
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical analysis of electronic cigarette liquids (e-liquids) and direct nicotine quantitation using surface-assisted flowing atmospheric-pressure afterglow desorption/ionization mass spectrometry (SA-FAPA-MS)†
Maximilian Heide a and
Carsten Engelhard‡*ab
a and
Carsten Engelhard‡*ab
aDepartment of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Str. 2, Siegen 57076, Germany. E-mail: engelhard@chemie.uni-siegen.de
bResearch Center of Micro- and Nanochemistry and (Bio)Technology, University of Siegen, Adolf-Reichwein-Str. 2, Siegen 57076, Germany
First published on 14th August 2023
Abstract
Ambient desorption/ionization mass spectrometry (ADI-MS) has been widely used for direct analysis of real samples without sample preparation or separation. Studies on the quantification of low molecular weight compounds in complex matrices with ADI-MS remain scarce. In this paper, we report the application of surface-assisted flowing atmospheric-pressure afterglow mass spectrometry (SA-FAPA-MS) for fast qualitative screening of electronic cigarette liquid (e-liquids) ingredients and direct quantification of nicotine. The quantification approach is rapid, uses a deuterated D4-nicotine standard spike, and does not require a preceding chromatography step or other methods to remove the complex sample matrix. Selected e-liquids were directly applied on thin-layer chromatography (TLC) plate surfaces (normal phase (NP) silica, reversed phase (RP) modified silica, cyano (CN) modified silica, and dimethyl (RP2) modified silica) after dilution and internal standard spiking. The plates served purely as sample carriers and no analyte separation was performed. Promising qualitative results were obtained, demonstrating the ability to detect nicotine alkaloids using this approach and the ability to differentiate e-liquids based on their flavor variations. In addition, dimethyl- (RP2-) and cyano-modified (CN-) silica surfaces were selected for quantification based on performance results of previous studies. It was shown that results were in high accordance with high-performance liquid chromatography (HPLC) experiments with lowest deviations <3% on dimethyl surfaces. Additional quantitative experiments including a certified reference material achieved equally satisfying results with lowest deviations of −1.1% from the certified nicotine content. For nicotine, detection limits down to the fmol range (96 fmol on CN and 20 fmol on RP2) were obtained. A detailed comparison of glass surfaces with functionalized surfaces showed that the functionalized surfaces were superior in terms of sample application reproducibility, mass spectra quality, sensitivity, and information density. Thus, functionalized thin-layer surfaces are considered promising tools for both qualitative and quantitative ADI-MS analysis of complex samples.
Introduction
Electronic cigarettes are increasingly used worldwide as an alternative to traditional tobacco smoking. Early efforts towards electronic cigarettes were made, for example, by H. A. Gilbert in 1965, who patented a smokeless non-tobacco cigarette.1 Today's electronic cigarettes typically work by vaporizing liquids containing nicotine (e-liquids). Details on the operating principles and on e-liquids have been covered in recent reviews.2–4 The main ingredients of e-liquids are propylene glycol, vegetable glycerin, and nicotine. While carcinogens present in classic tobacco smoke are typically not found in these e-liquids,5 e-liquid aerosols can reportedly contain other toxins including heavy metals, plasticizers, flame retardants, and nanoparticles, which can have adverse health effects.6–9Interestingly, some studies have shown that the nicotine content as stated on the product by the manufacturers deviated from the actual nicotine concentrations in e-liquids,3,10,11 a fact which warranted the development and application of new analytical methods, not only for consumer safety. Current methods for the characterization of e-liquids include gas chromatography (GC) coupled to flame ionization detection (FID), thermionic specific detection (TSD), and mass spectrometry (MS), respectively.7,12–21 Also, high-performance liquid chromatography (HPLC) methods with spectroscopic or mass spectrometric detection were used for nicotine quantification in e-liquids.10,22–28 For nicotine, limits of detection (LODs) ranging from 30 μg mL−1,23 50 μg g−1,7 down to 0.149 ng mL−1 were achieved.29 Sample preparation for the different GC and LC methods ranged from simple dilution and spiking with internal standard to time-consuming, more complex preparation with e.g. extraction steps. Separation times were not directly reported in many cases but ranged from a few minutes to over 25 minutes in studies where this was mentioned.7,12–14,17,18,23–25
Plasma spectrochemistry, particularly plasma-based ambient desorption/ionization mass spectrometry (ADI-MS), represents a promising analytical approach for the direct and fast analysis of e-liquid samples. In contrast to classical analytical methods, ADI-MS approaches can provide advantages in terms of time efficiency through rapid screening capabilities.30 A first plasma-based ADI-MS analysis of e-liquids using a Direct Analysis in Real-Time (DART) ionization source focused on qualitative analyte screening, while further quantification was performed by HPLC-MS/MS.26 An alternative candidate of plasma-based ionization sources first reported by Hieftje and coworkers is the flowing atmospheric-pressure afterglow (FAPA).31–33 In this type of ADI source, the helium afterglow is used to directly desorb and ionize the analytes. Compared to DART, the intrinsic thermal desorption capabilities are stronger due to the higher temperature of the afterglow, eliminating the need for additional heating.34 FAPA was optimized over time from the original pin-to-plate32,33 to the pin-to-capillary design which offers higher sensitivity and lower background interference.35 Further, Pfeuffer et al. designed the so-called halo-FAPA, which offers a direct sample introduction of solutions, vapors, or aerosols in addition to direct desorption and ionization from surfaces.36 Later, thin-layer chromatography (TLC) plates reportedly exhibited promising properties in direct screening applications of liquids and dried residues.37 It was found that TLC surfaces can be used merely as sample substrates in surface-assisted (SA) FAPA-MS and that a planar chromatography step was not required.38 Furthermore, the quantitative capabilities of this approach have been demonstrated for matrix- and concomitant-containing samples.38,39 In this study, a SA-FAPA-MS method for the rapid analysis of e-liquids is carefully optimized and different TLC materials are evaluated for best performance. High-resolution MS is used to confirm the presence of e-liquid ingredients and potentially harmful substances. In addition, a SA-FAPA-MS workflow based on a D4-nicotine spike, automated sample dosing, and mass spectral imaging of TLC plates is developed. Accurate quantification of nicotine in e-liquids is demonstrated and validated by HPLC.
Materials and methods
Reagents
Methanol (HPLC grade) was purchased from Fisher Chemicals (Hampton, NH, USA). Nicotine (>99% chemical purity) was acquired from Carl Roth GmbH + Co. KG (Karlsruhe, Germany). Tetra-deuterated (D4-)nicotine (100.1 ± 0.6 μg mL−1 in acetonitrile, isotopic purity 0.01% D0 vs. D4) was obtained from Sigma Aldrich (St. Louis, MO, USA). C18 reversed-phase (RP-)HPTLC LiChrospher® and normal-phase (NP-)HPTLC LiChrospher® plates were provided by Merck KGaA (Darmstadt, Germany), dimethyl (RP2-)TLC and cyano (CN-)HPTLC were purchased from MACHEREY-NAGEL (Düren, Germany). Three different e-liquids (12 mg mL−1 nicotine each, stated by manufacturer) were acquired at a local store for electronic cigarettes. The e-liquids only differed in their flavorings (“White Cake”, “Honey Fruits”, and “Eisbombe”).Preparation of e-liquid samples
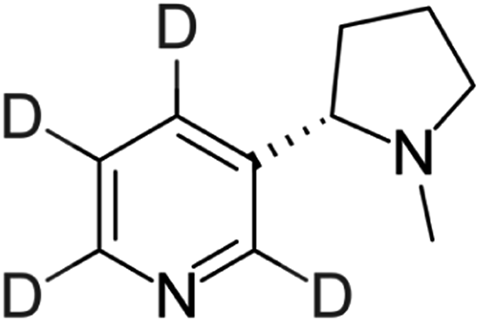
The e-liquid samples were diluted in methanol to obtain 10-fold, 100-fold, and 1000-fold diluted samples due to the high nicotine concentration (≈12 mg mL−1) specified by the manufacturer. Both higher dilution sample solutions (75 μL, respectively) were individually mixed with D4-nicotine solution (25 μL, 10 μg mL−1 ≙ 60.1 nmol mL−1) for quantitative analysis.For validation and additional quantitative data, a certified European reference material ERM®-DZ002a (“electronic cigarette liquid – nicotine & water”) for nicotine quantification in e-liquids was obtained from LGC Group (Teddington, United Kingdom). The certificate of analysis states a nicotine concentration of 18.39 ± 0.52 mg mL−1, which was determined by exact-matching liquid chromatography isotope dilution mass spectrometry. In addition, an artificial nicotine-free e-liquid matrix sample was prepared, containing 55% propylene glycol (>99.5% chemical purity, Fluka, now Sigma-Aldrich, St. Louis, MO, USA), 35% glycerol (>99.7% chemical purity Carl Roth GmbH + Co. KG, Karlsruhe, Germany), and 10% bidistilled water. Both samples were diluted 100-fold and 1000-fold in methanol. 65 μL of the respective dilutions were individually mixed with 35 μL of D4-nicotine solution (10 μg mL−1 ≙ 60.1 nmol mL−1) for quantitative SA-FAPA-MS analysis.
