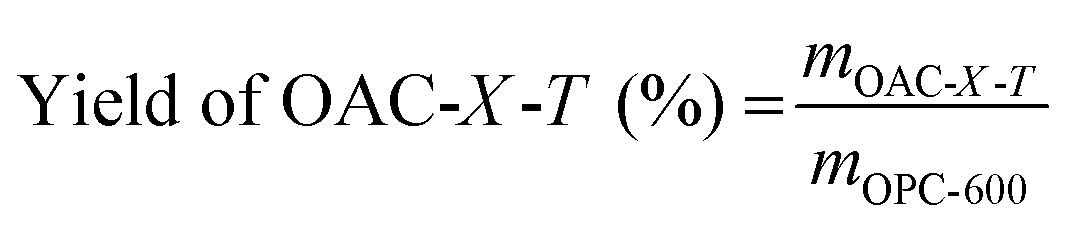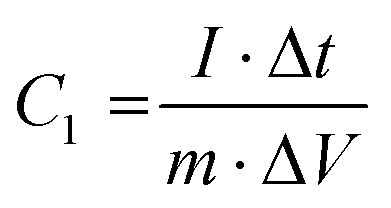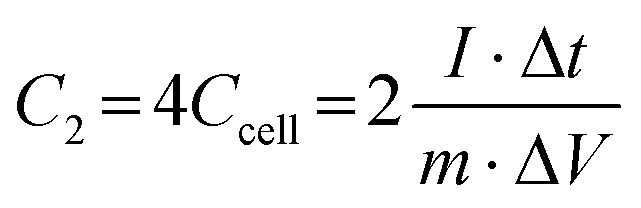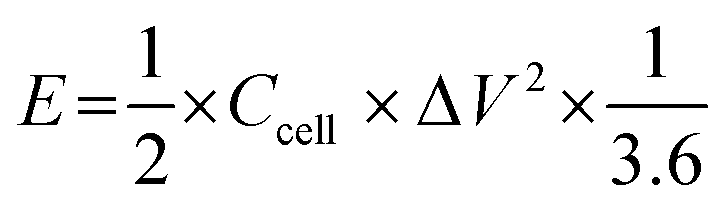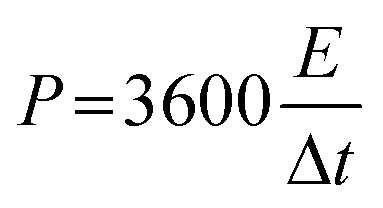DOI:
10.1039/D3RA04164F
(Paper)
RSC Adv., 2023,
13, 24140-24149
In situ N, O co-doped porous carbon derived from antibiotic fermentation residues as electrode material for high-performance supercapacitors†
Received
21st June 2023
, Accepted 30th July 2023
First published on 11th August 2023
Abstract
With the widespread use of antibiotics, the safe utilization of waste antibiotic fermentation residues has become an urgent issue to be resolved. In this study, in situ N, O co-doped porous carbon was prepared using fresh oxytetracycline fermentation residue under the mild activation of the green activator K2CO3. The optimal sample exhibited a 3D grid carbon skeleton structure, excellent specific surface area (SBET = 948 m2 g−1), and high nitrogen and oxygen content (N = 3.42 wt%, O = 14.86 wt%). Benefiting from its developed morphology, this sample demonstrated excellent electrochemical performance with a high specific capacitance of 310 F g−1 at a current density of 0.5 A g−1 in the three-electrode system. Moreover, it exhibited superior cycling stability with only a 5.32% loss of capacity after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in 6 M KOH aqueous electrolyte. Furthermore, the symmetric supercapacitor prepared from it exhibited a maximum energy density of 7.2 W h kg−1 at a power density of 124.9 W kg−1, demonstrating its promising application prospects. This study provided a green and facile process for the sustainable and harmless treatment of antibiotic fermentation residues.
000 cycles in 6 M KOH aqueous electrolyte. Furthermore, the symmetric supercapacitor prepared from it exhibited a maximum energy density of 7.2 W h kg−1 at a power density of 124.9 W kg−1, demonstrating its promising application prospects. This study provided a green and facile process for the sustainable and harmless treatment of antibiotic fermentation residues.
1. Introduction
Antibiotics are extensively deployed in therapeutic medicine, aquaculture, and livestock production.1,2 The worldwide consumption of antibiotics has surged by 46% over the past two decades.3 The problems of antibiotic resistance and environmental pollution are increasingly becoming serious due to the extensive use of antibiotic drugs. Christopher et al. developed a predictive estimation model that forecasts approximately 1.27 million deaths worldwide in 2019 being directly attributable to antibiotic resistance, posing a considerable threat to human health.4 Moreover, the production of antibiotics unavoidably generates massive amounts of antibiotic residues, including substances such as antibiotic fermentation residues (AFRs) and residual media, which can potentially harm the ecological environment.5 Therefore, AFRs are currently widely recognized as hazardous waste, and their proper disposal has become an urgent issue that must be addressed.
Traditional methods for treating AFRs involve incineration and landfill.6,7 Nonetheless, dioxins and other toxic substances can be easily generated during incineration, leading to secondary pollution via cross-media transfer.8 Direct landfilling often leads to difficulties such as a large area requirement, high disposal costs, and soil pollution due to the high water content and organic matter in AFRs.9 Many scholars have taken an interest in the pyrolytic carbonization treatment of antibiotic residues in recent years.5,10 As waste rich in nitrogen, oxygen, and organic matter, AFRs can be applied to a variety of fields by pyrolysis to produce functional porous carbon materials, which achieves the reduction and harmlessness of AFRs. Ahmad et al. utilized hydrothermal coupling pyrolytic carbonization methods to convert lincomycin fermentation residues into biochar, which was applied in the immobilization of lead ions in the soil.11 Wang et al. produced biochar from a mixture of penicillin fermentation residues and sludge, which was utilized for adsorbing penicillin from wastewater.12 Additionally, these residues have potential as raw materials for the preparation of carbon electrodes.13
Supercapacitors are rapidly emerging electronic energy storage devices that provide superior energy and power densities compared to conventional batteries.14 There are various potential applications of supercapacitors, including automotive power systems, consumer electronics, military equipment, and other fields.15–18 Porous carbon is often used as the electrode material for supercapacitors due to its high specific surface area, good chemical stability, low resistance, and high electrical conductivity.19 These properties make it an ideal material for optimizing the performance of supercapacitors. Moreover, doping nitrogen and oxygen into porous carbon can enhance its surface chemical activity and electron transport capabilities, thereby improving its electrochemical properties.20 An increasing number of biomass wastes containing high levels of heteroatomic (e.g., nitrogen or sulfur) elements and polyatomtic (e.g., nitrogen–oxygen or sulfur–oxygen) elements, such as eggshell membranes from food waste, tanning flakes from leather production waste, and livestock manure waste are continuously selected as precursors for porous carbon.21–23 Owing to the AFRs' rich organic composition of protein (nitrogen source) and polysaccharides (carbon source), there can serve ideal carbon precursors to prepare heteroatomic and polyatomic doped porous carbon material with high specific surface area and good conductivity.13 However, the research on utilization of AFRs as electrode material for supercapacitor is very limited.
In this work, nitrogen and oxygen-rich fresh oxytetracycline fermentation residue (OFR) was used to prepare a series of OFR activated carbons (OACs) with different specific surface areas and pore volumes by coupling pre-carbonization and pyrolytic activation by changing the ratio of activator and activation temperature. K2CO3 was selected here as a green activator compared with the traditionally used activators (e.g., KOH) because it is milder thus avoiding excessive activation and its non-toxic and low corrosive properties.24 Beside the investigation on the effects of activator ratio and activation temperature on the structure of OACs. The electrochemical performance of OACs were evaluated by cyclic voltammetry (CV), constant current charge/discharge (GCD), electrochemical impedance spectroscopy (EIS) and cycling performance tests. OAC-4-700 demonstrated the most excellent specific capacitance up to 310 F g−1 (0.5 A g−1, 6 M KOH) in the three-electrode system and good cycling stability with only 5.32% loss in capacity after 10000 cycles. This work displays that nitrogen–oxygen rich porous carbon materials made from OFR hold significant potential for supercapacitor applications.
2. Experimental
2.1 Materials and chemicals
The fresh OFR was obtained from Gansu Qilianshan Pharmaceutical Company (Jiuquan, Gansu Province, China). The chemical reagents used in this study included anhydrous potassium carbonate (K2CO3, 99%) purchased from Tianjin Kermel Chemical Reagent Co., Ltd, and hydrochloric acid (HCl, 38%) procured from Sinopharm Chemical Reagent Co., Ltd.
