
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(thienyl)ethenes with α-methoxymethyl groups. Syntheses, spectroscopic Hammett plots, and stabilities in PMMA films†
Thea Weingartza,
Sven Nagornya,
Jörg Adams b,
André Eitzerothb,
Marvin Schewec,
Christian Rembec and
Andreas Schmidt*a
b,
André Eitzerothb,
Marvin Schewec,
Christian Rembec and
Andreas Schmidt*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de
bClausthal University of Technology, Institute of Physical Chemistry, Arnold-Sommerfeld-Straße 4, D-38678 Clausthal-Zellerfeld, Germany. E-mail: adams@pc.tu-clausthal.de
cClausthal University of Technology, Institute for Electrical Information Technology, Leibnizstraße 28, D-38678 Clausthal-Zellerfeld, Germany
First published on 29th August 2023
Abstract
A series of bis(thienyl)ethenes (BTEs) possessing perfluorocyclopentene backbones and methoxymethyl groups (MOM) in the 2/2′-positions of the thiophenes was prepared and examined. The substitution pattern of the 5/5′-positions was varied, covering the range from electron-donating to electron-withdrawing. The substituent effects of the absorption wavelengths of the ring-opened and the ring-closed isomers, which are interconverted by reversible 6π-electrocyclizations and cycloreversions, are studied by means of the spectroscopic Hammett equation and the Hammett–Brown equation. Excellent correlations of these linear free energy relationships were found, when the σp values of the Hammett equation, which summarize inductive, mesomeric and field effects, were replaced to the Hammett–Brown σp+ and σp− values which also take direct conjugation into account. We studied solvent effects on the spectroscopic properties and embedded the BTEs into polymethylmethacrylate (PMMA) coatings to examine their fatigue resistance. By our studies, the spectroscopic properties of BTEs can be adjusted by variation of the substitution pattern to a desired excitation wavelength for switching processes.
Introduction
The phenomenon of photochromism encompasses several reversible light-induced processes resulting in the formation of species which differ in their spectroscopic properties.1 Photochromism can be based on reversible photoisomerism, or on the reversible formation of persistent radicals which reconstitute non-radical covalent forms. Some examples are shown in Scheme 1. cis–trans Isomerism causes the photochromism of azobenzenes 1,2 and structural isomerism results in photochromism of spiropyrans 2,3 furylfulgides 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and bis(thienyl)ethenes 4.1 Whereas the former reaction is a 6-endo-trig ring-closure under neutralization of the partial charges of the push–pull-chromophor, the two latter mentioned reactions are formal 6π-electrocyclizations to form ring-closed isomers. Punicines 5 and their derivatives are photochromic, because disproportionations produce radical cations and radical anions from the starting mesomeric betaine.5 Among the examples shown, 1,2-bis(thienyl)ethenes (BTEs)6 possessing perfluorocyclopentene backbones such as 4 undoubtedly are the most important. Depending on their substitution pattern they are thermally sufficiently stable and fatigue resistant,7 and their response rate is considered fast enough for potential applications.1 However, applications of BTEs usually require structural adjustments to find a compromise between desired absorption maxima for the switching processes and the other aforementioned properties. It is known that the C2/C2′ atoms of the thiophene rings of switchable BTEs need distances of 3.5–4.2 Å.8 Through-space conjugation between these positions cause considerably shorter distances (3.28 Å),9 which are also influenced by several other parameters such as the solvent polarity,10 the constitution,11 and substituent effects.12 In order to use BTEs in films, absorbances, quantum yields, stabilities and photoactivities are the most important parameters, because some BTEs fail to undergo reversible photo-induced isomerisations when the solutions are too concentrated.13 High concentrations of the dyes in the film, however, are desired to achieve high absorption contrasts. The 2-positions of the thiophene wings which are involved in the electrocyclization need to be substituted (R′ ≠ H) to prevent irreversible oxidations under formation of indeno[5,4-b:6,7-b′]dithiophenes 6 which are also described in a patent.14 In addition, photochemical decompositions to form dithiacyclopenta[a]acenaphthylenes 715 or 5-vinyl-indeno[5,4-b]thiophenes 816 result in unfavourably low fatigue resistencies scheme 2.
4 and bis(thienyl)ethenes 4.1 Whereas the former reaction is a 6-endo-trig ring-closure under neutralization of the partial charges of the push–pull-chromophor, the two latter mentioned reactions are formal 6π-electrocyclizations to form ring-closed isomers. Punicines 5 and their derivatives are photochromic, because disproportionations produce radical cations and radical anions from the starting mesomeric betaine.5 Among the examples shown, 1,2-bis(thienyl)ethenes (BTEs)6 possessing perfluorocyclopentene backbones such as 4 undoubtedly are the most important. Depending on their substitution pattern they are thermally sufficiently stable and fatigue resistant,7 and their response rate is considered fast enough for potential applications.1 However, applications of BTEs usually require structural adjustments to find a