
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assemblies in the molecular complexes of 4-cyanophenylboronic acid with different N-donor ligands †
Samina Easmin * and
Venkateswara Rao Pedireddi
* and
Venkateswara Rao Pedireddi
Solid State and Supramolecular Chemistry Laboratory, School of Basic Sciences, Indian Institute of Technology Bhubaneswar, Argul, Bhubaneswar 752 050, India. E-mail: se15@iitbbs.ac.in
First published on 2nd August 2023
Abstract
Molecular complexes of 4-cyanophenylboronic acid (CB) with various N-donor compounds having different conformational features, for example, rigid (1,10-phenanthroline (110phen), 4,7-phenanthroline (47phen), 1,7-phenanthroline (17phen) and acridine (acr)) and linear (1,2-bis(4-pyridyl)ethane (bpyea), 1,2-bis(4-pyridyl)ethene (bpyee) and 4,4′-azopyridine (azopy)), have been reported. In all complexes, the –B(OH)2 moiety is found to be in a syn–anti confirmation, with the exception of structures containing 110phen, bpyee, and azopy, wherein, syn–syn conformation is observed. Further, CB molecules remain intact in all structures except in the complexes with some linear N-donor ligands, wherein –B(OH)2 transforms to monoester (–B(OH)(OCH3)) prior to the formation of corresponding molecular complexes. In such boronic monoester complexes, the conformation of –B(OH)(OCH3) is syn–anti with respect to the –OH and –OCH3 groups. Also, complexes mediated by azopy and bpyee exist in both hydrated and anhydrous forms. In these anhydrous structures, the recognition pattern is through homomeric (juxtaposed –CN and –B(OH)2) as well as heteromeric (between hetero N-atom and –B(OH)2) O–H⋯N hydrogen bonds, while only heteromeric O–H⋯N hydrogen bonds hold co-formers in all other structures. Depending upon the conformational features of both co-formers, molecules are packed in crystal lattices in the form of stacked layers, helical chains, and crossed ribbons. All structures are fully characterized by single-crystal X-ray diffraction and phase purity is established by powder X-ray diffraction. Additionally, correlation among structures is explained by calculating a similarity index and performing a Hirshfeld surface analysis to quantify the strength and effectiveness of different types of intermolecular bonds that stabilize these structures along with the presentation of energy frameworks, representing the strength of the interactions in the form gradient cylinders. Also, the morphology of each complex was computed by BFDH methodology to correlate with the actual crystal morphology and packing arrangement.
1. Introduction
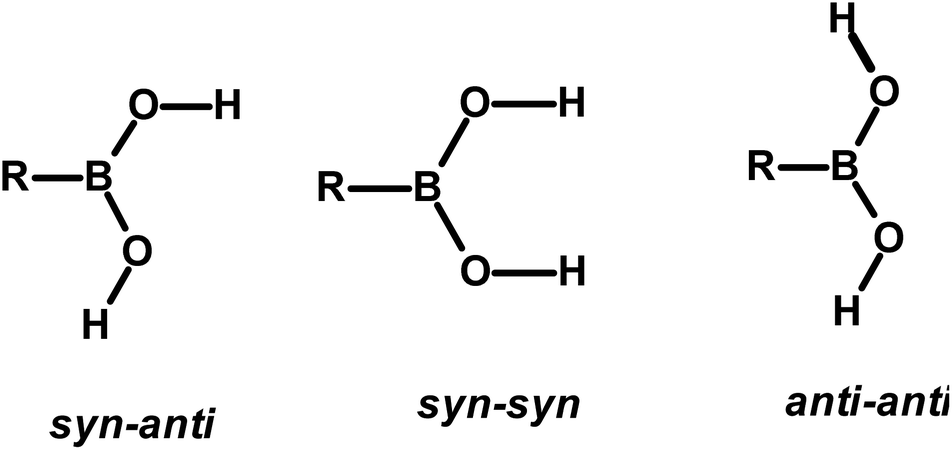