2.2 Preparation of N, O co-doped porous carbon
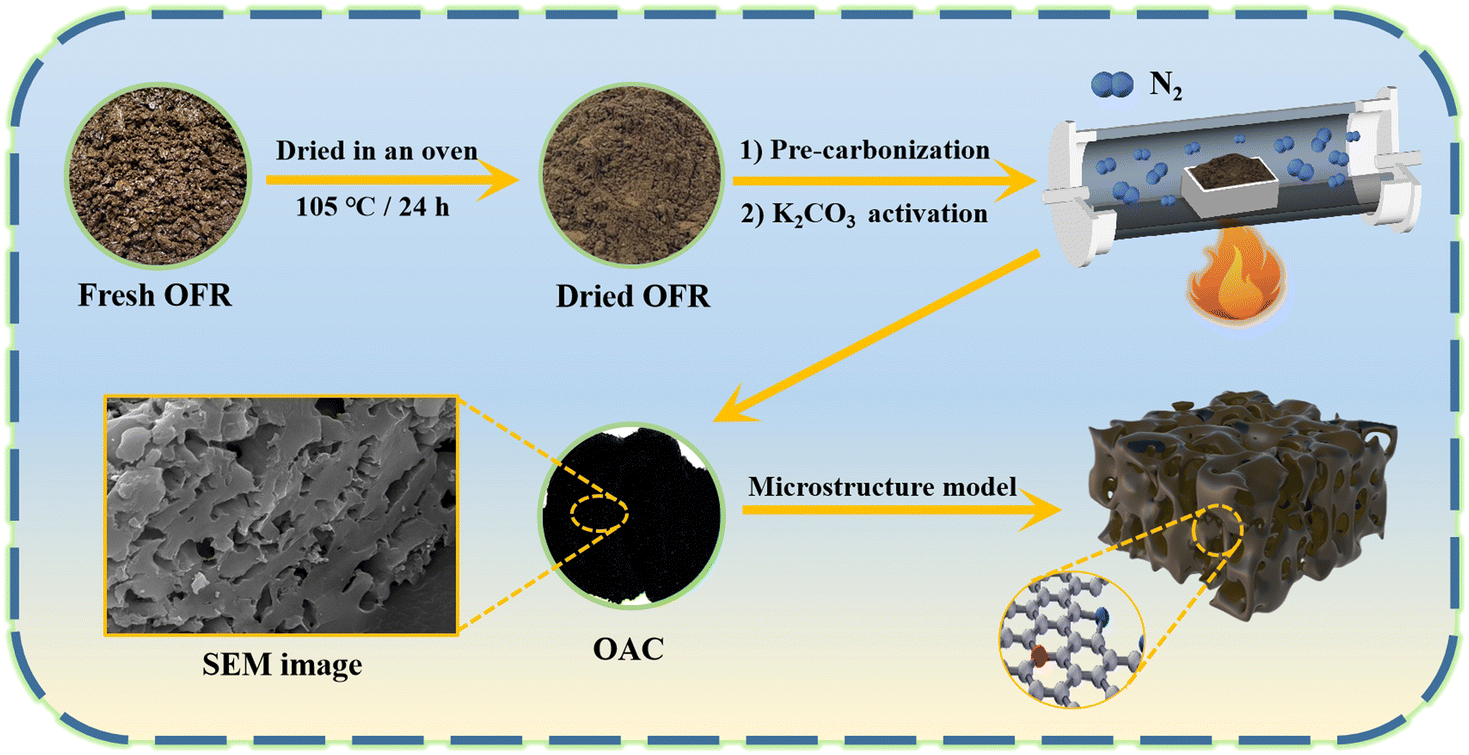
Fig. 1 shows the schematic of the synthesis process for the OAC. First, fresh OFR was dried in an oven at 105 °C to a constant weight for 24 h. The dried OFR was then ground into powder and passed through a 60 mesh sieve. Then, the sieved OFR was placed in an alumina boat and carbonized in a nitrogen atmosphere (25 mL min−1) at a heating rate of 2 °C min−1 until reaching 600 °C, which was maintained for 2 h. The product was then naturally cooled to room temperature in the tubular furnace under a nitrogen atmosphere. Next, the pre-carbonized product was filtered with deionized water three to four times and poured into a Petri dish, which was then dried to a constant weight at 80 °C for 12 h. The pre-carbonized sample after drying was named as OFR pre-carbonized carbon (OPC-600). Using K2CO3 as the activator, OPC-600 was mixed with K2CO3 at weight ratios of 1:X (X = 1, 2, or 4) in a mortar and ground into fine powder. The mixture was then placed in a nickel crucible and subjected to activation in a nitrogen atmosphere (25 mL min−1). The temperature was increased at a heating rate of 2 °C min−1 to a certain temperature and held for 1 h. Then, the sample was naturally cooled to room temperature in the tubular furnace under a nitrogen atmosphere. The activated sample was filtered with 0.1 M hydrochloric acid three to four times and washed with deionized water until neutralized. The washed sample was dried in a Petri dish at 80 °C to a constant weight. The activated samples were named as OAC-X-T, where X represented the weight ratio of K2CO3 and OPC-600 (X = 1, 2, or 4), and T represented the activation temperature (T = 600 °C, 700 °C, or 800 °C). The yield of OAC-X-T was determined by the ratio of the OPC-600 (eqn (1)).| |
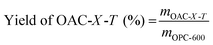 | (1) |
where mOAC-X-T is the quality of OFR activated carbon, mOPC-600 is the quality of OFR pre-carbonized carbon. From Table S1 (ESI†), it can be observed that the yield of OAC-X-T samples remained roughly around 50%. As the activation temperature and ratio increased, the yield of the samples generally decreased. Under the same reaction conditions, each experiment was conducted three times to ensure reproducibility. The results presented in Table S1 (ESI†) were the average values obtained from these three independent experiments.
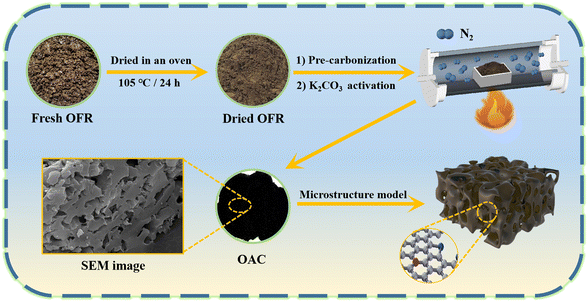 |
| | Fig. 1 Schematic of the synthesis process for the OAC. | |
2.3 Materials characterization
The morphological structure of samples was observed by field emission scanning electron microscopy (SEM, Gemini SEM 500, Germany) and 200 kV field emission transmission electron microscopy (TEM, JEOL JEM-F200, Japan). The specific surface area of porous carbon samples was analyzed using a multipoint Brunauer–Emmett–Teller (BET, V-Sorb 2800P, China) method. The total pore volume (Vt) was determined by the adsorption of nitrogen at a relative pressure of 0.99. The pore size distribution was analyzed by the nonlocal density functional theory (NLDFT) method. The elemental contents of C, H, and N of the samples were analyzed using an elemental analyzer (Vario EL cube, Germany). The surface elemental composition and chemical groups of porous carbon samples were analyzed by X-ray photoelectron spectroscopy (XPS, AXIS SUPRA+, UK). The crystal structure of the samples was characterized using an X-ray diffractometer (D8-ADVANCE, Germany). The Raman spectra of the samples were obtained using the laser micro-Raman spectrometer (Raman, Renishaw, UK).
2.4 Electrochemical measurements
2.4.1 Electrode preparation. In the three-electrode system, OAC-X-T, carbon black, and polytetrafluoroethylene binder (PTFE) were mixed in a mass ratio of 8:1:1, dispersed in the emulsion-breaking agent ethanol, and fully ground with a mortar and pestle to make the mixture homogeneous. The obtained composite after uniform grinding was coated on a piece of nickel foam sheet (1.0 × 1.0 cm2) and pressed into a thin sheet using a press (10–20 MPa). In the two-electrode system, the ratio of OAC-X-T, carbon black, and PTFE was kept constant, polypropylene diaphragm was selected for the assembly of symmetrical capacitors.
2.4.2 Electrochemical performance test. The electrochemical tests included CV, GCD, EIS and cyclic performance tests, which were conducted using an electrochemical workstation (CHI760E Chenhua, Shanghai, China). In the three-electrode system, Pt and HgO/Hg electrodes were used as counter and reference electrodes, respectively, and 6 M KOH was used as the electrolyte. In the two-electrode system, the simulated double-layer capacitor was composed of two identical carbon electrodes (1.0 × 1.0 cm2) sandwiched in the middle with a polypropylene diaphragm, and 6 M KOH was used as the electrolyte. EIS measurements were performed in the frequency range from 100 kHz to 10 MHz with an amplitude of 5 mV.The specific capacitance C1 (F g−1) for the supercapacitor constructed with a three-electrode system can be calculated by eqn (2) as shown below.
| |
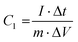 | (2) |
where
I (A) refers to the discharge current, Δ
t (s) refers to the discharge time,
m (g) is the mass of active material on the working electrode, and Δ
V (V) is the potential change range during the discharge.
