DOI:
10.1039/D3RA03841F
(Paper)
RSC Adv., 2023,
13, 25717-25728
Novel flurbiprofen clubbed oxadiazole derivatives as potential urease inhibitors and their molecular docking study†
Received
8th June 2023
, Accepted 16th August 2023
First published on 29th August 2023
Abstract
In this study, twenty eight novel oxadiazole derivatives (5–32) of the marketed available non-steroidal anti-inflammatory drug (NSAID), (S)-flurbiprofen (1), were synthesized via I2 mediated cyclo-addition reaction in better yields. The synthesized hydrazone-Schiff bases were cyclized with iodine by using potassium hydroxide as a base in DMSO solvent to obtain oxadiazole derivatives (5–32). Structures of the synthesized products were confirmed with HR-ESI-MS, 1H-NMR spectroscopy and CHN analysis. After structure confirmations all analogs were evaluated for urease (in vitro) inhibitory activity. Amongst the series, fourteen compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 were found to be excellent inhibitors of urease enzyme, having IC50 values of 12 ± 0.9 to 20 ± 0.5 μM, better than the standard thiourea (IC50 = 22 ± 2.2 μM), whereas the remaining fourteen derivatives displayed good to moderate activity. The in silico study was executed to analyse the interaction between the active site of the enzyme (urease) and the produced compounds. The docking study revealed that compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 had lower docking scores than the standard compound thiourea and revealed better interactions with the urease enzyme.
1. Introduction
Heterocyclic compounds, often known as heterocycles, are organic compounds that have at least one or more atoms in the ring other than carbon.1 These substances have wide-ranging biotic and pharmacological applications.2 Among heterocyclic compounds, 1,3,4-oxadiazole has emerged as an important framework for the synthesis of novel pharmaceuticals.3 Anti-hypertensive, anti-convulsant, anti-diabetic, anti-oxidant, anti-cancer, analgesic, anti-inflammatory, anti-bacterial, and anti-fungal qualities are only a few of the biological uses of compounds containing the 1,3,4-oxadiazole moiety.4–6 Because of its precious structure, which has significant biological potential, 1,3,4-oxadiazole containing heterocycles are crucial for molecular planning.7 These compounds can undergo a variety of processes. A number of medicinal molecules of the oxadiazole scaffold are utilized as medications in the market, including the well-known anti-hypertensive nesapidil, the strong PDF inhibitor furamizole, the HIV integrase inhibitor raltegravir, and the anti-cancer agent zibotentan.8,9 In addition, a number of oxadiazoles have been discovered to exhibit strong monoamine oxidase inhibitory potential and to be efficacious against lipoxygenase at the nano molar level.10
Non-steroidal anti-inflammatory drugs (NSAIDs) are important approved medications used throughout the world for the management of fever, pain, and inflammation because of their analgesic, anti-inflammatory and anti-pyretic properties.11 Flurbiprofen is a familiar NSAID identified by the trade term ANSAID and was sold for the first time by Pfizer in 1977 in Europe.12 This medication is well-known for its anti-inflammatory properties because it works by inhibiting the cyclooxygenase enzymes COX-I and COX-II, which block the production of prostaglandins.13 Flurbiprofen, a significant member of the NSAID group, is frequently utilized to diminish migraines, inflammation, osteoarthritis, acute gout, post-operative inflammation, soft tissue injuries, herpetic stromal keratitis, rheumatoid arthritis, and periodontal surgery.14–16
The primary objective of the current research study is to facilitate the transformation of a number of medications that are currently on the market into diverse molecules. Subsequently, these manufactured derivatives will be tested for a range of biological functions. In the past, a number of drug compounds were created and tested for their biological effects, and they were found to have strong enzyme inhibitory properties. For instance, compounds prepared from metronidazole exhibited outstanding α-glucuronidase and α-amylase inhibitory action, while derivatives of piroxicam were successfully tested for their anti-nociceptive activity. Similar to that, NSAIDs are also assessed for their actions that inhibit enzymes, which produce good to moderate results.17,18 Inhibitors of the SGLT2, α-amylase and α-glucosidase enzymes that include biphenyl rings have recently been identified as possible pointers for the cure of type-2 diabetes mellitus.19–22 Similarly Alam et al. reported the synthesis of flurbiprofen hydrazone derivatives as potent α-glucosidase inhibitors.11 However, Khan et al. reported the synthesis of 2-mercaptooxadiazole derivatives of flurbiprofen as excellent α-amylase inhibitors.6 Additionally, Taha et al. discovered that a variety of analogues containing fluorine atoms attached to the benzene rings had strong α-glucosidase inhibitory action and mentioned that the fluorine group is responsible for the potency of the active compounds (Fig. 1).23 The main scaffold of the flurbiprofen nucleus is fluoro substituted biphenyl rings. We, therefore, considered investigating the urease inhibitory potential of flurbiprofen derivatives.
 |
| | Fig. 1 Justification of the current study. | |
2. Results and discussion
2.1. Chemistry
Twenty eight oxadiazole derivatives (5–32) based on the (S)-flurbiprofen nucleus were successfully synthesized and purified through multi-step reactions as part of on-going efforts to find biologically/pharmacologically active and powerful urease inhibitors. Prior to being refluxed with excess hydrazine hydrate in ethanol to produce the desired hydrazide of flurbiprofen in noble to brilliant yields, commercially available flurbiprofen acid was first heated on continual stirring with sulfuric acid in absolute ethanol to acquire ester. To create the hydrazone Schiff base derivatives of flurbiprofen, the hydrazide was further refluxed with various substituted aromatic aldehydes in the presence of a catalytic amount of acetic acid in ethanol. Finally using iodine, potassium hydroxide and DMSO as a solvent the hydrazone Schiff bases were cyclized to oxadiazole (Scheme 1).24 After the reaction was completed, unreacted iodine was removed using 5% sodium thiosulphate (Na2S2O3). HR-ESI-MS and 1H-NMR spectroscopy were used to confirm the structures of the synthesized oxadiazole derivatives and finally, these products were analysed for urease (in vitro) inhibitory activity.
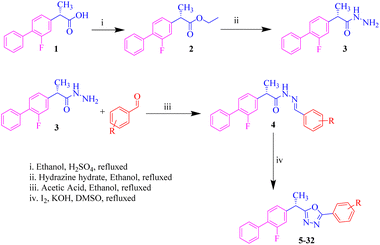 |
| | Scheme 1 Numerous replaced oxadiazole products based on flurbiprofen (5–32). | |
2.2. In vitro urease inhibitory activity
The synthesized products were evaluated for their in vitro urease inhibitory potential and compared to the standard thiourea (IC50 = 22 ± 2.2 μM). Among the synthesized series, fourteen compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 were found to be the most potent urease inhibitors better than the standard, having IC50 values of 12 ± 0.9, 12 ± 0.1, 12 ± 0.1, 13 ± 0.1, 13 ± 0.2, 13 ± 0.2, 14 ± 0.3, 15 ± 0.3, 15 ± 0.3, 16 ± 0.1, 16 ± 0.3, 16 ± 0.9, 20 ± 0.5, and 20 ± 0.5 μM, respectively. Furthermore, compounds 15 (IC50 = 31 ± 1.2 μM), 22 (IC50 = 31 ± 1.4 μM), 27 (IC50 = 34 ± 2.4 μM), 14 (IC50 = 35 ± 1.2 μM), 25 (IC50 = 35 ± 1.5 μM), 17 (IC50 = 36 ± 1.2 μM), 23 (IC50 = 37 ± 1.4 μM), 18 (IC50 = 38 ± 1.4 μM), and 5 (IC50 = 39 ± 1.2 μM) exhibited significant urease inhibitory activity, while five compounds 8, 11, 29, 9 and 12 showed moderate to less activity in the range of 41 ± 1.2 to 74 ± 1.3 μM (Table 1).
Table 1 Urease Inhibition Activity of Compounds (5–32)
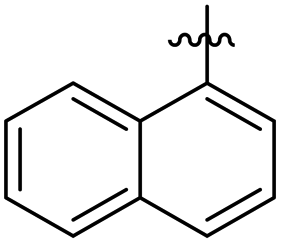
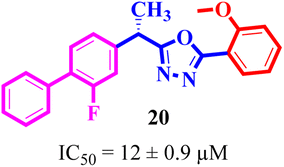
Structure activity relationship (SAR) study of the synthesized derivatives was performed by investigating the alteration in nature and position of involved groups or substituents (R) on the benzene rings. Compound 20 (IC50 = 12 ± 0.9 μM) was found to be the supreme effective inhibitor of urease enzyme. The potency of compound 20 might be due to the attachment of the methoxy (–OCH3) group at the ortho position of the benzene ring (Fig. 2).
