
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting pyrolysis decomposition of PFOA using computational nanoreactors: a thermodynamic study†
Elizabeth Serna-Sanchez and
Steven Pellizzeri *
*
Department of Chemistry and Biochemistry, Eastern Illinois University, 600 Lincoln Avenue, Charleston, IL 61920, USA. E-mail: spellizzeri@eiu.edu
First published on 30th August 2023
Abstract
Per- and polyfluoroalkyl substances (PFAS) are a large, complex, environmentally persistent, and ever-expanding group of manufactured chemicals. Disposal of these compounds could produce potentially dangerous products necessitating the need to quickly predict their decomposition products. This study focuses on the thermal decomposition of perfluorooctanoic acid (PFOA) using nanoreactor simulations to find the decomposition products and their respective energies. Applying the nanoreactor method, which is novel for this system, allows for rapid prediction of thermal decomposition pathways with minimal researcher bias and it predicted PFOA to decompose at ∼650 °C, consistent with previously reported experimental studies.
Per- and polyfluoroalkyl substances known as PFAS are a class of chemicals with fluorinated carbon moieties that have physical stability, chemical resistance, and are environmentally persistent.1–3 These chemicals are resistant to degradation because of their strong carbon–fluorine bonds.4,5 This strong bonding does not allow PFAS molecules to degrade naturally once they have been made. Many of these substances have been shown to cause harm to the human body, in particular perfluorooctanoic acid (PFOA), a kind of PFAS, which has been linked with pancreas, liver, and breast cancers.6,7 Its effect on humans has led to its restriction in consumer products; however, many industries such as aerospace, automotive, construction, electronics, and the military still use it.8 The continued use of these molecules have led to widespread contamination of PFAS within the soil, food, and water supply.8–10
PFAS waste in the water system is usually managed through a multitude of treatment. Activated carbon treatment is the most studied for this process and the most common because of its effectiveness in absorbing substances that interface between solids and liquids, such as PFAS.11,12 Removing the PFAS molecules is only the first step towards resolving the issue. To ensure no further contamination, the PFAS molecules have to be destroyed. Some current methods for PFAS degradation are electrochemical oxidation, incineration, ultrasonic sonication, mineralization, and UV treatments.11,13–17
The simplest way of destroying PFAS is through thermal decomposition via pyrolysis. Pyrolysis is the process of thermal decomposition of materials at high temperatures, often in an inert atmosphere.18 The thermal decomposition of PFAS is a complex process that has been the subject of much research in recent years,5,19–21 however little is still known about this process, its kinetics, and what process governs the final decomposition products. Computational chemistry provides a powerful tool for investigating the thermal decomposition of PFAS as it can provide detailed structural and kinetic information on the process. Several potential methods exist, such as ab initio molecular dynamics, which could be used to investigate the mechanisms and kinetics of the decomposition process. However, these methods require lengthy and complex calculations, a multitude of assumptions, or prior experimental guidance; therefore, we seek a simpler method to understand how PFAS thermally decomposes.
Herein, we provide a method to quickly investigate the thermodynamic processes involved in the decomposition of PFAS. We accomplished this using a computational nanoreactor under experimental conditions to promote the decomposition of PFOA using no researcher bias on the degradation products. In nanoreactor simulations, the molecules are kept within a spherical volume that is fixed at an origin by a time-dependent wall potential. Inside the wall potential, a meta-dynamics (MTD) simulation is used to explore the chemical compound, conformers, and the reaction space.22 This allows the molecule to impact the wall potential producing micro spikes in the potential energy (illustrated in Fig. 1) of the molecule which can be used to break bonds. This process would then provide information on what bond is the most likely to break under these conditions and with this information a decomposition pathway can be developed.
