
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasonic-induced synthesis of novel diverse arylidenes via Knoevenagel condensation reaction. Antitumor, QSAR, docking and DFT assessment†
Eman El-Sayed Ebeada,
Asmaa Aboelnagaa,
Ekhlass Nassara,
Mohamed M. Naguibb and
Mahmoud F. Ismail *c
*c
aChemistry Department, Faculty of Women for Arts, Science and Education, Ain Shams University, Heliopolis, Egypt
bDepartment of Biochemistry, Faculty of Science, Ain Shams University, 11566, Abbassia, Cairo, Egypt
cDepartment of Chemistry, Faculty of Science, Ain Shams University, 11566, Abbassia, Cairo, Egypt. E-mail: fawzy2010@sci.asu.edu.eg
First published on 10th October 2023
Abstract
A series of arylidenes derivatives was synthesized under ultrasonic methodology via Knoevenagel condensation reaction of cyanoacetohydrazide derivative with the appropriate aldehydes and/or ketone. The anticancer properties of the newly synthesized compounds were tested against four different human cancer cell lines (HEPG-2, MCF-7, HCT-116, and PC-3); compounds 5d and 6 demonstrated the greatest anticancer activity against all cancer cell lines. The MLR technique was used to create the QSAR model using five molecular descriptors (AATS6p, AATS7p, AATS8p, AATS0i, and SpMax4_Bhv). The examination of the constructed QSAR model equations revealed that the selected descriptors influence the tested compound's anti-proliferative activity. The descriptors identified in this work by QSAR models can be utilized to predict the anticancer activity levels of novel arylidenes derivatives. This will allow for significant cost savings in the drug development process and synthesis at pharmaceutical chemistry laboratories. According to the physicochemical properties, the results revealed that all of these compounds comply with Lipinski's Rule of Five, indicating that they may have high permeability across biological membranes and reveal drug-relevant properties. The Swiss Target Prediction webtool was used to assess the probable cellular mechanism for the promising candidate compounds (5d and 6), and the results revealed that adenosine A1 receptor (ADORA1) was a common target for both compounds. ADORA1 is involved in the regulation of cell metabolism and gene transcription. ADORA1 overexpression has been linked to a variety of cancers, including colon cancer, breast cancer, leukemia, and melanoma. The docking study of tested compounds 5d and 6 revealed that their binding scores to ADORA1 are more favorable than those of its co-crystalized ligand (DU172, selective ADORA1 antagonist) and adenosine (ADORA1 endogenous agonist), implying that they may hold great promise as an anti-cancer therapy. Density functional theory (DFT) with a (B3LYP)/6-31G (d,p) basis set was used to calculate the physicochemical parameters of these compounds. The theoretical data from the DFT computation was found to be in good agreement with the experimental values.
1. Introduction
In the field of heterocyclic compounds designation, there has been a markedly increased interest in the invention of new heterocyclic compounds, especially nitrogen-bearing ones, because of their prevalence in nature and also in pharmacology. Piperidine moiety is a generative basis of numerous therapeutically important compounds due to its numerous biological and pharmacological activities1,2 including anticancer3–5 anti-proliferative compounds against MCF-7 breast cancer and PC-3 prostate, HepG2 cancer cells,6 antidiabetic,7 antidepressant,8 H3R antagonistic,9 antibacterial,10 antifungal,11,12 antiviral, anti-HIV, antineoplastic,7 α2c antagonists,13 anesthetics,14 anti-inflammatory,15,16 hypotensive,17 antituberculosis18 and analgesic agent.19 Moreover, piperidine complexes represent one of the most ubiquitous heterocyclic moieties existing in food and drug administration (FDA-approved drugs) (Fig. 1).20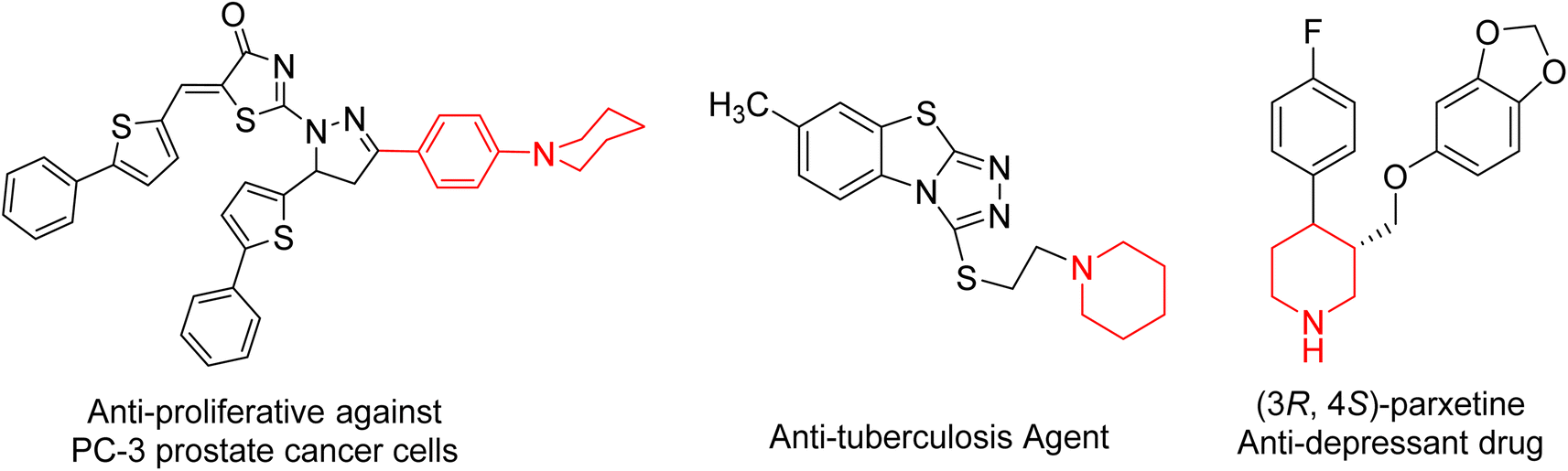 | ||
| Fig. 1 Selected pharmaceutical structures containing piperidine scaffold. | ||
On the other hand, cyanoacetohydrazide derivative is a particularly promising precursor in the combinatorial synthesis of functionalized heterocyclic compounds21 because cyanoacetohydrazide possesses a variety of reactive functional groups represented in different electrophilic and nucleophilic centers.22–26 So that, cyanoacetohydrazide is a privileged starting molecule for developing possible bioactive agents, and their derivatives represent an important class of heterocyclic compounds, they exhibited a wide spectrum of bioactivity. For example, antitumor,27–29 anti-fungal,30 anti-inflammatory,31,32 anti-microbial,33,34 as well as insecticidal35,36 and corrosion inhibitory activities.37
Noteworthy, it is well known that the ultrasonic energy has become widely used in pharmaceutical and industrial applications.38 Being a tool of green and sustainable chemistry, it is employed in organic synthesis39 as it enhances yield and purity of products, shortens reaction time by increasing reaction rate and mass transfer, and allows using milder reaction conditions in comparison to conventional thermal methods.40
Accordingly, we utilized an eco-friendly method; ultrasonic waves for synthesis of 2-cyano-N'-(4-(piperidin-1-yl)benzylidene)acetohydrazide 3 as a synthon to synthesize novel arylidenes and heteroarylidenes. The newly synthesized compounds were confirmed via spectral analyses such as FT-IR, 1H NMR, 13C NMR, and mass spectra. These compounds were evaluated for their antitumor activity. The chemical and physicochemical properties of the biochemical arrangements can be predicted using different computational performances41 by studying the stability of newly synthesized compounds and investigating the results of their biological evaluation by optimizing theoretical modeling with DFT/B3LYP/6-31G (d,p) density functional theory beside docking study.
2. Results and discussion
2.1. Chemistry
Initially, we documented the synthesis of 2-cyano-N′-(4-(piperidin-1-yl)benzylidene)acetohydrazide 3 via the forthright condensation of 4-(piperidin-1-yl)benzaldehyde 1 (ref. 42) with the indispensable cyanoacetohydrazide 2 (ref. 43) in absolute ethanol under both conventional42 and ultrasonic conditions (Scheme 1). Noticeably, the ultrasonic methodology gave a better yield with higher purity in a shorter time than the conventional method, as illustrated in Scheme 1.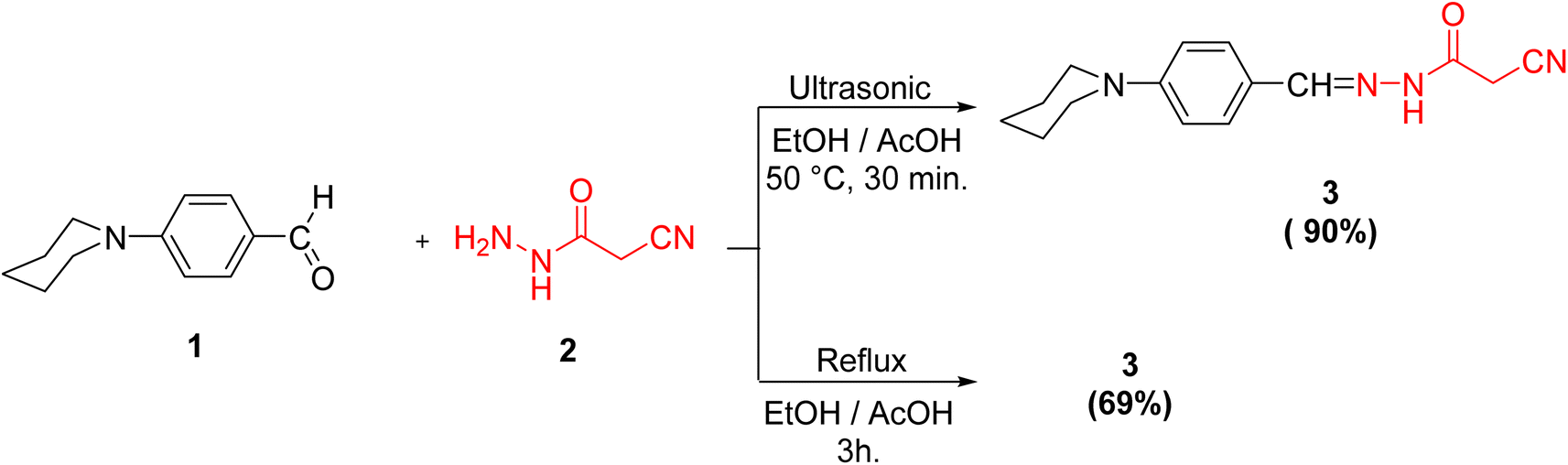 | ||
| Scheme 1 Synthesis of the target compound 3. | ||
Structurally, compound 3 was established by using different spectroscopic data. In the IR spectrum, there are three absorption bands for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, C
N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN functionalities at 2263, 1701, and 1610 cm−1, respectively. Additionally, the 1H NMR spectrum of 3 showed signals in accordance with NH, methylene, methine, piperidine and aromatic protons of the proposed structure. It was worthy of note that both 1H and 13C NMR spectra elucidated the presence of compound 3 in two diastereomeric isomers (Z-/E-) in the ratio 31
O and CN functionalities at 2263, 1701, and 1610 cm−1, respectively. Additionally, the 1H NMR spectrum of 3 showed signals in accordance with NH, methylene, methine, piperidine and aromatic protons of the proposed structure. It was worthy of note that both 1H and 13C NMR spectra elucidated the presence of compound 3 in two diastereomeric isomers (Z-/E-) in the ratio 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :69. The chemical shift values of the signals of NH and methylene protons of the E-isomer showed at lower field, but the chemical shift value of the signal of the imino methine (CHN) proton showed at higher field than the corresponding Z-isomer due to the coplanarity of 4-(N-piperidinyl)phenyl group and –NH–CO-CH2CN moiety in the E-isomer, which led to extending the conjugation about it (Fig. 2).
:69. The chemical shift values of the signals of NH and methylene protons of the E-isomer showed at lower field, but the chemical shift value of the signal of the imino methine (CHN) proton showed at higher field than the corresponding Z-isomer due to the coplanarity of 4-(N-piperidinyl)phenyl group and –NH–CO-CH2CN moiety in the E-isomer, which led to extending the conjugation about it (Fig. 2).
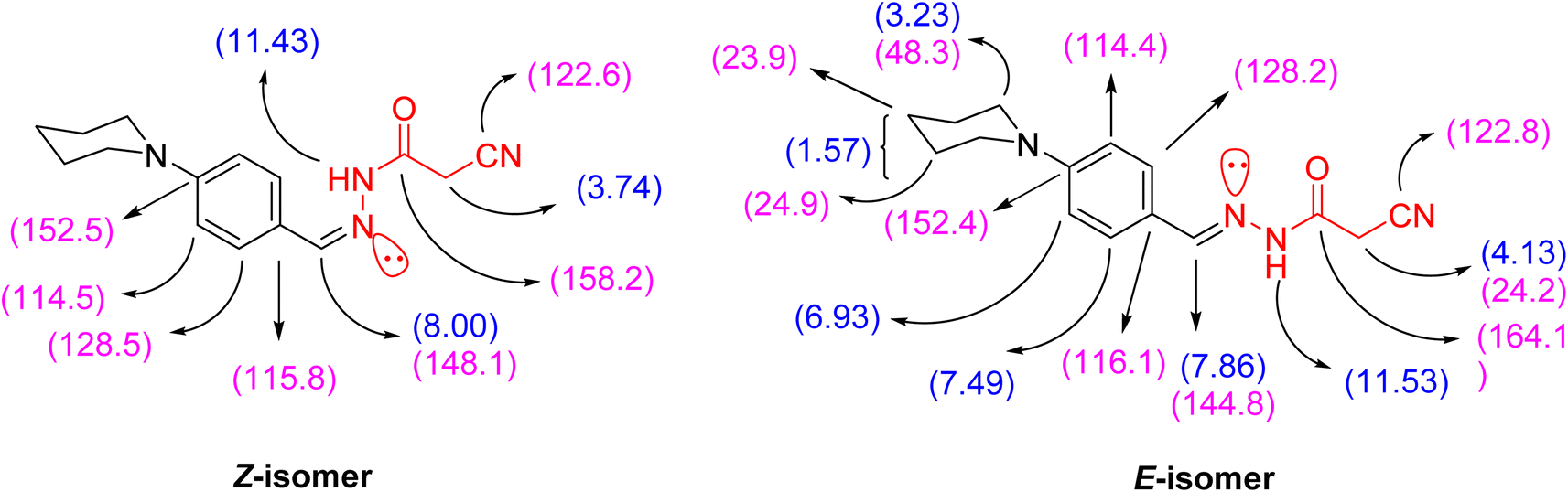 | ||
| Fig. 2 1H and 13C NMR values of the diastereomeric isomers of compound 3. | ||
The ratio between the two diastereomeric isomers (Z-/E-) was explicated computationally by the binding energy of both Z- and E-isomers; the E-isomer has a binding energy (ET = −876.690772 a.u) smaller than the corresponding Z-isomer (ET = −876.684965 a.u). Also, the energy gap of the E-isomer is 3.8887 eV is smaller than the energy gap of the corresponding Z-isomer is 4.1775 eV. It indicated that the E-isomer is more stable than the Z-one. The optimized geometry of both isomers clarified the coplanarity of the E-isomer and the letter has a dipole moment (μ = 11.696023 Debye) greater than the Z-isomer (μ = 8.552684 Debye) (Fig. 3).