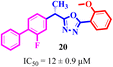 |
| | Fig. 2 Structure of the most potent urease inhibitor 20. | |
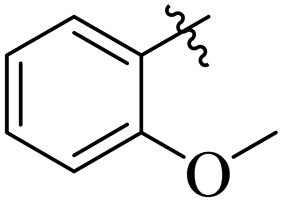
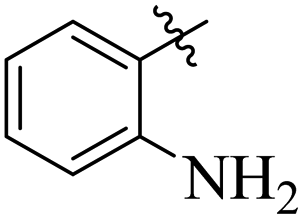
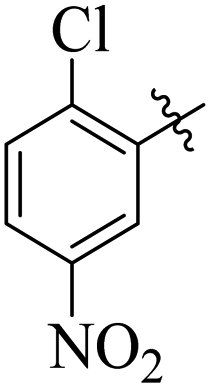
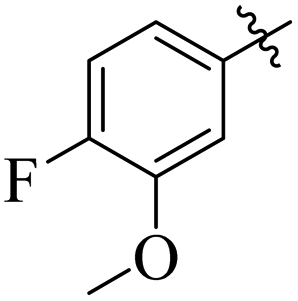
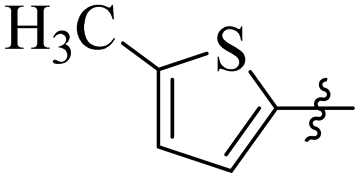
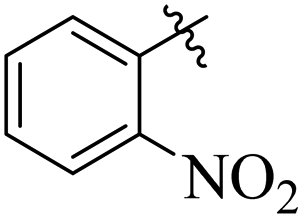
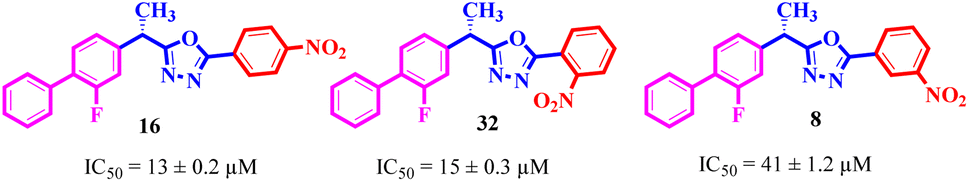
Comparing compounds 16 (IC50 = 13 ± 0.2 μM) and 32 (IC50 = 15 ± 0.3 μM) with 8 (IC50 = 41 ± 1.2 μM), the higher activity of 16 and 32 may perhaps be of nitro group at para and ortho positions attached to the ring of the benzene moiety. The compound 8 having less activity might be due to the nitro group attached to the meta position of the benzene ring (Fig. 3).
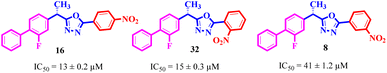 |
| | Fig. 3 Structure activity relationship comparison of compounds 16, 32, and 8. | |
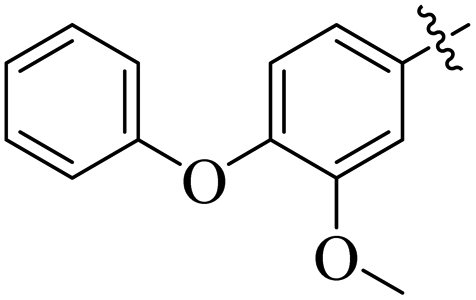
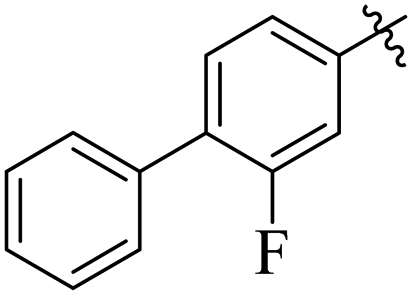
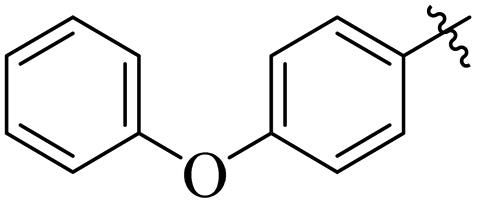
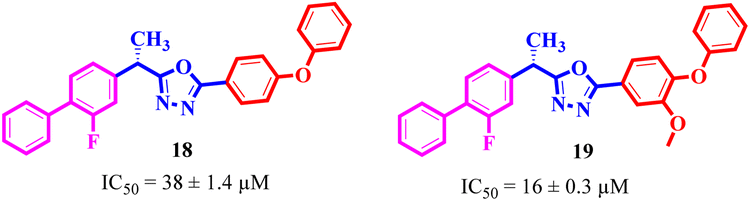
If we compare compounds 18 with 19 having IC50 values of 38 ± 1.4 and 16 ± 0.3 μM, respectively, the higher activity of compound 16 could be due to the presence of the methoxy group at the meta position of the benzene ring. Addition of the electron donating methoxy group to compound 19 at the meta position significantly increases the activity of the compound (Fig. 4).
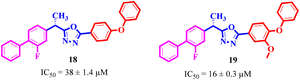 |
| | Fig. 4 Comparison of the structure activity relationship of compounds 18 and 19. | |
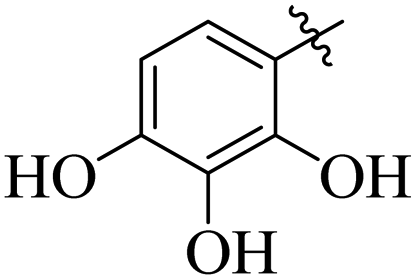
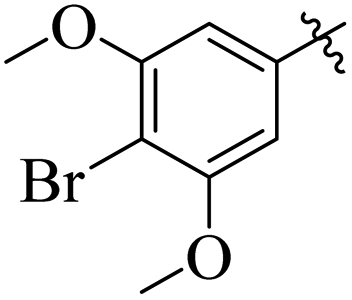
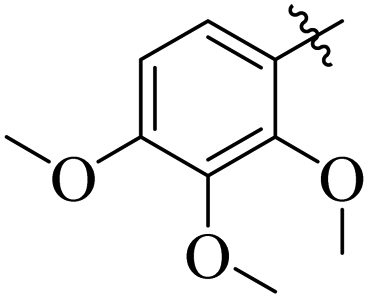
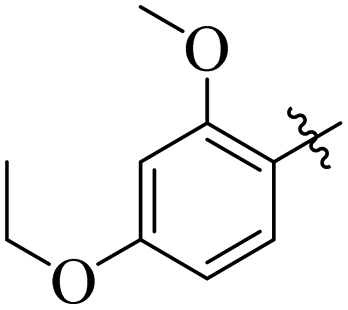
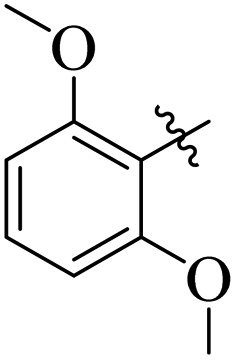
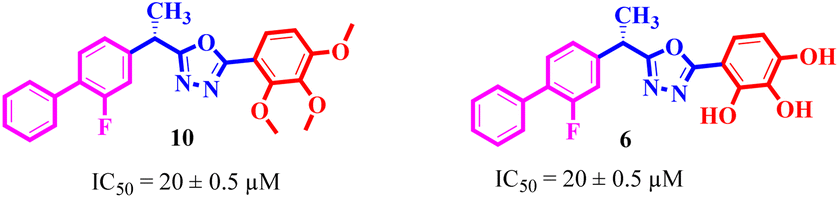
Similarly, by comparing compound 10 (IC50 = 20 ± 0.5 μM) with 6 (IC50 = 20 ± 0.5 μM), both derivatives showed excellent inhibitory potential better than the standard thiourea. The excellent activity of these compounds might be due to the presence of three electron donating groups attached at ortho, meta, and para positions of the benzene ring. A very slight decline occurs in the activity of compound 6 when the methoxy groups are replaced by hydroxyl groups at the same position (Fig. 5). It means that electron donating groups are responsible for enhancing the activity of the compounds against urease enzyme.
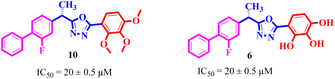 |
| | Fig. 5 Structure activity relationship study of compounds 10 and 6. | |
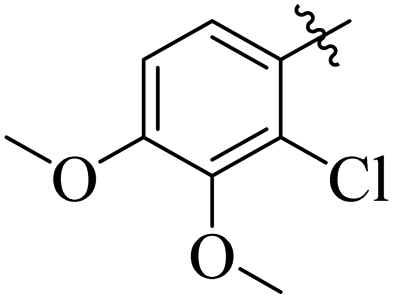
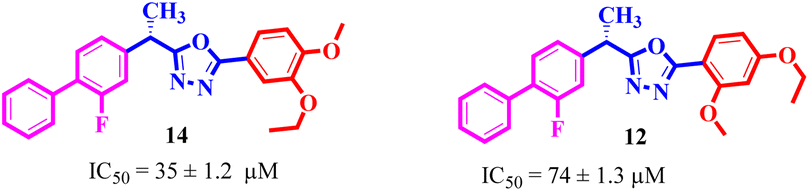
By comparing compound 14 with 12 with IC50 values of 35 ± 1.2 and 74 ± 1.3 μM, respectively, the nature of attached substituents is the same but the positions are different. Amongst them, compound 14 showed excellent activity compared to 12; the higher activity of this compound may be due to the presence of the ethoxy group at the meta position and the methoxy group at the para position of the benzene ring (Fig. 6). Transferring of the ethoxy group from meta to para and methoxy group from para to ortho group is responsible for the fall in activity of the compound.
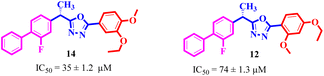 |
| | Fig. 6 Assessment of structure activity relationship study of compounds 14 and 12. | |
2.3. Docking analysis
The docking results indicated that compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 had lower docking scores than the reference compound and revealed better interactions. The compounds were docked against the urease and a total of ten positions were nominated for every compound. The top-ranked posture for each compound was further designated for the interaction analysis. Among all the compounds of the series compound 20 was predicted as the most potent inhibitor of urease, followed by compound 26 based on the docking score. The docking score of compound 20 was predicted as −5.53 and formed a total of five interactions with the urease enzyme. Compound 20 formed a total of three H-donor interactions with Gly279 and Cys321 residues. Compound 20 also formed two pi-H interactions with Cys321. Most of the interactions were found within the active site residues of the urease. The docking score of compound 26 was predicted to be −5.18 and compound 26 formed a total of four interactions with the urease enzyme. Compound 26 formed two H-donor interactions with the Cys321 residue. The 6-ring of compound 26 established two pi-H interactions with the Gln364 and Ala365 residues of the receptor. Compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 were found most potent compared to the reference compound thiourea in terms of docking score and interactions. Table 2 describes the docking scores and interactions of the most potent compounds as well as the reference compound. Fig. 7 displays the three dimensional interactions of the most potent compounds such as compounds 20 and 26 along with the reference compounds within the active site of the urease enzyme.
