate (N^N^N,33–35 N^N^C,36 N^O^O37,38), and tetradentate (N^N^N^N,39,40 N^N^O^O,41–45 N^N^N^O46). The N-heterocyclic carbene–iron complexes showed excellent activities, especially in the bulk polymerization of lactide. As an example, the complex 1 (Chart 1) produced PLA with molecular weight up to 50 kg mol−1 and narrow dispersity 1.6 at the molar ratio of lactide/iron of 10000:1;24 its polymerization rate constant kapp is up to 8.5 × 10−3 s−1, being an order of magnitude higher than that of the industrial Sn(Oct)2 system. The 4-arylimino-1,2,3-trihydroacridines N,N-bidentate iron(II) complexes (2, Chart 1) displayed high efficiency toward the ROP of ε-CL with the activation of LiCH2SiMe3 under mild conditions;30 meanwhile, the guanidine–iron complexes showed excellent activity for the ROP of lactides,31,32 also surpassing the performance of the Sn(Oct)2 system. In addition to N,N-bidentate ligands, N,O-bidentate ligands were useful; the N,O-iron complexes 3 (Chart 1) could polymerize both rac-LA and L-LA into long-chain polylactide in bulk with [M]/[I] ratios more than 5000:1.31 Bis(imino)pyridiyl–iron complexes with different oxidation states, 4 and 5 (Chart 1), similarly performed in the selective ROP of rac-LA,33 in which loading 0.2 mol% 4 (Fe(II), R = neopentyl), the conversion rate reached 94% after 10 min; meanwhile, the ROP of ε-CL by 5 (Fe(I), R = neophentyl) was achieved with 100% conversion within 10 min at 0.05 mol% catalyst loading. However, the highly oxidative iron(III) compound was inactive toward the ROP of rac-LA.35 The tridentate N^C^N iron(II) complex 6 showed a good activity toward the ROP of rac-LA, producing high molecular weight PLA (Mn = 3.5 × 105 g mol−1) and narrow dispersity (PDI = 1.2) with 85% conversion at the [LA]/[Fe] ratio of 5000:1;36 N^N^N 2,6-dipyrazolylpyridyliron(III) complex 7 (Chart 1) showed a good ROP of ε-CL, forming PCL with narrower dispersity (PDI as 1.18) with the [ε-CL]/[Fe] molar ratio of 300:1;34 in contrast, its analogue iron(II) complex was completely inactive toward the ROP of ε-CL.34 Using N^N^N^O tetradentate ligands, the tripodal ligated iron(III) complex 8 showed both high activity and stereoselectivity toward the ROP of rac-LA under mild conditions,46 extensively the N^N^O^O tetradentate Salen-iron(III) chlorides 9, which exhibited high catalytic activities toward the ROP of both lactide or ε-CL when using propylene oxide (PO) as the solvent, reaching 98% monomer conversion at the [ε-CL]/[Fe] molar ratio of 1000:1.44 Moreover, O^S^S^O-iron(III) complex (10, Chart 1) also efficiently promoted the ROP of both rac-LA and ε-CL using cyclohexene oxide (CHO) as the solvent, for example, at the [rac-LA]/[Fe] molar ratio of 10000:1, the 9700 h−1 turnover number (TON) was observed in 52 h.47
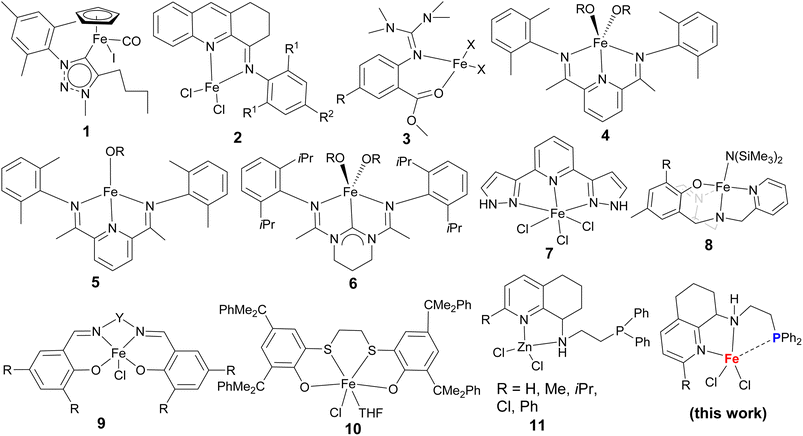 |
| | Chart 1 Efficient iron complexes for the ring opening polymerization of cyclic esters. | |
Pondering over the iron complex catalysts, the chelating heteroatoms are based on the hard biting atoms such as oxygen, carbon, or nitrogen donors. The ligands including soft phosphines have been commonly employed, for example, for the asymmetric transfer hydrogenation of ketones and imines in our group.48 However, there is no report of the iron complexes with P coordination for ROP of cyclic esters yet. Back in 1998, Dubois group reported enhanced polymerization with the Sn(Oct)2 system by the addition of triphenylphosphine, illustrating the positive influence of soft phosphine donor.49 Besides that, the phosphine-iminoquinoline iron(II) chlorides were reported to copolymerize ethylene with 1-hexene.50 Relying on the easily synthesized phosphorus groups,51,52 the N^N^P ligands were prepared for their zinc chlorides 11 (in Chart 1), which exhibited exceptionally high activity toward the ROP of ε CL and achieved its TOF 1.35 × 105 h−1 at a high [ε-CL]/[Zn] ratio of 5000:1.53 Subsequently, their iron(II) complexes (Chart 1) were prepared and used for the ROP of ε-CL in the presence of LiCH2SiMe3. Surprisingly, the outstanding activities have been observed herein for PCL production with unique high molecular weights.
2. Experimental section
2.1 General procedures
All operations were carried out under high purity nitrogen atmosphere using standard Schlenk or glove box techniques. Toluene, THF, n-hexane, and diethyl ether were dried by refluxing over sodium/benzophenone, distilling under nitrogen, and storing over activated molecular sieves (4 Å) for 24 h in a glove box prior to use. Dichloromethane was dried with CaH2. Diphenylphosphine was purchased from energy chemicals and used as received. 2-Chloroethylamine-hydrochloride and t-BuOK were purchased from Innochem, while LiCH2SiMe3 (0.55 M in n-hexane), while ultra-dried 1,2-dichloroethane and ε-CL were purchased from J&K Scientific. ε-CL was stirred over CaH2 for 24 h and used after vacuum distillation. NMR spectra were recorded on a Bruker DMX-400 instrument using TMS as an internal standard. IR spectra were recorded on a PerkinElmer System 2000 FT-IR spectrometer. Elemental analyses were carried out using a Flash EA 1112 microanalyzer. MALDI-TOF MS analysis was performed with a Bruker Ultraflex mass spectrometer. For MALDI MS analysis, mass spectra were acquired with a SmartBeam laser (355 nm) operating at 200 Hz and a laser focus of 50 μm. The device parameters for MALDI MS were chosen as follows: plate offset voltage, 19 kV; deflector detector voltage, 20 kV. Data were processed using DateAnalysis 3.0 (Bruker Daltonics). The GPC measurements were performed using a system composed of a 390-LC multidetector (MDS), 209-LC pump injection module (PIM), and a PL-GPC 50 plus instrument, with THF as the eluent (flow rate: 1 mL min−1, at 40 °C). Polystyrene was used as the standard to calculate the molecular weights and molecular weight distributions. L1–L4 were prepared according to the literature.53
2.1.1 2-(Diphenylphosphino)ethanamine. A mixture of potassium tert-butoxide (12.03 g, 105 mmol) and diphenylphosphine (9.63 g, 52.6 mmol) was added to a 250 mL double necked round bottom flask containing 100 mL THF and stirred for 1 h at ambient temperatures to form a red solution. 2-Chloroethylamine-hydrochloride (6.6 g, 55.6 mmol) was added to the reaction solution and refluxed at 80 °C for 20 h. The color of the solution gradually changed from red to yellow and finally to white. After removing the solvent, 10% HCl was added to make the solution acidic, washed three times with toluene, 10% NaOH was added to make the solution alkaline, extracted three times with toluene, and the final organic layer was washed three times with saturated NaCl. The extract was dried over MgSO4, filtered, and the solvent was removed under vacuum to give a yellow viscous product (9.32 g, 77%). 1H NMR: (400 MHz, CDCl3, TMS): δ 7.47–7.38 (m, 4H), 7.35–7.28 (m, 6H), 2.88–2.80 (m, 2H), 2.24 (t, J = 8.0 Hz), 1.57 (s, 2H). 13C NMR (100 MHz, CDCl3, TMS): δ 137.16, 13.64, 132.81, 132.62, 130.78, 130.66, 128.96, 128.84, 128.77, 128.58, 128.51, 38.81, 38.59, 30.91, 30.78. 31P NMR (162 MHz, CDCl3, TMS): δ −22.03.