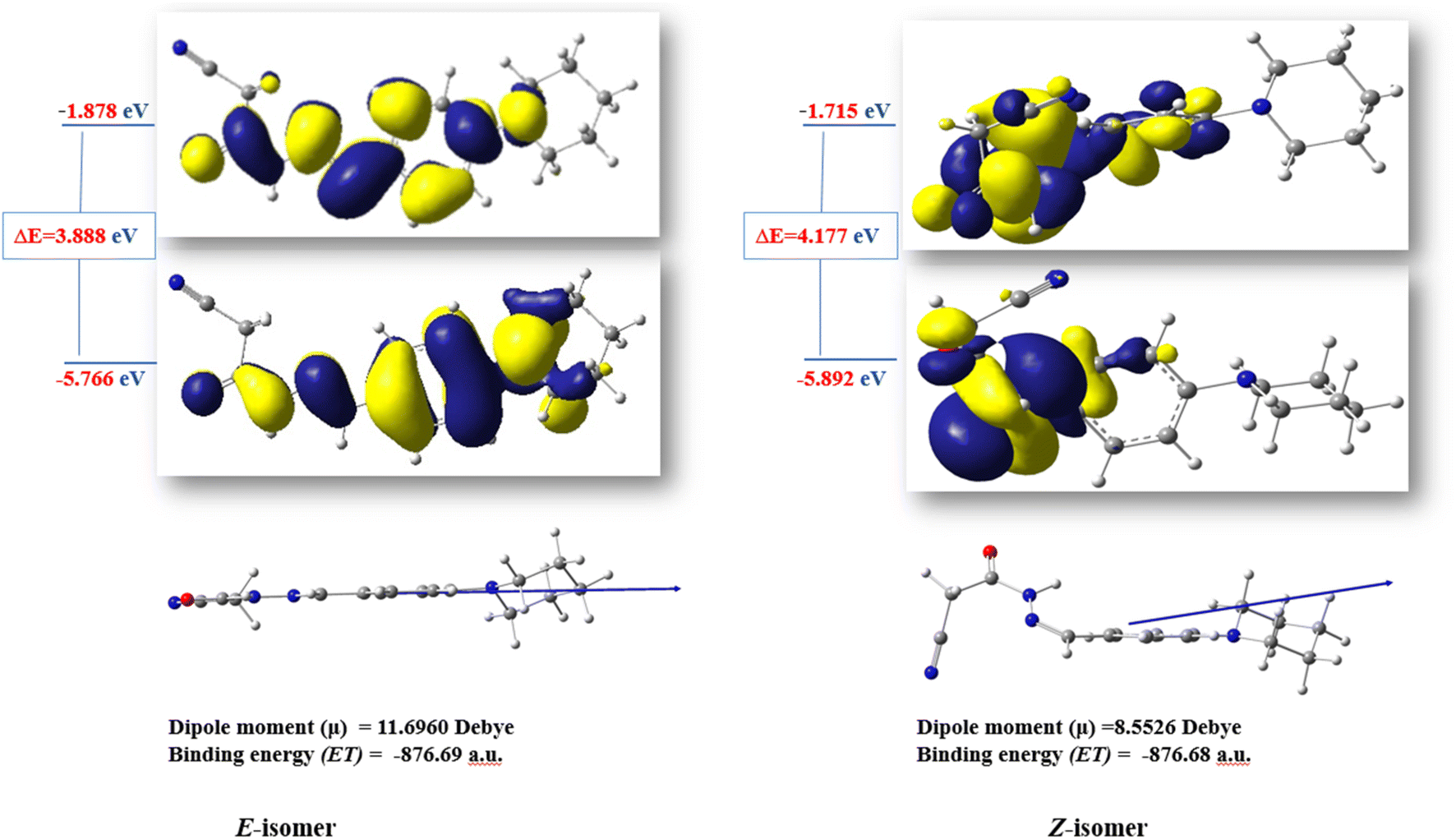 | ||
| Fig. 3 The optimized structures and HOMO–LUMO energy gap of the diastereomeric isomers of compound 3. | ||
Cyanoacetohydrazide moiety possesses manifold reaction sites, including electrophilic and nucleophilic sites. Therefore, let us dedicate our efforts to demonstrate the reactivity of the active methylene (as a nucleophilic center in the presence of a secondary base such as piperidine) of cyanoacetohydrazide derivative 3 under Knoevenagel condensation reaction using ultrasonic methodology. For example, sonication of compound 3 with a diverse range of aldehydes, like isonicotinaldehyde, 1H-pyrrole-2-carbaldehyde, 1H-indole-3-carbaldehyde and anthracene-9-carbaldehyde, beside a ketone, namely 4-chloroacetophenone, gives rise to the arylidene derivatives 4 and 5a–d, respectively.
Interestingly, the spectral analyses of the arylidene derivatives 4 and 5a–d unambiguously elucidated the predictable structures. For instance, the IR spectrum of 4 showed the stretching frequency of the conjugated cyano group at 2200 cm−1. Indisputably, the 1H NMR spectrum revealed two singlet peaks at 8.28 and 8.01 ppm compatible with HCN and HCC protons, and four doublet peaks: two peaks at 7.53 and 6.96 ppm with J = 8.0 Hz compatible with benzylidene protons and the other two doublet peaks at 7.91 and 7.04 ppm with J = 8.8 Hz compatible with C2,6–H(pyridine) and C3,5–H(pyridine) protons, respectively.
For arylidene derivative 5b, the vanish of the cyano group from the IR which indicated the partially hydrolyzed of cyano functionality. Also, the appearance of two peaks at 189.9 and 184.9 ppm in the 13C NMR spectrum indicated the presence of two carbonyl groups. Moreover, the 1H NMR spectrum visibly displayed three singlet peaks at 9.93, 9.67 and 8.27 ppm corresponding to HCN, HCC and C2–H(indole) protons, respectively. Furthermore, the mass spectrum of 5b showed the molecular ion peak at m/z = 415.97 (36.71%) which is in agreement with the molecular formula of the partially hydrolyzed arylidene derivative 5b (Scheme 2).
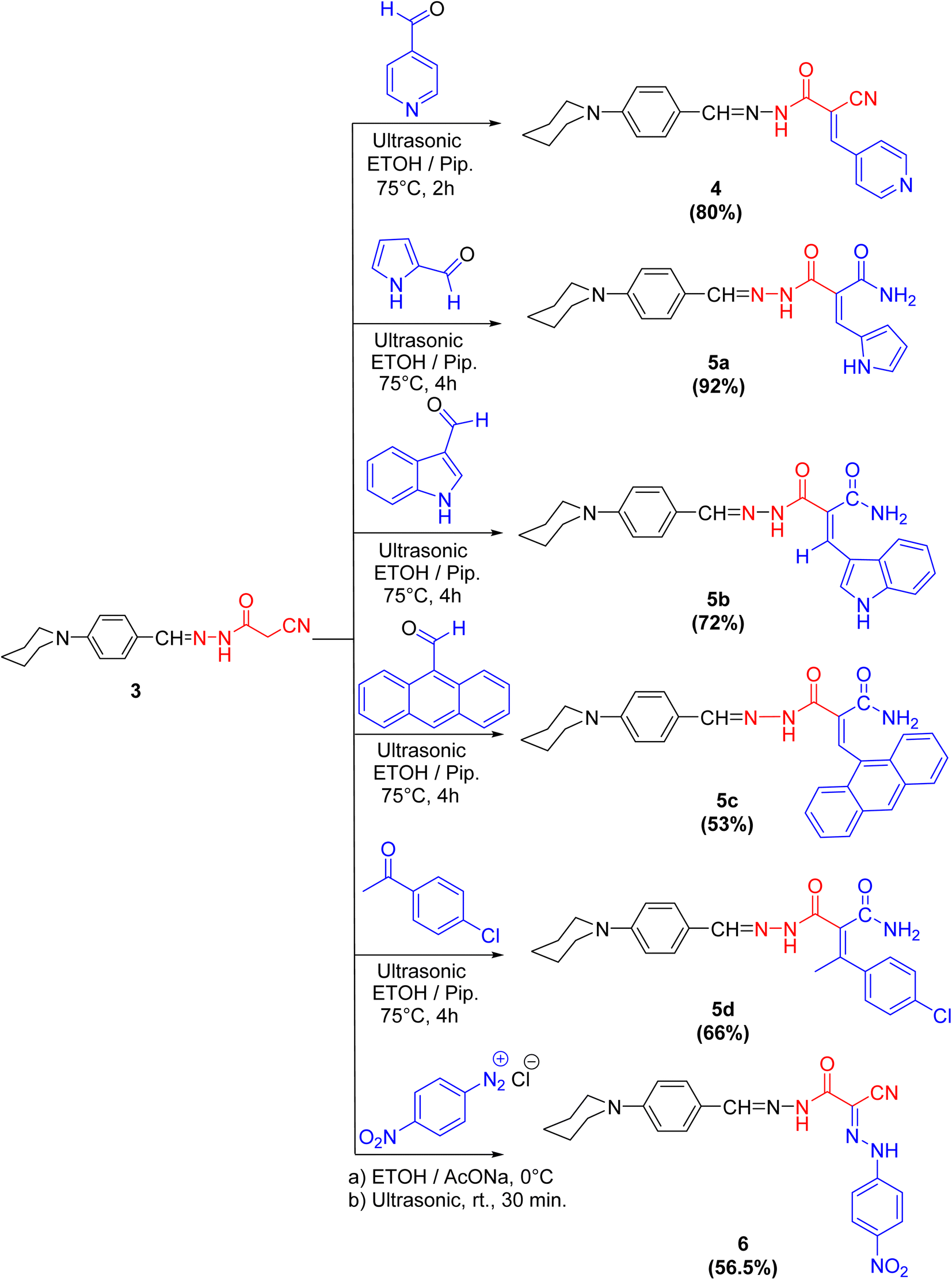 | ||
| Scheme 2 Synthetic pathway to compounds 4–6. | ||
In the coupling reaction, cyanoacetohydrazide derivative 3 was viable at the active methylene with 4-nitrobenzene diazonium chloride at ambient temperature under sonication conditions, leading to the formation of the hydrazone derivative 6 in a considerable yield. The IR spectrum displayed two bands characteristic for the asymmetric and symmetric vibrational coupling modes of the nitro group at 1515 and 1338 cm−1, respectively. Further, in 1H NMR spectrum of compound 6, two broad singlet peaks commutable with D2O at 9.66 and 6.58 ppm compatible with two NH protons were displayed.
It was notably that, in case of formation of compounds 5a–d, the nitrile group is partially hydrolyzed to the corresponding amide but through formation compounds 4 and 6, the nitrile group is preserved, because of isonicotinaldehyde is very reactive aldehyde due to pyridine ring act as an electron withdrawing group especially in position 2- and 4-, which accelerate the formation of compound 4 in a time not enough to hydrolyze the nitrile group. Also, the formation of compound 6 was done at ambient temperature and that was not enough to hydrolyze it.
2.2. Pharmacology
According to our initial screening results, compounds 5d and 6 had the highest cytotoxic activity, while compounds 3, 4, 5a, and 5b showed intermediate cytotoxic activity. The cytotoxicity of compound 5c was minimal (Table 1).
| Compound no. | In vitro cytotoxicity IC50b (μM) | |||
|---|---|---|---|---|
| HePG2 | HCT-116 | MCF-7 | PC3 | |
| a The data are expressed as the mean of three replicates ± standard deviation.b IC50 (μM): 1–10 (very strong). 11–20 (strong). 21–50 (moderate). 51–100 (weak) and above 100 (non-cytotoxic).c DOX: doxorubicin. | ||||
| DOX | 4.50 ± 0.2 | 5.23 ± 0.3 | 4.17 ± 0.2 | 8.87 ± 0.6 |
| 3 | 39.72 ± 3.5 | 25.34 ± 3.8 | 24.07 ± 3.6 | 38.60 ± 4.3 |
| 4 | 29.64 ± 3.0 | 23.79 ± 3.5 | 20.35 ± 2.5 | 31.38 ± 3.8 |
| 5a | 24.31 ± 2.6 | 14.11 ± 2.4 | 21.07 ± 2.3 | 34.76 ± 2.9 |
| 5b | 12.89 ± 0.9 | 17.70 ± 1.4 | 17.22 ± 0.6 | 14.65 ± 1.8 |
| 5c | 31.83 ± 2.1 | 44.06 ± 3.2 | 45.20 ± 2.0 | 41.34 ± 3.6 |
| 5d | 6.94 ± 0.4 | 11.62 ± 1.0 | 5.60 ± 0.4 | 10.51 ± 1.3 |
| 6 | 10.11 ± 3.1 | 8.23 ± 3.3 | 7.61 ± 3.4 | 13.83 ± 3.8 |
2.3. Structure activity relationship's (SAR's)
The structural activity relationships (SAR) were studied by comparing the experimental cytotoxicity of the newly synthesized compounds according to their structures as follows:(1) The structure of the parent compound 3 is based on the presence of the piperidine moiety, which is an important core of many drug molecules with antihistamine, anticancer, and antibacterial properties45 and hydrazone moiety, which is important for bioactivity as it undergoes azo reduction into toxic amine derivatives. In addition, the hydrazone functionality is known for its anticancer properties.46,47
(2) The cytotoxic activity of the parent compound 3 was found to be in a moderate value compared to the standard drug, doxorubicin which could be due to the presence of piperidine and hydrazone moieties.
(3) Different moieties possessing different lipophilic heteroaryl and phenyl rings with a variety of electron withdrawing and electron donating groups were added to the parent compound 3 in order to investigate their anticancer activities.
(4) Introduction of nitrogen bearing heterocyclic ring systems as pyridine, pyrrole and indole rings in compounds 4, 5a and 5b, respectively, improve the cytotoxic activity towards all cell lines as compared to compound 3 because the nitrogen bearing heterocycles and their products show several essential features and are being utilized in different ways as anticancer.
(5) Compounds 5d and 6 showed the highest anticancer activity towards all cell lines, this could be due to:
(a) The presence of hydrophobic chlorobenzene moiety (compound 5d) that may be essential for binding with the active site of many cellular enzymes, and electron donating group (Cl) that promotes the antitumor effect.48
(b) The presence of extra hydrazone moiety and nitro group (compound 6) that may be critical for anticancer activity.
(6) Incorporation of anthracene nucleus (compound 5c) showed lowest anticancer activity towards all cell lines as compared to the other tested compounds.
(7) Regarding the SAR of tested compounds (Fig. 4), there was a consistent relation between the lipophilicity and/or electronic property of the substituent groups and the antiproliferative activity. The introduction of a more nucleophilic and a lipophilic substituent to the parent compound 3 enhances the potency against cancer cell lines.