Table 2 Docking score and interactions of most potent compounds of the series
| C. no. |
Ligand |
Receptor |
Interacting residues |
Interaction type |
Distance |
Energy |
S score |
| 20 |
C 25 |
O |
GLY 279 |
H-Donor |
3.59 |
−1.1 |
−5.53 |
| O 32 |
SG |
CYS 321 |
H-Donor |
3.91 |
−0.1 |
| C 34 |
SG |
CYS 321 |
H-Donor |
3.05 |
−1.2 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
4.64 |
−0.1 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
3.76 |
−0.5 |
| 26 |
C 34 |
SG |
CYS 321 |
H-Donor |
3.92 |
−0.7 |
−5.18 |
| F 45 |
SG |
CYS 321 |
H-Donor |
3.71 |
−0.3 |
| 6-Ring |
CB |
GLN 364 |
Pi-H |
4.52 |
−0.5 |
| 6-Ring |
CA |
ALA 365 |
Pi-H |
4.54 |
−0.9 |
| 30 |
C 25 |
O |
GLY 279 |
H-Donor |
3.72 |
−0.1 |
−4.36 |
| N 29 |
NI |
NI 601 |
H-Acceptor |
2.67 |
−0.2 |
| N 30 |
NI |
NI 601 |
Metal |
2.49 |
−0.8 |
| C 14 |
5-Ring |
HIS 322 |
H-Pi |
4.49 |
−0.1 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
3.92 |
−0.6 |
| 24 |
C 14 |
OE1 |
GLU 222 |
H-Donor |
3.29 |
−0.3 |
−4.04 |
| C 22 |
OD1 |
ASP 223 |
H-Donor |
2.98 |
−0.3 |
| BR 43 |
O |
HIS 314 |
H-Donor |
3.59 |
−0.7 |
| 5-Ring |
CE1 |
HIS 221 |
Pi-H |
4.71 |
−0.2 |
| 21 |
BR 43 |
O |
HIS 314 |
H-Donor |
3.39 |
−0.4 |
−4.15 |
| N 29 |
CG2 |
THR 251 |
H-Acceptor |
4.16 |
−0.1 |
| N 30 |
CG2 |
THR 251 |
H-Acceptor |
4.26 |
−0.1 |
| 6-Ring |
CA |
CYS 321 |
Pi-H |
3.90 |
−0.3 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
3.80 |
−0.4 |
| 5-Ring |
CB |
CYS 321 |
Pi-H |
4.07 |
−0.1 |
| 16 |
C 11 |
SG |
CYS 321 |
H-Donor |
3.69 |
−0.3 |
−4.13 |
| C 14 |
SG |
CYS 321 |
H-Donor |
3.63 |
−0.5 |
| O 20 |
SG |
CYS 321 |
H-Donor |
3.53 |
−0.2 |
| C 26 |
SG |
CYS 321 |
H-Donor |
4.45 |
−0.2 |
| 28 |
C 22 |
OE1 |
GLU 222 |
H-Donor |
3.57 |
−0.3 |
−4.11 |
| C 25 |
OD2 |
ASP 223 |
H-Donor |
3.62 |
−0.2 |
| O 42 |
SD |
MET 317 |
H-Donor |
4.02 |
−0.2 |
| N 30 |
NH2 |
ARG 338 |
H-Acceptor |
3.06 |
−0.3 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
3.93 |
−0.2 |
| 6-Ring |
NE2 |
HIS 323 |
Pi-H |
3.74 |
−0.2 |
| 31 |
C 17 |
SG |
CYS 321 |
H-Donor |
3.59 |
−0.3 |
−3.97 |
| C 25 |
SG |
CYS 321 |
H-Donor |
3.95 |
−0.2 |
| N 29 |
NH2 |
ARG 338 |
H-Acceptor |
3.51 |
−0.5 |
| 6-Ring |
CB |
MET 317 |
Pi-H |
4.35 |
−0.3 |
| 32 |
O 32 |
SG |
CYS 321 |
H-Donor |
3.07 |
−0.3 |
−4.68 |
| C 37 |
O |
HIS 314 |
H-Donor |
3.42 |
−0.3 |
| N 30 |
NI |
NI 601 |
Metal |
2.61 |
−2.0 |
| O 44 |
NI |
NI 601 |
Metal |
2.57 |
−1.2 |
| 6-Ring |
CB |
ASP 165 |
Pi-H |
4.80 |
−0.3 |
| 7 |
N 43 |
SG |
CYS 321 |
H-Donor |
4.06 |
−0.5 |
−4.80 |
| N 29 |
NI |
NI 601 |
H-Acceptor |
2.70 |
−0.2 |
| N 30 |
NI |
NI 601 |
Metal |
2.51 |
−1.4 |
| 6-Ring |
CE1 |
HIS 221 |
Pi-H |
4.71 |
−0.4 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
4.07 |
−0.2 |
| 19 |
C 14 |
OE1 |
GLU 222 |
H-Donor |
3.46 |
−0.2 |
−4.25 |
| C 22 |
OE1 |
GLU 222 |
H-Donor |
3.63 |
−0.2 |
| C 25 |
OD1 |
ASP 223 |
H-Donor |
3.42 |
−0.2 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
4.28 |
−0.2 |
| 6-Ring |
NE2 |
HIS 323 |
Pi-H |
3.79 |
−0.4 |
| 13 |
C 25 |
SG |
CYS 321 |
H-Donor |
3.98 |
−0.3 |
−3.77 |
| C 36 |
OD2 |
ASP 223 |
H-Donor |
3.38 |
−0.9 |
| C 40 |
SG |
CYS 321 |
H-Donor |
3.65 |
−0.5 |
| 6-Ring |
CB |
MET 317 |
Pi-H |
4.66 |
−0.5 |
| 10 |
C 19 |
O |
GLN 364 |
H-Donor |
3.41 |
−0.2 |
−3.58 |
| C 52 |
O |
ASN 168 |
H-Donor |
3.43 |
−0.2 |
| O 41 |
NI |
NI 601 |
Metal |
2.51 |
−0.2 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
3.77 |
−0.2 |
| 6 |
O 32 |
SG |
CYS 321 |
H-Donor |
3.41 |
−0.2 |
−3.29 |
| N 29 |
CB |
ALA 169 |
H-Acceptor |
3.37 |
−0.2 |
| O 41 |
NH2 |
ARG 338 |
H-Acceptor |
2.84 |
−0.4 |
| 6-Ring |
CB |
CYS 321 |
Pi-H |
4.41 |
−0.1 |
| 6-Ring |
NE2 |
HIS 323 |
Pi-H |
3.71 |
−0.5 |
| Thiourea |
N 3 |
O |
ALA 278 |
H-Donor |
3.08 |
−1.6 |
−3.19 |
| S 1 |
CA |
GLY 279 |
H-Acceptor |
3.67 |
−1.0 |
| S 1 |
NH2 |
ARG 338 |
H-Acceptor |
3.78 |
−0.9 |
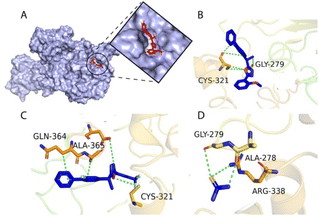 |
| | Fig. 7 (A) Active site of urease enzyme, and (B) 3D interaction of compound 20, (C) compound 26, and (D) reference compound with urease enzyme. Compounds are shown as blue sticks, while the green dotted lines represent bonds. | |
2.4. Docking validation
To validate the docking protocol of MOE software the co-crystallized ligand (PDB ID; 6ZO1) was removed from the active site and re-docked into the binding site of the urease enzyme. The RMSD value between the top-ranked docked conformation and the co-crystallized ligand was predicted to be 1.3 Å (Fig. 8), revealing the validity of the MOE docking protocol.
 |
| | Fig. 8 Superposition of co-crystallized and docked conformations of the ligand. The purple colour represents the native co-crystallized ligand and the cyan colour is the docked ligand. | |
3. Experimental
3.1. Materials and methods
Chemicals including reagents and solvents bought from BDH, Merck, and TCI were extra pure and utilized after receiving. Numerous substituted aldehydes (aromatic) and the starting material (S)-flurbiprofen having CAS No: 51543-39-6 and optical rotation were obtained from Sigma Aldrich. Dual wavelength ultra violet (UV) light of 254 and 365 nm was used for visualization of spots and aluminum cards (Merck, Kieselgel 60, GF254) were used for checking the direction of the desired reactions. Optical rotation was measured on an ATAGO, POLAX-2L polarimeter. Structure confirmation (1H and 13C NMR) was carried out on an Avance-Bruker 400 MHz spectrophotometer, whereas molecular masses of the obtained products were established via High Resolution Electro Spray Ionization Mass Spectrometry (HR-ESI-MS). CHN analyses were carried out on a Carlo Erba Strumentazione-Mod-1106, Italy.
