
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChalcone Mannich base derivatives: synthesis, antimalarial activities against Plasmodium knowlesi, and molecular docking analysis†
Jufrizal Syahri*a,
Rahmiwati Hilmaa,
Amatul Hamizah Alib,
Norzila Ismail c,
Ng Yee Lingd,
Nurlailia,
Beta Achromi Nurohmahe,
Hani Kartini Agustarf,
Lau Yee Lingd and
Jalifah Latip*b
c,
Ng Yee Lingd,
Nurlailia,
Beta Achromi Nurohmahe,
Hani Kartini Agustarf,
Lau Yee Lingd and
Jalifah Latip*b
aDepartment of Chemistry, Universitas Muhammadiyah Riau, Jalan Tuanku Tambusai Ujung, Pekanbaru, Indonesia. E-mail: jsyachri@umri.ac.id
bDepartment of Chemical Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia. E-mail: jalifah@ukm.edu.my
cDepartment of Pharmacology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
dDepartment of Parasitology, Faculty of Medicine, Universiti Malaya, 50603, Kuala Lumpur, Malaysia
eDepartment of Chemistry, Universitas Gadjah Mada, Jalan Kaliurang Sekip Utara Bulaksumur 21, 55281, Yogyakarta, Indonesia
fDepartment of Earth Science and Environment, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia
First published on 12th December 2023
Abstract
Development and discovery of new antimalarial drugs are needed to overcome the multi-resistance of Plasmodium parasites to commercially available drugs. Modifying the substitutions on the amine groups has been shown to increase antimalarial activities and decrease cross-resistance with chloroquine. In this study, we have synthesized several chalcone derivatives via the substitution of aminoalkyl groups into the aromatic chalcone ring using the Mannich-type reaction. The chalcone derivatives were evaluated for their antimalarial properties against Plasmodium knowlesi A1H1 and P. falciparum 3D7, as well as their molecular docking on Plasmodium falciparum dihydrofolate reductases-thymidylate synthase (PfDHFR-TS). Data from in vitro evaluation showed that chalcone Mannich-type base derivatives 2a, 2e, and 2h displayed potential antimalarial activities against P. knowlesi with EC50 of 2.64, 2.98, and 0.10 μM, respectively, and P. falciparum 3D7 with EC50 of 0.08, 2.69, and 0.15 μM, respectively. The synthesized compounds 2a, 2e, and 2h exerted high selectivity index (SI > 10) values on the A1H1 and 3D7 strains. The molecular docking analysis on PfDHFR-TS supported the in vitro assay of 2a, 2e, and 2h by displaying CDOCKER energy of −48.224, −43.292, and −45.851 kcal mol−1. Therefore, the evidence obtained here supports that PfDHFR-TS is a putative molecular target for the synthesized compound.
Introduction
Malaria is an infectious disease caused by protozoa parasites of the genus Plasmodium like P. falciparum and P. knowlesi, which can be transmitted through the bite of the female Anopheles spp. mosquito. According to the World Health Organization (WHO), malaria cases reached 247 million and approximately 619![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 malaria deaths occurred globally in 2021. Southeast Asia has the second highest cases of malaria globally.1,2 The high incidence rate of P. falciparum in Southeast Asia countries is expected due to gene mutation in the parasite which develops resistance to major antimalarial drugs such as chloroquine, artemisinin, and several new antimalarial compounds such as dihydroisoquinolones, spiroindolones, and pyrazoles.3–5 Although many studies have been conducted to assess the effectiveness of artemisinin derivatives6 as antimalarial agents, it is notable that N-sulfonylpiperidinedispiro-1,2,4,5-tetraoxane analogs demonstrate promising antimalarial properties during in vivo evaluations.7 Dispiro-tetraoxanes, synthetic peroxides, known for their efficacy against drug-resistant P. falciparum, have been discussed by Yadav et al.8 A significant drawback of relying on artemisinin derivatives as a sole treatment is the heightened occurrence of late-stage parasite recrudescence.4 Hence, exploring a single class of drugs with a specific mechanism of action to prevent mutation is insufficient in addressing malarial infection.
000 malaria deaths occurred globally in 2021. Southeast Asia has the second highest cases of malaria globally.1,2 The high incidence rate of P. falciparum in Southeast Asia countries is expected due to gene mutation in the parasite which develops resistance to major antimalarial drugs such as chloroquine, artemisinin, and several new antimalarial compounds such as dihydroisoquinolones, spiroindolones, and pyrazoles.3–5 Although many studies have been conducted to assess the effectiveness of artemisinin derivatives6 as antimalarial agents, it is notable that N-sulfonylpiperidinedispiro-1,2,4,5-tetraoxane analogs demonstrate promising antimalarial properties during in vivo evaluations.7 Dispiro-tetraoxanes, synthetic peroxides, known for their efficacy against drug-resistant P. falciparum, have been discussed by Yadav et al.8 A significant drawback of relying on artemisinin derivatives as a sole treatment is the heightened occurrence of late-stage parasite recrudescence.4 Hence, exploring a single class of drugs with a specific mechanism of action to prevent mutation is insufficient in addressing malarial infection.
One gene mutation that commonly occurs is at dihydrofolate reductase-thymidylate synthase of Plasmodium falciparum (PfDHFR-TS), a well-known target for the antifolate drugs such as pyrimethamine and cycloguanil. The targeted gene is crucial to produce folates and thymidylate (dTMP) required for DNA synthesis in the malarial parasite.9 The emergence of antifolate resistance in PfDHFR-TS is an urgent issue that needs to be encountered for antimalarial medication. Therefore, the rise in malaria cases linked to P. falciparum resistance highlights the importance of continual research, especially the heightened emphasis on discovering novel antimalarial drugs.
Chalcone is one of the natural compounds that are active as an antimalarial.10–12 The activity of the chalcone compound comes from the functional groups attached to the aromatic ring and the alpha–beta unsaturated ketone bridge that connects the two rings. The alpha–beta unsaturated ketone moiety in chalcone is considered a Michael acceptor. This conjugated double bond makes chalcone compounds have a mechanism for electron transfer. These properties also make chalcone-derived compounds possess many activities, such as antimalarial13–16 (Fig. 1), anticancer,17–19 anti-inflammatory,20,21 antibacterial,22–24 antioxidants,25,26 antidiabetic,27,28 antigiardial,29 and antileukemic.30 Chalcone derivatives with a substituent secondary amine group could be proposed to increase the antimalarial activity.31,32
 | ||
| Fig. 1 Chalcone derivatives possessed antimalarial properties.15,18 | ||
In the study of Structure–Activity Relationship (SAR) analysis, it is known that most of the commercially available antimalarial drugs contain the amine group.33 This fact justifies that the amine group plays an important role in antimalarial activity. According to Suwito et al.,13 amines can form electrostatic interactions with carbonyl groups in a protein of the Plasmodium parasite, which can kill the parasite.13 In our previous study, adding aminoalkylated such as morpholine, piperidine, and diethylamine on chalcone significantly enhanced the antimalarial activity with IC50 values in a range of 0.54 to 1.12 μM against P. falciparum 3D7, compared with the chalcone without amine group.34 A prenylated and allylated chalcone showed the best IC50 values of 1.08 and 1.73 μg mL−1, respectively against the P. falciparum 3D7 strain.14 In addition, we previously synthesized and assessed a series of chalcone derivatives in which one of them substituted with the piperidine moiety showed an IC50 value of 0.54 μM and was categorized as having potential antiplasmodial candidate.32
In this study, the substitution of aminoalkyl groups into the aromatic chalcone ring was prepared using the Mannich-type reaction and studied for the first time against P. knowlesi and P. falciparum. The aminoalkyl groups used are piperidine, methyl piperidine, morpholine, and dimethylamine. All chalcone Mannich-type base derivatives were tested for drug susceptibility test against the zoonotic parasite P. knowlesi, and non-zoonotic parasite P. falciparum in vitro evaluation and followed by molecular docking on PfDHFR-TS and ADMET analyses. We proposed that the presence of an amine group could increase the antimalarial activity of the chalcone compounds. The present study is crucial to obtain insights into the crucial role of the amine group in chalcone derivatives to prevent the emergence of antifolate resistance and malarial transmission.
