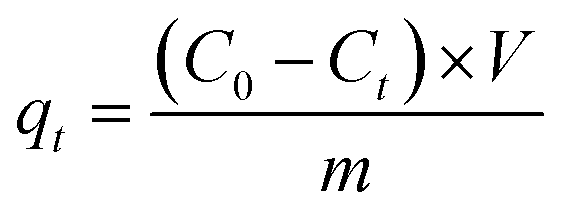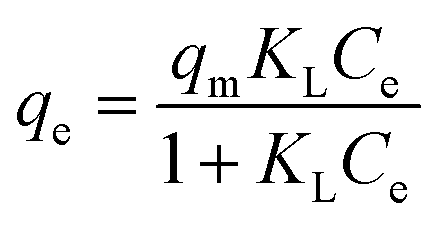DOI:
10.1039/D3RA05596E
(Paper)
RSC Adv., 2023,
13, 31002-31016
Poly(acrylic acid-co-2-acrylamido-2-methyl-1-propanesulfonic acid)-grafted chitosan hydrogels for effective adsorption and photocatalytic degradation of dyes
Received
16th August 2023
, Accepted 17th October 2023
First published on 23rd October 2023
1. Introduction
Wastewater contains harmful, carcinogenic, and stable organic pollutants1 that can endanger the health of living organisms if released into the environment. Organic dyes have broad industrial applications; nevertheless, direct dye discharge into water causes potential toxicity and reduced photosynthetic activity.2 Organic dyes can be removed from wastewater by various treatment methods. Commonly used methods include adsorption,3 physicochemical and membrane separation,4,5 electrochemical techniques, reverse osmosis, ion exchange, electrodialysis and advanced oxidation processes (AOPs).6 Adsorption processes have attracted a lot of attention due to their simplicity, versatility, and low cost.
Chitosan is an abundant natural polymer. The biocompatibility, biodegradability, polyfunctionality, and hydrophilicity of chitosan contribute to its potential as an adsorbent.7 However, it is unstable in acidic conditions and poor mechanical stability limits its application. To overcome these drawbacks, chitosan has been modified or composited with inorganic materials.8–12 Modification by crosslinking and grafting also improves the adsorption performance of chitosan.13 For dye adsorption, suitable functional groups can be added to chitosan by grafting with other polymers. Polymers used in this way include polyacrylamide,14 poly(methacrylic acid) (PMAA),15 poly(sodium 4-styrene sulfonate) (PSSA),16 poly(acrylic acid) (PAA),17 and poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS). Microspheres prepared from the graft polymerization of acrylic acid and 2-acrylamido-2-methyl-1-propanesulfonic acid (P(AA-co-AMPS)) onto chitosan demonstrated a high adsorption capacity towards methylene blue (MB).18,19 The presence of multifunctional groups including amino (–NH2), hydroxyl (–OH), carboxylic acid (–COOH), and sulfonic acid (–SO3H) groups in the microspheres accounted for their excellent removal ability. Hydrogels made from chitosan grafted with P(AA-co-AMPS) were also used to remove MB and rhodamine B (RhB) in aqueous solutions.20
Another method of treating wastewater is photocatalytic degradation. Typically, photocatalytic powders absorb energy from UV light or sunlight, and then reactive radical species are generated that oxidize toxic pollutants into non-toxic small molecules.21,22 Zinc oxide (ZnO) is a commonly used inorganic powder photocatalyst that exhibits high chemical stability and photocatalytic activity at an affordable price. Although powder photocatalysts are not easy to recover after use, supporting materials, including synthetic or natural polymers,23–27 can be impregnated with photocatalytic particles to facilitate the operation. Immobilizing photocatalysts in biodegradable natural polymers can reduce the generation of secondary waste.28,29
In this work, hydrogels of chitosan grafted with P(AA-co-AMPS) were synthesized via free radical polymerization with AA–AMPS at various molar ratios to investigate the effect of acidic functional groups on dye adsorption. Potassium persulfate (KPS) and N,N′-methylenebis(acrylamide) (MBA) were used as an initiator and a crosslinker, respectively. The morphology of the prepared hydrogels was investigated and their adsorption performances towards cationic and anionic dyes were explored. The effect of the AA–AMPS molar ratio on MB and CV adsorption was also studied. The most promising hydrogel was then further investigated in various CV adsorption conditions of contact time, adsorbent dose, pH, and initial concentration. Adsorption kinetics and isotherms were investigated. ZnO photocatalyst particles were synthesized and immobilized in the chitosan hydrogel grafted with the optimal P(AA-co-AMPS). The efficiency of photocatalytic dye degradation on ZnO and the potential of the hydrogel as a biodegradable adsorbent supporting material were studied.
2. Experimental
2.1 Materials
Chitosan (molecular weight 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–190000 Da, 75–85% deacetylated), AA (99%), AMPS (99%), reactive orange 16 (RO16), direct yellow 50 (DY50), orange G (OG), brilliant green (BG), and reactive black 5 (RB5) were purchased from Sigma-Aldrich (USA). MB (95%), CV (88%), malachite green (MG), and Congo red (CR) were from Loba Chemie (India). RhB was from UNILAB (Philippines). MBA (98%) was supplied by Fluka (USA). KPS (99.0%) was from VWR (USA). Sodium hydroxide (NaOH, 99%) was purchased from Ajax Finechem (Australia). Zinc acetate dihydrate (ZnAc2, 99.0%) was from EMSURE (USA). All chemicals were used as received.
000–190000 Da, 75–85% deacetylated), AA (99%), AMPS (99%), reactive orange 16 (RO16), direct yellow 50 (DY50), orange G (OG), brilliant green (BG), and reactive black 5 (RB5) were purchased from Sigma-Aldrich (USA). MB (95%), CV (88%), malachite green (MG), and Congo red (CR) were from Loba Chemie (India). RhB was from UNILAB (Philippines). MBA (98%) was supplied by Fluka (USA). KPS (99.0%) was from VWR (USA). Sodium hydroxide (NaOH, 99%) was purchased from Ajax Finechem (Australia). Zinc acetate dihydrate (ZnAc2, 99.0%) was from EMSURE (USA). All chemicals were used as received.
2.2 Synthesis of hydrogels
Chitosan-based hydrogels were prepared with AA–AMPS feed molar ratios of 1:5, 1:1, 5:1, 10:1 and 1:0. Free radical graft copolymerization of chitosan with partially neutralized AA and AMPS was conducted as follows: the desired amount of AA was dissolved in 25 mL of NaOH aqueous solution. The molar concentration of NaOH was fixed at 80% of the molar concentration of AA. The desired amount of AMPS and 0.45 g of chitosan were added to the solution and stirred for 20 min. The total weight of the feed monomers was fixed. MBA (0.1 mol% of the sum of moles of AA and AMPS) and KPS (0.1 mole% of the sum of moles of AA and AMPS) were then added. The mixture was stirred for 30 min, purged with nitrogen, and the reaction was then conducted at 50 °C for 90 min. The obtained rubbery gel was thoroughly washed with deionized water before being dried in an oven at 50 °C until a constant weight was achieved.
2.3 Synthesis of ZnO/P(AA-co-AMPS) hydrogel composite
The hydrogel fabricated with an AA–AMPS molar ratio of 10:1 was immersed in water until absorption equilibrium was reached. The swollen hydrogel was transferred to 5 mol% ZnAc2 solution and gently shaken at 100 rpm for 16 h at room temperature. The hydrogel was then soaked in 10 mol% NaOH aqueous solution and heated at 60 °C for 1 h. The hydrogel composite was soaked in water for 24 h to remove unreacted ZnAc2 and excess NaOH and then dried in an oven at 60 °C until completely dry.