The design and preparation of new materials, for example, co-crystals, with desired physical and chemical properties is a continuum process.1 The methodology involved in such experiments is essentially based on generating molecular recognition between the complementary molecular building blocks, invoking various intermolecular interactions such as hydrogen bonds, π–π interactions, etc.2 However, since the intermolecular interactions are so labile and facile to either re-orient or reorganize, the development of target-oriented materials is still elusive, despite a plethora of structural assemblies known in the literature.3Some functional groups like –COOH, –CONH2, and –CSNH2, form specific topological hydrogen bonding patterns that may determine the course of arrangement of molecules in crystal lattices; however, other auxiliary groups prevail on the molecules, and may also play a vital role in the ultimate topological arrangements.4 In addition, if the prime functional group shows high conformational flexibility to form distinct conformers, the degree of molecular aggregation further would expand and accordingly a diversity of archetypes may form.4c,5 Among such functional groups, an organoboronic acid, represented as –B(OH)2, shows three conformers in solid-state that can produce diverse recognition patterns, syn–anti, syn–syn, and anti–anti, as shown in Scheme 1, possibly leading to the formation more intriguing exotic supramolecular architectures.6
 | ||
| Scheme 1 | ||
Organoboronic acids are an important and significant class of compounds that have gained prominence due to their applications in organic synthesis, mainly in Suzuki coupling reaction7 and medicinal chemistry like enzyme inhibitors,8 saccharide, sensing agents,9 boron neutron capture therapy agents, etc.10 In fact, a recently emerging novel frontier research area, covalent organic frameworks (COFs), is also mainly due to the facile reactive features of boronic acids to form C–C bonds with ease in the presence of appropriate organic ligands.11 Apart from it, boronic acids have recently gained popularity in supramolecular chemistry due to the potency of –B(OH)2 moiety as a hydrogen bond donor as well as an acceptor, similar to carboxylic acid and amide functionalities.6a,12
Despite the demonstrated capability of the –B(OH)2 moiety to form O–H⋯O/O–H⋯N hydrogen bonds with various co-formers, similar to –COOH and –CONH2 groups, the number of structural investigations involving boronic acids is still significantly lower compared to carboxylic acid and amide complexes.3b,13
Thus, considering the significance of boronic acids in the general synthetic aspects and also effective applications in healthcare domains, as well as the most significant feature of the facile formation of different hydrogen bonding patterns and considerably a small number of studies only are available in the literature, different exercises have been carried out to develop a myriad of boronic acids in the form co-crystals.14
