DOI:
10.1039/D3RA04709A
(Paper)
RSC Adv., 2023,
13, 24450-24459
A novel cobalt(II) acetate complex bearing lutidine ligand: a promising electrocatalyst for oxygen evolution reaction†
Received
13th July 2023
, Accepted 7th August 2023
First published on 15th August 2023
Abstract
Developing cost-effective electrocatalysts using earth-abundant metal as an alternative to expensive precious metal catalyst remains a key challenge for researchers. Several strategies are being researched/tested for making low-cost transition metal complexes with controlled electron-density and coordination flexibility around the metal center to enhance their catalytic activity. Herein, we report a novel lutidine coordinated cobalt(II) acetate complex [(3,5-lutidine)2Co(OAc)2(H2O)2] (1) as a promising electrocatalyst for oxygen evolution reaction (OER). Complex 1 was characterized by FT-IR, elemental analysis, and single crystal X-ray diffraction data. The structure optimization of complex 1 was also done using DFT calculation and the obtained geometrical parameters were found to be in good agreement with the parameters obtained from the solid state structure obtained through single crystal X-ray diffraction data. Further, the molecular electrostatic potential (MEP) maps analysis of complex 1 observed electron rich centers that were found to be in agreement with the solid-state structure. It was understood that the coordination of lutidine as a Lewis base and acetate moiety as a flexible ligand will provide more coordination flexibility around the metal center to facilitate the catalytic reaction. Further, the electron rich centers around metal center will also support the enhancement of their catalytic activity. Complex 1 shows impressive OER activity, even better than the state-of-the-art IrO2 catalyst, in terms of turnover frequency (TOF: 0.05) and onset potential (1.50 V vs. RHE). The TOF for complex 1 is two and half times higher, while the onset potential is ca. 20 mV lower, than the benchmark IrO2 catalyst studied under identical conditions.
Introduction
The development of cost-effective and clean energy is of considerable scientific interest due to the rapid increase in energy consumption and environmental crisis associated with fossil fuels.1–3 Electrochemical water splitting is one of the promising strategies, which involves the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).4 However, the commercialization of these devices is largely limited due to the high over-potential and sluggish kinetics of the OER, which requires highly active electrocatalysts.5 Henceforth, in the last few decades, many research groups have been conducting their work in this area to develop efficient electrocatalysts for OER. Initially, precious metals (Pt, Ir and Ru) based catalysts were focused due to their robust nature and high activity.6–10 In the recent past, the demand for cost-effective earth-abundant transition metal based electrocatalysts has accelerated to overcome the high cost and lower abundance of precious metals.11–14 However, the OER activity of these transition metal-based catalysts has not been comparable to the Pt-group metals. Hence, the foremost challenge for chemists is to design and develop novel cost-effective, earth abundant and highly efficient catalysts as well as strategies to boost their activity for OER.
Cobalt-based catalysts have been significantly studied to facilitate the OER.15,16 For instance, cobalt oxides,17–20 hydroxides,21,22 sulfides,23–25 selenides,26,27 phosphates/phosphides,28,29 and nitrides30,31 have been established as advanced electrocatalyst for OER. Beside these materials, several molecular cobalt complexes have also been found as efficient electrocatalyst for OER in recent times.32–38 D. G. Nocera's group reported a β-octafluoro Co(III) xanthene hangman corrole as the effective OER catalyst under operating conditions at modest overpotential.32 J. T. Groves et al. reported a cationic cobalt–porphyrin complex as an efficient homogeneous electrocatalyst for water oxidation to study the role of buffer on the reactivity.33 C. N. Verani et al. designed a novel phenolate rich cobalt(III) complex which acts as an excellent water oxidation catalyst at moderate overpotential of 0.5 V.34 L. Sun et al. used cobalt porphyrin complexes as OER catalysts and found that the in situ generation of cobalt oxides thin film acts as a real catalyst.35 Recently, M. Nath et al. reported a cobalt complex bearing selelno-based ligand as OER electrocatalyst with very low overpotential of 320 mV.37 Zhen-Tao Yu et al. reported a cobalt(IV) diacetato complex bearing a substituted bipyridine dianionic ligand as highly active electrocatalyst for OER at an overpotential of only 360 mV at pH = 6.38 In case of molecular cobalt catalyst, many research groups reported that the in situ formation of Co(OH)2/cobalt oxide are the true catalyst.19,35,39
However, the nature of in situ generated catalyst depends upon the molecular nature of parent catalyst and hence the efficacy of the catalyst solely depends on role of parent complex. Overall, these representative examples of Co-complex based OER catalysts highlights the chemistry of ligands towards controlling the electron-density around the Co-metal center or an ability to partially oxidize the complex to make thin films of cobalt oxide. Hence, it was thought that the Lewis base ligand can control the electron-density around metal center to enhance their catalytic activity. Further, the coordination of Lewis base and acetate moiety will provide more coordination flexibility around metal center which supports the substrate coordination/removal during catalysis to facilitate the reaction cycle.38,40 In this view, we report a novel cost-effective lutidine coordinated cobalt(II) acetate complex [(3,5-lutidine)2Co(OAc)2(H2O)2] (1) as promising electrocatalytic activator for OER. The obtained data after OER electrocatalytic analysis indicates that complex 1 is better performer than the state-of-the-art IrO2 catalyst.
Experimental
Materials and methods
The details of materials and methods have been given in ESI.† The details of X-ray diffraction data collection and crystallographic information are also given in the ESI (see Table S1†).
Synthesis of complex 1
Co(OAc)2·4H2O (250 mg, 1 mmol) was dissolved in acetonitrile to get homogeneous solution, to which 3,5-lutidine (214 mg, 2 mmol in 5 mL acetonitrile) was added dropwise and stirred at room temperature for 3 h. The resulting solution was reduced using rotary evaporator and then kept at room temperature for crystallization. Crystals suitable for single crystal X-ray diffraction data of [(3,5-lutidine)2Co(OAc)2(H2O)2] (1) were obtained after five days. Yield: 94% (0.402 g, 0.940 mmol). FT-IR (KBr, ν in cm−1): 3425 (br) for ν(H2O); 1572 (s) for νasym(OCO); and 1419 (s) for νsym(OCO). Anal. calcd (%) for C18H28CoN2O6: C, 50.59; H, 6.60; N, 6.56. Found C, 51.02; H, 6.57; N, 6.71.
Electrochemical analysis
The electrochemical analysis for oxygen evolution reaction was performed in a three-electrode electrochemical setup, using Glassy carbon (GC) electrode with geometric area of 0.07 cm2 as working electrode, Hg/HgO (1 M NaOH) (E0 = 0.118 V vs. RHE) as reference electrode and graphite rod as counter electrode in 1 M KOH (pH = 14). The GC electrode was modified with catalyst ink prepared by dispersing the catalyst in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 100 μL isopropanol and water mixture using ultrasonicator. Carbon black acetylene was added as an additive in the catalyst to separate the catalyst layer with the ratio of catalyst to carbon black was 8:2. The loading of the catalyst on the GC surface was about 0.05 mg, which corresponds to normalized loading of 0.714 mg cm−2. Similarly, IrO2 obtained from Sigma Aldrich was also used to modify the electrode using same procedure for comparative study. All the potentials were converted to reversible hydrogen electrode (RHE) by using the following eqn (1):41
:1 100 μL isopropanol and water mixture using ultrasonicator. Carbon black acetylene was added as an additive in the catalyst to separate the catalyst layer with the ratio of catalyst to carbon black was 8:2. The loading of the catalyst on the GC surface was about 0.05 mg, which corresponds to normalized loading of 0.714 mg cm−2. Similarly, IrO2 obtained from Sigma Aldrich was also used to modify the electrode using same procedure for comparative study. All the potentials were converted to reversible hydrogen electrode (RHE) by using the following eqn (1):41| | |
ERHE = EHg/HgO/OH− + 0.118 + (0.059 pH)
| (1) |
The electrochemical measurements were performed on MetrohmAutolab 204 N instrument. The current density was normalized according to geometric surface area and electrochemical surface area. The overpotential (η) was calculated by subtracting the standard potential of water oxidation (1.23 V vs. RHE) from the experimental potential observed. The Double layer capacitance (Cdl) calculated from the slope of the plot of current density vs. scan rate, was used to estimate the electrochemically active surface area (ECSA) using eqn (2).
where
Cs, is the specific capacitance of the atomically smooth surface of the material and is taken to be 0.04 mF cm
−2.
42
The Tafel slope was calculated by using the eqn (3):
| | |
η = blogj + a
| (3) |
where
η is the overpotential and
b is the Tafel slope. The electrochemical impedance spectroscopy (EIS) measurements were performed by applying AC potential with 10 mV amplitude in a frequency range of 0.1 Hz to 10
5 Hz at 1.62 V
vs. RHE. The catalyst stability was analyzed by drop casting the catalyst material on glassy carbon plate electrode in 1 M KOH for 14 hours at a constant potential 1.65 V
vs. RHE. After the electrochemical stability, the spent catalyst material was used for post catalytic characterizations
i.e., FTIR, Raman, SEM, EDX and XPS.
Calculation of TOF
The values of TOF were evaluated by assuming all the metal ions as the active catalytic centers as per the following eqn (4).43| | |
TOF = J × A/4 × F × n
| (4) |
Here, J, A, F and n are the observed current density, geometrical surface area of the working electrode (0.07 cm2), Faraday constant (96485 C mol−1) and number of moles of active sites present in the catalyst. Since four electrons are needed to evolve one mole of O2, so the term ¼ is used. Further, n was determined by dividing the mass of material loaded (0.05 mg) onto the electrode surface to the molecular mass of the sample (eqn (5)).
