
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the way to potential antifungal compounds: synthesis and in vitro activity of 2-benzofuranylacetic acid amides†
Camilo Mahecha-Mahecha‡
a,
Paola Borrego-Muñoz b,
Luis M. Pombob and
Diego Gamba-Sánchez*a
b,
Luis M. Pombob and
Diego Gamba-Sánchez*a
aLaboratory of Organic Synthesis, Bio and Organocatalysis, Chemistry Department, Universidad de Los Andes, Cra 1 No. 18A-12 Q:305, Bogotá 111711, Colombia. E-mail: da.gamba1361@uniandes.edu.co
bEscuela de Medicina, Fundación Universitaria Juan N. Corpas, Bogotá 110311, Colombia
First published on 23rd August 2023
Abstract
Crop losses caused by microbial infections are a significant global issue, especially in tropical regions. The development of novel antimicrobial agents, particularly antifungal agents, has been explored from various perspectives, including chemical synthesis. However, conventional approaches typically involve synthesizing new and potent compounds on a small scale (a few milligrams), making the scale-up of the reaction a major challenge. In this manuscript, we present a method for the synthesis of new and active (against Fusarium oxysporum) benzofuranyl acetic acid amides. Our strategy allows us to synthesize the key precursor on the gram scale, enabling the production of sufficient quantities of other active compounds within short timeframes for conducting biological studies. All the reactions used in this manuscript are recognized by their industrial application.
Introduction
Microorganisms' resistance to current treatments poses a significant challenge for humans, not only from a medical perspective but also in various other fields such as veterinary, economy, biochemistry, and agriculture.1,2 The issue of crop losses due to microbial pathogenesis is particularly severe in tropical countries, but it affects the global community as a whole.3,4Fusarium oxysporum (FOX) is a highly infectious pathogen that poses a significant threat to global agricultural production. It is responsible for causing vascular wilt5 and contaminating crops such as wheat, barley, corn, bananas, and cotton, resulting in substantial worldwide losses estimated at approximately 80%.6–8 This invasive plant pathogen also contributes to the presence of mycotoxins in food, which not only jeopardizes food safety9 but also endangers the health of both humans and animals, making them susceptible to fungal infections.
In this context, remarkable progress is being made in the development of new bioactive compounds against phytopathogens such as FOX on a weekly basis.10–18 However, many of these advancements come with a significant drawback. Most active compounds are derived from natural sources, resulting in limited quantities that make it challenging to conduct thorough testing and field evaluations. While chemical synthesis has been regarded as a viable alternative for obtaining larger quantities of active compounds, recent developments often involve the use of expensive or complex materials, or techniques. Additionally, even if the active compounds are successfully synthesized, the amounts obtained are often insufficient for conducting field trials. Consequently, when a highly promising compound is discovered, the chemical industry faces new challenges in scaling up production, requiring the re-optimization of reaction conditions at each step. Therefore, it is crucial to provide potentially scalable protocols when synthesizing new compounds.
In light of these considerations, our research focused on the synthesis of benzofuranyl acetic acid amides using easily scalable processes. This investigation was inspired by the numerous active compounds that possess a benzofuran ring, analogue heterocycles and at least one amide bond,19–23 as illustrated in Fig. 1.
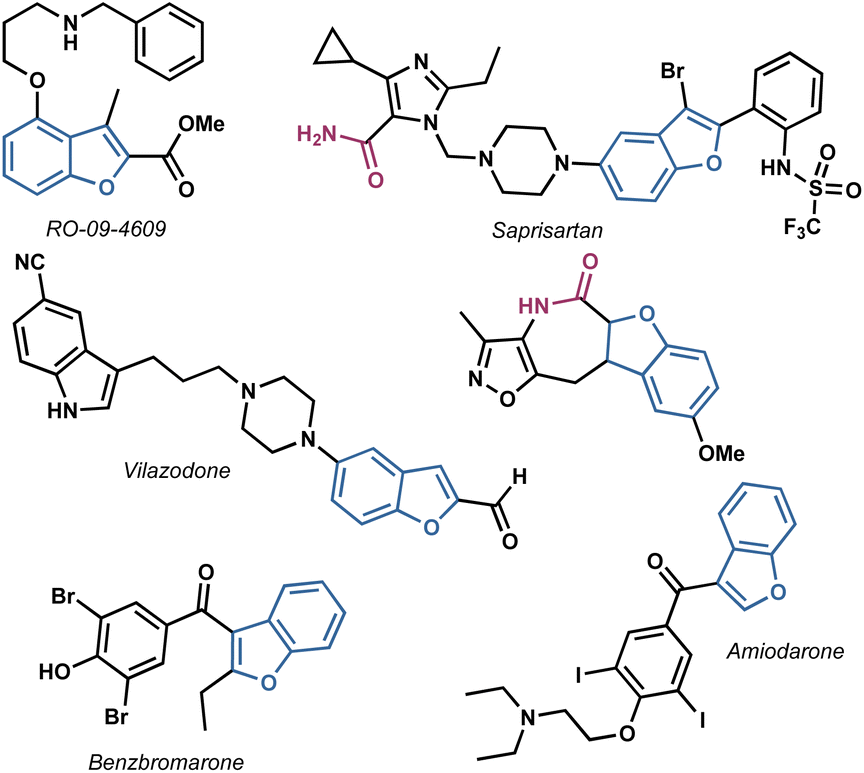 | ||
| Fig. 1 Structures of some active benzofuran-based compounds. | ||
The synthesis of benzofuranyl acetic acid amides is quite uncommon. Only a few reports can be found, which are based on different methods such as palladium-catalyzed aminocarbonylative cyclization,24 Pd-catalyzed alkyne cyclization,25 gold-catalyzed domino cyclization,26 and the coupling of benzofurane boronic acids with bromoketones.27
Alternatively, a plausible approach is the synthesis of benzofuranyl acetic acids and their esters, which provides a few more options. These options include cyclizations induced by radicals28,29 or transition metals,30–32 functionalization of benzofuran derivatives,33–36 or formal cycloadditions.37–39
Regarding the antifungal potential of benzofuran derivatives, the available literature is limited. Certain authors have assessed the activity against FOX of those derivatives and observed inhibition halos ranging from 28 to 32 mm, demonstrating better activity compared to the positive control nystatin, which exhibited a 23 mm halo.40 Moreover, Xie et al.41 have reported the in vivo and in vitro biological activity of some molecules containing a benzofuran ring against other phytopathogenic fungi such as Colletotrichum iagenarium and Erysiphe graminis. They obtained data comparable to the positive control enoxastrobin, showing inhibition up to 100%.