In this regard, an important feature derived from the literature is that 4-cyanophenylboronic acid (CB) is known to form only hydrated molecular complexes with some linear N-donor ligands, despite water is not used as a crystallization solvent.15 Thus, several co-crystallization experiments have been initiated to develop corresponding anhydrous complexes of CB and also by expanding the horizon of N-donors by considering rigid ligands as well, by employing 1,10-phenanthroline (110phen), 4,7-phenanthroline (47phen), 1,7-phenanthroline (17phen), acridine (acr) and 4,4′-azopyridine (azopy), 1,2-bis(4-pyridyl)ethane (bpyea) and 1,2-bis(4-pyridyl)ethene (bpyee). The three-dimensional structures of all co-crystals, thus, obtained have been determined by single crystal X-ray diffraction method to evaluate the exotic structural features. The asymmetric unit contents of each complex are presented (Table S1) in the form of ORTEP in the ESI.†
2. Experimental section
The chemicals used in this study were obtained from Sigma-Aldrich with >99% purity and have been used without further purification. The solvents employed for crystallization experiments were of the highest quality available (spectroscopy grade). The co-crystals were prepared by dissolving the corresponding reactants in an appropriate solvent and allowing the solution to evaporate either at ambient or low-temperature conditions. Within 72–96 hours, good-quality single crystals suitable for X-ray diffraction analysis were obtained in all instances.2.1. Crystal structure determination
Good-quality single crystals were carefully chosen after being viewed under a Leica microscope and glued to glass fiber by using an adhesive, and mounted on the goniometer of Bruker single-crystal X-ray diffractometer (D8 VENTURE), with Mo-Kα radiation (λ = 0.71073 Å) source, equipped with a PHOTON 100 CMOS detector. The crystals remained stable throughout the data collection, carried out in φ and ω scans, and the process was found to be smooth. The structures were determined by using the intrinsic phasing method followed by full-matrix least-squares refinement against F2 using SHELXTL, embedded within Bruker suite of programs.16 All non-hydrogen atoms were refined by anisotropic method and hydrogen atoms were either refined or placed in calculated positions (Table S2†). All the structural refinements converged to good R factors, as listed in Table 1 and the intermolecular interactions were computed by using PLATON (see Table 2).17 The packing diagrams were generated by using Diamond (version 4.6.3).18| Parameters | 1 | 2 | 3 | 4 | 5 | 6 | 6a | 6b | 7 | 7a | 7b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Formula | (C12H8N2):(C7H6BNO2):(CH3OH) | (C12H8N2):(C7H6BNO2) | (C12H8N2):(C7H6BNO2) | (C13H9N):(C7H6BNO2) | (C7H6BNO2):(C12H12N2):(H2O) | 2(C7H6BNO2):(C10H8N4) | (C10H8N4):(C7H6BNO2):(H2O) | 2(C8H8BNO2):(C10H8N4) | 2(C7H6BNO2):(C12H10N2) | 2(C7H6BNO2):(C12H10N2):4(O) | 2(C8H8BNO2):(C12H10N2) |
| Mr | 355.15 | 327.14 | 327.14 | 326.15 | 349.19 | 478.08 | 349.16 | 506.13 | 476.09 | 524.65 | 504.15 |
| Crystal color | Colorless | Brown | Brown | Yellow | Colorless | Orange | Orange | Orange | Colorless | Colorless | Colorless |
