
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(triNHC)-catalyzed polymerization of glycidol to produce ultralow-branched polyglycerol†
Kihyuk Sunga,
Jinsu Baek b,
Soonyoung Choic,
Byeong-Su Kimb,
Sang-Ho Leec,
In-Hwan Lee*d and
Hye-Young Jang*a
b,
Soonyoung Choic,
Byeong-Su Kimb,
Sang-Ho Leec,
In-Hwan Lee*d and
Hye-Young Jang*a
aDepartment of Energy Systems Research, Ajou University, Suwon 16499, Korea. E-mail: hyjang2@ajou.ac.kr; Tel: +82(031)-219-2555
bDepartment of Chemistry, Yonsei University, Seoul 03722, Korea
cCenter for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology (KRICT), Ulsan 44412, Korea
dDepartment of Chemistry, Ajou University, Suwon 16499, Korea. E-mail: ilee@ajou.ac.kr
First published on 11th August 2023
Abstract
We have successfully synthesized a novel form of polyglycerol with an unprecedentedly low degree of branching (DB = 0.08–0.18), eliminating the need for glycidol protection. Leveraging the remarkable efficiency and selectivity of our Cu(triNHC) catalyst, comprising copper(I) ions and NHC ligands, we achieved a highly selective polymerization process. The proposed Cu-coordination mechanisms presented the formation of linear L1,3 units while effectively suppressing dendritic units. Consequently, our pioneering approach yielded polyglycerol with an ultralow DB and exceptional yields. To comprehensively assess the physical properties and topology of the synthesized polyglycerol, we employed 1H diffusion-ordered spectroscopy, size-exclusion chromatography, and matrix-assisted laser desorption/ionization-time of flight spectrometry. Remarkably, the ultralow-branched cyclic polyglycerol (DB = 0.08) synthesized at 0 °C showcased extraordinary characteristics, exhibiting the lowest diffusion coefficient and the highest molecular weight. This achievement establishes the significant potential of our polyglycerol with a low degree of branching, revolutionizing the field of biocompatible polymers.
Introduction
Polyglycerol is a highly regarded alternative to commercial poly(ethylene glycol) (PEG) due to its biocompatibility, water-solubility, low toxicity, and hydroxyl groups.1–5 Compared to PEG, polyglycerol has a higher degree of water solubility and amorphous nature because of the hydroxy groups present in its polymer chain.6 Furthermore, these hydroxy groups enable the polymer to bind to important biological molecules and metal ions for catalysis.7–9 The tunable synthesis of polyglycerol has been widely investigated due to its versatile characteristics, including the control of its degree of branching (DB). By controlling the DB, the rheological and thermal properties of polyglycerol can be manipulated, which allows for a wide range of applications in the polymer industry.4,10–12A number of techniques for the synthesis of polyglycerol with a high DB have been reported. For example, anionic,13–15 cationic,16–22 frustrated Lewis pair-catalyzed,23 and heterogeneous double metal cyanide (DMC)-catalyzed polymerization24 have been investigated for the synthesis of hyperbranched polyglycerol (i.e., DB > 0.5). In contrast, the synthesis of low-branched polyglycerol has been more limited. To synthesize polyglycerol with an ultralow DB, protected glycidol ether has been utilized in anionic ring-opening polymerization, hydrolyzed diethylzinc catalysis, and tri-isobutyl aluminum/ammonium salts catalysis.11,25 However, the deprotection of the alcohol is required after polymerization in these methods. The polymerization of unprotected glycidol with a low DB is challenging due to the facile chain growth at each hydroxy group in the polymer chain. Although various methods, such as kinetically controlled reactions with Sn(II) catalysts,8 batch polymerization with DMC catalysts,24 and aqueous epoxide ring-opening (AEROP),26 have been employed to produce polyglycerol with a low DB (0.15–0.29), there is still a need for the development of a simple and efficient catalyst that can promote the synthesis of polyglycerol with an ultralow DB (Scheme 1).