It is worth noting that while numerous active compounds containing the benzofuran ring have been described and show promising bioactivities, only a few reports suggest that the mentioned antifungal activity can indeed be attributed to the benzofuran ring.42–44
Considering the privileged position of the amide bond in bioactive molecules and the scarcity of reports where a benzofuran served as the structural core of antifungal compounds, we identified benzofuranyl acetic acid amides as our target molecules (Fig. 2). Our hypothesis was that the amide bond would confer stability against hydrolysis (compared to esters), increase the molecules' polarity, thereby enhancing their solubility in water. Additionally, the introduction of polar functional groups in the amide aromatic ring would aid in this solubility enhancement. However, the ultimate activity would predominantly rely on the benzofuran core. The lack of reliable synthetic protocols for such molecules emphasizes the need to explore new synthetic strategies to develop them as promising agents for phytopathogenic control.
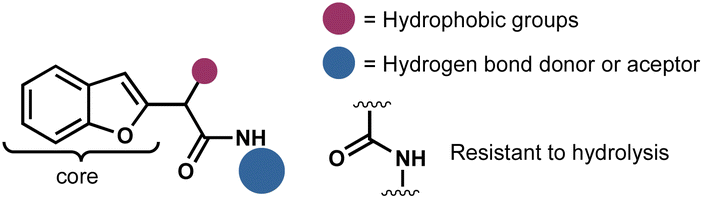 | ||
| Fig. 2 Schematical structure of target molecules. | ||
In light of these possibilities, we have conceived a method for synthesizing benzofuranyl acetic acid amides and the benzofuran core using either Sonogashira coupling-cyclization or rearrangements of methyl ketones. In this study, we present the results of employing these strategies and evaluate the in vitro activities of the obtained compounds against Fusarium oxysporum.
Results and discussion
We initiated our study by following the Sonogashira coupling method as described previously.38,45,46 It is well-known that when alkynes are coupled with o-iodophenols using the Sonogashira reaction, o-alkynyl alcohols are produced.47,48 These alcohols are in situ activated by the metal catalyst, leading to cyclizations and the formation of benzofurans. Based on this knowledge, we aimed to synthesize the desired amides using a one or two step process starting with 3-butynoic acid 2a, its methyl ester 2b, and the propynyl amide 2c. Unfortunately, all our attempts to carry out this reaction were unsuccessful. We utilized Pd(PPh3)2Cl2 as the catalyst, CuI as the cocatalyst, and Et3N in stoichiometric amounts. However, in all cases, we were able to recover the starting phenol unchanged. We hypothesize that the allene is formed in situ, which is somehow less reactive (Scheme 1a).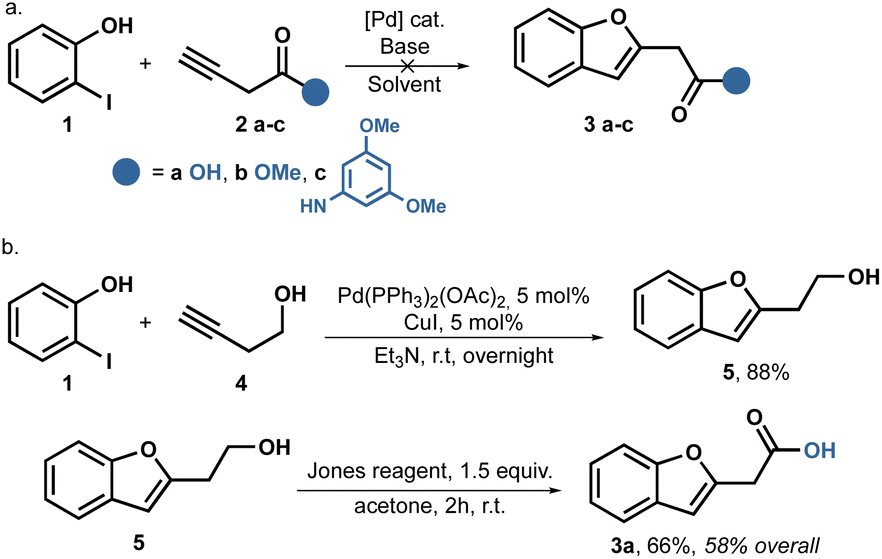 | ||
| Scheme 1 Sonogashira coupling to benzofuranyl acetic acid 3a. | ||
Despite attempting to change the base to Na2CO3, Cs2CO3, or the catalyst for Pd(AcO)2, we did not achieve better results. Fortunately, when we switched to Pd(PPh3)2(OAc)2 as the catalyst, we observed some conversion only when using the alcohol 4 as the substrate; no reaction was observed with the acid 2a, ester 2b, or amide 2c.
Consequently, we shifted our focus to a multi-step strategy and employed butynol 4 as the coupling partner. To our delight, the reaction proceeded smoothly, when using Et3N as the solvent and successfully achieved full conversion, ultimately isolating the product 5 with a yield of 88%. Furthermore, a Jones oxidation allowed us to obtain benzofuranyl acetic acid 3a in two steps, achieving an overall yield of 58%, as shown in Scheme 1b.
Despite the successful outcome of this sequence, we encountered difficulties when attempting to scale up the reaction. Increasing the reaction scale resulted in lower yields, and we were only able to replicate the yields up to 4 mmol scale.
Based on these findings, we conducted a Willgerodt–Kindler rearrangement.49 The reaction sequence begins by forming methyl ketone 8 through the combination of commercially available bromoacetone 6 and salicylic aldehyde 7. Under basic conditions, the reagents are heated, resulting in the production of methyl ketone 8 in a scale of up to 5.7 grams and a yield of 61%. For the Willgerodt–Kindler rearrangement, we employed sulfur as the reagent and morpholine as the base and solvent. To our satisfaction, the thioamide 9 was obtained at a scale of 4 grams with a yield of 66% after crystallization from ethanol. Subsequently, the hydrolysis of compound 9 proceeded smoothly using aqueous ethanol, achieving an 81% yield in the milligram scale and 90% in gram scale after acid-base extractions (Scheme 2).