| Crystal shape | Needle | Needle | Plate | Block | Blade | Plate | Block | Needle | Plate | Needle | Needle |
| T/K | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| λ(Mo-Kα)/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P212121 | P21 | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P |
P21/c | P |
P21/c | P21/c | P |
P21/c |
| a/Å | 3.8278(3) | 14.8728(7) | 7.1497(3) | 11.0840(8) | 7.5470(4) | 8.2777(5) | 8.8653(3) | 3.8043(3) | 8.4431(5) | 3.8665(2) | 3.9427(2) |
| b/Å | 17.1899(6) | 3.8786(2) | 8.7492(4) | 7.3311(5) | 11.5173(7) | 7.1087(4) | 9.0872(4) | 22.3944(13) | 7.3205(4) | 11.5022(5) | 22.6930(9) |
| c/Å | 26.2562(9) | 15.4857(8) | 13.6709(6) | 21.0913(13) | 11.9183(8) | 19.6723(9) | 12.2227(6) | 14.7329(8) | 19.6867(9) | 15.8177(8) | 14.9291(6) |
| α/° | 90 | 90 | 97.158(4) | 90 | 99.982(5) | 90 | 105.549(4) | 90 | 90 | 77.285(2) | 90 |
| β/° | 90 | 115.182(5) | 96.960(3) | 99.007(6) | 102.829(4) | 95.218(2) | 108.761(5) | 94.107(6) | 93.592(4) | 86.962(4) | 93.421(7) |
| γ/° | 90 | 90 | 95.413(4) | 90 | 105.296(7) | 90 | 90.865(7) | 90 | 90 | 87.577(5) | 90 |
| V/Å3 | 1727.64(16) | 808.40(8) | 837.04(6) | 1692.7(2) | 944.22(11) | 1152.79(11) | 892.67(7) | 1251.95(14) | 1214.40(11) | 684.91(6) | 1333.35(10) |
| Z | 4 | 2 | 2 | 4 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| Dc/g cm−3 | 1.365 | 1.344 | 1.298 | 1.280 | 1.228 | 1.377 | 1.299 | 1.343 | 1.302 | 1.272 | 1.256 |
| μ/mm−1 | 0.093 | 0.088 | 0.085 | 0.083 | 0.083 | 0.095 | 0.091 | 0.092 | 0.088 | 0.092 | 0.084 |
| F(000) | 736 | 340 | 340 | 680 | 368 | 496 | 364 | 528 | 496 | 274 | 528 |
| θ range [°] | 1.95–26.65 | 2.50–26.33 | 2.36–26.58 | 2.23–26.37 | 1.80–26.67 | 3.04–26.42 | 2.34–26.54 | 3.31–26.38 | 2.07–26.34 | 1.99–26.62 | 2.25–26.45 |
| Index ranges | −4 ≤ h ≤ 4 | −18 ≤ h ≤ 17 | −8 ≤ h ≤ 7 | −13 ≤ h ≤ 13 | −9 ≤ h ≤ 9 | −9 ≤ h ≤ 10 | −11 ≤ h ≤ 11 | −4 ≤ h ≤ 4 | −10 ≤ h ≤ 10 | −4 ≤ h ≤ 4 | −4 ≤ h ≤ 4 |
| −21 ≤ k ≤ 21 | −4 ≤ k ≤ 4 | −11 ≤ k ≤ 10 | −9 ≤ k ≤ 9 | −14 ≤ k ≤ 14 | −8 ≤ k ≤ 7 | −11 ≤ k ≤ 11 | −27 ≤ k ≤ 23 | −9 ≤ k ≤ 9 | −12 ≤ k ≤ 14 | −28 ≤ k ≤ 28 | |
| −33 ≤ l ≤ 33 | −15 ≤ l ≤ 19 | −17 ≤ l ≤ 17 | −20 ≤ l ≤ 26 | −15 ≤ l ≤ 14 | −22 ≤ l ≤ 24 | −15 ≤ l ≤ 15 | −18 ≤ l ≤ 18 | −24 ≤ l ≤ 24 | −19 ≤ l ≤ 19 | −18 ≤ l ≤ 13 | |
| Reflections | 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 424 424 |
8701 | 14761 |
11943 |
26286 |
6771 | 25898 |
9656 | 14399 |
14656 |
9015 |
| Unique reflections | 3595 | 3256 | 3431 | 3443 | 3941 | 2350 | 3672 | 2540 | 2462 | 2779 | 2719 |
| No. of parameters | 250 | 228 | 229 | 229 | 265 | 177 | 261 | 175 | 178 | 182 | 185 |
| GooF on F2 | 1.009 | 1.007 | 1.004 | 1.007 | 1.001 | 1.005 | 1.008 | 1.006 | 1.002 | 1.008 | 1.002 |
| R1, [I > 2σ(I)] | 0.053 | 0.057 | 0.055 | 0.051 | 0.064 | 0.045 | 0.055 | 0.058 | 0.046 | 0.073 | 0.060 |
| wR2, [I > 2σ(I)] | 0.125 | 0.139 | 0.138 | 0.117 | 0.165 | 0.113 | 0.1427 | 0.141 | 0.105 | 0.1975 | 0.137 |
| Largest diff. peak and hole (e Å−3) | 0.34/−0.38 | 0.18/−0.19 | 0.17/−0.14 | 0.19/−0.17 | 0.18/−0.18 | 0.29/−0.35 | 0.19/-0.25 | 0.35/−0.25 | 0.12/−0.15 | 0.32/−0.20 | 0.17/−0.16 |