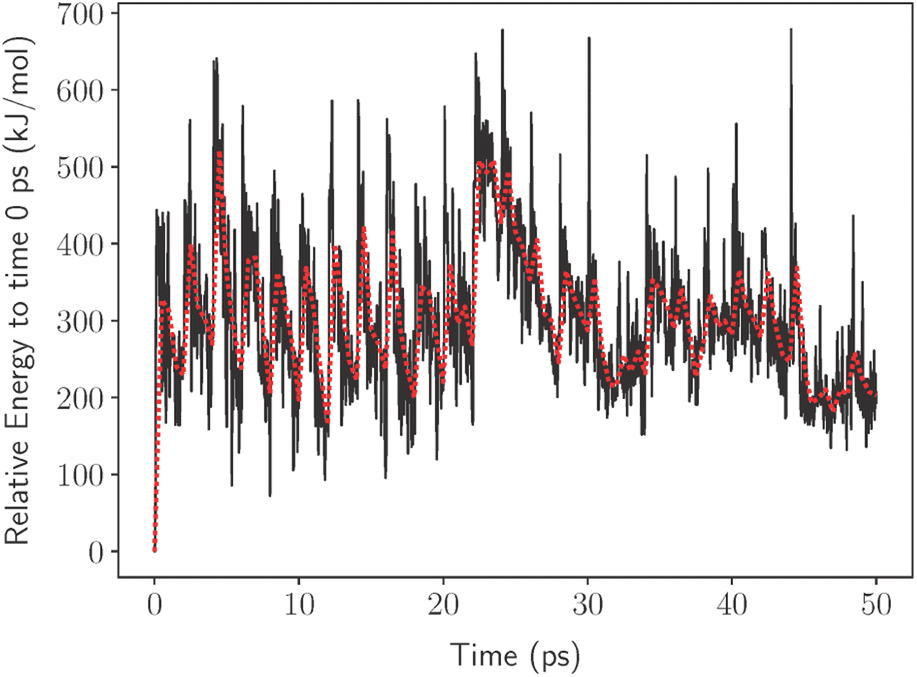 | ||
| Fig. 1 Total energy of the system for 50 ps relative to the energy at time 0 ps. The peaks are the result of rapid increases in temperature that promote the reaction to go forward. The dotted line shows the block average taken every 0.5 ps to provide clarity. The lengthening of the relaxation time back to the “average” energy at time ∼22 ps is due to bond breaking in the PFOA molecule. | ||
Using the reaction pathway found from the nanoreactor, the energies of all products were recalculated using density functional theory (DFT) to find the overall energies of the reaction. The benefit of using this method allows us to get a clearer understanding of a predicted thermodynamically preferred pathway without the need for prior experimental knowledge, researcher bias, or intensive user intervention for each possible step of the pathway.
The nanoreactor simulations were conducted using CREST version 2.11.1 (ref. 22 and 23) through XTB version 6.4.1.24 Simulations were conducted using the GFN2-xTB25 semi-empirical tight-binding method. Before running any of the MTD simulations the system was equilibrated by obtaining the structure of the molecule where the PFOA molecule reached a consistent energy level in the nanoreactor.
All structures obtained from the nanoreactor were re-optimized with Gaussian16, C.01.26 DFT calculations were performed using the hybrid meta density MPWB1K functional,27 6-31+G (d,p) basis set,28 and the GD3BJ empirical dispersion correction.29 The MPWB1K functional has been shown to provide good results in thermochemistry and thermochemical kinetics.27 Geometry relaxations were performed using the Berny algorithm using the GEDIIS optimization method with the RMS force convergence criterion set to 10−5 hartree Bohr−1. Geometry optimizations were performed for all atoms. Vibrational frequency analyses were performed on all structures to confirm their identities as stationary points by the lack of any imaginary frequencies. The resulting energies of the structures were used to find the energies of the whole reaction. A potential energy surface (PES) was constructed using the quasi-harmonic thermochemical data calculated from GoodVibes30 with Gaussian frequency calculations at a given temperature.