Results and discussions
Synthesis
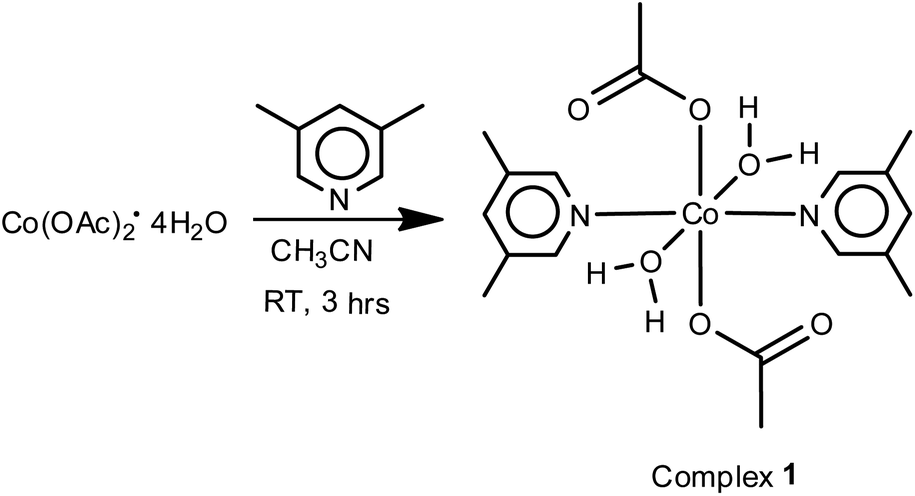
The schematic diagram for synthetic details of complex 1 is given in Scheme 1. The reactions of Co(OAc)2·4H2O with 3,5-lutidine in 1:2 ratio in acetonitrile at room temperature afforded complex 1 in quantitative yields. IR spectra of complex 1 (Fig. SI-3a in ESI†), shows bands at 1572 (s) and 1419 (s) for νasym(OCO) and νsym(OCO) characteristic for monodentate acetate moieties. The broad band at 3425 cm−1 confirms the presence of coordinated water molecules. Further, the bands at ρr(H2O) (710 cm−1) and ρw(H2O) (649 cm−1) also support the presence of coordinated water molecules.44
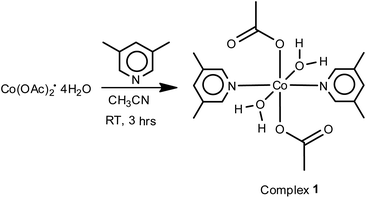 |
| | Scheme 1 Synthesis scheme of complex 1. | |
Crystal structure
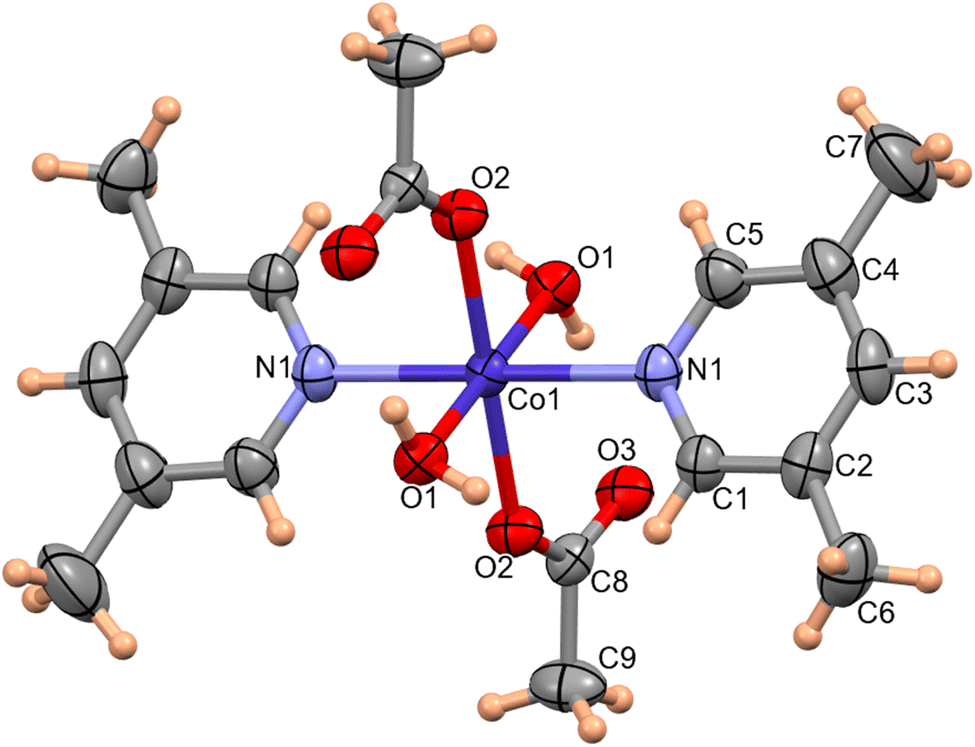
The molecular structure of complex 1 was determined by single-crystal X-ray diffraction data. The molecular structure of complex 1 with the atom labeling scheme is shown in Fig. 1. Complex 1 consists of a Co(II) atom coordinated by two 3,5-lutidine, two water molecules and two acetate moieties in monodentate mode to afford an octahedral geometry. The Co1–O1water, Co1–O2acetate and Co1–N1lutidine distances in complex 1 are 2.131(1), 2.068(1) and 2.153(2) Å, respectively.
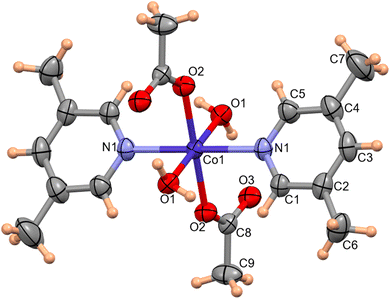 |
| | Fig. 1 ORTEP of complex 1 with 50% probability ellipsoid. Selected bond lengths (Å) and angles (°): Co1–O1 2.133(1), Co1–O2 2.068(1), Co1–N1 2.153(2); O1–Co1–O1 180.00(0), O2–Co1–O2 180.00(6), O2–Co1–N1–90.58(5), N1–Co1–N1 180.00(8), O2–Co1–O1 88.55(4), N1–Co1–O1 87.42(5). | |
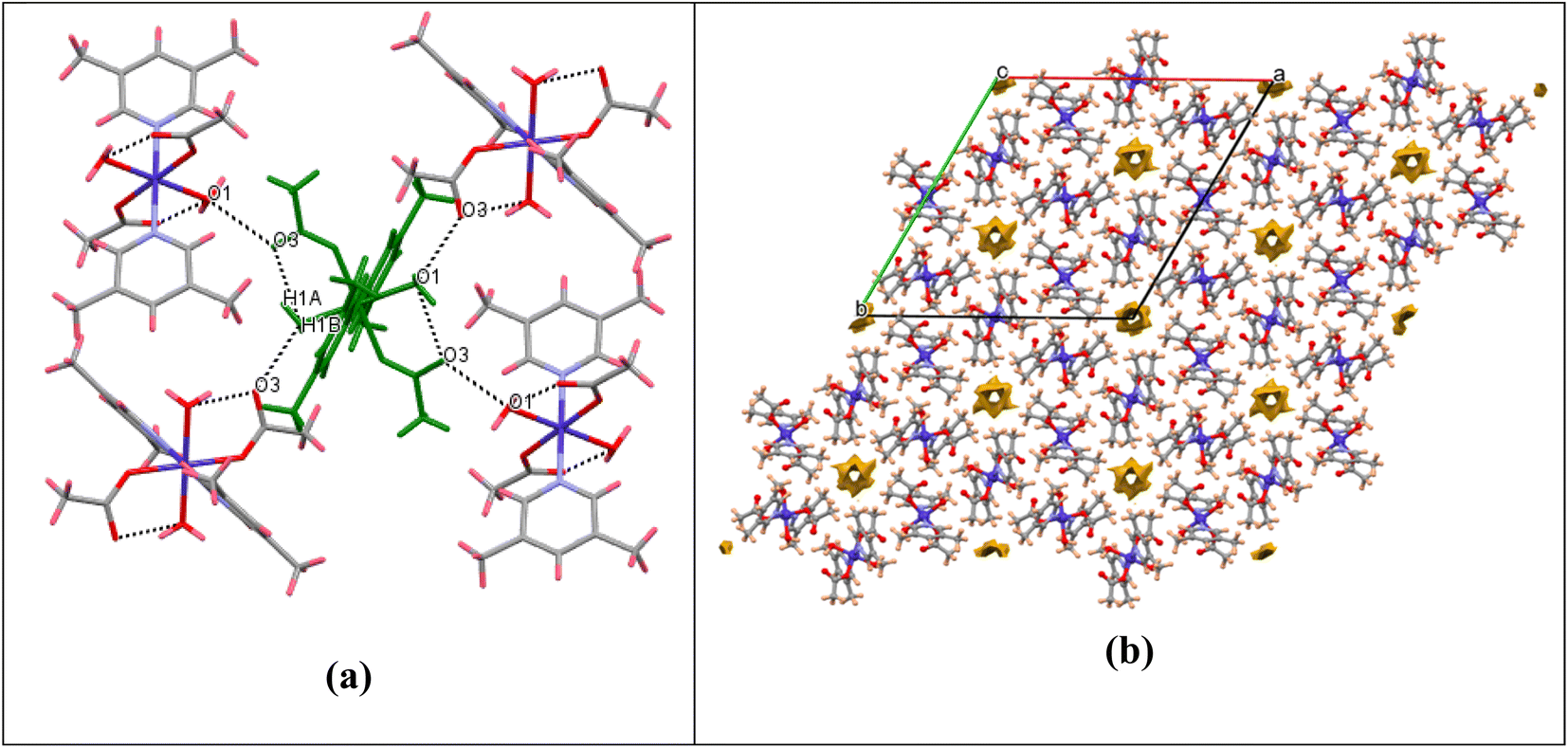
Significant hydrogen-bond parameters observed in complex 1 are listed in Table 1. The oxygen atom (O1) of water molecules in 1 act as hydrogen-bond donor, one to the carbonyl oxygen (O3) of the monodentate acetate within the molecule (intramolecular hydrogen bonding) and another with the carbonyl oxygen (O3) of the monodentate acetate of adjacent molecule (intermolecular hydrogen bonding) as depicted in Fig. 2a. The aforementioned hydrogen-bonding network grows to afford a three-dimensional supramolecular packing along the c-axis, as depicted in Fig. 2b. From packing diagram of complex 1, it was also observed that the crystal structure has various hexagonal vacant pores formed by the supramolecular assembly which could be useful for enhancing their catalytic, separation and gas storage properties. The void volume has also been calculated to be 114 Å3 per unit cell. Although the structural analysis shows a solvent accessible void volume of around 194 Å3 per unit cell, it is not apparent that water molecules can enter this void and the residual density value (0.7 Å) also do not imply the presence of water molecules inside this channel. The porous nature of complex 1 is also clearly visualized in FESEM image as depicted in Fig. SI-1 in ESI.†
Table 1 Significant hydrogen bond distances (Å) and bond angles (°) in the crystal structure of complex 1
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
∠D–H⋯A |
| O1–H1A⋯O3 |
0.860 |
1.946 |
2.708(2) |
148.70 |
| O1–H1B⋯O3 |
0.861 |
2.080 |
2.823(2) |
148.84 |
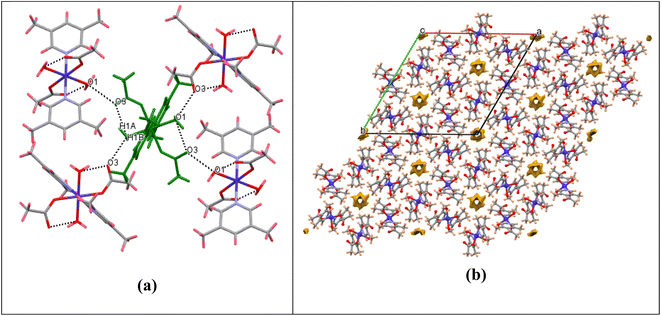 |