2.4 Batch adsorption experiment
A batch experiment was used to determine the adsorption capacity of fabricated hydrogels towards the cationic dyes MB, RhB, MG, CV, and BG, and the anionic dyes RB5, OG, CR, DY50, and RO16. The adsorption experiment was performed for 24 h using a hydrogel dose of 0.05 g in 50 mL of 1000 mg L−1 aqueous dye solution at ambient temperature. The concentration of the dye solution after adsorption was determined by UV-Vis spectrophotometry.
To investigate the effect of the AA–AMPS ratio on MB and CV adsorptions, 0.05 g of each preparation of the hydrogel was soaked in 50 mL of 1000 mg L−1 aqueous dye solution for 24 h. To optimize the CV adsorption conditions of the hydrogel fabricated with an AA–AMPS molar ratio of 10:1, 0.01–0.07 g of the hydrogel was soaked in 50 mL of CV aqueous solution at initial concentrations of 10–4000 mg L−1, in the pH range of 2.0–10.0, for 1–24 h, at ambient temperature. Solution pH was adjusted using 0.1 M HCl or 0.1 M NaOH solutions.
The amount of dye adsorbed at different times (qt, mg g−1) and removal efficiency (R, %) were calculated using eqn (1) and (2), respectively.
| |
 | (1) |
| |
 | (2) |
where
C0 and
Ct (mg L
−1) are the initial dye concentration and the dye concentration at a certain time, respectively.
V and
m represent the solution volume (L) and the weight (g) of the hydrogels, respectively.
2.5 Water absorption test
To measure water absorption capacity (WAC), dry samples were immersed in water for 24 h. The weight change of the hydrogels was measured, and WAC was calculated according to eqn (3).| |
 | (3) |
where Wo and Ws (g) are the weights of the hydrogel before and after water immersion, respectively.
2.6 Photocatalytic degradation test
The efficiency of photocatalytic degradation on the in situ synthesized ZnO particles was studied using a hydrogel composite dose of 0.05 g/50 mL of CV aqueous solution at a C0 = 10 mg L−1. After adsorbing CV for 24 h, the hydrogel composite was placed under a UV light source (39 mW cm−2) and natural sunlight (11 am–2 pm, Songkhla Province, Thailand), while soaking in water. The colors of the hydrogel composite before and after the photocatalytic degradation of the adsorbed dye were compared. The next cycle was repeated without further purification. At least three replicates were studied for each experiment.
2.7 Characterizations
Fourier transform infrared (FTIR) spectrometry (Tensor 27, Bruker, Germany) between 4000 and 600 cm−1 in attenuated total reflection mode was used to produce FTIR spectra that were processed by Opus 7.0 software. The concentration of dye solutions after adsorption was determined by using UV-Vis spectrophotometry (Agilent 8453, USA). Scanning electron microscopy (SEM, Quanta 400, FEI) was used to examine the morphology of fully swollen hydrogels. Samples were frozen in liquid nitrogen, dried under vacuum, and coated with gold. Chemical compositions were analyzed by energy-dispersive X-ray spectroscopy (EDS) coupled with SEM. The morphology of in situ synthesized ZnO particles in the 10:1 hydrogel matrix was studied using a transmission electron microscope (TEM, JEM-2010, JOEL). The X-ray diffraction (XRD) patterns of the 10:1 hydrogel and the ZnO/hydrogel composite were collected on an EMPYREAN X-ray diffractometer (PANalytical, United Kingdom) using Cu-Kα radiation. The thermal stability of the hydrogels and the hydrogel composite were evaluated by thermogravimetric analysis (TGA) (STA7200, Hitachi, Japan) in the temperature range of 35–650 °C at a heating rate of 10 °C min−1 under nitrogen gas.
3. Results and discussions
3.1 Synthesis of hydrogels
Chitosan-based hydrogels were prepared with five different AA–AMPS feed molar ratios. A possible chemical structure of these hydrogels is proposed in Scheme 1.30 Pale yellow gels were obtained with very high yields, with the 1:5 P(AA-co-AMPS) yielding 74% and the other four ratios yielding 91–98%. Grafting percentage (G, %) was determined gravimetrically and calculated using eqn (4):| |
 | (4) |
 |
| | Scheme 1 Synthetic route and possible chemical structure of P(AA-co-PAMPS)-grafted chitosan hydrogel. | |
The grafting percentages of the 1:5, 1:1, 5:1, 10:1 and 1:0 P(AA-co-AMPS) chitosan hydrogels were 1550, 2060, 2000, 2210, and 2140%, respectively.
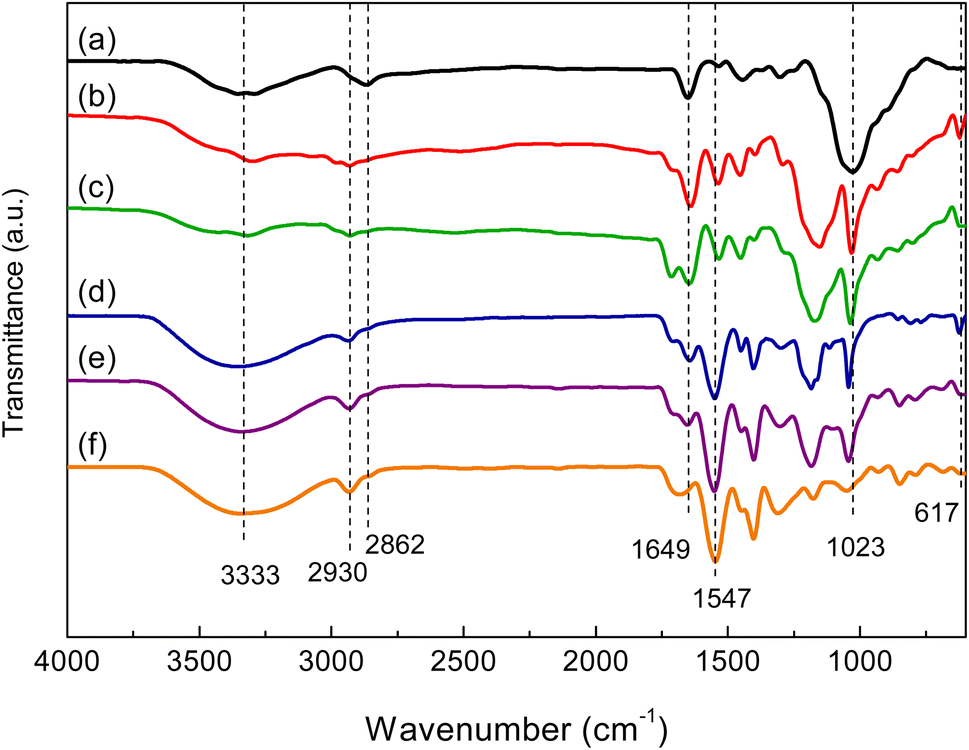
In the FTIR spectrum of chitosan (Fig. 1(a)), the broad absorption band located at 3000–3500 cm−1 was attributed to O–H and –NH2 groups. The band near 2862 cm−1 was ascribed to C–H stretching vibrations. The bands at 1649 and 1547 cm−1 corresponded to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of amide and the N–H bending vibration, respectively.20 In the FTIR spectra of hydrogels (Fig. 1(b)–(f)), the stretching vibrations of O–H at 3000–3500 cm−1 were significantly enhanced due to the presence of the O–H groups of chitosan and AA. The band at 2930 cm−1 was due to –CH2 groups formed by the copolymerization of AA and AMPS. The bands at 1023 and 617 cm−1 were ascribed to the valent oscillation of the S–O bond and the valent symmetric oscillation of the SO2 group of AMPS.20,30