(HP)TLC preparation
Glass-backed TLC plates measuring 4 cm × 5 cm were prepared from 10 cm × 20 cm plates for the surface-assisted mass spectrometry experiments. This procedure was maintained for all plate types to guarantee high comparability within the different experiments. A defined volume (1 μL, if not stated otherwise) of sample solution was deposited with a Linomat V spray-on application system (CAMAG, Muttenz, Switzerland) onto the plates with a dosing speed of 50 nL s−1. The spraying distance was 2.4 mm between the syringe tip and the plate surface. The step size (distance travelled between application of different sample spots) was kept constant at 5 mm for RP2-TLC and CN-HPTLC. For glass, the step size was increased to 10 mm. Samples were analyzed in triplicates, i.e., three 1 μL-samples were sprayed onto the plates in proximity. It is important to note that a time-consuming drying step was not required, and that no chromatographic step was conducted. The plates were only used as sample carriers. For mass spectrometric experiments, the prepared plates were placed in front of the MS instrument (see below for details).High-performance liquid chromatography (HPLC) validation method
HPLC with ultra-violet detection was used as a validation method. An RP-18 column (XBridge, 4.6 mm × 75 mm, 2.5 μm particle size, Waters, Eschborn, Germany) was used in an Agilent 1200 series system (Agilent Technologies, Santa Clara, CA, USA). For the selective detection of nicotine (λdet. = 260 nm), a gradient run was implemented. External calibration was performed for quantitative analysis. Details on the HPLC methods are given in the ESI.†Instrumentation
An Exactive HCD Orbitrap (Thermo Fisher Scientific, Bremen, Germany) was used for mass spectrometric analysis. A weekly instrument calibration was performed with conventional electrospray ionization (ESI†) and Pierce LTQ ESI positive ion calibration solution (Thermo Fisher Scientific, Bremen, Germany) containing caffeine (20 μg mL−1), Met-Arg-Phe-Ala (MRFA, 1 μg mL−1) and Ultramark 1621 (0.001%) in an aqueous solution of acetonitrile (50%), methanol (25%), and acetic acid (1%). The instrumental mass resolution was set to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (at 200 m/z). Day-to-day mass measurement uncertainty during the experiments was <2 ppm. Lock masses were included to increase mass accuracy (phthalic anhydride fragment at 149.2033 m/z; n-butyl benzenesulfonamide at 214.0896 m/z).
000 (at 200 m/z). Day-to-day mass measurement uncertainty during the experiments was <2 ppm. Lock masses were included to increase mass accuracy (phthalic anhydride fragment at 149.2033 m/z; n-butyl benzenesulfonamide at 214.0896 m/z).
A home-built pin-to-capillary FAPA source was used for the SA-FAPA-MS experiments. The ionization source consists of a Macor® ceramic (Schröder Spezialglas, Ellerau, Germany) discharge chamber, a stainless-steel pin cathode (1.6 mm outer diameter (o.d.), 100 mm length, sharpened tip) and a capillary anode (1.6 mm o.d., 1.0 mm inner diameter (i.d.), 15 mm length). The cathode-to-anode distance was 7.5 mm. A negative potential was applied to the cathode through a 5 kΩ resistor with a DC power supply (Kepco, Flushing, NY, USA) operated in current-controlled mode to generate the helium discharge. The current was set to 25 mA resulting in a potential of around 650 V. Grade 5 helium was acquired from Messer Industriegase GmbH (Siegen, Germany). A helium flow rate of 750 mL min−1 was adjusted with an EL-FLOW® Select mass flow controller (Bronkhorst Deutschland Nord GmbH, Kamen, Germany).
A home-built mounting fixture with suitable connectors was used to attach the ionization source to the MS. The mounting fixture also included a motorized stage (Newport Corporation, Irvine, CA, USA) for screening experiments and an acrylic housing for shielding from the laboratory environment. A LabVIEW-based (Version 11.0, 2011, National Instruments, Austin, TX, USA) software was used to control the stage. For SA-FAPA-MS experiment, a curved ion transfer capillary was used (0.6 mm inner diameter, 40 mm extension). The angle between the FAPA outlet and the probed surfaces was set to 70°. The distance between the FAPA outlet and the ion transfer capillary was set to 1.5 mm. The distance between the FAPA outlet and the plate surface was kept at 1 mm and the distance between the plate surface and the ion transfer capillary was kept at 0.5 mm. The TLC plates were held on the x–y stage by a clamp tensioned with a spring. Two-dimensional (2D) molecular maps were acquired using TLC surface line scans at a scan speed of 0.3 mm s−1 in x-direction and 0.5 mm line spacing in y-direction. As mentioned earlier, the LabVIEW software was used to control the table movement and to trigger the mass spectrometric data acquisition for automated MS imaging.
Data analysis
Mass spectrometric data were recorded using the Exactive Tune software (1.1 SP6, Thermo Scientific, Bremen, Germany). Analyte-selective data were acquired by extracted ion chronograms (XICs) based on a mass accuracy of ±4 ppm according to Table 1.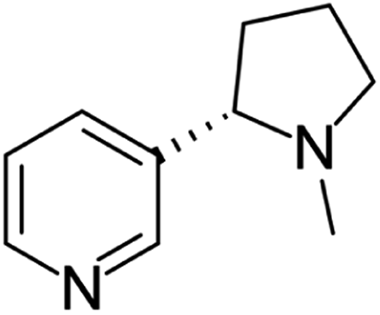
Additional data manipulation was completed with MZmine 2.53 (ref. 40) and Origin 2017 (OriginLab Corporation, Northampton, MA, USA). In addition to quantitative analysis, non-targeted analysis was also performed on nicotine-related alkaloids and other compounds (see Table 2). For the regions of interest (ROI) shown, the integration cut-off was set to 2% of the respective signal maximum.
| Analyte | Ionic species | Ion trace for XICb | Molecular structure |
|---|---|---|---|
| a Specification of listed sum formulas: M3 = C9H12N2; M4 = C10H12N2O; M5 = C10H12N2; M6 = C9H10N2; M7 = C6H5NO2; M8 = C6H6N2O; M9 = C10H10N2; M10 = C7H9N; M11 = C7H7NO.b Calculated with FreeStyle 1.7 (Thermo Scientific) based on sum formula of respective ionic species. | |||
| Nornicotine | [M3 + H]+ | 149.1067–149.1079 |  |
| Cotinine | [M4 + H]+ | 177.1015–177.1029 |  |
| Anatabine | [M5 + H]+ | 161.1067–161.1080 | 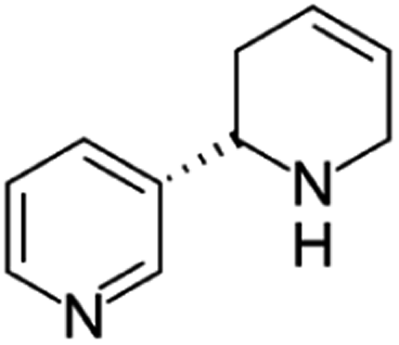 |
| Myosmine | [M6 + H]+ | 147.0911–147.0923 | 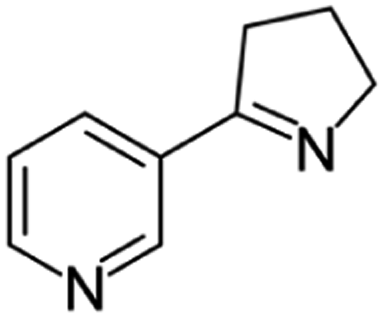 |
| Niacin | [M7 + H]+ | 124.0388–124.0398 | 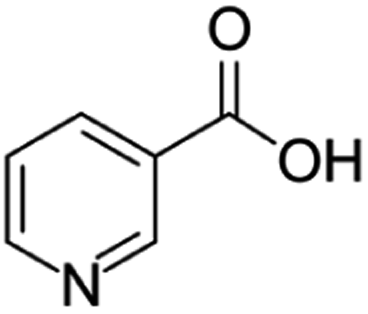 |