compromise between desired absorption maxima for the switching processes and the other aforementioned properties. It is known that the C2/C2′ atoms of the thiophene rings of switchable BTEs need distances of 3.5–4.2 Å.8 Through-space conjugation between these positions cause considerably shorter distances (3.28 Å),9 which are also influenced by several other parameters such as the solvent polarity,10 the constitution,11 and substituent effects.12 In order to use BTEs in films, absorbances, quantum yields, stabilities and photoactivities are the most important parameters, because some BTEs fail to undergo reversible photo-induced isomerisations when the solutions are too concentrated.13 High concentrations of the dyes in the film, however, are desired to achieve high absorption contrasts. The 2-positions of the thiophene wings which are involved in the electrocyclization need to be substituted (R′ ≠ H) to prevent irreversible oxidations under formation of indeno[5,4-b:6,7-b′]dithiophenes 6 which are also described in a patent.14 In addition, photochemical decompositions to form dithiacyclopenta[a]acenaphthylenes 715 or 5-vinyl-indeno[5,4-b]thiophenes 816 result in unfavourably low fatigue resistencies scheme 2.
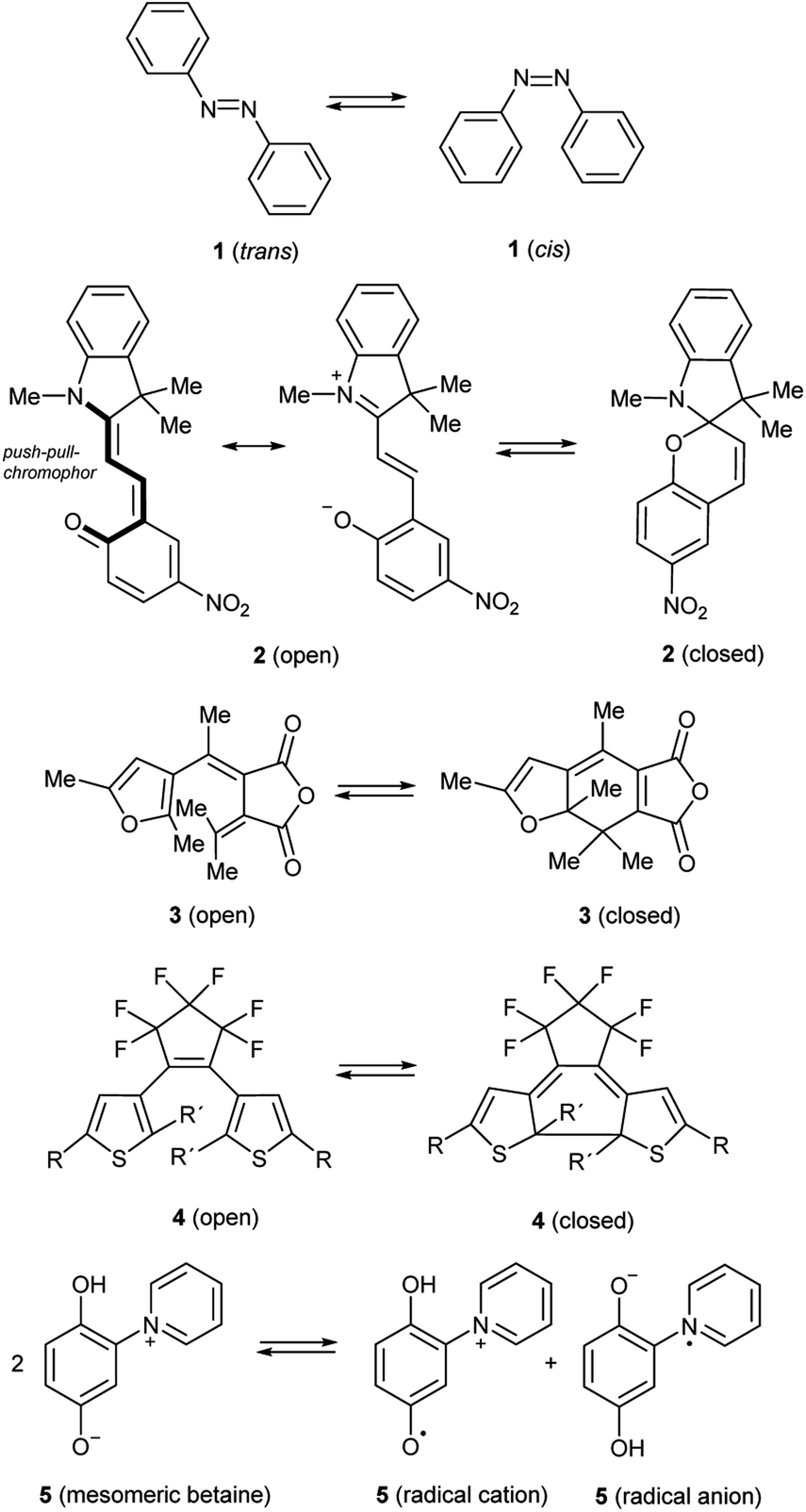 | ||
| Scheme 1 Examples of photochromic molecules. | ||
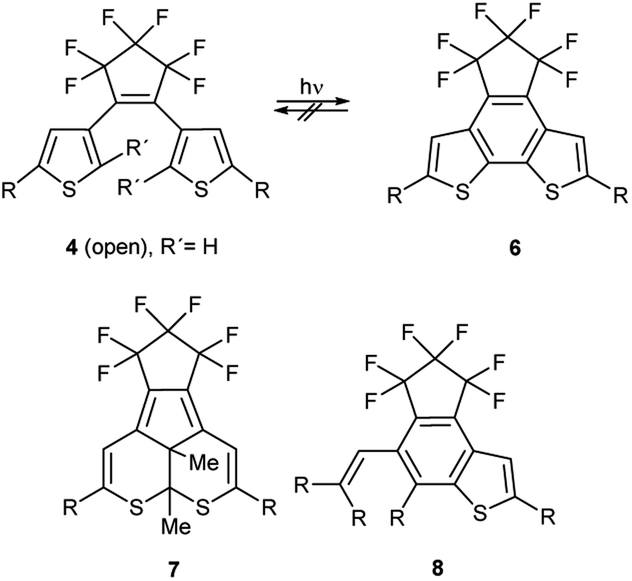 | ||
| Scheme 2 Decomposition products of BTEs. | ||
Only very few examples of BTEs which have other substituents than methyl groups in the 2-positions have been described to date.17 We report here on the syntheses of BTEs with 2-methoxymethyl groups and various substituents in the 5-position, covering the range from electron-donating to electron-withdrawing. We found a linear free enthalpy relationship between the Hammett–Brown substituent constants of the substituents on the absorption maxima of the ring-opened as well as the closed form, when a modified spectroscopic Hammett equation is applied. Furthermore, we studied the behavior of the BTEs in polymethylmethacrylate (PMMA) films and examined their photoresistencies.
Results and discussion
The series of BTEs was prepared starting from 4-bromothiophene-2-carbaldehyde 9 and 3-bromothiophene 14 (Scheme 3). Thiophene 14 was used as a starting material for the synthesis of the unsubstituted reference BTE 15a (X = H), and thiophene 9 served as starting material for the preparation of the BTEs 15b, 15c, 16, 22, and 23 with various substituents in their 5/5′-positions. First, 4-bromothiophene-2-carbaldehyde 9 was converted into the thiophene 10b [X = Me, method (A)] in very good yield by Wolf–Kizhner reduction of the aldehyde group with hydrazine, and into 10c [X = 1,3-dioxan-2-yl, method (B)] in almost quantitative yield by reaction with propanediol, respectively. Starting material 10b was mentioned in the literature, however, no experimental procedure was described.18 The formylation19 of 3-bromothiophene 14 as well as of 10b,c was performed under Vilsmeier–Haack conditions with lithium diisopropylamide and dimethylformamide [method (C)] to give 11a, 11b,20 and 11c in good to very good yields. Subsequent reduction to receive 12a–c was performed applying sodium borohydride in a mixture of water and THF [method (D)] in yields between 88% and 99%. The methoxy derivatives 13a–c were formed after treatment of 12a–c with sodium hydride and iodomethane in quantitative yields, respectively [method (E)]. We found that an exchange of the solvent methanol20 to THF reduced the reaction time considerably and increased the yields. The following reaction of 13a–c with octafluorocyclopentene gave the target BTEs 15a–c in low to moderate yields after bromide–lithium exchange with n-BuLi at −80 °C and substitution of fluorine of the substrate. Cleavage of the dioxane ring of 15c was accomplished by HCl and resulted in the formation of BTE 16 in 88% yield [method (G)]. Starting material 21 for the preparation of the target BTEs 22 and 23 was synthesized over several steps starting from 4-bromothiophene-2-carbaldehyde 9 which was first subjected to a reduction with sodium borohydride to give 17 according to modified literature procedures21 [method (D)], and then protected by a silyl group [method (H)]22 in quantitative yields, respectively. Performing the reaction under an inert atmosphere allowed a considerable shortened reaction time in comparison to the literature (1 h instead of 12 h22) and gave quantitative yields of thiophene 18. Formylation to form thiophene-aldehyde 19 [method (C)], followed by reduction to give 20 was followed by methylation to yield 21. The reaction of 21 with octafluorocyclopentene gave target BTE 22 in 50% yield. Because of the molecule's instability, the following reaction with TBAF [method (I)] was performed with a freshly prepared sample of 22 and gave BTE 23 in 30% yield.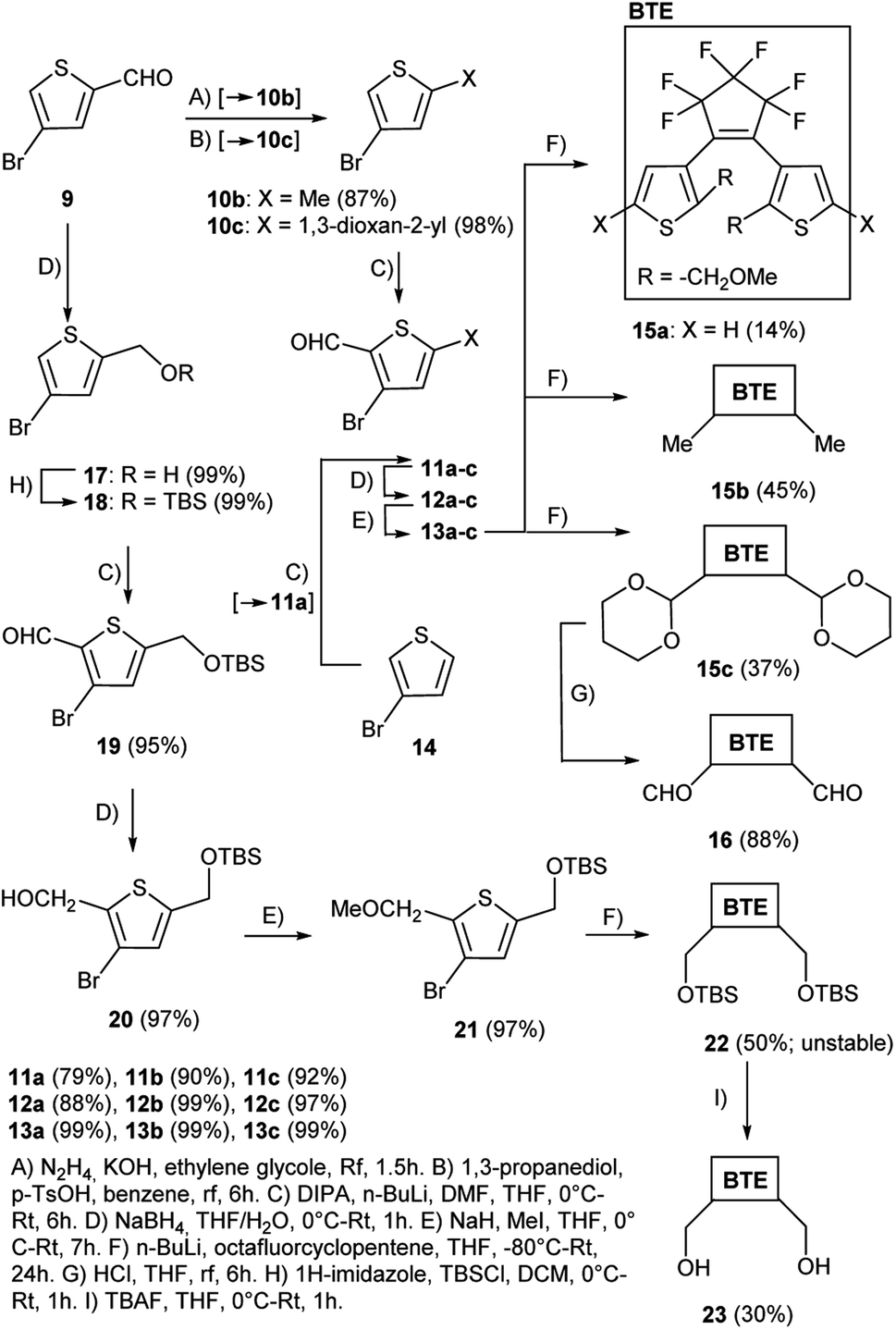 | ||
| Scheme 3 Synthesis of the target BTEs. | ||
The UV-vis absorption maxima of the 2-methoxymethyl-substituted BTEs described here measured in chloroform differ from those of the 2-methyl-substituted derivatives, as summarized in Table 1. All UV/vis absorption maxima of the open forms are slightly bathochromically shifted (Δλmax = −3.5 to −9.2 nm) in comparison to the methyl analogs. The absorptions of the closed forms differ by −8.6 nm to +8.1 nm. Changing the solvent from CHCl3 to THF causes shifts between −1.5 nm and −26.0 nm.