O stretching vibration of amide and the N–H bending vibration, respectively.20 In the FTIR spectra of hydrogels (Fig. 1(b)–(f)), the stretching vibrations of O–H at 3000–3500 cm−1 were significantly enhanced due to the presence of the O–H groups of chitosan and AA. The band at 2930 cm−1 was due to –CH2 groups formed by the copolymerization of AA and AMPS. The bands at 1023 and 617 cm−1 were ascribed to the valent oscillation of the S–O bond and the valent symmetric oscillation of the SO2 group of AMPS.20,30
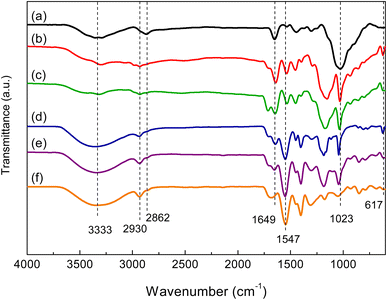 |
| | Fig. 1 FTIR spectra of chitosan (a) and P(AA-co-AMPS)-chitosan hydrogels at AA–AMPS ratios of (b) 1:5, (c) 1:1, (d) 5:1, (e) 10:1, and (f) 1:0. | |
The effect of the AA–AMPS ratio on the morphology and porosity of the hydrogels was observed under SEM. The micrographs showed variations in the pore size of the hydrogels (Fig. 2). At low AA content, the pores were very large compared with the samples that had higher AA content. Increasing the AA content decreased pore size and increased the uniformity of the pore-size distribution. This result was probably due to the reduced steric hindrance between repeating units of the polymer chains when AA content was higher. The smaller functional groups would promote closer hydrogen bonding, stronger interactions between interconnected porous structures, and higher crosslinking density.31–33
 |
| | Fig. 2 Micrographs show SEM cross-sections of chitosan-based hydrogels P(AA-co-AMPS) at AA–AMPS ratios of (a) 1:5, (b) 1:1, (c) 5:1, (d) 10:1, and (e) 1:0. The histogram shows the average pore size and pore size distribution of the hydrogels. | |
The AA–AMPS ratios affected water absorption of the hydrogels. The highest WAC (707.36 g g−1) was obtained from the 1:5 hydrogel (Fig. 3). However, the structural integrity of this sample was low compared with the other fully swollen hydrogels. This result was consistent with the thinner cell wall and the larger pore size of the 1:5 hydrogel. The hydrogels with smaller pores, absorbed less water but were more mechanically stable.
 |
| | Fig. 3 Water absorption capacity of chitosan-based P(AA-co-AMPS) hydrogels with various AA–AMPS ratios. | |
3.2 Adsorption performance of the 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 hydrogel towards various dyes
:1 hydrogel towards various dyes
In this study, the 5:1 hydrogel was used as a representative adsorbent. The adsorption of cationic dyes was superior (Fig. 4) due to the strong electrostatic interactions between carboxylic acid and sulfonic acid groups of the hydrogel and cationic dyes. The adsorption capacities towards MB and CV exceeded 860 mg g−1. The anionic dye adsorption capacities of this hydrogel were between 3 and 45 mg g−1.
 |
| | Fig. 4 Adsorption capacity of chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 5:1 towards various anionic and cationic dyes. (Co = 1000 mg L−1, dose = 0.05 g/50 mL, ambient temperature, contact time = 24 h, unadjusted pH). | |
3.3 Effect of AA–AMPS ratio on MB and CV adsorption by hydrogel
As the 5:1 hydrogel demonstrated higher adsorption capacities towards MB and CV, these two dyes were chosen to study the effect of the AA–AMPS ratio on dye adsorption. All hydrogels demonstrated comparable adsorption capacities of approximately 800 mg g−1 towards MB—about 24 times the adsorption capacity of pristine chitosan (Fig. 5). The influence of AA and AMPS contents on MB adsorption was also found to be negligible in a previous study that used P(AMPS-co-AA) particles as adsorbents.19 The effect of the AA–AMPS ratio in the hydrogel was more pronounced in CV adsorption. The adsorption capacity towards CV increased with increments of AA content. As the total weight of the feed monomers was fixed, the hydrogel with a higher AA content had a higher total number of moles of monomers. Therefore, the enhanced adsorption performance was due to the increase in the number of negatively-charged functional groups. The 10:1 hydrogel exhibited the highest adsorption capacity of 1032 mg g−1—about 23 times that of chitosan. The adsorption capacity of 1:5 hydrogel was not reported as the hydrogel broke into small pieces during stirring, possibly due to the low degree of crosslinking. This result was in good agreement with the water absorption test, in which the WAC of the 1:5 hydrogel was excessive. The low adsorption capacity of the 1:0 hydrogel was possibly due to the low WAC of the hydrogel.
 |
| | Fig. 5 Adsorptions of methylene blue (MB) and crystal violet (CV) by chitosan-based hydrogels containing P(AA-co-AMPS) at various AA–AMPS ratios. (Co = 1000 mg L−1, dose = 0.05 g/50 mL, ambient temperature, contact time = 24 h, unadjusted pH). | |
3.4 Effect of contact time on CV adsorption
Because the 10:1 hydrogel had the highest CV adsorption capacity, it was used for subsequent experiments. The adsorption rate of CV on the 10:1 hydrogel was initially fast due to the large number of adsorption sites, which quickly combined with CV in solution. As contact time progressed, more adsorption sites became occupied, and the electrostatic repulsion between adsorbed and unadsorbed CV molecules increased, the rate of adsorption slowed (Fig. 6).34 The hydrogel removed 91% of CV in 12 h and reached equilibrium at about 16 h. The adsorption capacity and removal efficiency at equilibrium were 1032 mg g−1 and 94%, respectively.