To find the best settings for the nanoreactor each of the parameters that would contribute to a change in the reaction were tested. The kpush controls the strength of the biasing potential, and it defines how much the system should be “pushed” to continue the reaction. If the value is too low no reaction takes place and if it is too high it could lead to chemically irrelevant reactions.31 The kpush value is limited by the α parameter which describes the size of the biasing potential and determines how much of the structure is still affected by the biasing potential.10,31
Values of kpush between 0.04 and 0.5 were tested with a constant α of 0.7 to see the effects the change in the value of kpush had on the molecule. Observations for changing the kpush are provided in Table S1.† Like the kpush, the α parameter was tested with values ranging from 0.3–1.6. Observations for changing the α parameter are provided in Table S2.† Optimal results for the nanoreactor were determined from the observations made from the changing kpush and α values. From the results, the higher values showed to have a higher success in activating PFOA, and through testing, the kpush value of 0.4 and the α value of 1.6 were determined as the optimal parameters. Additional observations with modified kpush and α values are provided in Table S3.† These values had to be adjusted for the nanoreactor simulations with the smaller molecular weight decomposition by-products since the values used for PFOA were too aggressive causing simulation failures.
The nanoreactor simulations have a PFOA molecule surrounded by helium atoms, as shown in Fig. 2. The surrounding helium does not interfere with the PFOA molecule, and it functions as a method to contain the molecule inside. When running the simulations, the temperature bath was at 1300 K and was run for 50 ps with a 0.5 fs time step. The cavity radius and size of the system corresponds to the mass density and was set at 0.2 g cm−3.22 Using the same parameter values, the decomposition by-products were isolated and left to decompose in their own nanoreactor. This continued until decomposition was no longer possible, resulting in the pathway shown in Scheme 1. Final kpush and α parameters for each decomposition product, as well as their observations, are provided in Table S4.†
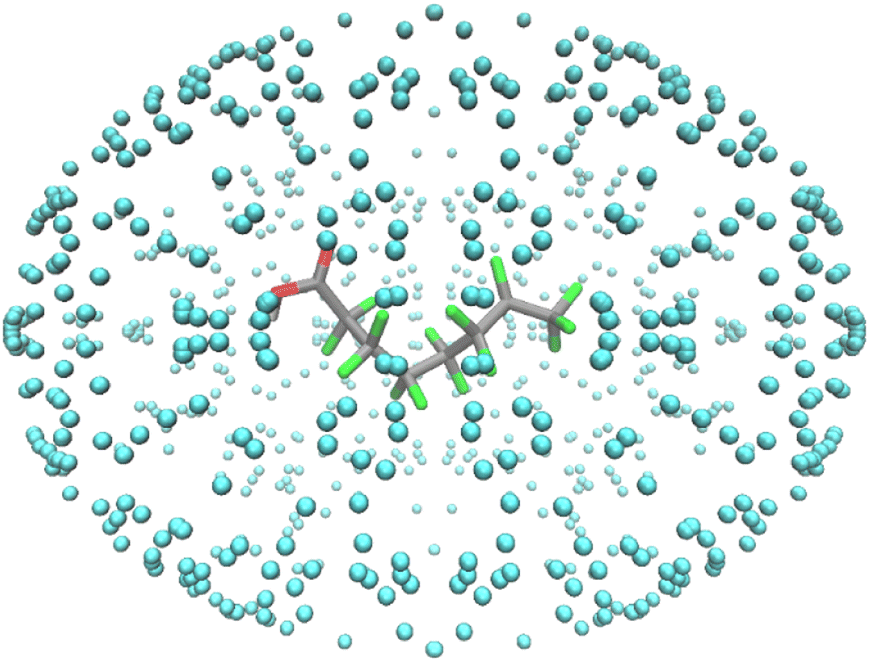 | ||
| Fig. 2 PFOA molecule surrounded by a wall of superheated helium atoms. | ||
 | ||
| Scheme 1 Lowest energy pathway for the total decomposition of PFOA determined using the nanoreactor simulations. | ||
Using the calculated energies (all numerical computational results are provided in Table S5†), a PES was plotted for temperatures from 298–1298 K at 100 K increments as shown in Fig. 3, for the reaction shown in Scheme 1. The first decomposition step was found to be the release of the carboxylic head group which quickly decomposes through the extraction of a fluorine to produce a terminal double bond in the carbon backbone to produce CO2 and HF. This process is illustrated in Fig. 4. C7F14 was then placed in its own nanoreactor and was left to decompose to produce CF3 and C6F11 from the terminal primary carbon. The next step involves the further decomposition of C6F11 through the loss of tetrafluoroethylene (C2F4) from the terminal primary carbon to produce C4F7. Lastly C4F7 further decomposed through the loss of tetrafluoroethylene to leave the final species C2F3, which was found to not decompose further.