| | Fig. 2 (a) Packing diagram showing O–H⋯O hydrogen bonding for complex 1. (b) Packing diagram showing the void surfaces in the crystal structure of complex 1, viewed down crystallographic c-axis. | |
Computational studies
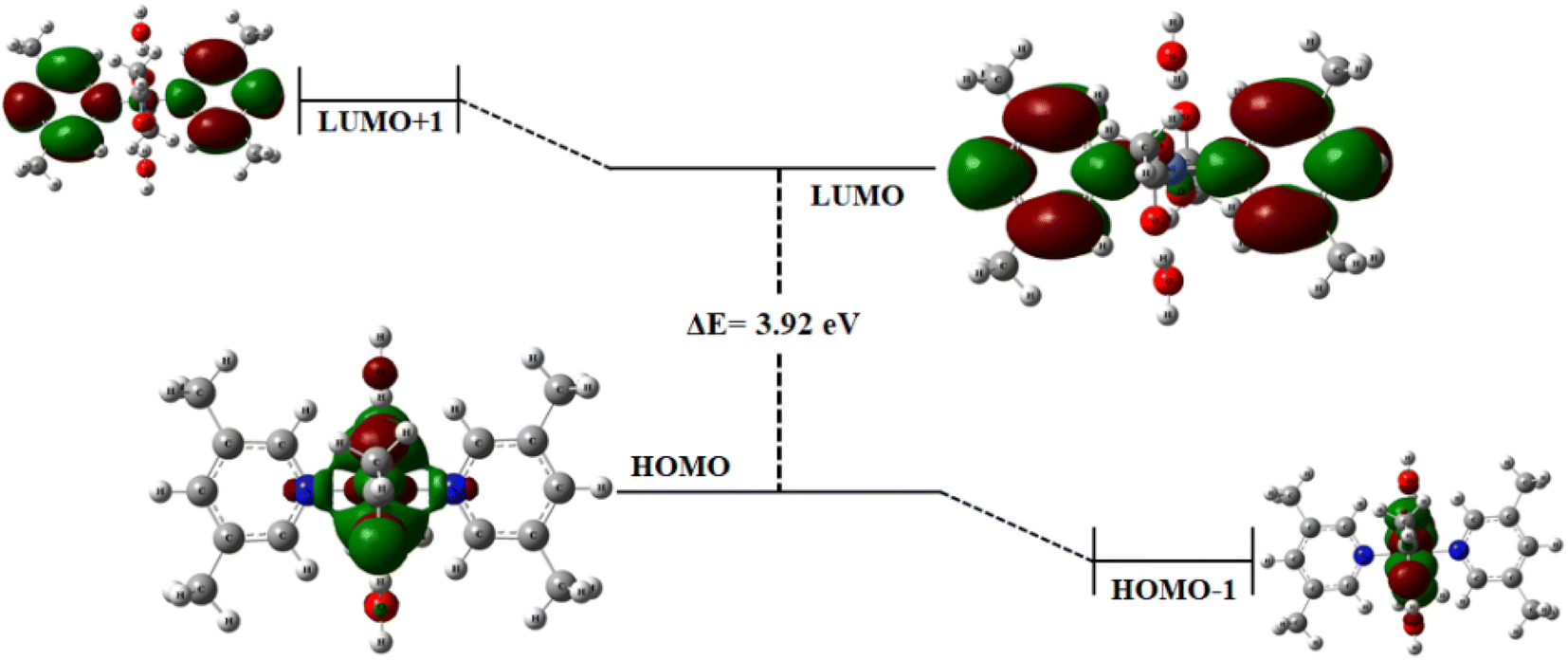
Frontier molecular orbitals. The optimized geometry of complex 1 is depicted in Fig. SI-2.† The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) along with HOMO−1 and LUMO+1 have been computed through DFT using GaussView 5.0 software. Generally, the electron donating orbital is regarded as HOMO and electron accepting is LUMO. These two orbitals are most interacting in the complex, and thus called frontier orbitals.45–48 The energy gaps were computed at LANL2DZ basis set. In complex 1, the HOMO and LUMO energies are −5.19 and −1.27 eV, respectively, leading to an energy gap of 3.92 eV (Fig. 3). Determining the energy gap is important because it displays the charge transfer behavior in a system,49 and the chemical reactivity and stability of the molecule as a whole depends largely on the energy gap. Larger the HOMO–LUMO energy gap, greater the stability and lower is the chemical reactivity of the molecule and vice versa.50,51 From the frontier molecular orbital (FMO) diagram, it has been observed that the electron density is densely accumulated around the metal ion and oxygen atoms in HOMO, while it is dispersed on and around the pyridine rings in LUMO. A slightly more gathered electronic density at the metal center is observed in HOMO−1. Thus, it is believed that the metal center is behaving as an active site for the incoming substrate to get adsorbed/attached. The subsequent steps for OER mechanism are expected to proceed from here.
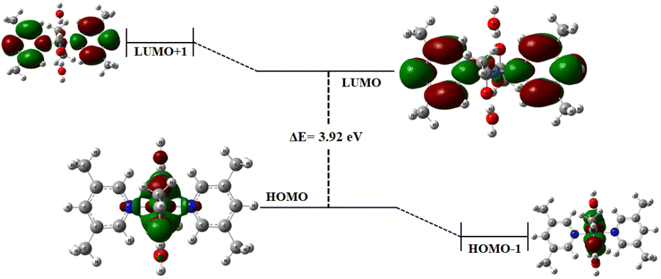 |
| | Fig. 3 Frontier molecular orbital diagram for complex 1 at iso-surface value = 0.02. | |
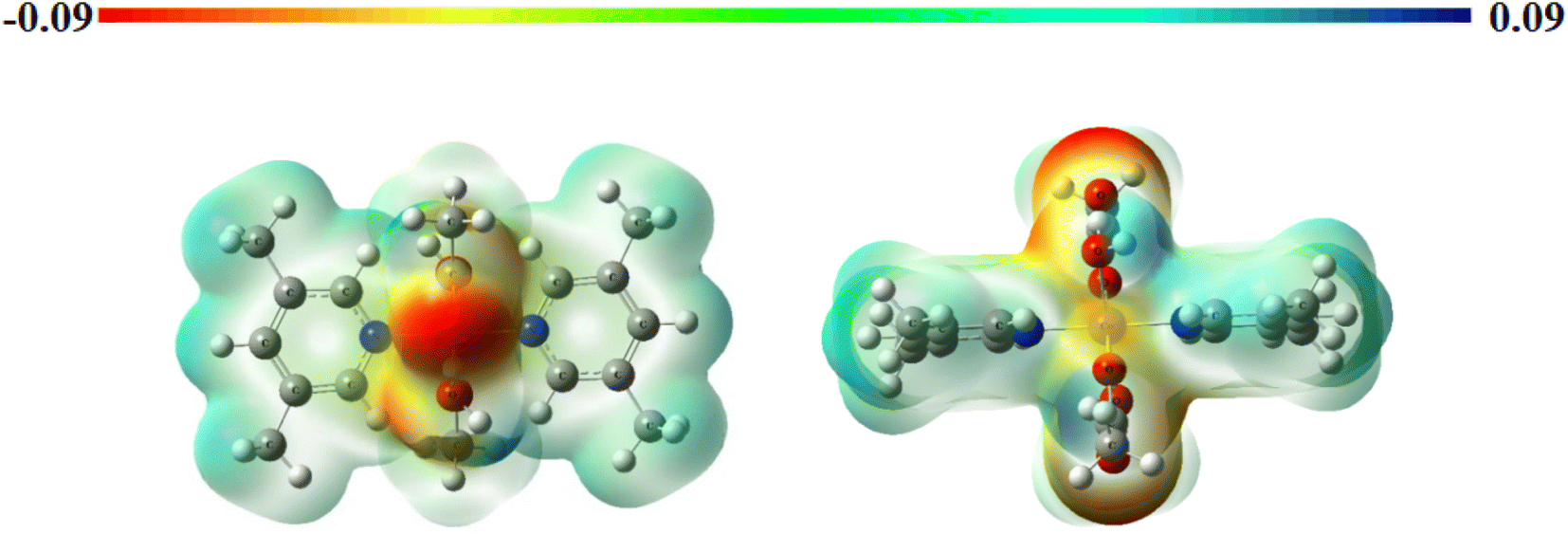
Molecular electrostatic potential surfaces. The molecular electrostatic potential (MEP) maps provide information about the relative polarity of a particular system. The MEP of complex 1 (Fig. 4) has been computed through DFT mode, using B3LYP level of theory and LANL2DZ as basis set. The scale having different colors here indicates varied electrostatic potentials within a molecular system.52 The order of potentials from low to high goes as red < orange < yellow < green < blue. On one end of the scale, the blue color suggests the presence of electropositive potential, while on the other end, the red color indicates the electronegative potentials, and the green color specifies zero potential. In this way, we can understand the relative polarity of a particular system.53,54 In the present case, we observe a deep red color at the oxygen centers of acetate ligand while a positive potential around the substituted pyridine ring. Lone pair of electrons is generally associated with a negative potential; thus, a reddish coloration is observed around the oxygen centers. In addition to this, the yellowish coloration around the metal center depicts the presence of electron density in this region. These observations can be well correlated with that of FMOs, as discussed above.