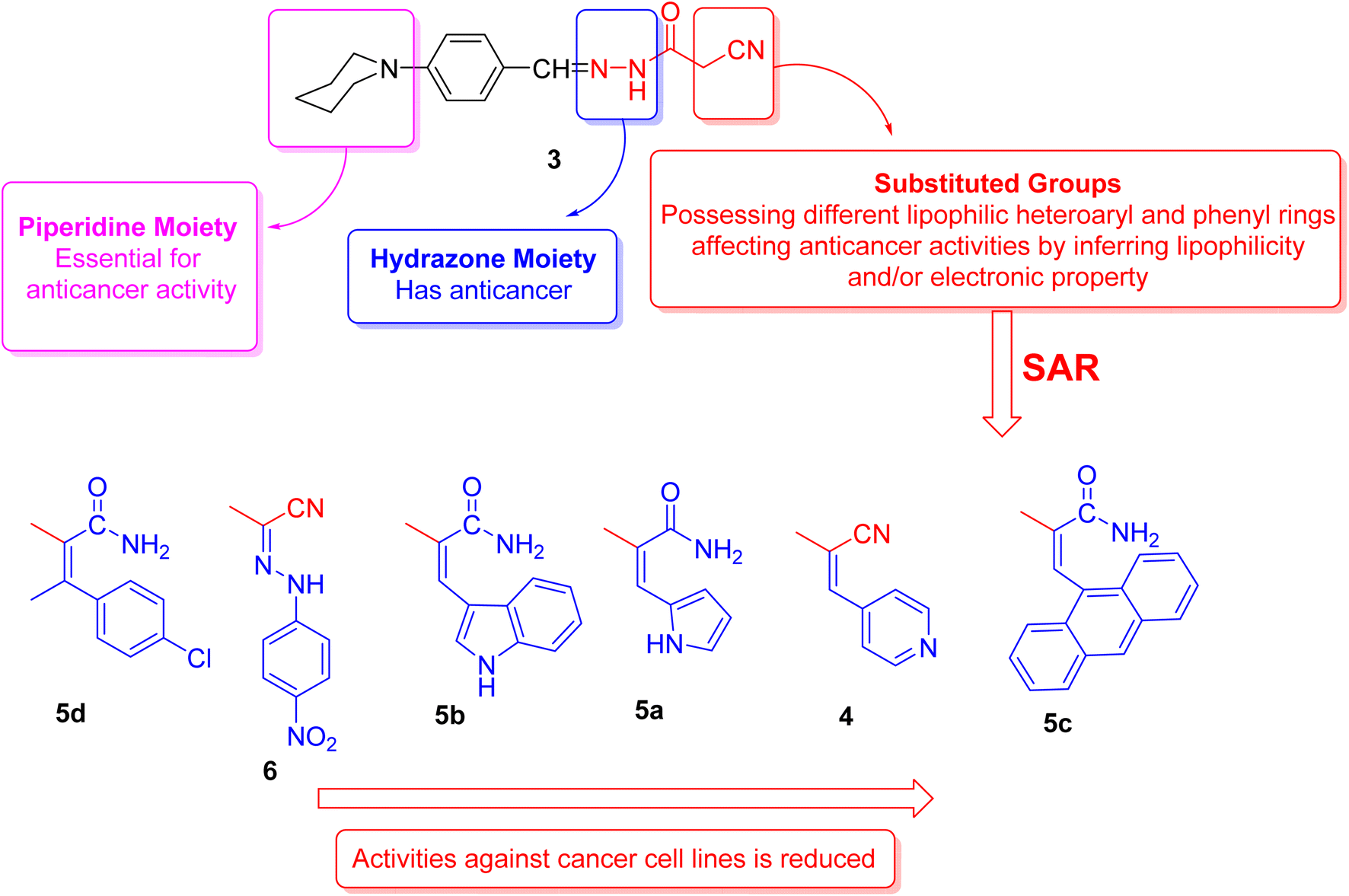 | ||
| Fig. 4 Structure activity relationship of piperidine derivatives. | ||
3. Quantitative structure–activity relationships (QSAR)
Quantitative structure–activity relationships (QSAR) are effective tools for drug discovery.49,50 We calculated multidimensional molecular descriptors (constitutional, topological, physicochemical, geometrical, and quantum) to find molecular descriptors associated to the anti-proliferative activity of the investigated compounds. We use statistical approaches to build QSAR models to achieve this goal.51 In this study, we employed the tested compounds to build QSAR models. Multiple linear regression (MLR) is the most widely used method in the development of QSAR due to its simplicity and robustness.52 Internal and external validations are used to test the predictive capacity of the produced models.3.1. The studied compounds
To develop the QSAR molecular modeling, we used the experimental IC50 values of the tested compounds that showed strong and moderate anti-cancer activities (IC50 < 35). In QSAR modeling, we don't use the activity in the form of IC50 (μM) but rather we need to convert it to the log scale to get pIC50 level (M) and this was done through the following equation (pIC50 = −log10(IC50 × 10−6)), which are presented in Table 2.| Compound no. | IC50 (μM) | pIC50 (M) |
|---|---|---|
| 3 | 24.07 | 4.6185 |
| 4 | 20.35 | 4.6914 |
| 5a | 14.11 | 4.8505 |
| 5b | 12.89 | 4.8897 |
| 5c | 31.83 | 4.491 |
| 5d | 5.6 | 5.2518 |
| 6 | 7.61 | 5.1186 |
3.2. Calculation of molecular descriptors
The studied molecules were initially subjected to energy minimization and then saved as SD files for later use in descriptor calculation. The calculation of molecular descriptors for the studied molecules began with the import of the various molecules in SD file format into the PaDEL-Descriptor software, which creates 1875 descriptors (1444 1D, 2D, and 431 3D descriptors).53 Data pretreatment was performed to eliminate uninformative descriptors from the generated descriptors pool (low variance descriptors, zeros and NA descriptors).3.3. Statistical methods
To construct QSAR model for the seven tested molecules obtained by in vitro synthesis, we used the statistical methods presented below.
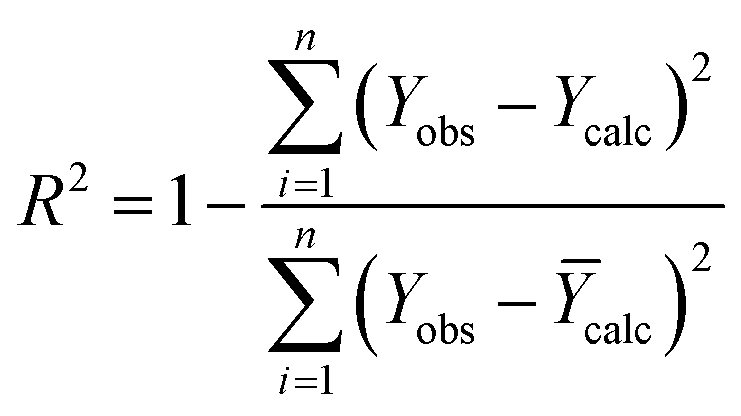 | (1) |
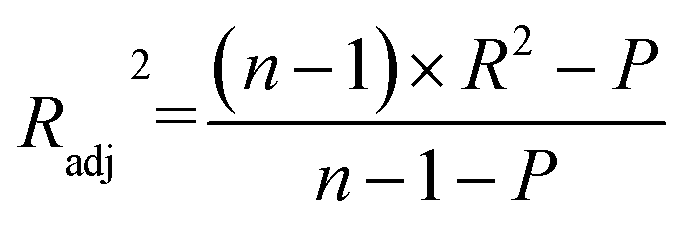 | (2) |
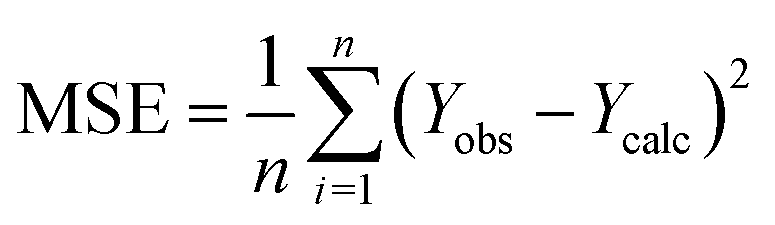 | (3) |
| Descriptors | Description | Class | MLR of the selected descriptors |
|---|---|---|---|
| a MLR: multiple linear regression; RSE: residual standard error. | |||
| AATS6p | Average Broto–Moreau autocorrelation – lag 6/weighted by polarizabilities | 2D | RSE: 0.02785 |
| AATS7p | Average Broto–Moreau autocorrelation – lag 7/weighted by polarizabilities | 2D | Multiple R-squared: 0.9982 |
| AATS8p | Average Broto–Moreau autocorrelation – lag 8/weighted by polarizabilities | 2D | Adjusted R-squared: 0.9894 |
| AATS0i | Average Broto–Moreau autocorrelation – lag 0/weighted by first ionization potential | 2D | F-Statistic: 112.8 |
| SpMax4_Bhv | Largest absolute eigenvalue of Burden modified matrix – n 4/weighted by relative van der Waals volumes | 2D | p-Value: 0.07135 |
| Compound no. | AATS6p | AATS7p | AATS8p | AATS0i | SpMax4_Bhv |
|---|---|---|---|---|---|
| 3 | 1.3495 | 1.3192 | 1.4282 | 160.1803 | 3.4539 |
| 4 | 1.2542 | 1.2236 | 1.3132 | 162.531 | 3.4523 |
| 5a | 1.3177 | 1.2991 | 1.2796 | 160.643 | 3.5643 |
| 5b | 1.3895 | 1.3419 | 1.2944 | 157.6286 | 3.5792 |
| 5c | 1.229 | 1.3354 | 1.3717 | 160.462 | 3.5364 |
| 5d | 1.4044 | 1.3469 | 1.309 | 163.3607 | 3.5039 |
| 6 | 1.3196 | 1.2695 | 1.2569 | 165.019 | 3.4937 |
The ChemMaster1.1 software (https://crescentsilico.wordpress.com/chemmaster/) was used for the model building based on MLR approach that establishes the relationship between the dependent variable (pIC50) and the independent variables (molecular descriptors)56 according to the following equation (eqn (4)).57
| Y = k1x1 + k2x2 + k3x3 + … C | (4) |
After identifying the most significant molecular descriptors, we divided the database into two sets (training and test). Thus, the training and test sets include 75% and 25% of the total data, respectively.58 The training set was used to create QSAR models, whereas the test set was used to assess the effectiveness of the developed model. The Kennard Stone method,59 which is supplied by the ChemMaster1.1 software, was used to divide the data set into training and test sets.
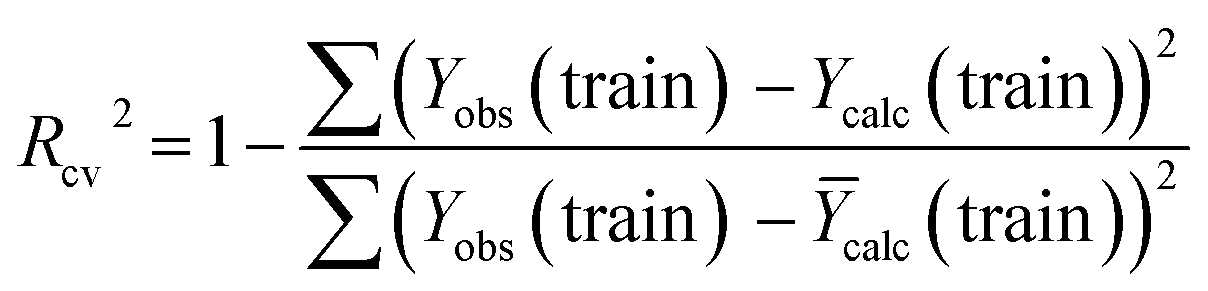 | (5) |
| R2 | RMSE | MSE | Rcv2 | |
|---|---|---|---|---|
| a RMSE: the root mean square error. | ||||
| Training set | 0.99 | 0 | 0 | 0.6 |
| Test set | 0.52 | 0.091 | 0.092 | |
The R2 defines the goodness of fit of the QSAR model. A QSAR model is considered acceptable when it has an R2 value > 0.6 for the training set. This model has an R2 of 0.99 for the training set. The value of Rcv2 should be more than 0.5 that indicates the accuracy of the obtained QSAR model through MLR technique.60 The value of Rcv2 less than R2 value indicates the fragility and weakness of the model when excluding any element of the training set. Therefore, it is apparent that the five descriptors in eqn (6) show a strong linear correlation with the biological activity of pIC50.
| pIC50 = +0.1495 × AATS6P + 0.04 × AATS7P − 0.0882 × AATS8P + 0.0162 × SPMAX4_Bhv + 0.1048 × AATS0i + 4.7819 | (6) |
An external validation assessment was performed to determine the developed model's ability to predict the activities of the external test set compounds. Compounds from the series of molecules investigated in this work are included in the test set, but they did not contribute to the construction of the QSAR models.
The external ability of the QSAR models to predict the activity of the test set molecules was assessed by calculating the correlation coefficient R2 test between the observed pIC50 values and the predicted pIC50 values after the inclusion of the test set61 (Table 6).
| Compound no. | pIC50 | Pred. pIC50 |
|---|---|---|
| 3 | 4.6185 | 4.62 |
| 4 | 4.6914 | 4.7 |
| 5a | 4.8505 | 4.84 |
| 5b | 4.8897 | 4.79 |
| 5c | 4.491 | 4.5 |
| 5d | 5.2518 | 5.3 |
| 6 | 5.1186 | 5.21 |
The model is statistically acceptable in prediction when the value of R2 test is greater than 0.5 and can be applied to new external data.62 This model has an R2 of 0.52 for the test set. As a result, external validation of the QSAR models ensures that these models have a high predictive potential for pIC50 values.
Fig. 5 illustrates the correlation between the observed and expected activity values of pIC50. The latter are derived for the molecules in both the test and training sets using a QSAR model based on the MLR approach. Fig. 5 shows that the distribution of observed and predicted pIC50 values is significantly correlated, which is attributable to the low MSE value achieved. As a result, it is obvious that the experimentally measured values and the QSAR model predictions are correlated.