3.1.1. Iodine mediated synthesis of oxadiazole derivatives (5–32) via cyclo addition reaction. Flurbiprofen hydrazides (3) (2 mmol) and a number of substituted aromatic aldehydes (2 mmol) were mixed in 10 ml pure ethanol used as a solvent and catalyst acetic acid in little quantity. The reaction (mixture) was stirred through continuous heating for 4–5 hours. At the end of the chemical reaction, products obtained after filtration were washed with hot water and n-hexane to eradicate extra aldehydes and dried out in reduced pressure.11 The obtained hydrazones of flurbiprofen were cyclized through iodine and potassium hydroxide in DMSO solvent. In a typical procedure, the obtained hydrazone Schiff base was dissolved in 15 ml DMSO and iodine (1.2 eq.) and KOH (3 eq.) were added to the reaction mixture. The reaction mixture was refluxed for 1–4 h with constant stirring. The improvement of reaction was tested with the help of thin layer chromatography (TLC) using a solvent system of n-hexane and ethyl acetate (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). After completion of the reaction, the mixture was poured into a beaker having icy distilled water. The mixture was treated with 5% Na2S2O3 solution to remove the remaining iodine and extracted three times (3 × 30 ml) with an organic solvent through a separating funnel. The organic layer was dried under air, precipitates were formed, and the products were recrystallized with absolute ethanol to get pure oxadiazole derivatives of flurbiprofen. The synthesized analogues were confirmed with the help of HR-ESI-MS and 1H-NMR spectroscopy.
:3). After completion of the reaction, the mixture was poured into a beaker having icy distilled water. The mixture was treated with 5% Na2S2O3 solution to remove the remaining iodine and extracted three times (3 × 30 ml) with an organic solvent through a separating funnel. The organic layer was dried under air, precipitates were formed, and the products were recrystallized with absolute ethanol to get pure oxadiazole derivatives of flurbiprofen. The synthesized analogues were confirmed with the help of HR-ESI-MS and 1H-NMR spectroscopy.
3.2. Urease inhibition assay (in vitro)
Urease inhibition potential of oxadiazole products was studied according to the described technique in the literature.25–27 Concisely, 40 μL buffers, 10 μL of the blended products and 10 μL of enzyme (5 U ml−1) were gestated for 10 minutes at 37 °C, in a 96 well plate. To each well, 40 μL of alkaline and phenolic reagents was added. Experiments were performed in triplicate. Absorbance was monitored at 625 nm with a microplate reader (Bio-TekELx 800, Instruments, Inc., USA). The standard inhibitor in this case was thiourea. The % inhibition was calculated by the use of the equation given below.
| % Inhibition = (absorbance control − absorbance sample)/absorbance control × 100 |
The IC50 values, and concentration needed for the inhibition of activity by 50% were determined using a non-linear regression graph, in which % inhibition of α-amylase (taken on the x-axis) was plotted against concentrations (taken on the y-axis). For this purpose GraphPad Prism Software (Version 7) was used.
3.3. Molecular docking
To carry out the molecular docking study low-resolution 3D structure of the urease enzyme [PDB code: 6ZO1] was retrieved from the PDB database.28 The water was removed from the crystal structure and hydrogen was added to the structure. Based on the Amber99 force field charges were assigned to all atoms.29 To draw the structures of compounds ChemDraw software was used and compounds were saved in the MDB database. The compounds were docked against the urease enzyme using the triangle matcher algorithm of the MOE software. For each compound, a total of ten poses were generated. The compounds were then ranked on the basis of the S score. Thiourea was employed as the positive control. PyMol software was then used to generate the 3D interactions30 of compounds in complex with urease.
3.4. Spectral data of the synthesized oxadiazole derivatives (5–32)
3.4.1. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(naphthalen-1-yl)-1,3,4-oxadiazole (5). Snowy amorphous solid; yield: 88%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.31 (d, J = 6.40 Hz, 3H), 3.83 (d, J = 7.12 Hz, 1H), 7.47–7.60 (m, 5H), 7.62–7.72 (m, 5H), 7.74 (t, J = 8.2 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.63 (s, 1H), 9.29 (d, J = 8.2 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 21.4 (1C), 42.7 (1C), 114.4 (1C), 121.3 (1C), 123.7 (1C), 125.9 (1C), 126.7 (2C), 127.1 (1C), 127.8 (5C), 128.2 (1C), 128.5 (3C), 130.0 (1C), 131.9 (1C), 133.8 (1C), 136.5 (1C), 141.2 (1C), 157.7 (1C), 166.8 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C26H19FN2O, 394.1481, found 394.4403; anal. calcd for C26H19FN2O (394.15): C, 79.17; H, 4.86; F, 4.82; N, 7.10; O, 4.06; found: C, 79.18; H, 4.84; N, 7.11.
3.4.2. 4-(5-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazol-2-yl) benzene-1,2,3-triol (6). White amorphous solid; yield: 85%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.63 (d, J = 7.1 Hz, 3H), 3.86 (d, J = 7.1 Hz, 1H), 7.22 (t, J = 7.9 Hz, 2H), 7.51–7.40 (m, 6H), 7.59 (d, J = 8.0 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 20.2 (1C), 40.7 (1C), 108.5 (1C), 114.4 (1C), 115.8 (1C), 126.2 (1C), 127.7 (4C), 128.2 (1C), 128.8 (2C), 130.9 (1C), 132.7 (1C), 136.5 (1C), 141.2 (1C), 146.8 (1C), 150.0 (1C), 160.1 (1C), 165.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C22H17FN2O4, 392.1172, found 392.1172; anal. calcd for C22H17FN2O4 (392.12) C, 67.34; H, 4.37; F, 4.84; N, 7.14; O, 16.31; found: C, 67.35; H, 4.39; N, 7.12.
3.4.3. 2-(5-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazol-2-yl) aniline (7). Silvery amorphous solid; yield: 82%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.31 (d, J = 7.1 Hz, 3H), 2.69 (q, J = 7.0 Hz, 1H), 7.41–7.45 (m, 3H), 7.51–7.58 (m, 4H), 7.80–7.87 (m, 3H), 8.05–8.10 (m, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 19.1 (1C), 42.3 (1C), 108.4 (1C), 113.4 (1C), 115.2 (1C), 126.2 (1C), 127.7 (3C), 128.1 (2C), 128.7 (3C), 129.3 (1C), 131.9 (1C), 136.5 (1C), 141.2 (1C), 150.1 (1C), 159.9 (1C), 165.2 (1C), 172.1 (1C); HR-ESI-MS [M + H]+ calculated for C22H18FN3O, 359.1434, found 359.3962; anal. calcd for C22H18FN3O (359.14) C, 73.52; H, 5.05; F, 5.29; N, 11.69; O, 4.45; found: C, 73.50; H, 5.03; N, 11.71.
3.4.4. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(3-nitrophenyl)-1,3,4-oxadiazole (8). Off-white amorphous solid; yield: 80%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.45 (d, J = 7.1 Hz, 3H), 3.61 (q, J = 7.1 Hz, 1H), 7.30–7.32 (m, 5H), 7.58 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 7.1 Hz, 1H), 7.85–7.83 (m, 2H), 8.14 (d, J = 8.3 Hz, 1H), 8.35 (t, J = 8.4 Hz, 1H), 8.61 (d, J = 8.3 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 21.9 (1C), 42.5 (1C), 113.4 (1C), 116.7 (1C), 118.3 (1C), 125.0 (1C), 126.1 (1C), 127.0 (1C), 127.8 (3C), 128.2 (1C), 128.8 (2C), 129.2 (1C), 130.9 (1C), 136.5 (1C), 140.2 (1C), 140.5 (1C), 161.7 (1C), 164.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C22H16FN3O3, 389.1176, found 389.3791; anal. calcd for C22H16FN3O3 (389.12) C, 67.86; H, 4.14; F, 4.88; N, 10.79; O, 12.33; found: C, 67.87; H, 4.17; N, 10.80.
3.4.5. 2-(5-Bromo-2-methoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (9). Ash-white amorphous solid; yield: 84%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.48 (d, J = 7.2 Hz, 3H), 3.51 (q, J = 7.2 Hz, 1H), 3.62 (s, 3H), 6.93–9.99 (m, 3H), 7.37–7.43 (m, 2H), 7.44–7.49 (m, 5H), 7.58–7.64 (m, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 18.9 (1C), 42.1 (1C), 58.0 (1C), 108.5 (1C), 112.1 (1C), 116.4 (1C), 119.4 (1C), 126.2 (1C), 127.8 (3C), 128.0 (1C), 128.4 (2C), 129.9 (1C), 131.0 (1C), 132.7 (1C), 136.5 (1C), 141.2 (1C), 158.7 (1C), 161.3 (1C), 162.2 (1C), 171.1 (1C); HR-ESI-MS [M + H]+ calculated for C23H18BrFN2O2, 452.0536, found 453.3036; anal. calcd for C23H18BrFN2O2 (552.05) C, 60.94; H, 4.00; Br, 17.63; F, 4.19; N, 6.18; O, 7.06; found: C, 60.97; H, 4.02; N, 6.19.