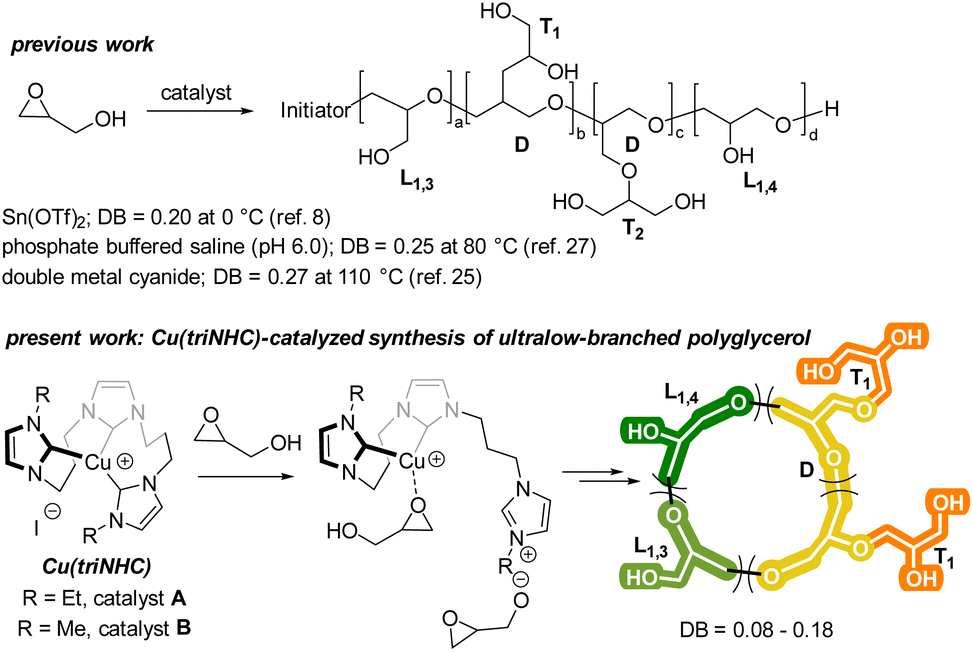 | ||
| Scheme 1 Synthesis of polyglycerol with low DBs. | ||
In a recent report, we described the synthesis of Cu(triNHC) (triNHC = tri-N-heterocyclic carbene) complexes and their use as catalysts in click chemistry and CO2 conversion.27,28 In this study, it is suggested that both the Cu ions and the dissociated NHC ligands in the Cu(triNHC) complex could facilitate the polymerization of glycidol with high tunability. The Cu ions coordinate with the oxygen in the epoxide, and the NHC ligand induces the deprotonation of the glycidol hydroxyl group, which initiates polymerization. The resulting polymer is an ultralow-branched polyglycerol with cyclic topology, as illustrated in Scheme 1. Its physical properties were evaluated using several techniques, including matrix-assisted laser desorption/ionization-time of flight (MALDI-ToF) spectrometry, size-exclusion chromatography (SEC), differential scanning calorimetry (DSC), and 1H diffusion-ordered spectroscopy (DOSY).
Experimental
General
Glycidol (96.0%), Cu(OTf)2 (98.0%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (98%), copper iodide (99.999%) were obtained from Sigma Aldrich. 1,3-Bis(2,6-diisopropylphenyl)imidazole-2-ylidene (98.0%) was obtained from Tokyo Chemical Industry. Toluene (99.9%) was sourced from Samchun Chemicals. Glycidol and toluene were dried with CaH2 and distilled prior to use. Proton nuclear magnetic resonance (1H NMR) spectra were recorded with a Jeol Resonance ECZ600R (600 MHz) spectrometer. Chemical shifts are reported in delta (δ) units, parts per million (ppm) relative to the center of a peak at 2.50 ppm for DMSO-d6. Coupling constants are reported in Hertz (Hz). Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded with a Jeol Resonance ECZ600R (150 MHz) spectrometer. Chemical shifts are reported in delta (δ) units, parts per million (ppm) relative to the center of a peak at 39.52 ppm for DMSO-d6. 1H diffusion-ordered spectroscopy (DOSY) was recorded on a Bruker AVANCE III HD 300 spectrometer at 25 °C. All samples were dissolved in DMSO-d6. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) measurement was performed using autoflex maX from Bruker. 2,5-Dihydroxybenzoic acid (DHB) was used as the matrix. For DMF–SEC, three polystyrene-gel columns [KD-802 (from Shodex); pore size, 150 Å; 8 mm i.d. × 300 mm, KD-803 (from Shodex); pore size, 500 Å; 8 mm i.d. × 300 mm, KD-804 (from Shodex); pore size, 1500 Å; 8 mm i.d. × 300 mm] were connected to a PU-4180 pump, a RI-4030 refractive-index detector, and a UV-4075 ultraviolet detector (JASCO); the flow rate was maintained at 1.0 ml min−1. The columns were calibrated against 13 standard poly(ethylene glycol) (PEO) samples (Agilent Technologies; Mp = 980–811 500; Mw/Mn = 1.03–1.11) to analyze the obtained polymer samples. Differential scanning calorimetry (DSC) was conducted on polymer samples under a dry nitrogen flow (40 ml min−1) in the temperature range of −70 to +70 °C at a heating or cooling rate of 10 °C min−1 on a Q2000 calorimeter (TA Instruments). Thermal gravimetric analysis (TGA) was conducted on the polymer sample under 20 ml min−1 of a dry nitrogen in the temperature range of +25 to +800 °C at a heating rate of 10 °C min−1 on STA 8122 High Temperature (Rigaku).Experimental procedure for the polymerization of glycidol