| Niacinamide | [M8 + H]+ | 123.0548–123.0558 | 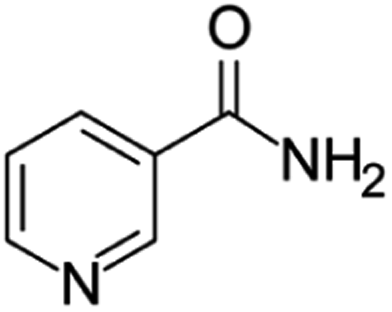 |
| Nicotyrine | [M9 + H]+ | 159.0910–159.0923 | 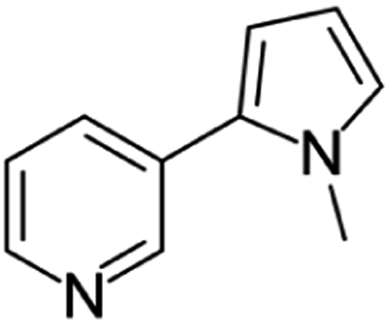 |
| N-Methylaniline | [M10 + H]+ | 108.0803–108.0812 | 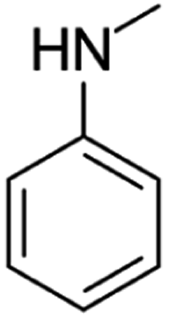 |
| 1-(3-pyridinyl)-ethanone | [M11 + H]+ | 122.0596–122.0605 | 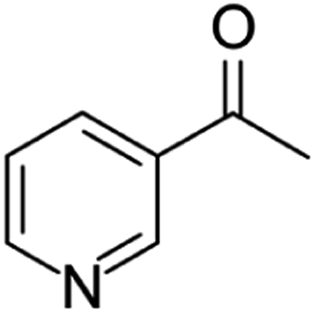 |
Safety considerations
The experimental setup for these experiments included high voltages and currents to power the FAPA source. All connections between the power supply and the ADI source were isolated to avoid electric shocks. An enclosure that covered the ionization source was used to prevent vaporized solvents and potentially toxic or corrosive chemicals and byproducts from entering the laboratory atmosphere. During an ongoing experiment all laboratory users were informed and made aware of hazards to avoid accidents or safety risks.Results and discussion
Quantification of nicotine in e-liquids
E-liquids are often advertised as alternative nicotine delivery systems for tobacco smokers. To assess the nicotine content in selected electronic cigarette liquids sold in Germany, a quantitative method based on SA-FAPA-MS with TLC plates as sample carriers was carefully developed and optimized. Because there is a broad variety of different TLC plates commercially available, we investigated the influence of the choice of TLC surface composition on the FAPA-MS response in earlier studies.37,38 As a general trend, we found that the use of dimethyl and cyanopropyl functionalized plates gave the best signal response for compounds of different polarities with low to medium molecular weight. This is why we decided to use RP2-TLC and CN-HPTLC plates (see materials and methods) as sample substrates for e-liquids in this study. In addition, an in-house developed RP-HPLC/UV method (see ESI† for details) was used to validate the FAPA-MS results. The quantitative results are summarized in Fig. 1 and Table S2.†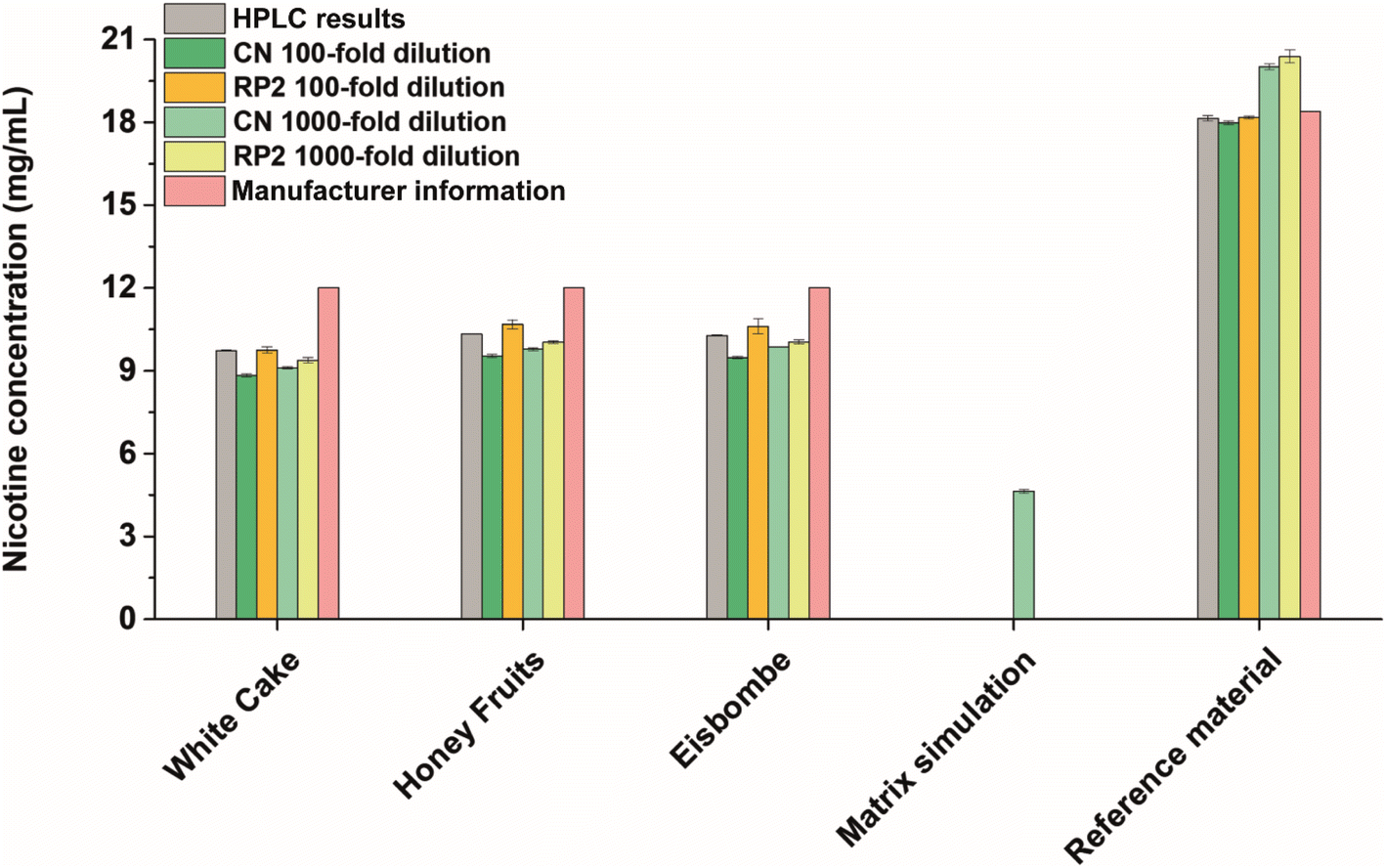 | ||
| Fig. 1 Nicotine concentrations determined by HPLC/UV (100-fold diluted samples, n = 3) and SA-FAPA-MS (RP2- and CN-modified, 100-fold and 1000-fold dilution, n = 3) compared to the manufacturer's indications. | ||
In all flavored e-liquid samples and with both FAPA-MS and RP-HPLC/UV, nicotine was found in amounts below 10.4 mg mL−1 and below the concentration stated on the product label (12 mg mL−1). We assume that these lower concentrations were found due to a poor quality control during manufacturing or due to the ageing of the e-liquids. Nicotine degradation products were also successfully detected (Fig. 3), including anatabine, cotinine, myosmine, nicotine-N-oxides, β-nicotyrine, and nornicotine, which is in accordance with the literature.41,42 These compounds are also known impurities in e-liquids as described in the European Pharmacopoeia.43
Within the experimentally obtained results, the values are in a similar range. Especially, the results based on the RP2-TLC surface and 100-fold dilution are in highest accordance with the HPLC/UV values. The best performance regarding the accuracy was achieved on RP2-TLC surfaces (0–3% deviation from validation values at 100-fold dilution, see Table S2†) while the best performance regarding the precision was achieved on CN-HPTLC plates (relative standard deviations (RSD) of 1‰ for 1000-fold dilution, see Table S2†). While this global trend is apparent, there are some small differences between HPLC/UV and FAPA-MS results. The highest deviation from the HPLC results was found for the samples applied on CN-HPTLC plates after 100-fold dilution with deviations up to −10% (“White Cake”). RP2-TLC plates gave the lowest deviations with a maximum of −4% (“White Cake”, 1000-fold dilution) with respect to the HPLC results. The RSDs as indicators for the repeatability were lowest for the HPLC/UV and CN-SA-FAPA-MS methods (all below 1%). RP2-SA-FAPA-MS showed RSDs between 0.5% and 2.5%. The SA-FAPA-MS methods LODs for nicotine were evaluated for CN- and RP2-surfaces and were found to be 96 fmol and 20 fmol of applied nicotine, respectively (see Fig. S5 and S6†). This corresponds to LODs of 15.5 ng mL−1 (CN) and 3.3 ng mL−1 (RP2) and a limit of quantitation (LOQ) of 46.5 ng mL−1 (CN) and 9.9 ng mL−1 (RP2).
For quality assurance and method validation, a European reference material with nicotine at a concentration of 18.39 ± 0.52 mg mL−1 and a nicotine-free simulated e-liquid matrix were analyzed both with HPLC/UV and SA-FAPA-MS. HPLC/UV analysis of the reference material resulted in a nicotine concentration of 18.14 ± 0.10 mg mL−1, which is in agreement with the certified value (−1.4%). Reference material analysis using SA-FAPA-MS was performed as well, with best results for 100-fold diluted samples on RP2-TLC and CN-HPTLC (−1.1% and −2.2% compared to certified value). A higher bias was observed when the sample was more dilute (1000-fold dilution) with recoveries of +8.8% with CN-HPTLC and +10.9% with RP2-TLC (see Table S2† for more details). It can be concluded here that the FAPA-MS method validation was successful with best results using 100-fold diluted samples.
Nicotine was not detected in the simulated matrix sample by HPLC/UV. Analysis of the same sample by FAPA-MS showed minimal nicotine amounts in the low μg mL−1 range (not visible in Fig. 1 due to the scaling). This can be explained by the presence of a small amount of unlabeled nicotine in the internal standard (isotopic purity 0.01% D0 vs. D4), which is added to each sample and during dilution. However, the apparent high nicotine value (4.64 ± 0.07 mg mL−1) measured at 1000-fold dilution on CN-HPTLC (cf. Fig. 1, “matrix simulation”) is too high for it to be an impurity in the standard. This is likely due to a contamination but could also be an isobaric interference. This observation and a small positive bias, which was also observed for the reference material at higher dilutions, should be kept in mind for possible future applications.
The results demonstrate the high precision and accuracy of the developed method, which is schematically illustrated in Fig. 2. At this point it should be mentioned again that no extensive sample preparation was performed, and yet such low RSDs and accurate results could be obtained.