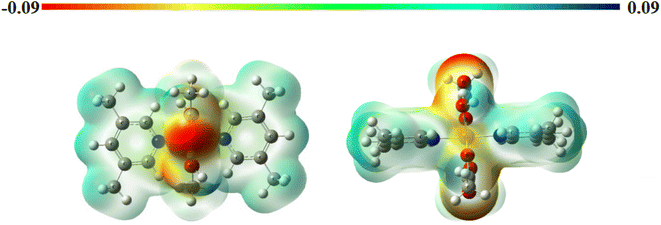 |
| | Fig. 4 Molecular electrostatic potential maps for complex 1 (iso-surface value = 0.002). | |
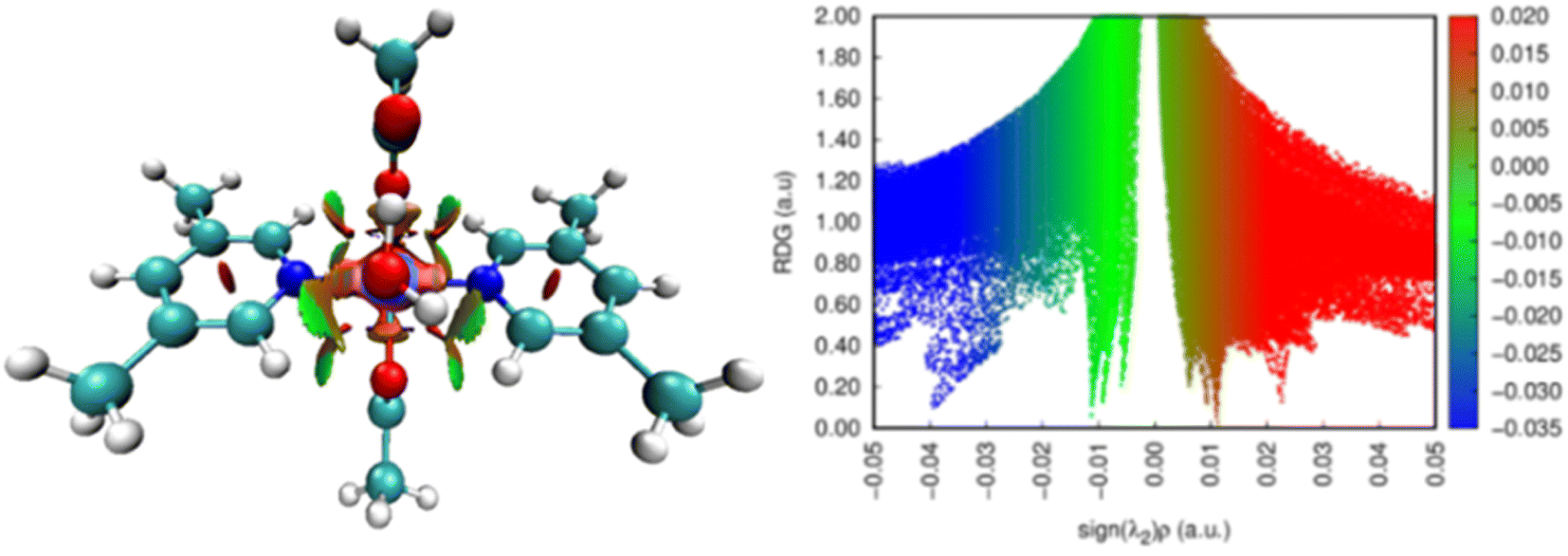
Non-covalent interactions. The reduced gradient of density (RDG) plots reveals information about weak, intramolecular, and intermolecular interactions.55,56 E. R. Johnson first introduced the RDG function, which is density and its first derivative together, representing the divergence from homogeneous electronic distribution.57 It is a dimensionless quantity. Based on the reduced density gradient, s and electron density, ρ, the NCI index plots are drawn as per eqn (6)| | |
s = 1/(2(3π2)1/3) × |∇ρ|/ρ4/3
| (6) |
The kind of interactions existing within a molecule is further understood by the Laplacian of density, ∇2ρ, which is the sum of three eigenvectors λ1, λ2 and λ3. In a certain system, λ3 differs along the internuclear direction, whereas the other two components (viz. λ1 and λ2) describe the variation of density in the plane normal to that of λ3 eigenvector. The value of eigenvector λ2 mainly suggests the type of interaction present in a system. For bonding interactions, such as H-bond, the value of λ2 < 0, while for non-bonding interactions λ2 is greater than zero. The value of λ2 is nearly zero for weak van der Waals interactions.57,58 The colors in RDG plot also suggest these interactions visually. For example, the blue color stands for H-bonds, green color suggests the presence of weak van der Waals interactions and the red color depicts the steric effect.54 Fig. 5 represents the non-covalent interactions in complex 1 and their two-dimensional RDG plots. In complex 1, the RDG plot displays the presence of H-bonds and steric repulsions co-existing within the complex.
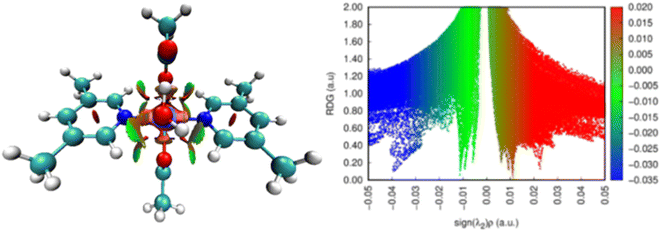 |
| | Fig. 5 Non-covalent interactions and RDG plots for complex 1 (iso-surface value = 0.5). | |
Oxygen evolution activity in alkaline electrolyte
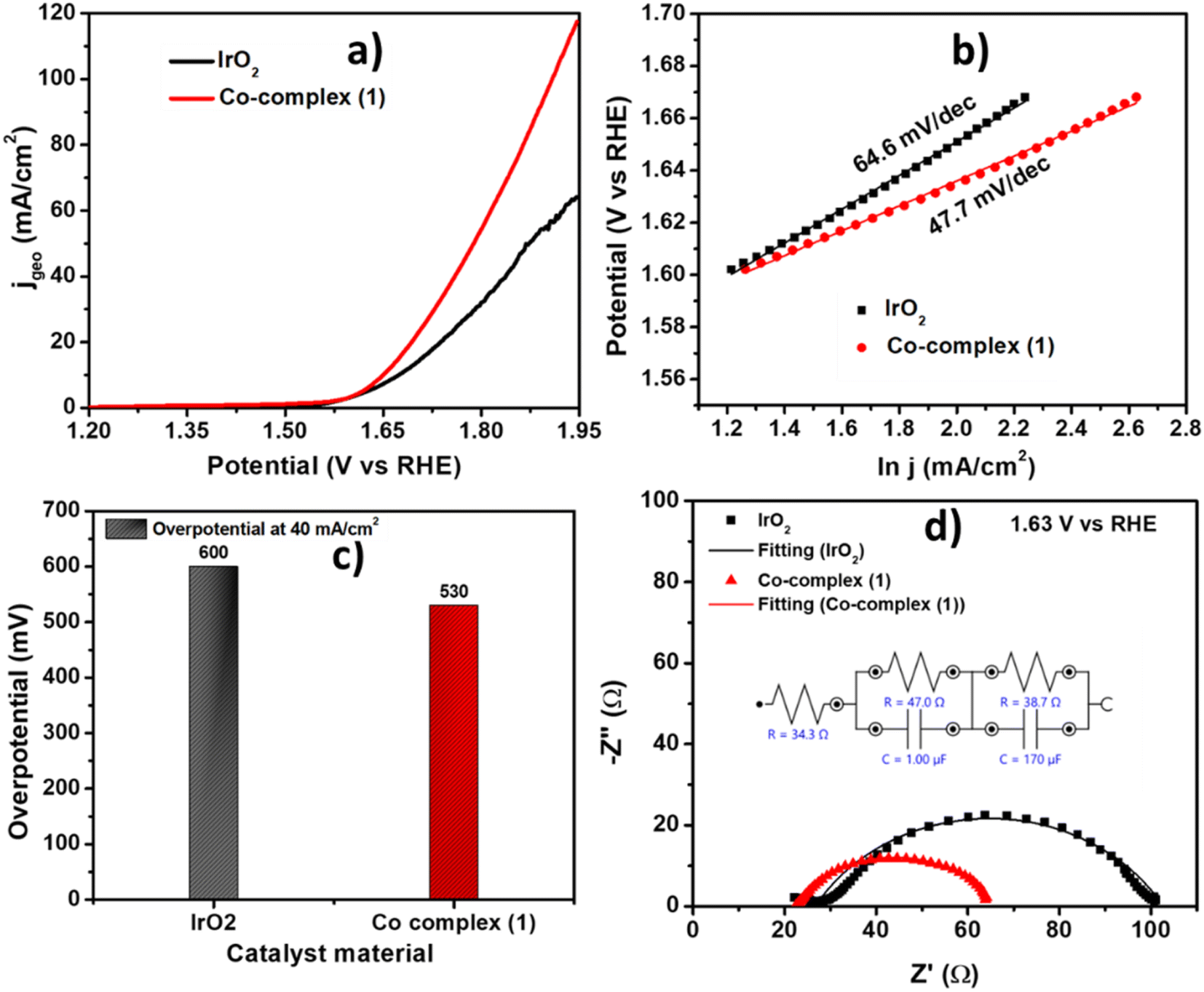
The electrocatalytic activity of the as prepared complex 1 was evaluated using Linear Sweep voltammetry (LSV) at scan rate 10 mV s−1 in 1 M KOH solution. The complex 1 showed onset potential of 1.50 V vs. RHE and high current density of 100 mA cm−2 at 1.87 V vs. RHE. The observed results towards OER electrocatalysis are even better than the state-of-the-art IrO2 catalyst which showed OER onset potential of 1.52 V i.e., 20 mV higher than complex 1. The comparative LSV is shown in Fig. 6a. The kinetics of the catalysis towards OER of the complex 1 was also compared to that of IrO2 catalyst using corresponding Tafel slope values. The commercial IrO2 exhibited a slope value of 64.6 mV dec−1, whereas the complex 1 showed 47.7 mV dec−1. Tafel slope indicating that our catalyst is showing facile kinetics towards OER, higher than commercial IrO2 