| BTE | 2/2′ = methoxymethyl | 2/2′ = methyl | ||
|---|---|---|---|---|
| λmax [nm] in THF | λmax [nm] in CHCl3 | λmax [nm] in CHCl3 | ||
| a n.d. = not determined | ||||
| 15a | Open | 231.0 | 243.0 | 233.8 |
| Closed | 511.0 | 519.0 | 514.0 | |
| 15b | Open | 214.0 | 242.0 | n.d.28 |
| Closed | 515.0 | 509.5 | 502.0 | |
| 15c | Open | 237.0 | 241.5 | 238.0 |
| Closed | 532.0 | 528.0 | 528.0 | |
| 16 | Open | 264.0 | 266.5 | 263.0 |
| Closed | 604.0 | 605.5 | 613.6 | |
| 22 | Open | Not stable | n.d. | n.d. |
| Closed | Not stable | n.d. | n.d. | |
| 23 | Open | 239.0 | 242.0 | 238.5 |
| Closed | 519.0 | 525.5 | 516.9 | |
In order to quantify the substituent effects in the 5-position on the spectroscopic properties, we performed Hammett correlations by means of the spectroscopic Hammett eqn 1, where R is the gas constant, T is the temperature, ρA is the absorption constant, and ET,R and ET,H are excitation energies of the substituted molecule and of its corresponding unsubstituted reference compound, respectively.23 The constant σ is characteristic of a substituent,24,25 and sum up the total electric effects such as inductive, mesomeric and field effects. These values were previously defined as substituents attached to substituted benzoic acids XC6H4COOH in para-position (σp) or meta-position (σm), the ionization of which was measured in water at 25 °C. The σ value is positive, when substituents exert electron-withdrawing properties on the ring, while negative values are indicative of electron-donating properties.25
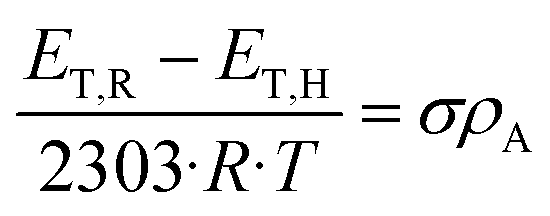 | (1) |
The energies are calculated as follows from the spectra:
| ET = h·c·ṽmax·NA | (2) |
Modified σ-values, σ+ and σ− values, have been introduced by H. C. Brown26 for substituents which can enter into direct resonance interaction with the reaction site in the transition state. They were determined as rate constants of the nucleophilic substitution of cumyl chlorides in 90% acetone/water (σp+) and from the deprotonation of substituted phenols as reference reaction (σp−). The UV/vis-spectra of the BTEs described here were measured in tetrahydrofurane under identical conditions (c = 0.05 mg mL−1) at rt and were used for the calculations of the excitation energies according to eqn 2 and Hammett as well as Hammett–Brown correlations according to eqn 1. Table 2 summarizes the results and the following Figures show the molar excitation energies plotted against the σp+ and σp− values of the different substituents. The value of the 1,3-dioxane-2-yl group was determined by us by application of the spectroscopic Hammett–Brown equation, as there is no constant available from the literature for this substituent.27 As it can be seen from the UV/vis-spectra, the electron-withdrawing groups, e.g. –CHO, lead to a bathochromic shift and the electron-donating groups lead to a hypsochromic shift. This results in a negative absorption constant and a negative slope of the correlations. Hammett plots employing the original σ values gave correlations which were rated with “satisfactory” according to a rating system by Jaffé,24c as a coefficient of determination of 0.9689 was found for the closed forms (Fig. S1 and S2, ESI†). Using σp+ values also resulted in satisfactory coefficients of determination of 0.9663 (Fig. 1). Employing Brown's σp− values, however, gave an excellent correlation according to the aforementioned classification, as a coefficient of determination of 0.9989 was calculated (Fig. 2).
| BTE | substituent | λmax [nm] | σp | σp+ | σp− | Exc. energy [kJ mol−1] |
|---|---|---|---|---|---|---|
| 15a | H | 243.0 | 0.00 | 0.00 | 0.00 | 492.29 |
| 519.0 | 230.49 | |||||
| 15b | Me | 242.0 | −0.17 | −0.31 | −0.17 | 494.32 |
| 509.5 | 234.79 | |||||
| 15c | 1,3-Dioxan-2-yl | 241.5 | 0.011 | 0.006 | 0.132 | 495.35 |
| 528.0 | 226.57 | |||||
| 16 | CHO | 266.5 | 0.42 | 0.73 | 1.03 | 448.88 |
| 605.5 | 197.57 | |||||
| 23 | CH2OH | 242.0 | 0.00 | −0.04 | 0.08 | 494.32 |
| 525.5 | 227.64 |
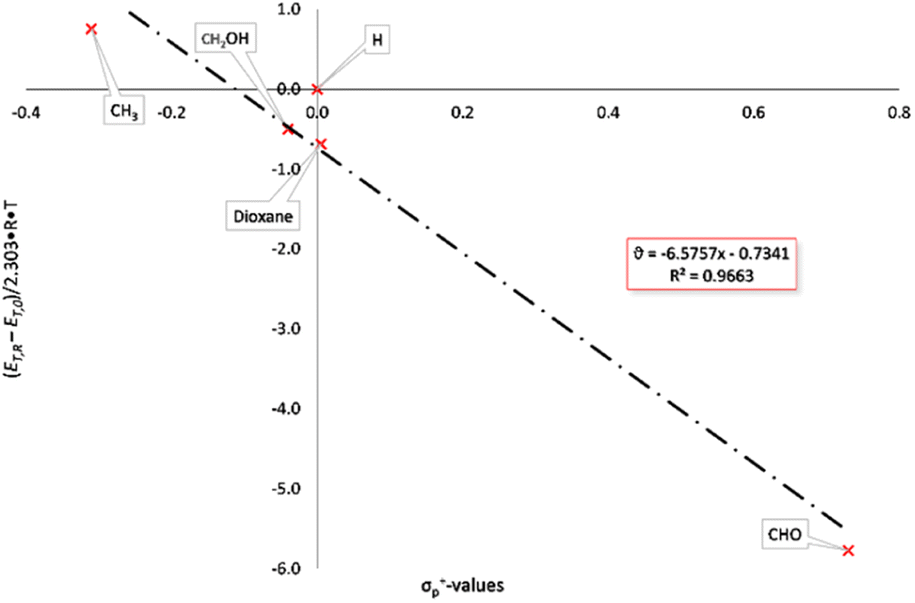 | ||
| Fig. 1 Hammett–Brown correlation of the BTEs (closed form) applying σp+ values. | ||
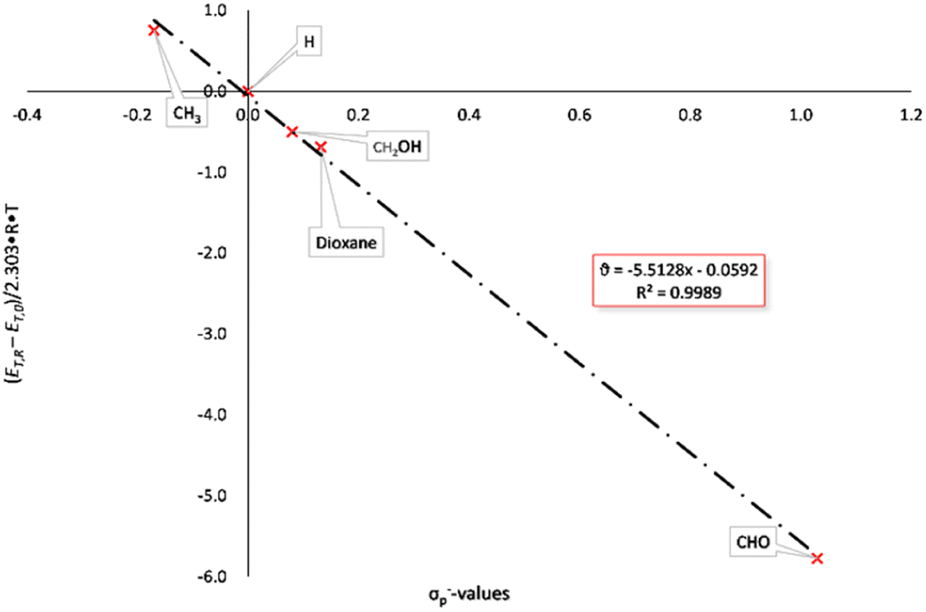 | ||
| Fig. 2 Hammett–Brown correlation of BTEs (closed form) with σp− values. | ||
As expected, changing the solvent from chloroform (Table 2) to THF (Table 3) leads to different values of the absorption maxima of the open as well as of the closed state, and therefore also affects the excitation energies. Thus, higher excitation energies are necessary in THF in comparison to chloroform to switch the molecule into the closed form. The choice of solvent also influences the quality of the correlation coefficients and coefficients of determination of the Hammett and Hammett–Brown plots. Table 4 summarizes values achieved in THF, dichloromethane, chloroform, and methanol. These solvents not only differ in their solvent polarities, as expressed in their ENT values (CH2Cl2: 0.309; CHCl3: 0.259; MeOH: 0.762; THF: 0.207),23a but also in their donicities and hydrogen bond donating properties. In contrast to DCM and chloroform, THF is an electron-pair-donating solvent (EPN solvent) which has a considerable donicity (DNN = 0.52).23a Apart from the strongest polarity within this series of solvents we applied, methanol is a strong hydrogen-bond donating solvent. All Hammett and Hammett–Brown correlations of open and closed forms in the different solvents (THF, CHCl3, CH2Cl2, MeOH) are shown in the ESI (Fig. S3–S24†). The correlations can be classified as “fair fit” when R is >0.90, “satisfactory” for R > 0.95 and “excellent” whenever R is >0.99).24c Table 4 summarizes correlation coefficients R and coefficients of determination R2 for all measurements. As can be seen from Table 4, best correlation coefficients and coefficients of determination were achieved with chloroform as solvent.