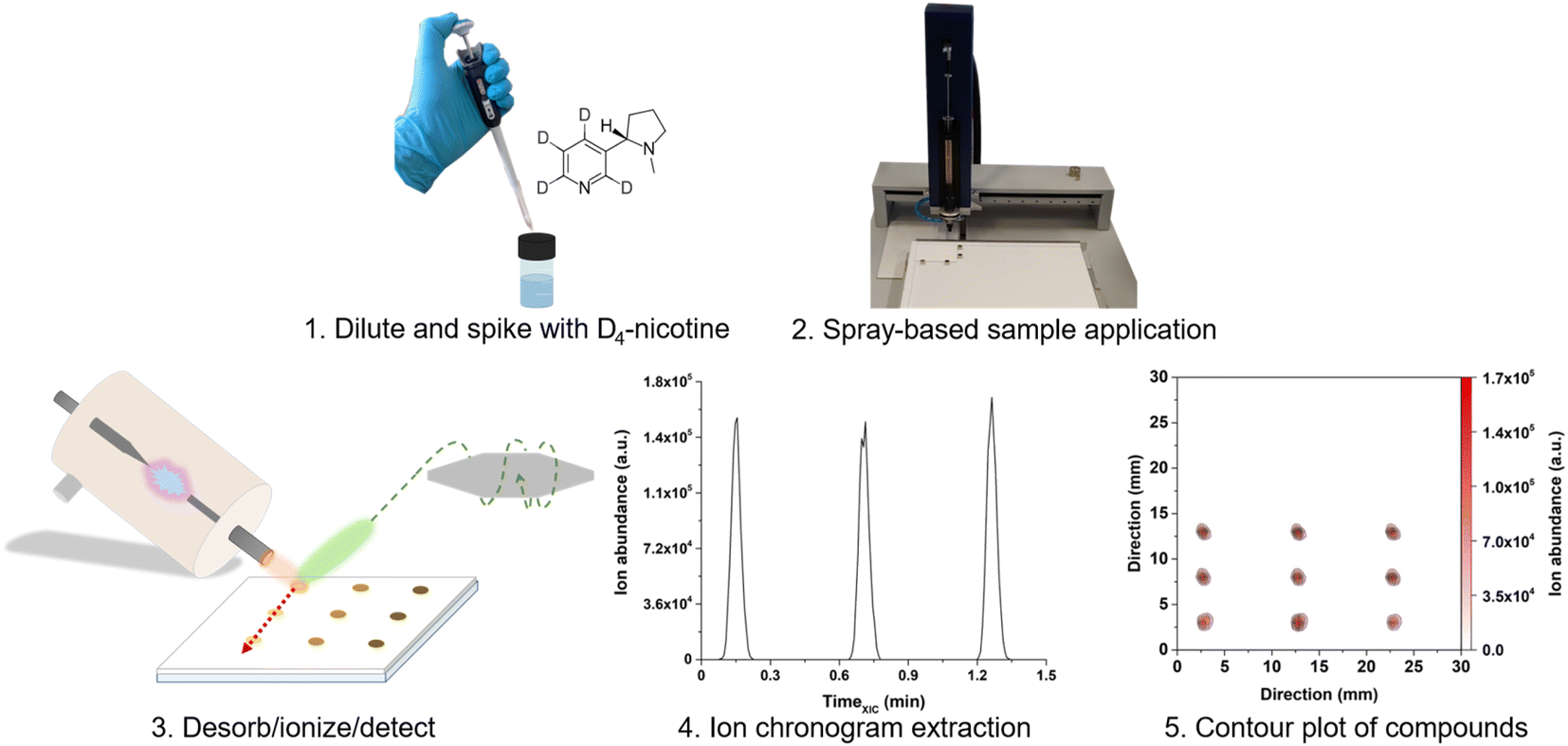 | ||
| Fig. 2 Schematic of the SA-FAPA-MS workflow for fast nicotine quantification in e-liquids. (1) Sample dilution and isotopologue spiking; (2) spray-based application of 1 μL of the liquid sample; (3) ambient desorption/ionization and mass spectrometric analysis; (4) extraction of the ion chronograms of the species of interest; (5) assembling the contour plots from the individual ion chronograms for further data evaluation. | ||
The FAPA-MS method is considered attractive for fast quantitative screening because sample preparation was rapid (∼15 min) and triplicate analysis of three samples by SA-FAPA-MS was completed in approximately 45 minutes. The analysis time can be further shortened by optimizing the sample spacing on the surfaces and by implementing more time-efficient scanning approaches like single line scans or spot-to-spot probing. In contrast, the HPLC/UV method (with four calibration standards and a run time of 6 min per injection) required approximately 126 min of analysis time. With sample preparation, system equilibration, and washing steps in-between each injection, triplicate analysis of three samples required circa 180 min.
Qualitative analysis of pyridine alkaloids and related compounds with SA-FAPA-MS
Potential health effects of e-cigarettes are related to the chemical composition of the e-liquid. While vendors mostly state the main product ingredients on the label (propylene glycol 55%, glycerol 35%, and flavorings 10% for the three e-liquids used in this study), e-liquids can contain potentially toxic substances. To assess the chemical composition of the e-liquids, a rapid qualitative screening approach based on SA-FAPA-MS was developed and used.Besides nicotine as the often desired active ingredient in e-liquids, numerous other structurally related compounds can be present, which can be categorized as pyridine or tobacco alkaloids. Here, rapid qualitative screening of the e-liquids by FAPA-MS was performed directly on several different surfaces without analyte extraction, matrix removal or preconcentration but only dilution. After merely 15 min of measurement time, the compounds listed in Table 2 were readily identified based on accurate mass measurements. In Fig. 3, the influence of the surface composition on the alkaloids' signal in the e-liquid termed “Eisbombe” is shown. All compounds listed in Table 2 were detectable with reasonable intensities (ranging from 1 × 108 to 1.4 × 104 a.u.) and signal stability (RSDs ranging from 60% to 0.3% over 1.5 min, line scan approach, 45 min total measurement time) on the different (HP)TLC surfaces. In contrast, analyte signals were lower by factors of 10–100, when uncoated TLC glass plates were used. Furthermore, N-methylaniline could not be detected from glass at all. The data for the other two e-liquids “White Cake” and “Honey Fruits” are shown in the ESI Fig. S2 and S3.† Qualitatively, all three e-liquids show similar results in terms of nicotine-related compounds, but minor differences in the data are apparent for individual flavoring compounds. For example, ethyl maltol and ethyl vanillin, which were reported as additives in e-liquids,44,45 were only detectable in the e-liquid “Eisbombe” (Fig. S4†). This confirms that qualitative differentiation of e-liquids is also possible with this rapid and direct screening method due to the high information density provided by high-resolution MS. Because most studies do not disclose the brand name of the e-liquids under investigation, it is not clear whether the three e-liquids used in this study have been under investigation before.
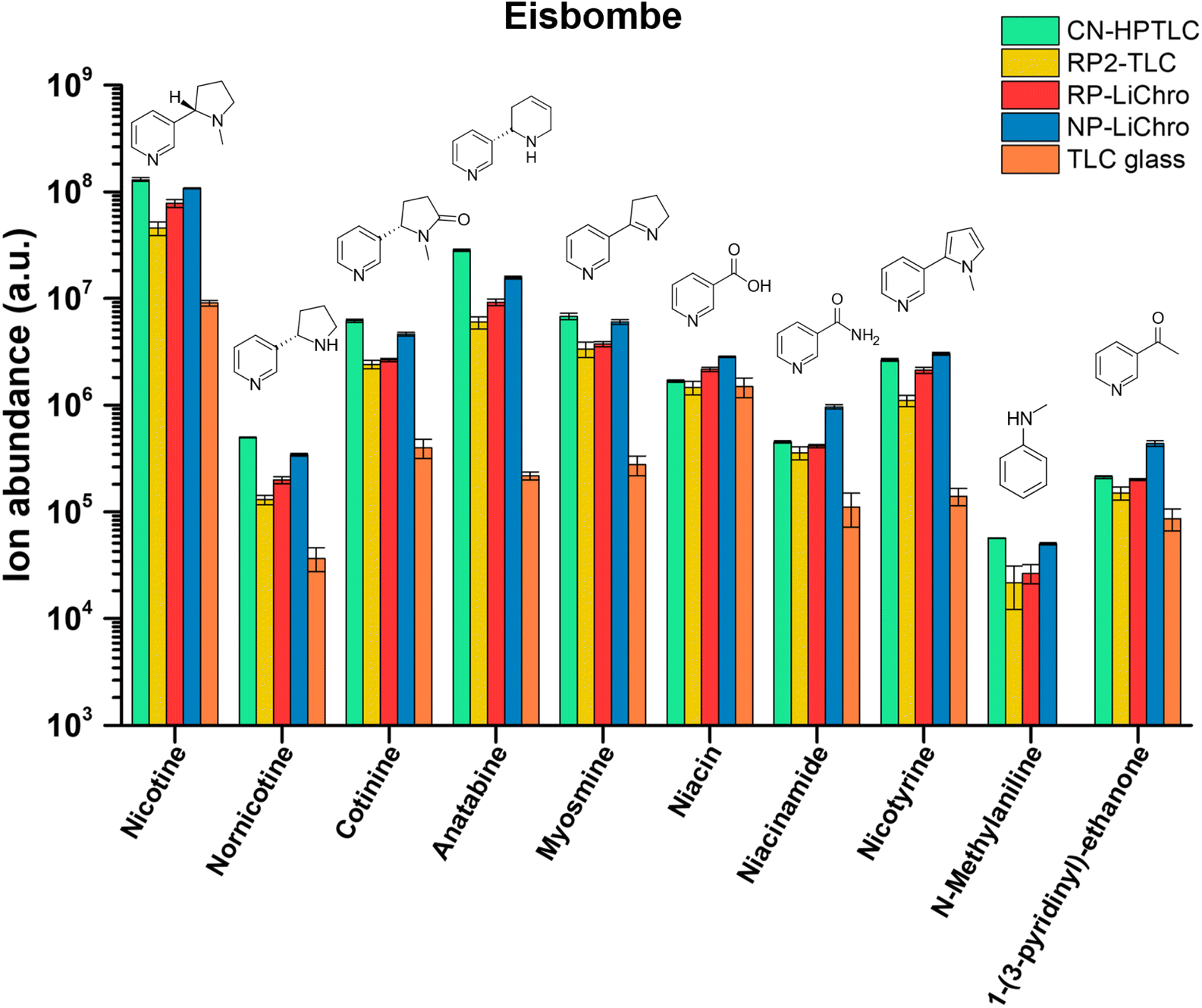 | ||
| Fig. 3 Intensities of nicotine-related compounds in e-liquid “Eisbombe” on the investigated surfaces based on signal integration of molecular contour plots of the respective [M + H]+ species according to Table 2. | ||
Overall, the use of a TLC substrate for ambient desorption/ionization resulted in a significantly higher ion yield compared to uncoated glass for the compound class discussed above. This finding is consistent with observations in our earlier studies,38 when we compared the performance of glass versus TLC plates for desorption/ionization of caffeine in beverage samples. Because these are qualitative data, no precise statement should be made on detection limits of individual analytes. However, given that these are nicotine-like compounds, analogues behavior regarding the sensitivity seems to be reasonable.