Glycidol (148.2 mg, 2 mmol), catalysts (21.2 mg, 2 mol%), and toluene (1.0 ml) were added to the reaction vial. The mixture was stirred at the indicated temperature for 16 h under the nitrogen atmosphere. Then, the reaction mixture was washed with diethyl ether to remove the unreacted monomer. The product was dried at 50 °C for 1 day before obtaining the yield and sample analysis.Experimental procedure for the catalyst recycling experiment
Glycidol (148.2 mg, 2 mmol), catalyst (21.2 mg, 2 mol%), and toluene (1.0 ml) were added to the reaction vial. The mixture was stirred at the indicated temperature for 16 h under the nitrogen atmosphere. After the reaction, glycidol (148.2 mg, 2 mmol) was added repeatedly in each cycle. Then, the reaction mixture was washed with diethyl ether to remove the unreacted monomer. The product was dried at 50 °C for 1 day before obtaining the yield and sample analysis.Results and discussion
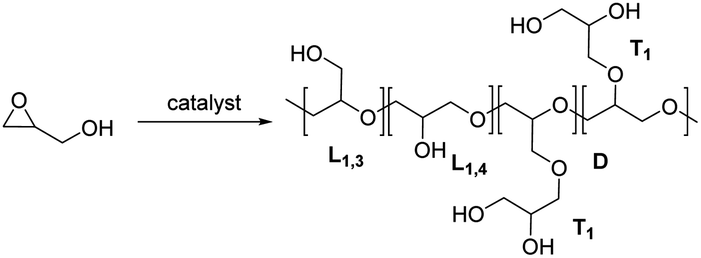
The polymerization of glycidol was conducted using catalyst A (2.0 mol%) in toluene at 25 °C, yielding a polymer with a 94% yield (entry 1 in Table 1). The DB of the resulting polymers was evaluated using the equation DB = 2D/(2D + L), where D is the fraction of dendritic units, and L is the fraction of linearly incorporated units (Table 1).29 The relative abundance for each unit within the synthesized polymers is also shown in Fig. 1. Polymerization with catalyst A at 25 °C produced a polymer with a very low DB (0.18). When the polymerization was carried out at 0 °C with catalyst A, a longer reaction time was required, resulting in an 87% yield with a DB of 0.08 (entry 2 in Table 1), the lowest DB reported to date for polyglycerol synthesized using glycidol without a protection group (see ESI, Table S1†). The polymer synthesized at 0 °C contained more L1,3 units and fewer D units than the polymer produced at 25 °C (Fig. 1). The higher number of L1,3 units was associated with the low DB of the polymer.30 The polymerization process with catalyst A was also investigated using different solvents (see ESI, Table S2†), and it was found that toluene produced the best results. Polyglycerol produced using this procedure exhibited insolubility in toluene, suppressing the growth of polymer chains with high molecular weight.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Temp (°C) | Time (h) | Yield (%) | DB | Mn (Da) | Mw/Mn | Tg (°C) | Diffusion coefficient (m2 s−1) |
| a Experimental conditions: the catalyst (2 mol%) and glycidol (2 mmol) in toluene (2.0 M) were stirred at the indicated temperature. DB was calculated by inverse-gated 13C NMR using the equation “2D/(2D + L)” (D = dendritic unit; L = linear unit). Mn was measured by size-exclusion chromatography (SEC) calibrated with PEO standard in DMF (45 °C, flow rate 1.0 ml min−1). Glass-transition temperature (Tg) was determined by differential scanning calorimetry (DSC) at a rate of 10 °C min−1. | |||||||||
| 1 | A | 25 | 16 | 94 | 0.18 | 2190 | 1.3 | −13 | 0.93 × 10−10 |
| 2 | A | 0 | 48 | 87 | 0.08 | 2660 | 1.6 | −16 | 0.67 × 10−10 |
| 3 | B | 25 | 16 | 85 | 0.12 | 2160 | 1.4 | −18 | 0.92 × 10−10 |
| 4 | IPr | 25 | 16 | 35 | 0.17 | 640 | 1.2 | −34 | 1.25 × 10−10 |
| 5 | TBD | 25 | 16 | 12 | 0.30 | 680 | 1.2 | −23 | 1.31 × 10−10 |
| 6 | TBD | 0 | 48 | 1.0 | — | — | — | — | — |
| 7 | CuI | 25 | 16 | — | — | — | — | — | — |
| 8 | Cu(OTf)2 | 25 | 16 | 98 | 0.41 | 1500 | 1.7 | −20 | — |
| 9 | Cu(OTf)2 | 0 | 48 | 98 | 0.34 | 1100 | 1.3 | −33 | — |
 |
|||||||||
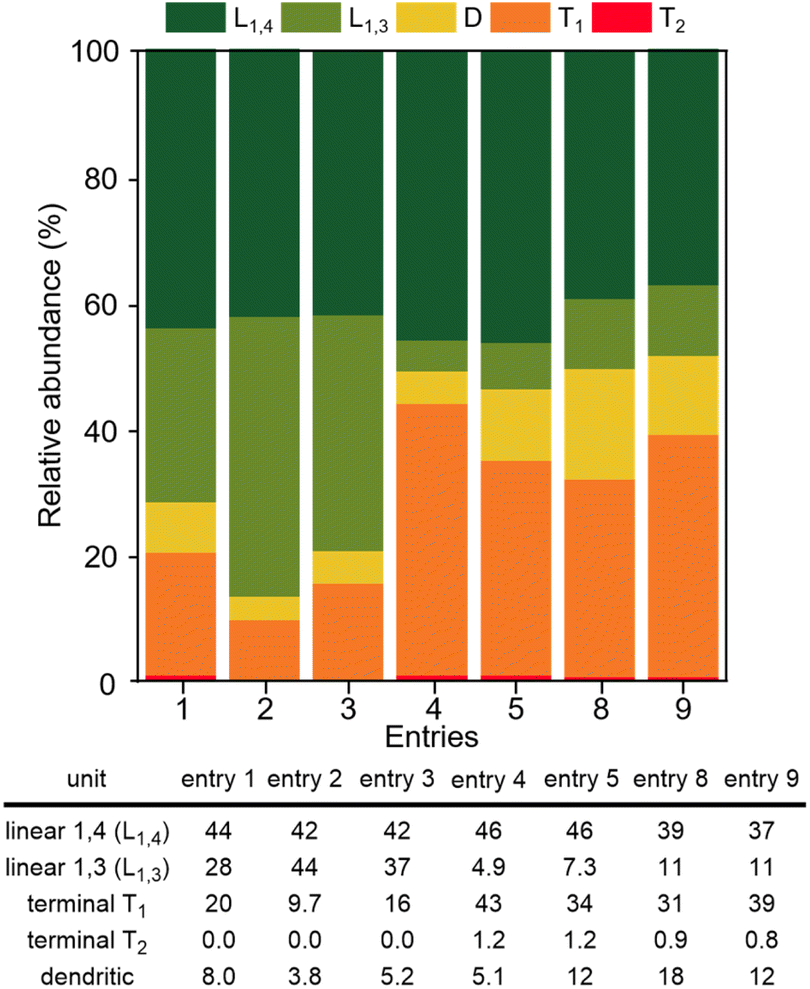 | ||