Two electrodes with the same active material loading are chosen in the two-electrode system to form a symmetrical supercapacitor. The single-electrode specific capacitance C2 (F g−1) and two-electrode specific capacitance Ccell (F g−1) of this system can be calculated by eqn (3).
| |
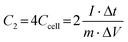 | (3) |
where
m (g) is the total mass of the electroactive material on the two working electrodes,
I (A) is the discharge current, Δ
t (s) refers to the discharge time, and Δ
V (V) denotes the voltage.
The calculation of energy density E (W h kg−1) and power density P (W kg−1) of supercapacitors can be realized on the basis of eqn (4) and (5), respectively.
| |
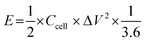 | (4) |
| |
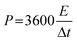 | (5) |
where
Ccell (F g
−1) is the specific capacitance of the symmetric supercapacitor, Δ
V (V) is the potential, and Δ
t (s) is the discharge time.
3. Results and discussion
3.1 Synthesis and physical characterization of porous carbon
The porous carbon's morphological structure is one of the important factors affecting the electrochemical properties. The surface morphology of OFR, OPC, and OACs prepared under different conditions was observed by SEM, which revealed the smooth surface of OFR with a uniform thickness distribution (Fig. 2a). After pre-carbonization by pyrolysis, the volatile organic compounds in OFR were released at high temperature, which increased surface roughness and showed a loose coral-like structure (Fig. 2b).13 The surface of carbon prepared under activation condition at 600 °C exhibited a dense coral-like structure after activation with K2CO3 but did not show any pores (Fig. 2c). This finding was due to the capability of K2CO3 to mainly remove some volatile substances at temperatures below 627 °C, resulting in considerably few pores.25 The reaction of K2CO3 + C ↔ K2O + 2CO can occur only when the activation temperature was increased to above 627 °C, which oxidized the carbon material with K2CO3 and converted it into gaseous CO to form a large number of pores. OAC-4-700 and OAC-4-800 prepared under activation conditions at 700 °C and 800 °C, respectively, had a large number of pores on their surface and exhibited a 3D grid carbon skeleton structure (Fig. 2d and e). In addition, the pore size and volume of the porous carbon further increased with the rise in proportion of the activating agent (Fig. 2d and f). As shown in Fig. 2g and h, the microstructure of OAC-4-700 was further examined using TEM, which showed that OAC-4-700 generated a large number of micropores and mesopores after mild activation with K2CO3. Furthermore, large pores could be observed in the magnified image of scanning electron microscopy (Fig. 2d). This multidimensional pore structure was advantageous for the penetration and diffusion of electrolyte and also provided additional reaction surface area, which enabled the effective combination of ions and charge carriers, thereby improving the reaction rate and efficiency of the electrochemical system.26 Dark field SEM mapping analysis was used to investigate the surface element distribution of OAC-4-700. The element mapping of C, N, and O on the surface of OAC-4-700 is shown in Fig. 2i, which indicated that their distributions on the carbon material surface were uniform.27 Combined with the element analysis in Table S1 (ESI†), the content of C, N, and O in OAC-4-700 was 79.47%, 3.42%, and 14.86%, respectively.
 |
| | Fig. 2 SEM images of (a) OFR, (b) OPC-600, (c) OAC-4-600, (d) OAC-4-700 with different magnifications. (e) OAC-4-800 with different magnifications. (f) OAC-2-700. (g and h) TEM images of OAC-4-700. (i) Dark field SEM mapping images of OAC-4-700 with C, N, and O elements. | |
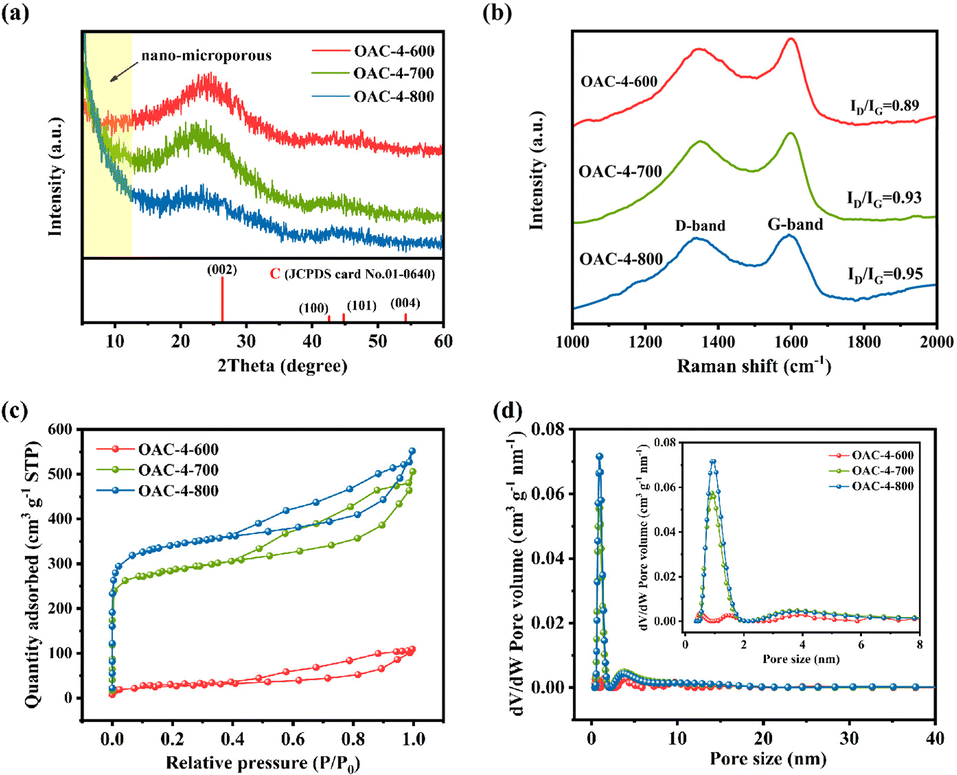
The lattice structure and graphitization degree of OACs were respectively analyzed through XRD and Raman spectra to further investigate the physical properties of OACs. Fig. 3a shows the XRD pattern of OAC-4-T, revealing that all samples had two broad diffraction peaks at positions of 23.5° and 44.8°. Notably, the low-angle region of OAC-4-700 and OAC-4-800 both exhibited high intensity characteristics, indicating the presence of well-developed nano-microporous structures.14 By comparing the obtained XRD pattern of OAC-4-T with the standard card of graphite (JCPDS card no. 01-0640), it was found that the broad peak at 23.5° corresponded to the diffraction peak of amorphous carbon (002), while the weak intensity diffraction peak at 44.8° (101) further revealed the graphitization degree of the sample, indicating that these samples comprised amorphous carbon with a low graphitization degree. Related studies had shown that this disordered amorphous carbon structure was advantageous for storing electrolyte ions in nanopores, which markedly improved the energy storage capacity.28
 |
| | Fig. 3 (a) XRD pattern, (b) Raman spectra, (c) N2 adsorption–desorption isotherms, and (d) pore size distribution of OAC-4-600, OAC-4-700, and OAC-4-800. | |
Raman spectra were used to provide a highly detailed characterization of the disorder degree and the graphitic structure of the samples. As shown in Fig. 3b, two characteristic bands were detected at 1348 cm−1 and 1590 cm−1, corresponding to the D and G-bands, respectively. The D and G-bands represented structural defects or disordered sp3 hybridized carbon and lattice vibrations or sp2 hybridized carbon, respectively.29,30 The intensity ratio of the D-band to G-band (ID/IG) was commonly used to evaluate the disorder degree and graphitization of samples. The ID/IG ratios of OAC-4-600, OAC-4-700, and OAC-4-800 were 0.89, 0.93, and 0.95, respectively. These gradually increased ratios with the elevated activation temperatures indicated that the lattice structural defects of the carbon material gradually raised and the disorder degree intensified. The existence of such lattice structural defects substantially increased the specific surface area and conductivity of carbon materials.31
The specific surface area and pore structure of the carbon material were analyzed by N2 adsorption–desorption isotherms. As shown in Fig. 3c, the samples of OAC-4-T exhibited a hysteresis loop, which belonged to the typical type IV adsorption isotherm according to the IUPAC classification standard.32 Specifically, according to Fig. 3c and S1a (ESI†), the N2 adsorption–desorption isotherm of OAC-4-600 did not significantly rise when the P/P0 is close to 0, indicating that the microporous of the sample was unsuccessfully developed. In addition, the H3 hysteresis loop represented an irregular pore structure with a small amount of non-uniformly distributed mesopores and macropores, which was consistent with the morphology shown in its SEM image (Fig. 2c).13 Simultaneously, all carbon materials, except for OAC-4-600, had similar N2 adsorption–desorption isotherms (Fig. 3c and S1a (ESI†)). Under low pressure conditions (P/P0 < 0.05), the N2 adsorption–desorption isotherm showed a sharp increase in adsorption volume, indicating that its abundance in a large number of micropores.33 A clear H4 hysteresis loop is observed under moderate pressure conditions (0.4 < P/P0 < 0.9), showing the presence of mesopores.34 Furthermore, the N2 adsorption–desorption isotherm slightly rose under high pressure conditions (0.95 < P/P0 < 1.0), indicating the presence of a small amount of macropores in the material.35 Second, the pore size distribution of the samples was calculated in accordance with the N2 adsorption–desorption isotherm using NLDFT (Fig. 3d and S1b (ESI†)), which showed that all carbon materials, except for OAC-4-600, were rich in micropores and mesopores. The micropore and mesopore sizes were generally distributed around 1 and 4 nm, respectively. The specific surface area and pore size distribution of different samples are summarized in Table 1, from which it can be seen that the specific surface area and pore volume of the samples were significantly improved with the increase of activation temperature and K2CO3 ratio. When the activation temperature was increased from 600 °C to 800 °C, the specific surface area of the porous carbon and the total pore volume increased from 109 m2 g−1 to 1133 m2 g−1 and from 0.18 m3 g−1 to 0.85 m3 g−1, respectively. When the K2CO3 ratio was increased from 1 to 4, the specific surface area of the porous carbon increased and the total pore volume respectively increased from 643 m2 g−1 to 948 m2 g−1 and from 0.53 m3 g−1 to 0.78 m3 g−1. Temperature was found to be the primary factor affecting the specific surface area and pore volume, while the impact of the K2CO3 ratio was comparatively minor.14 Overall, OACs had a multidimensional pore structure. It was recognized that micropores could store charges and ions and improve the surface reactivity of the electrode, mesopores could enhance the conductivity and transmission rate of the electrode, and macropores provided a wide space for the diffusion of electrolyte ions.33,36 The combined action of the three promoted a significant improvement in the electrode performance of OACs.