3.4.6. 2-(2,3,4-Trimethoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (10). Off-white amorphous solid; yield: 80%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 3.98 (q, J = 7.1 Hz, 1H), 3.93 (s, 3H), 3.96 (s, 3H), 3.97 (s, 3H), 4.12 (d, J = 7.1 Hz, 3H), 6.83 (d, J = 7.2 Hz, 1H), 7.40–7.44 (m, 2H), 7.45–7.52 (m, 4H), 7.57–7.59 (m, H), 7.69 (d, J = 7.2 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 21.3 (1C), 40.7 (1C), 57.0 (3C), 111.5 (1C), 111.8 (1C), 116.4 (1C), 126.2 (1C), 127.8 (4C), 128.0 (1C), 128.4 (2C), 131.9 (1C), 136.7 (1C), 138.2 (1C), 141.4 (1C), 144.4 (1C), 156.5 (1C), 160.7 (1C), 165.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C25H23FN2O4, 434.1642, found 434.4702; anal. calcd for C25H23FN2O4 (434.16) C, 69.11; H, 5.34; F, 4.37; N, 6.45; O, 14.73; found: C, 69.13; H, 4.37; N, 6.44.
3.4.7. 2-(2-Chloro-5-nitrophenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (11). Snowy white amorphous solid; yield: 85%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.34 (d, J = 7.2 Hz, 3H), 3.47 (q, J = 7.4 Hz, 1H), 7.47–7.60 (m, 9H), 7.63–7.78 (m, 1H), 7,87 (d, J = 8.5 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.5 (1C), 40.7 (1C), 115.2 (1C), 117.1 (1C), 119.5 (1C), 127.6 (1C), 127.7 (3C), 128.0 (1C), 128.2 (1C), 128.4 (2C), 130.9 (1C), 131.3 (1C), 133.2 (1C), 135.5 (1C), 139.2 (1C), 139.9 (1C), 159.3 (1C), 163.6 (1C), 171.1 (1C); HR-ESI-MS [M + H]+ calculated for C22H15ClFN3O3, 423.0786, found 423.8242; anal. calcd for C22H15ClFN3O3 (423.08) C, 62.35; H, 3.57; Cl, 8.37; F, 4.48; N, 9.91; O, 11.33; found: C, 62.33; H, 3.59; N, 9.90.
3.4.8. 2-(4-Ethoxy-2-methoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (12). White amorphous solid; yield: 78%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.49 (d, J = 6.8 Hz, 3H), 1.58 (t, J = 6.8 Hz, 3H), 4.01 (s, 3H), 4.24 (q, J = 6.8 Hz, 1H), 4.23 (q, J = 6.6 Hz, 2H), 7.02 (d, J = 7.2 Hz, 1H), 7.31 (s, 1H), 7.46–7.68 (m, 8H), 8.47 (m, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 14.2 (1C), 18.1 (1C), 42.7 (1C), 56.0 (1C), 64.3 (1C), 98.7 (1C), 110.5 (1C), 112.3 (1C), 114.4 (1C), 127.2 (1C), 127.6 (4C), 128.1 (1C), 128.4 (2C), 131.9 (1C), 134.5 (1C), 138.2 (1C), 155.3 (1C), 158.7 (1C), 160.9 (1C), 165.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C25H23FN2O3, 418.1693, found 418.4601; anal. calcd for C25H23FN2O3 (418.17) C, 71.76; H, 5.54; F, 4.54; N, 6.69; O, 11.47; found: C, 71.8; H, 5.55; N, 6.67.
3.4.9. 2-(4-Fluoro-3-methoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (13). Off white amorphous solid; yield: 88%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.35 (d, J = 7.0 Hz, 3H), 3.97 (q, J = 7.1 Hz, 1H), 4.01 (s, 3H), 7.26 (t, J = 7.4 Hz, 1H), 7.31 (s, 1H), 7.43–7.51 (m, 4H), 7.57 (d, J = 7.4 Hz, 1H), 7.67–7.71 (m, 1H), 7.70 (d, J = 8.0 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 20.3 (1C), 40.7 (1C), 56.0 (1C), 110.6 (1C), 115.4 (1C), 117.7 (1C), 126.0 (1C), 127.2 (1C), 127.5 (3C), 128.6 (1C), 128.8 (2C), 131.9 (1C), 133.4 (1C), 136.5 (1C), 140.2 (1C), 147.7 (1C), 153.9 (1C), 158.7 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C23H18F2N2O2, 392.1386, found 392.1342; anal. calcd for C23H18F2N2O2 (392.14) C, 70.40; H, 4.62; F, 9.68; N, 7.14; O, 8.15; found: C, 70.42; H, 4.63; N, 7.12.
3.4.10. 2-(3-Ethoxy-4-methoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (14). Off white amorphous solid; yield: 85%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.43 (d, J = 7.1 Hz, 3H), 1.81 (t, J = 6.9 Hz, 3H), 3.45 (q, J = 6.9 Hz, 1H), 3.61 (q, J = 7.1 Hz, 2H), 3.66 (s, 3H), 7.14–7.17 (m, 5H), 7.25 (d, J = 7.3 Hz, 1H), 7.32 (d, J = 7.3 Hz, 2H), 7.51–7.53 (m, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 16.2 (1C), 19.7 (1C), 41.7 (1C), 56.4 (1C), 64.5 (1C), 111.6 (1C), 112.2 (1C), 115.4 (1C), 125.0 (1C), 127.2 (1C), 127.8 (3C), 128.4 (2C), 129.2 (1C), 131.9 (1C), 136.5 (1C), 140.2 (1C), 148.4 (1C), 148.9 (1C), 160.7 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C25H23FN2O3, 418.1693, found 418.4601; anal. calcd for C25H23FN2O3 (418.17) C, 71.76; H, 5.54; F, 4.54; N, 6.69; O, 11.47; found: C, 71.77; H, 5.52; N, 6.71.
3.4.11. 2-(2,6-Dimethoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (15). White amorphous solid; yield: 88%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.42 (d, J = 7.0 Hz, 3H), 3.40 (q, J = 7.0 Hz, 1H), 3.69 (s, 6H), 7.10–7.12 (m, 5H), 7.27 (t, J = 7.0 Hz, 1H), 7.30 (d, J = 7.0 Hz, 2H), 7.52–7.55 (m, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 21.3 (1C), 40.7 (1C), 56.8 (2C), 114.4 (1C), 115.8 (2C), 120.9 (1C), 126.2 (1C), 127.8 (3C), 128.6 (1C), 128.4 (2C), 130.9 (1C), 131.2 (1C), 134.5 (1C), 138.2 (1C), 156.8 (2C), 159.7 (1C), 165.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C24H21FN2O3, 404.1536, found 404.4335; anal. calcd for C24H21FN2O3 (404.15) C, 71.27; H, 5.23; F, 4.70; N, 6.93; O, 11.87; found: C, 71.28; H, 5.25; N, 6.91.
3.4.12. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(4-nitrophenyl)-1,3,4-oxadiazole (16). White amorphous solid; yield: 80%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.44 (d, J = 6.7 Hz, 3H), 3.41 (q, J = 6.7 Hz, 1H), 7.11–7.13 (m, 5H), 7.52–7.55 (m, 3H), 7.73 (d, J = 8.5 Hz, 2H), 8.67 (d, J = 8.5 Hz, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 19.5 (1C), 40.9 (1C), 115.4 (1C), 116.6 (2C), 120.8 (1C), 127.0 (1C), 127.8 (3C), 128.2 (1C), 128.6 (2C), 129.4 (2C), 131.1 (1C), 136.3 (1C), 140.4 (1C), 139.3 (1C), 159.5 (1C), 163.8 (1C), 171.1 (1C); HR-ESI-MS [M + H]+ calculated for C22H16FN3O3, 389.1176, found 389.1176; anal. calcd for C22H16FN3O3 (389.12) C, 67.86; H, 4.14; F, 4.88; N, 10.79; O, 12.33; found: C, 67.88; H, 4.11; N, 10.80.
3.4.13. 2-(2-Chloro-3,4-dimethoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (17). Off white amorphous solid; yield: 90%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.65 (d, J = 6.9 Hz, 3H), 3.89 (q, J = 6.9 Hz, 1H), 3.94 (s, 3H), 3.98 (s, 3H), 6.96 (d, J = 7.5 Hz, 1H), 7.30–7.47 (m, 5H), 7.58 (d, J = 7.5 Hz, 1H), 7.77 (d, J = 7.2 Hz, 1H), 8.12 (s, 1H), 9.04 (s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 20.3 (1C), 41.1 (1C), 56.0 (2C), 110.2 (1C), 115.4 (1C), 120.0 (1C), 126.9 (1C), 127.9 (4C), 128.0 (1C), 128.2 (1C), 128.4 (2C), 130.9 (1C), 135.9 (1C), 137.2 (1C), 146.8 (1C), 155.1 (1C), 159.7 (1C), 165.2 (1C, s), 171.5 (1C); HR-ESI-MS [M + H]+ calculated for C24H20ClFN2O3, 438.1142, found 438.1140; anal. calcd for C24H20ClFN2O3 (438.11) C, 65.68; H, 4.59; Cl, 8.08; F, 4.33; N, 6.38; O, 10.94; found: C, 65.66; H, 4.58; N, 6.40.
3.4.14. 2-([1,1′-Biphenyl]-4-yl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (18). Off white amorphous solid; yield: 88%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.43 (d, J = 7.1 Hz, 3H), 3.44 (q, J = 7.1 Hz, 1H), 7.07–7.16 (m, 3H), 7.31 (d, J = 7.0 Hz, 2H), 7.40–7.63 (m, 5H), 7.91 (d, J = 8.0 Hz, 2H), 8.00–8.11 (m, 4H), 8.49 (d, J = 8.0 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.9 (1C), 40.1 (1C), 112.3 (2C), 115.2 (1C), 116.3 (2C), 122.8 (1C), 127.0 (1C), 127.5 (4C), 128.0 (1C), 128.4 (2C), 129.0 (2C), 129.6 (2C), 131.9 (1C), 136.5 (1C), 140.2 (1C), 156.1 (2C), 160.3 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C28H21FN2O2, 436.1638, found 436.4775; anal. calcd for C28H21FN2O2 (436.16) C, 77.05; H, 4.85; F, 4.35; N, 6.42; O, 7.33; found: C, 77.07; H, 4.82; N, 6.40.