Results and discussion
Synthesis of aminoalkylated chalcone derivatives
The prepared chalcones 1(a–d) were obtained in a good yield via Claisen–Schmidt aldol condensation. Meanwhile, we also successfully synthesized the aminoalkylated-chalcones 2(a–h) from chalcone 1(a–d) by term Mannich-type reaction to substitute some aminoalkyl groups (Fig. 2). The spectroscopy analysis (1H-NMR, 13C-NMR, and MS) defined the structure of the desired products. The chemical structures of the synthesized derivatives are depicted in Fig. 3. | ||
| Fig. 2 Reaction scheme of aminoalkylated chalcones derivatives 1(a–d) and 2(a–h) from substituted-benzaldehyde and substituted-acetophenone. (i) Kalium hydroxide (60%), ethanol, stir at room temperature (RT) overnight; (ii) aminoalkyl groups, formaldehyde, ethanol, stir for 20 hours. R, R1, R2 = functional group from aldehyde and ketone; R3 = aminoalkyl groups. | ||
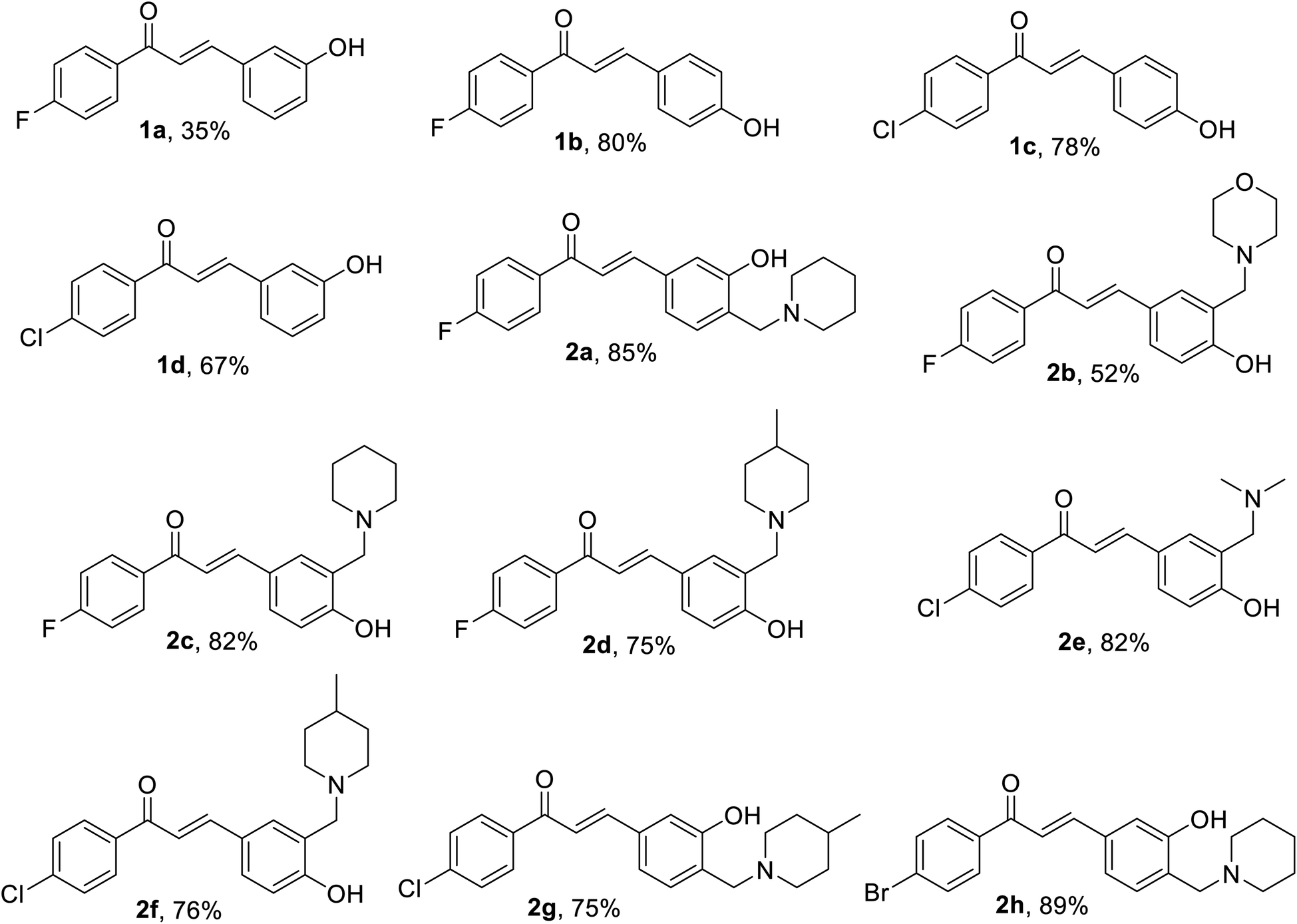 | ||
| Fig. 3 Molecular structures of synthesized chalcones (1a–1d, yield in %) and aminoalkylated chalcones derivatives (2a–2h, yield in %). | ||
Antimalarial activities against P. knowlesi A1H1 and P. falciparum 3D7
The in vitro antimalarial activity assay was performed against the P. knowlesi A1H1 and P. falciparum 3D7 strains using Plasmodium lactate dehydrogenase assay (pLDH). The findings in EC50 values (μM) against the P. knowlesi A1H1 and P. falciparum 3D7 are presented in Table 1. Based on the result in Table 1, from 12 synthesized derivatives, chalcone derivatives 2h possessed potent parasite inhibition against P. knowlesi A1H1 with the EC50 value in micromolar concentration (0.10 μM) as compared to other compounds. Chalcone derivatives 2a, 2d, 2e, and 2f exhibited active inhibition against the A1H1 strain with EC50 values of 2.64, 11.98, 2.98, and 19.91 μM respectively. The other seven chalcone derivatives were determined to have moderate antimalarial activities against the A1H1 strain. In addition, chalcone derivatives 2a and 2h were classified to have potential antimalarial activities with EC50 of 0.08 and 0.15 μM, respectively against P. falciparum 3D7, while 2d, 2e, and 2f exerted good antimalarial activities with EC50 values of 5.97, 2.69, and 17.03 μM respectively. The other synthesized chalcone derivatives gave moderate parasite inhibitory effects against the 3D7 strain. The antimalarial threshold activities are determined as follows, EC50 < 1 μM as potential activity, 2–20 μM as active/good activity, 21–100 μM as moderate activity, 101–200 as weak activity, and EC50 > 201 as inactive.34–36 The EC50 values for the reference antimalarial drug, chloroquine diphosphate (CQ) 0.04 μM for P. knowlesi A1H1 and 0.0018 μM for P. falciparum 3D7 in line with its usage as the frontline drug for treating malarial infection. The EC50 value of 2a against the A1H1 strain was about 66-fold higher than CQ, while the EC50 value of 2a against 3D7 was about 48-fold higher than CQ. In addition, the EC50 value of 2e against the A1H1 strain was about 75-fold higher than CQ, while the EC50 value of 2e against 3D7 was about 1400-fold higher than CQ. A high potency of 2h was seen as the EC50 value against A1H1 was about 3-fold higher than CQ, and 83-fold higher than CQ in 3D7 infection. Thus, chalcone derivatives 2a, 2e, and 2h possessed promising antimalarial activities against A1H1 and 3D7 strains.| Test compound | P. knowlesi A1H1 (CQ-sensitive) EC50 (μM) ± SD | P. falciparum 3D7 (CQ-sensitive) EC50 (μM) ± SD | WRL-68 cell (mammalian cell) IC50 (μM) ± SD |
|
|
|---|---|---|---|---|---|
| P. knowlesi | P. falciparum | ||||
| a CQ: chloroquine diphosphate, EC50: half-maximal effective concentration, IC50: half-maximal inhibition concentration, SD: standard deviation, nd: not determined. | |||||
| 1a | 29.00 ± 0.61 | 26.07 ± 0.03 | 38.94 ± 8.01 | 1.34 | 1.50 |
| 1b | 78.91 ± 0.36 | >41.28 | 11.10 ± 7.97 | 0.14 | 0.27 |
| 1c | 35.53 ± 3.88 | >38.66 | 18.50 ± 6.40 | 0.52 | 0.48 |
| 1d | 36.84 ± 6.27 | >38.66 | 11.44 ± 7.04 | 0.31 | 0.30 |
| 2a | 2.64 ± 1.74 | 0.08 ± 0.01 | 27.65 ± 6.47 | 10.50 | 345.63 |
| 2b | 24.64 ± 3.94 | >29.29 | 16.13 ± 6.76 | 0.65 | 0.55 |
| 2c | 21.06 ± 2.12 | >29.46 | 22.94 ± 5.97 | 1.09 | 0.78 |
| 2d | 11.98 ± 0.66 | 5.97 ± 0.02 | 10.37 ± 7.64 | 0.89 | 1.74 |
| 2e | 2.98 ± 0.14 | 2.69 ± 0.02 | 138.34 ± 7.81 | 46.42 | 51.42 |
| 2f | 19.91 ± 2.11 | 17.04 ± 0.02 | 80.65 ± 7.19 | 4.10 | 4.73 |
| 2g | 31.84 ± 0.30 | 27.54 ± 0.23 | 14.76 ± 6.37 | 0.47 | 0.54 |
| 2h | 0.10 ± 0.05 | 0.15 ± 0.03 | >99.00 | 990 | 660 |
| CQ | 0.04 ± 0.001 | 0.0018 ± 0.0008 | 102.82 ± 0.24 | >2500 | >2500 |
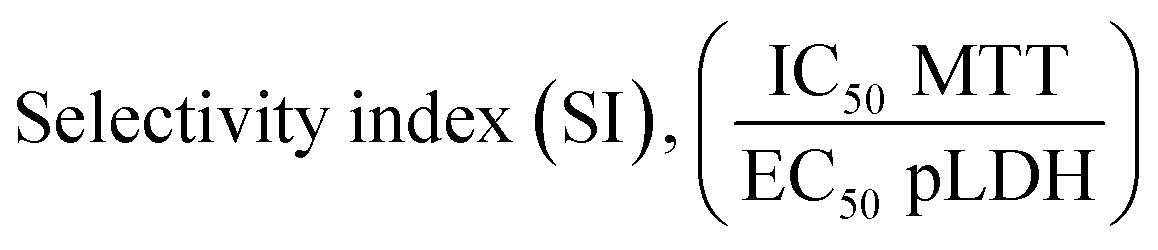
Structural-based analysis was also performed to determine the best substituents in chalcone derivatives that give the best candidate as antimalarial. In these findings, we postulated that the addition of an aminoalkyl group to chalcone could increase antimalarial efficacy. The addition of aminoalkyl groups such as piperidine, methyl piperidine, and diethylamine was proposed could increase antimalarial activity as discovered in this study. This can be seen from the presence of piperidine moiety in chalcone derivative 2a which has better antimalarial activity (EC50 of 0.08 μM) against P. falciparum 3D7 than compound 1a (EC50 of 26.06 μM). Similarly, the addition of the aminoalkyl group, dimethylamine in chalcone derivative 2e increased the antimalarial activity from chalcone derivative 1c with a reduction in EC50 value from 38.66 to 2.69 μM can be observed. In addition, the antimalarial effect can be better identified in chalcone derivative 2a (with piperidine moiety) against P. knowlesi A1H1 compared to 1a, which showed a lower EC50 value was observed in chalcone derivative 2a (2.64 μM). The addition of the aminoalkyl group, dimethylamine in chalcone derivative 2e also affected the antimalarial efficacy by lowering the EC50 value of chalcone derivative 1c from 35.5 to 2.98 μM (2e). The addition of piperidine moiety and bromide in chalcone derivative 2h enhanced the antimalarial efficacy of the compound against P. knowlesi A1H1 and P. falciparum 3D7. Thus, this result indicates that the aminoalkyl group does play a very important role in the antimalarial activity.