Table 1 Specific surface area and pore size distribution of different samples
| Sample |
SBETa (m2 g−1) |
Vtotalb (m3 g−1) |
Vmicroc (m3 g−1) |
Vmicro/Vtotal (%) |
Vmesod (m3 g−1) |
Vmeso/Vtotal (%) |
| SBET is specific surface area by the BET method at P/P0 = 0.001–0.05. Vtotal is total pore volume at P/P0 = 0.99. Vmicro is micropore volume and is evaluated by the t-plot method. Vmeso is mesopore volume and is evaluated by the BJH method. |
| OAC-4-600 |
109 |
0.18 |
0.01 |
5.56 |
0.16 |
88.89 |
| OAC-1-700 |
643 |
0.53 |
0.25 |
47.17 |
0.29 |
54.72 |
| OAC-2-700 |
915 |
0.70 |
0.34 |
48.57 |
0.36 |
51.43 |
| OAC-4-700 |
948 |
0.78 |
0.33 |
42.31 |
0.41 |
52.56 |
| OAC-4-800 |
1133 |
0.85 |
0.41 |
48.26 |
0.44 |
51.76 |
3.2 Chemical composition analysis of porous carbon
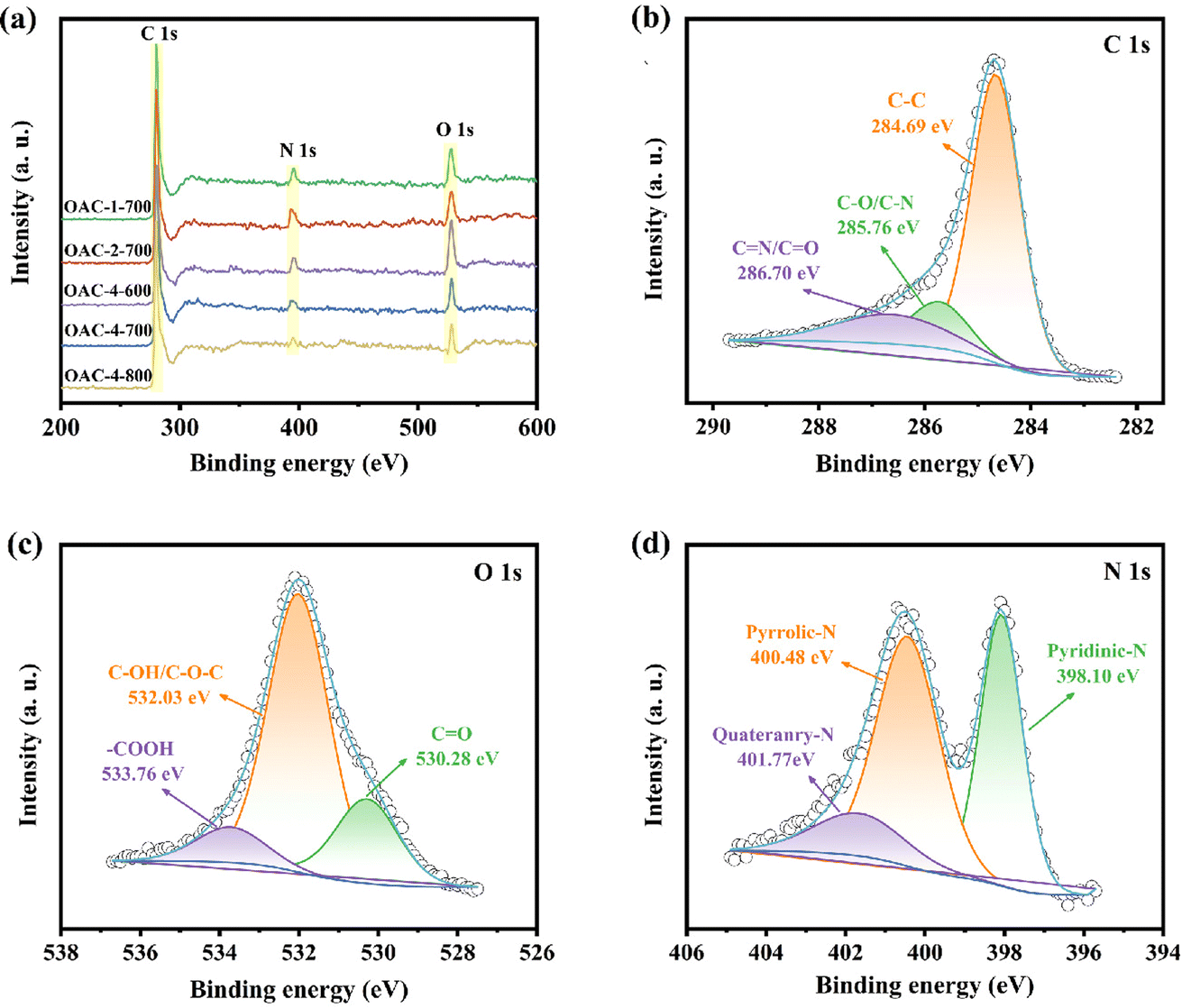
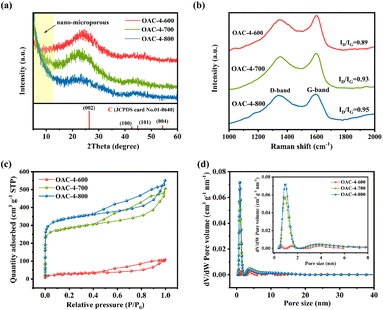
The chemical properties of the carbon material surface were investigated by XPS. The full-scan XPS spectra in Fig. 4a revealed a significant C 1s peak at 284 eV and two weak peaks at 400 and 530 eV, corresponding to N 1s and O 1s, respectively. Table S1† summarizes the elemental composition of different samples. The prepared OACs had a high oxygen content (12.47–29.28 wt%) due to the high oxygen content in the raw material OFR, and carbon materials with high oxygen content usually exhibited hydrophilicity, which is important for its wettability as the aqueous electrolyte.28 Meanwhile, the nitrogen groups abundant in OACs could increase the number of positive ion states, thereby improving the conductivity of the electrode.37 Furthermore, OAC-4-700 was selected to investigate the high-resolution XPS spectra of the C, N, and O elements. As shown in Fig. 4b, the high-resolution C 1s spectra were deconvoluted into three peaks: C–C (284.69 eV), C–O/C–N (285.76 eV), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/CO (286.70 eV). The O 1s spectra (Fig. 4c) can be deconvoluted into carbonyl (CO, 530.28 eV), phenyl hydroxide or ether (C–OH/C–O–C, 532.03 eV), and carboxyl (–COOH, 533.76 eV). The two later oxygen-containing groups could increase the interaction force between the carbon material and the aqueous electrolyte, thus improving the wettability and interfacial stability of the carbon material as the aqueous electrolyte.38 The N 1s spectra (Fig. 4d) could be deconvoluted into pyridinic-N (N-6, 398.10 eV), pyrrolic-N (N-5, 400.48 eV), and quaternary-N (N–Q, 401.17 eV). Related research had shown that N-5 and N-6 functional groups were effective in enhancing pseudocapacitance.39,40 On the one hand, N-5 demonstrated good electron-donating properties and high charge transfer mobility, which helped enhance the catalytic activity of carbon in electron transfer reactions. On the other hand, N-6 provided electron pairs coupled with π–π conjugated rings, endowing the carbon nanosheets with electron-donating properties, which is of considerable importance for improving the electrode capacitance. In addition, N-Q provided positive charges during the charging and discharging processes and participated in the reaction as an electron acceptor, which could effectively reduce the ion transfer resistance and improve the conductivity of the carbon electrode.36,41 The N/O elemental contents on the surface of the samples are shown in Table S1 and Fig. S2a (ESI†), which revealed that the N and O contents in the samples decrease with increasing activation temperature and K2CO3 ratio. Moreover, Fig. S2b (ESI†) shows that pyridinic-N was the most abundant type of nitrogen species in the OAC-4-700, with a proportion approximately three times higher than that of quaternary-N. On the whole, the high content of nitrogen-oxygen and a variety of nitrogen-oxygen functional groups were conducive to the superior electrochemical activity and cycling stability of OAC-4-700.42