catalyst (Fig. 6b). The complex 1 showed an overpotential of 530 mV to reach 40 mA cm−2 which is 70 mV lower than that of IrO2 (Fig. 6c). The comparison with similar materials in literature was also shown in Table 2 indicating the appreciable activity of our catalyst towards OER.34,36–38,59–61 The electrochemical impedance spectroscopy (EIS) was performed at a potential of 1.63 V vs. RHE to determine the charge transfer parameter for complex 1. The diameter of the Nyquist plot is related to the charge transfer resistance at the electrode–electrolyte interface. Nyquist plots shown in Fig. 6d, showed that the complex 1 has low charge transfer resistance of 43.5 Ω, also lower than IrO2 showing Rct of 63.8 Ω which is also consistent with the lower overpotentials required to reach higher current density from the LSV and Tafel analysis.
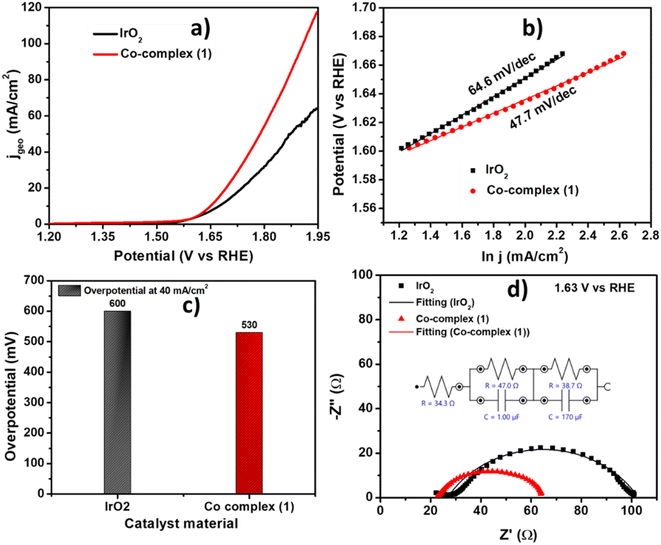 |
| | Fig. 6 (a) Comparison of LSV polarization curve for complex 1 and IrO2 at 10 mV s−1 in 1 M KOH, (b) the corresponding Tafel plots, (c) the overpotential comparison at 40 mA cm−2 and (d) the Nyquist plot comparison for complex 1 and IrO2. | |
Table 2 Comparison of the efficacy molecular cobalt electrocatalyst for OER
| Cobalt complexes |
Electrolyte |
Overpotential (mV)/current density (mA cm−2) |
Tafel slope (mV dec−1) |
TOF |
Ref. |
| Lmonoanionic = (E)-4-((2-hydroxynaphthalen-1-yl)diazenyl)benzenesulfonate), bipyalk = 2,2′-([2,2′-bipyridine]-6,6′-diyl)bis(propan-2-ol), CB[5] = cucurbit[5]uril, Ltrianionic = N′,N′′-(5-methyl-2-oxido-1,3-phenylene)bis(methan-1-yl-1-ylidene)bis(4-methoxybenzoylhydrazonate), Ldianionic = 4-chloro-1,2-bis[2-hydroxy-5-(phenylazo)benzylideneamino]benzene). |
| [(Lut)2Co(OAc)2(H2O)2] |
1 M KOH |
410 ± 20/10.0 |
45.7 |
0.05 s−1 |
This work |
| [Co(LN2O3)H2O] |
Sodium borate (pH = 11) |
500 |
— |
— |
34 |
| [(Lmonoanionic)2Co(CH3OH)4]a |
Phosphate buffer (pH = 9) |
520/0.5 |
— |
5 s−1 |
36 |
| [Co{(SePiPr2)2N}2 |
1 M KOH |
320/10.0 |
61.6 |
0.032 s−1 |
37 |
| [Co(bipyalk)(OAc)2]a |
Phosphate buffer (pH = 6) |
360/1.0 |
— |
1.5 s−1 |
38 |
| [Co@CB[5]]a/ITO |
Borate buffer (pH = 9.2) |
485/1.0 |
59.5 |
0.3 s−1 |
59 |
| [CoIICoIII(μ-OAc)(μ3-OH)(μ-Ltrianionic)]2a |
Sodium phosphate buffer of pH 7 |
768/1.0 |
320 |
1.1 × 10−3 h−1 |
60 |
| [CoLdianionic]a |
Buffer pH-11 |
360/10.0 |
135.9 |
— |
61 |
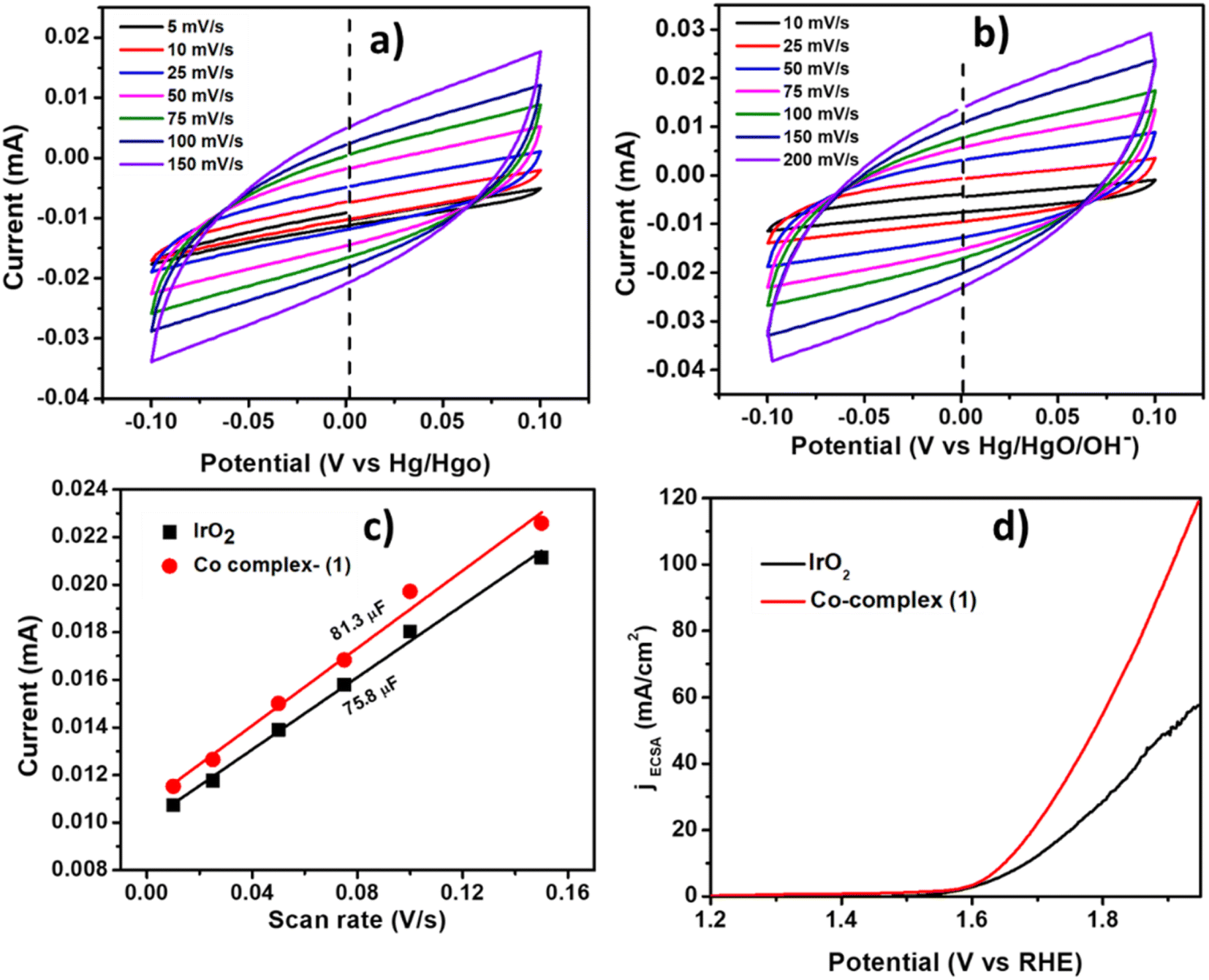
The Cdl was calculated by performing CV at different scan rates (Fig. 7a and b) and plotting the current density vs. scan rate (Fig. 7c) which was found to be 81.3 μF and 75.8 μF for complex 1 and IrO2 complex respectively. The current density normalized with ECSA was also shown in Fig. 7d, indicating the good electrocatalytic activity of Co-complex 1 in terms of high current density towards electrocatalytic OER. The summary of the OER electrochemical parameters is shown in Table 3.