 |
| | Fig. 6 Adsorption of CV at various contact times of a chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 10:1. (Co = 1000 mg L−1, dose = 0.05 g/50 mL, ambient temperature, unadjusted pH). | |
3.5 Kinetics of CV adsorption
To investigate the adsorption process of CV on the hydrogel, the pseudo-first-order (eqn (5)) and pseudo-second-order (eqn (6)) kinetics models were used:| | |
ln(qe − qt) = lnqe − k1t
| (5) |
| |
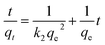 | (6) |
where qe and qt were the dye adsorbed (mg g−1) at equilibrium and at different times; k1 and k2 were the rate constants of the pseudo-first-order model, and the pseudo-second-order model, respectively.35
Fig. 7(a) and (b) show the pseudo-first-order linear fitting and the pseudo-second-order linear fitting, respectively. The kinetics parameters are listed in Table 1. The correlation coefficient of the pseudo-second-order model was closer to 1.0 and higher than that of the pseudo-first-order model, indicating that the adsorption rate was controlled by chemisorption. In addition, the calculated qe value was closer to the experimental qe value for the pseudo-second-order model. It was worth noting that the pseudo-second-order rate constant (k2) of this hydrogel was relatively lower than that of several hydrogel adsorbents for CV, such as modified rice bran/alginate hydrogel beads (5.69 × 10−4 g mg−1 min−1),36 husk of agarwood fruit/sodium alginate hydrogel (1.92 × 10−4 g mg−1 min−1),37 and cellulose/carrageenan magnetic hydrogel microbeads (6.8× 10−3 g mg−1 min−1).38
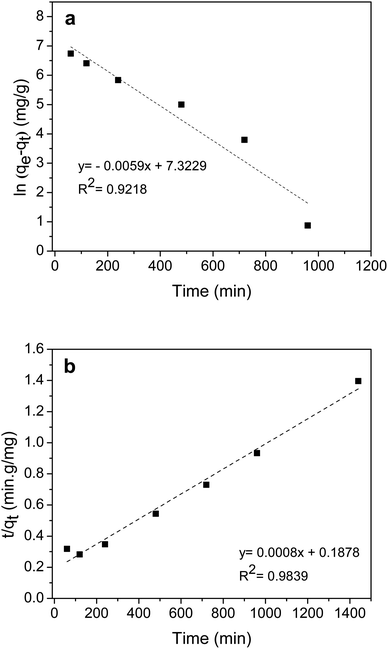 |
| | Fig. 7 Pseudo-first-order linear fitting (a) and pseudo-second-order linear fitting (b) of CV adsorption of a chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 10:1. (Co = 1000 mg L−1, dose = 0.05 g/50 mL, ambient temperature, unadjusted pH). | |
Table 1 Kinetics parameters
| |
Kinetics parameters |
| Experiment |
qe,exp (mg g−1) |
1032 |
| Pseudo-first-order |
qe,cal (mg g−1) |
1515 |
| k1 (min−1) |
0.0059 |
| R2 |
0.9218 |
| Pseudo-second-order |
qe,cal (mg g−1) |
1250 |
| k2 (g mg−1 min−1) |
3.408 × 10−6 |
| R2 |
0.9839 |
3.6 Effect of hydrogel dose on CV adsorption
As the adsorbent dose generally affected the efficiency of dye removal, the effect of the 10:1 hydrogel dose on CV adsorption was investigated. At a low dose of 0.01 g per 50 mL of CV solution, the adsorption capacity was only 322 mg g−1 and the removal efficiency was 6% (Fig. 8(a)). Upon increasing the hydrogel dose up to 0.05 g per 50 mL of CV solution, the adsorption capacity and removal efficiency increased due to the increase in the active sites to the dye molecules. At a dose of 0.07 g, the removal efficiency reached 98%, while the adsorption capacity decreased to 723 mg g−1. The decrease in the adsorption capacity was attributed to the limited number of dye molecules. Therefore, further studies were conducted with 0.05 g of hydrogel per 50 mL of CV solution.
 |
| | Fig. 8 Adsorption capacity and removal efficiency of a chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 10:1 towards aqueous crystal violet at (a) various doses (Co = 1000 mg L−1, V = 50 mL, ambient temperature, contact time = 24 h, unadjusted pH) and (b) at various pH values. (Co = 1000 mg L−1, dose = 0.05 g/50 mL, ambient temperature, contact time = 24 h). | |
3.7 Effect of pH on CV adsorption
As solution pH affects the surface charge and active sites of the adsorbent along with the degree of ionization and structure of the adsorbate, CV adsorption on the 10:1 hydrogel was investigated in the pH range of 2.0 to 10.0. The pH of the prepared CV solution was 4.3 and this was adjusted using 0.1 M HCl or 0.1 M NaOH solutions. In a strong acidic condition (pH = 2.0), adsorption capacity was low because the hydrogel network contracted, which prevented the adsorption of dye.39 At pH 4.3, a sharp increase in adsorption capacity and removal efficiency was observed (Fig. 8(b)). Since the pKa of AMPS = 1.5 and that of AA = 4.2,18 the sulfonic acid groups (–SO3H) of PAMPS and carboxyl groups (–COOH) of PAA were ionized at pH above 4.2.18 Therefore, electrostatic interactions with the protonated amine group of CV were stronger. A slight increase in capacity was observed at pH 6.0. Above pH 6.0, adsorption capacity decreased. The decrease in capacity could be attributed to a decrease in CV cationization, which reduced electrostatic interactions. Similar findings were reported in MB adsorption on chitosan-g-P(AA-co-AMPS).20
3.8 Effect of initial concentration of CV on adsorption and adsorption isotherm
A contact time of 24 h was allowed to elapse to ensure that adsorption reached equilibrium. Adsorption capacity increased with increments of initial concentration from 10 to 3000 mg L−1 (Fig. 9). At these concentrations, the number of available adsorption sites for bonding was greater than the number of dye molecules.40 The maximum adsorption capacity was 2023 mg g−1. The decrease in adsorption capacity at the initial concentration of 4000 mg L−1 resulted from the saturation of active sites on the hydrogel surface.41 The interaction between CV and the hydrogel was investigated using the Langmuir and Freundlich isotherm models. The Langmuir isotherm model describes the formation of a monolayer adsorption on a homogenous surface. The Freundlich model, on the other hand, assumes that adsorption sites are heterogenous. The stronger adsorption sites are occupied first, and as the coverage of the adsorbent surface increases, adsorption affinity decreases, resulting in multilayer adsorption.42 The Langmuir and Freundlich isotherm models can be expressed as eqn (7) and (8), respectively:| |
 | (7) |
where Ce (mg L−1) is the concentration of CV at equilibrium. qe (mg g−1) is the Langmuir maximum adsorption capacity at equilibrium and KL (L mg−1) is the Langmuir isotherm constant. KF (L mg−1) is the Freundlich isotherm constant, and n is the heterogeneity factor.
 |
| | Fig. 9 Adsorption capacity at various initial CV concentrations of a chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 10:1. (Dose = 0.05 g/50 mL, ambient temperature, contact time = 24 h, unadjusted pH). | |
The linear Langmuir isotherm and linear Freundlich isotherm of CV adsorption are shown in Fig. 10(a) and (b), respectively. The adsorption isotherm parameters are summarized in Table 2. The correlation coefficient of the Langmuir model was closer to 1.0 and higher than that of the Freundlich model, suggesting that the Langmuir model better described the adsorption of CV on the 10:1 hydrogel. The theoretical maximum adsorption capacity (qmax) of this hydrogel was 2500 mg g−1, which is higher than many other values reported for CV adsorbents (Table 3).
 |
| | Fig. 10 Linear Langmuir isotherm (a) and linear Freundlich isotherm (b) of CV adsorption of a chitosan-based P(AA-co-AMPS) hydrogel with an AA–AMPS ratio of 10:1. (Dose = 0.05 g/50 mL, ambient temperature, contact time = 24 h, unadjusted pH). | |
Table 2 Adsorption isotherm parameters
| Isotherm model |
Isotherm parameters |
| Calculated at the CV concentration of 1000 mg L−1. |
| Langmuir |
qm (mg g−1) |
2500 |
| KL (L mg−1) |
0.0054 |
| RLa |
0.1550 |
| R2 |
0.9666 |
| Freundlich |
KF (mg g−1) |
18.0185 |
| 1/n |
0.7799 |
| R2 |
0.8899 |
Table 3 Comparison with various adsorbents for CV
| Adsorbent |
qmax (mg g−1) |
KL (L mg−1) |
References |
| Polyaniline@TiO2 |
80 |
0.004 |
43 |
| p(NIPAM-co-MAA1.0)/b-CD0.4 |
1253.7 |
0.0136 |
44 |
| Gum arabic-cl-poly(acrylamide) nanohydrogel |
90.9 |
0.047 |
45 |
| Rarasaponin–bentonite-activated biochar composite |
518.6 |
0.0741 |
46 |
| Hydroxypropyl-β-cyclodextrin–polyurethane magnetic nanoconjugates |
1269 |
— |
47 |
| Zeolitic imidazolate framework-9 (Z9-600) |
26.8 |
— |
48 |
| Modified rice bran/alginate hydrogel beads |
454.55 |
0.011 |
36 |
| Husk of agarwood fruit/sodium alginate hydrogel |
370.37 |
0.022 |
37 |
| Cellulose/carrageenan magnetic hydrogel microbeads |
217.1 |
0.0159 |
38 |
| P(AA-co-AMPS)-grafted chitosan hydrogel |
2023 |
0.0054 |
This study |
In addition, the separation factor, RL, of the Langmuir model was calculated using eqn (9):
| |
 | (9) |
where
KL (L mg
−1) is the Langmuir isotherm constant, and
C0 (mg L
−1) is the initial dye concentration.