N/CO (286.70 eV). The O 1s spectra (Fig. 4c) can be deconvoluted into carbonyl (CO, 530.28 eV), phenyl hydroxide or ether (C–OH/C–O–C, 532.03 eV), and carboxyl (–COOH, 533.76 eV). The two later oxygen-containing groups could increase the interaction force between the carbon material and the aqueous electrolyte, thus improving the wettability and interfacial stability of the carbon material as the aqueous electrolyte.38 The N 1s spectra (Fig. 4d) could be deconvoluted into pyridinic-N (N-6, 398.10 eV), pyrrolic-N (N-5, 400.48 eV), and quaternary-N (N–Q, 401.17 eV). Related research had shown that N-5 and N-6 functional groups were effective in enhancing pseudocapacitance.39,40 On the one hand, N-5 demonstrated good electron-donating properties and high charge transfer mobility, which helped enhance the catalytic activity of carbon in electron transfer reactions. On the other hand, N-6 provided electron pairs coupled with π–π conjugated rings, endowing the carbon nanosheets with electron-donating properties, which is of considerable importance for improving the electrode capacitance. In addition, N-Q provided positive charges during the charging and discharging processes and participated in the reaction as an electron acceptor, which could effectively reduce the ion transfer resistance and improve the conductivity of the carbon electrode.36,41 The N/O elemental contents on the surface of the samples are shown in Table S1 and Fig. S2a (ESI†), which revealed that the N and O contents in the samples decrease with increasing activation temperature and K2CO3 ratio. Moreover, Fig. S2b (ESI†) shows that pyridinic-N was the most abundant type of nitrogen species in the OAC-4-700, with a proportion approximately three times higher than that of quaternary-N. On the whole, the high content of nitrogen-oxygen and a variety of nitrogen-oxygen functional groups were conducive to the superior electrochemical activity and cycling stability of OAC-4-700.42
 |
| | Fig. 4 (a) Full-scan XPS spectra of different samples. High-resolution XPS spectra of (b) C 1s, (c) O 1s, and (d) N 1s for OAC-4-700. | |
3.3 Electrochemical performance evaluation
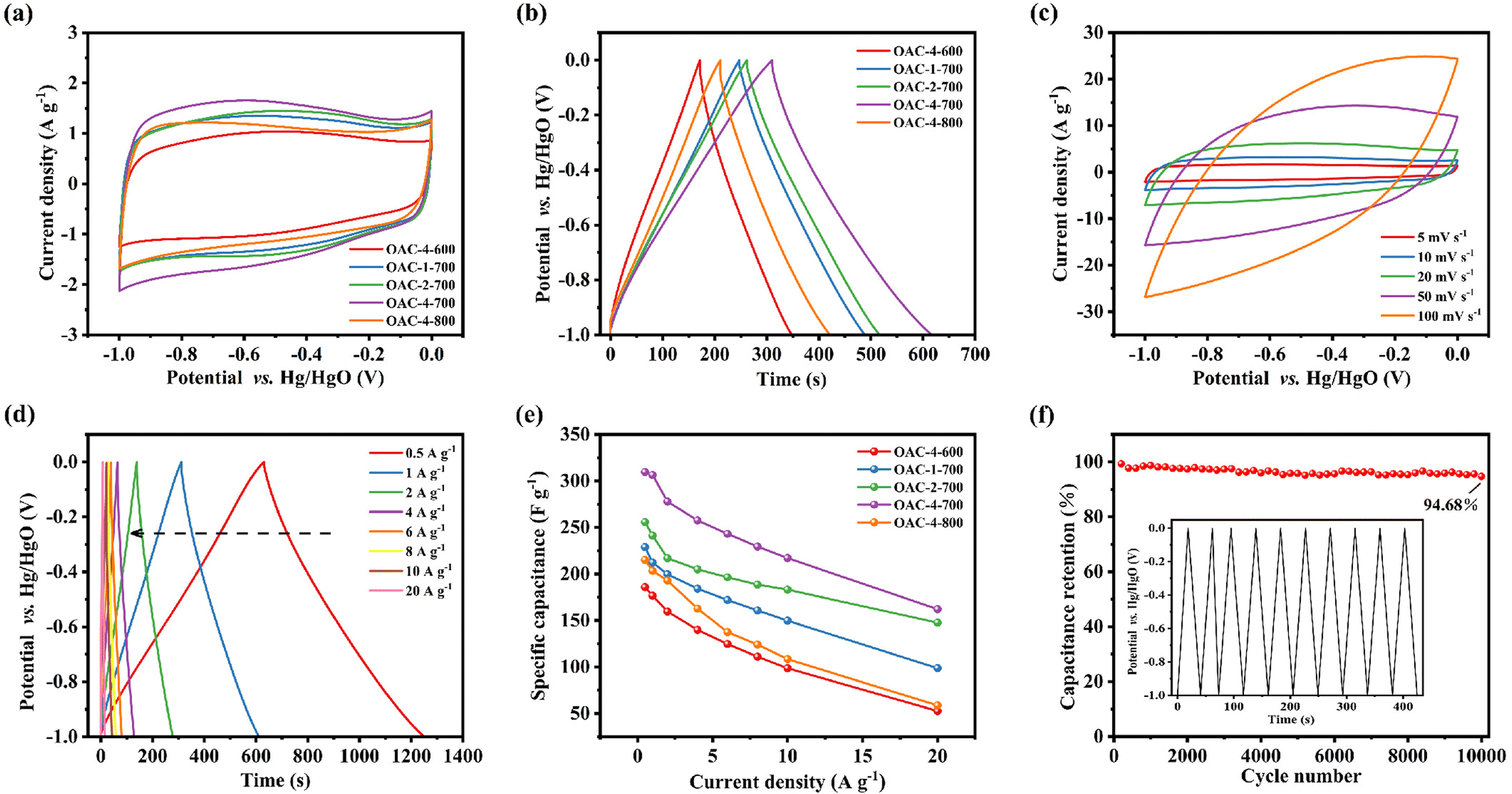
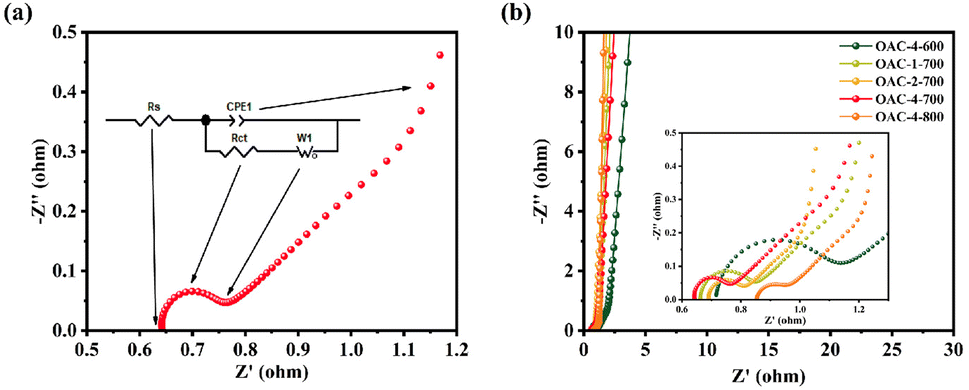
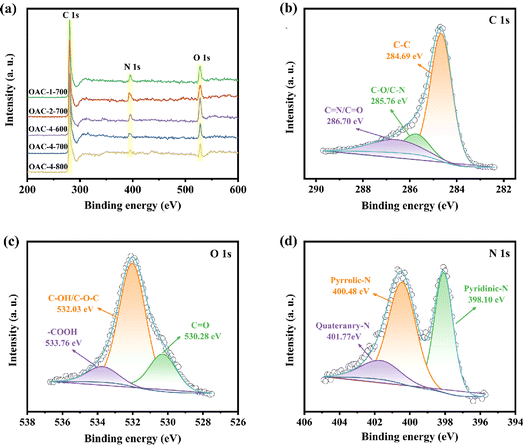
Electrochemical tests, including CV, GCD, and EIS, were conducted on a three-electrode system in a 6 M KOH electrolyte solution to explore the potential application of OACs in supercapacitors. At the scan rate of 5 mV s−1, the CV curves exhibited asymmetric rectangular shapes (Fig. 5a), with weak reversible peaks indicating the dominance of the EDLC in the electrochemical behavior and demonstrating a slight pseudocapacitive characteristic.43,44 This pseudocapacitive characteristic was mainly attributed to the reversible faradaic redox reaction of the nitrogen–oxygen functional groups contained in the sample.32 Among these carbon materials, OAC-4-700 exhibited the largest CV integral area, indicating that it had the highest charge storage capacity, which was directly related to its well-developed nanoporous structure and high nitrogen–oxygen content (N = 3.42 wt%, O = 14.86 wt%). Meanwhile, the GCD curves of OACs at a current density of 1 A g−1 are shown in Fig. 5b. All GCD curves exhibited an almost isosceles triangular shape, indicating excellent charge–discharge characteristics and electrochemical reversibility. The slight linear deviation in the curve further indicated a combination of EDLC and pseudocapacitive capacitance characteristics.45 Notably, compared with other samples, OAC-4-700 exhibited the longest charge–discharge time, confirming its highest charge storage capacity. Fig. 5c shows the CV curves of OAC-4-700 at different scan rates (5–100 mV s−1). The CV curve of OAC-4-700 approximately maintained a rectangular shape at low scan rates (5–20 mV s−1), indicating its fast ion transport capability. The CV curve exhibited a distorted trend as the scan rate increased because ion diffusion was restricted at high scan rates, resulting in uneven surface charges of the electrode and deformed CV curves.46 The GCD curves of OAC-4-700 at different current densities are shown in Fig. 5d. All GCD curves maintained high symmetry, revealing the effective transfer of ions at the electrode–electrolyte interface. The specific capacitance at different current densities could be calculated in accordance with the discharge curve and eqn (2). Fig. 5e summarizes the specific capacitance of different carbon materials at different current densities. Undoubtedly, OAC-4-700 exhibited the highest specific capacitance (310 F g−1) at a current density of 0.5 A g−1 and could still maintain 70% (217 F g−1) even under the high current density of 10 A