| CCDC No. | 2260224 | 2260226 | 2260225 | 2260227 | 2260237 | 2260231 | 2260236 | 2260234 | 2260235 | 2260233 | 2260232 |
| Co-crystals | O–H⋯O | O–H⋯N | C–H⋯O | C–H⋯N | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a For each structure, the three columns represent the distances of H⋯A, D⋯A and angles ∠D–H⋯A, respectively. | ||||||||||||
| 1 | 1.79 | 2.74(2) | 163 | 2.46 | 3.52(2) | 169 | 2.67 | 3.50(2) | 134 | |||
| 1.81 | 2.79(2) | 172 | 2.88 | 3.87(2) | 152 | 2.75 | 3.54(2) | 130 | ||||
| 2.54 | 3.60(2) | 168 | ||||||||||
| 2 | 1.85 | 2.79(1) | 158 | 2.39 | 3.43(3) | 164 | 2.54 | 3.59(2) | 164 | |||
| 1.98 | 2.88(2) | 151 | 2.53 | 3.58(1) | 162 | 2.56 | 3.51(1) | 147 | ||||
| 2.63 | 3.69(2) | 164 | ||||||||||
| 3 | 1.81 | 2.78(3) | 171 | 1.84 | 2.72(3) | 148 | 2.32 | 3.3(3) | 153 | 2.58 | 3.58(1) | 153 |
| 2.59 | 3.58(2) | 151 | ||||||||||
| 2.73 | 3.70(1) | 149 | ||||||||||
| 4 | 1.78 | 2.75(2) | 169 | 1.80 | 2.69(2) | 149 | 2.42 | 3.45(3) | 160 | 2.47 | 3.46(3) | 152 |
| 2.86 | 3.76(3) | 141 | ||||||||||
| 5 | 1.77 | 2.68(2) | 153 | 1.78 | 2.75(2) | 172 | 2.65 | 3.46(3) | 132 | |||
| 1.88 | 2.79(2) | 154 | 2.70 | 3.52(3) | 133 | |||||||
| 2.03 | 2.94(2) | 155 | 2.90 | 3.76(3) | 138 | |||||||
| 6 | 1.87 | 2.76(3) | 150 | 2.63 | 3.47(2) | 134 | 2.52 | 3.49(3) | 148 | |||
| 1.98 | 2.91(3) | 157 | 2.81 | 3.77(3) | 148 | 2.84 | 3.71(1) | 138 | ||||
| 6a | 1.97 | 2.72(1) | 131 | 1.81 | 2.77(3) | 164 | 2.49 | 3.45(3) | 149 | 2.45 | 3.51(3) | 167 |
| 1.84 | 2.72(1) | 148 | 1.87 | 2.79(2) | 156 | 2.83 | 3.67(2) | 134 | 2.71 | 3.67(2) | 148 | |
| 6b | 1.87 | 2.75(2) | 148 | 2.42 | 3.47(3) | 163 | 2.56 | 3.47(3) | 141 | |||
| 2.92 | 3.84(3) | 143 | ||||||||||
| 2.64 | 3.59(3) | 148 | ||||||||||
| 7 | 1.85 | 2.74(3) | 149 | 2.94 | 3.86(3) | 143 | 2.56 | 3.52(3) | 149 | |||
| 2.01 | 2.96(3) | 162 | 2.76 | 3.68(3) | 143 | |||||||
| 7a | 1.81 | 2.77(3) | 168 | 1.79 | 2.71(3) | 154 | 2.36 | 3.41(3) | 165 | 2.36 | 3.41(3) | 165 |
| 1.83 | 2.71(3) | 148 | 2.48 | 3.51(3) | 158 | 2.62 | 3.65(3) | 158 | ||||
| 1.84 | 2.71(3) | 147 | 2.79 | 3.77(3) | 151 | |||||||
| 2.82 | 3.71(3) | 140 | ||||||||||
| 7b | 1.87 | 2.74(2) | 147 | 2.71 | 3.74(2) | 160 | 2.64 | 2.60(2) | 148 | |||
| 2.70 | 3.52(2) | 133 | ||||||||||
2.2. Powder X-ray diffraction (PXRD)
PXRD patterns were recorded on a Bruker D8 Advance X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å). The voltage and current applied were 40 kV and 30 mA, respectively. Intensities were measured in the reflection mode in the 2θ range of 5–40°.2.3. Hirshfeld surface analysis
Hirshfeld surface analysis and 2D fingerprint calculations were performed using Crystal Explorer (version 21),19 by importing the atomic coordinates from the CIF files. Hirshfeld surfaces (separately for each co-former) are generated for each structure. The distance from the nearest nucleus inside and outside the surface was measured and represented by the di and de, respectively, while a normalized contact distance was represented as dnorm. The white, red, and blue colors have been selected for the visualization of the dnorm function with very high resolution.2.4. Energy framework