The RL value indicates the tendency of the adsorption process to be irreversible (RL = 0), favorable (0 < RL < 1), linear (RL = 1), or unfavorable (RL > 1).18 The Langmuir isotherm RL value of 0.1550 indicated a favorable adsorption. The KL value is related to the affinity of binding sites and is independent of qmax. The relatively low KL value of the 10:1 hydrogel suggests a low affinity for CV (Table 3).
3.9 ZnO/P(AA-co-AMPS) 10:1 hydrogel composite
Fig. 11(a) and (b) show photographs of the 10:1 hydrogel and ZnO/10:1 hydrogel composite. After the in situ synthesis of ZnO particles, the transparent hydrogel became partially white and opaque except in the outer area of the sample. Fig. 11(c) and (d) show TEM micrographs of ZnO particles in the 10:1 hydrogel matrix. The particles were well dispersed, and agglomerations were small. The average size of the particles was 16.91 ± 1.41 nm but the shape of the particles was not uniform. SEM-EDS (Fig. 11(e)) of the ZnO/10:1 hydrogel composite showed that the porosity of the hydrogel was maintained after the in situ synthesis of ZnO. In order to observe the distribution of ZnO particles, the image was taken at low magnification. The elemental mappings confirmed that Zn was evenly distributed and fully covered the surface of cell walls. Zn and O elements almost completely replaced C. SEM-EDS micrographs were further analyzed at higher magnification (Fig. 12) to determine the % element ratio which was summarized in Table 4. The results show that a very low content of Zn was observed on the outer surface of the hydrogel which was consistent with the clear layer as observed in Fig. 11(b). However, a much higher content of Zn was observed in the cross-section area indicating a dense incorporation of ZnO particles inside the hydrogel matrix. Na was detected on both the outer surface and the cross-section area, which was the residue of the precipitation.
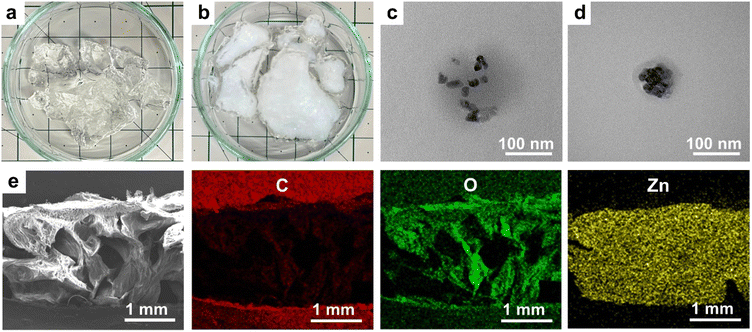 |
| | Fig. 11 Photographs of (a) 10:1 hydrogel and (b) ZnO/10:1 hydrogel composite, (c) and (d) TEM micrographs (magnification 150k×) of ZnO particles, and (e) SEM-EDS cross-section micrograph and the corresponding elemental maps of ZnO/10:1 hydrogel composite. | |
 |
| | Fig. 12 SEM-EDS micrographs and the corresponding elemental maps of ZnO/10:1 hydrogel composite (a) outer surface and (b) cross-section. | |
Table 4 Corresponding elemental weight percentages of ZnO/10:1 hydrogel composite
| Element |
Weight% |
| Outer surface |
Cross-section |
| C |
37.6 |
23.6 |
| O |
43.9 |
29.5 |
| Zn |
1.5 |
33.7 |
| Na |
17.0 |
13.2 |
Fig. 13(a) depicts XRD patterns of the 10:1 hydrogel and ZnO/10:1 hydrogel composite. The hydrogel showed a small broad peak at 20.1°, corresponding to the amorphous structure of PAA.49 The hydrogel composite showed additional peaks of the hexagonal phase of the wurtzite structure of ZnO at 30.9°, 33.6°, 35.4°, 46.7°, 55.7°, 62.0°, 65.2°, 67.0°, and 68.2° (JCPDS card no. 36-1451).50 The XRD analysis confirmed the successful conversion of Zn2+ ions to ZnO particles in the hydrogel matrix with high purity.
 |
| | Fig. 13 XRD patterns (a) and TGA thermograms (b) of 10:1 hydrogel and ZnO/10:1 hydrogel composite. | |
TGA thermograms of the 10:1 hydrogel and ZnO/10:1 hydrogel composite showed a continuous weight loss from 35 to 350 °C due to the loss of absorbed water molecules and the degradation of polymeric hydrogel via thermal scission of pendant groups, crosslinks, and backbones (Fig. 13(b)).51,52 However, the initial degradation temperature of the hydrogel composite was higher than that of the bare hydrogel (336 °C vs. 357 °C). The temperature shift indicated an increase in thermal stability. Moreover, because ZnO is stable at high temperatures, the thermal stability of the hydrogel composite could be used to determine the amount of ZnO incorporated in the hydrogel. The residual weight of the samples at 650 °C indicated the percentage of polymer char. Therefore, the 8.7% difference between the residual weight of the bare 10:1 hydrogel and that of the ZnO/10:1 hydrogel composite could be attributed to ZnO.
The performance of the hydrogel composite towards the photocatalytic degradation of adsorbed dye was studied under natural sunlight and artificial UV light. After 3 h under sunlight (Fig. 14(a)), the color of the hydrogel composite had changed from dark purple to almost colorless and whitish, indicating that the adsorbed dye molecules had been degraded. Under UV lamps (Fig. 14(c)), photocatalytic degradation took longer (6 h) due to the much lower power of irradiation. Although the photocatalytic degradation could not be quantitatively analyzed due to the irregular sample shapes, the reusability of the hydrogel composite was tested for adsorption and photocatalytic degradation. During photocatalytic degradation, the hydrogel composite was immersed in water to remove photodegraded products and simultaneously regenerate the hydrogel composite, thus no solvent desorption or washing step was required. In the first cycle, the hydrogel composite removed ∼95% of the dye from the solution by adsorption process (Fig. 14(e)). In the second cycle, the removal percentage of the sample regenerated by sunlight slightly decreased to 92% whereas the removal percentage of the sample regenerated by UV light decreased to 90%. The reduction in removal efficiency was due to the accumulation of degraded products on the surface of the hydrogel composite that hindered the activities of the free radical species.53–56
 |
| | Fig. 14 Photocatalytic degradation of CV adsorbed in ZnO/10:1 hydrogel composite under sunlight (a) cycle 1 and (b) cycle 2 and UV lamps (c) cycle 1 and (d) cycle 2, and (e) removal efficiency of the hydrogel composite. (Irradiation time is as indicated.) | |
Fig. 14(b)-3 h shows that, in the second cycle, the adsorbed dye was almost completely degraded by sunlight, however, the hydrogel composite was obviously less opaque. In the case of UV light, the darker color of the hydrogel composite was observed in the second cycle compared with the first cycle at the same irradiation time (6 h) (Fig. 14(c)-6 h and (d)-6 h). These results indicated that ZnO particles disappeared during irradiation and thus, the degradation performance of the photocatalytic degradation of CV was reduced due to the loss of ZnO particles. It has been reported that ZnO particles could undergo hydrolyzation and were dissolved into Zn2+ ions.57 In this work, the ZnO leaching increased with an increased exposure time. Besides, the loss of ZnO particles was probably due to the very small size of particles compared with the pore size of the hydrogel. This limitation will be our further study by reducing ZnO leaching by producing strong interactions between polymers and ZnO particles and using a powerful light source to decrease exposure time. Although the photocatalytic efficiency decreased and the reusability of the hydrogel composite was low, the adsorption efficiency of the hydrogel itself containing 2210% of grafting chitosan remained satisfactory. This composite material is easy to use, especially in material handling. The problem of photocatalytic powder recovery is avoided.