g−1, indicating its excellent ion transport rate. The cyclic stability of OAC-4-700 was tested at a current density of 10 A g−1 (Fig. 5f). The capacitance of OAC-4-700 could still maintain 94.68% after undergoing 10000 cycles, indicating its excellent cyclic performance.47 The electrochemical performance of OACs was further studied through EIS measurement (Fig. 6). The equivalent analog circuit diagram is shown in Fig. 6a (inset), where Rs and Rct represented the equivalent series and charge transfer resistances, respectively. A short line with a slope of 45° was found in the low-frequency region, which was described as the Warburg impedance (W). W was related to the diffusion coefficient of the charge substance in the solution.40 The Nyquist plots shown in Fig. 6b exhibited approximate vertical lines in the low-frequency region, indicating the good EDLC behavior of them.48 The values of Rs and Rct for different samples are summarized in Table S2 (ESI†). OAC-4-700 simultaneously demonstrated the smallest equivalent series resistance (Rs = 0.643 Ω) and charge transfer resistance (Rct = 0.092 Ω), indicating its low internal resistance and good conductivity.49 The small internal resistance also reduced the thermal losses, which could extend the service life of OAC-4-700.
 |
| | Fig. 5 Electrochemical performances of different samples tested via three-electrode system in 6 M KOH electrolyte. (a) CV curves at scan rates of 5 mV s−1. (b) GCD curves at 1 A g−1. (c) CV curves of OAC-4-700 at 5–100 mV s−1. (d) GCD curves of OAC-4-700 at 0.5–20 A g−1. (e) The specific capacitance of different samples at 0.5–20 A g−1. (f) The cycle stability measures at 10 A g−1 current density and the inner picture are the first 10 cycles. | |
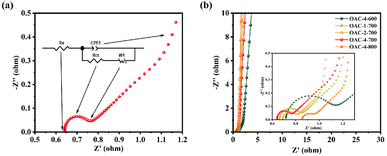 |
| | Fig. 6 (a) Nyquist plot and equivalent analog circuit diagram. (b) Nyquist plots of different samples. | |
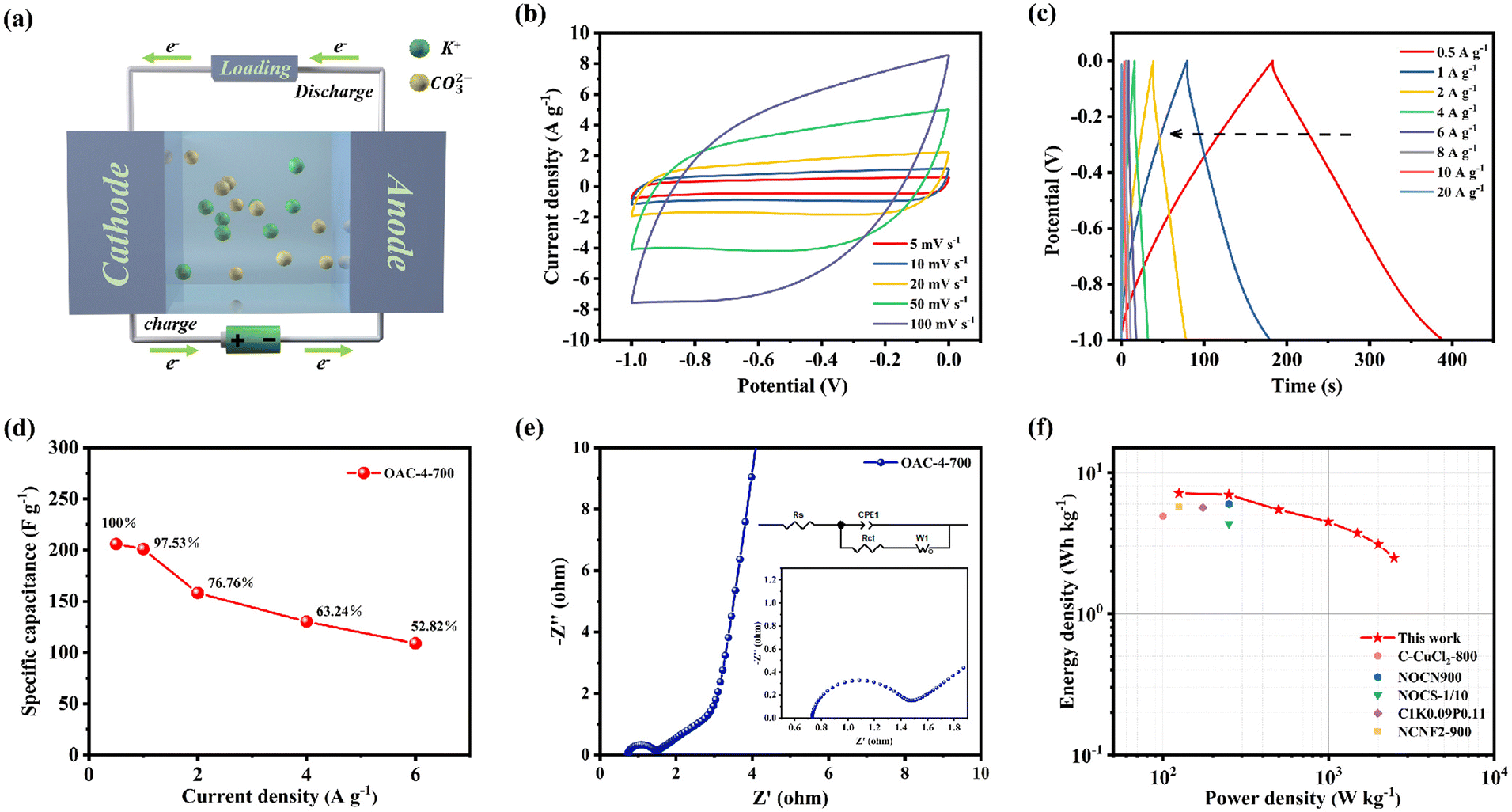
A symmetric supercapacitor was prepared with 6 M KOH as the electrolyte as shown in Fig. 7a to further evaluate the practical application of OAC-4-700 in supercapacitors. K+ and CO32− in the electrolyte could move to the cathode and anode, respectively, during charging of the symmetric supercapacitor, thus forming a double layer at the interface between the two electrodes and the electrolyte.50 The positive and negative charges on the electrodes were attracted to opposite charged ions in the solution after charging was completed, stabilizing the double layer and creating a relatively stable potential difference between the positive and negative electrodes. This potential difference lies in the energy stored in the symmetric supercapacitor.13,51 Simultaneously, charge migration on the electrodes generated current during discharge, and ions in the solution migrated to the electrolyte.18 The CV curve (Fig. 7b) reveals that the symmetric supercapacitor of OAC-4-700 was stable under different scanning rates, showing a nearly rectangular shape and exhibiting good EDLC properties.48 The GCD curve of OAC-4-700 in the two-electrode system is shown in Fig. 7c. Under different current densities, OAC-4-700 presented a symmetric triangular shape with a linear deviation, further confirming its typical EDLC properties and having partial pseudocapacitive characteristics. The specific capacitances of OAC-4-700, which were calculated by the GCD curves of various current densities, are summarized in Fig. 7d. At a current density of 1 A g−1, the specific capacitance of OAC-4-700 was 206 F g−1 and maintained 52.82% (109 F g−1) at 10 A g−1, indicating its good rate performance. The Nyquist plots of the symmetric supercapacitor with OAC-4-700 are shown in Fig. 7e, in which an almost vertical Warburg line could be observed in the low-frequency region, indicating its ideal capacitive performance. A semicircle could be observed in the amplified high-frequency region (Fig. 7e, inset) with Rs = 0.73 Ω and Rct = 0.66 Ω, indicating that the symmetric supercapacitor had good charge transfer kinetics. The cycle stability tests on OAC-4-700 based symmetric supercapacitor at 10 A g−1 is shown in Fig. S3 (ESI†). After 10000 cycles, it could still maintain a capacitance retention rate of 97.25%, demonstrating excellent cycling performance in a 6 M KOH electrolyte.