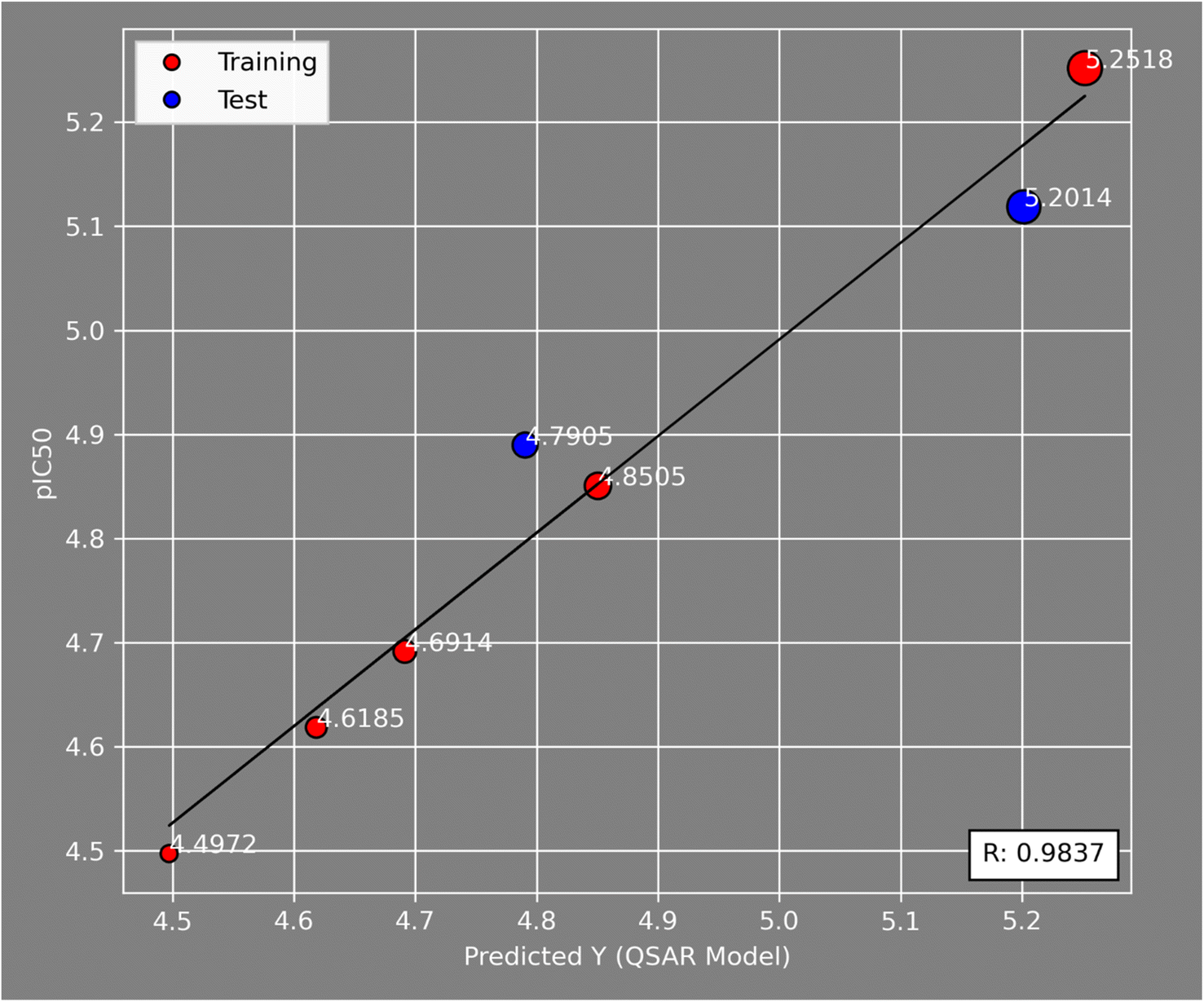 | ||
| Fig. 5 Correlations between the observed activity (pIC50) values and the predicted ones via the MLR model. | ||
4. Physicochemical properties
A successful drug candidate should achieve a precisely balanced combination of physicochemical properties and pharmacokinetics. ADMETlab 2.0, which is implemented as a publicly accessible web server with an easy-to-use interface, was used to calculate the overall physicochemical properties of the test compounds (Table 7).| Compound no. | MW | nHD | nHA | LogP |
Lipinski's rule | LogS |
QED |
|---|---|---|---|---|---|---|---|
| a Where; MW, molecular weight; nHA, number of hydrogen bond acceptor; nHD, number of hydrogen bond donor; logS, logarithm of aqueous solubility value, logP, logarithm of the n-octanol/water distribution coefficient; QED, a measure of drug-likeness based on the concept of desirability. |
|||||||
| 3 | 270.15 | 1 | 5 | 2.581 | Accepted | −4.048 | 0.672 |
| 4 | 359.17 | 1 | 6 | 3.551 | Accepted | −4.453 | 0.358 |
| 5a | 365.19 | 4 | 7 | 2.807 | Accepted | −4.213 | 0.24 |
| 5b | 415.2 | 4 | 7 | 3.715 | Accepted | −4.814 | 0.189 |
| 5c | 476.22 | 3 | 6 | 5.432 | Accepted | −6.462 | 0.102 |
| 5d | 424.17 | 3 | 6 | 4.451 | Accepted | −5.031 | 0.243 |
| 6 | 419.17 | 2 | 10 | 4.926 | Accepted | −6.068 | 0.402 |
To have robust membrane permeability, a drug candidate must have a molecular weight ≤ 500, partition coefficient values in the octanol/water system (logP) ≤ 5, and a number of hydrogen bond donors and acceptors ≤ 5 and ≤ 10, respectively.63 All of the compounds studied had hydrogen bond acceptors (nHA) of less than 8 and hydrogen bond donors (nHD) of less than 5. This is consistent with Lipinski's rule of five, indicating that these chemicals may have strong permeability or absorption properties across biological membranes. Furthermore, the majority of the compounds' lipophilicity, expressed as logP, was found to be less than 5, confirming their drug-relevant properties.
QED is a drug-likeness metric built on the concept of desirability. QED is determined by integrating the results of desirability functions based on eight drug-likeness-related variables, including MW, logP, nHA, nHD, the number of aromatic rings (nAr), and the number of alerts for undesirable functional groups. The average QED for attractive compounds is 0.67, 0.49 for unfavourable compounds, and 0.34 for unattractive compounds regarded as too complex.64 Compound 3 may have drug-like properties.
5. Molecular docking studies
Focused highlights and molecular docking studies are to be conducted for the most promising candidate compounds (compounds 5d and 6 that showed the highest cytotoxic activities towards the cancer cell lines) in this study to reveal the probable mechanism for their anticancer potency. The probable molecular targets for these compounds were investigated using SwissTargetPrediction (http://www.swisstargetprediction.ch/index.php) webtool provided by Swiss Institute of Bioinformatics (https://www.sib.swiss/) that allows the identification of the most probable macromolecular targets of a small molecule based on a combination of 2D and 3D similarity with a library of 370000 known actives on more than 3000 proteins from three different species (Homo sapiens, Mus musculus and Rattus norvegicus).65
The results of the target prediction revealed the presence of many targets for each compound from various categories including, G-protein coupled receptors (GPCRs), ion channels, kinases, and nuclear proteins. Adenosine A1 receptor (ADORA1; UniProt id: p30542) was found to be the common target for both compounds 5d and 6 (Table 8).
| Compound no. | The probable molecular targets |
|---|---|
| a The common probable molecular target for compounds 5d and 6. | |
| 5d | PIK3CB (PI3-kinase p110-beta subunit) |
| PIK3CA (PI3-kinase p110-alpha subunit) | |
| F9 (coagulation factor IX) | |
| PPID (peptidyl-prolyl cis–trans isomerase D) | |
| MAOB (monoamine oxidase B) | |
| GSK3B (glycogen synthase kinase-3 beta) | |
| CHRM4 (muscarinic acetylcholine receptor M4) | |
| ITK (tyrosine-protein kinase ITK/TSK) | |
| MMP13 (matrix metalloproteinase 13) | |
| ADORA1 (adenosine A1 receptor)a | |
| ADORA3 (adenosine A3 receptor) | |
| ABL1 (tyrosine-protein kinase ABL) | |
| GABRA2 (GABA receptor alpha-2 subunit) | |
| STK17B (serine/threonine-protein kinase 17B) | |
| STK17A (serine/threonine-protein kinase 17A) | |
|
|
| 6 | EDNRA/B (endothelin receptor ET-A/B) |
| SCN9A (sodium channel protein type IX alpha subunit) | |
| TBXA2R (thromboxane A2 receptor) | |
| EPHX1 (epoxide hydrolase 1) | |
| AURKA/B (serine/threonine-protein kinase Aurora-A/B) | |
| MAPK8 (c-Jun N-terminal kinase 1) | |
| MAPK10 (c-Jun N-terminal kinase 3) | |
| JAK1/2 (tyrosine-protein kinase JAK1/2) | |
| PTGS2 (cyclooxygenase-2) | |
| ESR2 (estrogen receptor beta) | |
| IMPDH2 (inosine-5′-monophosphate dehydrogenase 2) | |
| ADORA1 (adenosine A1 receptor)a | |
The adenosine A1 receptor is one member of the adenosine receptor group of G protein-coupled receptors with a high affinity for adenosine, which serves as its endogenous ligand.66 ADORA1 is known to block adenylate cyclase, lowering the level of cyclic adenosine monophosphate (cAMP) and regulating cell metabolism and gene transcription.67 ADORA1 has been found to be overexpressed in a variety of cancers, including colon cancer,68 breast cancer,69 leukemia,70 and melanoma.71
ADORA1 has been found to be upregulated in hepatocellular carcinoma (HCC) tissues, where it functions as an oncoprotein and a promoter of cell proliferation via the PI3K/AKT signaling pathway.72 ADORA1 has been found to be overexpressed in a variety of breast cancer cell lines,69 where it affects the transcriptional activity of the estrogen receptor-α (Erα) in breast cancer cells.73
In the present study, docking of the synthesized compounds 5d and 6 and co-crystalized reference ligands, DU172 (4-(3-(8-cyclohexyl-2,6-dioxo-1-propyl-1,2,6,7-tetrahydro-3H-purin-3-yl)propyl)carbamoyl)benzenesulfonyl fluoride; one of the selective antagonist of ADORA1 and receptor endogenous agonist adenosine (Fig. 6) with ADORA1 (PDB: 5uen) were performed using AutoDock vina modeling simulation software, by placing the small molecules into the extracellular ligand binding pocket of the target in order to preliminarily predict the protein-binding affinity, as well as the preferred orientation of the docking pose. The docking results, presented as binding free energy and the interacting ligand binding pocket residues, are tabulated in (Table 9).
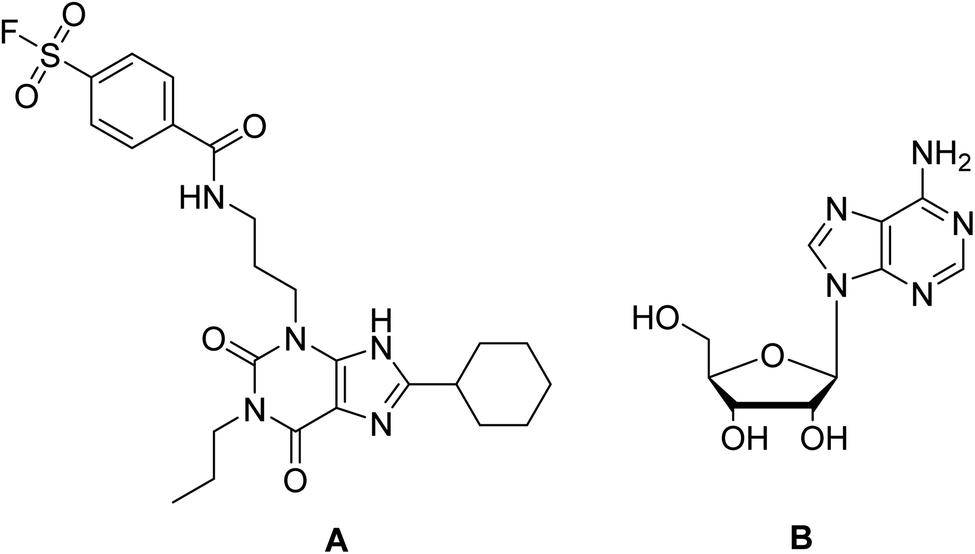 | ||
| Fig. 6 Reference compounds used in molecular docking (A) DU172, selective A1 receptor antagonist. (B) Adenosine, A1 receptor endogenous agonist. | ||
| Compounds | ΔG (kcal mol−1) | Interacted ligand binding pocket residues |
|---|---|---|
| 5d | −9.834 | Val62, Leu65, Ala66, Ile69, Val83, Val87, Leu88, Cys169, Phe171, Glu172, Met180, Leu250 |
| 6 | −9.17 | Tyr12, Asn70, Phe171, Glu172, Leu250, Tyr271, Ile274 |
| Adenosine (endogenous agonist) | −6.578 | Ala66, Ile69, Val83, Val87, Cys169 |
| DU172 (co-crystalized ligand & receptor antagonist) | −8.763 | Ala66, Phe171, Glu172, Leu250, Leu253, Ile274, His278 |
The proposed binding mode of co-crystalized ligand, DU172 and endogenous agonist, adenosine showed an affinity value of −8.763 and −6.578 kcal mol−1, respectively while the studied compounds 5d and 6 showed binding energies −9.834 and −9.17 kcal mol−1, respectively (Table 9).
Comparing to the binding mode of DU172 and adenosine (Fig. 7), we found that the compound 5d form extensive hydrophobic interactions with non-polar amino acid residues that form the binding pocket due to the presence of hydrophobic chlorobenzene moiety and electron donating group (Cl), therefore it binds more strongly to the target as compared to that of the co-crystalized ligand (Fig. 8). The presence of nitrobenzene moiety in compound 6 favors the formation of a number of nonpolar and hydrogen bonds with the amino acid side chains in the binding pockets, making the binding energy is more favorable than that in case of the co-crystalized ligand (Fig. 9).