| Fig. 1 Relative abundance of the linear, terminal, and dendritic units for the polymers in Table 1 (calculated using inverse-gated 13C NMR). | ||
Catalyst B, which contains methyl-substituted NHC ligands, exhibited similar activity to catalyst A (entry 3 in Table 1). Using catalyst B, the relative abundance of D, L1,3, L1,4, and T1 was 5.2%, 37%, 42%, and 16%, respectively (Fig. 1). Catalyst B produced more L1,3 units and fewer D and T1 units than catalyst A. Similar high yields and low DBs of polyglycerol were observed for both catalysts A and B, indicating that the influence of the terminal substituent of NHC ligands is negligible. Cu(triNHC) catalysts contain Cu ions and NHC ligands, where Cu ions induce the Cu-coordination mechanism, and the carbene ligand dissociated from Cu(triNHC) catalysts is basic enough to initiate deprotonation of glycidol. The function of the carbene ligand of Cu(triNHC) in the polymerization was evaluated as follows. The organic carbene compound, 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene (IPr) and the organic base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were tested at room temperature, resulting in low polymer yields (entries 4 and 5 in Table 1).31 The reaction using TBD at 0 °C produced a polymer with a yield of only 1.0% (entry 6 in Table 1). IPr produced polyglycerol with a relatively low DB but high relative abundance of terminal units (43% for T1 and 1.2% for T2 units), low molecular weight (Mn = 640), and low yield, indicating that IPr was not as effective as Cu(triNHC) catalysts (catalysts A and B) in promoting chain growth. In contrast, the reaction using TBD produced a polymer with a DB of 0.30, higher than that of catalyst A or B (entry 5 of Table 1). The Cu(I) catalyst without carbene ligands did not induce the desired polymerization (entry 7 in Table 1), while Cu(OTf)2-catalyzed polymerization resulted in good yields at 25 °C and 0 °C (entries 8 and 9 of Table 1). Cu(OTf)2-induced polymerization at 25 °C and 0 °C led to a DB of 0.41 and 0.34, respectively, with more D and T units observed than with the Cu(triNHC) catalyst. Overall, both Cu(I) ions and carbene ligands are essential for the low DB of polyglycerol and chain growth while maintaining high catalytic activity.
The topology of the polyglycerol was analyzed through MALDI-ToF analysis, as shown in Fig. 2. Polyglycerol obtained using catalyst A at 25 °C (entry 1 in Table 1) displayed signals that corresponded to Mobserved (Da) = n × 74.04 + Na+ with regularly spaced peaks of 74.04 Da, which represents the mass of glycidol (Fig. 2a). The signals could be attributed to cyclic polymers or acyclic polymers with an epoxy group at the chain end (Fig. 2c). To determine the polymer topology, polyglycerol (entry 1 in Table 1) was reacted with N3−. The addition of azide was not observed in the reaction mixture (Fig. 2b and S2†). The polymer synthesized at 0 °C with catalyst A showed major peaks that corresponded to Mobserved (Da) = n × 74.04 + Na+ (see ESI, Fig. S3†). On the other hand, the polyglycerol obtained using catalyst B at 25 °C displayed signals indicating the presence of cyclic polymers (see ESI, Fig. S4†). The polymers synthesized using IPr and TBD exhibited peaks of Mobserved (Da) = n × 74.04 + Na+, which were further confirmed to have cyclic topology through N3− addition reactions (see ESI, Fig. S5 and S6†). MALDI-ToF analysis of the polymer produced using Cu(OTf)2 revealed the presence of both cyclic and acyclic structures (see ESI, Fig. S7 and S8†).