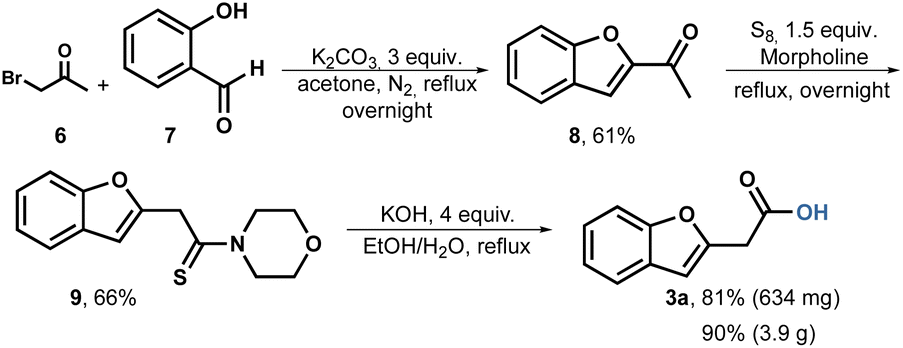 | ||
| Scheme 2 Willgerodt–Kindler rearrangement to benzofuranyl acetic acid 3a. | ||
Up to this point, we have successfully employed two methods to achieve the synthesis of benzofuranyl acetic acid 3a, yielding satisfactory overall results on a multi-gram scale. Subsequently, our focus shifted towards obtaining N-aryl amides.
We selected aminophenols as the reacting amines, with the hope of enhancing the water solubility of the resulting products. Since the primary goal of this research was to establish a dependable method for obtaining potential antifungal agents suitable for field trials, water solubility played a crucial role. Therefore, incorporating hydroxy groups into the aromatic ring could aid in achieving this essential feature.
Our attempts to perform a transamidation of thioamide50–52 9 with aromatic amines proved unsuccessful. Consequently, we explored alternative traditional amidation conditions,53,54 including the direct amidation of the carboxylic acid;55 however, the use of coupling reagents proved to be relatively straightforward. Initially, we conducted the amidations using dicyclohexylcarbodiimide (DCC) as the coupling reagent. While the reaction itself proceeded well, we encountered several challenges in purifying the product. Regardless of the solvent used, separating the product from the cyclohexyl urea by-product proved difficult. To overcome this issue, we switched to EDCI, as the urea by-product is water-soluble. With this modification, we successfully obtained compounds 10a–c in acceptable to good yields (Scheme 3).
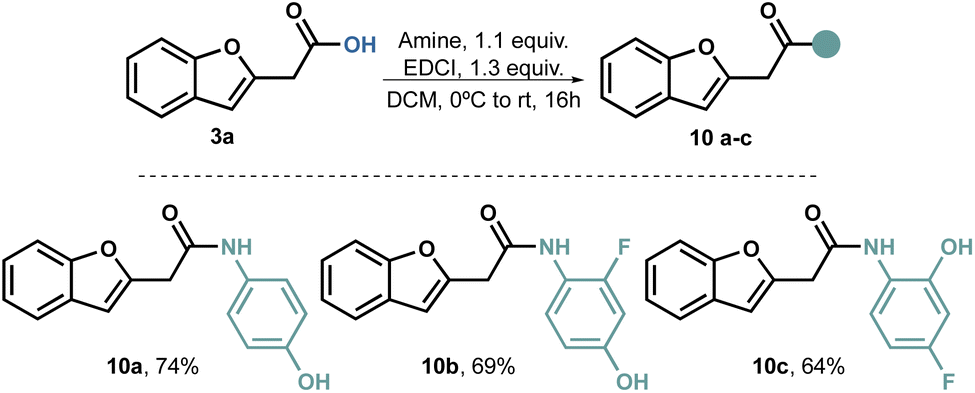 | ||
| Scheme 3 Amidation of 3a with hydroxyanilines. | ||
One noteworthy aspect of this amidation process is its reliability, as we found no preconceived limitations. Consequently, the same protocol can be applied to various types of amines.
With the aim of establishing a dependable approach for acquiring amides with a benzofuranyl acetic core that possess structural diversity, our objective was to introduce alkyl groups in α positions.
It is widely acknowledged that the alkylation of primary and secondary amides poses a challenge primarily due to the deprotonation occurring at the more acidic nitrogen. Therefore, we revisited the deprotonation process of methyl ester 11, which had been obtained by esterifying 3a with excellent yield.
Extensive optimization was required for the deprotonation–alkylation process. Initially, we used n-BuLi as the deprotonating agent, but it resulted in virtually no conversion. Upon switching to LDA, we managed to isolate 35% of product 12a. To improve the yield, we introduced TMEDA as an additive, which increased the yield to 51%. However, it was only when we used HMPA as a additive that we achieved full conversion and were able to isolate the desired product 12a in a 75% yield. Through this process, we stablished that the combination of LDA and HMPA proved to be the optimal choice for achieving alkylation of 11 with alkyl iodides. Consequently, we extended the method to EtI, obtaining 12b with a 73% yield.
The objective of introducing these alkyl groups was to evaluate their potential impact on the antifungal properties of the target compound. As we did not observe any significant effects in this case (vide infra), we did not carry out additional alkylations. Nonetheless, it is plausible to assume that this reaction may be limited to primary unhindered alkyl iodides. Subsequent hydrolysis under traditional conditions yielded alkylated acids 13a and 13b in excellent yields.
To complete the amidation, we followed the previously described procedure, resulting in the synthesis of four additional amides (14a–d) with acceptable to good yields (Scheme 4).