 | ||
| Fig. 3 Gibbs free energy reaction profile from temperatures of 298–1298 K for the thermal decomposition of PFOA. Energies were determined using the MPWB1K-D3 functional and 6-31+G(d,p) basis set. | ||
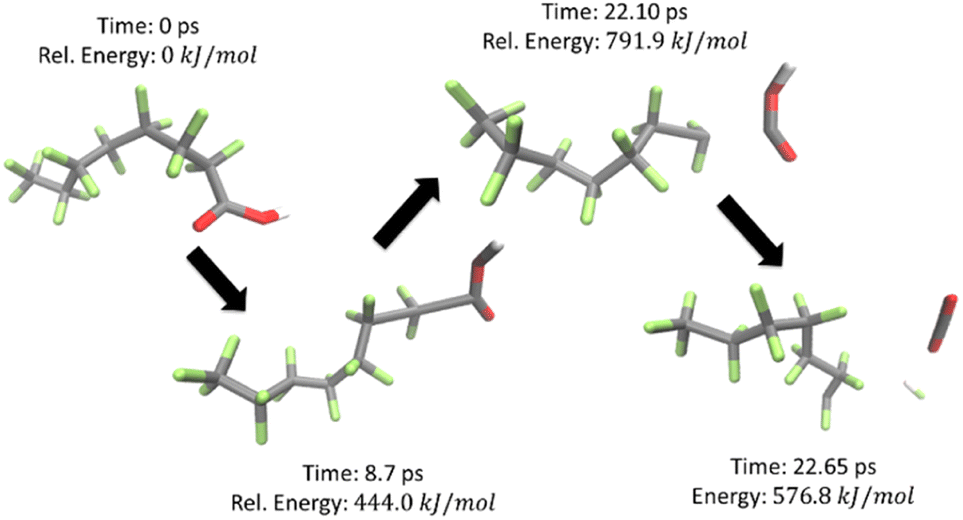 | ||
| Fig. 4 Snapshots of the nanoreactor simulation for the decomposition of PFOA. Simulation time of 50 ps, timestep of 0.5 fs, and bath temperature of 1300 K. Energies were determined using the GFN2-xTB method. | ||
Over the course of the nanoreactor simulations only radical elimination reactions were observed with no intramolecular rearrangements or radical insertion for the steps to produce branched products. Additionally, no cycloaddition steps were observed. We attribute this to the low density inside of the nanoreactor which promoted migration of the species away from each other. Lastly, we attribute the stability of the highly reactive radical products in the nanoreactor to the high thermal bath which promoted only bond breaking events over recombination events during the course of the simulation.
As the temperature increases the reaction goes towards spontaneity. From 298 K to 698 K the Gibbs free energy for the reaction (ΔGrxn) increases at every step showing the overall reaction to be nonspontaneous. Between 798 K and 898 K the first four reaction steps start to be spontaneous, however the final decomposition of C4H7 is still non-spontaneous. Above 989 K, each reaction step becomes spontaneous, which implies that complete decomposition can only be achieved at or above this temperature.
As stated above, the CF3 and C2F3 radicals were found to not decompose further, therefore, one could hypothesize that these two radicals could recombine, if allowed, to form the saturated fluorocarbon C3F6. This was found to be unsurprisingly highly exergonic with ΔGrxn ranging from −404.9 kJ mol−1 at 298 K, decreasing steadily to −218.1 kJ mol−1 at 1298 K. The complete table of values over the tested temperature range is given in Table S6.† The reduction in ΔGrxn is due to the loss of entropy from the recombination of the two radical species.
Fig. 5 shows the overall ΔGrxn (not including the formation of C3F6 from radical recombination) decreasing with an increase of temperature producing the linear relationship shown in eqn (1). Using this trend, the temperature right before the reaction becomes spontaneous was calculated to be 907.79 K.