2.1.2 N-(2-(Diphenylphosphino)ethyl)-2-mesityl-5,6,7,8-tetrahydroquinolin-8-amine (L5): synthesis of ligands L5–L7. Following the processes reported in the literature,53,54 2-mesityl-6,7-dihydroquinolin-8(5H)-one (2.65 g, 10 mmol), 2-(diphenylphosphino)ethanamine (2.98 g, 13 mmol), and sodium triacetoxyborohydride (4.45 g, 21 mmol), dissolved in 50 mL of 1,2-dichloroethane, were added to a 200 mL Schlenk flask and stirred at room temperature for 6 h. During the reaction, the solid disappeared gradually and the color of the solution was yellow. After the reaction, the mixture was quenched by saturated NaHCO3, the yellow organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The crude product was dried over MgSO4, and the solvent was removed using a rotary evaporator. Pure yellow oily product was obtained by basic alumina column chromatography with petroleum ether/ethyl acetate (1/1, v/v). Yield: 1.91 g, 40%. 1H NMR: (400 MHz, CDCl3, TMS): δ 7.81–7.64 (m, 1H), 7.52–7.38 (m, 5H), 7.36–7.25 (m, 5H), 6.98 (d, J = 7.7 Hz, 1H), 6.93 (s, 2H), 3.81 (t, J = 4.0 Hz, 1H), 2.95–2.72 (m, 4H), 2.38–2.25 (m, 5H), 2.11 (s, 1H), 2.05–1.94 (m, 8H), 1.82–1.66 (m, 2H). 13C NMR (100 MHz, CDCl3, TMS): δ 156.80, 137.80, 137.30, 137.07, 135.94, 132.89, 132.70, 132.67, 132.49, 130.06, 128.68, 128.56, 128.43, 128.40, 128.36, 128.33, 122.88, 57.91, 44.55, 44.32, 28.68, 28.47, 21.05, 20.37, 19.77. 31P NMR (162 MHz, CDCl3, TMS): δ −20.25. FT-IR (cm−1): 3305 (w), 3054 (w), 3007 (w), 2926 (m), 2858 (w), 1610 (w), 1586 (w), 1564 (m), 1456 (s), 1432 (s), 1382 (w), 1342 (w), 1259 (m), 1184 (w), 1148 (w), 1099 (m), 1024 (m), 850 (m), 802 (m), 738 (s), 694 (s). Anal. calcd for C32H35N2P (1/6 CH2Cl2): C, 78.40; H, 7.23; N, 5.68. Found: C, 78.41; H, 7.39; N, 5.56.
2.1.3 N-(2-(Diphenylphosphino)ethyl)-2-(2,4,6-triisopropyl-phenyl)-5,6,7,8-tetrahydroquinolin-8-amine (L6). Using the procedure similar to that described for L5, L6 was obtained as a yellow oily product (4.75 g, 84%). 1H NMR: (400 MHz, CDCl3, TMS): δ 7.48–7.37 (m, 5H), 7.35–7.26 (m, 6H), 7.08 (d, J = 2.0 Hz, 2H), 7.04 (d, J = 7.7 Hz, 1H), 3.80 (t, J = 5.6 Hz, 1H), 3.02–2.72 (m, 5H), 2.67–2.45 (m, 2H), 2.29 (t, J = 7.8 Hz, 2H), 2.12–1.98 (m, 2H), 1.86–1.68 (m, 2H), 1.31 (d, J = 6.9 Hz, 6H), 1.12–1.02 (m, 12H). 13C NMR (100 MHz, CDCl3, TMS): δ 156.98, 156.63, 148.48, 146.58, 146.26, 138.72, 138.56, 138.43, 136.49, 136.42, 132.87, 132.68, 132.63, 132.44, 129.97, 128.53, 128.41, 128.37, 128.34, 128.30, 123.28, 120.79, 120.68, 57.79, 44.62, 44.39, 34.36, 30.23, 30.21, 29.16, 29.04, 28.73, 28.41, 24.29, 24.21, 24.08, 24.05, 19.53. 31P NMR (162 MHz, CDCl3, TMS): δ −20.38. FT-IR (cm−1): 3312 (w), 3051 (w), 2957 (s), 2925 (m), 2865 (m), 1608 (w), 1591 (w), 1564 (m), 1456 (s), 1432 (s), 1381 (m), 1361 (m), 1313 (w), 1260 (w), 1153 (w), 1064 (m), 1025 (m), 994 (w), 875 (m), 737 (s), 694 (s). Anal. calcd for C38H47N2P (1/10 CH2Cl2): C, 80.10; H, 8.33; N, 4.90. Found: C, 80.35; H, 8.41; N, 4.92.
2.1.4 N-Butyl-5,6,7,8-tetrahydroquinolin-8-amine (L7). Using the procedure similar to that described for L5, L7 was obtained as a red oily liquid (1.24 g, 61%). 1H NMR: (400 MHz, CDCl3, TMS): δ 8.37 (d, J = 4.3 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.05–6.98 (m, 1H), 3.75 (t, J = 6.2 Hz, 1H), 2.85–2.63 (m, 4H), 2.53 (s, 1H), 2.18–1.89 (m, 2H), 1.81–1.64 (m, 2H), 1.61–1.47 (m, 2H), 1.46–1.33 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 157.73, 146.79, 136.73, 132.32, 121.63, 63.22, 58.16, 47.56, 32.57, 28.89, 28.72, 20.61, 19.61, 13.99.
2.1.5 N-Butyl-2-mesityl-5,6,7,8-tetrahydroquinolin-8-amine (L8). Using the procedure similar to that described for L7, L8 was obtained as a red oily liquid (1.9 g, 60%). 1H NMR: (400 MHz, CDCl3, TMS): δ 7.31 (d, J = 7.8 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.83 (m, 2H), 3.73 (t, J = 6.0 Hz, 1H), 2.83–2.50 (m, 5H), 2.23 (d, J = 8.5 Hz, 3H), 2.15–2.04 (m, 1H), 1.95 (s, 6H), 1.80–1.63 (m, 2H), 1.50–1.35 (m, 2H), 1.33–1.21 (m, 2H), 0.81 (t, J = 7.3 Hz, 3H).
2.2 Synthesis of iron(II) complexes Fe1–Fe8
2.2.1 Synthesis of Fe1. In the glove box, L1 (0.39 g, 1.08 mmol), dissolved in 10 mL THF, was added to a 50 mL Schlenk flask. FeCl2·4H2O (0.21 g, 1.08 mmol) dissolved in 5 mL THF was then added dropwise to the ligand solution, and the mixture was stirred overnight. After that, the solution was concentrated under vacuum, and diethyl ether was added to form a precipitate. The precipitate was filtered and washed with diethyl ether to obtain a yellow solid. Yield: 0.36 g, 69%. FT-IR (cm−1): 3223 (w), 3049 (w), 2938 (w), 2862 (w), 1621 (w), 1587 (m), 1481 (m), 1435 (s), 1366 (w), 1321 (m), 1278 (w), 1209 (w), 1182 (w), 1128 (m), 1081 (s), 1029 (w), 998 (w), 934 (w), 905 (w), 873 (m), 794 (m), 746 (s), 696 (s). Anal. calcd for C23H25Cl2FeN2P (2/3 CH2Cl2): C, 52.27; H, 4.88; N, 5.15. Found: C, 52.56; H, 4.91; N, 5.28.
2.2.2 Fe2. Using a procedure similar to synthesize Fe1, Fe2 was synthesized as a yellow powder (0.44 g, 80%). FT-IR (cm−1): 3125 (m), 3057 (w), 2951 (w), 2865 (w), 1597 (w), 1573 (w), 1475 (m), 1433 (m), 1409 (w), 1346 (w), 1309 (w), 1258 (w), 1190 (w), 1092 (m), 1028 (m), 966 (m), 941 (w), 817 (w), 744 (s), 694 (s). Anal. calcd for C24H27Cl2FeN2P (2/11 CH2Cl2): C, 56.22; H, 5.34; N, 5.42. Found: C, 56.59; H, 5.42; N, 5.49.
2.2.3 Fe3. Using a procedure similar to synthesize Fe1, Fe3 was prepared as a yellow powder (0.39 g, 71%). FT-IR (cm−1): 3174 (m), 3054 (w), 2936 (w), 2865 (w), 1598 (w). 1571 (m), 1478 (m), 1433 (m), 1405 (m), 1354 (w), 1312 (w), 1270 (w), 1214 (w), 1184 (w), 1094 (m), 1049 (m), 1024 (m), 968 (m), 939 (m), 861 (m), 828 (w), 745 (s), 696 (s). Anal. calcd for C26H31Cl2FeN2P (1/4 CH2Cl2): C, 57.27; H, 5.77; N, 5.09. Found: C, 57.19; H, 5.49; N, 5.42.
2.2.4 Fe4. Using a procedure similar to synthesize Fe1, Fe4 was prepared as a brown powder (0.53 g, 78%). FT-IR (cm−1): 3185 (m), 3049 (w), 2948 (w), 2913 (w), 2847 (w), 1572 (m), 1480 (w), 1431 (s), 1346 (w), 1313 (w), 1254 (w), 1219 (m), 1184 (w), 1143 (m), 1086 (s), 1024 (m), 966 (m), 940 (m), 894 (w), 865 (m), 831 (w), 746 (s), 696 (s), 656 (m). Anal. calcd for C23H24Cl3FeN2P (1/2 CH2Cl2): C, 50.04; H, 4.47; N, 4.97. Found: C, 50.24; H, 4.32; N, 5.27.
2.2.5 Fe5. Using a procedure similar to synthesize Fe1, Fe5 was prepared as a light-yellow powder (0.25 g, 69%). FT-IR (cm−1): 3209 (m), 3074 (w), 2961 (w), 2878 (w), 1611 (w), 1572 (m), 1465 (m), 1434 (m), 1379 (w), 1317 (w), 1255 (m), 1185 (w), 1092 (m), 1067 (m), 1030 (w), 989 (m), 946 (m), 915 (w), 854 (m), 794 (w), 736 (s), 697 (s). Anal. calcd for C32H35Cl2FeN2P (1/7 CH2Cl2): C, 62.52; H, 5.76; N, 4.54. Found: C, 62.50; H, 5.74; N, 4.71.