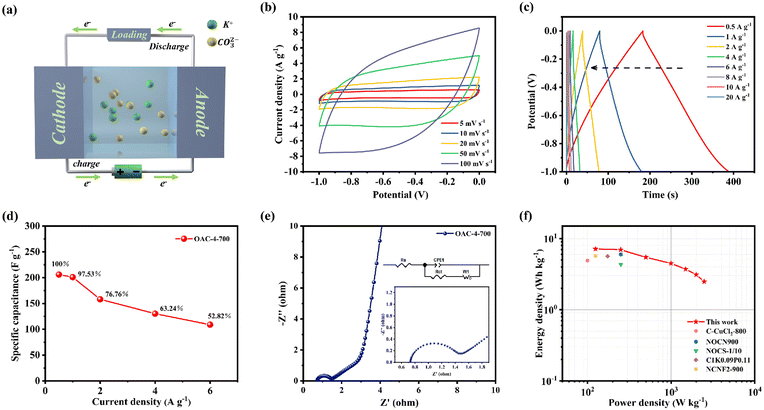 |
| | Fig. 7 Electrochemical performance of OAC-4-700 based supercapacitor tested in a two-electrode configuration in an aqueous electrolyte of 6 M KOH. (a) Schematic illustration of the OAC-4-700//OAC-4-700 symmetric supercapacitor. (b) CV curves at 5–100 mV s−1. (c) GCD curves at 0.5–20 A g−1. (d) Specific capacitances at 0.5–20 A g−1. (e) Nyquist plots. (f) Ragone plots. | |
Energy and power densities are also important indicators to evaluate the performance of supercapacitors in practical applications. Energy and power densities of OAC-4-700 at different specific capacitance values were calculated by combining the specific capacitance values of the symmetric supercapacitor under different current densities with eqn (4) and (5), respectively (Fig. 7f). The Ragone plots (Fig. 7f) show that the power density of the OAC-4-700 symmetric supercapacitor in a 6 M KOH electrolyte was 124.9 W kg−1 and its maximum energy density was 7.2 W h kg−1. Compared with previously reported carbonaceous materials, such as N-doped porous carbons derived from nanorod-shaped N-rich polymers (C–CuCl2-800, 4.91 W h kg−1 at a power density of 100 W kg−1),17 two-dimensional oxygen-containing carbon nanosheets with layered pores (NOCN900, 6 W h kg−1 at a power density of 250 W kg−1),29 N-self-doped carbon nanofiber aerogels (NCNF2-900, 5.7 W h kg−1 at a power density of 125 W kg−1),33 N/O co-doped carbon sponges (NOCS-1/10, 4.33 W h kg−1 at a power density of 250 W kg−1),52 and activated carbon microspheres prepared from xylose as a precursor (C1K0.09P0.11, 5.65 W h kg−1 at a power density of 173.88 W kg−1),53 the OAC-4-700 symmetric supercapacitor demonstrated good energy storage and release performance.
4. Conclusions
Overall, waste OFR was converted to in situ N, O co-doped porous carbon with multidimensional pore structures. The optimal sample (OAC-4-700) had a unique 3D grid carbon skeleton structure, high specific surface area (SBET = 948 m2 g−1), and high nitrogen–oxygen content (N = 3.42 wt%, O = 14.86 wt%). OAC-4-700 demonstrated a high specific capacitance of 310 F g−1 at 0.5 A g−1 in a three-electrode system due to the high electrical conductivity provided by the three-dimensional pore structure and the electrochemical activity endowed by the high nitrogen–oxygen content. Even under high current density conditions of 10 A g−1, OAC-4-700 could still maintain 70% of its capacitance (217 F g−1). In addition, OAC-4-700 retained 94.68% of its high capacitance even after 10000 cycles at 10 A g−1, indicating its excellent cycling stability. More significantly, OAC-4-700 exhibited outstanding electrochemical performance when it was assembled as an electrode material for a symmetrical supercapacitor. The maximum energy density of the supercapacitor reached 7.2 W h kg−1 at the power density of 124.9 W kg−1. These results demonstrated the good potential of OFR-based in situ N, O co-doped porous carbon as favorable electrode material for supercapacitor. The present study provided a green and facile process for dealing with antibiotic fermentation residues and demonstrated the potential commercial applications of their prepared electrode materials in the energy storage field.
Author contributions
Shumeng Qin: Methodology, Conceptualization, Data curation, Writing – original draft. Peiliang Liu: Conceptualization, Writing – review and editing. Jieni Wang: Investigation, Validation. Chenxiao Liu: Methodology, Visualization. Qizhao Wang and Xuanyu Chen: Data Curation and Editing. Shuqin Zhang and Yijun Tian: Software, Data curation. Fangfang Zhang: Supervision, Writing – review and editing. Lin Wang: Conceptualization, Supervision, Resources. Zhangdong Wei: Writing – review and editing, Language editing. Leichang Cao: Project administration, Conceptualization, Supervision, Language editing. Jinglai Zhang: Investigation, Writing – review and editing. Shicheng Zhang: Supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (No. 52100164), the China Postdoctoral Science Foundation (No. 2023M731169), the Key Scientific Research Projects of Universities in Henan Province (No. 23A610006), the Key Science and Technology Department Project of Henan Province (No. 222102320252), the Science and Technology Development Plan of Kaifeng City – Science and Technology Research Project, and Yellow River Scholar Program of Henan University. The authors are also grateful for the provision of a scholarship to Leichang Cao by Shanghai Tongji Gao Tingyao Environmental Science & Technology Development.
References
- R. Daghrir and P. Drogui, Environ. Chem. Lett., 2013, 11, 209–227 CrossRef CAS.
- M. Kumar, S. Jaiswal, K. K. Sodhi, P. Shree, D. K. Singh, P. K. Agrawal and P. Shukla, Environ. Int., 2019, 124, 448–461 CrossRef CAS PubMed.
- A. J. Browne, M. G. Chipeta, G. Haines-Woodhouse, E. P. A. Kumaran, B. H. K. Hamadani, S. Zaraa, N. J. Henry, A. Deshpande, N. P. J. Day, A. D. Lopez, S. Dunachie, C. E. Moore, A. Stergachis, S. I. Hay and C. Dolecek, Lancet Planet. Health, 2021, 5, 893–904 CrossRef PubMed.
- J. L. M. Christopher, S. I. Kevin, S. Fablina, S. Lucien, R. A. Gisela and G. Authia, Lancet, 2022, 399, 629–655 CrossRef PubMed.
- X. Zhang, W. Cai, S. Zhao, X. Li, F. Jia, F. Ma and H. Yao, Environ. Res., 2021, 192, 110261 CrossRef CAS PubMed.
- X. Jiang, Y. Feng, G. Lv, Y. Du, D. Qin, X. Li, Y. Chi, J. Yan and X. Liu, Environ. Sci. Technol., 2012, 46, 13539–13544 CrossRef CAS PubMed.
- X. Liu, S. Yang, Y. Wang, H. Zhao and L. Song, Sci. Total Environ., 2018, 634, 1231–1237 CrossRef CAS PubMed.