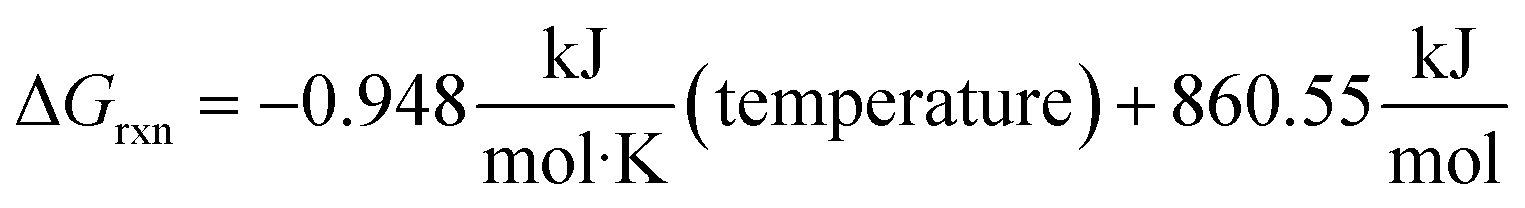 | (1) |
 | ||
| Fig. 5 Gibbs free reaction energy as a function of temperature for the thermal decomposition of PFOA. Energies were determined using the MPWB1K-D3 functional and 6-31+G(d,p) basis set. | ||
Any temperature higher than 907.79 K (634.64 °C) would then result in a negative ΔGrxn resulting in a spontaneous reaction. This result is in agreement with previously published experimental pyrolysis temperatures for PFOA which starts to decompose around 600 °C to 700 °C.4,5,32–34 Additionally the first mechanistic step we propose (formation of C7F14) is also consistent with literature experimental work looking at the thermal decomposition of PFOA using gas-phase NMR and GC-MS.35,36
In summary, the goal of this work was to find the thermal decomposition pathway of PFOA through a computational process that required little user input and minimal assumptions. We have shown that using a computationally inexpensive nanoreactor we were able to study the decomposition of PFOA with an experimentally consistent decomposition temperature, of around 650 °C, and similar mechanistic steps to available experimental characterization. This implies that the proposed pathway is a likely thermodynamic decomposition route. This procedure could be expanded to the ever-growing field of PFAS degradation with minimal adjustment making this a valuable tool to study this important process. Further studies in our group are currently underway with this in mind. One observation made of this decomposition pathway was the production of two tetrafluoroethylene molecules per PFOA molecule. Since tetrafluoroethylene is heavily used in the production of polytetrafluoroethylene polymers such as Teflon and Fluon there could be an economic interest in this collection if reaction conditions can be modified to preferentially make this product.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Eastern Illinois University through generous start-up funds is gratefully acknowledged.Notes and references
- C. Ng, I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, S. Patton, M. Scheringer, X. Trier and Z. Wang, Environ. Sci. Technol., 2021, 55, 12755–12765 CAS.
- X. Lyu, F. Xiao, C. Shen, J. Chen, C. M. Park, Y. Sun, M. Flury and D. Wang, Rev. Geophys., 2022, 60, e2021RG000765 CrossRef.
- A. J. Williams, L. G. T. Gaines, C. M. Grulke, C. N. Lowe, G. F. B. Sinclair, V. Samano, I. Thillainadarajah, B. Meyer, G. Patlewicz and A. M. Richard, Front. Environ. Sci., 2022, 10, 850019 CrossRef PubMed.
- M. Altarawneh, M. H. Almatarneh and B. Z. Dlugogorski, Chemosphere, 2022, 286, 131685 CrossRef CAS PubMed.
- R. Kumar, T. K. Dada, A. Whelan, P. Cannon, M. Sheehan, L. Reeves and E. Antunes, J. Hazard. Mater., 2023, 452, 131212 CrossRef CAS PubMed.
- PFOA Called Likely Human Carcinogen, https://cen.acs.org/articles/83/i27/PFOA-CALLED-LIKELY-HUMAN-CARCINOGEN.html, accessed December 19, 2021 Search PubMed.