2.2.6 Fe6. Using a procedure similar to synthesize Fe1, Fe6 was prepared as a light-yellow powder (0.36 g, 75%). FT-IR (cm−1): 3216 (m), 3050 (w), 2960 (m), 2925 (w), 2867 (w), 1602 (m), 1568 (m), 1457 (m), 1430 (m), 1383 (m), 1359 (m), 1250 (m), 1182 (m), 1095 (m), 1066 (m), 1022 (w), 985 (m), 945 (m), 917 (w), 867 (m), 836 (w), 776 (w), 737 (s), 697 (s). Anal. Calcd for C38H47Cl2FeN2P (1/8 CH2Cl2): C, 65.40; H, 6.80; N, 4.00. Found: C, 65.49; H, 6.82; N, 4.19.
2.2.7 Fe7. Using a procedure similar to synthesize Fe1, Fe7 was prepared as an orange powder (0.20 g, 77%). FT-IR (cm−1): 3201 (m), 3083 (w), 2952 (m), 2867 (m), 1594 (m), 1453 (s), 1383 (w), 1336 (w), 1281 (w), 1241 (w), 1219 (m), 1189 (m), 1128 (m), 1077 (m), 1017 (m), 960 (m), 910 (w), 860 (s), 799 (m), 776 (m), 719 (m).
2.2.8 Fe8. Using a procedure similar to synthesize Fe1, Fe8 was prepared as a light-yellow powder (0.35 g, 78%). FT-IR (cm−1): 3199 (w), 2933 (m), 2865 (m), 2706 (w), 1612 (w), 1593 (w), 1566 (m), 1461 (s), 1380 (m), 1307 (w), 1253 (m), 1228 (w), 1193 (m), 1055 (s), 985 (m), 943 (w), 898 (w), 859 (s), 792 (m), 756 (w), 731 (m).
2.3 X-ray crystallographic studies
The method followed for obtaining a single crystal of the iron complexes by solvent diffusion was as follows: diethyl ether was diffused into a dichloromethane solution. X-ray single crystal data was collected using Cu-Kα radiation (λ = 1.54184 (Å)) on a Rigaku RAXIS Fast IP diffractometer at 170(11) K. The cell parameters were obtained by the global optimization of the positions of all the reflected signals collected. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least squares on F2. All hydrogen atoms were placed in the calculated positions. Structure solution and refinement were performed using the SHELXTL-97 package.55–57 Details of the X-ray structure determinations and refinements for Fe1, Fe2, Fe4, Fe5, and Fe6 are provided in Table 1. The details of the X-ray structure determinations and refinements for Fe4′ are provided in Table S1.†
Table 1 Crystal data and structure refinements for Fe1, Fe2, and Fe4–Fe6
| |
Fe1 |
Fe2 |
Fe4 |
Fe5 |
Fe6 |
| Empirical formula |
C46H50Cl4Fe2N4P2 |
C24H27Cl2FeN2P |
C23H24Cl3FeN2P |
C32H35Cl2FeN2P |
C38H47Cl2FeN2P |
| Formula weight |
974.34 |
501.19 |
521.61 |
605.34 |
689.49 |
| Temperature/K |
169.98(11) |
169.99(10) |
170.00(11) |
170.15 |
170.00(11) |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P21/n |
P21/n |
P21 |
| a/Å |
8.5815(5) |
12.08330(10) |
11.2884(3) |
10.8311(2) |
10.48430(10) |
| b/Å |
10.7155(4) |
15.72850(10) |
19.7047(3) |
11.3008(2) |
12.94410(10) |
| c/Å |
14.7219(8) |
12.54790(10) |
11.5379(3) |
24.9795(4) |
14.17160(10) |
| α/° |
71.656(4) |
90 |
90 |
90 |
90 |
| β/° |
78.877(5) |
99.9600(10) |
115.527(3) |
91.011(2) |
108.9370(10) |
| γ/° |
85.997(4) |
90 |
90 |
90 |
90 |
| Volume/Å3 |
1260.76(12) |
2348.81(3) |
2315.90(11) |
3057.02(9) |
1819.13(3) |
| Z |
1 |
4 |
4 |
4 |
2 |
| ρcalc/g cm3 |
1.283 |
1.417 |
1.496 |
1.315 |
1.259 |
| μ/mm−1 |
7.422 |
7.984 |
9.159 |
6.229 |
5.294 |
| F(000) |
504.0 |
1040.0 |
1072.0 |
1264.0 |
728.0 |
| Crystal size/mm3 |
0.2 × 0.15 × 0.1 |
0.12 × 0.12 × 0.12 |
0.2 × 0.1 × 0.05 |
0.200 × 0.050 × 0.050 |
0.4 × 0.25 × 0.2 |
| Radiation |
CuKα (λ = 1.54184) |
CuKα (λ = 1.54184) |
CuKα (λ = 1.54184) |
CuKα (λ = 1.54184) |
CuKα (λ = 1.54184) |
| 2θ range for data collection/° |
6.43 to 154.35 |
7.428 to 154.722 |
8.976 to 153.326 |
7.078 to 154.762 |
6.594 to 154.082 |
| Index ranges |
−10 ≤ h ≤ 10 |
−15 ≤ h ≤ 15 |
−14 ≤ h ≤ 13 |
−13 ≤ h ≤ 13 |
−13 ≤ h ≤ 13 |
| −13 ≤ k ≤ 13 |
−19 ≤ k ≤ 19 |
−24 ≤ k ≤ 23 |
−10 ≤ k ≤ 14 |
−15 ≤ k ≤ 16 |
| −18 ≤ l ≤ 18 |
−14 ≤ l ≤ 15 |
−11 ≤ l ≤ 14 |
−25 ≤ l ≤ 31 |
−17 ≤ l ≤ 17 |
| Reflections collected |
24265 |
32335 |
16218 |
22759 |
28114 |
| Data/restraints/parameters |
5177 [Rint = 0.0434, Rsigma = 0.0297] |
4890 [Rint = 0.0271, Rsigma = 0.0166] |
4709 [Rint = 0.0325, Rsigma = 0.0311] |
6284 [Rint = 0.0506, Rsigma = 0.0442] |
7360 [Rint = 0.0294, Rsigma = 0.0239] |
| Goodness-of-fit on F2 |
5177/30/317 |
4890/0/272 |
4709/0/271 |
6284/0/346 |
7360/1/403 |
| Final R indices [I ≥ 2σ(I)] |
1.063 |
1.025 |
1.048 |
1.064 |
1.065 |
| Largest diff. Peak/hole/e Å−3 |
R1 = 0.1022, wR2 = 0.2587 |
R1 = 0.0289, wR2 = 0.0736 |
R1 = 0.0362, wR2 = 0.0881 |
R1 = 0.0697, wR2 = 0.2064 |
R1 = 0.0636, wR2 = 0.1646 |
2.3 General procedure for the ring opening polymerization of ε-caprolactone under nitrogen atmosphere
The precatalyst Fe6 (0.013 g, 0.02 mmol) and toluene (1 mL) were added to 25 mL Schlenk flask. Then, 2 equivalents of LiCH2SiMe3 were added dropwise to the solution, the color immediately changed from yellow to red-brown, and the mixture was stirred 30 min at room temperature. After the reaction, it was immediately injected into ε-CL (0.456 g, 4 mmol) and then put into the oil bath with a set temperature to react for different times. Finally, methanol (20 mL) was added to terminate the polymerization, and the resulting polymer was filtered and dried in a vacuum drying oven at 50 °C for 24 h.
3 Results and discussion
3.1 Syntheses and characterization of ligands L1–L6 and iron(II) complexes Fe1–Fe6
The series of N-(2-(diphenylphosphino)ethyl)-5,6,7,8-tetrahydroquinolin-8-amines, N-(Ph2PCH2CH2)-2-RC9H9N-8-NH (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H L1, Me L2, iPr L3, Cl L4, 2,4,6-MeC6H2 L5, 2,4,6-iPrC6H2 L6) was prepared according to the literature.53 Among these, new ligands of L5 and L6 were characterized by 1H/13C/31P NMR and FT-IR spectroscopy and elemental analysis. Then, iron(II) complexes Fe1–Fe6 were synthesized by the treatment of the corresponding L1–L6 with 1 equivalent FeCl2·4H2O in THF at room temperature under nitrogen atmosphere (Scheme 1). All iron(II) complexes were identified by IR and elemental analysis, and the crystal structures of Fe1, Fe2, and Fe4–Fe6 were further determined by single-crystal X-ray diffraction.
H L1, Me L2, iPr L3, Cl L4, 2,4,6-MeC6H2 L5, 2,4,6-iPrC6H2 L6) was prepared according to the literature.53 Among these, new ligands of L5 and L6 were characterized by 1H/13C/31P NMR and FT-IR spectroscopy and elemental analysis. Then, iron(II) complexes Fe1–Fe6 were synthesized by the treatment of the corresponding L1–L6 with 1 equivalent FeCl2·4H2O in THF at room temperature under nitrogen atmosphere (Scheme 1). All iron(II) complexes were identified by IR and elemental analysis, and the crystal structures of Fe1, Fe2, and Fe4–Fe6 were further determined by single-crystal X-ray diffraction.