3.4.15. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(3-methoxy-4-phenoxyphenyl)-1,3,4-oxadiazole (19). White amorphous solid; yield: 90%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.64 (d, J = 7.0 Hz, 3H), 3.98 (s, 3H), 5.29 (q, J = 7.0 Hz, 1H), 6.95 (d, J = 7.6 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 7.24–7.27 (m, 2H), 7.34–7.7.59 (m, 10H), 7.66 (s, 1H), 7.57–7.59 (m, 6H), 7.87 (t, J = 7.0 Hz, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 19.9 (1C, s), 39.9 (1C), 56.6 (1C), 109.6 (1C), 111.4 (1C), 115.0 (1C), 116.3 (2C), 126.0 (1C), 127.3 (1C), 127.4 (4C), 128.0 (1C), 128.4 (2C), 129.0 (1C), 129.6 (2C), 131.9 (1C), 134.5 (1C), 140.2 (1C), 147.0 (1C), 148.2 (1C), 156.5 (1C), 159.9 (1C), 164.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C29H23FN2O2, 466.1693, found 466.5035; anal. calcd for C29H23FN2O2 (466.17) C, 74.66; H, 4.97; F, 4.07; N, 6.00; O, 10.29; found: C, 74.69; H, 4.99; N, 6.02.
3.4.16. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(2-methoxyphenyl)-1,3,4-oxadiazole (20). White amorphous solid; yield: 92%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.34 (d, J = 7.0 Hz, 3H), 3.46 (q, J = 7.0 Hz, 1H), 3.94 (s, 3H), 7.17–7.19 (m, 4H), 7.27 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 7.6 Hz, 2H), 7.45 (d, J = 7.6 Hz, 1H), 7.58–7.60 (m, 4H); 13C-NMR (150 MHz, DMSO-d6) δ 19.1 (1C), 41.9 (1C), 56.4 (1C), 110.9 (1C), 115.8 (1C), 117.8 (1C), 125.9 (1C), 127.0 (1C), 127.6 (3C), 128.2 (1C), 128.5 (3C), 129.6 (1C), 131.9 (1C), 136.5 (1C), 139.6 (1C), 160.7 (1C), 162.1 (1C), 165.2 (1C), 171.9 (1C); HR-ESI-MS [M + H]+ calculated for C23H19FN2O2, 374.1431, found 374.4076; anal. calcd for C23H19FN2O2 (374.14) C, 73.78; H, 5.11; F, 5.07; N, 7.48; O, 8.55; found: C, 73.80; H, 5.10; N, 7.51.
3.4.17. 2-(4-Bromophenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (21). White amorphous solid; yield: 85%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.44 (d, J = 6.8 Hz, 3H), 3.40 (q, J = 6.8 Hz, 1H), 7.42–7.70 (m, 11H), 7.94 (d, J = 8.0 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.9 (1C), 41.9 (1C), 114.4 (1C), 121.3 (1C), 122.8 (1C), 127.4 (1C), 127.9 (3C), 128.3 (1C), 128.7 (2C), 129.3 (2C), 130.7 (1C), 132.3 (2C), 135.5 (1C), 138.2 (1C), 160.5 (1C), 164.2 (1C), 172.1 (1C); HR-ESI-MS [M + H]+ calculated for C22H16BrFN2O, 422.0430, found 423.2776; anal. calcd for C22H16BrFN2O (422.04) C, 62.43; H, 3.81; Br, 18.88; F, 4.49; N, 6.62; O, 3.78; found: C, 62.44; H, 3.83; N, 6.64.
3.4.18. 2-(2-Fluoro-4-methoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (22). Yellowish amorphous solid; yield: 90%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.38 (d, J = 7.3 Hz, 3H), 3.46 (q, J = 7.3 Hz, 1H), 3.91 (s, 3H), 7.25–7.27 (m, 5H), 7.45 (d, J = 7.4 Hz, 1H), 7.49 (d, J = 7.4 Hz, 2H), 7.51 (s, 1H), 7.59–7.61 (m, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 20.1 (1C), 41.7 (1C), 56.6 (1C), 102.5 (1C), 114.3 (1C), 115.4 (1C), 127.0 (1C), 127.5 (4C), 128.0 (1C), 128.2 (1C), 128.4 (2C), 131.3 (1C), 136.1 (1C), 139.4 (1C), 160.3 (1C), 160.6 (2C), 164.4 (1C), 171.5 (1C, s); HR-ESI-MS [M + H]+ calculated for C23H18F2N2O2, 392.1336, found 392.3980; anal. calcd for C23H18F2N2O2 (392.13) C, 70.40; H, 4.62; F, 9.68; N, 7.14; O, 8.15; found: C, 70.43; H, 4.61; N, 7.12.
3.4.19. 2-(4-Bromo-3,5-dimethoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (23). Pale yellow amorphous solid; yield: 92%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.43 (d, J = 7.0 Hz, 3H), 3.44 (q, J = 7.0 Hz, 1H), 3.91 (s, 3H), 3.92 (s, 3H), 7.29–7.32 (m, 5H), 7.38 (s, 2H), 7.60–7.62 (m, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 20.5 (1C), 39.7 (1C), 55.8 (2C), 106.0 (1C), 110.6 (2C), 114.4 (1C), 125.6 (1C), 127.0 (1C), 127.5 (3C), 128.0 (1C), 128.4 (2C), 131.1 (1C), 136.7 (1C), 138.9 (1C), 156.4 (2C), 159.5 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C24H19BrF2N2O3, 482.0641, found 482.3296; anal. calcd for C24H19BrF2N2O3 (482.06) C, 59.64; H, 4.17; Br, 16.53; F, 3.93; N, 5.80; O, 9.93; found: C, 59.66; H, 4.19; N, 5.77.
3.4.20. 2-(4-Bromo-2-fluorophenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (24). White amorphous solid; yield: 75%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.31 (d, J = 6.6 Hz, 3H), 3.49 (q, J = 6.6 Hz, 1H), 7.45–7.53 (m, 7H), 7.61–7.63 (m, 2H), 8.03 (t, J = 7.5 Hz, 1H), 8.59 (s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.3 (1C), 40.3 (1C), 114.4 (1C), 119.5 (1C), 121.5 (1C), 127.0 (1C), 127.8 (4C), 128.0 (1C), 128.2 (1C), 128.4 (2C), 130.9 (1C), 131.5 (1C), 135.7 (1C), 140.4 (1C), 160.5 (1C), 161.9 (1C), 164.2 (1C), 172.5 (1C); HR-ESI-MS [M + H]+ calculated for C22H15BrF2N2O, 440.0336, found 441.2681; anal. calcd for C22H15BrF2N2O (440.03) C, 59.88; H, 3.43; Br, 18.11; F, 8.61; N, 6.35; O, 3.63; found: C, 59.89; H, 3.45; N, 6.35.
3.4.21. 2-(2,5-Dimethoxyphenyl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl) ethyl)-1,3,4-oxadiazole (25). White amorphous solid; yield: 78%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.23 (d, J = 6.7 Hz, 3H), 3.54 (q, J = 6.7 Hz, 1H), 3.74 (s, 3H), 3.90 (s, 3H), 6.90 (d, J = 7.0 Hz, 1H), 7.01–7.03 (m, 5H), 7.17 (s, 1H), 7.26 (d, J = 7.0 Hz, 1H), 7.46–7.49 (m, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 19.7 (1C), 40.7 (1C), 56.0 (2C), 110.5 (2C), 112.5 (1C), 115.4 (2C), 127.2 (1C), 127.7 (3C), 128.4 (1C), 128.6 (2C), 131.7 (1C), 137.5 (1C), 141.4 (1C), 157.8 (1C), 160.65 (1C), 160.1 (1C), 164.2 (1C), 172.5 (1C); HR-ESI-MS [M + H]+ calculated for C24H21FN2O3, 404.1536, found 404.1536; anal. calcd for C24H21FN2O3 (404.15) C, 71.27; H, 5.23; F, 4.70; N, 6.93; O, 11.87; found: C, 71.29; H, 5.24; N, 4.67.
3.4.22. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(3-(trifluoromethyl) phenyl)-1,3,4-oxadiazole (26). White amorphous solid; yield: 92%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.58 (d, J = 6.9 Hz, 3H), 2.69 (q, J = 6.9 Hz, 1H), 7.31–7.62 (m, 5H), 7.68–7.80 (m, 3H), 7.78 (d, J = 7.5 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H), 8.19 (t, J = 8.5 Hz, 1H), 8.29 (d, J = 8.5 Hz, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.3 (1C), 40.7 (1C), 115.4 (1C), 123.7 (2C), 126.2 (1C), 126.5 (1C), 127.2 (1C), 127.4 (1C), 127.8 (3C), 128.0 (1C), 128.2 (1C), 128.4 (2C), 130.9 (1C), 131.4 (1C), 135.1 (1C), 139.8 (1C), 158.7 (1C), 164.2 (1C), 170.9 (1C); HR-ESI-MS [M + H]+ calculated for C23H16F4N2O, 412.1199, found 412.3796; anal. calcd for C23H16F4N2O (412.12) C, 66.99; H, 3.91; F, 18.43; N, 6.79; O, 3.88; found: C, 67.01; H, 3.90; N, 6.77.