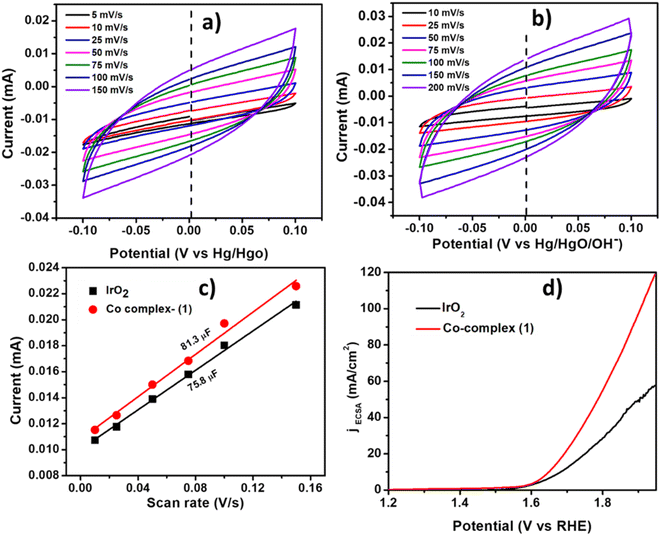 |
| | Fig. 7 The CVs (cyclic voltammograms) at different scan rates in the capacitive region for (a) IrO2 and (b) complex 1, (c) the plot of current vs. scan rates for Cdl calculation, (d) the comparison of LSV curves normalized by ECSA for complex 1 and IrO2. | |
Table 3 The comparison of activity parameters for OER for Co-complex (1) and IrO2
| Parameters |
Complex 1 |
IrO2 |
| Eonset (V vs. RHE) |
1.51 ± 0.01 |
1.52 |
| η10mA/cm2 (mV vs. RHE) |
410 ± 20 |
440 |
| Tafel slope (mV dec−1) |
45.7 |
64.6 |
| Mass activity (mA mg−1) @ 1.64 |
14 |
8.5 |
| Rct (Ω) |
43.5 |
63.8 |
| Cdl (μF) |
81.3 |
78.5 |
| TOF (1.73 V vs. RHE) |
0.05 |
0.02 |
| α |
0.59 |
0.56 |
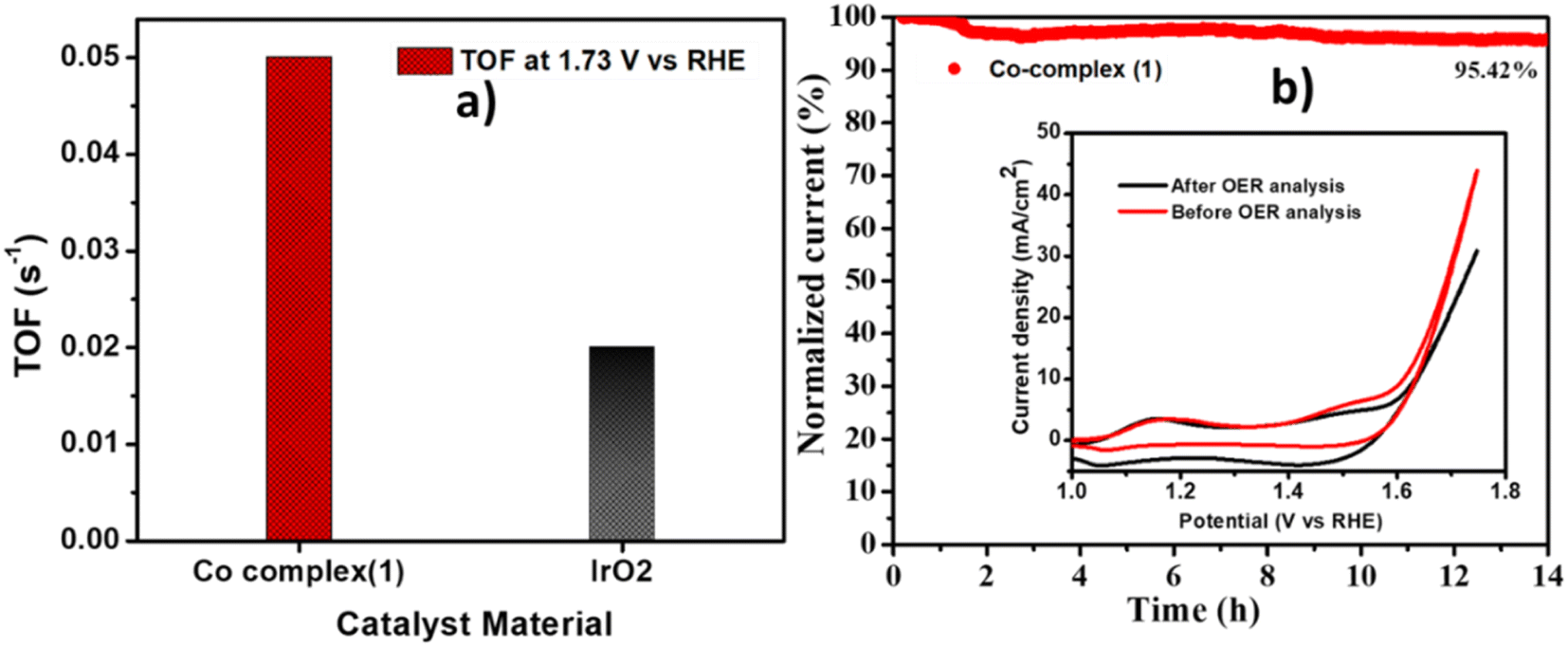
The turn over frequency, which indicates the intrinsic activity of the catalyst was calculated by using the equation given in the experimental section. The complex 1 showed the two times higher TOF (0.05 s−1) at 1.73 V as compared to IrO2 (0.02 s−1) (Fig. 8a). The long term stability of complex 1 was also checked using chronoamperometry method, by applying a constant potential of 1.65 V for 14 hours. It was observed (Fig. 8b) that the catalyst showed high stability with retention of 95% current. The CV curves before and after the OER stability are given in the inset of Fig. 8b, showing a slight decrement in the current density after 14 hours of stability, indicating robustness of the catalyst.
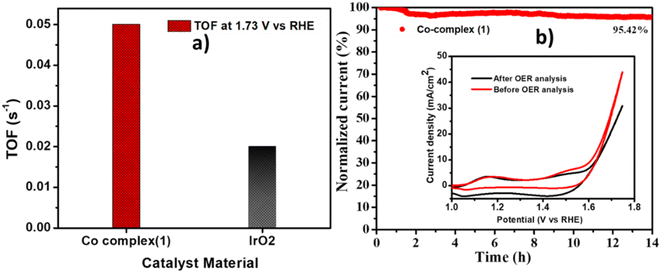 |
| | Fig. 8 (a) The comparison of TOF at 1.73 V vs. RHE for Co-complex (1) and IrO2, (b) chronoamperometry curve at 1.65 V vs. RHE for 14 hours for Co-complex (1). | |
The post catalytic characterisation of the spent electrodes (after OER analysis) were performed after the durability test on the Co-complex (1). The post catalytic FTIR (Fig. SI-3a in ESI†) shows the peak at 3431 cm−1 corresponding to the –OH vibration, 2921 cm−1 for Co–OH stretching. The peak present at 1633 cm−1 corresponds to Co–O.62,63 These peaks in the post OER FTIR suggests that the formation of Co(OH)2/CoOOH could act as the main active sites for the OER. However, along with these, some peaks matching with the initial spectra at 1038 and 1153 cm−1 indicate that the complex 1 is not totally degraded/decomposed after catalysis. The spectra suggests that the oxidation of Co(II) to Co(III) occurs at high oxidation potential and Co act as the catalytic centre for OER. The FESEM image after catalysis shows agglomeration of the particles (Fig. SI-3b in ESI†). The EDX spectra as shown in Fig. SI-4a in ESI†) indicates the presence of C, N, Co and O in the Co-complex (1). After catalysis (Fig. SI-4b in ESI†), the EDX of the sample displays an increase in the O content as well as presence of Co, C, N and K (due to KOH as electrolyte) indicating the presence of Co(OH)2 at the surface of the catalyst. The elemental mapping (Fig. SI-5 in ESI†) shows the uniform distribution of the elements on the surface before and after catalysis which indicates that the complex is not totally changed to hydroxide species. This suggests that formation of thin layer of Co(OH)2/CoOOH can be the reason for high activity of Co-complex (1) towards OER.