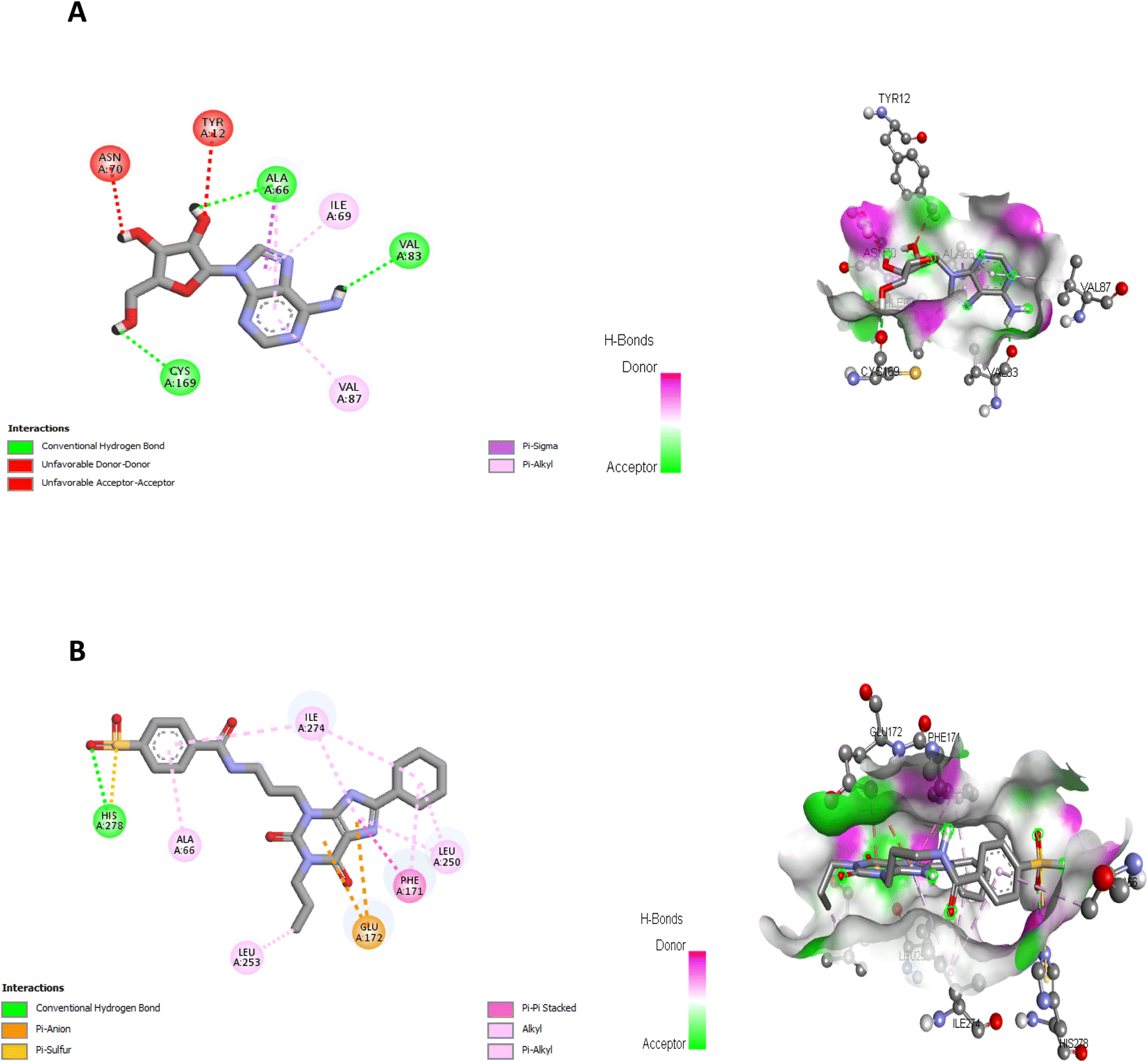 | ||
| Fig. 7 (A) Interactions of ligand binding pocket residues with adenosine (3D (right) and 2D (left)). (B) Interactions of ligand binding pocket residues with DU172 (3D (right) and 2D (left)). | ||
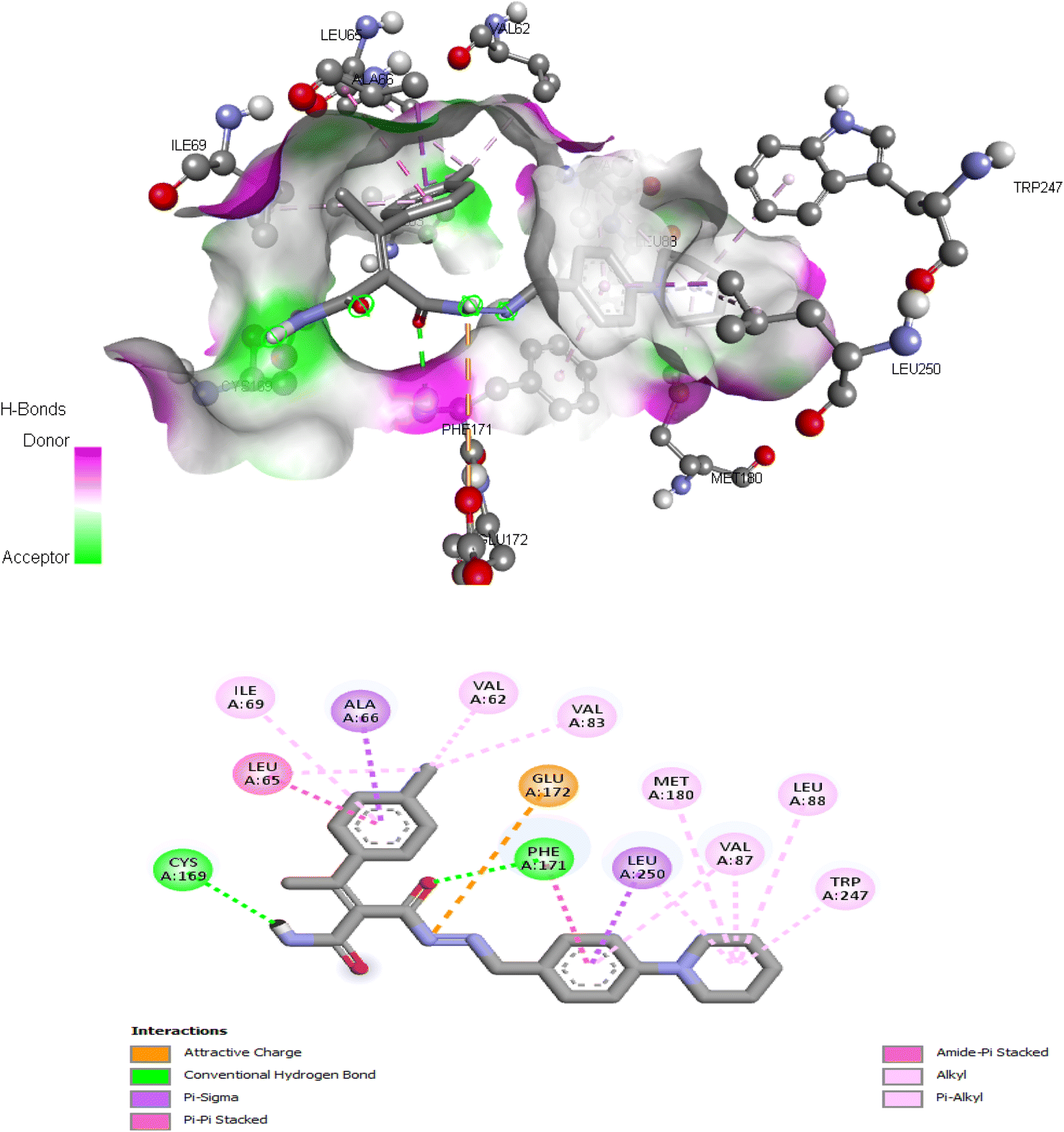 | ||
| Fig. 8 Interactions of ligand binding pocket residues with compound 5d (3D (up) and 2D (down)). | ||
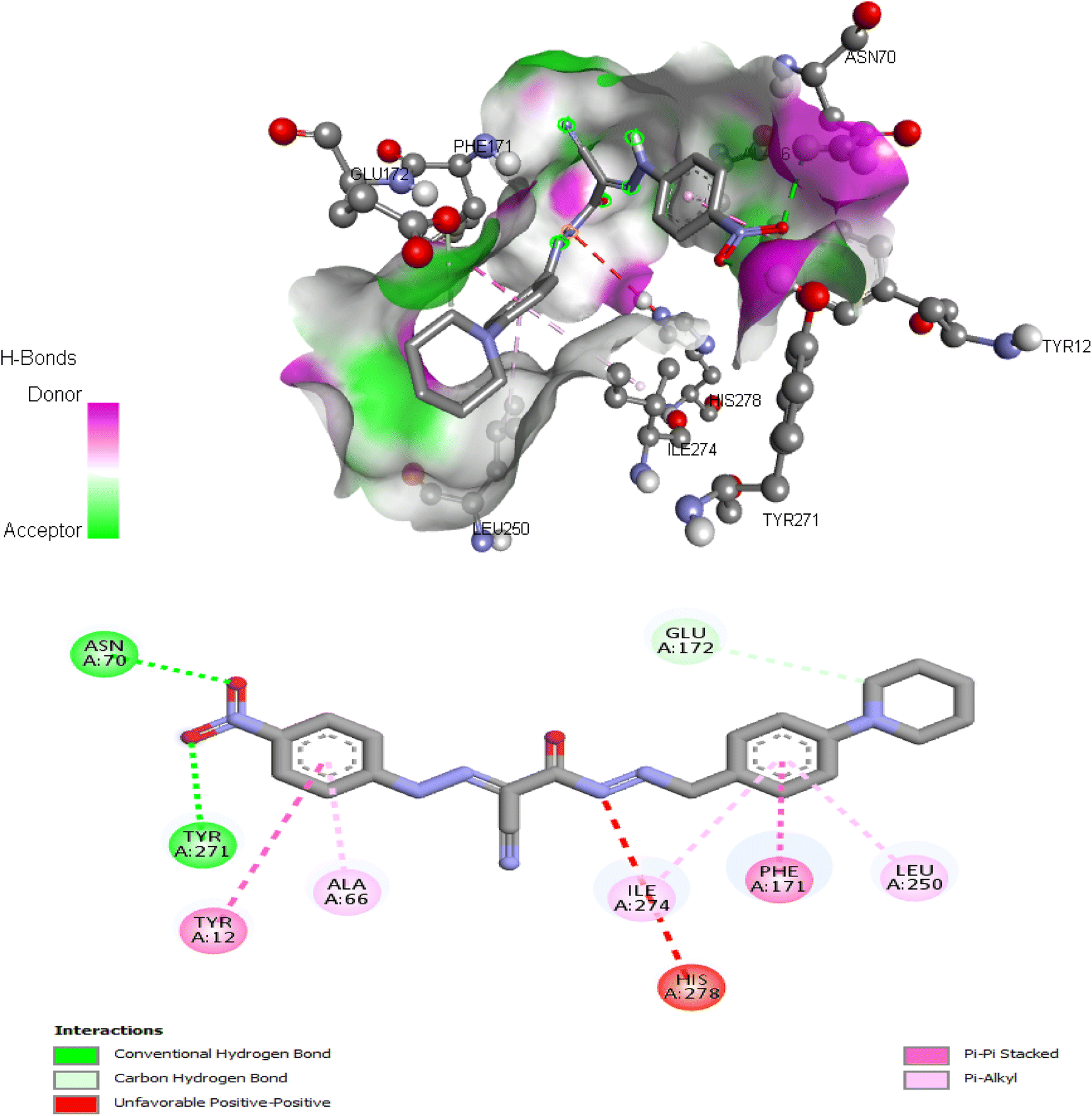 | ||
| Fig. 9 Interactions of ligand binding pocket residues with compound 6 (3D (up) and 2D (down)). | ||
Finally, the binding affinities correlates with the cytotoxicity activity of the tested compounds suggesting that the anticancer activities of the tested compounds may be related to their ability to antagonize and block the action of the extracellular adenosine on ADORA1 receptor signaling in cancer cells, leading to cell death.
6. Theoretical investigation
6.1. Optimization geometry
Density functional theory at the (B3LYP)/6-31G (d,p) level, was used for theoretical analysis of the energies and the geometry of both 3 Z- and E-isomers and its derivatives 4, 5a–d and 6 (Fig. 1 and 10).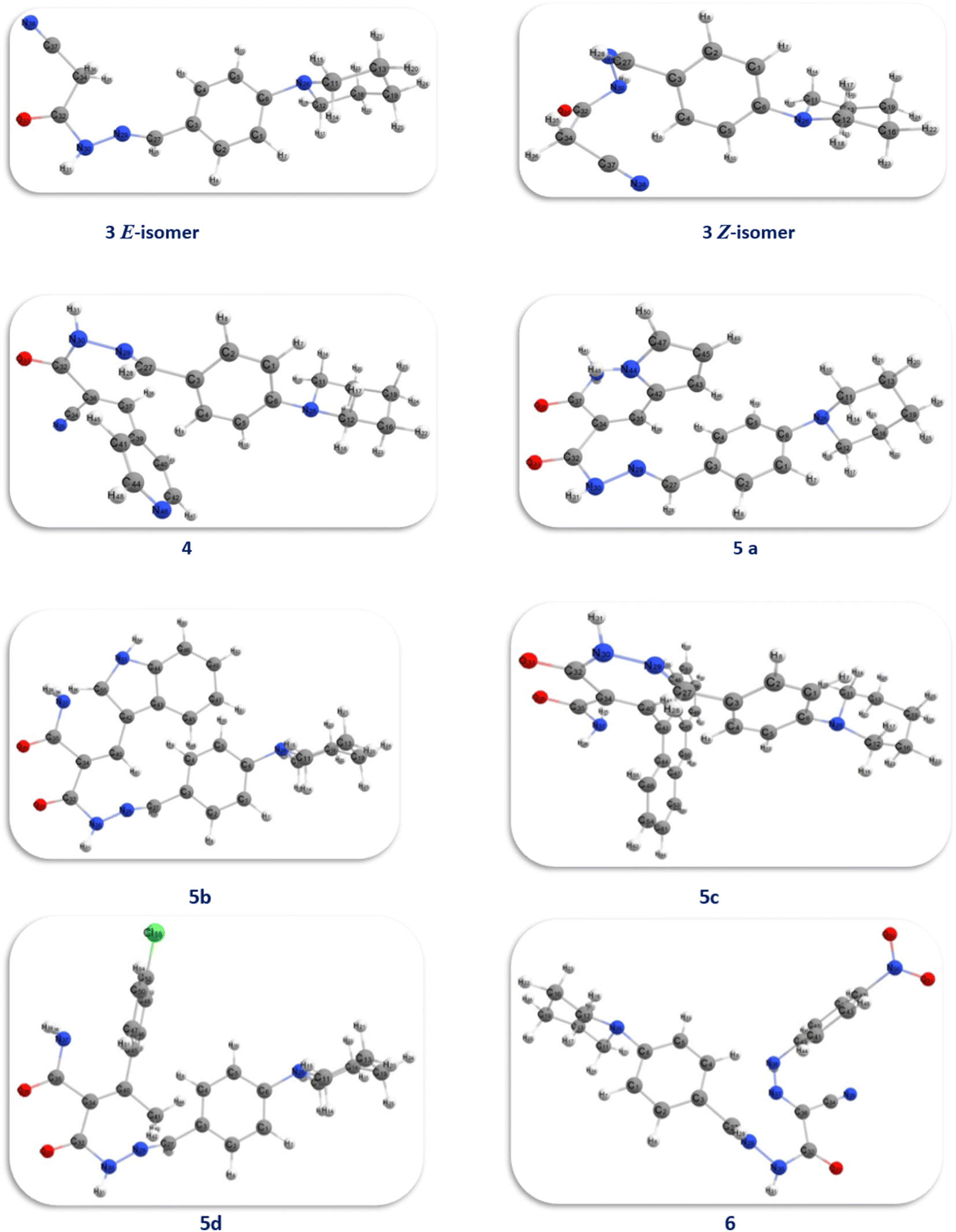 | ||
| Fig. 10 Optimized model of compounds 3, 4, 5a–d and 6. | ||
By studying the optimum geometry of the investigated compounds, it found that the presence of planner benzene ring attached to the piperidine ring which acquires a slightly warped chair shape and the cyanoacetohydrazide moiety making these compounds non planner.74 This non-planarity is a key factor in both its activity and biological activity, as all investigated compounds showed high and moderate antitumor activity, especially compounds 5b, 5d, and 6.