3.4.23. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(4-(methylthio)phenyl)-1,3,4-oxadiazole (27). White amorphous solid; yield: 78%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.42 (d, J = 7.1 Hz, 3H), 2.01 (s, 3H), 3.52 (q, J = 7.1 Hz, 1H), 7.25–7.28 (m, 5H), 7.29 (d, J = 7.8 Hz, 2H), 7.49–7.51 (m, 3H), 7.86 (d, J = 7.8 Hz, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 16.2 (1C), 20.3 (1C), 41.1 (1C), 116.4 (1C), 121.8 (1C), 125.0 (2C), 126.8 (1C), 127.6 (5C), 128.2 (1C), 128.8 (2C), 131.9 (1C), 136.1 (1C), 140.2 (1C), 143.3 (1C), 160.7 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C23H19FN2OS, 390.1202, found 390.4732; anal. calcd for C23H19FN2OS (390.12) C, 70.75; H, 4.90; F, 4.87; N, 7.17; O, 4.10; S, 8.21; found: C, 70.76; H, 4.91; N, 7.14.
3.4.24. 2-Bromo-4-(5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazol-2-yl)phenol (28). Brownish amorphous solid; yield: 75%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.33 (d, J = 6.8 Hz, 3H), 3.42 (q, J = 6.8 Hz, 1H), 7.23–7.25 (m, 5H), 7.29 (d, J = 7.0 Hz, 1H), 7.32 (d, J = 7.0 Hz, 1H), 7.89–7.91 (m, 3H), 8.22 (d, J = 1.2 Hz, 1H), 10.55 (br.s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.3 (1C), 40.7 (1C), 109.7 (1C), 115.4 (1C), 116.9 (1C), 126.0 (1C), 127.2 (1C), 127.6 (3C), 128.2 (1C), 128.4 (2C), 129.0 (1C), 130.9 (1C), 131.0 (1C), 135.5 (1C), 139.2 (1C), 152.3 (1C), 159.7 (1C), 163.2 (1C), 171.3 (1C); HR-ESI-MS [M + H]+ calculated for C22H16BrFN2O2, 438.9535, found 439.1737; anal. calcd for C22H16BrFN2O2 (438.95) C, 60.15; H, 3.67; Br, 18.19; F, 4.32; N, 6.38; O, 7.28; found: C, 60.16; H, 3.65; N, 6.39.
3.4.25. 2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-5-(1-(2-fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazole (29). Brown amorphous solid; yield: 72%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.25 (d, J = 7.0 Hz, 3H), 3.06 (q, J = 7.0 Hz, 1H), 7.47–7.53 (m, 8H), 7.59–7.88 (m, 5H), 7.78 (d, J = 7.5 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 8.31 (s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.7 (1C), 41.7 (1C), 116.4 (1C), 124.7 (1C), 126.2 (1C), 126.8 (1C), 127.2 (1C), 127.4 (1C), 127.8 (7C), 128.7 (4C), 129.9 (1C), 131.9 (1C), 133.1 (1C), 136.5 (1C), 139.3 (2C), 159.5 (1C), 164.2 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C28H20F2N2O, 438.1649, found 438.4940; anal. calcd for C28H20F2N2O (438.16) C, 76.70; H, 4.60; F, 8.67; N, 6.39; O, 3.65; found: C, 76.73; H, 4.57; N, 6.40.
3.4.26. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(5-methylthiophen-2-yl)-1,3,4-oxadiazole (30). White amorphous solid; yield: 82%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.31 (d, J = 7.1 Hz, 3H), 2.41 (s, 3H), 3.41 (q, J = 7.1 Hz, 1H), 6.88–6.90 (m, 6H), 7.13 (d, J = 7.6 Hz, 1H), 7.56–7.60 (m, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 16.4 (1C), 20.3 (1C), 41.7 (1C), 113.4 (1C), 124.7 (1C), 127.2 (1C), 127.4 (1C), 127.8 (3C), 128.2 (1C), 128.4 (2C), 130.9 (1C), 133.1 (1C), 136.5 (1C), 141.2 (1C), 142.5 (1C), 160.7 (1C), 162.2 (1C), 173.3 (1C); HR-ESI-MS [M + H]+ calculated for C21H17FN2OS, 364.1046, found 364.4359; anal. calcd for C21H17FN2OS (364.10) C, 69.21; H, 4.70; F, 5.21; N, 7.69; O, 4.39; S, 8.80; found: C, 69.22; H, 4.73; N, 7.67.
3.4.27. 3-(5-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-1,3,4-oxadiazol-2-yl)benzene-1,2-diol (31). Off white amorphous solid; yield: 92%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.25 (d, J = 6.4 Hz, 3H), 3.42 (q, J = 6.4 Hz, 1H), 6.87 (d, J = 7.0 Hz, 1H), 7.16–7.20 (m, 5H), 7.25 (t, J = 7.1 Hz, 1H), 7.37 (d, J = 7.1 Hz, 1H), 7.81–7.84 (m, 3H), 10.52 (br.s, 2H); 13C-NMR (150 MHz, DMSO-d6) δ 18.9 (1C), 41.1 (1C), 108.5 (1C), 113.1 (1C), 115.4 (1C), 125.9 (1C), 127.0 (1C), 127.4 (3C), 128.2 (1C), 128.8 (2C), 129.8 (1C), 131.9 (1C), 136.3 (1C), 140.2 (1C), 146.6 (1C), 148.9 (1C), 160.7 (1C), 165.2 (1C), 173.5 (1C); HR-ESI-MS [M + H]+ calculated for C22H17FN2O3, 376.1223, found 376.3804; anal. calcd for C22H17FN2O3 (376.12) C, 70.20; H, 4.55; F, 5.05; N, 7.44; O, 12.75; found: C, 70.22; H, 4.53; N, 7.46.
3.4.28. 2-(1-(2-Fluoro-[1,1′-biphenyl]-4-yl)ethyl)-5-(2-nitrophenyl)-1,3,4-oxadiazole (32). White amorphous solid; yield: 83%; 1H-NMR (400 MHz, CDCl3; δ, ppm): 1.31 (d, J = 6.4 Hz, 3H), 2.69 (q, J = 6.4 Hz, 1H), 7.46–7.51 (m, 3H), 7.53–7.65 (m, 2H), 7.82–7.86 (m, 3H), 8.02 (d, J = 8.2 Hz, 2H), 8.14 (d, J = 8.6 Hz, 1H), 8.59 (s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ 19.3 (1C), 42.7 (1C), 107.4 (1C), 116.4 (1C), 119.7 (1C), 126.2 (1C), 127.3 (3C), 128.1 (2C), 128.5 (3C), 129.3 (1C), 130.9 (1C), 136.5 (1C), 138.2 (1C), 139.7 (1C), 158.7 (1C), 163.0 (1C), 172.3 (1C); HR-ESI-MS [M + H]+ calculated for C22H16FN3O3, 389.1176, found 389.3791; anal. calcd for C22H16FN3O3 (389.12) C, 67.86; H, 4.14; F, 4.88; N, 10.79; O, 12.33; found: C, 67.85; H, 4.16; N, 4.90.
4. Conclusion
Twenty-eight novel oxadiazole derivatives (5–32) based on the commercially available drug (S)-flurbiprofen have been synthesized in very good yields through multistep reactions. The synthesized analogues were confirmed by way of HR-MS (ESI+), and 1H-NMR spectroscopic procedures. As a final point, the compounds were tested for their in vitro urease inhibitory activity. All the compounds showed varying degrees of inhibition activity. Fourteen compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 were found as the supreme powerful urease inhibitors having IC50 values of 12 ± 0.9 to 20 ± 0.5 μM, better than the standard thiourea (IC50 = 22 ± 2.2 μM). On the other hand, nine compounds exhibited significant activity, while five compounds showed moderate to less urease inhibitory activity. The docking results indicated that compounds 20, 26, 30, 24, 21, 16, 28, 31, 32, 7, 19, 13, 10, and 6 had lower docking scores than the reference compound and revealed better interactions with the urease enzyme, which was in agreement with the experimental study.
Author contributions
S. A. and A. S. synthesized the compounds. A. A. and M. K. wrote the original draft of the manuscript. A. W. and A. K. conducted activity and docking study. A. F. A. and M. A. performed structural elucidation. M. K. and A. Al supervised the project and assisted in reviewing and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Authors are thankful to researchers supporting project number (RSP2023R335), King Saud University, Riyadh, Saudi Arabia.
References
- K. Biernacki, M. Daśko, O. Ciupak, K. Kubiński, J. Rachon and S. Demkowicz, Novel 1,2,4-oxadiazole derivatives in drug discovery, Pharmaceuticals, 2020, 13(6), 111 CrossRef CAS PubMed
 .
. - I. O. Alisi, A. Uzairu and S. E. Abechi, Free radical scavenging mechanism of 1,3,4-oxadiazole derivatives: thermodynamics of O–H and N–H bond cleavage, Heliyon, 2020, 6(3), e03683 CrossRef CAS .
- A. S. Alp, G. Kilcigil, E. D. Özdamar, T. Çoban and B. Eke, Synthesis and evaluation of antioxidant activities of novel 1,3,4-oxadiazole and imine containing 1H-benzimidazoles, Turk. J. Chem., 2015, 39(1), 42–53 CrossRef CAS .
- M. M. G. El-Din, M. I. El-Gamal, M. S. Abdel-Maksoud, K. H. Yoo and C.-H. Oh, Synthesis and in vitro antiproliferative activity of new 1,3,4-oxadiazole derivatives possessing sulfonamide moiety, Eur. J. Med. Chem., 2015, 90, 45–52 CrossRef .