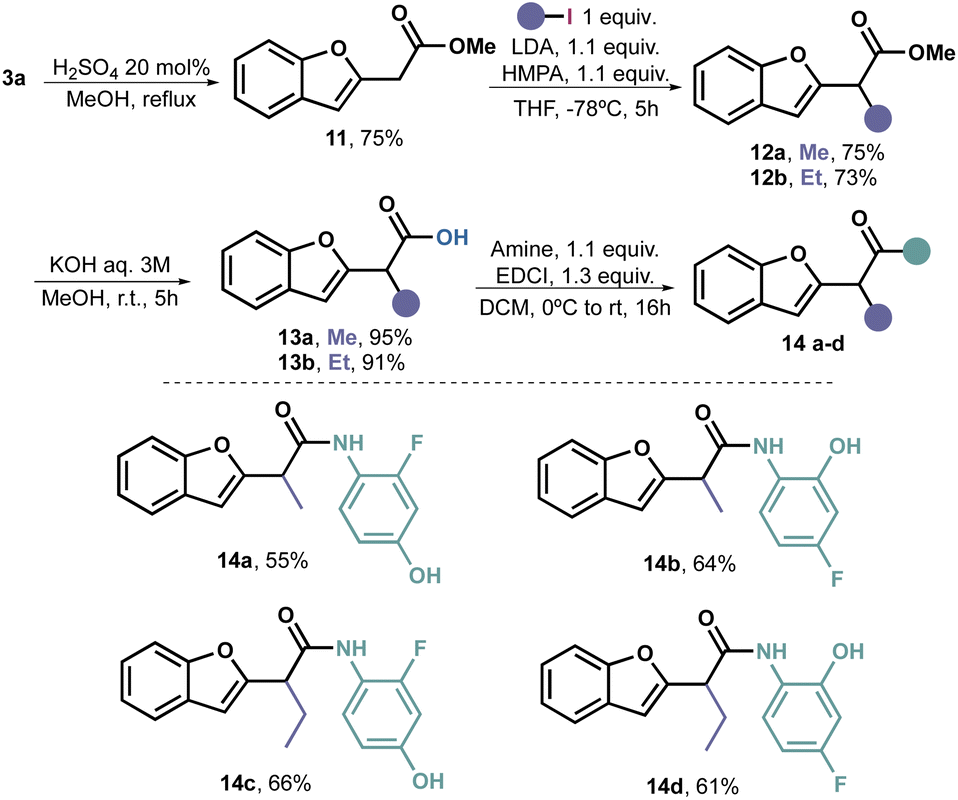 | ||
| Scheme 4 Synthesis of α-alkyl benzofuranyl acetic acid amides 14. | ||
Armed with these results, we decided to conduct biological activity tests using F. oxysporum for this purpose (Table 1).
FOX is a pathogenic fungus that affects crops such as bananas, tomatoes, tropical fruits, and flowers, among others.7 Therefore, it has been used as a model system for testing antifungal activities for many years.
Initially, we tested the tioamide 9 as an analogue of the target molecules, but it showed no activity. Among compounds 10a–c, only 10b exhibited some inhibition on F. oxysporum growth (IC50 = 0.42 mM). This suggests that the substitution pattern of 2-fluoro-4-hydroxy in the amine allows for some activity. This hypothesis was confirmed by the results obtained with compounds 14a and 14c (IC50 = 1.27 and 0.70 mM, respectively). Additionally, when comparing compounds 14a with 14c and 14b with 14d, there appears to be an apparent effect of the size of the alkyl group.
Furthermore, we decided to prepare an analogue amide from phenylacetic acid 16 (Scheme 5a) and an o-benzyloxy derivative 17 (Scheme 5b). Compound 16 exhibited an unusual growth of fungus, rendering it impossible to determine the IC50. However, this behaviour was solely observed at high concentrations, thus indicating that benzofurans have indeed potential antifungal activity and confirming our working hypothesis. On the other hand, the decision to use the benzyl group is supported by literature reports, which often highlight the presence of large hydrophobic groups in the structure of antifungal agents. Therefore, our aim was to determine whether the position 2 of the aromatic ring played a significant role in the compounds' activity. Since compound 17 did not exhibit any activity, it is reasonable to assume that small hydrogen bond acceptors would be more promising as substituents for this position.
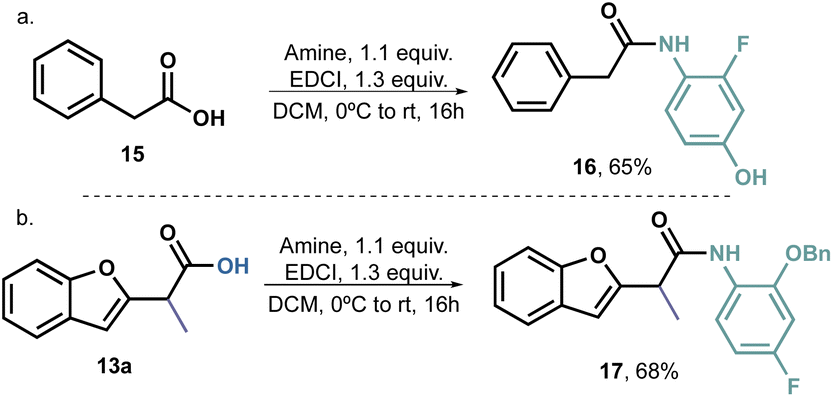 | ||
| Scheme 5 Amidation of 15 and 13a to 16 and 17 respectively. | ||
Although the activities of compounds 10b (IC50 = 0.42 mM) and 14c are not comparable to the positive control or other previously described potent antifungal compounds, their inhibition suggests that modifying the alkyl chain or functionalizing the benzofuran ring may lead to the discovery of new and potent inhibitors of F. oxysporum. The synthetic method described in this paper can be utilized for both types of functionalization and modifications. It should be relatively easy to functionalize the benzofuran ring at least in three different stages, and alkylating with longer chains is plausible.
It is worth noting that when evaluated for antibacterial activity against Agrobacterium tumefaciens and Pectobacterium carotovorum, the compounds showed no activity.