Cytotoxic activities against WRL-68 mammalian cells and selectivity indexes
The findings of the cytotoxicity assay (MTT) against the WRL-68 mammalian cell line revealed that all twelve chalcone derivatives did not show any strong toxicity effect in vitro (Table 1). The IC50 values of all compounds were determined in the range of 10 to 80 μM, suggesting the compounds were moderately toxic to the cell line. Chalcone derivatives 2e and 2h were considered non-toxic with IC50 more than 100 μM. The threshold level for cytotoxic effect was referred to as compounds with IC50 values lower than 10 μM were classified as highly toxic to mammalian cells.37,38 Based on the cytotoxic and antimalarial data, we further calculated the selectivity indexes (SI) of the chalcone derivatives to test the efficacy of the synthesized compounds as antimalarials and their pharmacological properties as safe drugs. For in vitro evaluation against P. knowlesi A1H1, chalcone derivatives 2a, 2e, and 2h exerted SI values of more than 10. The high SI values (SI = 10 to 990) shown by these compounds explained the antimalarial efficacy of these compounds as selective inhibition toward the P. knowlesi A1H1. In addition, for in vitro assessment against P. falciparum 3D7, 2a, 2e, and 2h had SI values greater than 10 (SI = 51 to 660), indicating that their pharmacological potential exceeded that of selective and safe drugs. Other compounds exhibited low SI values (SI < 10) indicating that the compounds had higher cytotoxic properties and lower parasite inhibition effect as compared to 2a, 2e, and 2h. The antimalarial properties of 2a, 2e, and 2h are almost comparable for both parasite strains. This might be caused by both parasite strains being sensitive strains that do not develop into multidrug-resistant varieties.From all tested chalcone derivatives, we would like to suggest 2a, 2e, and 2h are the most promising antimalarial candidate against both the zoonotic parasite, P. knowlesi, and non-zoonotic parasite, P. falciparum due to their active inhibition of both strains according to their low EC50 antimalarial values, high IC50 cytotoxic values as well as high SI values. These findings are in accordance with our postulation that the addition of an aminoalkyl group to chalcone could increase the parasite inhibitory effect. The presence of piperidine moiety in chalcone derivatives 2a, and 2h, and the addition of the aminoalkyl group, dimethylamine in chalcone derivative 2e increased the antimalarial activities of both compounds against P. knowlesi and P. falciparum. These findings further justify that the substitution of the amine group in chalcone derivatives could enhance the antimalarial efficacy of the compounds. A series of synthesized chalcone derivatives in our previous study such as chalcone derivatives added with morpholine, diethylamine, and piperidine moieties produced almost similar parasite inhibitory effect against P. falciparum 3D7.11,32,33 However, the antimalarial properties of (E)-1-(4-fluorophenyl)-3-(3-hydroxy-4-(piperidin-1-ylmethyl)phenyl)prop-2-en-1-one (2a), (E)-1-(4-chlorophenyl)-3-(3-dimethylamino)-(4-hydroxyphenyl)prop-2-en-1-one (2e), and (E)-1-(4-bromophenyl)-3-(3-hydroxy-4-(piperidin-1-ylmethyl)phenyl)prop-2-en-1-one (2h) against P. knowlesi, and P. falciparum are the first report so far.
Molecular docking analysis
The molecular docking result showed that chalcones 2a, 2e, and 2h have CDOCKER energy of −48.224, −43.292, and −45.851 kcal mol−1 (Table 2) (Fig. 4a–c), apparently higher than the co-crystal inhibitor WR9910 with −54.320 kcal mol−1.39 The re-docking process of the PfDHFR-TS–WR99210 complex showed interaction with Ala16, Ile164, Phe58, Tyr170, Ser108, Ile14, Asp54, Cys15, Leu164, and Met55. Meanwhile, the molecular docking result of chalcones 2a, 2e, and 2h in Table 2 displayed interaction with the common active site residues of the re-docking of the co-crystal inhibitor WR9910. Compounds 2a, 2e, and 2h have functional groups are –OH; halogen substituent (F, Cl, and Br); enolates; amines; and two benzene rings. The –OH group plays an important role in the formation of interaction via hydrogen bonds. In addition, the substituted halogen and aminoalkyl groups also take part in the interaction by forming hydrogen bonds. The presence of enolates and benzene rings will result in π-bonds with amino acids in PfDHFR-TS resistance (Fig. 5).| Compounds | CDOCKER energy (kcal mol−1) | Hydrogen-bonding |
|---|---|---|
| 2a | −48.224 | Ile164, Ala16, Cys15, Ser108, Gly166 |
| 2e | −43.292 | Ile164, Ala16, Cys15, Tyr170 |
| 2h | −45.851 | Ile164, Tyr170, Ser111, Ile112 |
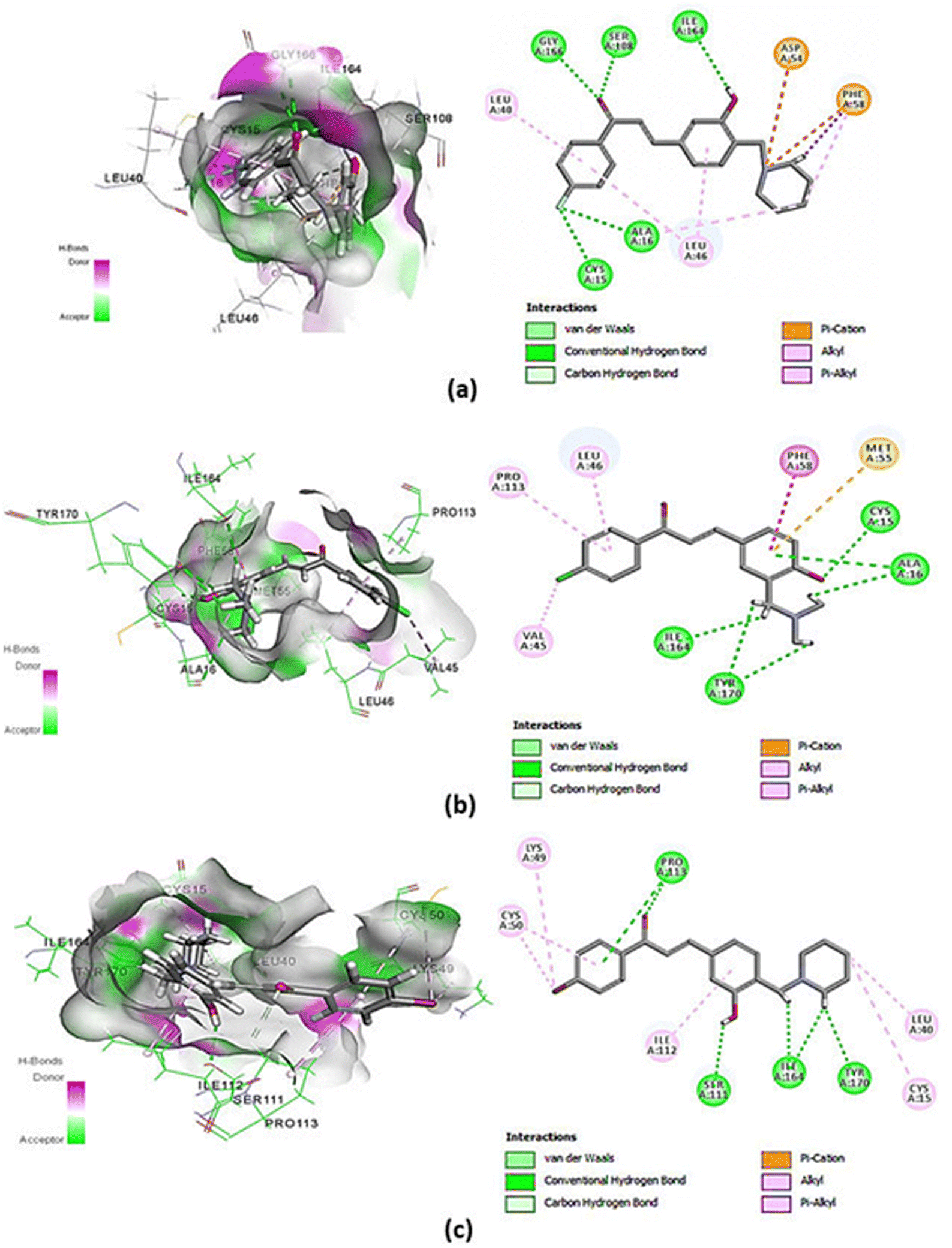 | ||
| Fig. 4 Visualization of the interaction (2-dimensional and 3-dimensional) among (a) chalcone derivative 2a, (b) chalcone derivative 2e, and (c) chalcone derivative 2h with PfDHFR-TS. The atom coloring scheme is as follows: carbons in grey, oxygen in red, and nitrogen in blue. Green lines represent hydrogen-bonding interactions, while the pink line represents π-bonding interactions. | ||
 | ||
| Fig. 5 A plausible mechanism of action of the synthesized chalcone derivatives on malarial parasite. | ||
In our previous study, we mentioned that a previously synthesized chalcone derivative with piperidine moiety showed not only in vitro activity but also in silico computational properties proven by its best hydrogen bond interaction with Ile112, Ile64, Ser111, Ser108, Asp54, Tyr170, and Pro113 residues on PfDHFR-TS (CDOCKER energy = −48.84 kcal mol−1).32,33 Not only that, the most active compound (E)-3-(3,4-dimethoxy phenyl)-1-(2-hydroxy-4-methoxy-5-(prenyl)phenyl)-prop-2-en-1-one was employed in the docking simulation showing perfect binding with amino acids Ala16, Ser108, Ile164, Trp48, and Phe58 in PfDHFR-TS.11 The in silico properties of these synthesized chalcone derivatives 2a, 2e, and 2h on PfDHFR-TS supported the in vitro antimalarial effect. A similar effect can be observed in this study where in silico properties of chalcone derivatives 2a, 2e, and 2h also corroborated with their in vitro antimalarial activities.