The proposed mechanism of photocatalytic degradation of CV adsorbed in the hydrogel composite is shown in Fig. 15. When ZnO photocatalyst particles were exposed to light, they gained energy and generated electrons (e−) in the conduction band (CB) and holes (h+) in the valence band (VB). These e− and h+ further reacted with oxygen (O2) and water (H2O) molecules, respectively, producing superoxide radical anions O2˙− and hydroxyl radicals (˙OH) which decomposed CV molecules into non-toxic small molecules.
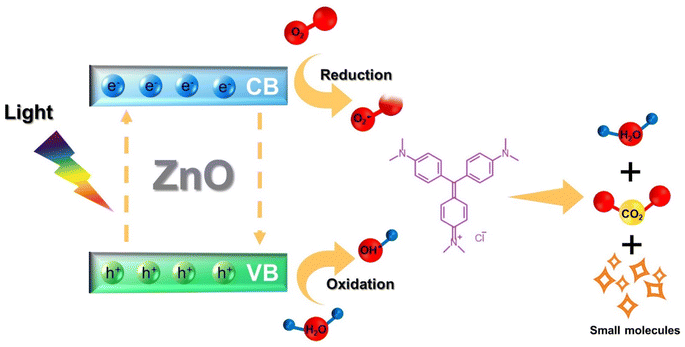 |
| | Fig. 15 The proposed photocatalytic degradation mechanism of CV by ZnO encapsulated in 10:1 hydrogel. | |
4. Conclusions
P(AA-co-AMPS)-grafted hydrogels were successfully synthesized via free radical polymerization with high yields (>90%). Grafting P(AA-co-AMPS) enhanced the adsorption capacity of chitosan, and the hydrogels showed superior adsorption capacity towards cationic dyes compared to anionic dyes. The effect of AA and AMPS contents on adsorption was observed in CV adsorption. The 10:1 hydrogel removed 91% of CV in 12 h and reached equilibrium after 16 h. The maximum adsorption capacity of this hydrogel towards CV was 2023 mg g−1, which is higher than the reported values of most other CV adsorbents. The adsorption kinetics and adsorption isotherm were described by the pseudo-second-order kinetics model and the Langmuir isotherm model, respectively. ZnO particles incorporated in the 10:1 hydrogel by in situ synthesis yielded a stable hydrogel composite with photocatalytic properties. The hydrogel composite could almost completely photodegrade adsorbed CV dye under sunlight in less than 3 h. The adsorption capacity of the hydrogel composite in the 2nd cycle remained higher than 90% but photocatalytic degradation decreased. This composite material could be used for wastewater treatment, adsorbing, and degrading dye pollutants without requiring a photocatalyst recovery procedure.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Fundamental Fund of Khon Kaen University (KKU), and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation. The authors would like to thank Dr Chonnakarn Panawong for instrumental assistance.
References
- Y. Hong, C. Li, B. Yin, D. Li, Z. Zhang, B. Mao, W. Fan, W. Gu and W. Shi, Promoting visible-light-induced photocatalytic degradation of tetracycline by an efficient and stable beta-Bi2O3@g-C3N4 core/shell nanocomposite, Chem. Eng. J., 2018, 338, 137–146, DOI:10.1016/j.cej.2017.12.108.
- M. Mathur, D. Gola, R. Panja, A. Malik and S. Z. Ahammad, Performance evaluation of two Aspergillus spp. for the decolourization of reactive dyes by bioaccumulation and biosorption, Environ. Sci. Pollut. Res., 2018, 25, 345–352, DOI:10.1007/s11356-017-0417-0.
- M. Bilal, I. Ihsanullah, M. U. Hassan Shah, A. V. Bhaskar Reddy and T. M. Aminabhavi, Recent advances in the removal of dyes from wastewater using low-cost adsorbents, J. Environ. Manage., 2022, 321, 115981, DOI:10.1016/j.jenvman.2022.115981.
- R. Abejón, M. de Cazes, M. P. Belleville and J. Sanchez-Marcano, Large-scale enzymatic membrane reactors for tetracycline degradation in WWTP effluents, Water Res., 2015, 73, 118–131, DOI:10.1016/j.watres.2015.01.012.
- Z. Liu, M. Zhu, L. Zhao, C. Deng, J. Ma, Z. Wang, H. Liu and H. Wang, Aqueous tetracycline degradation by coal-based carbon electrocatalytic filtration membrane: effect of nano antimony-doped tin dioxide coating, Chem. Eng. J., 2017, 314, 59–68, DOI:10.1016/j.cej.2016.12.093.
- A. G. Varghese, S. A. Paul and M. S. Latha, Remediation of heavy metals and dyes from wastewater using cellulose-based adsorbents, Environ. Chem. Lett., 2019, 17, 867–877, DOI:10.1007/s10311-018-00843-z.
- G. Crini and P. M. Badot, Application of chitosan, a natural aminopolysaccharide, for dye removal from aqueous solutions by adsorption processes using batch studies: a review of recent literature, Prog. Polym. Sci., 2008, 33, 399–447, DOI:10.1016/j.progpolymsci.2007.11.001.
- S. Ma, M. Zhang, J. Nie, J. Tan, S. Song and Y. Luo, Lightweight and porous cellulose-based foams with high loadings of zeolitic imidazolate frameworks-8 for adsorption applications, Carbohydr. Polym., 2019, 208, 328–335, DOI:10.1016/j.carbpol.2018.12.081.
- Y. Wang, X. Dai, Y. Zhan, X. Ding, M. Wang and X. Wang, In situ growth of ZIF-8 nanoparticles on chitosan to form the hybrid nanocomposites for high-efficiency removal of Congo Red, Int. J. Biol. Macromol., 2019, 137, 77–86, DOI:10.1016/j.ijbiomac.2019.06.195.
- Y. Wang, G. Xia, C. Wu, J. Sun, R. Song and W. Huang, Porous chitosan doped with graphene oxide as highly effective adsorbent for methyl orange and amido black 10B, Carbohydr. Polym., 2015, 115, 686–693, DOI:10.1016/j.carbpol.2014.09.041.
- S. Kang, L. Qin, Y. Zhao, W. Wang, T. Zhang, L. Yang, F. Rao and S. Song, Enhanced removal of methyl orange on exfoliated montmorillonite/chitosan gel in presence of methylene blue, Chemosphere, 2020, 238, 124693, DOI:10.1016/j.chemosphere.2019.124693.
- K. Phonlakan, B. Khamsuk, N. Soontonhong, C. Panawong, P. Kongseng, S. Chantarak and S. Budsombat, Composite beads from chitosan and zeolitic imidazolate framework-8 for the adsorption and photocatalytic degradation of reactive red 141, RSC Adv., 2023, 13, 12295–12308, 10.1039/d3ra01187a.
- M. Vakili, M. Rafatullah, B. Salamatinia, A. Z. Abdullah, M. H. Ibrahim, K. B. Tan, Z. Gholami and P. Amouzgar, Application of chitosan and its derivatives as adsorbents for dye removal from water and wastewater: a review, Carbohydr. Polym., 2014, 113, 115–130, DOI:10.1016/j.carbpol.2014.07.007.