Conclusions
In conclusion, we have described two distinct methods for synthesizing benzofuranyl acetic acid using commercially available materials. Both methods are applicable on a 2 mmol scale, with one of them being suitable for multigram quantities. Additionally, we have presented the alpha alkylation of these substrates and subsequent amidation under mild reaction conditions. In total, we have synthesized eight new benzofuranyl acetic acid amides and provided preliminary results on their biological activity against Fusarium oxysporum. Our ongoing research focuses on further functionalizing the molecules described in this study and testing their biological activity against other fungal strains. The results will be presented in due course.Experimental section
General information
All the solvents were used as received from the supplier without any modifications. All reagents were used as received from commercial suppliers. Reactions progress was monitored by thin layer chromatography (TLC) performed on aluminium plates coated with silica gel F254 with 0.2 mm thickness TLC plates were visualized using ultraviolet (UV) light at 254 nm or stained with p-anisaldehyde, vanillin, ninhydrin, or phosphomolybdic acid solutions. Flash column chromatography was performed using silica gel 60 (230–400 mesh). Neat infrared spectra were recorded using a Thermo Nicolet-Nexus (FT-IR) with PIKE MIRacle ATR cell. Wavenumbers (nmax) are reported in cm−1. High-resolution mass spectrometry was recorded using an Agilent 5973 (70 eV) spectrometer using electrospray ionization (ESI). GC-MS was recorded in a Thermo Scientific Trace 1300. NMR spectra were recorded using a BRUKER Advance III HD Ascend 400 spectrometer. Chemical shifts are given in parts per million (ppm, δ), referenced to TMS (1H and 13C) and trifluoroacetic acid (19F) (δ = −75.39), the solvent peak of CDCl3 defined at δ = 7.26 ppm (1H NMR) and δ = 77.16 (13C NMR), DMSO-d6 defined at δ = 2.5 ppm (1H NMR) and δ = 39.70 (13C NMR). Coupling constants are quoted in Hz (J). 1H NMR splitting patterns were designated as singlet (s), doublet (d), triplet (t), quartet (q), (td) triplet of doublets and multiplet (m). Splitting patterns that could not be interpreted or easily visualized were designated as multiplet (m) or broad (br). Melting points were measured on solids after chromatography using a 1101D-MEL-TEMP melting point apparatus.Experimental procedures and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) to afford 2-(benzofuran-2-yl) acetic acid (3a) as a brown solid (0.272 g, 66%). (Method B): 2-(benzofuran-2-yl)-1-morpholinoethane-1-thione 9 (21.8 mmol, 5.705 g) were dissolved in 30.0 mL of ethanol and subsequently KOH (87.2 mmol, 4.89 g) solution in 20.0 mL of a mixture 1–1 ethanol–H2O was added; the resulting mixture was heated to reflux, and the reaction was followed by TLC. The crude was concentrated, and the basic aqueous solution washed three times with EtOAc to remove organic impurities and morpholine; the aqueous phase was acidified with HCl, the mixture extracted with EtOAc (3 × 20 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to produce pure 2-(benzofuran-2-yl) acetic acid (3a) as a brown solid (3.9 g, 90%). 1H NMR (400 MHz, CDCl3) δ: 7.53 (dd, J = 7.6, 0.8 Hz, 1H), 7.46–7.43 (m, 1H), 7.28–7.16 (m, 2H), 6.66 (d, J = 0.6 Hz, 1H), 3.88 (s, 2H). 13C NMR (100 MHz, CDCl3) δ: 174., 155.1, 149.8, 128.5, 124.3, 123.0, 121.0, 111.3, 105.7, 34.4. FT-IR (neat) ν(cm−1): 3105, 2607, 1716, 1608, 1454, 1384, 1253, 1103, 959, 813, 740, 659. HRMS (ESI): calc. for C10H8NaO3 [M + Na]+ 199.0371; found: 199.0374, mp: 96–99 °C.:5) to afford 1-(benzofuran-2-yl)ethan-1-one (8) as a white solid (5.72 g, 61%), alternatively the product may be recrystallized from 95% ethanol obtaining 54% yield. 1H NMR (400 MHz, CDCl3) δ: δ 7.72 (d, J = 7.9 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.53–7.44 (m, 2H), 7.32 (t, J = 7.5 Hz, 1H), 2.62 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 188.8, 155.7, 152.7, 128.4, 127.1, 124.0, 123.4, 113.2, 112.5, 26.6. FT-IR (neat) ν(cm−1): 3120, 3086, 1670, 1612, 1554, 1292, 1172, 1076, 972, 748, 636. HRMS (ESI): calc. for C10H9O2 [M + H]+ 161.0597; found 161.0602, mp: 67–71 °C.:10) to obtain 2-(benzofuran-2-yl)-1-morpholinoethane-1-thione (9) as a brown solid (4.03 g, 61%). 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.35–7.18 (m, 2H), 6.69 (s, 1H), 4.51 (s, 2H), 4.42 (t, J = 5.0 Hz, 2H), 3.93 (d, J = 4.9 Hz, 2H), 3.84 (t, J = 5.0 Hz, 2H), 3.74 (t, J = 4.9 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 196.3, 154.7, 152.4, 128.3, 124.1, 123.0, 120.9, 111.0, 104.6, 66.4, 51.2, 50.3, 43.9. FT-IR (neat) ν(cm−1): 3086, 2858, 1608, 1481, 1273, 1111, 1026, 956, 887, 817, 759, 686. HRMS (ESI): calc. for C14H16NO2S [M + H]+, 262.0902; found, 262.0903, mp: 156–160 °C.