Physicochemical properties and ADMET analysis for chalcone derivatives
Investigation of pharmacokinetic parameters is crucial in the evaluation of drug candidates. It has been stated that the development of chalcone derivatives as drug candidates has some issues related to decreased bioavailability, low distribution, accelerated metabolism, and elimination.40 In this work, further studies related to the drug-likeness and ADMET properties were only carried out on chalcones 2a, 2e, and 2h with potential antimalarial activities against P. knowlesi and P. falciparum. Physicochemical properties of the prepared compounds were revealed by following the Lipinski rule based on the molecular weight (MW), number of hydrogen bond acceptors (nHA), number of hydrogen bond donors (nHD), and the logarithm of the n-octanol/water distribution (logP). Therefore, without any violations of the Lipinski rule, chalcone 2a, 2e, and 2h were predicted to be orally bioavailable. Nevertheless, logP values that indicate lipophilicity parameters were considered to be higher than the optimal values (0–3). This result proposes that it could later affect the membrane permeability and hydrophobic binding to macromolecules.
The human colon adenocarcinoma cell lines (Caco-2) have been used to predict drug permeability. The prepared chalcones 2a, 2e, and 2h were considered to have a proper Caco-permeability, as they have a predictive value of ≥5.15log cm s−1. P-glycoprotein (P-gp) is an efflux transmembrane protein that is partly responsible for the clearance of medicines through the hepatic and renal systems. The transport system of this protein is possible to be induced or inhibited and might lead to the interaction of medicine–medicine and medicine–food. Therefore, it is important to assess the P-gp parameter, both for the inhibitory and substrate of a compound in drug discovery. According to Table 3, chalcone derivatives 2a, 2e, and 2h were predicted to be a non-substrate of the P-glycoprotein (P-gp). In addition, only chalcone 2h was the P-gp inhibitor.
| Parameters | 2a | 2e | 2h |
|---|---|---|---|
| a The symbol * indicates prediction using admetSAR 2.0 (https://lmmd.ecust.edu.cn/admetsar2/, accessed on 1 January 2022). | |||
| Drug-likeness | |||
| • Molecular weight | 339.160 | 315.100 | 399.080 |
| • H-bond acceptor | 3 | 3 | 3 |
| • H-bond donor | 1 | 1 | 1 |
| • LogP |
3.982 | 3.692 | 4.568 |
| • TPSA | 40.540 | 40.540 | 40.540 |
|
|||
| A (absorption) | |||
| • Human intestinal absorption (HIA) | – – (0.012) | – – – (0.005) | – – – (0.021) |
| • Caco-2 permeability (log cm s−1) | −4.889 | −4.6 | −4.933 |
| • P-glycoprotein inhibitor | – (0.488) | – – – (0.017) | ++ (0.786) |
| • P-glycoprotein substrate | – – – (0.001) | – – – (0.002) | – – – (0.001) |
| • F20% | – – (0.293) | – – – (0.001) | – – – (0.009) |
| • F30% | – (0.467) | – – – (0.000) | – – – (0.042) |
|
|||
| D (distribution) | |||
| • Plasma protein binding (PPB) (%) | 98.109 | 96.399 | 97.796 |
| • Blood–brain barrier penetration (BBB) (cm s−1) | + (0.502) | + (0.604) | – (0.478) |
| • Volume distribution (L kg−1) | 1.274 | 2.128 | 1.334 |
| • Fu (%) | 1.869 | 3.275 | 1.535 |
|
|||
| M (metabolism) | |||
| • CYP1A2 substrate | + (0.687) | +++ (0.867) | – (0.356) |
| • CYP1A2 inhibitor | ++ (0.759) | ++ (0.960) | ++ (0.817) |
| • CYP2C19 substrate | – – – (0.066) | ++ (0.765) | – – – (0.069) |
| • CYP2C19 inhibitor | + (0.591) | – (0.308) | ++ (0.757) |
| • CYP2C9 substrate | – (0.353) | – (0.455) | – (0.375) |
| • CYP2C9 inhibitor | – – (0.137) | – – – (0.065) | – – (0.156) |
| • CYP2D6 substrate | +++ (0.912) | +++ (0.926) | +++ (0.906) |
| • CYP2D6 inhibitor | +++ (0.960) | +++ (0.969) | +++ (0.970) |
| • CYP3A4 substrate | – – (0.255) | – (0.445) | – – (0.254) |
| • CYP3A4 inhibitor | – – (0.183) | – – – (0.047) | – – (0.172) |
|
|||
| E (excretion)* | |||
| • Half-time (t1/2) | 0.093 | 0.612 | 0.128 |
| • Clearance (mL min−1 kg−1) | 11.936 | 12.620 | 4.691 |
|
|||
| T (toxicity) | |||
| • Human hepatotoxicity (H-HT) | – (0.330) | + (0.512) | – – (0.126) |
| • hERG blockers | +++ (0.978) | – (0.426) | +++ (0.980) |
| • Rat oral acute toxicity | ++ (0.734) | + (0.562) | – (0.327) |
| • AMES toxicity | – – (0.108) | – – (0.174) | – – – (0.044) |
| • Drug-induced liver injury (DILI) | ++ (0.723) | ++ (0.851) | ++ (0.857) |
| • Carcinogenicity | – (0.350) | – – (0.184) | – – (0.278) |
| • Acute oral toxicity (kg mol−1)* | 2.751 | 2.723 | 2.765 |
Human oral bioavailability was assessed for both in 20 and 30%. Theoretically, bioavailability is presented as 100% when a medication is administered straight into the bloodstream and is usually completely used by the body.41 However, an orally administrated drug might have decreased bioavailability. Chalcone derivatives 2e and 2h were predicted to have a good result by showing bioavailability ≥ 20 and 30% with predictive values in the range of 0–0.3. Meanwhile, chalcone derivative 2a was assessed to have good bioavailability of ≥20% and moderate bioavailability of 30% (in the range of 0.3–0.7).
The ADMET analysis showed that chalcone derivatives 2a, 2e, and 2h were categorized to have medium BBB parameters with an empirical value between 0.3–0.7, indicating a little BBB penetration. Meanwhile, the empirical VD value of chalcone derivatives 2a, 2e, and 2h was in the range of 0.04 to 20 L kg−1, which was classified as good. Theoretically, the VD is needed to explain the in vivo distribution of drugs, for example, the tendencies to bind to plasma protein, measurement of the distribution amount in body fluid, or the uptake amount in issues. Lastly, the fraction unbound in plasma (Fu) parameter of chalcone derivatives 2a, 2e, and 2h resulted in less than 5%, indicating a poor unbound state of drugs in plasma.