| BTE | Substituent | λmax [nm] | σp | σp+ | σp− | Exc. energy [kJ mol−1] |
|---|---|---|---|---|---|---|
| 15a | H | 231.8 | 0.00 | 0.00 | 0.00 | 516.08 |
| 510.2 | 234.47 | |||||
| 15b | Me | 236.1 | −0.17 | −0.31 | −0.17 | 506.68 |
| 511.2 | 243.01 | |||||
| 15c | 1,3-Dioxan-2-yl | 235.0 | 0.011 | 0.006 | 0.132 | 509.05 |
| 532.0 | 224.86 | |||||
| 16 | CHO | 264.4 | 0.42 | 0.73 | 1.03 | 452.10 |
| 600.6 | 199.18 | |||||
| 23 | CH2OH | 237.8 | 0.00 | −0.04 | 0.08 | 503.06 |
| 517.0 | 231.39 |
| σp | Solvent | |||
|---|---|---|---|---|
| THF | CHCl3 | DCM | MeOH | |
| σp Open | R = 0.9073 | R = 0.9392 | R = 0.9226 | R = 0.9170 |
| R2 = 0.8232 | R2 = 0.8821 | R2 = 0.8511 | R2 = 0.8409 | |
| σp Closed | R = 0.9494 | R = 0.9843 | R = 0.9540 | R = 0.9579 |
| R2 = 0.9013 | R2 = 0.9689 | R2 = 0.9102 | R2 = 0.9175 | |
| σp+ Open | R = 0.9039 | R = 0.9429 | R = 0.9126 | R = 0.9081 |
| R2 = 0.8171 | R2 = 0.8890 | R2 = 0.8328 | R2 = 0.8246 | |
| σp+ Closed | R = 0.9496 | R = 0.9830 | R = 0.9561 | R = 0.9606 |
| R2 = 0.9018 | R2 = 0.9663 | R2 = 0.9141 | R2 = 0.9228 | |
| σp− Open | R = 0.9546 | R = 0.9647 | R = 0.9581 | R = 0.9513 |
| R2 = 0.9112 | R2 = 0.9306 | R2 = 0.9179 | R2 = 0.9050 | |
| σp− Closed | R = 0.9836 | R = 0.9994 | R = 0.9857 | R = 0.9866 |
| R2 = 0.9674 | R2 = 0.9989 | R2 = 0.9716 | R2 = 0.9734 | |
To understand the effect of the methoxymethyl groups we compared the spectrocopic data with those of the reference compound shown in Scheme 4, which was prepared as described earlier.27 Table 5 shows the quantum yields and the molar absorption coefficients. Comparing 15a and the reference BTE reveals that the MOM group increases the molar absorption coefficient εA of the ring-opened form, whereas the quantum yields φA,B decrease slightly. Best quantum yields φA,B were achieved with molecule 15c containing the 1,3-dioxan-2-yl rings and 23 possessing –CH2OH substituents in the 5/5′-positions, and the best absorption coefficient with the dialdehyde BTE 16. Interestingly, BTE 16 reaches a value of 49.4% quantum yield in the transferring process to the closed state, despite needing the lowest excitation energy compared to the other molecules.
 | ||
| Scheme 4 Reference BTE. | ||
| BTE | λmax,UV [nm] | φA,B [%] | εA/103 | λmax,Vis [nm] | φB,A | εB/103 |
|---|---|---|---|---|---|---|
| a These spectra were measured in THF with a concentration of 0.025 g mol−1 ε/103 is given in [L mol−1 cm−1]. | ||||||
| 15a | 231.8 | 56.7 | 15.11 | 510.2 | 0.1004 | 2.30 |
| 15b | 236.1 | 50.9 | 19.78 | 511.2 | 0.0596 | 3.72 |
| 15c | 235.0 | 66.4 | 18.52 | 532.0 | 0.0720 | 2.99 |
| 16 | 264.4 | 49.4 | 27.48 | 600.6 | 0.0129 | 3.13 |
| 23 | 237.8 | 61.8 | 16.69 | 517.0 | 0.0736 | 4.31 |
| Ref. | 233.1 | 55.3 | 11.86 | 512.4 | 0.2362 | 3.29 |
As expected, the signals of the thiophenes in the 1H NMR spectrum were shifted to smaller resonance frequencies δ, when the sample was irradiated (Table 6). Even after irradiation of the BTEs over a period of 8 h, it was not possible to convert them completely into their closed forms. As there were still signals of the open form in all cases, it was only possible to determine the conversion rate using the same irradiation time. As already mentioned, BTE 16 had the highest conversion during this experiment. The BTEs 15b and 15c reached the same conversion rate of 38%, whereas the BTEs 15a and 23 have lower conversion rates of 21% and 28%.
| BTE | δarom (open) [ppm] | δarom (closed) [ppm] | Conversion [%] |
|---|---|---|---|
| a The NMR-spectra were measured in CDCl3 and the samples were irradiated at 254 nm for 2 h with a mercury lamp. | |||
| 15a | 7.38 (d) | 7.04 (d) | 21 |
| 7.12 (d) | 6.32 (d) | ||
| 15b | 6.77 (s) | 6.06 (s) | 38 |
| 15c | 7.11 (s) | 6.37 (s) | 38 |
| 16 | 7.73 (s) | 7.01 (s) | 45 |
| 23 | 6.98 (s) | 6.29 (s) | 28 |
In the next step we determined the effect of a polymethyl methacrylate (PMMA) matrix on the spectroscopic properties of the embedded BTEs and on their stabilities. PMMA is a preferred polymer to form matrices because of its high transparency and photostability.29 The films were formed on a quartz surface by spin coating. The rotation speed was varied to adjust the thickness of the film, and UV/vis measurements confirmed the correlation between the film thickness and their absorptions. The thickness of the films was determined by confocal laser scanning microscopy (CLSM), which scanned an artificial scratch on the surface of the film by a laser to determine the distance between film surface and quartz surface. The software uses a method, where two points are set across the scratch with an average of 100 lines to determine the film thickness. For the films produced at 2500 rpm, the CLSM method gave a thickness of 400 nm, whereas a higher rotation speed of 3000 and 3500 rpm gave thinner films. The results of the stability tests in PMMA matrices of selected BTEs are shown below. The corresponding test setup is described in the experimental section. Fig. 3 shows a cyclization of BTE 15c over 99 cycles. We used a yellow laser with 565 nm for the measurement and a reset beam, and a UV laser with 365 nm to induce the 6π-electrocyclization. For this kind of measurement a mixture of 3 mg of the corresponding BTE and 9 mg of solid PMMA was dissolved in 44.5 μL anisol which corresponds to a solid concentration of 24%.