Influence of the surfaces on molecular contour plots
Data presented in Fig. 3 are based on the mean signal and relative standard deviation of XICs signals from three analyte spots (ROI) in 2-dimensional molecular maps (contour plots). While Fig. 3 aids in the mean signal comparison of different compounds across different substrates, differences in the spot size and shape cannot be assessed. Exemplarily, Fig. 4 shows the influence of different surfaces on the spot appearance and signal response for anatabine ([M + H]+, m/ztheo 161.1073, positive ion mode, measured m/z of 161.1073–161.1074 maximum deviation of 0.59 ppm from m/ztheo) in the three e-liquids under investigation.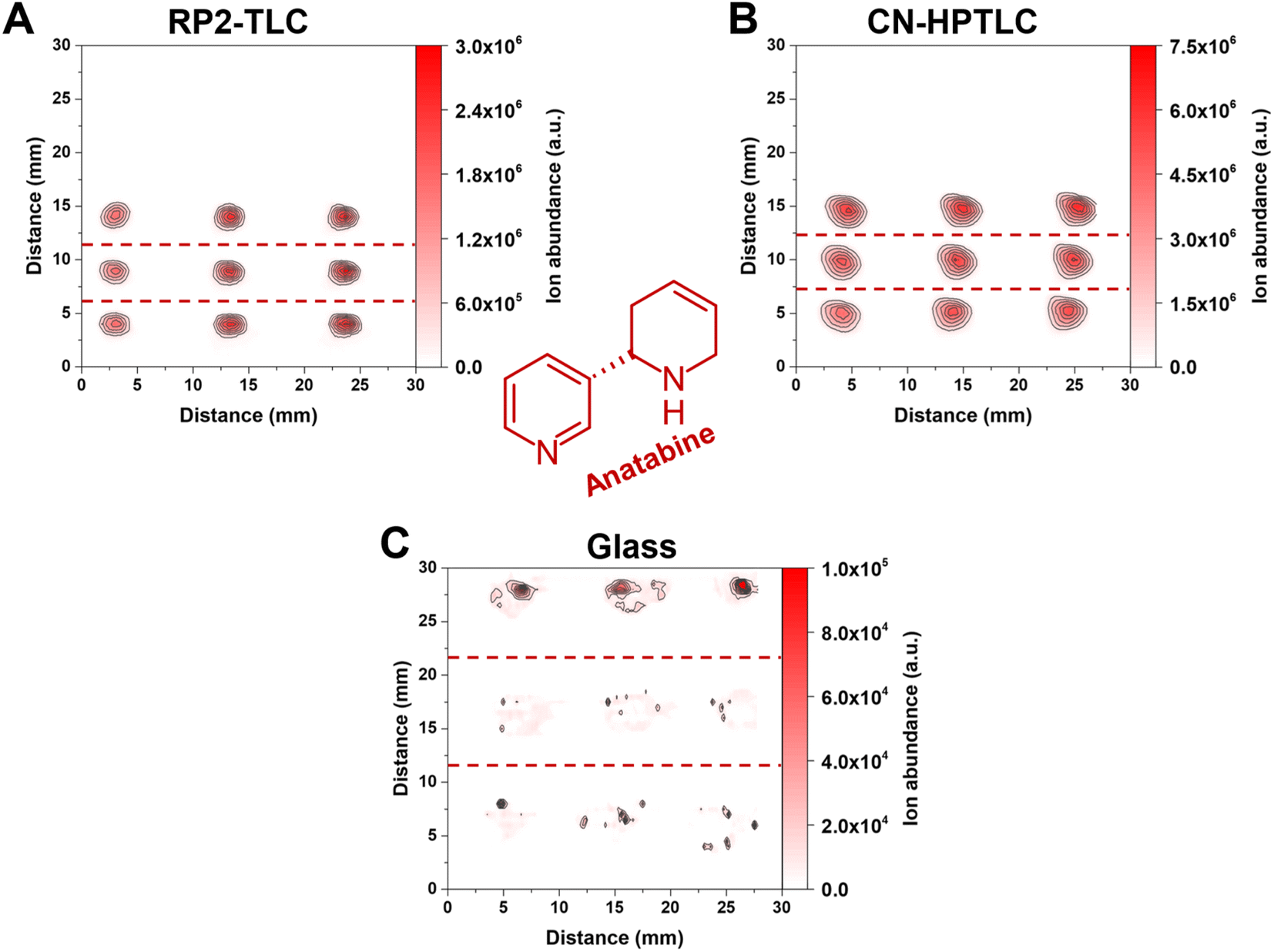 | ||
| Fig. 4 Contour plots of anatabine in the three e-liquid samples on the three different surfaces. (A) Represents RP2-TLC, (B) CN-HPTLC and (C) glass. The deposited sample volume was 1 μL per spot (n = 3). From top to bottom in the individual 2D-plots, the first row of signal points correspond to “White Cake”, the second row to “Honey Fruits” and the third row to “Eisbombe”. The FAPA source was operated at a helium flow rate of 750 mL min−1 and a discharge current of 25 mA. The used ion trace for protonated anatabine is listed in Table 2. | ||
Most importantly, a significantly lower signal of anatabine was recorded with FAPA-MS when glass served as the sampling substrate (for “Eisbombe” −99.23% and −96.35% ROI signal compared to RP2-TLC and CN-HPTLC, for “White Cake” −99.62% and −98.49% ROI signal compared to RP2-TLC and CN-HPTLC, and for “Honey Fruits” −99.60% and −98.22% ROI signal compared to RP2-TLC and CN-HPTLC, respectively). The use of CN-HPTLC plates served best and resulted in a signal approximately twice as high as the one with RP2 (local signal maximum on CN-HPTLC 6.45 × 106 a.u., ROI signal 2.83 × 107 a.u.). Differences in sample spot size and shape were observed as well. Please note that all three contour plots in Fig. 4 have the same dimensions (3 cm × 3 cm) and sample application conditions were identical as described in the materials and methods section (1 μL, 10-fold diluted e-liquids with methanol, 50 nL s−1 dosing speed). Overall, it was found that the anatabine signal areas on RP2 were circular in shape, less broad and more focused (ca. 2.5 mm diameter) compared to signals on CN-HPTLC plates. A similar trend was also observed for other compounds under investigation (e.g., cotinine, myosmine, and nornicotine, data not shown). In contrast, anatabine spots on glass were found to be less symmetrical and appeared smeared across a larger distance (ca. 7 mm and 5 mm spatial spread in x- and y-direction, respectively) on the glass surface. This observation was also made for other compounds in the three e-liquids (e.g., cotinine, myosmine, and nornicotine, data not shown). As a result of the spot shape irregularities and reduced signal response for the nicotine alkaloids, glass is considered less attractive as a sample support for e-liquids analysis by SA-FAPA-MS. This leads to significantly lower signal intensities and lower reproducibility. Only for the protonated niacin similar but still lower signal yields were obtained on glass. A similar conclusion was drawn earlier but for the quantitation of caffeine in beverages by FAPA-MS.38 Clearly, the TLC-based surfaces studied here work well with the spray-on sample loading system and result in fit-for-purpose molecular maps after FAPA-MS analysis.
Influence of the sampling surface on the mass spectra of diluted e-liquids
During SA-FAPA-MS method optimization it was observed that surfaces with different properties (glass vs. TLC plates) may lead to differences in signal height and signal distribution across the monitored mass spectral range (m/z 100–450) for otherwise identical samples. The differences of the XIC signal and the appearance of the contour plots for the different surfaces has already been discussed above. Here, we turn to changes in the appearance (skyline) and the dominant ions in the respective mass spectra themselves.Because dimethyl- and cyano-modified surfaces gave the most promising quantitative results compared to other surfaces, mass spectra using these surfaces and compared to glass are shown in Fig. 5.