- Toxicological profile for perfluoroalkyls, U.S. Department of Health and Human Services, Centers for Disease Control and Prevention (U.S.), Agency for Toxic Substances and Disease Registry, 2018 Search PubMed.
- Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS), https://www.niehs.nih.gov/health/topics/agents/pfc/index.cfm, accessed December 20, 2021 Search PubMed.
- M. L. Brusseau, R. H. Anderson and B. Guo, Sci. Total Environ., 2020, 740, 140017 CrossRef CAS PubMed.
- B. C. Crone, T. F. Speth, D. G. Wahman, S. J. Smith, G. Abulikemu, E. J. Kleiner and J. G. Pressman, Crit. Rev. Environ. Sci. Technol., 2019, 49, 2359–2396 CrossRef PubMed.
- S. Das and A. Ronen, Membranes, 2022, 12, 662 CrossRef CAS PubMed.
- M. N. Nadagouda and T. Lee, Acc. Mater. Res., 2021, 2, 129–133 CrossRef CAS PubMed.
- J. N. Meegoda, B. Bezerra de Souza, M. M. Casarini and J. A. Kewalramani, Int. J. Environ. Res. Public Health, 2022, 19, 16397 CrossRef PubMed.
- S. Yang, S. Fernando, T. M. Holsen and Y. Yang, Environ. Sci. Technol. Lett., 2019, 6, 775–780 CrossRef CAS.
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2019, 53, 3718–3728 CrossRef CAS PubMed.
- B. Trang, Y. Li, X.-S. Xue, M. Ateia, K. N. Houk and W. R. Dichtel, Science, 2022, 377, 839–845 CrossRef CAS PubMed.
- T. Shende, G. Andaluri and R. Suri, Chem. Eng. J. Adv., 2023, 15, 100509 CrossRef CAS.
- H. Zhou, Combustible Solid Waste Thermochemical Conversion: A Study of Interactions and Influence Factors, Springer, Singapore, 1st edn, 2017 Search PubMed.
- G. K. Longendyke, S. Katel and Y. Wang, Environ. Sci.: Processes Impacts, 2022, 24, 196–208 RSC.
- J. Wang, M. Song, I. Abusallout and D. Hanigan, Environ. Sci. Technol., 2023, 57, 6179–6187 CrossRef CAS PubMed.
- J. Wang, Z. Lin, X. He, M. Song, P. Westerhoff, K. Doudrick and D. Hanigan, Environ. Sci. Technol., 2022, 56, 5355–5370 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11, e1493 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- A. Modelli, L. Mussoni and D. Fabbri, J. Phys. Chem. A, 2006, 110, 6482–6486 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000Research, 2020, 9, 291 Search PubMed.
- M. Koerstz, M. H. Rasmussen and J. H. Jensen, SciPost Chem., 2021, 1, 003 CrossRef.
- F. Xiao, P. C. Sasi, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and D. Soli, Environ. Sci. Technol. Lett., 2020, 7, 343–350 CrossRef CAS.
- N. Watanabe, M. Takata, S. Takemine and K. Yamamoto, Environ. Sci. Pollut. Res., 2018, 25, 7200–7205 CrossRef CAS PubMed.
- S. Kundu, S. Patel, P. Halder, T. Patel, M. H. Marzbali, B. K. Pramanik, J. Paz-Ferreiro, C. C. de Figueiredo, D. Bergmann, A. Surapaneni, M. Megharaj and K. Shah, Environ. Sci.: Water Res. Technol., 2021, 7, 638–649 RSC.
- A. Alinezhad, P. Challa Sasi, P. Zhang, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and F. Xiao, ACS EST Eng., 2022, 2, 198–209 CrossRef CAS.
- P. J. Krusic, A. A. Marchione and D. C. Roe, J. Fluorine Chem., 2005, 126, 1510–1516 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: This includes observations on changing metadynamic parameters, relative energies for the decomposition products, and optimized structure files. See DOI: https://doi.org/10.1039/d3ra05187k |
| This journal is © The Royal Society of Chemistry 2023 |