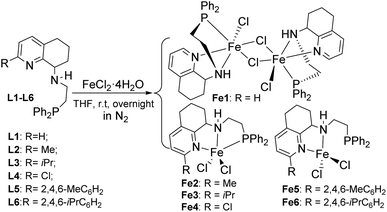 |
| | Scheme 1 Synthesis of iron(II) complexes Fe1–Fe6, LFeCl2 (L: L1–L6). | |
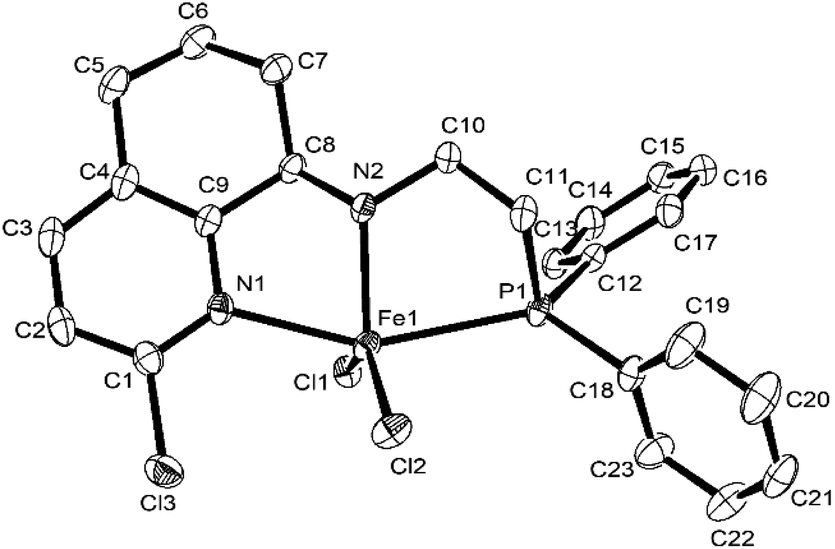
Single crystals of Fe1, Fe2, Fe4, Fe5, and Fe6 suitable for X-ray diffraction were individually obtained at room temperature by diffusing diethyl ether into their dichloromethane solutions under nitrogen atmosphere. Their molecular structures are shown in Fig. 1–5; their selected bond lengths and angles are collected in Tables 2 and 3. Interestingly, the molecular structure of Fe1 showed a dimer that was composed of two iron metal centers with a Cl bridge, which is different from other complexes. Fig. 1 showed that each iron atom is six-coordinated by two N atoms, one P atom, and three Cl atoms, forming a distorted octahedral geometry around Fe, similar to that in the literature. In that case, in different solvent, monomeric or chlorine-bridged dinuclear iron(II) complexes were formed.58 The bond length of the Fe–Npy (2.224(6) (Å)) bond is shorter than that of the Fe–Nimino (2.255(7) (Å)) bond, which is consistent with that of the previous analogs.59 The bond length of Fe–Cl1 (2.302(3) (Å)) (Table 2) is similar to that previously reported,60 but the bond length of Fe1–Cl2 (2.5093(18) (Å)) and Fe1–Cl2i (2.5129(18) (Å)) is much longer than that of Fe1–Cl1, which may be attributed to the formation of the bridging structure of Cl atoms. The bond length of Fe–P [2.5268(18) Å] falls in the normal range reported in the literature.50
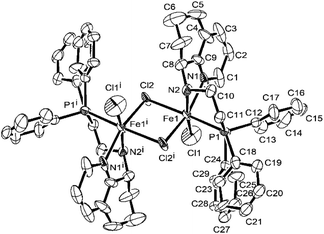 |
| | Fig. 1 ORTEP drawing of Fe1 with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. The superscript ‘i’ denotes a symmetry-generated atom. | |
Table 2 Selected bond lengths (Å) and angles (°) for Fe1
| Bond length (Å) |
| Fe1–N1 |
2.224(6) |
Fe1–Cl2 |
2.5093(18) |
| Fe1–N2 |
2.255(7) |
Fe1–Cl2i |
2.5149(18) |
| Fe1–P1 |
2.5268(18) |
N2–C8 |
1.460(9) |
| Fe1–Cl1 |
2.302(3) |
P1–C11 |
1.850(7) |
| Bond angles (°) |
| N1–Fe1–Cl1 |
99.6(3) |
N2–Fe1–P1 |
80.34(14) |
| N1–Fe1–Cl2 |
90.48(15) |
Cl1–Fe1–Cl2 |
92.43(12) |
| N1–Fe1–P1 |
95.05(14) |
Cl1–Fe1–P2 |
98.41(11) |
| N1–Fe1–N2 |
72.0(3) |
C8–N2–Fe1 |
107.4(6) |
| N2–Fe1–Cl1 |
171.3(2) |
C11–P1–Fe1 |
122.1(2) |
| N2–Fe1–Cl2 |
90.06(15) |
|
|
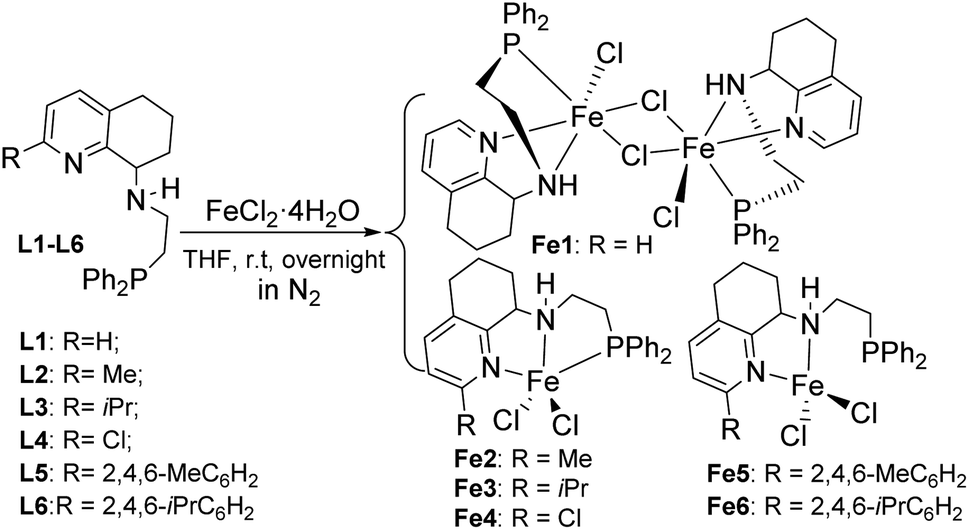
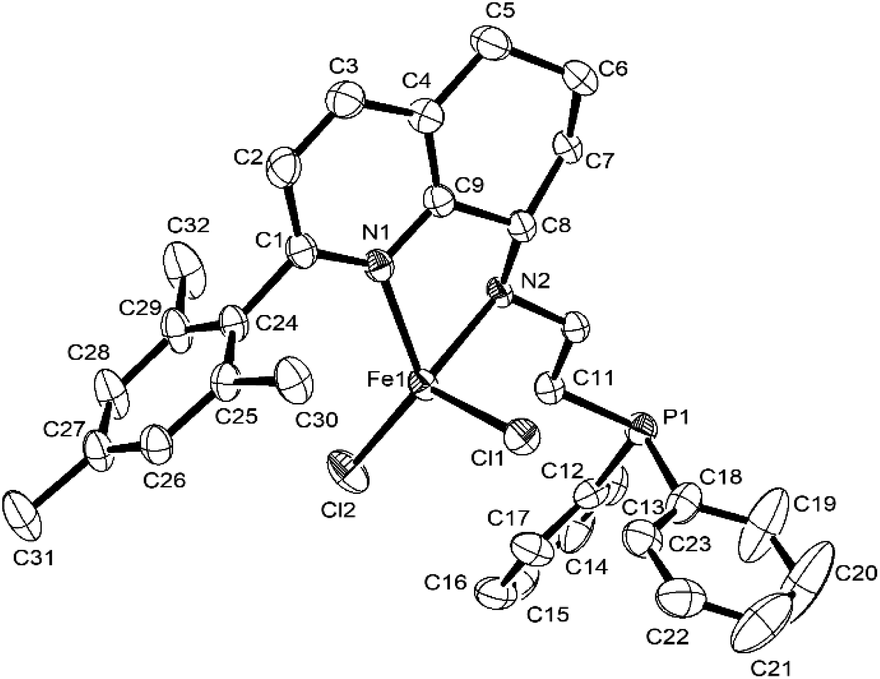
Fig. 2 and 3 showed that both Fe2 and Fe4 are monomeric species with a similar structure, in which iron is five-coordinated by Npy, Nimine, and P from the ligand and two Cl, forming a trigonal bipyramidal geometry around iron, similar to the structure in the literature.50,58 The difference is that the Fe atom moves slightly outward by 0.174 Å relative to the normal plane formed by N1, N2, and P1 in Fe2, while the Fe atom almost falls in the same plane with N1, N1, and P1 in Fe4. Moreover, the bond length of Fe–P in Fe1 (2.5268(18) Å), Fe2 (2.6180 Å), and Fe4 (2.6691(7) Å) followed the order Fe1 (H) < Fe2 (Me) < Fe4 (Cl), indicating the effect of the electron withdrawing ability of R substituents, which led to different molecular structures. Especially, in Fe2 and Fe4, the bond lengths of Fe–Npy bond are much longer than that of Fe–Nimino (2.2242 vs. 2.1630; 2.2912(19) vs. 2.149(2)) in both the cases (Table 3), which showed that the coordination effect of Fe–Nimino is stronger than that of Fe–Npy, unlike that in Fe1 complexes.