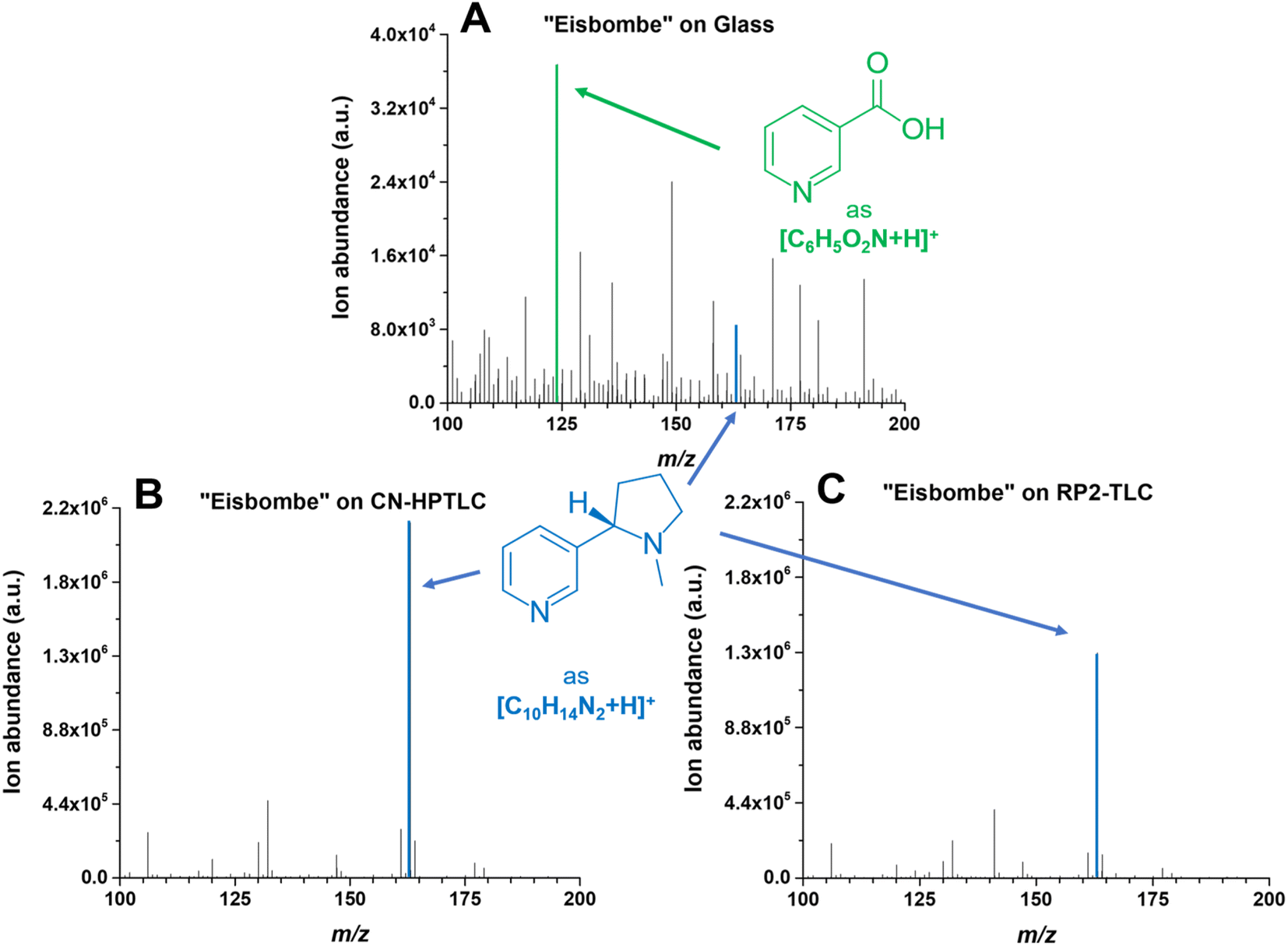 | ||
| Fig. 5 Mass spectra of “Eisbombe” flavored e-liquid 10-fold diluted in methanol. The spectrum labeled with (A) is based on desorption and ionization from the pure glass surface, the lower two spectra are based on cyano- (B) and dimethyl-modified (C) surfaces. The FAPA source was operated at a helium flow rate of 750 mL min−1 and a discharge current of 25 mA. Mass traces for the two depicted protonated analytes nicotine (C10H14N2) and niacin (C6H5O2N) are listed in Table 2. The extracted data are based on the respective chronograms with the highest total ion yields. | ||
The predominant ionic species in mass spectra from SA-FAPA-MS analysis with cyano and dimethyl surfaces was found to be protonated nicotine ([M + H]+, m/ztheo 163.1230, positive ion mode) with a measured m/z of 163.1228–163.1229 (maximum deviation of −1.01 ppm from the theoretical m/z). When the same e-liquid (“Eisbombe”) was probed by the FAPA source from a glass substrate, protonated niacin was found to be the most abundant species ([M + H]+, m/ztheo 124.0393, positive ion mode) with a measured m/z of 124.0393 (deviation of 0.04 ppm from the theoretical m/z). While the glass surface does not exhibit selectivity for analyte desorption, both functionalized surfaces appear to allow selective desorption. It is hypothesized here that the applied sample solutions can be embedded in the surface structure due to the porous and adsorbent morphology and interact differently compared to a bare glass surface. This influences the desorption and subsequent ionization process for the observed substances, as can be seen in the case of nicotine and related compounds. Fig. 4 shows that the analytes are spatially more confined and exhibit less spot broadening on the functionalized surfaces. In contrast, relatively more smearing occurs on glass, which also induces a lower desorption rate and, thus, lower mass spectral signal. However, because not only the intensities themselves but also the coverage of the detected species differ significantly in the mass spectra, direct analyte–surface interactions may also take place. Several factors including surface polarity and morphology may play a role here. For example, the moderate polarity of RP2- and CN-functionalized surfaces compared to polar silica or non-polar C18-alkyl functionalized silica is believed to be important. In addition, the porous, permeable surface morphology allows analytes to be embedded after application and drying, which is not possible on impermeable surfaces such as glass. Here, the spatial analyte density during spot sampling from porous versus non-porous materials is likely to be different. These effects will be studied in more detail in the future.
In general, it can be observed that the spectra for the (HP)TLC surfaces are dominated by nicotine-based species, whereas glass shows a higher number of characteristic sample related species at similar intensities as nicotine. A logarithmic scaling of the functionalized surface-based spectra would of course reveal numerous other species as depicted in Fig. 3.
In Fig. 6, a zoom-in into m/z range 100–200 is provided to illustrate the abundance of other low-molecular-weight species in the e-liquid probed by FAPA-MS on a pure glass surface.
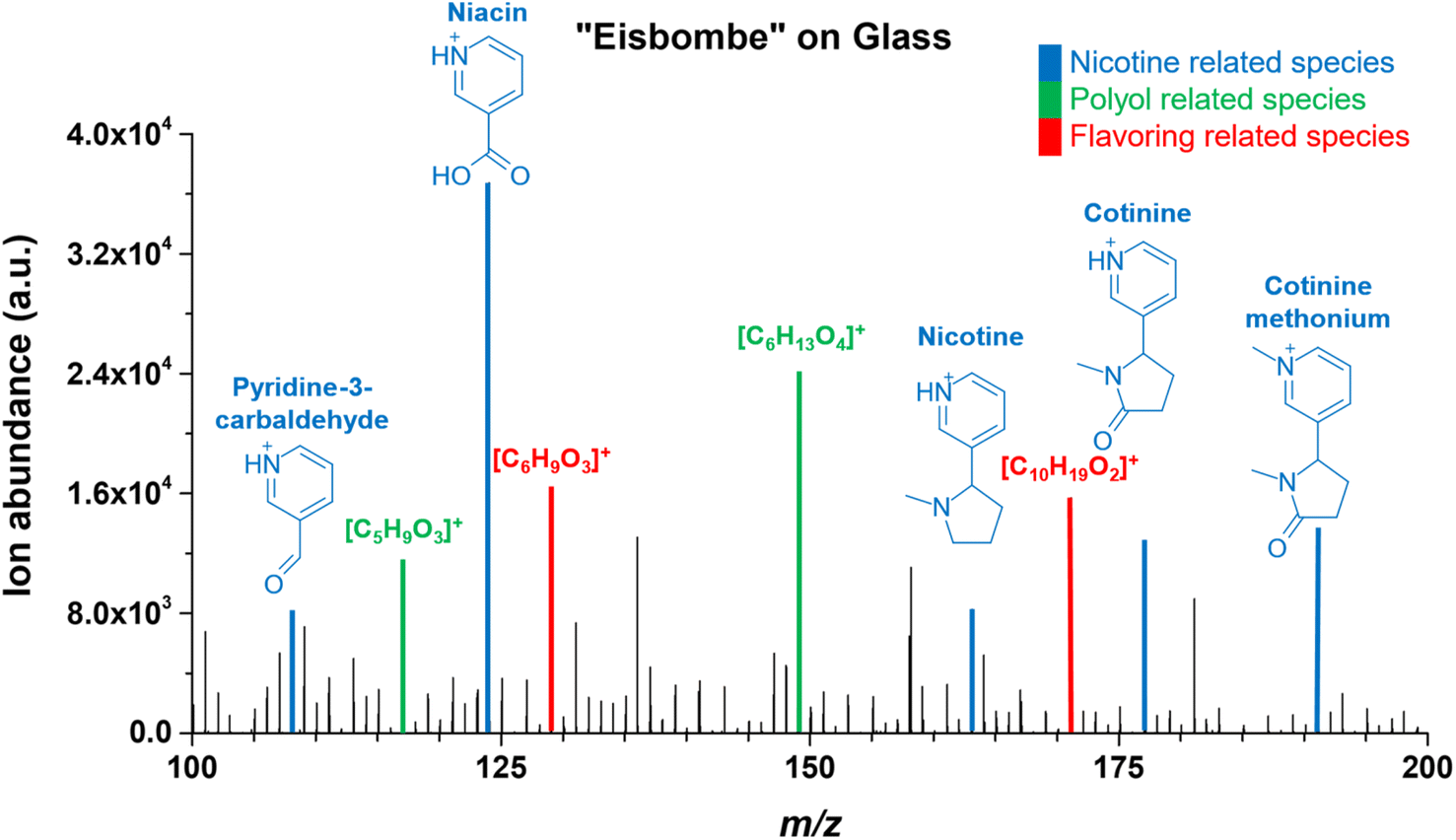 | ||
| Fig. 6 Mass spectrum of “Eisbombe” flavored e-liquid 10-fold diluted in Methanol on glass. The mass range is limited from 100–200 m/z, as this is where most species occur with significant intensity. Blue shows signals for nicotine related species that can be assigned to molecular structures. Green shows signals for polyol related species and red shows signals for flavoring related species, in both cases possible molecular formulae for the ionic species are given. The FAPA source was operated at a helium flow rate of 750 mL min−1 and a discharge current of 25 mA. The extracted data are based on the respective chronogram with the highest total ion yields. | ||
In addition to nicotine, other nicotine related substances such as pyridine-3-carbamaldehyde, niacin, cotinine, and cotinine methonium were detected at similar or even higher signal intensities. Cotinine methonium ion (M+, m/ztheo 191.1179, positive ion mode) with a measured m/z of 191.1179 (deviation of −0.10 ppm from the theoretical m/z) is a quaternary ammonium compound directly related to nicotine (also described for the metabolism of nicotine).46