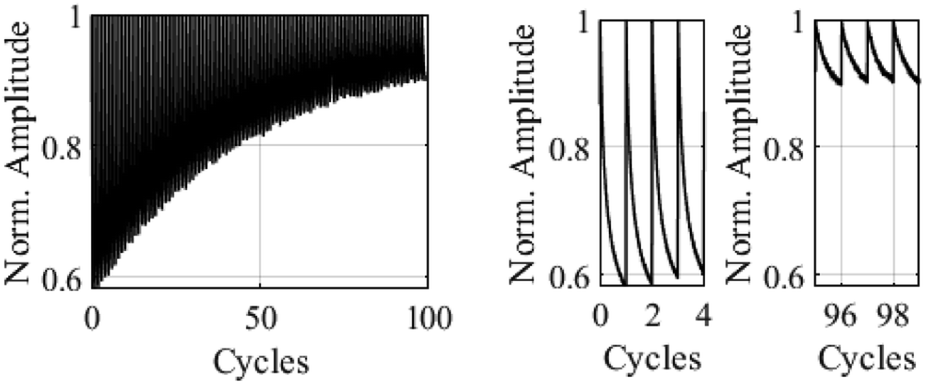 | ||
| Fig. 3 Measured light power normalized to 1 as a function of the transmitted laser power over 99 cycles for BTE 15c. One cycle is 3 s. Full plot (left) and two sections of 4 cycles at the start and at the end (right). Data is sampled with 5000 Hz and averaged over 10 samples. | ||
The sections on the right in Fig. 3 show that the measured yellow laser power returns to a similar value at the start of each cycle. As the cycle progresses, the thin film gradually switches to the closed form and absorbs more yellow light, leading to a gradual decrease in the measured light power. However, after multiple cycles, an irreversible degradation of the dye molecules was observed, resulting in increasingly smaller changes in the measured light power.
Fig. 4 displays the absorptions of the examined BTEs, which can be determined from the negative decadic logarithm of the measured normalized light powers.
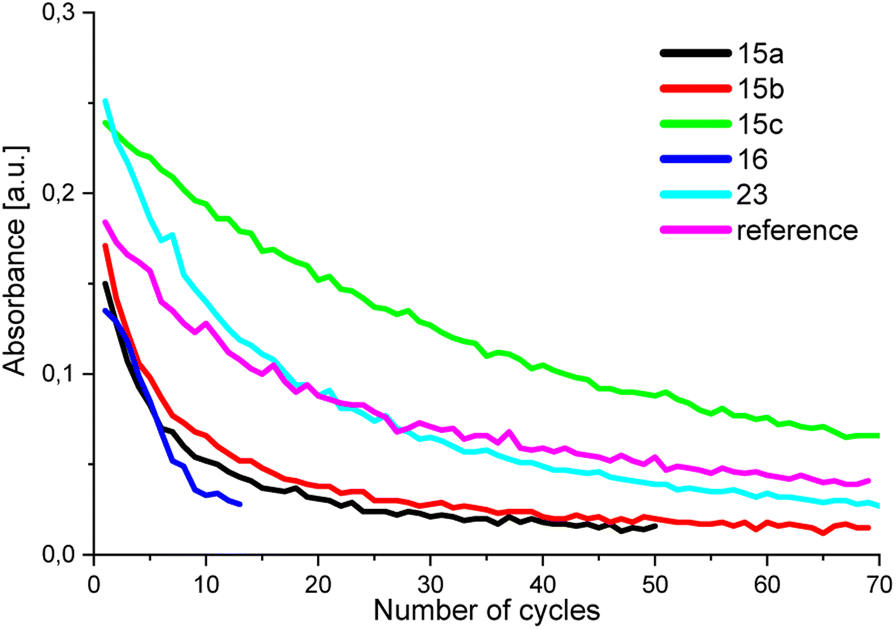 | ||
| Fig. 4 Absorption of the BTEs in the closed form. The curves show a clear reduction in absorption due to degradation processes. | ||
After around 35 cycles, the absorption of BTE 15c has degraded by approximately 50%, while BTE 15a, BTE 15b, and BTE 23 take about 9, 10, and 15 cycles, respectively, to reach the same level of degradation. The absorption of the reference has degraded by 50% after around 22 cycles. As expected, BTE 16 shows a less stable behaviour as it degrades faster than the other BTEs and reaches 50% at already 6 cycles. Due to the electron-withdrawing effects of the aldehyde functions the system of BTE 16 becomes less stable. The comparison of BTE 15a and the reference molecule shows that the methyl group of the reference has a stabilizing effect on the molecule system, as it degrades less fast. To increase the stability of this BTE system, different substituents can be introduced. As shown in Fig. 4, BTE 15c with 1,3-dioxanyl substituents in 5-position reaches a degradation of 50% after approximately 35 cycles. This leads to a short conclusion that the 1,3-dioxan-2-yl substituents have a stabilizing effect on the molecule in comparison to the BTEs 15a and BTE 23 containing a –CH2OH group is also more stable than the BTEs with only H and Me as substituents. It can be shown that the molecules are able to be stabilized by introducing electron-donating substituents.
In summary we present first 2/2′-MOM-substituted bis(thienyl)ethenes and linear free enthalpy relationships according the spectroscopic Hammett equation modified by Brown's substituent constants σ+ and σ−, which take direct conjugation into account. We examined the stabilities of these bis(thienyl)ethenes and found that the BTE with 1,3-dioxan-2-yl substituents in the 5/5′-positions and 2/2′-MOM groups is the most stable of the series in PMMA films.
Experimental section
General
All reactions were carried out under an atmosphere of nitrogen in oven-dried glassware. All chemicals were purchased and used without further purification unless otherwise mentioned. Anhydrous solvents were dried according to standard procedures before usage. Melting points were determined with a PerkinElmer DSC6. The ATR-IR spectra were obtained on a Bruker Alpha in the range of 400 to 4000 cm−1. 1H NMR spectra were recorded at 400 MHz or 600 MHz with a Bruker Avance/Avance III. 13C NMR spectra were recorded at 100 MHz or 150 MHz, with the solvent peak used as the internal reference. Multiplicities are described by using the following abbreviations: s = singlet, d = doublet, t = triplet, and m = multiplet. Signal orientations in DEPT experiments were described as follows: o = no signal; + = up (CH, CH3); − = down (CH2). The electrospray ionization mass spectra (ESI-MS) were measured with a Bruker Impact-II mass spectrometer. Samples were sprayed from MeCN. Chromatography: The reactions were traced by thin layer chromatography with silica gel 60 (F254, MERCK KGAA). For the detection of substances, quenching was used at either 254 nm or 366 nm with a mercury lamp. The preparative column chromatography was conducted through silica gel 60 (230–400 mesh).The films were produced on a quartz substrate with a size of 10 × 10 mm (1 mm thickness, PLANO). For the photostability measurements we used sapphire substrates (10.0 mm Ø, 1 mm thickness). For building up a matrix we used poly(methyl methacyrylate) (PMMA, average Mw 120000 by GPC, SIGMA-ALDRICH), of which were dissolved 1000 mg in 10 mL of tetrahydrofurane (THF, 99.9%, anhydrous, inhibitor-free). The BTE (10 mg) was dissolved in 1 mL of THF (anhydrous, inhibitor-free, Sigma Aldrich). After evaporating of the THF, the different BTEs and PMMA were dissolved in anisole (99%, SIGMA-ALDRICH) within 1–7 days and these solutions were used for the coating of the quartz or sapphire substrate. As standard conditions we used 12% concentration of the solid, whereas the utilized ratio of BTE and PMMA was 50:50. For the photostability measurements we used a 24% solid concentration and a ratio of BTE and PMMA of 25:75. For producing the coatings a Spin-coater (LAURELL WS-650 MZ-23MPP) was used. The produced coatings were investigated by using CLSM (Keyence VK-X210), for the scratch was used a canula (Sterican®, 0.40 × 20 mm). For taking pictures and determining the thickness was used the software “observation application”.
The UV-vis spectra were recorded with the UV-vis-NIR spectrophotometer (type UV-670 JASCO GMBH, Germany), and the reviewed wavelengths were in the range of 200 and 800 nm. The baseline was recorded using solving-filled cuvettes as a reference. The concentration of the used solutions was 0.05 mg mL−1. The used quartz cuvettes had a path length of 1 cm. Absorbance spectra of open and closed form derivatives were documented. For the switching procedure the light of a fluorescence spectrometer (FP-8500, JASCO) was used and guided via a liquid light guide (250-series, Lumatec) into the UV-vis-spectrometer. A self-made cell holder allows irradiate the solutions while simultaneously acquire spectra. The bandwidth of switching light was set to 5 nm, resulting in a light intensity of 0.5 mW.