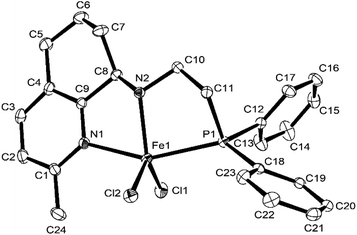 |
| | Fig. 2 ORTEP drawing of Fe2 with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. | |
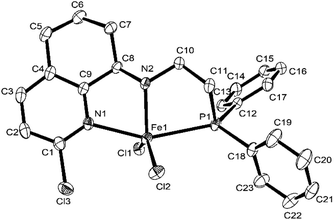 |
| | Fig. 3 ORTEP drawing of Fe4 with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. | |
Table 3 Selected bond lengths (Å) and angles (°) for Fe2, Fe4, Fe5, and Fe6
| |
Fe2 |
Fe4 |
Fe5 |
Fe6 |
| Bond length (Å) |
| Fe1–N1 |
2.2242(14) |
2.2912(19) |
2.107(4) |
2.105(5) |
| Fe1–N2 |
2.1630(14) |
2.149(2) |
2.148(3) |
2.166(5) |
| Fe1–Cl1 |
2.2873(5) |
2.2819(6) |
2.2645(13) |
2.2244(16) |
| Fe1–Cl2 |
2.3384(5) |
2.3108(6) |
2.2146(14) |
2.2693(18) |
| Fe1–P1 |
2.6180(5) |
2.6691(7) |
|
|
| N2–C8 |
1.479(2) |
1.482(3) |
1.489(6) |
1.486(7) |
| P1–C11 |
1.8408(17) |
1.838(3) |
1.850(5) |
1.850(6) |
|
| Bond angles (°) |
| N1–Fe1–Cl1 |
89.84(4) |
91.22(5) |
103.94(11) |
129.14(14) |
| N1–Fe1–Cl2 |
101.79(4) |
100.98(5) |
126.08(11) |
104.43(14) |
| N1–Fe1–P1 |
154.71(4) |
153.45(6) |
|
|
| N2–Fe1–N1 |
75.11(5) |
73.23(7) |
77.92(13) |
76.74(18) |
| N2–Fe1–Cl1 |
129.54(4) |
116.36(5) |
100.80(10) |
116.91(14) |
| N2–Fe1–Cl2 |
106.39(4) |
106.93(5) |
116.37(11) |
103.16(15) |
| N2–Fe1–P1 |
80.44(4) |
80.22(5) |
|
|
| Cl1–Fe1–Cl2 |
123.89(2) |
136.71(3) |
121.43(6) |
117.50(7) |
| Cl1–Fe1–P1 |
100.964(17) |
100.42(2) |
|
|
| C8–N2–Fe1 |
108.03(10) |
106.12(14) |
103.4(2) |
102.6(3) |
| C11–P1–Fe1 |
92.77(5) |
90.61(8) |
|
|
In contrast, the molecular structure of Fe5 and Fe6 (Fig. 4 and 5) showed that iron was coordinated by two nitrogen atoms and two chlorine atoms, possessing a tetrahedron geometry, in which the phosphorus atom dissociated with iron. In the molecular structures of Fe5 and Fe6, the plane of the benzene ring of the substituent is almost perpendicular to the plane of the three atoms of N, N, and Fe, and the dihedral angles are 83.17° and 73.6°, respectively.
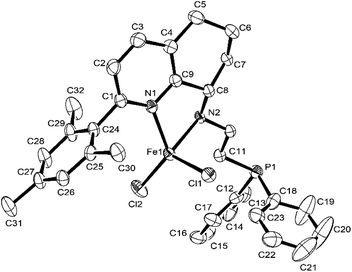 |
| | Fig. 4 ORTEP drawing of Fe5 with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. | |
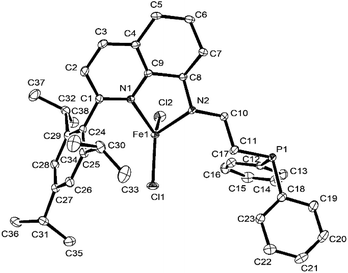 |
| | Fig. 5 ORTEP drawing of Fe6 with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. | |
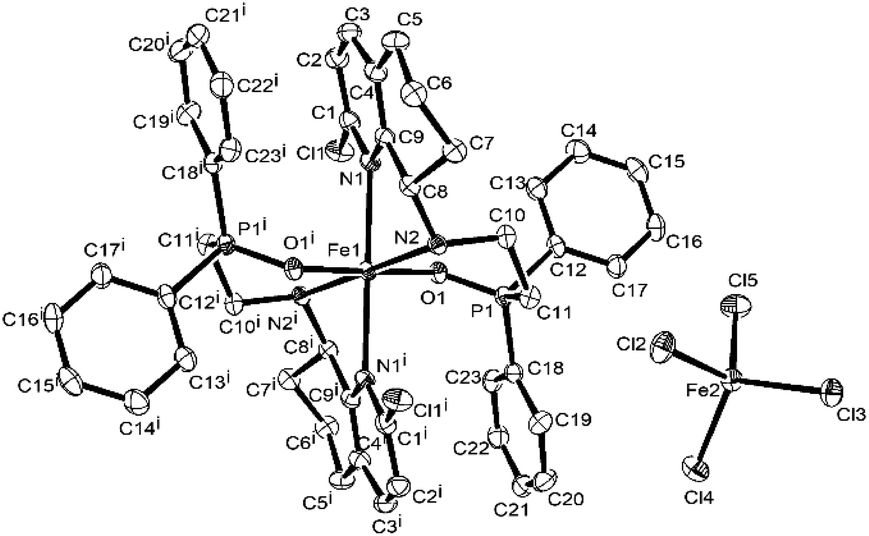
Considering the possible oxidization reaction of –PPh2 by O2 in the air, the monitoring 31P NMR of L4 and L1 in air was conducted, and the results showed that the ligand in the solid state was quite stable and there was no change in the 31P NMR spectrum after several weeks. However, the L4 and L1 in CDCl3 solution was very unstable, and there was a new peak in the 31P NMR spectrum increasing with time (shown in Fig. S9 and S10†), indicating the easy oxidation of P(III) by O2 in the air. Then, the reaction of ligand L4 and FeCl3 under the atmosphere of N2 was conducted in dry EtOH and a tiny precipitate was observed. After filtration under N2 atmosphere, a part of the orange mother solution was layered by diethyl ether, and the single crystals suitable for X-ray diffraction were obtained after two days. Surprisingly, the crystal structure showed that it possessed the same structure as that of Fe4 (L4FeCl2) but contained different bond lengths and bond angles. Subsequently, when the remaining mother solution was kept for additional one week under N2, yellow crystals Fe4′ were obtained (Scheme 2). The structure of Fe4′ is shown in Fig. 6, and the selected bond lengths and bond angles are shown in Table S2.† The X-ray diffraction of these yellow crystal showed that Fe4′ is a bisligated iron(II) salt with the anion [FeCl4]−, in which iron in the cation is six-coordinated with distorted octahedral geometry; the apical sites are occupied by two O atoms, with the N atoms in the equatorial sites. Besides, the bond lengths of the PO bond are 1.512(3) (Å) and 1.5083(14) (Å), respectively, which falls in the range of the bond length of the PO (+5) reported in the literature,61–63 further indicating that phosphorus(III) is oxidized into phosphorus(V). The reason for this phenomenon is not clear and needs to be further explored.
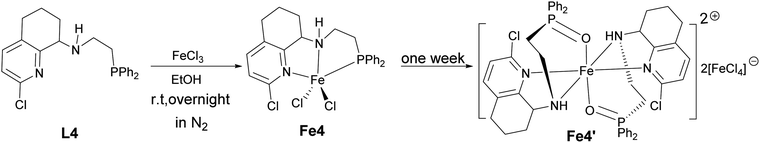 |
| | Scheme 2 Synthesis of bisligated iron complex Fe4′ in N2. | |
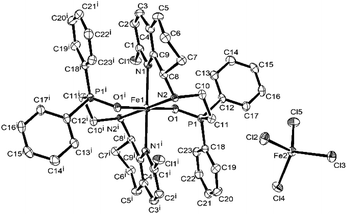 |
| | Fig. 6 ORTEP representation of Fe4′ with the thermal ellipsoids set at the 30% probability level. All the hydrogen atoms have been omitted for clarity. The superscript ‘i’ and ‘ii’ denote a symmetry-generated atom. | |
3.2 Ring opening polymerization of ε-CL using iron(II) complexes of Fe1–Fe6 with LiCH2SiMe3 in situ
As our previous work showed that zinc complexes with the same ligand exhibited remarkable activity for the ring opening polymerization of ε-CL, the iron complexes of Fe1–Fe6 were also evaluated for the ROP of ε-CL under nitrogen atmosphere. Firstly, Fe6 was employed to optimize the polymerization condition. The results showed that either without or with one equivalent of LiCH2SiMe3 activation, it cannot catalyze the polymerization of ε-CL, similar to the result of their zinc analogues. In contrast, when 2 equivalent LiCH2SiMe3 was used, the conversion of ε-CL reached up to 100% with [ε-CL]/[Fe] = 200 and 30 °C in 10 min. Therefore, Fe6 with two equivalents of LiCH2SiMe3 were used as initiators to investigate the effects of temperature, polymerization time, and molar ratio of monomer to iron on the ROP of ε-CL. The results are collected in Table 4.