Mass spectral peaks color-coded in green were assigned to ionic species of polyols, which are one of the main ingredients in e-liquids. Because a variety of isobaric polyols with different isomeric structures exists, only the sum formulas are given in Fig. 6. Mass spectral peaks color-coded in red were assigned to the sum formulas [C10H19O2]+ (m/ztheo 171.1380, positive ion mode) with a measured m/z of 171.1379 (deviation of −0.21 ppm from the theoretical m/z) and [C6H9O3]+ (m/ztheo 129.0546, positive ion mode) with a measured m/z of 129.0546 (deviation of −0.08 ppm from the theoretical m/z). The species can be assigned to protonated furaneol (C6H8O3) and protonated decalactones (C10H18O2) which are usually present as the two isomers γ-decalactone and δ-decalactone. These compounds are also mentioned in the literature as flavoring agents in e-liquids.44
Contrary to the glass surface, sampling from functionalized surfaces results in significantly different mass spectra. For comparison, a more detailed look at an exemplary mass spectrum on a CN-HPTLC surface is shown in Fig. 7. Nicotine ([M + H]+, m/ztheo 163.1230, positive ion mode) with a measured m/z of 163.1228 (deviation of −1.01 ppm from the theoretical m/z) shows the highest signal intensity on the CN-HPTLC surface. In addition, nicotine fragmentation products (also highlighted in blue) can be readily identified at m/z 132.0807 ([M + H]+, m/ztheo 132.0808, deviation of −0.50 ppm from the theoretical m/z) and 106.0652 ([M + H]+, m/ztheo 106.0651, deviation of 0.70 ppm from the theoretical m/z), which are also reported in the literature.47
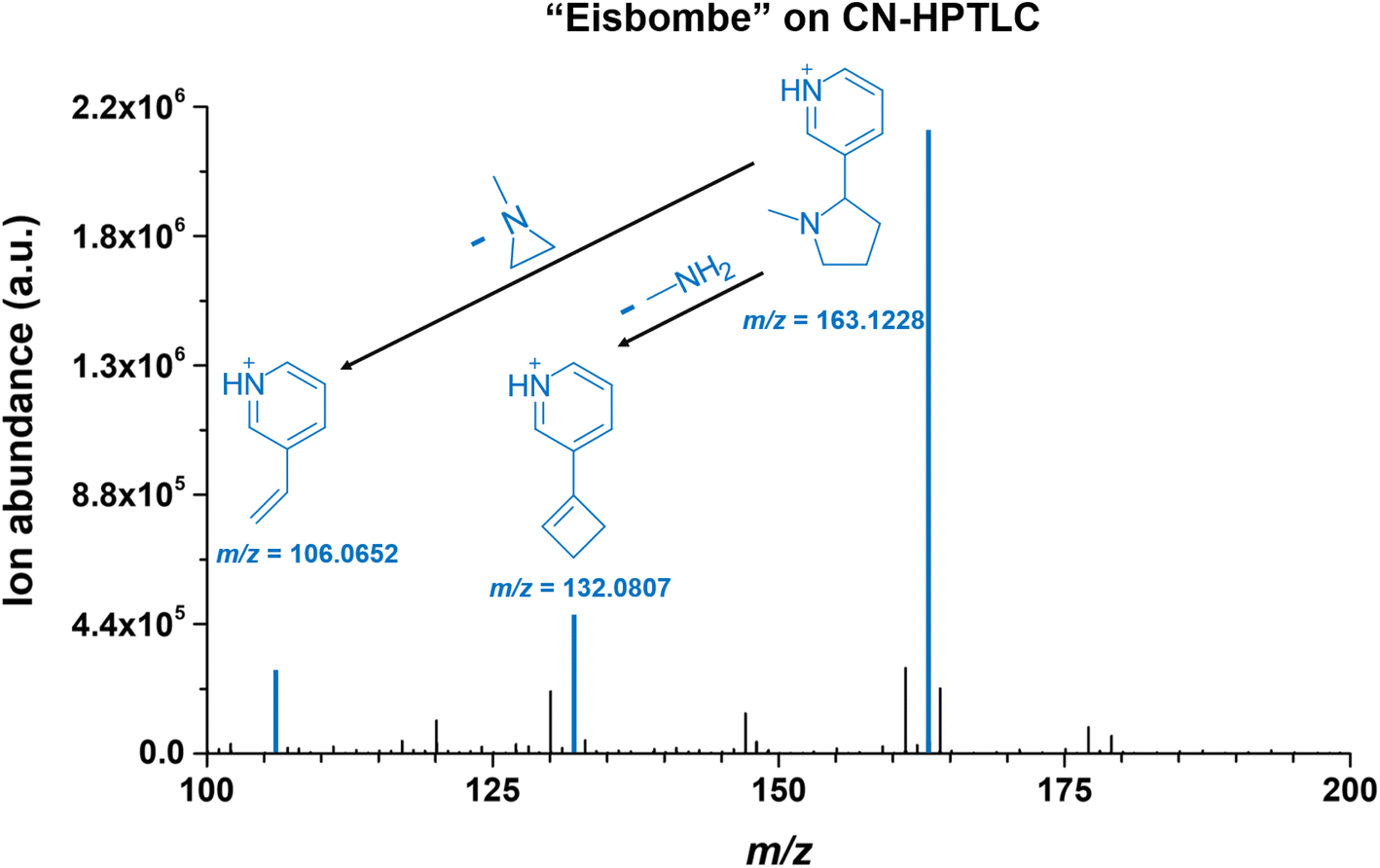 | ||
| Fig. 7 Mass spectrum of “Eisbombe” flavored e-liquid 10-fold diluted in methanol on CN-HPTLC. The mass range is limited from 100–200 m/z as the range with the occurrence of the most dominant species. The blue highlighted signals show nicotine (m/z 163.1228, Δppm of −1.01 ppm from theoretical m/z) and two fragment ions (m/z 132.0807, Δppm of −0.50 from theoretical m/z and m/z 106.0651, Δppm of 0.70 from theoretical m/z) originating from neutral losses. | ||
Furthermore, other reported fragmentation products can be detected with lower intensity at m/z 130.0651 and m/z 120.0808 (data not shown), both assignable to the nicotine fragmentation pathway.47 A comparative look at Fig. 5 shows that similar mass spectra are generated on the two functionalized surfaces. Only a signal at m/z 141.0545 is additionally detected with higher intensity on the RP2 surface. The species can be assigned to ethyl maltol ([M + H]+, m/ztheo 141.0546, deviation of −0.64 ppm from the theoretical m/z), which, as already mentioned, is also used as a flavoring in e-liquids.45 On glass, only the fragmentation product at m/z 132.0808 can be detected with a significantly lower signal intensity.
Conclusion
Surface-assisted ambient desorption/ionization high-resolution mass spectrometry proves to be a suitable method for direct analysis of electronic cigarette liquids. Functionalized thin-layer surfaces were used as sample support. Significantly improved results for nicotine and nicotine-related substances were obtained compared to simple glass surfaces. Differentiation of three different e-liquids was demonstrated based on qualitative differences in flavor additives. In addition to direct target analysis, this also opens up the future possibility to perform multivariate data analyses.The workflow required only very small amounts of sample (1 μL) and can be automated. Sample dosing on TLC plates was found to be very reproducible and resulted in spatially defined sample spots when silica, octadecyl, cyano, and dimethyl surfaces were used. Similarly, ion yields for all target analytes were found to be significantly higher compared to glass when silica, octadecyl, cyano, or dimethyl surfaces were used.
Sample preparation for quantitative analysis with SA-FAPA-MS was rapid and required only sample dilution and the addition of an isotopologue standard. Such standards are nowadays available and affordable for a wide range of analytes. The instrumental analysis itself was significantly faster than a comparative HPLC-UV method. Low detection limits in the fmol range were achieved for nicotine in the e-liquid matrix and demonstrate the significantly improved performance compared to results when glass surfaces were used. Interestingly, the measured concentrations of nicotine (with both FAPA-MS and HPLC/UV) diverged from those stated on the e-liquid label (approximately −16%). All e-liquids contained one or more potentially harmful substances, including N-methylaniline and 1-(3-pyridinyl)-ethanone.