:20) to afford 2-(benzofuran-2-yl) acetic acid (3a) as a brown solid (0.272 g, 66%). (Method B): 2-(benzofuran-2-yl)-1-morpholinoethane-1-thione 9 (21.8 mmol, 5.705 g) were dissolved in 30.0 mL of ethanol and subsequently KOH (87.2 mmol, 4.89 g) solution in 20.0 mL of a mixture 1–1 ethanol–H2O was added; the resulting mixture was heated to reflux, and the reaction was followed by TLC. The crude was concentrated, and the basic aqueous solution washed three times with EtOAc to remove organic impurities and morpholine; the aqueous phase was acidified with HCl, the mixture extracted with EtOAc (3 × 20 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to produce pure 2-(benzofuran-2-yl) acetic acid (3a) as a brown solid (3.9 g, 90%). 1H NMR (400 MHz, CDCl3) δ: 7.53 (dd, J = 7.6, 0.8 Hz, 1H), 7.46–7.43 (m, 1H), 7.28–7.16 (m, 2H), 6.66 (d, J = 0.6 Hz, 1H), 3.88 (s, 2H). 13C NMR (100 MHz, CDCl3) δ: 174., 155.1, 149.8, 128.5, 124.3, 123.0, 121.0, 111.3, 105.7, 34.4. FT-IR (neat) ν(cm−1): 3105, 2607, 1716, 1608, 1454, 1384, 1253, 1103, 959, 813, 740, 659. HRMS (ESI): calc. for C10H8NaO3 [M + Na]+ 199.0371; found: 199.0374, mp: 96–99 °C.:5) to afford 1-(benzofuran-2-yl)ethan-1-one (8) as a white solid (5.72 g, 61%), alternatively the product may be recrystallized from 95% ethanol obtaining 54% yield. 1H NMR (400 MHz, CDCl3) δ: δ 7.72 (d, J = 7.9 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.53–7.44 (m, 2H), 7.32 (t, J = 7.5 Hz, 1H), 2.62 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 188.8, 155.7, 152.7, 128.4, 127.1, 124.0, 123.4, 113.2, 112.5, 26.6. FT-IR (neat) ν(cm−1): 3120, 3086, 1670, 1612, 1554, 1292, 1172, 1076, 972, 748, 636. HRMS (ESI): calc. for C10H9O2 [M + H]+ 161.0597; found 161.0602, mp: 67–71 °C.:10) to obtain 2-(benzofuran-2-yl)-1-morpholinoethane-1-thione (9) as a brown solid (4.03 g, 61%). 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.35–7.18 (m, 2H), 6.69 (s, 1H), 4.51 (s, 2H), 4.42 (t, J = 5.0 Hz, 2H), 3.93 (d, J = 4.9 Hz, 2H), 3.84 (t, J = 5.0 Hz, 2H), 3.74 (t, J = 4.9 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 196.3, 154.7, 152.4, 128.3, 124.1, 123.0, 120.9, 111.0, 104.6, 66.4, 51.2, 50.3, 43.9. FT-IR (neat) ν(cm−1): 3086, 2858, 1608, 1481, 1273, 1111, 1026, 956, 887, 817, 759, 686. HRMS (ESI): calc. for C14H16NO2S [M + H]+, 262.0902; found, 262.0903, mp: 156–160 °C.General procedure for amidation
To a stirred solution of the corresponding acid (1.0 equiv.), and aniline derivative (1.1 equiv.), in DCM at 0 °C; was added EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide 1.3 equiv.), the mixture is stirred and left under stirring until reaching r.t. gradually and then stirred for 16 h at r.t. The mixture was poured into 10% HCl solution and extracted with EtOAc (3 × 20 mL); and the combined organic extracts were combined, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude mixture was purified using flash chromatography to afford the pure product.:50/DCM 100%. 1H NMR (400 MHz, DMSO-d6) δ: 10.38 (s, 1H), 9.46 (s, 1H), 7.78 (dd, J = 8.9, 6.4 Hz, 1H), 7.55 (dd, J = 25.9, 7.5 Hz, 2H), 7.37–7.10 (m, 2H), 6.77 (s, 1H), 6.67 (dd, J = 10.3, 2.8 Hz, 1H), 6.60 (td, J = 8.7, 2.8 Hz, 1H), 4.01 (s, 2H). 13C NMR (100 MHz, DMSO-d6): 166.7, 159.1 (d, J = 240.4 Hz), 154.4, 153.5, 149.7 (d, J = 11.1 Hz), 128.6, 123.9, 123.7 (d, J = 9.9 Hz), 122.9, 122.8 (d, J = 2.9 Hz), 120.9, 111.0, 105.2 (d, J = 21.9 Hz), 104.9, 102.7 (d, J = 24.9 Hz), 36.3. 19F-NMR (374 MHz, DMSO-d6) δ: −117.4. FT-IR (neat) ν(cm−1): 3371, 2897, 2750, 1654, 1527, 1423, 1238, 1157, 1103, 979, 933, 759. HRMS (ESI): calc. for C16H13FNO3 (M + H+), 286.0869; found, 286.0869, mp: 165–168 °C.:10) to afford methyl 2-(benzofuran-2-yl)propanoate (12a) as a colorless liquid (0.80 g, 75%). 1H NMR (400 MHz, CDCl3) δ: 7.52 (d, J = 7.7 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.22 (dt, J = 19.6, 7.4 Hz, 2H), 6.57 (s, 1H), 3.96 (q, J = 7.3 Hz, 1H), 3.72 (s, 3H), 1.62 (d, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 172.6, 156.3, 154.8, 128.4, 124.0, 122.8, 120.8, 111.2, 103.2, 52.5, 39.9, 15.8. FT-IR (neat) ν(cm−1): 2989, 1743, 1610, 1455, 1380, 1201, 1095, 945, 740. HRMS (ESI): calc. for C12H12O3Na [M + Na]+: 227.0679; found: 227.0675.:10). 1H NMR (400 MHz, CDCl3) δ: 8.80 (s, 1H), 7.74 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.35–7.18 (m, 2H), 6.85 (dd, J = 8.7, 5.7 Hz, 1H),6.72 (d, J = 10.5 Hz, 2H), 6.59–6.49 (m, 1H), 4.06 (q, J = 7.3 Hz, 1H), 1.75 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz CDCl3) δ: 171.7, 161.7 (d, J = 245.2 Hz), 155.3, 154.9, 150.4 (d, J = 12.3 Hz), 127.9, 124.8, 123.3, 123.2 (d, J = 10.3 Hz), 121.6 (d, J = 3.1 Hz), 121.2, 111.2, 107.3 (d, J = 23.1 Hz), 107.0 (d, J = 24.6 Hz), 104.6, 41.5, 15.8.19F-NMR (374 MHz, CDCl3) δ: −114.2. FT-IR (neat) ν(cm−1): 3379, 2931, 1647, 1539, 1431, 1280, 1149, 968, 840, 752, 655, 459. HRMS (ESI): calc. for C17H15FNO3 (M + H+), 300.1036; found, 300.1040, mp: 138–142 °C.:20. 