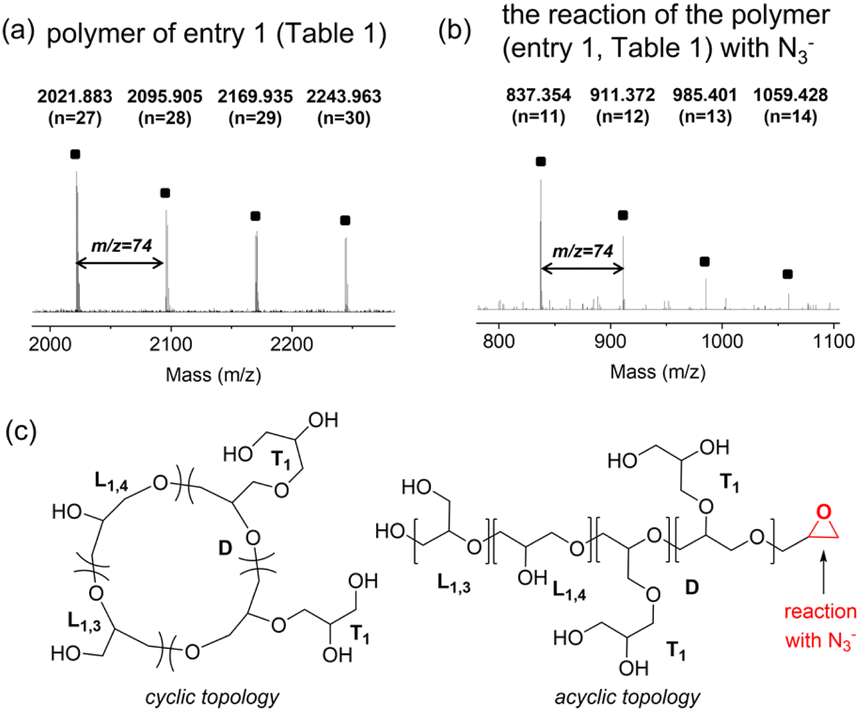 | ||
| Fig. 2 (a) MALDI-ToF analysis of the polyglycerol produced using catalyst A (entry 1 in Table 1), (b) MALDI-ToF analysis of the polyglycerol after the reaction with N3−, and (c) cyclic and acyclic polyglycerol structures. | ||
DOSY and SEC were employed to investigate the behavior of selected polyglycerol samples in solution. The diffusion coefficient obtained from DOSY provides information on the mobility of polymer molecules in fluid (Table 1 and Fig. S9–S15†), which is closely related to the hydrodynamic volume of the polymer. As the molecular weights obtained from SEC were closely related to the hydrodynamic volume, the correlation between the diffusion coefficient and the molecular weight (the hydrodynamic volume) of the polymers was examined. The polyglycerol samples for entries 1, 2, and 3 in Table 1 were analyzed using DMF–SEC, revealing an Mn of 2190, 2660, and 2160, respectively (see ESI, Fig. S16–S18†). The diffusion coefficients for these polymers were 0.93 × 10−10 m2 s−1, 0.67 × 10−10 m2 s−1, and 0.92 × 10−10 m2 s−1, respectively (Table 1). Entries 1 and 3 in Table 1 exhibited similar molecular weights and diffusion coefficients, while entry 2 had the lowest diffusion coefficient and the highest molecular weight. Previous research has shown that diffusion coefficients are influenced by several factors, including the DB, molecular weight, and topology of a polymer.23 The low diffusion coefficient for the polymer listed as entry 2 in Table 1 resulted from its high molecular weight and low DB. Additionally, chain-end analysis using MALDI-ToF revealed that a small proportion of acyclic chains were present for this polymer (see ESI, Fig. S3a†). Thus, the effect of the topology of polyglycerol in explaining the low diffusion coefficient cannot be completely ruled out. The polyglycerol listed as entry 4 in Table 1, which had a low molecular weight according to SEC analysis (Mn = 637), had a higher diffusion coefficient (1.25 × 10−10 m2 s−1) than entry 1 (Mn = 2190 and diffusion coefficient = 0.93 × 10−10 m2 s−1), though they exhibited similar DBs (0.18 for entry 1 and 0.17 for entry 4) and the same cyclic topology. Therefore, this comparison confirmed the close correlation between the molecular weight and the diffusion coefficient.32 Overall, there is a correlation that can be established between the diffusion coefficient, molecular weight, and hydrodynamic volume of polymers that have similar DB and topology. The glass transition temperature (Tg) of the polyglycerols was also measured (Table 1 and Fig. S33†). The Tg of polyglycerols can be affected by the intra- and intermolecular hydrogen bonding of the polymer chains arising from the DB, molecular weight, and topology. A comparison of Tg values for entries 1 and 4 showed that Tg was correlated with the molecular weights when the DBs were similar. The thermal property of polyglycerol was assessed using thermal gravimetric analysis (TGA), which revealed decomposition occurring at 393 °C (Fig. S34†).
The Cu(triNHC) catalyst exhibited a significant polyglycerol yield with remarkably low branching. The coordination of the three NHC ligands enhances the stability of Cu(I) catalysts, effectively preventing oxidation to Cu(II). Accordingly, we conducted accumulation experiments by adding glycidol to the reaction mixture to assess the recyclability of catalyst A (Fig. 3a). The yields for the second and third cycles remained high. The analysis of the unit distribution was conducted using the reaction mixture (Fig. 3b). Interestingly, the proportion of L1,3 units increased and that of T units decreased in the second and third cycles. Additionally, the SEC traces displayed a shift toward a higher molecular weight region and exhibited a bimodal distribution with an increase in the number of accumulation cycles (Fig. 3c). This finding indicates that the initiation process from the freshly added glycidol monomer may compete with the chain-extension process from the alcohol end-group of the pre-existing polyglycerol during the accumulation cycles. However, because polyglycerol is insoluble, additional glycidols were reacted with Cu(triNHC) catalysts to induce new chain growth from glycidol. The polymer chain growth from the pre-existing polyglycerol was not as facile as the formation of new chains from glycidol.