Materials and methods
Chemicals and instrumentation
All chemicals were purchased from Sigma-Aldrich, and Merck and used without further purification. All the solvents and reagents used in the synthesis were analysis and synthesis grade. For molecular docking: the protein used for molecular docking studies was the crystal protein of chloroquine-sensitive Plasmodium falciparum dihydrofolate reductases-thymidylate synthase (PfDHFR-TS) (PDB ID: 1J3I). This study used a PC with Intel® Core™ i7 CPU M 350 4.54 GHz; RAM 8.00 GB. Molecular docking using Discovery Studio 2016 (Accelrys, San Diego, USA). Melting points of the prepared compounds were determined in an open capillary tube on Electrothermal 9100. The molecular weight of the compounds was determined based on the MS from Shimadzu QP2010S. The 1H- and 13C-NMR spectra were recorded using tetramethylsilane as an internal standard on JEOL JNMECA (500 MHz).Synthesis procedures
:ethyl acetate in 3:1 ratio). After the completion of the reaction, the solvent was evaporated under a reduced pressure rotary evaporator, and the solid product obtained was purified using column chromatography with hexane:ethyl acetate mixture (0–50% gradient) as the eluent.32,42
(E)-1-(4-Fluorophenyl)-3-(3-hydroxyphenyl)prop-2-en-1-one (1a). Compound 1a with the molecular formula of C15H11FO2 was produced as an orange-yellow solid with a melting point of 112–113 °C, molecular ion (m/z) of 242.07, and yield of 35%. 1H-NMR (CD3OD, 600 MHz) δ (ppm): 7.91 (m, 2H, H-2, H-6), 7.19 (d, J = 9.0 Hz, 2H, H-3, H-5), 7.50 (d, J = 15.6 Hz, 1H, Hα), 7.68 (d, J = 15.6 Hz, 1H, Hβ), 7.17 (m, 1H, H-6′), 7.29 (m, 1H, H-5′), 6.91 (dd, J = 9.6 and J = 1.8 Hz, 1H, H-4′), 7.05 (s, 1H, H-2′), and 4.82 (s, 1H, O–H). 13C-NMR (CD3OD, 150 MHz) δ (ppm): 135.8 (1C, C-1), 132.6 (2C, C-2, and C-6), 116.7 (2C, C-3, and C-5), 166.4 (1C, C-4/C–F), 121.5 (1C, C-α), 146.9 (1C, C-β), 191.1 (1C, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.4 (1C, C-1′), 122.6 (1C, C-2′), 131.2 (1C, C-3′), 119.4 (1C, C-4′), 159.4 (1C, C-5′), and 116.9 (1C, C-6′).
O), 137.4 (1C, C-1′), 122.6 (1C, C-2′), 131.2 (1C, C-3′), 119.4 (1C, C-4′), 159.4 (1C, C-5′), and 116.9 (1C, C-6′).
(E)-1-(4-Fluorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (1b). Compound 1b with the molecular formula of C15H11FO2 was obtained as a bright yellow solid with a melting point of 149.2–151 °C, molecular ion (m/z) of 242.25, and yield of 80%. 1H-NMR (CD3OD, 600 MHz) δ (ppm): 7.92 (d, J = 8.4 Hz, 2H, H-2, H-6), 7.17 (m, 2H, H-3, H-5), 7.40 (d, J = 15 Hz, 1H, Hα), 7.63 (d, J = 15 Hz, 1H, Hβ), 7.49 (m, 2H, H-2′, H-6′), 6.87 (d, J1 = 9.0 Hz, 2H, H-3′, H-5′), and 9.95 (s, 1H, O–H). 13C-NMR (CD3OD, 150 MHz) δ (ppm): 134.7 (1C, C-1), 131.3 (2C, C-2, C-6), 115.6 (2C, C-3, C-5), 166.3 (1C, C-4/C–F), 122.3 (1C, C-α), 144.1 (1C, C-β), 185.4 (1C, C
O), 138.1 (1C, C-1′), 122.1 (2C, C-2′, C-6′), 116.5 (2C, C-3′, C-6′), and 158.2 (1C, C-4′).
(E)-1-(4-Chlorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (1c). Compound 1c with the molecular formula of C15H11ClO2 was afforded a bright yellow solid with a melting point of 135.3–137.8 °C, molecular ion (m/z) of 258.04, and yield of 78%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.99 (d, J = 8.4 Hz, 2H, H-2, H-6), 7.52 (m, 2H, H-3, H-5), 7.40 (d, J = 15 Hz, 1H, Hα), 7.63 (d, J = 15 Hz, 1H, Hβ), 7.49 (m, 2H, H-2′, H-6′), 6.87 (d, J = 9.0 Hz, 2H, H-3′, H-5′), and 9.95 (s, 1H, O–H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 137.6 (1C, C-1), 131.0 (2C, C-2, C-6), 129.7 (2C, C-3, C-5), 139.1 (1C, C-4/C–Cl), 120.4 (1C, C-α), 144.9 (1C, C-β), 188.7 (1C, C
O), 127.7 (1C, C-1′), 129.5 (2C, C-2′, C-6′), 116.4 (2C, C-3′, C-5′), and 159.9 (1C, C-4′).
(E)-1-(4-Chlorophenyl)-3-(3-hydroxyphenyl)prop-2-en-1-one (1d). Compound 1d with the molecular formula of C15H11ClO2 was obtained as a brownish yellow solid with a melting point of 135.2–137.9 °C, molecular ion (m/z) of 258.70, and yield of 67%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.46 (m, 2H, H-2, H-6), 8.04 (d, J = 9.0 Hz, 2H, H-3, H-5), 7.74 (d, J = 15 Hz, 1H, Hα), 8.02 (d, J = 15 Hz, 1H, Hβ), 7.28 (m, 1H, H-2′), 7.10 (brs, 1H, H-3′), 7.02 (m, 1H, H-4′), 7.00 (m, 1H, H-6′), and 4.84 (s, 1H, O–H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 136.7 (1C, C-1), 130.0 (2C, C-2, C-6), 129.1 (2C, C-3, C-5), 139.2 (1C, C-4/C–Cl), 121.2 (1C, C-α), 145.6 (1C, C-β), 189.4 (1C, C
O), 135.3 (1C, C-1′), 124.9 (1C, C-2′), 131.2 (1C, C-3′), 125.0 (1C, C-4′), 158.6 (1C, C-5′), and 115.0 (1C, C-6′).
(E)-1-(4-Fluorophenyl)-3-(3-hydroxy-4-(piperidin-1-ylmethyl)phenyl)prop-2-en-1-one (2a). Compound 2a with the molecular formula of C21H22FNO2 was obtained as a yellow solid with a melting point of 155–156 °C, and a yield of 85%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.97 (m, 2H, H-2, H-6), 7.09 (d, J = 9.0 Hz, 2H, H-3, H-5), 7.39 (d, J = 15.6 Hz, 1H, Hα), 7.65 (d, J = 15.6 Hz, 1H, Hβ), 7.24 (m, 1H, H-2′), 7.03 (m, 1H, H-6′), 7.05 (m, 1H, H-3′), 4.84 (s, 1H, O–H), 3.68 (s, 2H, CH2), and 1.66–1.13 (m, 10H, H-2′′, H-3′′, H-4′′, H-5′′, H-6′′). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 134.6 (1C, C-1), 131.1 (2C, C-2, C-6), 115.7 (2C, C-3, C-5), 165.6 (1C, C-4/C–F), 192.2 (1C, C
O), 120.1 (1C, C-α), 158.9 (1C, C-β), 158.5 (1C, C-1′), 115.1 (1C, C-2′), 188.9 (1C, C-3′), 120.5 (1C, C-4′), 121.3 (1C, C-5′), 116.8 (1C, C-6′), and 53.9–23.7 (CH2, C-2′′, C-3′′, C-4′′, C-5′′, C-6′′).
(E)-1-(4-Fluorophenyl)-3-(4-hydroxy-3-(morpholinomethyl)phenyl)prop-2-en-1-one (2b). Compound 2b with the molecular formula of C20H20FNO3 and molecular ion (m/z) of 341.38 was obtained as a brown solid, and a yield of 52%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.96 (m, 2H, H-2, H-6), 7.24 (m, 2H, H-3, H-5), 7.42 (d, J = 15 Hz, 1H, Hα), 7.76 (d, J = 15 Hz, 1H, Hβ), 7.48 (m, 1H, H-2′), 6.85 (d, J = 5 Hz, 1H, H-3′), 7.29 (m, 1H, H-6′), 4.84 (s, 1H, O–H), 3.71 (s, 2H, CH2), 2.67 (m, 2H, H-2′′, H-6′′), and 3.69 (m, 2H, H-3′′, H-5′′). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 134.7 (1C, C-1), 131.3 (2C, C-2, C-6), 115.9 (2C, C-3, C-5), 165.4 (1C, C-4/C–F), 122.2 (1C, C-α), 144.7 (1C, C-β), 190.2 (1C, C
O), 127.0 (1C, C-1′), 130.0 (1C, C-2′), 126.6 (1C, C-3′), 157.2 (1C, C-4′), 116.2 (1C, C-5′), 131.3 (1C, C-6′), 59.6 (CH2), 54.2 (2C, C-2′′, C-6′′), and 66.4 (2C, C-3′′, C-5′′).
(E)-1-(4-Fluorophenyl)-3-(4-hydroxy-3-(piperidin-1-ylmethyl)phenyl)prop-2-en-1-one (2c). Compound 2c with the molecular formula of C21H22FNO2 and molecular ion (m/z) of 339.41 was afforded as a light brown solid, and yield of 82%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 8.05 (d, J = 8.4 Hz, 2H, H-2, H-6), 7.17 (m, 2H, H-3, H-5), 7.34 (d, J = 15 Hz, 1H, Hα), 7.75 (d, J = 15 Hz, 1H, Hβ), 7.50 (m, 2H, H-2′, H-6′), 6.85 (d, J = 10 Hz, 1H, H-3′), 7.28 (m, 1H, H-6′), 4.84 (s, 1H, O–H), 3.73 (s, 2H, CH2), and 2.51–1.25 (10H, H-2′′, H-3′′, H-4′′, H-5′′, H-6′′). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 134.9 (1C, C-1), 130.9 (2C, C-2, C-6), 115.6 (2C, C-3, C-5), 166.4 (1C, C-4/C–F), 118.2 (1C, C-α), 145.4 (1C, C-β), 188.8 (1C, C
O), 125.8 (1C, C-1′), 129.5 (2C, C-2′, C-6′), 122.0 (1C, C-3′), 161.3 (1C, C-4′), 116.8 (1C, C-5′), 61.9 (1C, C-1′′), 53.8 (2C, C-2′′, C-6′′), 23.8 (1C, C-4′′), and 25.7 (2C, C-3′′, C-5′′).