In the future, SA-FAPA-MS could be helpful as a fast screening tool for quality control during manufacturing and to ensure accurate nicotine content information on the product label. In addition, it could be used to detect accidental contamination of “nicotine-free” products to ensure safe consumption for users who want to reduce or avoid nicotine intake altogether.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
This work was supported with selected TLC plates from Merck KGaA, Darmstadt, Germany. The authors have no other relevant financial or non-financial interests to disclose.Acknowledgements
The authors thank Merck KGaA, Darmstadt, Germany, for providing selected TLC plates and MACHEREY-NAGEL, Düren, Germany, for helpful discussions. All members from the mechanical workshop in the Department of Chemistry and Biology at the University of Siegen are acknowledged for their continued support and manufacturing of custom pieces. The University of Siegen is acknowledged for funding.References
- H. A. Gilbert, US Pat.3200819A, 1965 Search PubMed
.
- R. M. Strongin, Annu. Rev. Anal. Chem., 2019, 12, 23–39 CrossRef PubMed
- A. Taylor, K. Dunn and S. Turfus, Drug Test. Anal., 2021, 13, 242–260 CrossRef CAS PubMed
- R. O'Connor, L. M. Schneller, N. J. Felicione, R. Talhout, M. L. Goniewicz and D. L. Ashley, Tob. Control, 2022, 31, 175–182 CrossRef PubMed
- Y. J. Lee, C. J. Na, L. Botao, K. H. Kim and Y. S. Son, Sci. Total Environ., 2020, 703, 134567 CrossRef CAS PubMed
- A. Bhatnagar, Curr. Cardiovasc. Risk Rep., 2016, 10, 1–10 CrossRef PubMed
- J. G. Lisko, H. Tran, S. B. Stanfill, B. C. Blount and C. H. Watson, Nicotine Tob. Res., 2015, 17, 1270–1278 CrossRef PubMed
- J. A. Oh and H. S. Shin, J. Chromatogr. Sci., 2015, 53, 841–848 CAS
- C. Hutzler, M. Paschke, S. Kruschinski, F. Henkler, J. Hahn and A. Luch, Arch. Toxicol., 2014, 88, 1295–1308 CrossRef CAS PubMed
- B. Davis, M. Dang, J. Kim and P. Talbot, Nicotine Tob. Res., 2015, 17, 134–141 CrossRef PubMed
- M. L. Trehy, W. Ye, M. E. Hadwiger, T. W. Moore, J. F. Allgire, J. T. Woodruff, S. S. Ahadi, J. C. Black and B. J. Westenberger, J. Liq. Chromatogr. Relat. Technol., 2011, 34, 1442–1458 CrossRef CAS
- M. Srbinoska, Z. Kavrakovski, V. Rafajlovska and J. Simonovska, Arh. Hig. Rada Toksikol., 2019, 70, 130–133 CrossRef CAS PubMed
- A. El-Hellani, R. El-Hage, R. Baalbaki, R. Salman, S. Talih, A. Shihadeh and N. A. Saliba, Chem. Res. Toxicol., 2015, 28, 1532–1537 Search PubMed
- M. L. Goniewicz, R. Gupta, Y. H. Lee, S. Reinhardt, S. Kim, B. Kim, L. Kosmider and A. Sobczak, Int. J. Drug Policy, 2015, 26, 583–588 CrossRef PubMed
- S. Kim, M. L. Goniewicz, S. Yu, B. Kim and R. Gupta, Int. J. Environ. Res. Public Health, 2015, 12, 4859–4868 CrossRef CAS PubMed
- N. J. Leigh, R. I. Lawton, P. A. Hershberger and M. L. Goniewicz, Tob. Control, 2016, 25, ii81–ii87 CrossRef PubMed
- J. Dai, K. H. Kim, J. E. Szulejko, S. H. Jo, K. Kwon and D. W. Choi, Microchem. J., 2018, 140, 262–268 CrossRef CAS
- E. Chivers, M. Janka, P. Franklin, B. Mullins and A. Larcombe, Med. J. Aust., 2019, 210, 127–128 CrossRef PubMed
- K. E. Farsalinos, I. G. Gillman, M. S. Melvin, A. R. Paolantonio, W. J. Gardow, K. E. Humphries, S. E. Brown, K. Poulas and V. Voudris, Int. J. Environ. Res. Public Health, 2015, 12, 3439–3452 CrossRef CAS PubMed
- J. F. Etter and A. Bugey, Addict. Behav., 2017, 73, 137–143 CrossRef PubMed
- M. L. Goniewicz, T. Kuma, M. Gawron, J. Knysak and L. Kosmider, Nicotine Tob. Res., 2013, 15, 158–166 CrossRef CAS PubMed
- B. H. Raymond, K. Collette-Merrill, R. G. Harrison, S. Jarvis and R. J. Rasmussen, J. Addict. Med., 2018, 12, 127–131 CrossRef CAS PubMed
- J. F. Etter, E. Zather and S. Svensson, Addiction, 2013, 108, 1671–1679 CrossRef PubMed
- J. M. Cameron, D. N. Howell, J. R. White, D. M. Andrenyak, M. E. Layton and J. M. Roll, Tob. Control, 2014, 23, 77–78 CrossRef PubMed
- M. Famele, J. Palmisani, C. Ferranti, C. Abenavoli, L. Palleschi, R. Mancinelli, R. M. Fidente, G. de Gennaro and R. Draisci, J. Sep. Sci., 2017, 40, 1049–1056 CrossRef CAS PubMed
- M. R. Peace, T. R. Baird, N. Smith, C. E. Wolf, J. L. Poklis and A. Poklis, J. Anal. Toxicol., 2016, 40, 403–407 CrossRef CAS PubMed
- M. Bansal, M. Sharma, C. Bullen and D. Svirskis, Int. J. Environ. Res. Public Health, 2018, 15, 1–11 Search PubMed
- R. Jackson, M. Huskey and S. Brown, Int. J. Pharm. Pract., 2020, 28, 290–294 CrossRef PubMed
- M. D. Crenshaw, M. E. Tefft, S. S. Buehler, M. C. Brinkman, P. I. Clark and S. M. Gordon, Magn. Reson. Chem., 2016, 54, 901–904 CrossRef CAS PubMed
- J. T. Shelley, S. P. Badal, C. Engelhard and H. Hayen, Anal. Bioanal. Chem., 2018, 410, 4061–4076 CrossRef CAS PubMed
- F. J. Andrade, W. C. Wetzel, G. C. Y. Chan, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, J. Anal. At. Spectrom., 2006, 21, 1175–1184 RSC
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2646–2653 CrossRef CAS PubMed
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2654–2663 CrossRef CAS PubMed
- J. T. Shelley, J. S. Wiley, G. C. Y. Chan, G. D. Schilling, S. J. Ray and G. M. Hieftje, J. Am. Soc. Mass Spectrom., 2009, 20, 837–844 CrossRef CAS PubMed
- J. T. Shelley, J. S. Wiley and G. M. Hieftje, Anal. Chem., 2011, 83, 5741–5748 CrossRef CAS PubMed
- K. P. Pfeuffer, J. N. Schaper, J. T. Shelley, S. J. Ray, G. C. Y. Chan, N. H. Bings and G. M. Hieftje, Anal. Chem., 2013, 85, 7512–7518 CrossRef CAS PubMed
- C. Kuhlmann, M. Heide and C. Engelhard, Anal. Bioanal. Chem., 2019, 411, 6213–6225 CrossRef CAS PubMed
- M. Heide, C. C. Escobar-Carranza and C. Engelhard, Anal. Bioanal. Chem., 2022, 414, 4481–4495 CrossRef CAS PubMed
- M. Guc, S. Rutecka and G. Schroeder, Biomolecules, 2020, 10, 1–25 Search PubMed
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Oresic, BMC Bioinf., 2010, 11, 11 CrossRef PubMed
- J. W. Flora, C. T. Wilkinson, K. M. Sink, D. L. McKinney and J. H. Miller, J. Liq. Chromatogr. Relat. Technol., 2016, 39, 821–829 CrossRef CAS
- Y. Son, O. Wackowski, C. Weisel, S. Schwander, G. Mainelis, C. Delnevo and Q. Y. Meng, Chem. Res. Toxicol., 2018, 31, 861–868 Search PubMed
- Council of Europe, European Pharmacopoeia 7.0, 2012, pp. 2567–2568 Search PubMed
- N. Eddingsaas, T. Pagano, C. Cummings, I. Rahman, R. Robinson and E. Hensel, Int. J. Environ. Res. Public Health, 2018, 15, 1–14 Search PubMed
- A. Larcombe, S. Allard, P. Pringle, R. Mead-Hunter, N. Anderson and B. Mullins, Med. J. Aust., 2022, 216, 27–32 Search PubMed
- H. Mckennis, E. R. Browman and L. B. Turnbull, J. Biol. Chem., 1963, 238, 719–723 CrossRef CAS PubMed
- C. Medana, V. Santoro, F. Dal Bello, C. Sala, M. Pazzi, M. Sarro and P. Calza, Rapid Commun. Mass Spectrom., 2016, 30, 2617–2627 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03931e |
| ‡ Federal Institute of Materials Research and Testing (BAM), Richard-Willstätter-Str. 11, 12489 Berlin, Germany |
| This journal is © The Royal Society of Chemistry 2023 |