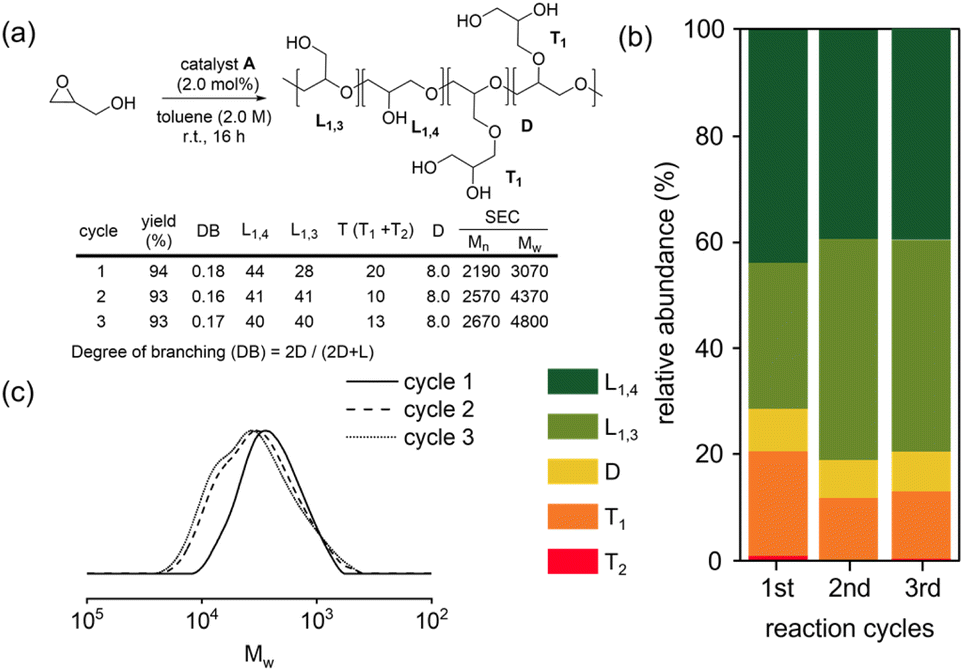 | ||
| Fig. 3 (a) Recycling experiments, (b) the relative abundance of the linear, terminal, and dendritic units for each cycle calculated using inverse-gated 13C NMR, and (c) SEC traces of the polyglycerols. | ||
Polyglycerol synthesis can occur via several mechanisms, including the activated chain end (ACE), activated monomer (AM), and metal-coordination mechanisms.16,19,24,26 In the case of the Cu(triNHC)-catalyzed reaction, the reaction mechanism was investigated based on the catalyst structure and the polymer chain units (Scheme 2). In our previous studies, it was found that Cu(triNHC) dissociates a carbene ligand to deprotonate the alcohol for the reaction.28 Similarly, in this study, the reaction begins with the deprotonation of glycidol by the dissociated carbene ligand from the Cu catalyst. The proposed Cu complex having a protonated imidazolium group was observed by 1H NMR of the reaction mixture (Fig. S35†). The catalytic reaction in dioxane which did not exhibit the polymer formation showed no dissociation of carbene ligand in the reaction mixture (Fig. S35†). When the glycidol alkoxide, located proximal to the imidazolium ion, adds to Cu-coordinated glycidol without proton exchange of resulting alkoxides, it leads to the production of L1,3 units.24 The Cu complex coordinated with the epoxide intermediate was observed by ESI-MS (see the ESI, Fig. S36†). On the other hand, the addition of glycerol alkoxide to the epoxide, followed by proton exchange forming terminal alkoxides, results in the formation of L1,4 units. Finally, the Cu(triNHC)-catalyzed intramolecular reaction of the epoxide and the alkoxide at each end of the polymer chain produces a cyclic polymer. The Cu-coordination mechanism suppressing proton exchange is more strongly favored at 0 °C than at 25 °C, resulting in more L1,3 units and a lower DB than the reaction at room temperature. Because this reaction is initiated by catalysts without additional anionic initiators, and the proton exchange is limited compared to anionic polymerization, the formation of dendritic units is suppressed, resulting in lower DB values. Poor polymerization results with the use of TBD at 0 °C suggest that base-induced anionic polymerization is likely to be suppressed at low temperatures. Overall, the superior activity of the Cu(triNHC) catalyst in producing polyglycerol with an ultralow DB and a cyclic topology was the result of the Cu-coordinated reaction of glycidol and glycidol alkoxide at the proximal carbenes inside the catalyst reaction sphere.24,33,34