(E)-1-(4-Fluorophenyl)-3-((4-hydroxy-3-(4-methylpiperidin-1-yl)methyl)phenyl)prop-2-en-1-one (2d). Compound 2d with the molecular formula of C22H24FNO2 and molecular ion (m/z) of 353.44 was produced as a dark yellow solid, and a yield of 75%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 8.05 (d, J = 8.4 Hz, 2H, H-2, H-6), 7.15 (m, 2H, H-3, H-5), 7.43 (d, J = 15 Hz, 1H, Hα), 7.75 (d, J = 15 Hz, 1H, Hβ), 7.50 (m, 2H, H-2′, H-6′), 6.84 (d, J = 10 Hz, 1H, H-3′), 4.84 (s, 1H, O–H), 3.73 (s, 2H, CH2), 2.97–1.72 (m, 10H, H-2′′, H-3′′, H-4′′, H-5′′, H-6′′), and 1.30 (m, 3H, CH3). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 134.9 (1C, C-1), 130.9 (2C, C-2, C-6), 115.6 (2C, C-3, C-5), 166.6, 164.4 (1C, C-4/C–F), 122.0 (1C, C-α), 145.4 (1C, C-β), 188.8 (1C, C
O), 129.2 (1C, C-1′), 130.6 (1C, C-2′), 125.8 (1C, C-3′), 161.3 (1C, C-4′), 116.8 (1C, C-5′), 129.5 (1C, C-6′), 61.5 (1C, C-1′′), 53.3 (2C, C-2′′, C-6′′), 34.0 (2C, C-3′′, C-5′′), 30.4 (1C, C-4′′), and 21.6 (1C, CH3).
(E)-1-(4-Chlorophenyl)-3-(3-dimethylamino)-(4-hydroxyphenyl)prop-2-en-1-one (2e). Compound 2e with the molecular formula of C17H16ClNO2 and molecular ion (m/z) of 301.77 was produced as a brown solid with a yield of 82%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.96 (m, 2H, H-2, H-6), 7.42 (m, 2H, H-3, H-5), 7.45 (d, J = 15 Hz, 1H, Hα), 7.76 (d, J = 15 Hz, 1H, Hβ), 6.94 (m, 1H, H-2′), 6.70 (d, J = 10 Hz, 1H, H-3′), 7.48 (m, 1H, H-6′), 4.84 (s, 1H, O–H), 3.75 (s, 2H, CH2), and 2.35 (s, 6H, CH3). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 136.6 (1C, C-1), 130.6 (2C, C-2, C-6), 129.7 (2C, C-3, C-5), 138.8 (1C, C-4/C–Cl), 122.3 (1C, C-α), 145.8 (1C, C-β), 189.3 (1C, C
O), 126.6 (1C, C-1′), 130.6 (1C, C-2′), 125.7 (1C, C-3′), 161.3 (1C, C-4′), 116.9 (1C, C-5′), 128.9 (1C, C-6′), 44.4 (3C, CH3), and 62.5 (1C, CH2).
(E)-1-(4-Chlorophenyl)-3-(4-hydroxy-3-((4-methylpiperidin-1-yl)methyl)phenyl)prop-2-en-1-one (2f). Compound 2f with the molecular formula of C22H24ClNO2 and molecular ion (m/z) of 369.89 was obtained as a brick red solid with a yield of 76%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.96 (d, J = 8.4 Hz, 2H, H-2, H-6), 7.49 (m, 2H, H-3, H-5), 7.32 (d, J = 15 Hz, 1H, Hα), 7.76 (d, J = 15 Hz, 1H, Hβ), 7.51 (m, 2H, H-2′, H-6′), 6.85 (d, J = 10 Hz, 1H, H-3′), 4.84 (s, 1H, O–H), 3.74 (s, 2H, CH2), 2.98–3.0 (m, 2H, H-2′′, H-6′′), 1.33–1.71 (m, 2H, H-3′′, H-5′′), 2.14 (m, 1H, H-4′′), and 1.31 (s, 3H, CH3). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 137.9 (1C, C-1), 129.8 (2C, C-2, C-6), 129.3 (2C, C-3, C-5), 138.8 (1C, C-4/C–Cl), 122.0 (1C, C-α), 145.7 (1C, C-β), 189.2 (1C, C
O), 129.3 (1C, C-1′), 129.8 (1C, C-2′), 129.6 (1C, C-6′), 128.8 (1C, C-3′), 116.8 (1C, C-5′), 61.5 (1C, C-1′′), 53.0 (2C, C-2′′, C-6′′), 34.0 (2C, C-3′′, C-5′′), 30.0 (1C, C-4′′), and 21.5 (3C, CH3).
(E)-1-(4-Chlorophenyl)-3-(3-hydroxy-4-((4-methylpiperidin-1-yl)methyl)phenyl)prop-2-en-1-one (2g). Compound 2g with the molecular formula of C22H24ClNO2 and molecular ion (m/z) of 369.89 was obtained as a brick red solid with a yield of 75%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.96 (d, J = 3.51 Hz, 2H, H-2, H-6), 7.49 (d, J = 10 Hz, 2H, H-3, H-5), 7.32 (d, J = 15 Hz, 1H, Hα), 7.76 (d, J = 15 Hz, 1H, Hβ), 7.51 (m, 1H, H-2′), 6.85 (d, J = 10 Hz, 1H, H-3′), 6.92 (d, J = 5 Hz, 1H, H-6′), 4.84 (s, 1H, O–H), 3.74 (s, 2H, CH2), 2.98–3.0 (m, 2H, H-2′′, H-6′′), 1.33–1.71 (m, 2H, H-3′′, H-5′′), 2.14 (m, 1H, H-4′′), and 1.31 (s, 3H, CH3). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 137.9 (1C, C-1), 129.8 (2C, C-2, C-6), 129.3 (2C, C-3, C-5), 138.8 (1C, C-4/C–Cl), 122.0 (1C, C-α), 145.7 (1C, C-β), 189.2 (1C, C
O), 129.3 (1C, C-1′), 129.8 (1C, C-2′), 129.6 (1C, C-6′), 128.8 (1C, C-3′), 116.8 (1C, C-5′), 61.5 (1C, C-1′′), 53.0 (2C, C-2′′, C-6′′), 34.0 (2C, C-3′′, C-5′′), 30.0 (1C, C-4′′), and 21.5 (3C, CH3).
(E)-1-(4-Bromophenyl)-3-(3-hydroxy-4-(piperidin-1-ylmethyl)phenyl)prop-2-en-1-one (2h). Compound 2h with the molecular formula of C21H22BrNO2 and molecular ion (m/z) of 399.83 was produced as a bright yellow amorphous solid, and a yield of 89%. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 7.81 (d, J = 8.5 Hz, 2H, H-2, H-6), 7.77 (d, J = 15.6 Hz, 1H, Hβ), 7.57 (m, 2H, H-3′, H-5′), 7.34 (d, J = 15.6 Hz, 1H, Hα), 7.04 (m, 1H, H-2′), 6.96 (m, 1H, H-6′), 4.84 (s, 1H, O–H), 3.63 (s, 2H, CH2), and 2.61–1.58 (m, 10H, H-2′′, H-3′′, H-4′′, H-5′′, H-6′′). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 189.4 (1C, C-1), 131.9 (2C, C-2, C-6), 130.0 (C-3, C-5), 129.0 (C-4/C–Br), 120.1 (1C, C-α), 158.9 (1C, C-β), 135.1 (1C, C-1′), 115.0 (1C, C-2′), 158.6 (1C, C-3′), 124.9 (1C, C-4′), 131.8 (1C, C-5′), 120.2 (1C, C-6′), 62.0 (CH2), 54.0 (C-2′′, C-6′′), 25.8 (C-3′′, C-5′′), and 23.9 (C-4′′).