Table 4 Ring opening polymerization of ε-CL by Fe1–Fe6/2LiCH2SiMe3
| Run |
Cat |
ε-CL:Fe |
T/°C |
t min−1 |
Conv.b (%) |
Mnc (×104 g mol−1) |
PDIc |
TOF (×103 h−1) |
| Reaction conditions: 1.0 mL toluene, 20 μmol Fe + 40 μmol LiCH2SiMe3. Determined by 1H NMR spectroscopy. GPC data were recorded in THF vs. polystyrene standards, using a correcting factor of 0.56. 1 mL THF. 1 mL CH2Cl2. 1 mL n-hexane. |
| 1 |
Fe6 |
200:1 |
30 |
10 |
100 |
1.21 |
2.24 |
1.20 |
| 2 |
Fe6 |
400:1 |
30 |
10 |
88 |
1.25 |
2.43 |
2.11 |
| 3 |
Fe6 |
600:1 |
30 |
10 |
68 |
1.16 |
2.53 |
2.45 |
| 4 |
Fe6 |
700:1 |
30 |
10 |
55 |
3.49 |
2.76 |
2.31 |
| 5 |
Fe6 |
800:1 |
30 |
10 |
48 |
3.13 |
2.88 |
2.30 |
| 6 |
Fe6 |
900:1 |
30 |
10 |
37 |
4.80 |
1.89 |
2.00 |
| 7 |
Fe6 |
1000:1 |
30 |
10 |
28 |
4.24 |
3.90 |
1.68 |
| 8 |
Fe6 |
1000:1 |
40 |
10 |
30 |
6.67 |
3.38 |
1.80 |
| 9 |
Fe6 |
1000:1 |
50 |
10 |
47 |
5.12 |
2.33 |
2.82 |
| 10 |
Fe6 |
1000:1 |
60 |
10 |
51 |
5.50 |
2.77 |
3.06 |
| 11 |
Fe6 |
1000:1 |
70 |
10 |
56 |
8.12 |
2.53 |
3.36 |
| 12 |
Fe6 |
1000:1 |
80 |
10 |
88 |
8.06 |
1.69 |
5.28 |
| 13 |
Fe6 |
1000:1 |
90 |
10 |
100 |
7.43 |
2.25 |
6.00 |
| 14 |
Fe6 |
1500:1 |
90 |
3 |
17 |
4.20 |
2.18 |
5.10 |
| 15 |
Fe6 |
1500:1 |
90 |
5 |
41 |
6.46 |
3.24 |
7.38 |
| 16 |
Fe6 |
1500:1 |
90 |
7 |
68 |
7.52 |
2.81 |
8.74 |
| 17 |
Fe6 |
1500:1 |
90 |
10 |
87 |
11.24 |
1.93 |
7.83 |
| 18 |
Fe6 |
1500:1 |
90 |
20 |
93 |
18.47 |
1.97 |
4.18 |
| 19 |
Fe6 |
1800:1 |
90 |
10 |
72 |
26.09 |
1.66 |
7.78 |
| 20 |
Fe6 |
2000:1 |
90 |
10 |
54 |
23.94 |
1.85 |
6.48 |
| 21 |
Fe6 |
2300:1 |
90 |
10 |
32 |
23.49 |
1.69 |
4.42 |
| 22 |
Fe6 |
200:1 |
30 |
10 |
76 |
4.87 |
3.14 |
0.91 |
| 23 |
Fe6 |
200:1 |
30 |
10 |
0 |
— |
— |
— |
| 24 |
Fe6 |
200:1 |
30 |
10 |
>99 |
6.05 |
2.16 |
1.19 |
| 25 |
Fe1 |
1500:1 |
90 |
10 |
93 |
12.18 |
2.24 |
8.37 |
| 26 |
Fe2 |
1500:1 |
90 |
10 |
89 |
9.21 |
3.04 |
8.01 |
| 27 |
Fe3 |
1500:1 |
90 |
10 |
88 |
11.20 |
2.53 |
7.92 |
| 28 |
Fe4 |
1500:1 |
90 |
10 |
55 |
21.93 |
2.77 |
4.95 |
| 29 |
Fe5 |
1500:1 |
90 |
10 |
98 |
24.35 |
1.85 |
8.82 |
| 30 |
Fe7 |
1500:1 |
90 |
10 |
89 |
11.01 |
2.20 |
8.01 |
| 31 |
Fe8 |
1500:1 |
90 |
10 |
15 |
12.2 |
2.25 |
|
Firstly, the effect of the molar ratio of monomer to iron on the ring opening polymerization of ε-CL was investigated when the temperature was kept at 30 °C, and the reaction time was 10 min (runs 1–7, Table 4). As the molar ratio of [ε-CL]:[Fe] increases from 200:1 to 1000:1, the monomer conversion decreased from 100% to 28%. However, there was a small variation of the turnover frequency (TOF) in the range of 2.00–2.45 × 103 h−1 when the molar ratio of ε-CL to iron changed from 400 to 900, suggesting the similar polymerization rate under these conditions. At the same time, the molecular weight significantly increased from 1.16 to 4.80 × 104 g mol−1, indicating that higher monomer concentration led to faster coordination and higher propagation rate.64
Secondly, because temperature has an important influence on the catalytic efficiency,65 parallel experiments were carried out at different temperatures with the molar ratio of [ε-CL]:[Fe] = 1000:1 within 10 min (runs 7–13, Table 4). It is obvious that the conversion rate gradually increased with the temperature from 28% at 30 °C to 100% at 90 °C, and the TOF values also increased from 1.68 × 103 h−1 at 30 °C to 6.00 × 103 h−1 at 90 °C, indicating the good thermal stability of the active species to some extent. However, the molecular weight distribution of the obtained polymer is very broad (PDI = 1.69–3.90), which can be explained by more side reactions of the transesterification reaction at higher temperature or multisite active species.66 Then, at 90 °C, the molar ratio of [ε-CL]:[Fe] was further improved from 1000 to 2300 by fixing the polymerization time to 10 min; the monomer conversion gradually decreased from 100% to 32%, while the TOF values varied between 4.42 and 7.83 × 103 h−1 without any trend (runs 17, 19–21, Table 4). The molecular weight of the polymer changed from 11.24 × 104 g mol−1 to 26.09 × 104 g mol−1 without any trend, and molecular weight distribution was broad (PDI = 1.66–1.93).
The effect of reaction time on the polymerization of ε-CL was studied at 90 °C with a molar ratio of [ε-CL]:[Fe] = 1500:1 (runs 14–18, Table 4). The results in Table 4 show that monomer conversion increases with time from 17% in 3 min to 93% in 20 min. In the meantime, the TOF value increases from 5.10 × 103 h−1 (3 min) to 8.74 × 103 h−1 (7 min), suggesting the introduction time of the active species. Further extending the polymerization time from 7 to 20 min led to a decrease in the TOF value from 8.74 × 103 h−1 to 3.60 × 103 h−1, suggesting the partial deactivation of the active species over time.67 Moreover, the molecular weight of the obtained polymer increased rapidly from 4.20 × 104 g mol−1 to 18.47 × 104 g mol−1 with a broad molecular weight distribution (PDI = 1.93–2.81).
Finally, the effect of the solvent on the ring opening polymerization of ε-CL at 30 °C and 10 min was also investigated respectively (runs 1, 22–24, Table 4). The results showed that the conversion rate in toluene (100%) and n-hexane (>99%) was significantly higher than that in THF (76%) and CH2Cl2 (0%) under the same conditions. This may be due to the higher activation free energy required to initiate the reaction in polar solvents, according to DFT calculation.68 According to the optimized conditions by Fe6, the ring opening polymerization of ε-caprolactone by other iron(II) complexes Fe1–Fe5 was also conducted at the molar ratio of [ε-CL]:[Fe] = 1500:1 and at 90 °C within 10 min (runs 18, 25–29, Table 4). All these iron(II) complexes with different substituents showed high catalytic activity except for Fe4 with low conversion, and the monomer conversions were in the order Fe1 (H, 93%) > Fe2 (Me, 89%) > Fe3 (iPr, 88%) > Fe4 (Cl, 55%); Fe5 (MeC6H2, 98%) > Fe6 (iPrC6H2, 87%). In addition, Fig. 7 shows that Fe5 has the highest activity (TOF = 8.82 × 103 h−1) among these precatalysts (Fe1, Fe2, Fe3, Fe4, Fe6) (TOF range: 4.95–8.37 × 103 h−1), and the Mn of the polymer obtained by Fe5 (Mn = 24.35 × 104 g mol−1) is much higher than that by other procatalysts (Mn range: 9.21–21.93 × 104 g mol−1). These results indicated that the difference in the catalytic efficiency is related to the size of the substituents, in which the electron withdrawing group will lead to lower efficiency. However, the reason for the large difference in the catalytic efficiency by Fe4 and other iron(II) complexes may be that the electron-withdrawing chlorine group reduces the electron cloud density of the iron metal center. Recently, we have reviewed the progress of iron compound for the ring opening polymerization of cyclic esters and found that very limited examples showed high TOF over 1000 h−1 toward the ROP of ε-CL, but many iron compounds exhibited high TOF (>1000 h−1) toward lactides.10,47
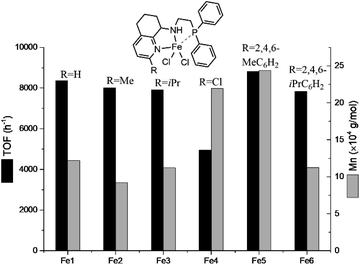 |
| | Fig. 7 Comparing the catalytic activity and molecular weight of the PCL for different iron(II) procatalysts. Conditions: ε-CL:[Fe/2LiCH2SiMe3] = 1500:1, 1 mL toluene, 90 °C. | |
The above iron(II) complexes Fe1–Fe6 showed good catalytic activity for the ring opening polymerization of ε-CL. Considering the potential influence of coordination or dissociation of phosphine(III) to iron that varies with substituents, we replaced 2-(diphenylphosphino)ethanamine with n-butylamine to prepare the L7 and the corresponding Fe7 (Scheme 3) without phosphine. Simultaneously, the Fe7/2LiCH2SiMe3 system was used to catalyze the ROP of ε-CL, and the result showed that its catalytic efficiency and the molecular weight of polymer are lower than that by Fe1 (conversion: 89% vs. 94%, Mn: 11.0 vs. 12.1 × 104 g mol−1) (runs 17 and 30, Table 4), further indicating that the –PPh2 moiety plays an important role in the polymerization, which is different from the results of the zinc analogues.53 In addition, the analogue of Fe5, Fe8 without phosphine was also prepared but showed much lower efficiency and produced much lower molecular weight polymer (run 29 vs. 31, Table 4).