To determine the quantum yields, the UV-vis-NIR spectrophotometer mentioned above was used. As solvent, THF (99.9%, anhydrous, inhibitor-free) was used. A concentration of 0.025 mg mL−1 was applied. The measurements concerning the switching process (transfer to the closed state) were done according to literature.30 The quantum yields of the reverse reaction (close to open) were determined using a procedure described by Stranius and Börjesson.31 For the irradiation process the setup described above was used with a bandwidth of irradiation of 2.5 nm (open to close) and 10 nm (close to open) and a light intensity of 0.5 mW. For the irradiation of molecule 16 was used a 3 watt LED (630–640 nm, Bridgelux/Epistar)
To determine the stabilities of the BTEs in films, we used a setup as shown in Fig. 5. We applied a yellow laser with a wavelength of 565 nm and a power of approx. 150 mW (MPB Communications 565 nm laser, Model F-04306-2) and a UV laser with a wavelength of 325 nm (Kimmon Koha HeCd Laser IK 3083 R-D). To control the intensity of the yellow laser, we employed a rotation stage with a half wave plate and a polarizing beamsplitter. An acousto-optic modulator (MQ110-A3-UV) is used to regulate the light power of the UV laser. A photosensor (Hamamatsu H10722-20 with 740 mV gain) is employed as detector, with an edge filter positioned in front to block UV light.
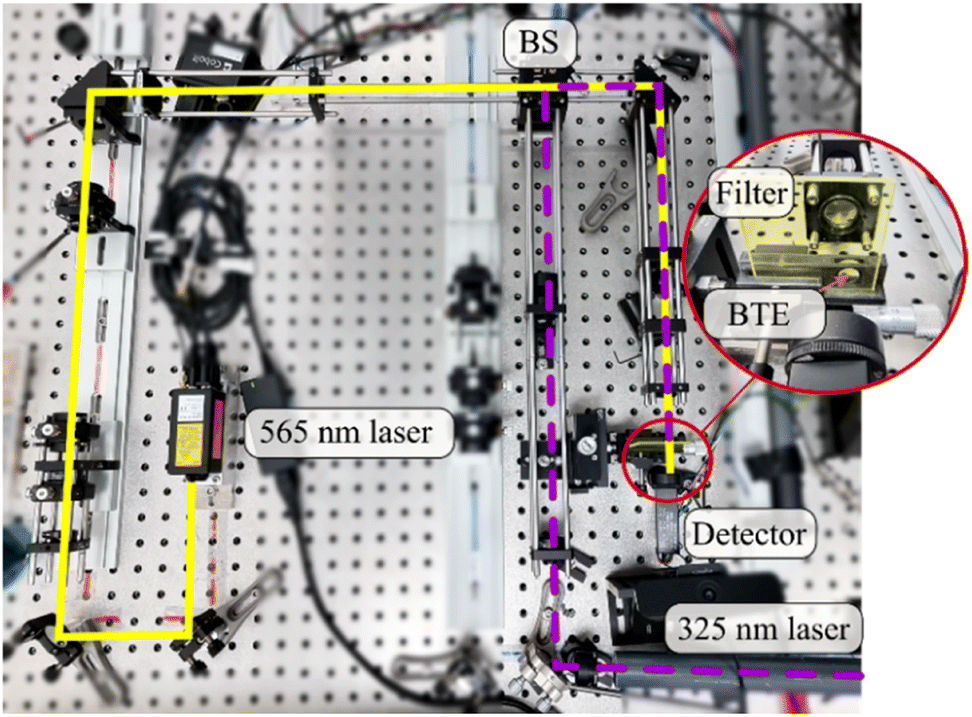 | ||
| Fig. 5 Experimental setup for the determineation of stabilities. The yellow and UV laser are superimposed by a beam splitter (BS) and focused on the film by a lens. The transmitted yellow light is detected by a detector, while the transmitted UV light is blocked by an edge filter. | ||
The beams were aligned using a pellicle beam splitter (BS) and positioned on the film by a mirror. An achromat is used to focus the beams to a diameter of approx. 200 μm.
A cycle consisted of two steps:
(I) Switching: the film was exposed to both the UV laser and the yellow laser simultaneously. The yellow laser power is set low enough to not interfere with the switching to the closed form. During this process, the absorption for yellow light increases, and the detected power decreases. For this switching process, the UV laser power on the target was set to approx. 0.5 mW and the yellow laser power was set to 10 μW for 2 s. To measure only the yellow laser power, an edge filter is used to block most of the UV laser power.
(II) Reset, the film is illuminated only by the yellow laser to reverse the switching process to the open form. A high laser power of about 10 mW is used for 4 seconds. During the reset, the switching process is almost completely reversed. The detected laser power at the beginning of each cycle is therefore approximately the same. To determine the photostationary state, the film was exposed to UV light until the transmitted yellow laser power was nearly constant and did not decrease further. This time Tmax is defined as the time to fully complete the switching process. A switching time of about 90% of Tmax is then selected for the cyclization. Note that the aforementioned times were specified for BTE 15a and BTE 15b. For BTE 15c and BTE 23 6 seconds to reset and 3 seconds for the switching was used, while the laser powers were the same.
General procedure for the formylation [method (C)]
Diisopropylamine (1.2 eq.) was dissolved in 30 mL of anhydrous THF. At 0 °C was slowly added n-butyllithium (24% in cyclohexane, 1.1 eq.). The reaction mixture was stirred at this temperature for 45 min. The thiophene derivative (1.0 eq.) was dissolved in 10 mL of anhydrous THF and added to the reaction mixture at 0 °C. The reaction was stirred for 30 min at this temperature and then three hours at room temperature. The reaction was then quenched with 40 mL of a saturated ammonium chloride solution. The aqueous phase was extracted two times with 50 mL of diethyl ether. The organic phase was washed once with 20 mL of 1M HCl, two times with 50 mL of water, and dried over MgSO4. Finally, the solvent was removed in vacuo.General procedure for the synthesis of the alcohols [method (D)]
The aldehyde (1.0 eq.) was dissolved in a mixture of THF and water (10:1). At 0 °C, sodium borohydride (0.5 eq.) was slowly added. The reaction mixture was stirred for 30 min at 0 °C and another 30 min at room temperature. Afterwards the reaction was quenched with 50 mL of water followed by an aqueous workup. The aqueous phase was extracted with 50 mL of diethyl ether (two times). The combined organic phases were washed with 100 mL of water (four times). Finally, the organic phase was dried over MgSO4 and the solvent was removed in vacuo.
General procedure for the methylation [method (E)]
The alcohol was dissolved in 30 mL of anhydrous THF. Sodium hydride (60%, 1.1 eq.) was slowly added at 0 °C. Iodomethane (1.1 eq.) was then added after 50 min at 0 °C. The reaction mixture was stirred for 30 min at this temperature and another four hours at room temperature. The reaction was then quenched with 50 mL of water followed by an aqueous workup. The aqueous phase was extracted twice with 50 mL of diethyl ether, and the combined organic phases were washed three times with 50 mL of water. The organic phase was dried over MgSO4 and the solvent was removed in vacuo.General procedure for the synthesis of the BTEs [(method F)]
The reaction was carried out under a nitrogen atmosphere. 1 eq. of the halogenated thiophene was dissolved in anhydrous THF and the mixture was then cooled to −80 °C. To this solution 1.05 eq of a solution of 2.8 M n-BuLi in hexane was added. After 30 min, octafluorocyclopentene (0.5 eq.) was added in small portions over a period of 30 min. After 2 h the reaction was allowed to warm to room temperature and was then stirred overnight. A portion of 30 mL of water was then added to quench the reaction. The aqueous phase was extracted two times with 50 mL diethyl ether, and the organic phase was washed once with 20 mL of 1M HCl, and two times with 50 mL of water. Then, it was dried over MgSO4 and the solvent was removed in vacuo. The crude product was purified by column chromatography to afford the BTEs.Synthesis of BTE 15a
Synthesis of BTE 15b
Synthesis of BTE 15c
Synthesis of BTE 16
Synthesis of BTE 22
Synthesis of BTE 23
:1) gave the product. Yield 0.599 g (87%), orange oil. 1H NMR (CDCl3, 600 MHz): δ = 6.96 (s, 2H, H3, H3′), 4.75 (s, 4H, H8, H8′), 3.91 (s, 4H, H6, H6′), 3.17 (s, 6H, H7, H7′) ppm. 13C NMR (CDCl3, 150 MHz): δ = 145.2 (o, C2, C2′), 143.2 (o, C5, C5′), 136.6–136.2 (m, o, C9, C9′), 124.7 (+, C3, C3′), 123.9 (o, C4, C4′), 117.8–114.2 (m, o, C10, C10′), 113.0–108.8 (m, o, C11), 68.1 (−, C6, C6′), 59.9 (−, C8, C8′), 58.2 (+, C7, C7′) ppm. IR (ATR): ν = 3373, 2934, 2829, 1635, 1560, 1496, 1455, 1364, 1334, 1268, 1190, 1120, 1087, 1038, 1023, 987, 970, 952, 933, 854, 826, 745, 712, 684, 654, 645, 575, 551, 523, 414 cm−1. HR-ESI-MS: calc. [M]+: 488.0551, calc. [M + Na+]: 511.0443; found [M + Na+]: 511.0446 (Δ = 0.0003).Author contributions
Thea Weingartz performed all syntheses, spectroscopic characterizations and produced all PMMA films. Sven Nagorny prepared the reference compound and investigated the Hammett–Brown correlations together with Thea Weingartz. André Eitzeroth determined the absorption coefficients and quantum yields. Marvin Schewe constructed the experimental setup and investigated the stabilities of the films. Jörg Adams, Christian Rembe and Andreas Schmidt are group leaders. Together with other cooperation partners (Alexander Egner, Vladimir Belov, Stefan Hell), they have a common project funded by the German Research Foundation (Deutsche Forschungsgemeinschaft DFG), called “NanoVidere – Photochromic Layers for Nanoscopy”.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The German Research Foundation is gratefully acknowledged for funding these studies (project numbers 504588119 (A. S.) and 412988268 (C. R.)).Notes and references
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Recent examples: (a) A. Kunz, N. Oberhof, F. Scherz, L. Martins, A. Dreuw and H. A. Wegner, Chem. –Eur. J., 2022, 28, e202200972 CrossRef CAS PubMed; (b) F. Gao, Z. Bi, S. Wang, Z. Zhao, Y. Dong and X. Li, Colloids Surf., A, 2022, 647, 129088 CrossRef CAS; (c) B. Xu, J. Ding, A. Caliari, N. Lu, F. Han, Y. Xia, J. Xu and T. Yomo, Biochem. Biophys. Res. Commun., 2022, 618, 113–118 CrossRef CAS PubMed.