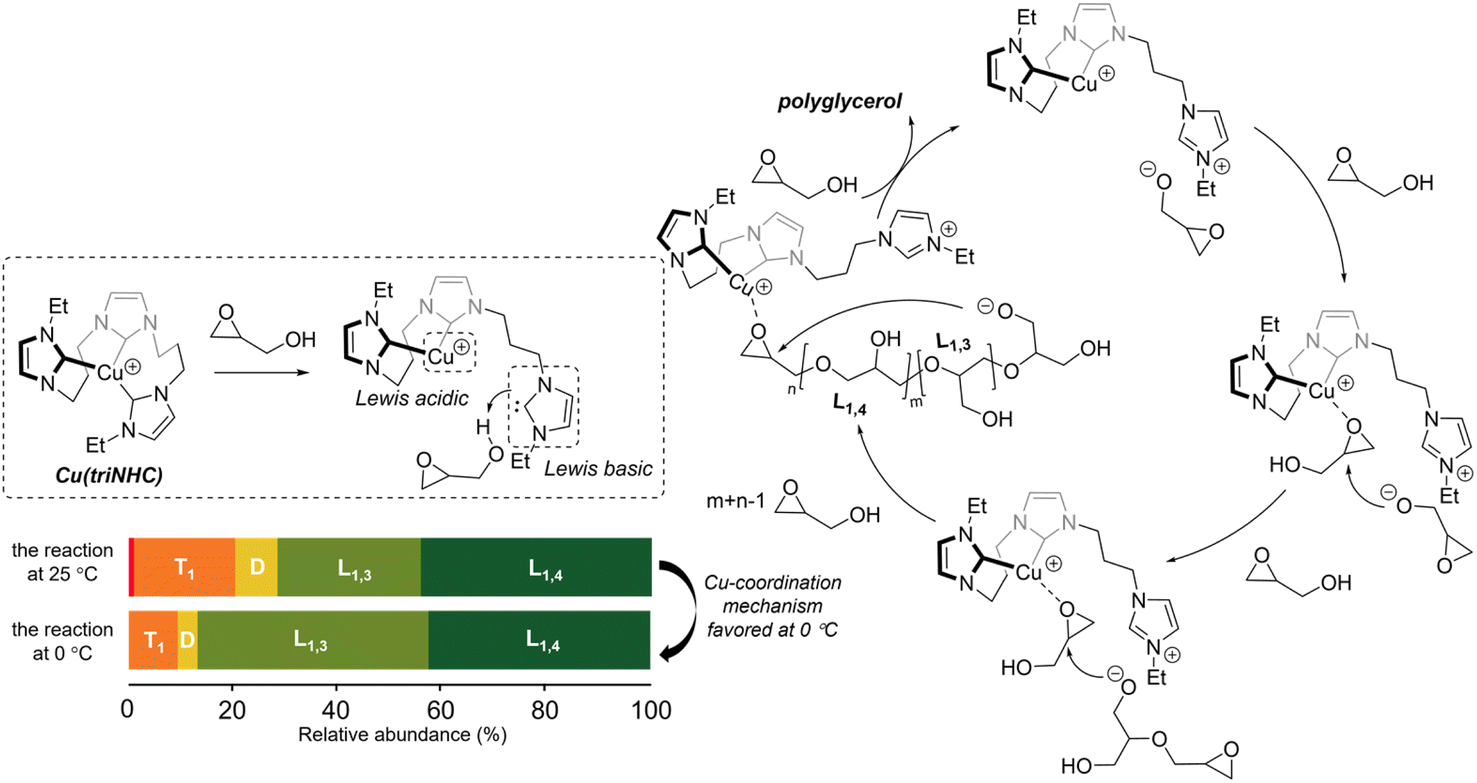 | ||
| Scheme 2 Cu-coordinated mechanisms for polyglycerol synthesis. | ||
Conclusions
We demonstrated the synthesis of polyglycerol with a low degree of branching and a cyclic topology using Cu(triNHC) catalysis. The unique structure of Cu(triNHC), which contains Cu ions and NHC ligands, facilitated the polymerization of glycidol with an extremely low DB. Glycidol alkoxides generated by the NHC ligand were provided to the Cu-coordinated glycidol, which promoted efficient polymerization with suppressed proton transfer and favored the formation of L1,3 units. Consequently, the higher number of L1,3 units and the lower number of L1,4 and D units resulted in polyglycerol with an ultralow DB. The high stability of the Cu(triNHC) catalyst was demonstrated in a recycling experiment, with high catalytic activity observed up to a third cycle. The obtained polyglycerol's topology, diffusion coefficient, molecular weight, and Tg were analyzed using MALDI-ToF, DOSY, SEC, and DSC. In comparison to previous research that primarily focused on synthesizing highly branched polyglycerols, the development of practical methods to produce cyclic ultralow-branched polyglycerols without glycidol protection is relatively rare. Therefore, our findings of Cu(triNHC)-catalyzed protocols expand the possibilities for using polyglycerol in the polymer and biomaterial industries.Author contributions
H.-Y. Jang: conceptualization, supervision, writing original draft, and review & editing. K. Sung: investigation and formal analysis. J. Baek, S. Choi: formal analysis. B.-S. Kim, S.-H. Lee, I.-H. Lee: review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Carbon to X Program (No. 2020M3H7A1098283) and National Research Foundation Program (No. 2022R1A2C1004387) by the Ministry of Science and ICT, and Basic Science Research Program (No. 2021R1A6A1A10044950) by the Ministry of Education, Republic of Korea.Notes and references
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- C. Siegers, M. Biesalski and R. Haag, Chem.–Eur. J., 2004, 10, 2831–2838 CrossRef CAS PubMed.