- B. B. Kashid, P. H. Salunkhe, B. B. Dongare, K. R. More, V. M. Khedkar and A. A. Ghanwat, Synthesis of novel of 2, 5-disubstituted 1,3,4-oxadiazole derivatives and their in vitro anti-inflammatory, anti-oxidant evaluation, and molecular docking study, Bioorg. Med. Chem. Lett., 2020, 30(12), 127136 CrossRef CAS PubMed .
- M. Khan, A. Alam, K. M. Khan, U. Salar, S. Chigurupati, A. Wadood, F. Ali, J. I. Mohammad, M. Riaz and S. Perveen, Flurbiprofen derivatives as novel α-amylase inhibitors: Biology-oriented drug synthesis (BIODS), in vitro, and in silico evaluation, Bioorg. Chem., 2018, 81, 157–167 CrossRef CAS PubMed .
- S. Shah, M. Khan, M. Ali, A. Wadood, A. Ur Rehman, Z. Shah, M. Yousaf, U. Salar and K. M. Khan, Bis-1,3,4-Oxadiazole Derivatives as Novel and Potential Urease Inhibitors; Synthesis, In Vitro, and In Silico Studies, Med. Chem., 2022, 18(7), 820–830 CrossRef CAS PubMed .
- A. R. Nesaragi, R. R. Kamble, S. Dixit, B. Kodasi, S. R. Hoolageri, P. K. Bayannavar, J. P. Dasappa, S. Vootla, S. D. Joshi and V. M. Kumbar, Green synthesis of therapeutically active 1,3,4-oxadiazoles as antioxidants, selective COX-2 inhibitors and their in silico studies, Bioorg. Med. Chem. Lett., 2021, 43, 128112 CrossRef CAS PubMed .
- S. G. Musharraf, A. Bibi, N. Shahid, M. Najam-ul-Haq, N. Ambreen, M. Khan, K. M. Khan and M. I. Choudhary, Benzimidazole, coumrindione and flavone derivatives as alternate UV laser desorption ionization (LDI) matrices for peptides analysis, Chem. Cent. J., 2013, 7(1), 1–13 CrossRef PubMed .
- K. Mohammed Khan, M. Khan, N. Ambreen, F. Rahim, S. Naureen, S. Perveen, M. Iqbal Choudhary and W. Voelter, Synthesis and β-glucuronidase inhibitory potential of benzimidazole derivatives, Med. Chem., 2012, 8(3), 421–427 CrossRef PubMed .
- A. Alam, M. Ali, N. U. Rehman, S. Ullah, S. A. Halim, A. Latif, Zainab, A. Khan, O. Ullah and S. Ahmad, Bio-oriented synthesis of novel (S)-flurbiprofen clubbed hydrazone Schiff's bases for diabetic management: In vitro and in silico studies, Pharmaceuticals, 2022, 15(6), 672 CrossRef CAS PubMed .
- S. Ahmad, M. Khan, M. I. A. Shah, M. Ali, A. Alam, M. Riaz and K. M. Khan, Synthetic Transformation of 2-{2-Fluoro [1,1′-biphenyl]-4-yl} Propanoic Acid into Hydrazide–Hydrazone Derivatives: In Vitro Urease Inhibition and In Silico Study, ACS Omega, 2022, 7(49), 45077–45087 CrossRef CAS PubMed .
- P. Çıkla, E. Tatar, İ. Küçükgüzel, F. Şahin, D. Yurdakul, A. Basu, R. Krishnan, D. B. Nichols, N. Kaushik-Basu and Ş. G. Küçükgüzel, Synthesis and characterization of flurbiprofen hydrazide derivatives as potential anti-HCV, anticancer and antimicrobial agents, Med. Chem. Res., 2013, 22, 5685–5699 CrossRef .
- A. Khullar, A review on Flurbiprofen: Therapeutic challenges in Emerging Active Metabolizing Biphenyl, Bull. Pure Appl. Sci., 2020, 39, 77–89 CrossRef .
- P. Çıkla, E. Tatar and İ. Küçükgüzel, et al., Synthesis and characterization of flurbiprofen hydrazide derivatives as potential anti-HCV, anticancer and antimicrobial agents, Med. Chem. Res., 2013, 22, 5685–5699 CrossRef .
- S. Aydin, N. Kaushik-Basu, P. Arora, B. Amartya, D. Nichols, T. T TALELE, M. Akkurt, İ. Çelik, O. Büyükgüngör and Ş. G. Küçükgüzel, Microwave assisted synthesis of some novel Flurbiprofen hydrazidehydrazones as anti-HCV NS5B and anticancer agents, Marmara Pharm. J., 2013, 17(1), 26–34 CrossRef CAS .
- K. A. Musa and L. A. Eriksson, Photochemical and photophysical properties, and photodegradation mechanism, of the non-steroid anti-inflammatory drug Flurbiprofen, J. Photochem. Photobiol., A, 2009, 202(1), 48–56 CrossRef CAS .
- S. Sagdinc and H. Pir, Spectroscopic and DFT studies of flurbiprofen as dimer and its Cu (II) and Hg (II) complexes, Spectrochim. Acta, Part A, 2009, 73(1), 181–194 CrossRef PubMed .
- M. Taha, N. H. Ismail, S. Imran, A. Wadood, M. Ali, F. Rahim, A. A. Khan and M. Riaz, Novel thiosemicarbazide–oxadiazole hybrids as unprecedented inhibitors of yeast α-glucosidase and in silico binding analysis, RSC Adv., 2016, 6(40), 33733–33742 RSC .
- M. Taha, T. Noreen, S. Imran, F. Nawaz, S. Chigurupati, M. Selvaraj, F. Rahim, N. Hadiani Ismail, A. Kumar and A. Mosaddik, Synthesis, α-amylase inhibition and molecular docking study of bisindolylmethane sulfonamide derivatives, Med. Chem. Res., 2019, 28, 2010–2022 CrossRef CAS .
- F. Rahim, H. Ullah, M. T. Javid, A. Wadood, M. Taha, M. Ashraf, A. Shaukat, M. Junaid, S. Hussain and W. Rehman, Synthesis, in vitro evaluation and molecular docking studies of thiazole derivatives as new inhibitors of α-glucosidase, Bioorg. Chem., 2015, 62, 15–21 CrossRef CAS PubMed .
- M. Taha, N. H. Ismail, S. Imran, A. Wadood, F. Rahim, S. M. Saad, K. M. Khan and A. Nasir, Synthesis, molecular docking and α-glucosidase inhibition of 5-aryl-2-(6′-nitrobenzofuran-2′-yl)-1,3,4-oxadiazoles, Bioorg. Chem., 2016, 66, 117–123 CrossRef CAS PubMed .
- M. Nawaz, M. Taha, F. Qureshi, N. Ullah, M. Selvaraj, S. Shahzad, S. Chigurupati, S. A. Abubshait, T. Ahmad and S. Chinnam, Synthesis, α-amylase and α-glucosidase inhibition and molecular docking studies of indazole derivatives, J. Biomol. Struct. Dyn., 2022, 40(21), 10730–10740 CrossRef CAS PubMed .
- W. Yu, G. Huang, Y. Zhang, H. Liu, L. Dong, X. Yu, Y. Li and J. Chang, I2-mediated oxidative C–O bond formation for the synthesis of 1,3,4-oxadiazoles from aldehydes and hydrazides, J. Org. Chem., 2013, 78(20), 10337–10343 CrossRef CAS PubMed .
- W.-J. Mao, P.-C. Lv, L. Shi, H.-Q. Li and H.-L. Zhu, Synthesis, molecular docking and biological evaluation of metronidazole derivatives as potent Helicobacter pylori urease inhibitors, Bioorg. Med. Chem., 2009, 17(21), 7531–7536 CrossRef CAS PubMed .
- G. Akyüz, Synthesis and urease inhibition studies of some new quinazolinones, J. Heterocycl. Chem., 2021, 58(5), 1164–1170 CrossRef .
- Z. Amtul, R. Siddiqui and M. Choudhary, Chemistry and mechanism of urease
inhibition, Curr. Med. Chem., 2002, 9(14), 1323–1348 CrossRef CAS PubMed .
- L. Mazzei, U. Contaldo, F. Musiani, M. Cianci, G. Bagnolini, M. Roberti and S. Ciurli, Inhibition of Urease, a Ni-Enzyme: The Reactivity of a Key Thiol With Mono-and Di-Substituted Catechols Elucidated by Kinetic, Structural, and Theoretical Studies, Angew. Chem., 2021, 133(11), 6094–6100 CrossRef .
- A. Wadood, A. Ajmal, M. Junaid, A. U. Rehman, R. Uddin, S. S. Azam, A. Z. Khan and A. Ali, Machine Learning-based Virtual Screening for STAT3 Anticancer Drug Target, Curr. Pharm. Des., 2022, 28(36), 3023–3032 CrossRef CAS PubMed .
- B. Bano, K. M. Khan, B. Fatima, M. Taha, N. H. Ismail, A. Wadood, M. Ghufran and S. Perveen, Synthesis, in vitro β-glucuronidase inhibitory potential and molecular docking studies of quinolines, Eur. J. Med. Chem., 2017, 139, 849–864 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Aftab Alamb,
Amar Ajmalc,
Abdul Wadoodc,
Azim Khand,
Abdullah F. AlAsmarie,
Metab Alharbie,
Abdulrahman Alshammarie and
Abdul Shakoora
*a,
Aftab Alamb,
Amar Ajmalc,
Abdul Wadoodc,
Azim Khand,
Abdullah F. AlAsmarie,
Metab Alharbie,
Abdulrahman Alshammarie and
Abdul Shakoora