- Z. Yang, H. Yang, Z. Jiang, T. Cai, H. Li, H. Li, A. Li and R. Cheng, Flocculation of both anionic and cationic dyes in aqueous solutions by the amphoteric grafting flocculant carboxymethyl chitosan-graft-polyacrylamide, J. Hazard. Mater., 2013, 254–255, 36–45, DOI:10.1016/j.jhazmat.2013.03.053.
- J. Maity and S. K. Ray, Enhanced adsorption of methyl violet and congo red by using semi and full IPN of polymethacrylic acid and chitosan, Carbohydr. Polym., 2014, 104, 8–16, DOI:10.1016/j.carbpol.2013.12.086.
- J. Men, H. Shi, C. Dong, Y. Yang, Y. Han, R. Wang, Y. Zhang, T. Zhao and J. Li, Preparation of poly(sodium 4-styrene sulfonate) grafted magnetic chitosan microspheres for adsorption of cationic dyes, Int. J. Biol. Macromol., 2021, 181, 810–823, DOI:10.1016/j.ijbiomac.2021.04.079.
- L. Wang, J. Zhang and A. Wang, Fast removal of methylene blue from aqueous solution by adsorption onto chitosan-g-poly(acrylic acid)/attapulgite composite, Desalination, 2011, 266, 33–39, DOI:10.1016/j.desal.2010.07.065.
- B. Xu, C. Zheng, H. Zheng, Y. Wang, C. Zhao, C. Zhao and S. Zhang, Polymer-grafted magnetic microspheres for enhanced removal of methylene blue from aqueous solutions, RSC Adv., 2017, 7, 47029–47037, 10.1039/c7ra06810g.
- X. Huang, D. Zhou, Y. Wang, C. He, W. Zhao, S. Sun and C. Zhao, A green approach towards functional hydrogel particles from synthetic polymers via spherical capsule mini-reactors, Chem. Eng. J., 2019, 359, 1360–1371, DOI:10.1016/j.cej.2018.11.042.
- Y. Tang, T. He, Y. Liu, B. Zhou, R. Yang and L. Zhu, Sorption behavior of methylene blue and rhodamine B mixed dyes onto chitosan graft poly(acrylic acid-co-2-acrylamide-2-methyl propane sulfonic acid) hydrogel, Adv. Polym. Technol., 2018, 37, 2568–2578, DOI:10.1002/adv.21932.
- J. Guo, H. Sun, X. Yuan, L. Jiang, Z. Wu, H. Yu, N. Tang, M. Yu, M. Yan and J. Liang, Photocatalytic degradation of persistent organic pollutants by Co-Cl bond reinforced CoAl-LDH/Bi12O17Cl2 photocatalyst: mechanism and application prospect evaluation, Water Res., 2022, 219, 118558, DOI:10.1016/J.WATRES.2022.118558.
- S. Sharma and S. Basu, Highly reusable visible light active hierarchical porous WO3/SiO2 monolith in centimeter length scale for enhanced photocatalytic degradation of toxic pollutants, Sep. Purif. Technol., 2020, 231, 115916, DOI:10.1016/j.seppur.2019.115916.
- Y. Li, H. Zhang, X. Hu, X. Zhao and M. Han, Efficient Visible-Light-Induced Photocatalytic Activity of a 3D-Ordered Titania Hybrid Photocatalyst with a Core/Shell Structure of Dye-Containing Polymer/Titania, J. Phys. Chem. C, 2008, 112, 14973–14979, DOI:10.1021/jp8055152.
- C. Ma, J. Zhou, Z. Cui, Y. Wang and Z. Zou, In situ growth MoO3 nanoflake on conjugated polymer: an advanced photocatalyst for hydrogen evolution from water solution under solar light, Sol. Energy Mater. Sol. Cells, 2016, 150, 102–111, DOI:10.1016/j.solmat.2016.02.010.
- D. Ponnamma, J.-J. Cabibihan, M. Rajan, S. S. Pethaiah, K. Deshmukh, J. P. Gogoi, S. K. K. Pasha, M. B. Ahamed, J. Krishnegowda, B. N. Chandrashekar, A. R. Polu and C. Cheng, Synthesis, optimization and applications of ZnO/polymer nanocomposites, Mater. Sci. Eng., C, 2019, 98, 1210–1240, DOI:10.1016/j.msec.2019.01.081.
- Y. Zhang, S. Wei, H. Zhang, S. Liu, F. Nawaz and F.-S. Xiao, Nanoporous polymer monoliths as adsorptive supports for robust photocatalyst of Degussa P25, J. Colloid Interface Sci., 2009, 339, 434–438, DOI:10.1016/j.jcis.2009.07.050.
- P. Maijan, P. Amornpitoksuk and S. Chantarak, Synthesis and characterization of poly(vinyl alcohol-g-acrylamide)/SiO2@ZnO photocatalytic hydrogel composite for removal and degradation of methylene blue, Polymer, 2020, 203, 122771, DOI:10.1016/j.polymer.2020.122771.
- B. Zhang, H. Yu, Y. Zhang, Z. Luo, W. Han, W. Qiu and T. Zhao, Bacterial cellulose derived monolithic titania aerogel consisting of 3D reticulate titania nanofibers, Cellulose, 2018, 25, 7189–7196, DOI:10.1007/s10570-018-2073-z.
- M. Alavi and A. Nokhodchi, An overview on antimicrobial and wound healing properties of ZnO nanobiofilms, hydrogels, and bionanocomposites based on cellulose, chitosan, and alginate polymers, Carbohydr. Polym., 2020, 227, 115349, DOI:10.1016/j.carbpol.2019.115349.
- A. M. K. Najjar, W. M. Z. W. Yunus, M. B. Ahmad and M. Z. A. Rahman, Preparation and characterization of poly(2-acrylamido-2-methylpropane-sulfonic acid) grafted chitosan using potassium persulfate as redox initiator, J. Appl. Polym. Sci., 2000, 77, 2314–2318, DOI:10.1002/1097-4628(20000906)77:10<2314::AID-APP25>3.0.CO;2-7.
- Y. Liu, Y. Zheng and A. Wang, Enhanced adsorption of methylene blue from aqueous solution by chitosan-g-poly (acrylic acid)/vermiculite hydrogel composites, J. Environ. Sci., 2010, 22, 486–493, DOI:10.1016/s1001-0742(09)60134-0.
- K. Varaprasad, K. Vimala, S. Ravindra, N. Narayana Reddy, G. Siva Mohana Reddy and K. Mohana Raju, Biodegradable chitosan hydrogels for in vitro drug release studies of 5-flurouracil an anticancer drug, J. Polym. Environ., 2012, 20, 573–582, DOI:10.1007/s10924-012-0412-y.
- Y. Zhao, J. Kang and T. Tan, Salt-, pH- and temperature-responsive semi-interpenetrating polymer network hydrogel based on poly(aspartic acid) and poly(acrylic acid), Polymer, 2006, 47, 7702–7710, DOI:10.1016/j.polymer.2006.08.056.
- H. Zhang, Q. Dang, C. Liu, D. Yu, Y. Wang, X. Pu, Y. Liu, Y. Liang and D. Cha, Fabrication of methyl acrylate and tetraethylenepentamine grafted magnetic chitosan microparticles for capture of Cd(II) from aqueous solutions, J. Hazard. Mater., 2019, 366, 346–357, DOI:10.1016/j.jhazmat.2018.12.006.
- E. D. Revellame, D. L. Fortela, W. Sharp, R. Hernandez and M. E. Zappi, Adsorption kinetic modeling using pseudo-first order and pseudo-second order rate laws: a review, Cleaner Engineering and Technology, 2020, 1, 100032, DOI:10.1016/j.clet.2020.100032.