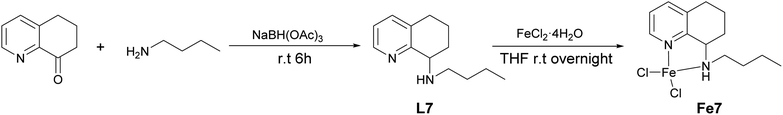 |
| | Scheme 3 Synthetic routes for L7 and Fe7. | |
In order to explore the mechanism of the ring opening polymerization of ε-CL by these iron(II) complexes, the obtained polymer (run 3, Table 4, [ε-CL]/[Fe] = 600:1) was characterized by MALDI-TOF mass spectrometry and 1H NMR spectroscopy (Fig. 8 and 9). In the MALDI-TOF mass spectrum, there are two families of peaks (A/A* and B), in which A/A* was clearly assigned to a linear structure (CH3O[C6H10O2]nH + Na+/K+), and B could be assigned to a cyclic structure ([C6H10O2]n + K+) or linear structure (CH3O[C6H10O2]nH + Li+). Considering that the polymer structure may contain Li+, its structure should be CH3O[C6H10O2]nH + Li+, similar with that in the literature.69 In the meantime, the 1H NMR spectrum showed the typical peak of methoxyl group at 3.67 ppm, which was consistent with the analysis of the MALDI-TOF spectrum. According to our previous reports,70 the methoxyl group is derived from methanol, which terminates polymerization. The polymer obtained at the molar ratio of [ε-CL]/[Fe] = 200:1 (run 1, Table 4) was also characterized by MALDI-TOF and 1H NMR spectroscopy (Fig. 10 and 11). The MALDI-TOF mass spectrum showed three families of peaks (A, B/B*, and C/C*), in which A was assigned to a linear structure ([C6H10O2]n + CH2SiMe3 + K+ + H), and the minor peaks B/B* and C/C* were attributed to the cyclic structure ([C6H10O2]n + Na+/Li+) and linear structure (CH3O[C6H10O2]nH + Na+/Li+), respectively. In the 1H NMR spectrum, the signal of OCH3 and CH2SiMe3 appeared at about 3.67 ppm and 0.1 ppm, which was consistent with the above MALDI-TOF analysis. Also, the appearance of CH2SiMe3 indicated that the polymerization probably followed the coordination insertion mechanism via the intermediate Fe–CH2SiMe3.
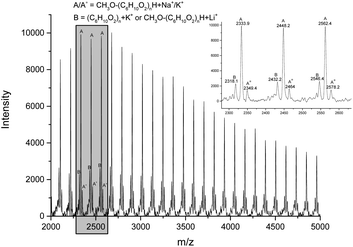 |
| | Fig. 8 MALDI-TOF mass spectrum of the PCL obtained using Fe6 (run 3, Table 4). | |
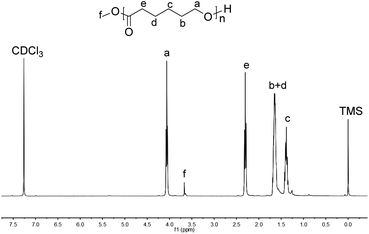 |
| | Fig. 9 1H NMR spectrum of the PCL obtained using Fe6 (run 3, Table 4). | |
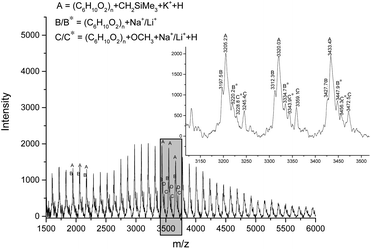 |
| | Fig. 10 MALDI-TOF mass spectrum of the PCL obtained using Fe6 (run 1, Table 4). | |
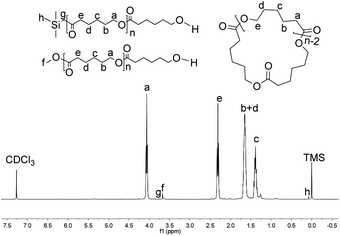 |
| | Fig. 11 1H NMR spectrum of the PCL obtained using Fe6 (run 1, Table 4). | |
As the difference structure found with different monomer/iron molar ratio, in order to deeply investigate the effect of monomer ratio on the structure, the polymers obtained in the molar ratio of [ε-CL]/[Fe] at 100:1, 400:1, and 800:1 were characterized by MALDI-TOF mass spectrometry and 1H NMR spectroscopy (shown in Fig. S11–16†). When [ε-CL]/[Fe] = 100:1, the MALDI-TOF mass spectrum (Fig. S11†) showed three families of peaks (A/A*, B, and C), in which A/A* corresponds to the cyclic species ([C6H10O2]n + K+/Na+), while the minor peak B may be assigned to the cyclic structure ([C6H10O2]n + K+) or linear structure ([C6H10O2]n + CH2SiMe3 + Li+ + H). Because of the presence of a little CH2SiMe3 signal near 0.1 ppm in the 1H NMR spectrum (Fig. S12†), peak B could be assumed to be ([C6H10O2]n + CH2SiMe3 + Li+ + H). In addition, there is no signal of methoxyl in the 1H NMR spectrum; thus, there will be no linear structure with methoxy group as the end group. In contrast, according to MAIDI-TOF and 1H NMR spectrum analysis with molar ratio of [ε-CL]/[Fe] = 400:1 (Fig. S13 and S14†) and 800:1 (Fig. S15 and S16†), the obtained PCL mainly has a linear structure with methoxyl as the end group. These results further indicate that the microstructure of the polymer is greatly affected by the molar ratio of the monomer to iron: at low molar ratio of [ε-CL]/[Fe] ([ε-CL]/[Fe] = 100:1), the structure of PCL is mainly a cyclic structure; at 200 molar ratio of [ε-CL]/[Fe], the linear structure with CH2SiMe3 end group was the major one; at higher ratios ([ε-CL]/[Fe] = 400:1–800:1), the linear structure with methoxy as the end group was the major one.
Therefore, based on the analysis of the polymer structure, we proposed the polymerization proceeded in two paths (as shown in Scheme 4). Fe6 reacts with two equivalent LiCH2SiMe3 to form the intermediates of iron dialkyl. At a low molar ratio of monomer to iron, polymerization proceeds via path I. Firstly, the LFe(CH2SiMe3)2 dialkyl intermediate was generated from the iron complexes with LiCH2SiMe3. Subsequently, the intermediate was coordinated with ε-CL, and then the ring opening polymerization occurs to form linear PCL with the CH2SiMe3 group as the end group. Cyclic PCLs were assumed from intramolecular transesterification. When increasing the molar ratio of [ε-CL]/[Fe] to 200, polymerization proceeds by two paths, and the obtained polymer have three type microstructures, in which the linear polymer capped with CH2SiMe3 group was the major one. Further increasing the molar ratio over 400, polymerization proceeded via path II, in which Fe–CH2SiMe3 does not directly initiate the polymerization because there is no –CH2SiMe3 signal in the polymer spectrum. According to the literature, the iron–carbon bond is loose enough to coordinate with the ε-caprolactone monomer and then the insertion propagation step proceeds through the zwitterionic intermediate.71,72 After methanol quenching, its end group is capped with the methoxy group.73
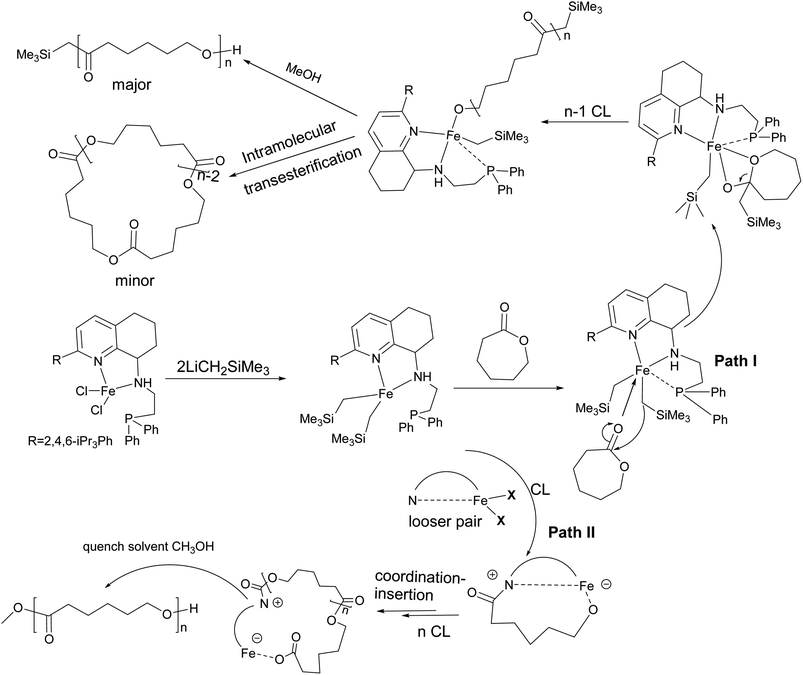 |
| | Scheme 4 Proposed mechanism for ROP of ε-CL by Fe6/2LiCH2SiMe3. | |
4 Conclusions
In this paper, a series of iron(II) dichloride complexes Fe1–Fe6 bearing 8-aminotetrahydroquinolines was successfully synthesized and characterized by FT-IR spectroscopy and elemental analysis. X-ray diffraction study showed that Fe1–Fe6 have diverse structures such as dimer, mononuclear with or without phosphine coordination that varied with the R substituent. In addition, the catalytic system in situ consisting of iron complexes and LiCH2SiMe3 showed a high activity [TOF: 4.95–8.82 × 103 h−1] for the ring opening polymerization of ε-caprolactone, producing the high molecular weight polymer (Mn: 9.21–24.3 × 104 g mol−1) despite the broad molecular weight distribution, which is a rare example of iron catalysts producing the high molecular weight PCL under mild conditions.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51973005). The project was also funded by State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University.
Notes and references
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- I. D'Auria, V. Ferrara, C. Tedesco, W. Kretschmer, R. Kempe and C. Pellecchia, ACS Appl. Polym. Mater., 2021, 3, 4035–4043 CrossRef.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- J. Wu, T.-L. Yu, C.-T. Chena and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144–1164 CrossRef CAS.