1H NMR (400 MHz, CDCl3) δ: 8.79 (s, 1H), 7.87 (s, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.48 (d, J = 8.2 Hz, 1H), 7.36–7.15 (m, 2H), 6.91 (d, J = 5.3 Hz, 1H), 6.88–6.59 (m, 2H), 6.61–6.43 (m, 1H), 4.09–3.70 (m, 1H), 2.33–2.21 (m, 1H), 2.20–2.06 (m, 1H), 1.05 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz CDCl3) δ: 171.4, 161.6 (d, J = 246.2 Hz), 155.0, 154.2, 150.3 (d, J = 9.9 Hz), 128.0, 124.7, 123.3, 123.2 (d, J = 10.3 Hz), 121.7 (d, J = 3.2 Hz), 121.2, 111.3, 107.4 (d, J = 23.0 Hz), 106.8 (d, J = 22.7 Hz), 105.4, 49.4, 24.5, 12.2. 19F-NMR (374 MHz, CDCl3) δ = −114.3. FT-IR (neat) ν(cm−1) = 2970, 1654, 1608, 1454, 1254, 1153, 976, 802. HRMS (ESI): calc. for C18H17FNO3 [M + H]+ 314.1186; found, 314.1181.:10. 1H NMR (400 MHz, CDCl3) δ: 8.32 (t, J = 7.5 Hz, 1H), 8.16 (s, 1H), 7.69–7.02 (m, 9H), 6.73–6.51 (m, 3H), 4.97 (s, 2H), 4.15–3.71 (m, 1H), 1.68 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz CDCl3) δ: 169.1, 159.0 (d, J = 242.9 Hz), 156.5, 154.9, 148.1 (d, J = 9.9 Hz), 135.6, 128.8, 128.2, 127.6, 124.0 (d, J = 3.3 Hz), 120.7 (d, J = 9.2 Hz), 111.1, 120.9, 123.0, 124.3, 128.5, 107.2 (d, J = 21.7 Hz), 103.8, 100.1 (d, J = 27.1 Hz), 71.0, 42.5, 15.7. 19F-NMR (374 MHz, CDCl3) δ: −115.8. FT-IR (neat) ν(cm−1): 3271, 3032, 1874, 1654, 1535, 1284, 1222, 972, 825, 732. HRMS (ESI): calc. for C24H21FNO3 (M + H+), 390.1505; found, 390.1499, mp: 118–122 °C.Author contributions
C. M.-M. investigation, methodology, validation, writing-review and editing and visualisation; P. B.-M. investigation biological activity, validation, writing-review and editing and visualisation; L M. P writing-review and editing and funding acquisition and D. G.-S. conceptualisation, project administration, resources, writing-original draft, writing-review and editing, funding acquisition, supervision, and visualisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Universidad de Los Andes and Chemistry Department for financial support. C. M.-M. acknowledges Chemistry Department at Universidad de Los Andes for his fellowship. We are kindly grateful to Laura Ibarra, Laura Lopez, and Sandra Ortiz for the measurements of IR and Mass spectra. We kindly acknowledge research group in Bacteriología Agrícola y Ambiental (BA&A) from Universidad de Antioquia in Colombia for isolation and donation of Fusarium oxysporum, Agrobacterium tumefaciens and Pectobacterium carotovorum strains, and Vicerrectoría Académica from Fundación Universitaria Juan N. Corpas for financial support for biological studies.Notes and references
- L. Serwecińska, Water, 2020, 12, 3313 CrossRef.
- C. Manyi-Loh, S. Mamphweli, E. Meyer and A. Okoh, Molecules, 2018, 23, 795 CrossRef PubMed.
- J. B. Ristaino, P. K. Anderson, D. P. Bebber, K. A. Brauman, N. J. Cunniffe, N. V. Fedoroff, C. Finegold, K. A. Garrett, C. A. Gilligan, C. M. Jones, M. D. Martin, G. K. MacDonald, P. Neenan, A. Records, D. G. Schmale, L. Tateosian and Q. Wei, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022239118 CrossRef CAS PubMed.
- M. Tudi, H. Daniel Ruan, L. Wang, J. Lyu, R. Sadler, D. Connell, C. Chu and D. T. Phung, Int. J. Environ. Res. Public Health, 2021, 18, 1112 CrossRef CAS PubMed.
- T. R. Gordon, Annu. Rev. Phytopathol., 2017, 55, 23–39 CrossRef CAS PubMed.
- F. Shabani and L. Kumar, PLoS One, 2013, 8, e83404 CrossRef PubMed.
- L. Burgess and W. Bryden, Microbiol. Aust., 2012, 33, 22–25 CrossRef.
- D. H. M. Alkhalifah, E. Damra, M. B. Melhem and W. N. Hozzein, Microorganisms, 2023, 11, 468 CrossRef PubMed.
- E. Sandulachi, A. Ghendov-Mosanu, D. Cojocari and R. Sturza, Adv. Microbiol., 2021, 11, 541–553 CrossRef CAS.
- G. B. Eshun, F. J. Osonga, T. Erdogan, A. Gölcü and O. A. Sadik, RSC Adv., 2023, 13, 21781–21792 RSC.
- A. Akhmedov, R. Gamirov, Y. Panina, E. Sokolova, Y. Leonteva, E. Tarasova, R. Potekhina, I. Fitsev, D. Shurpik and I. Stoikov, Org. Biomol. Chem., 2023, 21, 4863–4873 RSC.
- L. Tai, S.-X. Sun, K. Yue, J.-Q. Chai, S.-T. Hou, G.-Y. Dai, C.-L. Yang and M. Chen, New J. Chem., 2023, 47, 12850–12860 RSC.
- I. I. Ozturk, K. Turk, A. M. Grześkiewicz, M. Kubicki, C. N. Banti and S. K. Hadjikakou, New J. Chem., 2023, 47, 12779–12789 RSC.
- L. Du, S. Haldar, J. B. King, A. O. Mattes, S. Srivastava, K. L. Wendt, J. You, C. Cunningham and R. H. Cichewicz, J. Nat. Prod., 2023 DOI:10.1021/acs.jnatprod.3c00382.
- S. Y. Jeong, A. Alishir, S. Zhang, Y. Zhang, S. Choi, C. Pang, H. Y. Bae, W. H. Jung and K. H. Kim, J. Nat. Prod., 2023 DOI:10.1021/acs.jnatprod.3c00193.
- X.-y. Zhi, Y. Zhang, Y.-f. Li, Y. Liu, W.-p. Niu, Y. Li, C.-r. Zhang, H. Cao, X.-j. Hao and C. Yang, J. Agric. Food Chem., 2023, 71, 11680–11691 CrossRef CAS.
- C. Yang, S. Sun, W. Li, Y. Mao, Q. Wang, Y. Duan, R. Csuk and S. Li, J. Agric. Food Chem., 2023, 71, 11341–11349 CrossRef CAS.
- M. Noman, Azizullah, T. Ahmed, Y. Gao, H. Wang, X. Xiong, J. Wang, J. Lou, D. Li and F. Song, J. Agric. Food Chem., 2023, 71, 11667–11679 CrossRef CAS PubMed.