Antimalarial assay of chalcone derivatives using in vitro P. knowlesi A1H1 and P. falciparum 3D7 cultures
Plasmodium knowlesi A1H1 strain was collected from Malaria Culture Laboratory, Department of Parasitology, Faculty of Medicine, Universiti Malaya (contributed by Robert W. Moon, London School of Hygiene and Tropical Medicine, London, UK) and P. falciparum 3D7 (chloroquine-sensitive) (MRA-102) were originally obtained from BEI Resources, NIAID, NIH (P. falciparum, strain 3D7, MRA-102, contributed by Daniel J. Carucci). The parasites were cultivated in fresh human blood (group O rhesus positive) suspended in RPMI 1640 (Invitrogen Life Technologies, NY, USA) supplemented with glucose (3 g L−1), hypoxanthine (45 μg L−1), and gentamicin (50 μg L−1). Plasmodium knowlesi was cultivated in RPMI 1640 medium supplemented with 10% horse serum (Gibco, New Zealand)43 and P. falciparum culture medium was supplemented with 10% AlbuMAX I as described by Trager and Jensen.44 Plasmodium lactate dehydrogenase (pLDH) assays for P. knowlesi and P. falciparum were performed based on a method by Makler and Hinrichs45 with slight modifications.46,47 Parasites (2% hematocrit and 2% parasitemia) were cultivated in a synchronized phase in 96-well plates. A total of 1 μL of the test compound in various concentrations was added with 99 μL of the parasite culture. Parasitized red blood cells (no treatment) were set as the controls while unparasitized red blood cells (no treatment) were set as the blanks in the assay. Chloroquine diphosphate served as a reference drug in the assay. The well plate was then put into the chamber and given mixed gas (O2 5%, CO2 5%, and N2 90%) and incubated for 24 hours at 37 °C for P. knowlesi and 72 hours at 37 °C for P. falciparum. Afterward, the plates were frozen at −20 °C for 24 hours and thawed at 37 °C for 30 minutes to lyse the red blood cells. Malstat reagent (100 μL) and nitro blue tetrazolium salt/phenazine ethosulfate (NBT/PES) (25 μL) were added to a new plate containing the treated cultures (25 μL). The color development of the plates was monitored at 650 nm using a SpectraMax Paradigm® Multi-Mode microplate reader. Data were analyzed through a non-linear regression via GraphPad Prism software to obtain EC50 values (half-maximal effective concentration).Cytotoxicity assay of chalcone derivatives using in vitro WRL-68 mammalian cell line
A human normal liver WRL68 cell line (ATCC: CL-48) was purchased from the American Type Culture Collection (ATCC).48 The cell lines were cultivated in MEM medium (supplemented with 5% of FBS and 1% of penicillin–streptomycin) in a tissue culture flask (T25) (Nunc, USA) at 37 °C and 5% CO2 in a sterile incubator. The culture medium was changed every day until cells became confluent and ready to be sub-culture. When the cells reached confluency, the media were removed and washed with phosphate buffer saline (PBS, Gibco, USA). Cells were detached by adding trypsin (Gibco, USA) and incubated for 5–10 minutes at 37 °C. The cell suspension was transferred into a 15 mL sterile tube and centrifuged at 300 g for 5 minutes. The cells were resuspended in a culture medium and maintained in a continuous culture before being used for the cytotoxic assay.The WRL-68 cell lines were used in the colorimetric assay of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) to study in vitro cytotoxicity activity.49 About 100 μL cell suspension (5 × 104 cells) were seeded in 96-well microplates for 24 hours. The culture medium was removed from each well and 200 μL of complete medium was added. The cells then were treated with 2 μL of chalcone derivatives diluted in dimethyl sulfoxide (DMSO) at various concentrations (3–990 μM). After 72 hours of incubation at 37 °C, the medium was discarded, and the treated cells were mixed with 20 μL of MTT solution (5 mg mL−1). Absorbance was recorded at 570 nm using enzyme-linked immune sorbent assay (ELISA) SpectraMax Paradigm® Multi-Mode plate reader. Cell viability (%) was determined using the following formula:

Selectivity indexes calculation
The selectivity index (SI) was calculated as the ratio between the inhibitory activity against the normal liver cell line (IC50 WRL-68) and the Plasmodium parasites, P. knowlesi (EC50 PkA1H1) or P. falciparum (EC50 Pf3D7). The SI value is calculated based on the formula i.e. SI = (IC50 for normal cell line WRL-68)/(EC50 for Plasmodium spp.). The higher SI value (SI > 10) for a test compound theoretically indicates the effectiveness and safety of a drug.50–53Molecular docking
Molecular docking analysis of the synthesized chalcone derivatives 2a, 2e, and 2h on PfDHFR-TS was conducted referring to the standard docking protocol CDOCKER method from Discovery Studio software.32 Molecular docking was carried out toward the crystal protein structure of the CQ-sensitive Plasmodium falciparum dihydrofolate reductase-thymidylate synthase (PfDHFR-TS) with Protein Database Bank (PDB) ID of IJ3I (resolution of 2.33 Å) (https://www.rcsb.org/, accessed on 1 January 2022). Before the analysis, the ligands and the receptor protein were constructed using Discovery Studio software. Hydrogen atoms were added to the protein molecule, and the pH of the ionizable amino acids (residues) was adjusted to 7.4. The receptor was maintained rigid during the docking analysis. The docking tolerance of the ligand–receptor was set as 0.25 Å with a few nonpolar or polar hotspots in the receptor. The conformations of the ligands were set at 500 within the relative energy threshold of 20.54ADMET parameters
The physicochemical and pharmacokinetic analysis i.e. absorption, distribution, metabolism, excretion, and toxicology (ADMET) parameters were predicted using the online webserver of admetSAR 2.0 (https://lmmd.ecust.edu.cn/admetsar2/admetopt/, accessed on 1 January 2022)55 and ADMETlab 2.0 (https://admetmesh.scbdd.com/service/evaluation/index, assessed on 1 January 2022).56 Drug-like properties were assessed based on the prediction of Lipinski's ‘rule of 5’ including molecular weight (MW) < 500 g mol−1, number of hydrogen bond acceptors (nHA) ≤ 10, number of hydrogen bond donors (nHD) ≤ 5, and the logarithm of the n-octanol/water distribution (logP) ≤ 5.57 Lipinski's rule predicts that if molecules have two or more violations of the rules, consequently they would potentially not be orally bioavailable as a drug (unacceptable). Meanwhile, the parameters of the absorption, distribution, metabolism, excretion, and toxicology properties are listed in Table 3.
Conclusions
The results of this study indicated that the addition of aminoalkyl groups to chalcone derivatives can increase the antimalarial activity of chalcone Mannich-type base derivatives. The best antimalarial activities were shown by compounds 2a, 2e, and 2h against P. knowlesi and P. falciparum. The results of in vitro antimalarial activities were also supported by the results of analysis using molecular docking suggesting that PfDHFR-TS is a promising target for these compounds. Thus, the findings in this study suggest that the substitution of the amine group in chalcone derivatives is crucial as an alternative therapy to combat drug resistance in P. falciparum and the high incidence of malarial infection in P. knowlesi. However, in vivo trials need to be carried out to further verify the antimalarial action and mechanism of these chalcone derivatives. This study may provide insights into alternative drug discovery efforts to combat antifolate resistance and malaria transmission worldwide.Author contributions
Conceptualization, JS, JL; methodology, JS, AHA, NYL, NI; investigation, JS, AHA, NYL, NI, RH, NL, BAN; data curation, JS, HKA, LYL, JL, BAN; writing—original draft preparation, JS, AHA, JL; writing—review and editing, JS, JL, HKA; supervision, JS, HKA, JL, LYL; funding acquisition, JS, JL. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial assistance from the Deputy of Research and Development Strengthening of The Ministry of Education, Culture, Research, and Technology/National Agency for Research and Innovation through LLDIKTI Region X with grant number 074/LL10/PG-PDTT/2021 is highly appreciated and Universiti Kebangsaan Malaysia (GUP-2021-047). We appreciate the great support from the staff and students from the Department of Parasitology, Faculty of Medicine, University of Malaya.Notes and references
- World Health Organization (WHO), World Malaria Report, Geneva, 2022, https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 Search PubMed.
- Ministry of Health (MOH), Tren Kasus Malaria Menurun, Indonesia, 2022, https://www.kemkes.go.id/article/view/21042300004/tren-kasus-malaria-menurun.html, accessed on 30 June 2022 Search PubMed.
- N. J. Spillman, R. J. Allen, C. W. McNamara, B. K. Yeung, E. A. Winzeler, T. T. Diagana and K. Kirk, Cell Host Microbe, 2013, 13, 227–237 CrossRef CAS PubMed.
- E. A. Ashley, M. Dhorda, R. M. Fairhurst, C. Amaratunga, P. Lim, S. Suon, S. Sreng, J. M. Anderson, S. Mao and B. Sam, N. Engl. J. Med., 2014, 371(5), 411–423 CrossRef PubMed.
- C. H. Sibley, Science, 2015, 347, 373–374 CrossRef CAS PubMed.
- B. Sharma, P. Singh, A. K. Singh and S. K. Awasthi, Eur. J. Med. Chem., 2021, 219, 113408 CrossRef CAS PubMed.
- P. Singh, C. Sharma, B. Sharma, A. Mishra, D. Agarwal, D. Kannan, J. Held, S. Singh and S. K. Awasthi, Eur. J. Med. Chem., 2022, 244, 114774 CrossRef CAS PubMed.
- P. Yadav, B. Sharma, C. Sharma, P. Singh and S. K. Awasthi, ACS Omega, 2020, 5(12), 6472–6480 CrossRef CAS PubMed.
- N. Chaianantakul, R. Sirawarapornm and W. Sirawaraporn, Malar. J., 2013, 12(1), 1–13 CrossRef PubMed.
- R. Reeta, V. Rajendran, T. M. Rangarajan, R. P. Ayushee Singh and M. Singh, Bioorg. Chem., 2019, 86, 631–640 CrossRef CAS PubMed.
- J. Syahri, K. Rullah, R. Armunanto, E. Yuanita, B. A. Nurohmah, M. F. F. Mohd Aluwi, L. K. Wai and B. Purwono, Asian Pac. J. Trop. Dis., 2017, 7, 9–14 Search PubMed.