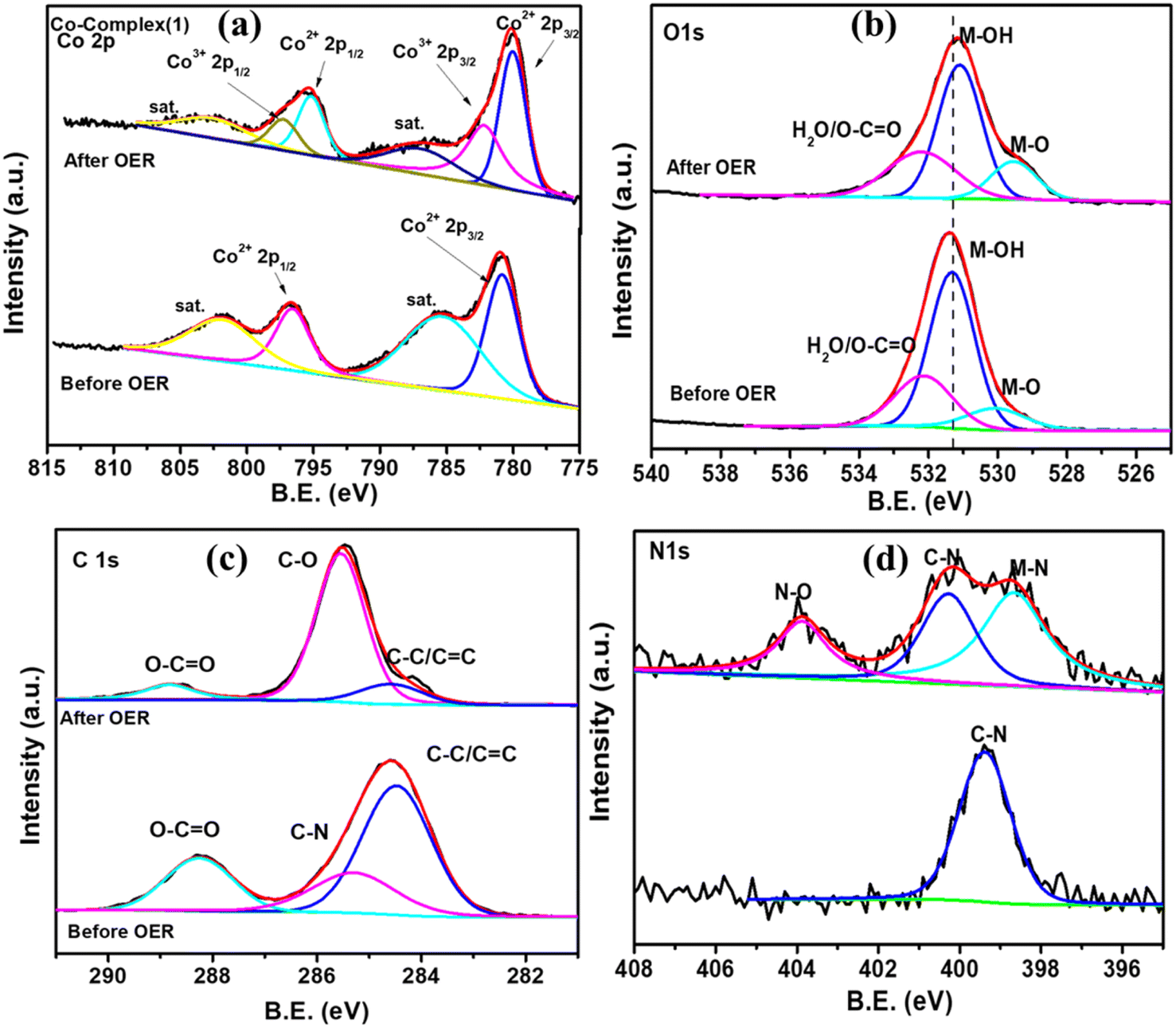
The X-ray photoelectron spectroscopy was performed to further understand the chemical composition and oxidation state in the pre and post catalytic OER samples as shown in Fig. 9. The Co 2p core level spectra of Co-complex (1) (Fig. 9a) shows two peak at 780.85 eV (Co2+ 2p3/2) and 796.49 eV (Co2+ 2p1/2) and two strong satellite peaks confirming the presence of Co2+ in the sample. After OER analysis, the slight shift of the Co2+ peak to 780.19 eV and the appearance of Co3+ peak at 782.18 eV is in accordance with the FTIR analysis, confirming that the formation of Co(OH)2 and CoOOH at the surface.64 The M–O and M–OH peak intensity increases in the post catalytic O 1s spectra (Fig. 9b) also showing the formation of Co(OH)2 at the surface. The C 1s XPS spectra shows the presence of C–C (284.6), C–N (285.4) and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.2) in the pristine sample. After catalysis, the relative composition of C–C decreases and C–O increases. This indicates the oxidation of C due to the oxidative environment provided by OER. Similarly the occurance of N–O peak in N 1s spectra is also due to oxidation. Thus, the post catalytic XPS, FTIR and EDX analysis confirmed the retention of the electrocatalyst in terms of bulk composition, and formation of Co2+ (Co(OH)2) and Co3+ (CoOOH) plays a crucial role during OER.
O (288.2) in the pristine sample. After catalysis, the relative composition of C–C decreases and C–O increases. This indicates the oxidation of C due to the oxidative environment provided by OER. Similarly the occurance of N–O peak in N 1s spectra is also due to oxidation. Thus, the post catalytic XPS, FTIR and EDX analysis confirmed the retention of the electrocatalyst in terms of bulk composition, and formation of Co2+ (Co(OH)2) and Co3+ (CoOOH) plays a crucial role during OER.
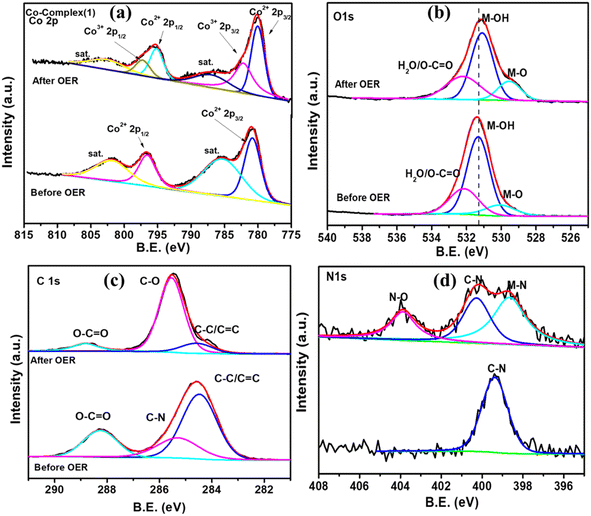 |
| | Fig. 9 XPS core level spectra comparison of (a) Co 2p, (b) O 1s, (c) C 1s and (d) N 1s before and after catalysis of Co-complex (1). | |
The high OER performance of the Co-complex (1) may have been resulted due to its highly porous connected network, high accessible Co active centres and high surface area. The interconnected network arising from the OH⋯O hydrogen bonding leads to a 3-D structure with hexagonal vacant pores which helped inenhancing mass transfer and ion transport at the electrode–electrolyte interface leading to high electrocatalytic performance of the Co-complex 1.
Conclusion
We have the novel lutidine coordinated cobalt(II) acetate complex [(3,5-lutidine)2Co(OAc)2(H2O)2] (1) by simplest condensation reaction. The structure of complex 1 was determined by single crystal X-ray diffraction data and found distorted octahedral geometry around the metal center. The non-covalent interaction found in complex 1 resulted in O–H⋯O hydrogen-bonding network that grown in a three-dimensional structure with various hexagonal vacant pore which could be useful for enhancing their catalytic activity. The comparative study of geometrical parameters obtained from the XRD data and DFT calculations was found in good agreement. The MEP maps analysis depicted the electron rich centers in the complex 1 are also found well in agreement with the solid-state structure. It was thought that the coordination flexibility and electron rich centers around metal center will also support in the enhancement of their catalytic activity. The Co-complex 1 showed excellent electrocatalytic OER activity better than state-of-the-art IrO2, which may be due to the 3D porous structure, rapid transport of ions and high accessible Co active sites, higher electron density, and coordination flexibility around metal center. Further, the post catalytic XPS, FTIR and EDX analysis also confirmed the retention of the electrocatalyst in terms of bulk composition, and formation of Co2+ (Co(OH)2) and Co3+ (CoOOH) OER.
Data availability
All data generated or analyzed during this study are included in this published article and its ESI.†.
Author contributions
DT: investigation, writing, PJ: electrochemical study and writing, DA: computational studies, Athul Sudheendranath and Sajesh P. Thomas: SCXRD, UK: supervision, writing, review & editing, PPI: supervision, writing, review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
The authors UK and PPI acknowledge the Science and Engineering Board (SERB) New Delhi, India for funding [TAR/2020/000232]. DT and PJ acknowledge University Grant Commission and IIT Delhi for research fellowship. Authors also acknowledge the Department of Chemistry and University Science Instrumentation Center, University of Delhi, and Department of Chemistry, IIT Delhi for instrumental facilities.
References
- T. Yao, X. An, H. Han, J. Q. Chen and C. Li, Adv. Energy Mater., 2018, 8, 1800210 CrossRef.
- D. Fa, J. Yuan, G. Feng, S. Lei and W. Hu, Angew. Chem., Int. Ed., 2023, 62, e202300532 CrossRef CAS PubMed.