- Recent examples: (a) H. Zhao, J. Huai, C. Weng and H. Han, J. Mol. Struct., 2022, 1263, 133146 CrossRef CAS; (b) D. Mukherjee, G. Chakraborty, N. Md. Hasan, U. Pal, P. Singh, T. Rakshit, R. I. Alsantali, D. Saha, A. Tanusri, A. Saleh, R. Das and S. K. Pal, J. Photochem. Photobiol., A, 2022, 430, 113958 CrossRef CAS.
- Recent example: S. Padgaonkar, C. T. Eckdahl, J. K. Sowa, R. Lopez-Arteaga, D. E. Westmoreland, E. F. Woods, S. Irgen-Gioro, B. Nagasing, T. Seideman, M. C. Hersam, J. A. Kalow and E. A. Weiss, Nano Lett., 2021, 21, 854–860 CrossRef CAS PubMed.
- (a) M. Albrecht, M. Gjikaj and A. Schmidt, Tetrahedron, 2010, 66, 7149–7154 CrossRef CAS; (b) M. Albrecht, M. Yulikov, T. Kohn, G. Jeschke, J. Adams and A. Schmidt, J. Mater. Chem., 2010, 20, 3025–3034 RSC; (c) M. Albrecht, O. Schneider and A. Schmidt, Org. Biomol. Chem., 2009, 7, 1445–1453 RSC; (d) A. Schmidt, M. Albrecht, T. Mordhorst, M. Topp and G. Jeschke, J. Mater. Chem., 2007, 17, 2793–2800 RSC.
- (a) K. Matsuda and M. Irie, J. Photochem. Photobiol., C, 2004, 5, 169–182 CrossRef CAS; (b) H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC; (c) T. J. Wigglesworth, A. J. Myles and N. R. Branda, Eur. J. Org Chem., 2005, 1233–1238 CrossRef CAS; (d) C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129 CrossRef CAS; (e) M. Irie and M. Morimoto, Pure Appl. Chem., 2009, 81, 1655–1665 CrossRef CAS; (f) T. Tsujioka and M. Irie, J. Photochem. Photobiol., C, 2010, 11, 1–14 CrossRef CAS; (g) A. Bianco, S. Perissinotto, M. Garbugli, G. Lanzani and C. Bertarelli, Laser Photonics Rev., 2001, 5, 711–736 CrossRef; (h) C. Bertarelli, A. Bianco, R. Castagna and G. Pariani, J. Photochem. Photobiol., C, 2011, 12, 106–125 CrossRef CAS; (i) G. Szalóki and J.-L. Pozzo, Chem.–Eur. J., 2013, 19, 11124–11132 CrossRef PubMed; (j) A. Bianchi, E. Delgado-Pinar, E. Garcia-Espana, C. Giorgi and F. Pina, Coord. Chem. Rev., 2014, 260, 156–215 CrossRef CAS; (k) E. C. Harvey, B. L. Feringa, J. G. Vos, W. R. Browne and M. T. Pryce, Coord. Chem. Rev., 2015, 282–283, 77–86 CrossRef CAS; (l) A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44(11), 3719–3759 RSC; (m) C. Xiao, W.-Y. Zhao, D.-Y. Zhou, Y. Huang, Y. Tao, W.-H. Wu and C. Yang, Chin. Chem. Lett., 2015, 26(7), 817–824 CrossRef CAS; (n) M. Irie, Pure Appl. Chem., 2015, 87(7), 617–626 CrossRef CAS; (o) S.-Z. Pu, Q. Sun, C.-B. Fan, R.-J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4(15), 3075–3093 RSC; (p) A. G. Lvov and V. Z. Shirinyan, Chem. heterocycl. compounds, 2016, 52(9), 658–665 CrossRef CAS; (q) L.-N. Fu, B. Leng, Y.-S. Li and S.-K. Gao, Chin. Chem. Lett., 2016, 27(8), 1319–1329 CrossRef CAS; (r) S.-L. Huang, T. S. A. Hor and G.-X. Jin, Coord. Chem. Rev., 2017, 346, 112–122 CrossRef CAS.
- (a) M. Herder, B. M. Schmidt, L. Grubert, M. Paetzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed; (b) M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–750 RSC; (c) T. Hirose, Y. Inoue, J.-y. Hasegawa, K. Higashiguchi and K. Matsuda, J. Phys. Chem. A, 2014, 118, 1084–1093 CrossRef CAS PubMed; (d) E. C. Harvey, J. Areephong, A. A. Cafolla, C. Long, W. R. Browne, B. L. Feringa and M. T. Pryce, Organometallics, 2014, 33, 447–456 CrossRef CAS; (e) T. Sakano, Y. Imaizumi, T. Hirose and K. Matsuda, Chem. Lett., 2013, 42, 1537–1539 CrossRef CAS; (f) H. Shoji and S. Kobatake, Chem. Commun., 2013, 49, 2362–2364 RSC.
- (a) H. Wang, H. Lin, W. Xu and D. Zhu, Chem. –Eur. J., 2013, 3366–3373 CrossRef CAS PubMed; (b) H. Lin, W. Xu and D. B. Zhu, J. Mater. Chem., 2010, 20, 884–890 RSC; (c) S. Kobatake, U. Kingo, E. Tsuchida and M. Irie, Chem. Commun., 2002, 2804–2805 RSC; (d) K. Uchida, E. Tsuchida, Y. Aoi, S. Nakamura and M. Irie, Chem. Lett., 1999, 63, 64 Search PubMed; (e) M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed; (f) K. Morimitsu, S. Kobatake and M. Irie, Tetrahedron Lett., 2004, 45, 1155–1158 CrossRef CAS.
- S. Nagorny, F. Lederle, V. Udachin, T. Weingartz, E. G. Hübner, S. Dahle, W. Maus-Friedrichs, J. Adams and A. Schmidt, Eur. J. Org Chem., 2021, 3178–3189 CrossRef CAS.
- S. Kobatake, Y. Terakawa and H. Imagawa, Tetrahedron, 2009, 65, 6104–6108 CrossRef CAS.
- (a) S. Fukumoto, T. Nakashima and T. Kawai, Eur. J. Org Chem., 2011, 5047–5053 CrossRef CAS; (b) S. Fukumoto, T. Nakashima and T. Kawai, Angew. Chem., Int. Ed., 2011, 50, 1565–1568 CrossRef CAS PubMed; (c) W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS PubMed.
- (a) Y. Ishibashi, H. Miyasaka, S. Kobatake, M. Irie and Y. Yokoyama, Pacific Rim Conference on Lasers and Electrooptic, 2007, vol. 1–4, p. 1364 Search PubMed; (b) Y. Ishibashi, M. Mukaida, M. Falkenstrom, H. Miyasaka, S. Kobatake and M. Irie, Phys. Chem. Chem. Phys., 2009, 11, 2640–2648 RSC; (c) Y. Ishibashi, K. Okuno, C. Ota, T. Umesato, T. Katayama, M. Murakami, S. Kobatake, M. Irie and H. Miyasaka, Photochem. Photobiol. Sci., 2010, 9, 172–180 CrossRef CAS PubMed; (d) S. P. Aloïse, M. Sliwa, Z. Pawlowska, J. Réhault, J. Dubois, O. Poizat, G. Buntinx, A. Perrier, F. Maurel and S. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 7379–7390 CrossRef PubMed.