- G.-B. Hong, T.-J. Yu, H.-C. Lee and C.-M. Ma, Using rice bran hydrogel beads to remove dye from aqueous solutions, Sustainability, 2021, 13, 5640, DOI:10.3390/su13105640.
- C. M. Ma, B.-Y. Yang and G.-B. Hong, Husk of agarwood fruit-based hydrogel beads for adsorption of cationic and anionic dyes in aqueous solutions, Molecules, 2021, 26, 1437, DOI:10.3390/molecules26051437.
- S. Park, Y. Oh, J. Yun, E. Yoo, D. Jung, K. K. Oh and S. H. Lee, Cellulose/biopolymer/Fe3O4 hydrogel microbeads for dye and protein adsorption, Cellulose, 2020, 27, 2757–2773, DOI:10.1007/s10570-020-02974-5.
- L. Zhu, C. Guan, B. Zhou, Z. Zhang, R. Yang, Y. Tang and J. Yang, Adsorption of dyes onto sodium alginate graft poly(acrylic acid-co-2-acrylamide-2-methyl propane sulfonic acid)/kaolin hydrogel composite, Polym. Polym. Compos., 2017, 25, 627–634, DOI:10.1177/096739111702500808.
- P. Kongseng, P. Amornpitoksuk and S. Chantarak, Development of multifunctional hydrogel composite based on poly(vinyl alcohol-g-acrylamide) for removal and photocatalytic degradation of organic dyes, React. Funct. Polym., 2022, 172, 105207, DOI:10.1016/j.reactfunctpolym.2022.105207.
- Y. Wang, L. Zhao, H. Peng, J. Wu, Z. Liu and X. Guo, Removal of anionic dyes from aqueous solutions by cellulose-based adsorbents: equilibrium, kinetics, and thermodynamics, J. Chem. Eng. Data, 2016, 61, 3266–3276, DOI:10.1021/acs.jced.6b00340.
- H. Yan, H. Yang, A. Li and R. Cheng, pH-tunable surface charge of chitosan/graphene oxide composite adsorbent for efficient removal of multiple pollutants from water, Chem. Eng. J., 2016, 284, 1397–1405, DOI:10.1016/j.cej.2015.06.030.
- M. Maruthapandi, L. Eswaran, J. H. T. Luong and A. Gedanken, Sonochemical preparation of polyaniline@TiO2 and polyaniline@SiO2 for the removal of anionic and cationic dyes, Ultrason. Sonochem., 2020, 62, 104864, DOI:10.1016/j.ultsonch.2019.104864.
- S. Jia, D. Tang, J. Peng, Z. Sun and X. Yang, β-Cyclodextrin modified electrospinning fibers with good regeneration for efficient temperature-enhanced adsorption of crystal violet, Carbohydr. Polym., 2019, 208, 486–494, DOI:10.1016/j.carbpol.2018.12.075.
- G. Sharma, A. Kumar, M. Naushad, A. García-Peñas, A. H. Al-Muhtaseb, A. A. Ghfar, V. Sharma, T. Ahamad and F. J. Stadler, Fabrication and characterization of gum arabic-cl-poly(acrylamide) nanohydrogel for effective adsorption of crystal violet dye, Carbohydr. Polym., 2018, 202, 444–453, DOI:10.1016/j.carbpol.2018.09.004.
- L. Laysandra, F. H. Santosa, V. Austen, F. E. Soetaredjo, K. Foe, J. N. Putro, Y.-H. Ju and S. Ismadji, Rarasaponin-bentonite-activated biochar from durian shells composite for removal of crystal violet and Cr(VI) from aqueous solution, Environ. Sci. Pollut. Res., 2018, 25, 30680–30695, DOI:10.1007/s11356-018-3104-x.
- S. Nasiri and N. Alizadeh, Synthesis and adsorption behavior of hydroxypropyl-β-cyclodextrin–polyurethane magnetic nanoconjugates for crystal and methyl violet dyes removal from aqueous solutions, RSC Adv., 2019, 9, 24603–24616, 10.1039/c9ra03335a.
- T.-T. Han, H.-L. Bai, Y.-Y. Liu and J.-F. Ma, Synthesis of nanoporous cobalt/carbon materials by a carbonized zeolitic imidazolate framework-9 and adsorption of dyes, New J. Chem., 2018, 42, 717–724, 10.1039/c7nj03745g.
- S. Moulay and N. Bensacia, Removal of heavy metals by homolytically functionalized poly(acrylic acid) with hydroquinone, Int. J. Ind. Chem., 2016, 7, 369–389, DOI:10.1007/s40090-016-0097-5.
- S. Li, P. C. Chiang, L. Ding, K. J. Shah, Q. Chen and S. Chen, ZnO-chitosan/rectorite nanocomposite exhibiting high photocatalytic activities under visible-light irradiation, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2020, 35, 310–319, DOI:10.1007/s11595-020-2258-9.
- M. A. Abu-Saied, E. A. Soliman and E. A. A. Desouki, Development of proton exchange membranes based on chitosan blended with poly(2-acrylamido-2-methylpropane sulfonic acid) for fuel cells applications, Mater. Today Commun., 2020, 25, 101536, DOI:10.1016/j.mtcomm.2020.101536.
- M. Mohammadian, R. Sahraei and M. Ghaemy, Synthesis and fabrication of antibacterial hydrogel beads based on modified-gum tragacanth/poly(vinyl alcohol)/Ag0 highly efficient sorbent for hard water softening, Chemosphere, 2019, 225, 259–269, DOI:10.1016/j.chemosphere.2019.03.040.
- G. Sharma, A. Kumar, M. Naushad, B. Thakur, D.-V. N. Vo, B. Gao, A. A. Al-Kahtani and F. J. Stadler, Adsorptional-photocatalytic removal of fast sulphon black dye by using chitin-cl-poly(itaconic acid-co-acrylamide)/zirconium tungstate nanocomposite hydrogel, J. Hazard. Mater., 2021, 416, 125714, DOI:10.1016/j.jhazmat.2021.125714.
- N. Kumar, H. Mittal, S. M. Alhassan and S. S. Ray, Bionanocomposite hydrogel for the adsorption of dye and reusability of generated waste for the photodegradation of ciprofloxacin: a demonstration of the circularity concept for water purification, ACS Sustain. Chem. Eng., 2018, 6, 17011–17025, DOI:10.1021/acssuschemeng.8b04347.
- M. Thomas, G. A. Naikoo, M. U. D. Sheikh, M. Bano and F. Khan, Effective photocatalytic degradation of Congo red dye using alginate/carboxymethyl cellulose/TiO2 nanocomposite hydrogel under direct sunlight irradiation, J. Photochem. Photobiol., A, 2016, 327, 33–43, DOI:10.1016/j.jphotochem.2016.05.005.
- L. Zhang, P. Ma, L. Dai, S. Li, W. Yu and J. Guan, In situ crystallization and growth of TiO2 nanospheres between MXene layers for improved adsorption and visible light photocatalysis, Catal. Sci. Technol., 2021, 11, 3834–3844, 10.1039/D1CY00239B.
- M. Malekkiani, A. Heshmati Jannat Magham, F. Ravari and M. Dadmehr, Facile fabrication of ternary MWCNTs/ZnO/chitosan nanocomposite for enhanced photocatalytic degradation of methylene blue and antibacterial activity, Sci. Rep., 2022, 12, 5927, DOI:10.1038/s41598-022-09571-5.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Panjalak Meetam
a,
Panjalak Meetam