The presence of indole, 4-chlorophenyl, and 4-nitrophenyl rings in 5b, 5d, and 6 respectively, provides structural diversity and electronic effects in these compounds since these rings have distinct electronic properties; for example, the non-planarity of the indole pyrrole ring can affect their interactions with biomolecules. Also, the withdrawal groups, 4-chlorophenyl and 4-nitrophenyl, positively affect the biological activities of the investigated compounds.75
The dipole moment values of the investigated compounds were analyzed from the DFT results, the values of the dipole moment were between 6.2518-11.6960 Debye. The dipole moments tell us about the charge separation in a molecule. The lower the difference in electronegativity of bonded atoms, the lower the dipole moment. Compound 5a demonstrated the lowest value dipole moment of 6.2518 Debye due to the presence of pyrrole rings that cause the generation of weak dipole moment.76
Moreover, the polarizability increases as the volume occupied by the electrons increases. This occurs in compounds containing electron-withdrawing groups, which makes the movement of electrons looser, resulting in a high dipole moment value in both compounds 5d and 6, this explained the highest activity of both compounds.76
Additionally, HOMO and LUMO are crucial properties for figuring out the reactivity of the precursor compound 3, the essential cyanoacetohydrazide scaffold, and their produced products 4, 5a–d, and 6 and they are useful tools for exploring several crucial parameters for calculating a variety of significant parameters like the absolute electronegativity (χ), the chemical potential (Pi), the chemical hardness (η), the chemical softness (S) and the global electrophilicity (ω), these parameters have been under the spotlight for the purpose of interpreting several chemical reactions, as well as structure–activity relationships.77,78 Table 10 and Fig. 11 showed that the energy difference between HOMO and LUMO (energy gap, ΔE) for compounds 4–6 in the range of 2.7859–3.8746 eV according to the following order of 6 < 4< 5c < 5a< 5b < 5d and the hardness values η ranging from 1.3929 for compound 6 to 1.937 for compound 5d, these values make the electronic transition within these compounds easy depending on the behavior of each moiety. For example, compound 5d has a relatively higher energy gap (3.8746 eV) than other compounds due to 4-Cl phenyl, which reduces the tendency of HOMO electrons to transfer to LUMO orbitals, whereas compound 6 has the lowest energy gap (ΔE = 2.7859 eV) and the maximum softness (0.7179 eV−1) according to the theoretical calculations, it may illustrated by the presence of CN group and the 4-nitro phenyl ring this two withdrawing group have an excellent inductive properties which polarize the aromatic ring more strongly, making this aromatic-compound less effective against anticancer.79,80
| No. | (ET) (a.u) | Dipole moment | EHOMO (eV) | ELUMO (eV) | ΔE (eV) | χ (eV) | η (eV) | S (eV)−1 | Pi (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 (E) | −876.69 | 11.6960 | −5.7668 | −1.8781 | 3.8887 | 3.8225 | 1.9443 | 0.5143 | −3.8225 | 3.7573 |
| 3 (Z) | −876.68 | 8.5526 | −5.8926 | −1.7151 | 4.1774 | 3.8038 | 2.0887 | 0.4787 | −3.8038 | 3.4636 |
| 4 | −1161.87 | 7.5834 | −5.6868 | −2.616 | 3.0707 | 4.1514 | 1.5353 | 0.6512 | −4.1514 | 5.6125 |
| 5a | −1200.26 | 6.2518 | −5.5043 | −1.9246 | 3.5796 | 3.7144 | 1.7898 | 0.5587 | −3.7144 | 3.8543 |
| 5b | −1353.90 | 6.2859 | −5.4477 | −1.7366 | 3.7110 | 3.5921 | 1.8555 | 0.5389 | −3.5921 | 3.4770 |
| 5c | −1529.60 | 7.2529 | −5.4936 | −2.2062 | 3.2874 | 3.8499 | 1.6437 | 0.6083 | −3.8499 | 4.5088 |
| 5d | −1721.22 | 7.6637 | −5.6354 | −1.7608 | 3.8746 | 3.6981 | 1.9373 | 0.5161 | −3.6981 | 3.5297 |
| 6 | −1421.57 | 8.3540 | −6.4183 | −3.6324 | 2.7859 | 5.0253 | 1.3929 | 0.7179 | −5.0253 | 9.0650 |
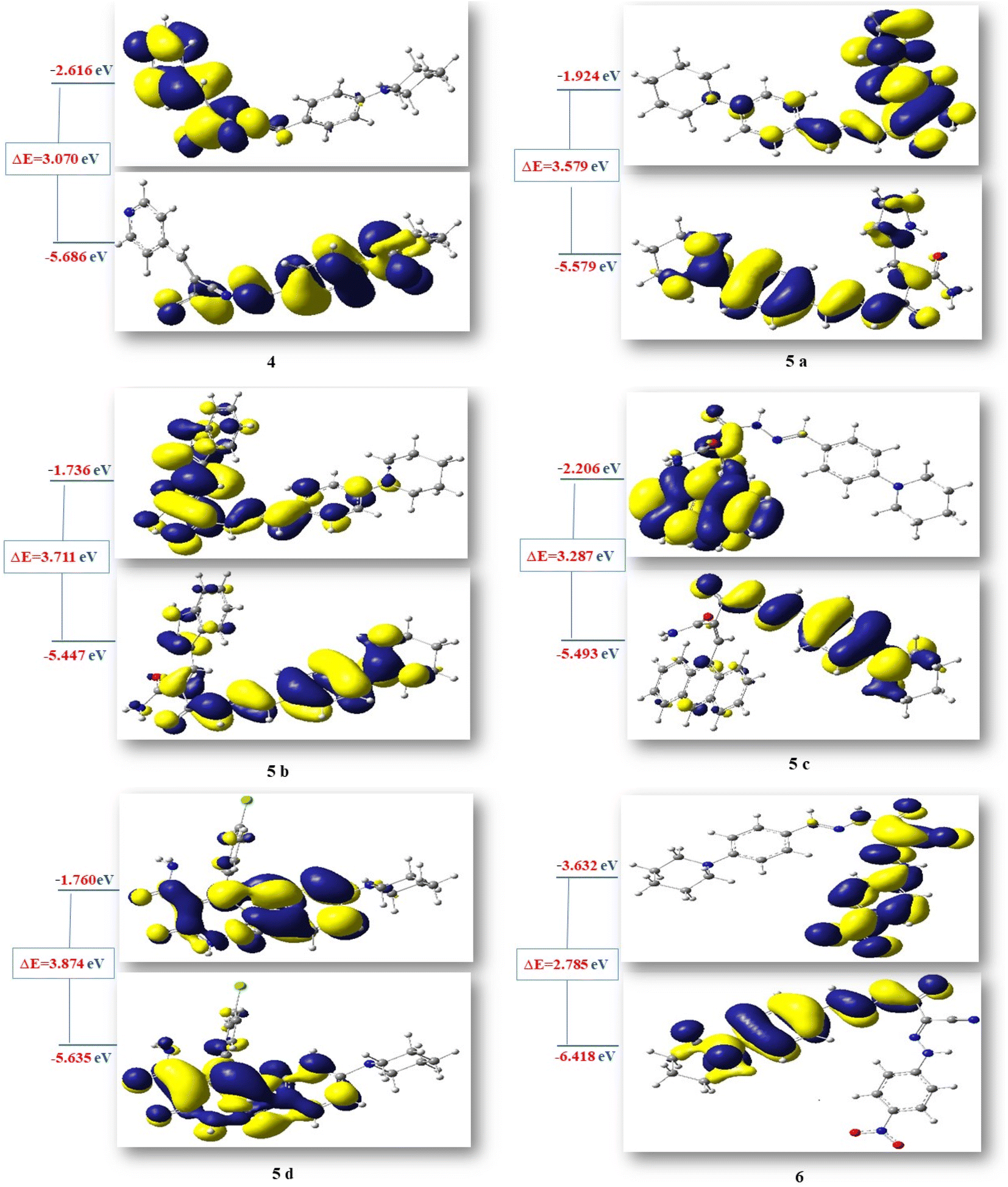 | ||
| Fig. 11 HOMO–LUMO energy gap (eV) of compounds 4, 5a–d and 6. | ||
Due to the specified electrophilicity and electron flow between donor and acceptor, compounds 4 and 6 exhibited the highest electrophilicity index (ω) values of 5.6125 eV and 9.0650 eV, respectively. In addition, the distribution of atomic charges affects the determination of the electronic chemical vector potential (Pi) in compounds, which is built on the position of negative and positive charges. The (Pi) values of all investigated compounds were between −3.5921 and −5.0253 eV. This high negative chemical potential value means that these compounds are a soft molecule with a high polarizability.81
Physicochemical properties of the molecular structure are taught through the MEP. DFT calculation using the optimized structure with the same basis set was used to determine MEP surface analysis of the compound for prediction of relative reactivity positions in a species to resist nucleophilic and electrophilic attacks as shown in Fig. 12. The color code of the compound is between 6.687 × 10−2 (blue region) and −6.687 × 10−2 (red region), these two regions in the MEP structure indicate an electron-rich region, which are localized over the electronegative atoms (oxygen, and nitrogen), and an electron-poor region, respectively.
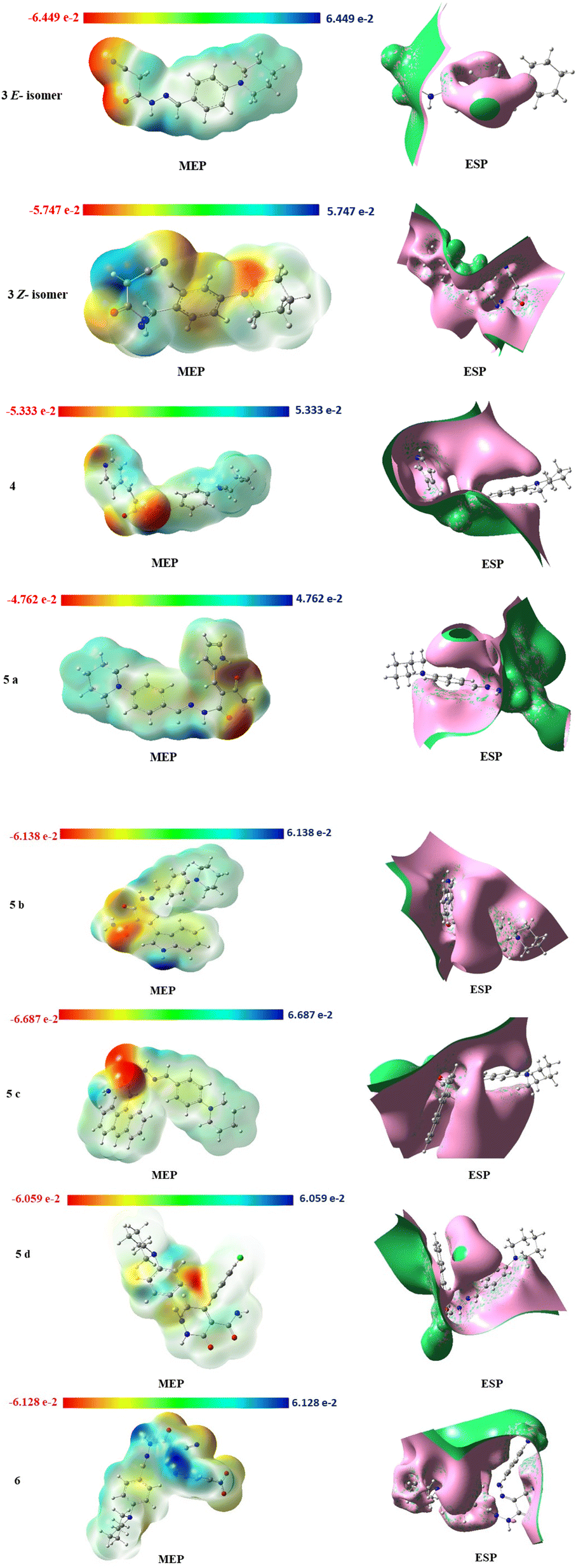 | ||
| Fig. 12 MEP isosurfaces and ESP isosurfaces of compounds 3, 4, 5a–d and 6. | ||
The electrostatic potential surface ESP (Fig. 12) indicates the distance from the molecule where the (+) charge experiences a predetermined attraction or repulsion that defines the local (−) and (+) potential regions in the molecule to predict the interaction of the charge with both the electron cloud and fixed nuclei. In our investigated compounds, the most of the interaction bonds between compounds 5b, 5c and 5d are due to electron charges of the cyanoacetohydrazide moiety which connected to different aromatic rings.
7 Conclusion
Under Knoevenagel condensation reaction, by using ultrasonic methodology, a series of arylidenes derivatives 4 and 5a–d was synthesized through reaction of cyanoacetohydrazide derivative 3 with different aldehydes such as isonicotinaldehyde, 1H-pyrrole-2-carbaldehyde, 1H-indole-3-carbaldehyde and anthracene-9-carbaldehyde, beside 4-chloroacetophenone as a ketone, respectively. Further, reaction of cyanoacetohydrazide derivative 3 with 4-nitrobenzene diazonium chloride gave the hydrazone derivative 6. All the synthesized compounds were conspicuously interpreted using different spectroscopic analyses such as IR, 1H and 13C NMR and mass spectra. The anti-proliferative screening results of the synthesized compounds against four different human cancer cell lines; hepatocellular carcinoma (HEPG-2), mammary gland breast cancer (MCF-7), colorectal carcinoma colon cancer (HCT-116) and human prostate cancer (PC-3), the majority of tested compounds showed a significant cytotoxic activity towards certain cancer cell and have been targeted for further studies. Gratifyingly, compounds 5d and 6 showed the highest cytotoxic activity. Meanwhile, compound 5c proved to have the weakest activity.Based on the correlation coefficient values, five chemical descriptors were chosen as inputs for creating the QSAR model using the MLR technique: AATS6p, AATS7p, AATS8p, AATS0i, and SpMax4_Bhv. Internal and external validations were used to assess and evaluate the generated QSAR models for statistical significance and predictive capability. The examination of the constructed QSAR model equations revealed that the selected descriptors influence the tested compound's anti-proliferative activity. The descriptors identified in this work by QSAR models can be utilized to predict the anticancer activity levels of novel arylidenes derivatives. This will allow for significant cost savings in the drug development process and synthesis at pharmaceutical chemistry laboratories.
According to the physicochemical properties, all compounds follow Lipinski's Rule of Five and they may exhibit drug-relevant features that should be investigated further in future studies. The results of the probable molecular mechanism for the promising candidate compounds (5d and 6) revealed the presence of adenosine A1 receptor (ADORA1) as a common target for both compounds. ADORA1 receptor activation can be involved in multiple types of cancer cells including colon cancer, breast cancer, leukemia, and melanoma. The docking study of tested compounds 5d and 6 revealed that their binding scores to the ADORA1 ligand binding site were more favorable as compared to that of its selective antagonist (DU172) and endogenous agonist (adenosine) suggesting that the anticancer activities of the tested compounds may be related to their ability to block the action of the ADORA1 receptor signaling in cancer cells, leading to cell death. Noteworthy, the computational study via DFT and MEP indicated that the theoretical data was found to be in good agreement with the experimental values of compounds in vitro.
8. Experimental
8.1. Chemistry
4-(Piperidin-1-yl)benzaldehyde 1 (ref. 42) and cyanoacetohydrazide 2 (ref. 43) were previously prepared according to literature methods.
2-Cyano-N′-(4-(piperidin-1-yl)benzylidene)acetohydrazide 3.
Method A. A mixture of 4-(piperidin-1-yl)benzaldehyde 1 (1.89 g, 0.01 mol) and cyanoacetohydrazide 2 (0.99 g, 0.01 mol) in ethanol (30 ml) containing drops of AcOH was heated under reflux for 3 h. The solid product which formed on hot was collected by filtration, dried well and then recrystallized from ethanol to afford 3, yield: 69%.