- H.-L. Qin, Z.-W. Zhang, R. Lekkala, H. Alsulami and K. Rakesh, Eur. J. Med. Chem., 2020, 193, 112215 CrossRef CAS PubMed.
- H. Suwito, J. Jumina, M. Mustofa, P. Pudjiastuti, M. Z. Fanani, Y. Kimata-Ariga, R. Katahira, T. Kawakami, T. Fujiwara, T. Hase, H. M. Sirat and N. N. Puspaningsih, Molecules, 2014, 19, 21473–21488 CrossRef PubMed.
- R. Jyoti, R. Gaur, Y. Kumar, H. S. Cheema, D. S. Kapkoti, M. P. Darokar, F. Khan and R. S. Bhakuni, Nat. Prod. Res., 2021, 35(19), 3261–3268 CrossRef CAS PubMed.
- N. Tadigoppula, V. Korthikunta, S. Gupta, P. Kancharla, T. Khaliq, A. Soni, R. K. Srivastava, K. Srivastava, S. K. Puri, K. S. R. Raju, D. Wahajuddin, P. S. Sijwali, V. Kumar and I. S. Mohammad, J. Med. Chem., 2013, 56, 31–45 CrossRef CAS PubMed.
- A. Wilhelm, P. Kendrekar, A. E. M. Noreljaleel, E. T. Abay, S. L. Bonnet, L. Wiesner, C. de Kock, K. J. Swart and J. H. Westhuizen, J. Nat. Prod., 2015, 78, 1848–1858 CrossRef CAS PubMed.
- B. Ngameni, K. Cedric, A. T. Mbaveng, M. Erdoğan, I. Simo, V. Kuete and A. Daştan, Bioorg. Med. Chem. Lett., 2021, 35, 127827 CrossRef CAS PubMed.
- B. Rioux, A. Pinon, A. Gamond, F. Martin, A. Laurent, Y. Champavier, C. Barette, B. Liagre, C. Fagnere and V. Sol, Eur. J. Med. Chem., 2021, 222, 113586 CrossRef CAS PubMed.
- S. Sivapriya, K. Sivakumar and H. Manikandan, Chem. Data Collect., 2021, 35, 100762 CrossRef CAS.
- H. Ur-Rashid, Y. Xu, N. Ahmad, Y. Muhammad and L. Wang, Bioorg. Chem., 2019, 87, 335–365 CrossRef CAS PubMed.
- L. Wang, X. Yang, Y. Zhang, R. Chen, Y. Cui and Q. Wang, J. Nat. Prod., 2019, 82, 2761–2767 CrossRef CAS PubMed.
- W. Dan and J. Dai, Eur. J. Med. Chem., 2020, 187, 111980 CrossRef CAS PubMed.
- T. S. Freitas, J. C. Xavier, R. L. Pereira, J. E. Rocha, F. F. Campina, J. B. de Araújo Neto, M. M. Silva, C. R. Barbosa, E. S. Marinho and C. E. Nogueira, Microb. Pathog., 2021, 161, 105286 CrossRef CAS PubMed.
- M. A. Garcia, R. S. Theodoro, J. C. Sardi, M. B. Santos, G. M. Ayusso, F. R. Pavan, A. R. Costa, L. M. Santa Cruz, P. L. Rosalen and L. O. Regasini, Bioorg. Chem., 2021, 116, 105279 CrossRef CAS PubMed.
- S. K. Konidala, V. Kotra, R. C. S. R. Danduga, P. K. Kola, R. R. Bhandare and A. B. Shaik, Arabian J. Chem., 2021, 14(6), 103154 CrossRef CAS.
- G. Prabakaran, S. Manivarman and M. Bharanidharan, Mater. Today: Proc., 2022, 48, 400–408 CAS.
- A. Rammohan, B. V. Bhaskar, N. Venkateswarlu, W. Gu and G. V. Zyryanov, Bioorg. Chem., 2020, 95, 103527 CrossRef CAS PubMed.
- T. I. Adelusi, L. Du, A. Chowdhury, G. Xiaoke, Q. Lu and X. Yin, Life Sci., 2021, 284, 118982 CrossRef CAS PubMed.
- D. Cáceres-Castillo, R. M. Carballo, R. Quijano-Quiñones, G. Mirón-López, M. Graniel-Sabido, R. E. Moo-Puc and G. J. Mena-Rejón, Med. Chem. Res., 2020, 29, 431–441 CrossRef.
- D. Quaglio, N. Zhdanovskaya, G. Tobajas, V. Cuartas, S. Balducci, M. S. Christodoulou, G. Fabrizi, M. Gargantilla, E.-M. Priego and A. l. Carmona Pestaña, ACS Med. Chem. Lett., 2019, 10(4), 639–643 CrossRef CAS PubMed.
- J. Syahri, H. Nasution, B. A. Nurohmah, B. Purwono and E. Yuanita, J. Appl. Pharm. Sci., 2020, 10, 001–005 CAS.
- J. Syahri, H. Nasution, B. A. Nurohmah, B. Purwono, E. Yuanita, N. H. Zakaria and N. I. Hassan, Sains Malays., 2020, 49, 2667–2677 CrossRef CAS.
- M. Mushtaque, Eur. J. Med. Chem., 2015, 90, 280–295 CrossRef CAS PubMed.
- M. F. Dolabela, S. G. Oliveira, J. M. Nascimento, J. M. Peres, H. Wagner, M. M. Póvoa and A. B. de Oliveira, Phytomedicine, 2008, 15(5), 367–372 CrossRef CAS PubMed.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. Van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. Slingsby, Nat. Rev. Drug Discovery, 2015, 14(11), 751–758 CrossRef CAS PubMed.
- A. H. Ali, H. K. Agustar, N. I. Hassan, J. Latip, N. Embi and H. M. Sidek, Data Brief, 2020, 33, 106592 CrossRef PubMed.
- A. M. Burger and H. H. Fiebig, Handbook of Anticancer Pharmacokinetics and Pharmacodynamics, 2014, pp. 23–38 Search PubMed.
- P. Wiji Prasetyaningrum, A. Bahtiar and H. Hayun, Sci. Pharm., 2018, 86, 25 CrossRef PubMed.
- B. Purwono, B. Nurohmah, P. Fathurrohman and J. Syahri, Rasayan J. Chem., 2021, 14(1), 94–100 CrossRef CAS.
- S. Sinha, A. Prakash, B. Medhi, A. Sehgal, D. I. Batovska and R. Sehgal, BMC Res. Notes, 2021, 14, 264 CrossRef CAS PubMed.
- R. Kumar, A. Sharma and P. K. Varadwaj, J. Nat. Sci., Biol. Med., 2011, 2(2), 168 CrossRef CAS PubMed.
- N. Nurlaili, H. Saputri, S. Z. Nasution, R. Hilma and J. Syahri, AIP Conf. Proc., 2021, 2370(1), 1–9 Search PubMed.
- A. Amir, B. Russell, J. W. K. Liew, R. W. Moon, M. Y. Fong, I. Vythilingam, V. Subramaniam, G. Snounou and Y. L. Lau, Sci. Rep., 2016, 6(1), 24623 CrossRef CAS PubMed.
- W. Trager and J. B. Jensen, Int. J. Parasitol., 1997, 27, 989–1006 CrossRef CAS PubMed.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48(2), 205–210 CrossRef CAS PubMed.
- S. Nkhoma, M. Molyneux and S. Ward, Am. J. Trop. Med. Hyg., 2007, 76(6), 1107–1112 CrossRef CAS PubMed.
- M. A. Shamsuddin, A. H. Ali, N. H. Zakaria, M. F. Mohammat, A. S. Hamzah, Z. Shaameri, K. W. Lam, W. F. Mark-Lee, H. K. Agustar and M. R. Mohd Abd Razak, Pharmaceuticals, 2021, 14(11), 1174 CrossRef CAS PubMed.
- American Type Culture Collection (ATCC), Virginia, United States, 2022, https://www.atcc.org/products/cl-48, accessed on 1 January 2022.
- T. Mosmann, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- D. P. Iwaniuk, E. D. Whetmore, N. Rosa, K. Ekoue-Kovi, J. Alumasa, A. C. de Dios, P. D. Roepe and C. Wolf, Bioorg. Med. Chem., 2009, 17(18), 6560–6566 CrossRef CAS PubMed.
- S. O. Sarr, S. Perrotey, I. Fall, S. Ennahar, M. Zhao, Y. M. Diop, E. Candolfi and E. Marchioni, Malar. J., 2011, 10, 1–10 CrossRef PubMed.
- N. H. M. Radzuan, N. A. Z. Norazmi, A. H. Ali and M. Abu, Sains Malays., 2021, 50(10), 2945–2956 CrossRef.
- N. A. Z. Norazmi, N. H. Mukhtar, A. H. Ali, N. H. Abd and I. Hassan, Sains Malays., 2022, 51(4), 1123–1130 CrossRef CAS.
- J. Yuvaniyama, P. Chitnumsub, S. Kamchonwongpaisan, J. Vanichtanankul, W. Sirawaraporn, P. Taylor, M. D. Walkinshaw and Y. Yuthavong, Nat. Struct. Mol. Biol., 2003, 10, 357–365 CrossRef CAS PubMed.
- C.-Y. Jia, J.-Y. Li, G.-F. Hao and G.-F. Yang, Drug Discovery Today, 2020, 25(1), 248–258 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, w5–w14 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05361j |
| This journal is © The Royal Society of Chemistry 2023 |