- J. Guo, P. Haquette, J. Martin, K. Salim and C. M. Thomas, Angew. Chem., Int. Ed., 2013, 52, 13584–13587 CrossRef CAS PubMed.
- W. F. McDonough and S.-S. Sun, Chem. Geol., 1995, 120, 3–4 CrossRef.
- Y. Wang, X. Wang, W. Zhang and W.-H. Sun, Organometallics, 2023, 42, 1680–1692 CrossRef CAS.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- D. S. Belov, L. Mathivathanan, M. Beazley, W. B. Martin and K. Bukhryakov, Angew. Chem., Int. Ed., 2021, 60, 2934–2938 CrossRef CAS PubMed.
- B. Li, C. Hu, X. Pan and X. Chen, Chem.-Asian J., 2023, 18, e202201031 CrossRef CAS PubMed.
- H. R. Kricheldorf, T. Mang and J. M. Jonte, Macromolecules, 1984, 17, 2173–2181 CrossRef CAS.
- H. R. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497–502 CrossRef CAS.
- A. Södergård and M. Stolt, Macromol. Symp, 1998, 130, 393–402 CrossRef.
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2009, 301, 84–92 CrossRef CAS.
- C. S. Hege and S. M. Schiller, Green Chem., 2014, 16, 1410–1416 RSC.
- M. Stolt and A. Södergård, Macromolecules, 1999, 32, 6412–6417 CrossRef CAS.
-
(a) B. J. O'Keefe, S. M. Monnier, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 123, 339–340 CrossRef PubMed;
(b) B. Yu, X. Xu, Z.-N. Huang, K. Tao, H.-L. Sun, Q.-Z. Yang and W. Wang, Organometallics, 2023, 42, 565–570 CrossRef CAS.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem., Soc., 2002, 124, 4384–4393 CrossRef PubMed.
- A. Arbaoui, C. Redshaw, M. R. J. Elsegood, V. E. Wright, A. Yoshizawa and T. Yamato, Chem.–Asian J., 2010, 5, 621–633 CrossRef CAS PubMed.
- P. V. S. Nylund, B. Monney, C. Weder and M. Albrecht, Catal. Sci. Technol., 2022, 12, 996–1004 RSC.
- V. C. Gibson, E. L. Marshall, D. Navarro-Llobet, A. J. P. White and D. J. A. Williams, J. Chem. Soc., Dalton Trans., 2002, 4321–4322 RSC.
- Y. Li, Q. Wu and M. Gao, Adv. Mat. Res., 2011, 393–395, 1346–1349 Search PubMed.
- L. A. Brown, F. S. Wekesa, D. K. Unruh, M. Findlater and B. K. Long, Polym. Chem., 2017, 55, 2824–2830 CrossRef CAS.
- C. M. Manna, A. Kaur, L. M. Yablon, F. Haeffner, B. Li and J. A. Byers, J. Am. Chem. Soc., 2015, 137, 14232–14235 CrossRef CAS PubMed.
- O. H. Hashmi, F. Capet, M. Visseaux and Y. Champouret, Eur. J. Inorg. Chem., 2022, e202200073 CrossRef CAS.
- J. Dou, D. Zhu, W. Zhang, R. Wang, S. Wang, Q. Zhang, X. Zhang and W.-H. Sun, Inorg. Chim. Acta, 2019, 488, 299–303 CrossRef CAS.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemenn, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS PubMed.
- R. D. Rittinghaus, A. Karabulut, A. Hoffmannn and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2021, 60, 21795–21800 CrossRef CAS PubMed.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Lovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- Y.-Y. Fang, W.-J. Gong, X.-J. Shang, H.-X. Li, J. Gao and J.-P. Lang, Dalton Trans., 2014, 43, 8282–8289 RSC.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- C. M. Manna, H. Z. Kaplan, B. Li and J. A. Byers, Polyhedron, 2014, 84, 160–167 CrossRef CAS.
- Y. Kang, H.-R. Park, M. H. Lee, J. An, Y. Kim and J. Lee, Polyhedron, 2015, 95, 24–29 CrossRef CAS.
- A. C. Silvino, A. L. C. Rodrigues and A. L. C. Resende, Inorg. Chem. Commun., 2015, 55, 39–42 CrossRef CAS.
- H. R. Kricheldorf and C. Boettcher, Makromol. Chem., 1993, 194, 463–473 CrossRef CAS.
- U. Herber, K. Hegner, D. Wolters, R. Siris, K. Wrober, A. Hoffmann, C. Lochenie, B. Weber, D. Kuckling and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2017, 1341–1354 CrossRef CAS.
- B. B. Idage, S. B. Idage, A. S. Kasegaonkar and R. V. Jadhav, Mater. Sci. Eng. B, 2010, 168, 193–198 CrossRef CAS.
- M. Cozzolino, V. Leo, C. Tedesco, M. Mazzeo and M. Lamberti, Dalton Trans., 2018, 47, 13229–13238 RSC.
- O. J. Driscoll, C. K. C. Leung, M. F. Mahon, P. Mckeown and M. D. Jones, Eur. J. Inorg. Chem., 2018, 5129–5135 CrossRef CAS.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 50, 9188–9195 CrossRef CAS.
- Y. Liang, R. Duan, C.-Y. Hu, L.-L. Li, X. Pang, W.-X. Zhang and X.-S. Chen, Chin. J. Polym. Sci., 2018, 36, 185–189 CrossRef CAS.
- P. Marin, M. J.-L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L. M. Chamoreeau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 CrossRef CAS PubMed.
- S. Impemba, F. D. Monica, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2020, 13, 141–145 CrossRef CAS PubMed.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed.
- P. Degee, P. Dubois, S. Jacobsen, H. G. Fritz and R. Jerome, J, Polym. Sci., Part A: Polym. Chem., 1999, 37, 2413–2420 CrossRef CAS.
- D. Zhang, Y. Zhang, W. Hou, Z. Guan and Z. Huang, Organometallics, 2017, 36, 3758–3764 CrossRef CAS.
- R. G. Goel and W. O. Ogini, Inorg. Chem., 1977, 16, 1968–1972 CrossRef CAS.
- R. G. Goel, W. P. Henry and N. K. Jha, Inorg. Chem., 1982, 21, 2551–2555 CrossRef CAS.
- F. Cao, Y. Wang, X. Wang, W. Zhang, G. A. Solan, R. Wang, Y. Ma, X. Hao and W.-H. Sun, Catal. Sci. Technol., 2022, 12, 6687–6703 RSC.
- B. Pan, E. Yue, Q. Liu, X. Yang, Z. Wang and W.-H. Sun, ACS Catal., 2016, 6, 1247–1253 CrossRef CAS.
- G. M. Sheldrick, Acta Cryst, 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Cryst, 2015, C71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
- M. V. Gradiski, B. T. H. Tsui, A. J. Lough and R. H. Morris, Dalton Trans., 2019, 48, 2150–2159 RSC.
- S. Du, X. Wang, W. Zhang, Z. Flisak, Y. Sun and W.-H. Sun, Polym. Chem., 2016, 7, 4188–4197 RSC.
- Y. Zhang, H. Suo, F. Huang, T. Liang, X. Hu and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 830–884 CrossRef CAS.
- L. Ling, J. Hu, Y. Huo and H. Zhang, Tetrahedron, 2017, 73, 86–97 CrossRef CAS.
- M. M. Ibrahim, Inorg. Chem. Commun., 2006, 9, 1215–1218 CrossRef CAS.
- T. Luster, H. J. Van de Roovaart, K. J. Korman, G. G. Sands, K. M. Dunn, A. Spyker, R. J. Staples, S. M. Biros and J. E. Bender, Dalton Trans., 2022, 51, 9103–9115 RSC.
- D. Zhu, L. Guo, W. Zhang, X. Hu, K. Nomura, A. Vignesh, X. Hao, Q. Zhang and W.-H. Sun, Dalton Trans., 2019, 48, 4157–4167 RSC.
- Y. Jiang, W. Zhang, M. Han, X. Wang, G. A. Solan, R. Wang, Y. Ma and W.-H. Sun, Polymer, 2022, 242, 124602 CrossRef CAS.
- Q. Zhang, W. Zhang, N. M. Rajendran, T. Liang and W.-H. Sun, Dalton Trans., 2017, 46, 7833–7843 RSC.
- Q. Zhang, W. Zhang, G. A. Solan, T. Liang and W.-H. Sun, Polymers, 2018, 10, 764 CrossRef PubMed.
- J. Jitonnom and W. Meelua, Chem. Phys. Lett., 2020, 750, 137482 CrossRef CAS.
- E. Kober, R. Petrus, P. Kocięcka, Z. Janas and P. Sobota, Polyhedron, 2015, 85, 814–823 CrossRef CAS.
- J. Gao, W. Zhang, F. Cao, G. A. Solan, Q. Zhang, Y. Jiang, X. Hao and W.-H. Sun, Mol. Catal., 2020, 498, 111280 CrossRef CAS.
- M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS.
- J. A. Castro-Osma, C. Alonso-Moreno, J. C. García-Martinez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez and A. Otero, Macromolecules, 2013, 46, 6388–6394 CrossRef CAS.
- M. Bhunia, G. Vijaykumar, D. Adhikari and S. K. Mandal, Inorg. Chem., 2017, 56, 14459–14466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H/13C/3lP NMR spectra of ligand L5–L7; MALDI-TOF and 1H NMR spectrum of PCL. CCDC 2288930–2288935 (Fe1, Fe2, Fe4, Fe4′, Fe5 and Fe6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra05867k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Xing Wangbc,
Weiwei Zuo
*ab,
Xing Wangbc,
Weiwei Zuo