Method B. A mixture of 4-(piperidin-1-yl)benzaldehyde 1 (1.89 g, 0.01 mol) and cyanoacetohydrazide 2 (0.99 g, 0.01 mol) in ethanol (30 ml) containing drops of AcOH was subjected to ultrasonic energy for 30 min. at 50 °C. The precipitated solid was filtered off, dried well and then recrystallized from ethanol to afford 3, yield: 90%.
3: as pale-yellow crystals; m.p.: 200–202 °C. IR (KBr, ν/cm−1): 3201 (NH), 2968, 2932, 2832 (C–Haliph), 2263 (CN), 1701 (CO), 1610 (CN). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.49 (d, 2H, Ar-H, Ho, J = 8.8 Hz), 6.93 (d, 2H, Ar-H, Hm, J = 9.2 Hz), 3.23 (br. s, 4H, N(CH2)2), 1.57 (br. s, 6H, CH2–(CH2)3–CH2), for E-isomer [11.53 (s, 1H, NH, exchangeable with D2O), 7.86 (s, 1H, CHN), 4.13 (s, 2H, CH2CN), for Z-isomer [11.43 (s, 1H, NH, exchangeable with D2O), 8.00 (s, 1H, (HCN), 3.74 (s, 2H, CH2CN)]. 13C NMR (100 MHz, DMSO-d6) δ (ppm): 164.1, 158.2, 152.5, 152.4, 148.1, 144.8, 128.5, 128.2, 122.8, 122.6, 116.1, 115.8, 114.5, 114.4, 48.3 (2), 24.9 (2), 24.2, 23.9. MS m/z (%): 270.58 (M˙+; 62.95). Anal. Calcd. for C15H18N4O (270.34): C, 66.64; H, 6.71; N, 20.73. Found: C, 66.59; H, 6.68; N, 20.83.
General method for synthesis of arylidene derivatives 4 and 5a–d. A mixture of compound 3 (2.7 g, 0.01 mol) and appropriate aldehydes and/or ketone, namely isonicotinaldehyde, 1H-pyrrole-2-carbaldehyde, 1H-indole-3-carbaldehyde, anthracene-9-carbaldehyde, and 4-chloroacetophenone (0.01 mol), in ethanol (30 ml) containing a few drops of piperidine was sonicated for 2 h (for isonicotinaldehyde) and/or 4 h (for the others) at 75 °C. After cooling the resulting mixture at ambient temperature was poured into ice/water and acidified with dilute acetic acid. The precipitated solid was filtered off, washed several times with cold water, dried well and then recrystallized from the appropriate solvent to afford the arylidene derivatives 4 and 5a–d.
2-Cyano-N′-(4-(piperidin-1-yl)benzylidene)-3-(pyridin-4-yl)acrylohydrazide 4. As dark yellow crystals (ethanol); m.p.: 280–282 °C, yield: 80%. IR (KBr, ν/cm−1): 3283 (NH), 2938, 2851 (C-Haliph), 2200 (C
N), 1682 (CO), 1604 (CN or CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (s, 1H, NH, exchangeable with D2O), 8.28 (s, 1H, HCN), 8.01 (s, 1H, HCC), 7.91 (br. s, 2H, C2,6–H(pyridine)), 7.53 (d, 2H, Ar-H, Ho, J = 8.0 Hz), 7.04 (d, 2H, C3,5–H(pyridine), J = 8.8 Hz), 6.96 (d, 2H, Ar-H, Hm, J = 8.0 Hz), 3.46 (t, 4H, N-(CH2)2, J = 5.2 Hz, J = 4.8 Hz), 1.58 (br.s, 6H, –CH2–(CH2)3–CH2–). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 167.4, 152.8, 150.2, 148.3 (2), 140.5, 137.8, 132.1, 132.0 (2), 129.1 (2), 115.0 (2), 113.8, 112.3, 48.8 (2), 30.2 (2), 28.8. MS m/z (%): 359.21 (M˙+; 35.57). Anal. Calcd. for C21H21N5O (359.43): C, 70.17; H, 5.89; N, 19.48. Found: C, 70.20; H, 5.97; N, 19.44.
2-(2-(4-(Piperidin-1-yl)benzylidene)hydrazine-1-carbonyl)-3-(1H-pyrrol-2-yl)acrylamide 5a. As brown crystals (water); m.p.: 258–260 °C, yield: 92%. IR (KBr, ν/cm−1): 3355, 3220, 3190 (NH2, NH), 2928, 2850 (C-Haliph), 1684, 1663 (C
O), 1598 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.1 (s, 1H, NH, exchangeable with D2O), 10.47 (s, 1H, NH, exchangeable with D2O), 9.68 (s, 1H, HCN), 7.68 (d, 2H, Ar-H, Ho, J = 8.8 Hz), 7.63 (s, 1H, HCC), 7.47 (br. s, 1H, C5–Hpyrrole), 7.01 (d, 2H, Ar-H, Hm, J = 8.8 Hz), 6.86 (t, 1H, C4–Hpyrrole), 6.41 (br. s, 1H, C3-Hpyrrole), 5.69 (br. s, 2H, NH2, exchangeable with D2O), 3.42 (t, 4H, N-(CH2)2, J = 5.6 Hz, J = 4.4 Hz), 1.59–1.57 (m, 6H, –CH2–(CH2)3–CH2–). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 165.1, 164.9, 154.8, 153.3, 131.9, 129.6, 129.4, 128.5, 128.3, 125.5, 123.0, 113.2 (2), 112.7, 112.0, 47.8 (2), 25.0 (2), 24.1. MS m/z (%): 347.38 (M˙+–H2O; 35.89). Anal. Calcd. for C20H23N5O2 (365.44): C, 65.73; H, 6.34; N, 19.16. Found: C, 65.74; H, 6.31; N, 19.18.
3-(1H-Indol-3-yl)-2-(2-(4-(piperidin-1-yl)benzylidene)hydrazine-1-carbonyl)acrylamide 5b. As dark red crystals (ethanol); m.p.: 123–125 °C, yield: 72%. IR (KBr, ν/cm−1): 3367, 3320, 3216 (NH2, NH), 2932, 2852 (C-Haliph), 1650 (C
O), 1594 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.06 (br. s, 2H, 2NH, exchangeable with D2O), 9.93 (s, 1H, HCN), 9.67 (s, 1H, HCC), 8.27 (s, 1H, C2–H(indole)), 8.09 (d, 1H, C4–H(indole), J = 7.2 Hz), 7.67 (d, 2H, Ar-H, Ho, J = 8.8 Hz), 7.51 (d, 1H, C7–H(indole), J = 7.6 Hz), 7.27–7.19 (m, 2H, 2C5,6–H(indole)), 7.00 (d, 2H, Ar-H, Hm, J = 8.8 Hz), 3.40 (br. s, 4H, N-(CH2)2), 3.10 (br. s, 2H, NH2, exchangeable with D2O), 1.58 (br. s, 6H, –CH2–(CH2)3–CH2–). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 189.9, 184.9, 154.6 (2), 138.4, 137.0, 131.6 (2), 125.4 (2), 124.1, 123.4, 122.0, 120.8, 118.1, 113.8, 113.0 (2), 112.4, 47.6 (2), 23.9 (2), 22.2. MS m/z (%): 415.97 (M˙+; 36.71). Anal. Calcd. for C24H25N5O2 (415.50): C, 69.38; H, 6.07; N, 16.86. Found: C, 69.42; H, 6.20; N, 16.83.
3-(Anthracen-9-yl)-2-(2-(4-(piperidin-1-yl)benzylidene)hydrazine-1-carbonyl)acrylamide 5c. As red crystals (ethanol/water); m.p.: 122–124 °C, yield: 53%. IR (KBr, ν/cm−1): 3453, 3338, 3229 (NH2, NH), 2953, 2874, 2809 (C–Haliph), 1658 (C
O), 1623 (CN or CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.50 (br. s, 1H, NH, exchangeable with D2O), 9.04 (d, 2H, Ar-H, J = 9.6 Hz), 9.02 (s, 1H, HCN), 8.78 (s, 1H, C10–Hanthracene), 8.4 (s, 1H, HCC), 7.96 (d, 2H, C1,8–Hanthracene, J = 9.2 Hz), 7.76 (d, 2H, Ar-H, J = 8 Hz), 7.75–7.59 (m, 6H, Ar-H), 4.27 (s, 2H, NH2, exchangeable with D2O), 3.35 (br. s, 4H, N-(CH2)2), 1.58 (br. s, 6H, –CH2–(CH2)3–CH2–). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 189.9, 162.5, 154.6, 150.9, 149.2, 138.5, 135.2 (2), 131.6, 131.3 (2), 130.6 (2), 129.3 (2), 128.7, 126.3, 125.8 (2), 125.4, 124.4, 123.4 (2), 113.0 (2), 48.2 (2), 24.8 (2), 23.9. MS m/z (%): 477.15 (M˙+ + 1; 34.19), 476.20 (M˙+; 5.19). Anal. Calcd. for C30H28N4O2 (476.58): C, 75.61; H, 5.92; N, 11.76. Found: C, 75.59; H, 5.93; N, 11.80.
3-(4-Chlorophenyl)-2-(2-(4-(piperidin-1-yl)benzylidene)hydrazine-1-carbonyl)but-2-enamide 5d. As brown crystals (ethanol/water); m.p.: 285–287 °C, yield: 66%. IR (KBr, ν/cm−1): 3196, 3163 (br.) (NH, NH2), 2933, 2854 (C–Haliph), 1686 (C
O), 1592 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.13 (br. s, 1H, NH, exchangeable with D2O), 10.86 (br. s, 2H, NH2, exchangeable with D2O), 8.21 (d, 2H, Ar-H, J = 8.4 Hz), 7.77 (d, 2H, Ar-H, J = 8.4 Hz), 7.57 (s, 1H, HCN), 7.56 (d, 2H, Ar-H, J = 7.2 Hz), 7.02 (d, 2H, Ar-H, J = 8.4 Hz), 3.28 (br.s, 4H, N-(CH2)2), 2.57 (s, 3H, CH3), 1.61 (br. s, 6H, –CH2–(CH2)3–CH2–). MS m/z (%): 424.11 (M˙+; 27.44). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 189.9, 159.9, 154.6, 151.7, 144.5, 137.8, 134.0, 131.6 (2), 129.5 (2), 128.7 (2), 125.4, 114.1, 113.0 (2), 47.6 (2), 24.8 (2), 23.9, 22.2. Anal. Calcd. for C23H25ClN4O2 (424.93): C, 65.01; H, 5.93; N, 13.19. Found: C, 65.11; H, 5.94; N, 13.27.
N-(4-Nitrophenyl)-2-oxo-2-(2-(4-(piperidin-1-yl)benzylidene)hydrazinyl)acetohydrazonoyl cyanide 6. To p-nitroaniline (1.38 g, 0.01 mol) concentrated HCl (3 ml) was added and cooled to 0–5 °C in ice bath then cooled sodium nitrite solution (1.0 g in 10 ml water) was added dropwise to the acidic solution of p-nitroaniline. The reaction mixture was then stirred for 10 min to form the diazotized salt of p-nitroaniline. To a cold mixture of compound 3 (2.7 g, 0.01 mol) and sodium acetate (4.10 g, 0.05 mol) in ethanol (50 ml), the prepared diazotized salt of p-nitroaniline was added dropwise with stirring. The mixture was ultrasonicated at ambient temperature for 30 min. The reaction mixture was left overnight. The precipitated solid was filtered off, dried well and then recrystallized from ethanol to give 6 as black crystals; m.p.: 128–130 °C, yield: 56.5%. IR (KBr, ν/cm−1): 3419, 3195 (NH), 2928, 2855 (C–Haliph), 2215 (C
N), 1680 (CO), 1642 (CN), 1596 (CC), 1515, 1338 (NO2). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.66 (br. s, 1H, NH, exchangeable with D2O), 8.41 (s, 1H, HCN), 8.32–6.98 (m, 8H, Ar-H), 6.58 (br. s, H, NH, exchangeable with D2O), 3.38 (br.s, 4H, N-(CH2)2), 1.59 (br. s, 6H, –CH2–(CH2)3–CH2–). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 155.7, 153.5, 147.7, 142.6, 133.6, 126.4 (2), 125.2 (2), 119.2, 115.9 (2), 112.4 (2), 111.8, 110.8, 47.7 (2), 24.8 (2), 23.9. MS m/z (%): 419.18 (M˙+; 21.64). Anal. Calcd. for C21H21N7O3 (419.45): C, 60.13; H, 5.05; N, 23.38. Found: C, 60.07; H, 5.24; N, 23.22.
8.2. Cytotoxicity and antitumor evaluation
Antitumor activity was tested at the drugs department, Faculty of Pharmacy, Mansoura University, Egypt.8.2.1.1 Cell line. Four human tumor cell lines namely, hepatocellular carcinoma (HEPG-2), mammary gland breast cancer (MCF-7), colorectal carcinoma colon cancer (HCT-116) and human prostate cancer (PC-3). The cell lines were obtained from ATCC via Holding company for biological products and vaccines (VACSERA), Cairo, Egypt. For comparison, doxorubicin was utilized as a standard anticancer drug.
The cell lines were seeded in a 96-well plate at a density of 104 cells per well and incubated at 37 °C for 48 hours with 5% CO2. The cells were cultured for 24 hours after being exposed to various chemical concentrations. After a 24-hours incubation period, 20 μl of a 5 mg ml−1 MTT solution was added and incubated for 4 hours. To dissolve the generated purple formazan, each well receives 100 μl of dimethyl sulfoxide (DMSO) (Sigma Co., St. Louis, USA). The colorimetric assay is measured and recorded at wavelength of 570 nm using a plate reader (EXL 800, USA). The proportion of relative cell viability was estimated as (A570 of treated samples/A570 of untreated sample) × 100.
8.3. Docking studies
AutoDock Vina modeling simulation software (AutoDock Vina v.1.2.0) was used to predict the protein-ligand binding affinity, as well as the preferred orientation of the docking pose between the amino acid residues that form the ligand binding pocket of the ADORA1 receptor and the studied compounds 5d and 6, in addition to the receptor endogenous agonist (adenosine) and DU172 (selective ADORA1 antagonists) that were used as reference ligand. The 3D-structure of ADORA1 protein bound to the co-crystalized ligand, DU172, was downloaded from Protein Data Bank (PDB) (https://www.rcsb.org) at a resolution of 3.2 (PDB: 5uen).PyMOL molecular visualization tool (PyMOL v.2.5.4) (Schrödinger, Inc.) was used to extract the ADORA1 receptor from its co-crystalized DU172, after adding hydrogen bonds to both. The extracted files were in PDB format.84 Auto-Dock (MGL-tools) was used to determine the docking site and the grid box dimensions of ligand binding pocket.85 The grid box dimensions were selected by centering grid box on the Du172 ligand, included in crystal structure (Table 11). Moreover, the target protein and the tested compounds were exported in PDBQT format (AutoDock format) using Open Babel v.2.3.1.86
| Spacing | n.pts (x; y; z) | Center (x; y; z) |
|---|---|---|
| 42; 30; 40 | 55.151; 58.867; 143.833 |
A maximum of 9 poses was considered for each molecule where the target protein was kept as the rigid receptor while keeping the conformation of the ligands as flexible.87 Finally, the most favorable pose was selected according to the minimum free energy of the protein–ligand complex and for analyzing the type of interactions between the ligand and the protein, BIOVIA Discovery Studio (DS) Visualizer v.4.5. was used.