- (a) S.-J. Lim, B.-K. An and S. Y. Park, Macromolecules, 2005, 38, 6236–6239 CrossRef CAS; (b) A. Fernandez and J.-M. Lehn, Adv. Mater., 1998, 10, 1519–1522 CrossRef; (c) T. Kawai, T. Sasaki and M. Irie, Chem. Commun., 2001, 711–712 RSC; (d) A. Osuka, D. Fujikane, H. Shinmori, S. Kobatake and M. Irie, J. Org. Chem., 2001, 66, 3913–3923 CrossRef CAS PubMed; (e) K. Yagi, C. F. Soong and M. Irie, J. Org. Chem., 2001, 66, 5419–5423 CrossRef CAS PubMed; (f) T. B. Norsten and N. R. Branda, J. Am. Chem. Soc., 2001, 123, 1784–1785 CrossRef CAS PubMed; (g) B. Chen, M. Wang, Y. Wu and H. Tian, Chem. Commun., 2002, 1060, 1061 Search PubMed; (h) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed; (i) S. Murase, M. Teramoto, H. Furukawa, Y. Miyashita and K. Horie, Macromolecules, 2003, 36, 964–966 CrossRef CAS; (j) G. Pariani, R. Castagna, R. Menon, C. Bertarelli and A. Bianco, Opt. Lett., 2013, 38, 3024–3027 CrossRef CAS PubMed.
- T. Ago, T. Kubota and H. Fukumoto, 2020, JP2020050618.
- (a) M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–750 RSC; (b) T. Hirose, Y. Inoue, J.-y. Hasegawa, K. Higashiguchi and K. Matsuda, J. Phys. Chem. A, 2014, 118, 1084–1093 CrossRef CAS PubMed; (c) E. C. Harvey, J. Areephong, A. A. Cafolla, C. Long, W. R. Browne, B. L. Feringa and M. T. Pryce, Organometallics, 2014, 33, 447–456 CrossRef CAS; (d) T. Sakano, Y. Imaizumi, T. Hirose and K. Matsuda, Chem. Lett., 2013, 42, 1537–1539 CrossRef CAS; (e) H. Ikeda, A. Sakai, A. Kawabe, H. Namai and K. Mizuno, Tetrahedron Lett., 2008, 49, 4972–4976 CrossRef CAS; (f) G. Sevez, J. Gan, J. Pan, X. Sallenave, A. Colin, H. Saadoui, A. Saleh, F. Vögtle and J.-L. Pozzo, J. Phys. Org. Chem., 2007, 20, 888–893 CrossRef CAS; (g) A. Peters and N. R. Branda, Adv. Mater. Opt. Electron., 2000, 10, 245–249 CrossRef CAS; (h) K. Higashiguchi, K. Matsuda, S. Kobatake, T. Yamada, T. Kawai and M. Irie, Bull. Chem. Soc. Jpn., 2000, 73, 2389–2394 CrossRef CAS.
- (a) H. Shoji and S. Kobatake, Chem. Commun., 2013, 49, 2362–2364 RSC; (b) H.-H. Liu and Y. Chen, New J. Chem., 2012, 36, 2223–2227 RSC; (c) K. Higashiguchi, K. Matsuda, T. Yamada, T. Kawai and M. Irie, Chem. Lett., 2000, 29, 1358–1359 CrossRef.
- (a) K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS PubMed; (b) C. Zheng, S. Pu, J. Xu, M. Luo, D. Huang and L. Shen, Tetrahedron, 2007, 63, 5437–5449 CrossRef CAS.
- I. E. Nifant'ev, A. A. Vinogradov, A. V. Churakov, V. V. Bagrov, I. A. Kashulin, V. A. Roznyatovsky, Y. K. Grishin and P. V. Ivchenko, Appl. Catal., A, 2019, 571, 12–24 CrossRef.
- M. Manuela, M. Raposo, C. Herbivo, V. Hugues, G. Clermont, M. C. R. Castro, A. Comel and M. Blanchard-Desce, Eur. J. Org Chem., 2016, 2016, 5263–5273 CrossRef.
- M. Fukagawa, I. Kawamura, T. Ubukata and Y. Yokoyama, Chem. –Eur. J., 2013, 19, 9434–9437 CrossRef CAS PubMed.
- C. Jagusch, M. Negri, U. E. Hille, Q. Hu, M. Bartels, K. Jahn-Hoffmann, M. A. P.-B. Mendieta, B. Rodenwaldt, U. Müller-Vieira, D. Schmidt, T. Lauterbach, M. Recanatini, A. Cavalli and R. W. Hartmann, Bioorg. Med. Chem., 2008, 16, 1992–2010 CrossRef CAS PubMed.
- S. P. Langston, S. Grossman, D. England, R. Afroze, N. Bence, D. Bowman, N. Bump, R. Chau, B.-C. Chuang, C. Claiborne, L. Cohen, K. Connolly, M. Duffey, N. Durvasula, S. Freeze, M. Gallery, K. Galvin, J. Gaulin, R. Gershman, P. Greenspan, J. Grieves, J. Guo, N. Gulavita, S. Hailu, X. He, K. Hoar, Y. Hu, Z. Hu, M. Ito, M.-S. Kim, S. W. Lane, D. Lok, A. Lublinsky, W. Mallender, C. McIntyre, J. Minissale, H. Mizutani, M. Mizutani, N. Molchinova, K. Ono, A. Patil, M. Qian, J. Riceberg, V. Shindi, M. D. Sintchak, K. Song, T. Soucy, Y. Wang, H. Xu, X. Yang, A. Zawadzka, J. Zhang and S. M. Pulukuri, J. Med. Chem., 2021, 64, 2501–2520 CrossRef CAS PubMed.
- (a) C. Reichardt, Solvent and Solvent Effects in Organic Chemistry, Wiley-VCH, 2003, vol. 3 Search PubMed; (b) C. Reichardt and R. Müller, Liebigs Ann. Chem., 1976, 1937–1952 CrossRef CAS; (c) H. Wamhoff and A. Schmidt, J. Org. Chem., 1993, 58, 6976–6984 CrossRef CAS; (d) C. Reichardt and W. Grahn, Chem. Ber., 1970, 103, 1072–1087 CrossRef CAS; (e) E. M. Kosower, D. Hofmann and K. Wallenfels, J. Am. Chem. Soc., 1962, 84, 2755–2757 CrossRef CAS.
- (a) L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS; (b) L. P. Hammett, Trans. Faraday Soc., 1938, 34, 156–165 RSC; (c) H. H. Jaffé, Chem. Rev., 1953, 53, 191–261 CrossRef; (d) O. Exner, Adv. Linear Free Energy Relat., ed. N. B. Chapman, 1972, pp. 1–69 Search PubMed; (e) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. B. Smith, March's Advanced Organic Chemistry, Wiley, 7th edn, 2013, pp. 352–355 Search PubMed.
- (a) Y. Okamoto, T. Inukai and H. C. Brown, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef; (b) J. Shorter, Chem. Unserer Zeit, 1985, 19, 197–208 CrossRef CAS.
- S. Nagorny, T. Weingartz, J. C. Namyslo, J. Adams and A. Schmidt, Eur. J. Org Chem., 2023, 26, e202200996 CrossRef CAS.
- S. Kobatake, T. Yamada, K. Uchida, N. Kato and M. Irie, J. Am. Chem. Soc., 1999, 121, 2380–2386 CrossRef CAS.
- (a) Q.-T. Fu, X. Yan, T. Li, X.-Y. Zhang, Y. He, W.-D. Zhang, Y. Liu, Y. Li and Z.-G. Gu, New J. Chem., 2019, 43, 15797–15803 RSC; (b) U. Ali, K. J. B. A. Karim and N. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef CAS.
- M. Maafi, Molecules, 2008, 13, 2260–2302 CrossRef CAS PubMed.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- H. Zhao, J. W. Dankwardt, S. G. Koenig and S. P. Singh, Tetrahedron Lett., 2012, 53, 166–169 CrossRef CAS.
- E. Kianmehr, M. Fardpour and K. M. Khan, Eur. J. Org Chem., 2017, 18, 2661–2668 CrossRef.
- A. de Meijere, L. Zhao, V. N. Belov, M. Bossi, M. Noltemeyer and S. W. Hell, Chem. –Eur. J., 2007, 13, 2503–2516 CrossRef CAS PubMed.
- S. Thayumanavan, J. Mendez and S. R. Marder, J. Org. Chem., 1999, 64, 4289–4297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Hammett–Brown correlations, 1H NMR, 13C NMR and UV spectra. See DOI: https://doi.org/10.1039/d3ra04444k |
| This journal is © The Royal Society of Chemistry 2023 |