- Y. Qiu, Z. Liu, X. Zhang, A. Sun and J. Liu, J. Colloid Interface Sci., 2022, 625, 50–58 CrossRef CAS PubMed.
- Y. Chen, M. Wang, S. Xiang, J. Liu, S. Feng, C. Wang, N. Zhang, T. Feng, M. Yang, K. Zhang and B. Yang, ACS Sustainable Chem. Eng., 2019, 7, 10912–10919 CrossRef CAS.
- T. Ghosh and G. Maayan, Angew. Chem., Int. Ed., 2019, 58, 2785–2790 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Duand and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- B. Deng, Y. Long, C. Yang, P. Du, R. Wang, K. Huang and H. Wu, Chem. Commun., 2021, 57, 7830–7833 RSC.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- Q. Liang, J. Chen, F. Wang and Y. Li, Coord. Chem. Rev., 2020, 424, 213488 CrossRef CAS.
- J. Yu, F. A. Garcés-Pineda, J. González-Cobos, M. Peña-Díaz, C. Rogero, S. Giménez, M. C. Spadaro, J. Arbiol, S. Barja and J. R. Galán-Mascarós, Nat. Commun., 2022, 13, 4341 CrossRef CAS PubMed.
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- W. Zhang, L. Cui and J. Liu, J. Alloys Compd., 2020, 821, 153542 CrossRef CAS.
- A. Badruzzaman, A. Yuda, A. Ashok and A. Kumar, Inorg. Chim. Acta, 2020, 511, 119854 CrossRef CAS.
- L. Reith, C. A. Triana, F. Pazoki, M. Amiri, M. Nyman and G. R. Patzke, J. Am. Chem. Soc., 2021, 143, 15022–15038 CrossRef CAS PubMed.
- B. Paul, P. Bhanja, S. Sharma, Y. Yamauchi, Z. A. Alothman, Z. L. Wang and R. B. A. Bhaumik, J. Colloid Interface Sci., 2021, 582, 322–332 CrossRef CAS PubMed.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef CAS.
- Y. Zhang, Q. Fu, B. Song and P. Xu, Acc. Mater. Res., 2022, 3, 1088–1100 CrossRef CAS.
- F. Lyu, Y. Bai, Q. Wang, L. Wang, X. Zhanga and Y. Yin, Dalton Trans., 2017, 46, 10545–10548 RSC.
- B. Zhang, J. Zhang, X. Tan, D. Tan, J. Shi, F. Zhang, L. Liu, Z. Su, B. Han, L. Zheng and J. Zhang, Chem. Commun., 2018, 54, 4045 RSC.
- S. Y. Chae, Y. J. Hwang, J. H. Choi and O. S. Joo, Electrochim. Acta, 2013, 114, 745–749 CrossRef CAS.
- S. B. Kale, A. C. Lokhande, R. B. Pujari and C. D. Lokhande, J. Colloid Interface Sci., 2018, 532, 491–499 CrossRef CAS PubMed.
- Y. Kang, Y. He, D. Pohl, B. Rellinghaus, D. Chen, M. Schmidt, V. Süβ, Q. Mu, F. Li, Q. Yang, H. Chen, Y. Ma, G. Auffermann and G. L. C. Felser, ACS Appl. Mater. Interfaces, 2022, 14, 19324–19331 CrossRef CAS PubMed.
- M. Liaoa, G. Zenga, T. Luoa, Z. Jina, Y. Wanga, X. Koua and D. Xiao, Electrochim. Acta, 2016, 194, 59–66 CrossRef.
- Q. S. Zhong, W. Y. Xia, B. C. Liu, C. W. Xu and N. Li, Int. J. Hydrogen Energy, 2019, 44, 10182–10189 CrossRef CAS.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- M. Zhu, Y. Zhou, Y. Sun, C. Zhu, L. Hu, J. Gao, H. Huang, Y. Liu and Z. Kang, Dalton Trans., 2018, 47, 5459–5464 RSC.
- P. Chen, K. Xu, Y. Tong, X. Li, S. Tao, Z. Fang, W. Chu, X. Wua and C. Wu, Inorg. Chem. Front., 2016, 3, 236–242 RSC.
- X. Peng, C. Pi, X. Zhang, S. Li, K. Huo and P. K. Chu, Sustainable Energy Fuels, 2019, 3, 366 RSC.
- D. K. Dogutan, R. McGuire Jr and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579 CrossRef CAS PubMed.
- S. Gonawala, H. Baydoun, L. Wickramasinghe and C. N. Verani, Chem. Commun., 2016, 52, 8440–8443 RSC.
- Q. Daniel, R. B. Ambre, B. Zhang, B. Philippe, H. Chen, F. Li, K. Fan, S. Ahmadi, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 1143–1149 CrossRef CAS.
- H. T. Shi, X. X. Li, F. H. Wu and W. B. Yu, Dalton Trans., 2017, 46, 16321–16326 RSC.
- I. M. Abdullahi, J. Masud, P. C. Ioannou, E. Ferentinos, P. Kyritsis and M. Nath, Molecules, 2021, 26, 945 CrossRef CAS PubMed.
- Y. F. Su, W. Z. Luo, W. Q. Lin, Y. B. Su, Z. J. Li, Y. J. Yuan, J. F. Li, G. H. Chen, Z. Li, Z. T. Yu and Z. Zou, Angew. Chem., Int. Ed., 2022, 61, e202201430 CAS.
- A. Valizadeh, R. Bikas, S. Nandy, T. Lis, K. H. Chae and M. M. Najafpour, Dalton Trans., 2022, 51, 220–230 RSC.
- U. Kumar, J. Thomas and N. Thirupathi, Inorg. Chem., 2010, 49, 62–72 CrossRef CAS PubMed.
- P. Jain, S. Jha and P. P. Ingole, Sustainable Energy Fuels, 2022, 6, 1094–1107 RSC.
- Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS PubMed.
- R. K. Tripathy, A. K. Samantaraand and J. N. Behera, Dalton Trans., 2019, 48, 10557–10564 RSC.
- S. C. Manna, S. Mistria and A. D. Jana, CrystEngComm, 2012, 14, 7415–7422 RSC.
- C. Quintana, G. Silva, A. H. Klahn, V. Artigas, M. Fuentealba, C. Biot, I. Halloum, L. Kremer, N. Novoa and R. Arancibia, Polyhedron, 2017, 134, 166–172 CrossRef CAS.
- C. Soriano-Correa, C. Barrientos-Salcedo, M. Francisco-Marquez and C. I. Sainz-Díaz, J. Mol. Graphics Modell., 2018, 81, 116–124 CrossRef CAS PubMed.
- N. Elangovan, R. Thomas, S. Sowrirajan, K. P. Manoj and A. Irfan, Polycyclic Aromat. Compd., 2021, 42, 6818–6835 CrossRef.
- S. M. Hiremath, A. Suvitha, N. R. Patil, C. S. Hiremath, S. S. Khemalapure, S. K. Pattanayak, V. S. Negalurmath and K. Obelannavar, J. Mol. Struct., 2018, 1171, 362–374 CrossRef CAS.
- K. Karrouchi, S. A. Brandán, Y. Sert, H. El-marzouqi, S. Radi, M. Ferbinteanu, M. E. A. Faouzi, Y. Garcia and M. Ansa, J. Mol. Struct., 2020, 1219, 128541 CrossRef CAS.
- M. Alizadeh, Z. Mirjafary and H. Saeidian, J. Mol. Struct., 2020, 1203, 127405 CrossRef CAS.
- N. Elangovan, R. Thomas and S. Sowrirajan, J. Mol. Struct., 2022, 1250, 131762 CrossRef CAS.
- M. Shahid, M. Salim, M. Khalid, M. N. Tahir, M. U. Khan and A. A. C. Braga, J. Mol. Struct., 2018, 1161, 66–75 CrossRef CAS.
- M. Thirunavukkarasu, G. Balaji, S. Muthu, B. R. Raajaraman and P. Ramesh, Chem. Data Collect., 2021, 31, 100622 CrossRef CAS.
- K. P. Manoj, N. Elangovan and S. Chandrasekar, Inorg. Chem. Commun., 2022, 139, 109324 CrossRef CAS.
- A. Prabakaran, V. Vijayakumar, N. Radhakrishnan, R. Chidambaram and S. Muthu, Polycycl. Aromat. Compd., 2020, 42, 925–941 CrossRef.
- J. M. del Campo, J. L. Gázquez, R. J. Alvarez-Mendez and A. Vela, Int. J. Quantum Chem., 2012, 112, 3594–3598 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. C. García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- F. Li, H. Yang, Q. Zhuo, D. Zhou, X. Wu, P. Zhang, Z. Yao and L. Sun, Angew. Chem., 2021, 133, 2004–2013 CrossRef.
- A. K. Srivastava, A. Mondal, S. Konar and S. Pal, Dalton Trans., 2022, 51, 4510–4521 RSC.
- Z. Shaghaghi, P. S. Kouhsangini and R. Mohammad-Rezaei, Appl. Organomet. Chem., 2021, 35, e6103 CAS.
- A. D. Jagadale, D. P. Dubal and C. D. Lokhande, Mater. Res. Bull., 2012, 47, 672–676 CrossRef CAS.
- M. Adeel, S. Parisi, M. Mauceri, K. Asif, M. Bartoletti, F. Puglisi, I. Caligiuri, M. M. Rahman, V. Canzonieri and F. Rizzolio, ACS Omega, 2021, 43, 28611–28619 CrossRef PubMed.
- P. Jain, S. Jha and P. P. Ingole, ACS Appl. Energy Mater., 2023, 6, 3278–3290 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data for the complex 1 CCDC 2204239. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04709a |
| ‡ Deepika Tanwar and Priya Jain contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Pravin P. Ingole
c,
Pravin P. Ingole