- Solvay Chemicals, Polyglycerols in Industrial Applications, 2005, http://www.solvaychemicals.com/ Search PubMed.
- M. Schömer, C. Schüll and H. Frey, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 995–1019 CrossRef.
- S. Abbina, S. Vappala, P. Kumar, E. M. J. Siren, C. C. La, U. Abbasi, D. E. Brooks and J. N. Kizhakkedathu, J. Mater. Chem. B, 2017, 5, 9249–9277 RSC.
- M. Schömer and H. Frey, Macromolecules, 2012, 45, 3039–3046 CrossRef.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 12, 190–218 CrossRef.
- B. R. Spears, J. Waksal, C. McQuade, L. Lanier and E. Harth, Chem. Commun., 2013, 49, 2394–2396 RSC.
- K. R. Kumar, K. N. Kizhakkedathu and D. Brooks, Macromol. Chem. Phys., 2004, 205, 567–573 CrossRef CAS.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed.
- A. Thomas, S. S. Müller and H. Frey, Biomacromolecules, 2014, 15, 1935–1954 CrossRef CAS PubMed.
- S. Y. Lee, M. Kim, T. K. Won, S. H. Back, Y. Hong, B.-S. Kim and D. J. Ahn, Nat. Commun., 2022, 13, 34300 Search PubMed.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- R. K. Kainthan, E. B. Muliawan, S. G. Hatzikiriakos and D. E. Brooks, Macromolecules, 2006, 39, 7708–7717 CrossRef CAS.
- C. Schubert, M. Schömer, M. Steube, S. Decker, C. Friedrich and H. Frey, Macromol. Chem. Phys., 2018, 219, 1700376 CrossRef.
- R. Tokar, P. Kubisa, S. Penczek and A. Dworak, Macromolecules, 1994, 27, 320–322 CrossRef CAS.
- A. T. Royappa, M. L. Vogt and V. Sharma, J. Appl. Polym. Sci., 2004, 91, 1344–1351 CrossRef CAS.
- I. Asenjo-Sanz, A. Veloso, J. I. Miranda, J. A. Pomposo and F. Barroso-Bujans, Polym. Chem., 2014, 5, 6905–6908 RSC.
- E. Mohammadifar, A. Bodaghi, A. Dadkjajtehrani, A. N. Kharat, M. Adeli and R. Haag, ACS Macro Lett., 2017, 6, 35–40 CrossRef CAS.
- M. Dadkhah, H. Shamlooei, E. Mohammadifar and M. Adeli, RSC Adv., 2018, 8, 217–221 RSC.
- S. E. Kim, H. J. Yang, S. Choi, E. Hwang, M. Kim, H.-J. Paik, J.-E. Jeong, Y. I. Park, J. C. Kim, B.-S. Kim and S.-H. Lee, Green Chem., 2022, 24, 251–258 RSC.
- M. A. A. Assiri, E. G. Urreizti, C. A. Pagnacco, E. Gónzalez de San Román and F. Barroso-Bujans, Eur. Polym. J., 2022, 171, 111194 CrossRef.
- S. E. Kim, Y.-R. Lee, M. Kim, E. Seo, H.-J. Paik, J. C. Kim, J.-E. Jeong, Y. I. Park, B.-S. Kim and S.-H. Lee, Polym. Chem., 2022, 13, 1243–1252 RSC.
- C. H. Tran, M. W. Lee, S. A. Kim, H. B. Jang and I. Kim, Macromolecules, 2020, 53, 2051–2060 CrossRef CAS.
- A. Haouet, M. Sepulchre and N. Spassky, Eur. Polym. J., 1983, 19, 1089–1098 CrossRef CAS.
- B. R. Spears, M. A. Marin, J. Montenegro-Burke, B. C. Evans, J. McLean and E. Harth, Macromolecules, 2016, 49, 2022–2027 CrossRef CAS.
- C. Seo, Y.-J. Cheong, W. Yoon, J. Kim, J. Shin, H. Yun, S.-J. Kim and H.-Y. Jang, Organometallics, 2021, 40, 16–22 CrossRef CAS.
- C. Seo, S. E. Kim, H. Kim and H.-Y. Jang, ACS Sustainable Chem. Eng., 2022, 10, 5643–5650 CrossRef CAS.
- D. Hölter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- C. Schubert, M. Schömer, M. Steube, S. Decker, C. Friedrich and H. Frey, Macromol. Chem. Phys., 2018, 219, 1700376 CrossRef.
- N. E. Kamber, W. Jeong, S. Gonzalez, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2009, 42, 1634–1639 CrossRef CAS.
- W. Li, H. Chung, C. Daeffler, J. A. Johnson and R. H. Grubbs, Macromolecules, 2012, 45, 9595–9603 CrossRef CAS PubMed.
- Y.-J. Huang, X.-H. Zhang, Z.-J. Hua, S.-L. Chen and G.-R. Qi, Macromol. Chem. Phys., 2010, 211, 1229–1237 CrossRef CAS.
- J. Ai, X. Min, C.-Y. Gao, H.-R. Tian, S. Dang and Z.-M. Sun, Dalton Trans., 2017, 46, 6756–6761 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04422j |
| This journal is © The Royal Society of Chemistry 2023 |