- H. Van Luong, P. T. Tai, M. Nishijo, D. M. Trung, P. N. Thao, P. Van Son, N. Van Long, N. T. Linh and H. Nishijo, Sci. Total Environ., 2018, 628–629, 484–489 CrossRef PubMed.
- W. Zhong, Z. Li, J. Yang, C. Liu, B. Tian, Y. Wang and P. Chen, Bioresour. Technol., 2014, 151, 436–440 CrossRef CAS PubMed.
- Y. Liu, X. Zhu, X. Wei, S. Zhang, J. Chen and Z. J. Ren, Chem. Eng. J., 2018, 334, 1101–1107 CrossRef CAS.
- S. Ahmad, X. Zhu, J. Luo, S. Zhou, C. Zhang, J. Fan, J. H. Clark and S. Zhang, J. Hazard. Mater., 2020, 393, 122446 CrossRef CAS PubMed.
- Q. Wang, Z. Zhang, G. Xu and G. Li, J. Hazard. Mater., 2021, 413, 125385 CrossRef CAS PubMed.
- J. Hu, C. Hong, C. Zhao, Y. Si, Y. Xing, W. Ling, B. Zhang, Z. Li, Y. Wang, L. Feng and J. Yang, J. Alloys Compd., 2022, 918, 165453 Search PubMed.
- F. Wu, J. Gao, X. Zhai, M. Xie, Y. Sun, H. Kang, Q. Tian and H. Qiu, Carbon, 2019, 147, 242–251 CrossRef CAS.
- F. Naseri, S. Karimi, E. Farjah and E. Schaltz, Renewable Sustainable Energy Rev., 2022, 155, 111913 CrossRef CAS.
- N. A. Rashidi and S. Yusup, J. Energy Storage, 2020, 32, 101757 CrossRef.
- L. Zheng, B. Tang, X. Dai, T. Xing, Y. Ouyang, Y. Wang, B. Chang, H. Shu and X. Wang, Chem. Eng. J., 2020, 399, 125671 CrossRef CAS.
- D. P. Chatterjee and A. K. Nandi, J. Mater. Chem. A, 2021, 9, 15880–15918 RSC.
- J. Mu, S. Wong, Q. Li, P. F. Zhou, J. Zhou, Y. Zhao, J. Sunarso and S. P. Zhuo, J. Alloys Compd., 2020, 832, 154950 CrossRef CAS.
- A. Amiri, Y. J. Chen, C. B. Teng and M. Naraghi, Energy Storage Mater., 2020, 25, 731–739 CrossRef.
- Z. Li, L. Zhang, B. S. Amirkhiz, X. Tan, Z. Xu, H. Wang, B. C. Olsen, C. M. B. Holt and D. Mitlin, Adv. Energy Mater., 2012, 2, 431–437 CrossRef CAS.
- P. Liu, Z. Xing, X. Wang, S. Diao, B. Duan, C. Yang and L. Shi, J. Mater. Sci.: Mater. Electron., 2022, 33, 4887–4901 CrossRef CAS.
- M. Sandhiya, M. P. Nadira and M. Sathish, Energy Fuels, 2021, 35, 15094–15100 CrossRef CAS.
- L. J. Wang, F. Sun, F. Hao, Z. B. Qu, J. H. Gao, M. J. Liu, K. F. Wang, G. B. Zhao and Y. K. Qin, Chem. Eng. J., 2020, 383, 123205 CrossRef CAS.
- Y. T. Li, Y. T. Pi, L. M. Lu, S. H. Xu and T. Z. Ren, J. Power Sources, 2015, 299, 519–528 CrossRef CAS.
- S. Zhao, D. W. Wang, R. Amal and L. Dai, Adv. Mater., 2019, 31, e1801526 CrossRef PubMed.
- X. Wang, A. Dong, Y. Hu, J. Qian and S. Huang, Chem. Commun., 2020, 56, 10809–10823 RSC.
- P. Han, M. Cheng, D. Luo, W. Cui, H. Liu, J. Du, M. Wang, Y. Zhao, L. Chen, C. Zhu and J. Xu, Energy Storage Mater., 2020, 24, 486–494 CrossRef.
- F. Ran, X. Yang, X. Xu, S. Li, Y. Liu and L. Shao, Chem. Eng. J., 2021, 412, 128673 CrossRef CAS.
- G. Byatarayappa, V. Guna, K. Venkatesh, N. Reddy, N. Nagaraju and K. Nagaraju, J. Environ. Chem. Eng., 2021, 9, 106525 CrossRef CAS.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- C. Ma, J. Bai, M. Demir, X. Hu, S. Liu and L. Wang, Fuel, 2022, 326, 125119 CrossRef CAS.
- H. Chen, T. Liu, J. Mou, W. Zhang, Z. Jiang, J. Liu, J. Huang and M. Liu, Nano Energy, 2019, 63, 103836 CrossRef CAS.
- Y. Lu, S. Zhang, J. Yin, C. Bai, J. Zhang, Y. Li, Y. Yang, Z. Ge, M. Zhang, L. Wei, M. Ma, Y. Ma and Y. Chen, Carbon, 2017, 124, 64–71 CrossRef CAS.
- A. Tripathy, S. Mohanty, S. K. Nayak and A. Ramadoss, J. Environ. Chem. Eng., 2021, 9, 106398 CrossRef CAS.
- X. J. Wei, Y. B. Li and S. Y. Gao, J. Mater. Chem. A, 2017, 5, 181–188 RSC.
- J. Huang, N. Wang, J. Wang, N. Huang, M. Bayati and T. Xiaoteng Liu, J. Electroanal. Chem., 2021, 894, 115362 CrossRef CAS.
- H. Ma, C. Li, M. Zhang, J. D. Hong and G. Shi, J. Mater. Chem. A, 2017, 5, 17040–17047 RSC.
- Y. Li, K. Ye, K. Cheng, D. Cao, Y. Pan, S. Kong, X. Zhang and G. Wang, J. Electroanal. Chem., 2014, 727, 154–162 CrossRef CAS.
- Z. Shang, X. An, H. Zhang, M. Shen, F. Baker, Y. Liu, L. Liu, J. Yang, H. Cao, Q. Xu, H. Liu and Y. Ni, Carbon, 2020, 161, 62–70 CrossRef CAS.
- S. Dai, Z. Liu, B. Zhao, J. Zeng, H. Hu, Q. Zhang, D. Chen, C. Qu, D. Dang and M. Liu, J. Power Sources, 2018, 387, 43–48 CrossRef CAS.
- Y. Li, G. Wang, T. Wei, Z. Fan and P. Yan, Nano Energy, 2016, 19, 165–175 CrossRef CAS.
- X. L. Zhou, L. Y. Zhu, Y. Yang, L. J. Xu, X. J. Qian, J. Zhou, W. L. Dong and M. Jiang, Chemosphere, 2022, 300, 134552 CrossRef CAS PubMed.
- J. Qian, X. Wang, L. Chai, L. Liang, T. Li, Y. Hu and S. Huang, Cryst. Growth Des., 2018, 18, 2358–2364 CrossRef CAS.
- Y. Song, W. Qu, Y. He, H. Yang, M. Du, A. Wang, Q. Yang and Y. Chen, Journal of Energy Storage, 2020, 32, 101877 CrossRef.
- W. J. Liu, K. Tian, L. L. Ling, H. Q. Yu and H. Jiang, Environ. Sci. Technol., 2016, 50, 12421–12428 CrossRef CAS PubMed.
- S. Xu, A. Dong, Y. Hu, Z. Yang, S. Huang and J. Qian, J. Mater. Chem. A, 2023, 11, 9721 RSC.
- R. Zou, H. Y. Quan, W. X. Wang, W. M. Gao, Y. H. Dong and D. Z. Chen, J. Environ. Chem. Eng., 2018, 6, 258–265 CrossRef CAS.
- Y. Wang, R. Liu, Y. Tian, Z. Sun, Z. Huang, X. Wu and B. Li, Chem. Eng. J., 2020, 384, 123263 CrossRef CAS.
- S. X. Yan, Q. Wang, S. H. Luo, Y. H. Zhang, X. Liu, Y. G. Liu, Z. Y. Wang, A. M. Hao and T. F. Yi, J. Power Sources, 2020, 461, 228151 CrossRef CAS.
- M. Bora, S. M. Benoy, J. Tamuly and B. K. Saikia, J. Environ. Chem. Eng., 2021, 9, 104986 CrossRef CAS.
- R. Zhang, X. Jing, Y. Chu, L. Wang, W. Kang, D. Wei, H. Li and S. Xiong, J. Mater. Chem. A, 2018, 6, 17730–17739 RSC.
- P. Waribam, S. D. Ngo, T. T. V. Tran, S. Kongparakul, P. Reubroycharoen, N. Chanlek, L. Wei, H. Zhang, G. Guan and C. Samart, Waste Manag., 2020, 105, 492–500 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ba,
Jinglai Zhang
*ba,
Jinglai Zhang