8.4. DFT theoretical calculations
The investigated compounds were theoretically calculated using Gaussian 09 software (2009, Gaussian 09, Revision A.1, Gaussian. Inc.: Wallingford, CT, USA).88 The DFT computations were carried out using the B3LYP 6-31G (d,p) basis set. The structural geometry was strengthened by evading the molecular symmetry restrictions in contrast to all geometrical variables and by minimizing its energy. The optimized compounds' molecular structure was sketched using Gauss View.89Conflicts of interest
There are no conflicts to declare.References
- E. Nassar, Y. A.-M. El-Badry and H. El Kazaz, Chem. Pharm. Bull., 2016, 64, 558–563 CrossRef CAS PubMed.
- A. M. Fahim, H. S. Magar, E. Nasar, F. M. Abdelrazek and A. Aboelnaga, J. Inorg. Organomet. Polym. Mater., 2022, 32, 240–266 CrossRef CAS.
- M. M. Abdelshaheed, I. M. Fawzy, H. I. El-Subbagh and K. M. Youssef, Future Journal of Pharmaceutical Sciences, 2021, 7, 1–11 CrossRef.
- P. Goel, O. Alam, M. J. Naim, F. Nawaz, M. Iqbal and M. I. Alam, Eur. J. Med. Chem., 2018, 157, 480–502 CrossRef CAS PubMed.
- S. N. Mokale, P. N. Dube, S. A. Bhavale, I. Sayed, A. Begum, M. C. Nevase, V. R. Shelke and A. Mujaheed, Med. Chem. Res., 2015, 24, 1842–1856 CrossRef CAS.
- A. Aboelnaga, E. Mansour, A. M. Fahim and G. H. Elsayed, J. Mol. Struct., 2022, 1251, 131945 CrossRef CAS.
- R. N. Asha, M. Sankarganesh, N. Bhuvanesh and B. R. D. Nayagam, J. Mol. Struct., 2022, 1250, 131692 CrossRef CAS.
- Z.-S. Gu, W.-T. Wang, H. Qian, A.-N. Zhou, H.-B. Sun, Q.-W. Zhang and J.-Q. Li, Bioorg. Med. Chem. Lett., 2019, 29, 126703 CrossRef CAS PubMed.
- F. Xiao, R. Yan, Y. Zhang, S. Wang, S. Chen, N. Zhou and X. Deng, Arch. Pharm., 2021, 354, 2000298 CrossRef CAS PubMed.
- J. Jayabharathi, A. Manimekalai, T. C. Vani and M. Padmavathy, Eur. J. Med. Chem., 2007, 42, 593–605 CrossRef CAS PubMed.
- G. Aridoss, S. Balasubramanian, P. Parthiban and S. Kabilan, Eur. J. Med. Chem., 2007, 42, 851–860 CrossRef CAS PubMed.
- M. G. Abouelenein, A. E.-H. A. Ismail, A. Aboelnaga, M. A. Tantawy, N. M. El-Ebiary and S. A. El-Assaly, J. Mol. Struct., 2023, 1275, 134587 CrossRef CAS.
- S. Wang, A. Haikarainen, A. Pohjakallio, J. Sipilä, J. Kaskinoro, S. Juhila, N. Jalava, M. Koskinen, M. Vesajoki and E. Kumpulainen, Bioorg. Med. Chem. Lett., 2022, 69, 128783 CrossRef CAS PubMed.
- S. McElvain and T. P. Carney, J. Am. Chem. Soc., 1946, 68, 2592–2600 CrossRef CAS PubMed.
- S. A. Khanum, V. Girish, S. Suparshwa and N. F. Khanum, Bioorg. Med. Chem. Lett., 2009, 19, 1887–1891 CrossRef CAS PubMed.
- M. N. Yousif, Mini-Rev. Org. Chem., 2022, 19, 125–135 CrossRef CAS.
- S. Sivakumar, Chem. Sci. Rev. Lett., 2016, 5, 99–105 Search PubMed.
- M. SV, S. Belagali and M. Bhat, Anti-Infect. Agents, 2020, 18, 362–374 CAS.
- S. Deekonda, J. Cole, S. Sunna, D. Rankin, T. M. Largent-Milnes, P. Davis, N. M. BassiriRad, J. Lai, T. W. Vanderah and F. Porecca, Bioorg. Med. Chem. Lett., 2016, 26, 222–227 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. R. Mahmoud, S. A. Shiba, A. K. El-Ziaty, F. S. M. Abu El-Azm and M. F. Ismail, Synth. Commun., 2014, 44, 1094–1102 CrossRef CAS.
- M. F. Ismail and A. A. El-sayed, J. Iran. Chem. Soc., 2018, 16, 921–937 CrossRef.
- S. A. Shaker and M. I. Marzouk, Molecules, 2016, 21, 155 CrossRef PubMed.
- M. F. Ismail and G. A. Elsayed, Synth. Commun., 2018, 48, 892–905 CrossRef CAS.
- M. R. Mahmoud, A. K. El-Ziaty, F. S. A. El-Azm, M. F. Ismail and S. A. Shiba, J. Chem. Res., 2013, 37, 80–85 CrossRef CAS.
- M. F. Ismail, A. F. Aly, S. S. Abdel-Wahab and A. A. El-Sayed, Polycyclic Aromat. Compd., 2023, 43, 1288–1308 CrossRef CAS.
- N. A. Hamed, M. I. Marzouk, M. F. Ismail and M. H. Hekal, Synth. Commun., 2019, 49, 3017–3029 CAS.
- M. H. Hekal, Y. M. Ali and F. S. M. Abu El-Azm, Synth. Commun., 2020, 50, 2839–2852 CrossRef CAS.
- M. A. Gouda, M. A. Salem, M. I. Marzouk, N. F. Mahmoud and M. F. Ismail, Chem. Biodiversity, 2023, 20, e202300706 CrossRef CAS PubMed.
- H. M. Refat and A. A. Fadda, Eur. J. Med. Chem., 2013, 70, 419–426 CrossRef CAS PubMed.
- H. Hosseini and M. Bayat, Top. Curr. Chem., 2018, 376, 40 CrossRef PubMed.
- D. Priya, P. Gopinath, L. S. Dhivya, A. Vijaybabu, M. Haritha, S. Palaniappan and M. K. Kathiravan, ChemistrySelect, 2022, 7, e202104429 CrossRef CAS.
- M. R. Mahmoud, F. S. M. Abu El-Azm, A. T. Ali and Y. M. Ali, Synth. Commun., 2017, 47, 1591–1600 CrossRef CAS.
- M. A. Salem, S. Y. Abbas, M. H. Helal and A. Y. Alzahrani, J. Heterocycl. Chem., 2021, 58, 2117–2123 CrossRef CAS.
- A. M. M. Mohamed, M. F. Ismail, H. M. F. Madkour, A. F. Aly and M. S. Salem, Synth. Commun., 2020, 50, 3424–3442 CrossRef CAS.
- M. F. Ismail, A. I. Hashem, R. A. Sleem and A. I. Hassaballah, ChemistrySelect, 2023, 8, e202204946 CrossRef CAS.
- A. N. El-hoshoudy, M. A. El-Raouf, M. M. Attya, A. F. Ahmed and M. Waly, J. Chem. Appl., 2021, 33–53, DOI:10.36811/jca.2021.110009.
- A. Aboelnaga, M. Hagar and S. M. Soliman, Molecules, 2016, 21, 848 CrossRef PubMed.
- C. Hazra, S. Tonde, B. Dhanvijay, D. Kundu, A. Satdive, S. Tayde, B. Toksha, J. Naik and A. Chatterjee, Chem. Eng. J., 2023, 451, 138996 CrossRef CAS.
- S. Soror, A. M. Fahim, S. Elabbady, E. Nassar and A. Aboelnaga, J. Biomol. Struct. Dyn., 2022, 40, 5409–5426 CrossRef CAS PubMed.
- A. M. Fahim, E. H. I. Ismael, G. H. Elsayed and A. M. Farag, J. Biomol. Struct. Dyn., 2022, 40, 9177–9193 CrossRef CAS PubMed.
- M. S. A. El-Gaby, J. Chin. Chem. Soc., 2004, 51, 125–134 CrossRef CAS.
- S. H. Doss, W. W. Wardakhan and N. A. Louca, Arch. Pharmacal Res., 2001, 24, 377–384 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- C. Y. Hong, Y. K. Kim, J. H. Chang, S. H. Kim, H. Choi, D. H. Nam, Y. Z. Kim and J. H. Kwak, J. Med. Chem., 1997, 40, 3584–3593 CrossRef CAS PubMed.
- S. Şenkardeş, M. İ. Han, N. Kulabaş, M. Abbak, Ö. Çevik, İ. Küçükgüzel and Ş. G. Küçükgüzel, Mol. Diversity, 2020, 24, 673–689 CrossRef PubMed.
- N. Hristova-Avakumova, K. Yoncheva, B. Nikolova-Mladenova, T. Traykov, G. Momekov and V. Hadjimitova, Redox Rep., 2017, 22, 408–417 CrossRef CAS PubMed.
- S. Benaka Prasad, K. Vinaya, C. Ananda Kumar, S. Swarup and K. Rangappa, Med. Chem. Res., 2010, 19, 220–235 CrossRef CAS.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- S. Chtita, M. Ghamali, A. Ousaa, A. Aouidate, A. Belhassan, A. I. Taourati, V. H. Masand, M. Bouachrine and T. Lakhlifi, Heliyon, 2019, 5, 1–26 CrossRef PubMed.
- A. Tropsha, Mol. Inf., 2010, 29, 476–488 CrossRef CAS PubMed.
- K. Roy, S. Kar and R. N. Das, Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment, Academic press, 2015 Search PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- T. Lumley and A. Miller, _leaps: Regression Subset Selection_. R package version 3.1, 2020, Retrieved from https://CRAN.R-project.org/package=leaps Search PubMed.
- R. Kiralj and M. Ferreira, J. Braz. Chem. Soc., 2009, 20, 770–787 CrossRef CAS.
- D. E. Arthur, A. Uzairu, P. Mamza, S. E. Abechi and G. Shallangwa, J. King Saud Univ., Sci., 2020, 32, 324–331 CrossRef.
- S. N. Adawara, G. A. Shallangwa, P. A. Mamza and A. Ibrahim, Beni-Suef University Journal of Basic and Applied Sciences, 2020, 9, 1–17 CrossRef.
- A. Lagunin, A. Zakharov, D. Filimonov and V. Poroikov, Mol. Inf., 2011, 30, 241–250 CrossRef CAS PubMed.
- R. W. Kennard and L. A. Stone, Technometrics, 1969, 11, 137–148 CrossRef.
- V. Catalani, M. Botha, J. M. Corkery, A. Guirguis, A. Vento, N. Scherbaum and F. Schifano, Pharmaceuticals, 2021, 14, 720 CrossRef CAS PubMed.
- Y. Isyaku, A. Uzairu, S. Uba, M. T. Ibrahim and A. B. Umar, Bulletin of the National Research Centre, 2020, 44, 1–11 CrossRef.
- S. Chtita, A. Belhassan, M. Bakhouch, A. I. Taourati, A. Aouidate, S. Belaidi, M. Moutaabbid, S. Belaaouad, M. Bouachrine and T. Lakhlifi, Chemom. Intell. Lab. Syst., 2021, 210, 104266 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2019, 47, W357–W364 CrossRef CAS PubMed.
- A. Townsend-Nicholson, P. Schofield and E. Baker, Genomics, 1995, 26, 423–425 CrossRef CAS PubMed.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, K.-N. Klotz and J. Linden, Pharmacol. Rev., 2001, 53, 527–552 CAS.
- H.-E. Khoo, C.-L. Ho, V. J. Chhatwal, S. T. Chan, S.-S. Ngoi and S. M. Moochhala, Cancer Lett., 1996, 106, 17–21 CrossRef CAS PubMed.
- A. Mirza, A. Basso, S. Black, M. Malkowski, L. Kwee, J. A. Patcher, J. E. Lachowicz, Y. Wang and S. Liu, Cancer Biol. Ther., 2005, 4, 1355–1360 CrossRef CAS PubMed.
- S. Gessi, K. Varani, S. Merighi, A. Morelli, D. Ferrari, E. Leung, P. G. Baraldi, G. Spalluto and P. A. Borea, Br. J. Pharmacol., 2001, 134, 116–126 CrossRef CAS PubMed.
- S. Merighi, K. Varani, S. Gessi, E. Cattabriga, V. Iannotta, C. Ulouglu, E. Leung and P. A. Borea, Br. J. Pharmacol., 2001, 134, 1215–1226 CrossRef CAS PubMed.
- S. Ni, Q. Wei and L. Yang, OncoTargets Ther., 2020, 13, 12409–12419 CrossRef CAS PubMed.
- Z. Lin, P. Yin, S. Reierstad, M. O'Halloran, J. V. Coon, E. Pearson, G. Mutlu and S. Bulun, Oncogene, 2010, 29, 1114–1122 CrossRef CAS PubMed.
- D. S. Shin, K. S. Min and B. G. Kim, World J. Adv. Res. Rev., 2018, 6, 262712 Search PubMed.
- S. Biswal, U. Sahoo, S. Sethy, H. Kumar and M. Banerjee, Asian J. Pharm. Clin. Res., 2012, 5, 1–6 CAS.
- J. Rahman, A. M. Tareq, M. M. Hossain, S. A. Sakib, M. N. Islam, M. H. Ali, A. N. Uddin, M. Hoque, M. S. Nasrin and T. B. Emran, Pharmaceuticals, 2020, 13, 232 CrossRef CAS PubMed.
- A. A. El-Sayed, G. A. Elsayed, S. A. Rizk and M. F. Ismail, Org. Prep. Proced. Int., 2023, 16, 1–14 CrossRef.
- A. A. El-Sayed, M. F. Ismail, A. E.-G. E. Amr and A. M. Naglah, Molecules, 2019, 24, 3787 CrossRef PubMed.
- H. E. Tolan, A. M. Fahim and E. H. Ismael, J. Mol. Struct., 2023, 1283, 135238 CrossRef CAS.
- A. Attia, A. Aboelnaga and A. M. Fahim, Egypt. J. Chem., 2023, 66, 95–111 Search PubMed.
- M. A. Mumit, T. K. Pal, M. A. Alam, M. A.-A.-A.-A. Islam, S. Paul and M. C. Sheikh, J. Mol. Struct., 2020, 1220, 128715 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS PubMed.
- L. Schrödinger and W. DeLano, PyMOL, 2020, http://www.pymol.org/pymol Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- N. O'Boyle, J. Cheminf., 2011, 3, 33 Search PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- R. A. Gaussian09, Inc., Wallingford CT, 2009, vol. 121, 150–166.
- R. Dennington, T. Keith, J. Millam and V. GaussView, GaussView, Version, Shawnee Mission KS, 2009, vol. 5 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05799b |
| This journal is © The Royal Society of Chemistry 2023 |