- A. A. Abbas and K. M. Dawood, RSC Adv., 2023, 13, 11096–11120 RSC.
- H. X. Ngo, S. Garneau-Tsodikova and K. D. Green, MedChemComm, 2016, 7, 1285–1306 RSC.
- Y. Liu, Y. Wang, G. Dong, Y. Zhang, S. Wu, Z. Miao, J. Yao, W. Zhang and C. Sheng, MedChemComm, 2013, 4, 1551–1561 RSC.
- M. Masubuchi, H. Ebiike, K.-i. Kawasaki, S. Sogabe, K. Morikami, Y. Shiratori, S. Tsujii, T. Fujii, K. Sakata, M. Hayase, H. Shindoh, Y. Aoki, T. Ohtsuka and N. Shimma, Bioorg. Med. Chem., 2003, 11, 4463–4478 CrossRef CAS PubMed.
- X. Cao, M. Wang, Z. Li and X. Xu, J. Agric. Food Chem., 2023, 71, 8757–8768 CrossRef CAS PubMed.
- B. Gabriele, R. Mancuso, G. Salerno and M. Costa, J. Org. Chem., 2007, 72, 9278–9282 CrossRef CAS PubMed.
- N. Thirupathi, M. Hari Babu, V. Dwivedi, R. Kant and M. Sridhar Reddy, Org. Lett., 2014, 16, 2908–2911 CrossRef CAS PubMed.
- Y. He, Z. Li, K. Robeyns, L. Van Meervelt and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2018, 57, 272–276 CrossRef CAS.
- B. Li, T. Li, M. A. Aliyu, Z. H. Li and W. Tang, Angew. Chem., Int. Ed., 2019, 58, 11355–11359 CrossRef CAS PubMed.
- K.-O. Kim and J. Tae, Synthesis, 2005, 2005, 387–390 CrossRef.
- E. J. Enholm and G. Prasad, Tetrahedron Lett., 1989, 30, 4939–4942 CrossRef.
- B. Gabriele, R. Mancuso, G. Salerno and M. Costa, Adv. Synth. Catal., 2006, 348, 1101–1109 CrossRef CAS.
- Y. Fang and C. Li, J. Org. Chem., 2006, 71, 6427–6431 CrossRef CAS PubMed.
- T. Huang, Y. Shao, S. Tang and J. Sun, Org. Biomol. Chem., 2023, 21, 5752–5756 RSC.
- V. Ramella, Z. He, C. G. Daniliuc and A. Studer, Eur. J. Org Chem., 2016, 2016, 2268–2273 CrossRef CAS.
- A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem. –Eur. J., 2013, 19, 7691–7695 CrossRef CAS PubMed.
- Y. Yamane, K. Yoshinaga, M. Sumimoto and T. Nishikata, ACS Catal., 2019, 9, 1757–1762 CrossRef CAS.
- W. Tian, B. Li, D. Tian and W. Tang, Chin. Chem. Lett., 2022, 33, 197–200 CrossRef CAS.
- M. Chakravarty and K. C. Kumara Swamy, J. Org. Chem., 2006, 71, 9128–9138 CrossRef CAS PubMed.
- P. K. Mandali and D. K. Chand, Synthesis, 2015, 47, 1661–1668 CrossRef CAS.
- C. E. Castro, E. J. Gaughan and D. C. Owsley, J. Org. Chem., 1966, 31, 4071–4078 CrossRef CAS.
- P. Borrego-Muñoz, F. Ospina and D. Quiroga, Molecules, 2021, 26, 3997 CrossRef PubMed.
- Y.-Q. Xie, Y.-B. Huang, J.-S. Liu, L.-Y. Ye, L.-M. Che, S. Tu and C.-L. Liu, Pest Manage. Sci., 2015, 71, 404–414 CrossRef CAS PubMed.
- H. Xu, Z. Hou, Z. Liang, M.-B. Guo, X. Su and C. Guo, Chin. J. Chem., 2019, 37, 1245–1250 CrossRef CAS.
- B. Shankar, P. Jalapathi, M. Ramesh, A. K. Kumar, M. Ragavender and G. Bharath, Russ. J. Gen. Chem., 2016, 86, 1711–1721 CrossRef CAS.
- S.-S. Du, X.-F. Luo, J.-X. An, Z.-J. Zhang, S.-Y. Zhang, Y.-R. Wang, Y.-Y. Ding, W.-Q. Jiang, B.-Q. Zhang, Y. Ma, Y. Zhou, Y.-M. Hu and Y.-Q. Liu, Pest Manage. Sci., 2023, 79, 2748–2761 CrossRef CAS PubMed.
- M. Csékei, Z. Novák and A. Kotschy, Tetrahedron, 2008, 64, 8992–8996 CrossRef.
- M. J. Moure, R. SanMartin and E. Domínguez, Adv. Synth. Catal., 2014, 356, 2070–2080 CrossRef CAS.
- M. Nakamura, L. Ilies, S. Otsubo and E. Nakamura, Angew. Chem., Int. Ed., 2006, 45, 944–947 CrossRef CAS PubMed.
- M. Nakamura, L. Ilies, S. Otsubo and E. Nakamura, Org. Lett., 2006, 8, 2803–2805 CrossRef CAS PubMed.
- D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 7870–7880 RSC.
- A. Ojeda-Porras and D. Gamba-Sánchez, Tetrahedron Lett., 2015, 56, 4308–4311 CrossRef CAS.
- G. Li, Y. Xing, H. Zhao, J. Zhang, X. Hong and M. Szostak, Angew. Chem., Int. Ed., 2022, 61, e202200144 CrossRef CAS PubMed.
- D. R. Goud and U. Pathak, Synthesis, 2012, 44, 3678–3682 CrossRef CAS.
- A. Ojeda-Porras and D. Gamba-Sánchez, J. Org. Chem., 2016, 81, 11548–11555 CrossRef CAS PubMed.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- A. Ojeda-Porras, A. Hernández-Santana and D. Gamba-Sánchez, Green Chem., 2015, 17, 3157–3163 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data, copies of NMR spectra and details of biological activity measurement. See DOI: https://doi.org/10.1039/d3ra04737g |
| ‡ Current address: Escuela de Medicina, Fundación Universitaria Juan N. Corpas, Bogotá 110311, Colombia. |
| This journal is © The Royal Society of Chemistry 2023 |