The energy frameworks for all the co-crystals have been estimated based on the interaction energies by using Crystal Explorer 21.5 software.19 The energy calculations are performed at the B3LYP/6-31G(d,p) level of theory and using the crystal geometry of the respective co-crystals. The energy framework was constructed with an equal tube size (scale factor) of 100 and the energy threshold (cut-off) value was set to 5 kJ mol−1.3. Results and discussion
Good-quality single crystals of CB with various N-donor compounds as listed in Chart 1 are obtained upon co-crystallization from an appropriate solvent (Chart 1). Structure determination of these complexes reveals that each adduct forms distinctly unique molecular arrangements, especially the conformation of –B(OH)2 moiety and the nature of hydrogen bonds (homomeric and heteromeric), which are discussed in detail for all complexes.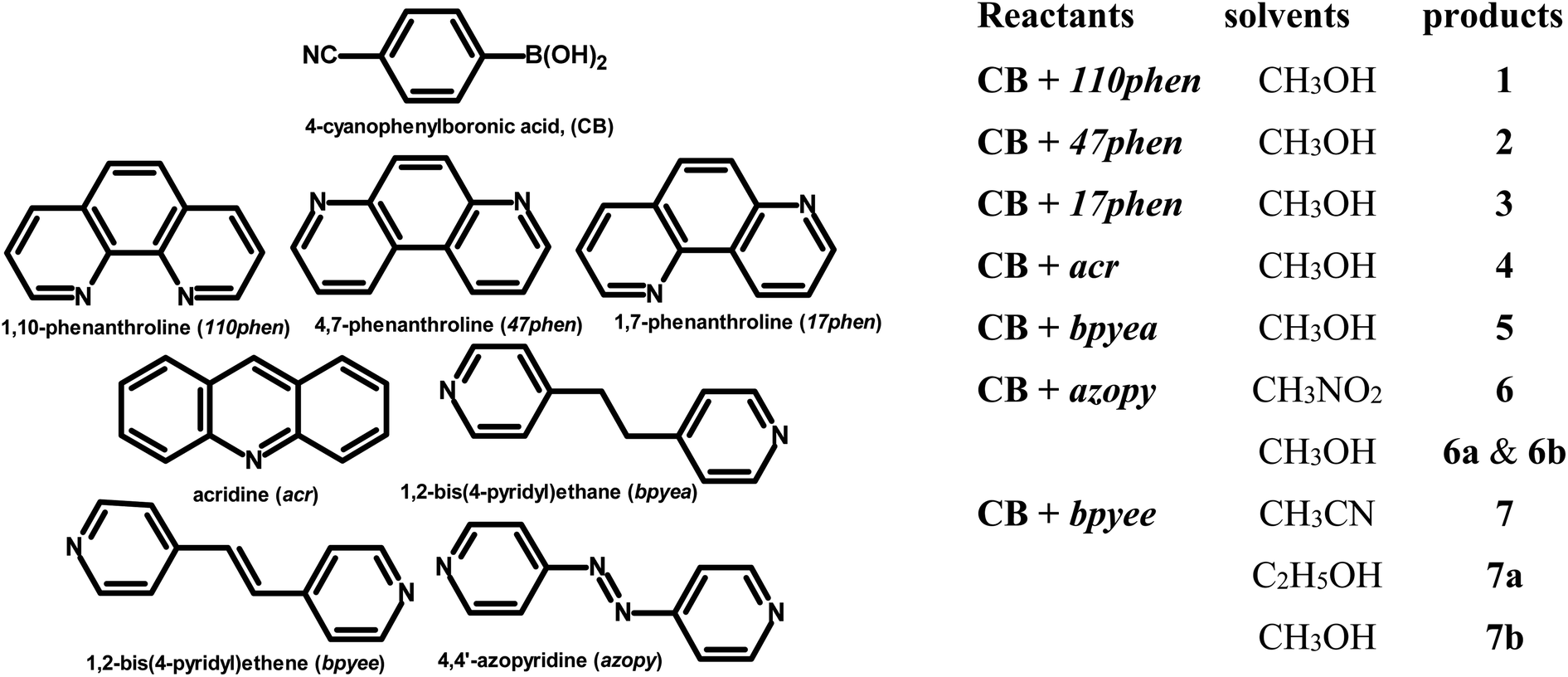 | ||
| Chart 1 | ||
3.1. Molecular complexes of CB with rigid N-donor ligands
:1 ratio, gives needle-shaped good-quality single crystals within 72 h, which are labeled as 1 for ease of discussion. X-ray diffraction analysis reveals that the components of 1, crystallize in an orthorhombic non-centrosymmetric (chiral) P212121 space group, with an asymmetric unit consisting of 1:1 molecule of co-formers and molecules of crystallization solvent (CH3OH). The pertinent crystallographic information is given in Table 1. The constituents of an asymmetric unit are shown in Table S1,† in the form of ORTEP.The molecules in complex 1 arrange in the form of sheets with voids (7 × 6 Å2) that are inhabited by molecules of crystallization solvent (CH3OH), as shown in Fig. 1a. The guest molecules (CH3OH), indeed, form infinite chains by holding each other through hydrogen bonds. Within each sheet, the co-formers interact with each other through O–H⋯N hydrogen bonds formed between –B(OH)2 moiety prevails on CB and the N-atoms of 110phen. It is pertinent to mention that such a recognition process involves –B(OH)2 moiety in a syn–syn conformation, which is a commonly known conformation in similar complexes.6d,20 The hydrogen bonds, thus, formed appear in the form of an unsymmetrical Etter's pattern, R22(9), with the corresponding H⋯N distances being 1.79 Å (O⋯N, 2.74 Å) and 1.81 Å (O⋯N, 2.79 Å) as shown in Fig. 1b. Furthermore, the assemblages self-assemble to constitute quartet ensembles through the formation of C–H⋯O (H⋯O, 2.46 Å; C⋯O, 3.52 Å) hydrogen bonds (homomeric, between the molecules of CB) in the form of a symmetrical pattern, R22(12) and a single C–H⋯N (heteromeric, between the co-formers) hydrogen bond, with the corresponding H⋯N distance being 2.75 Å (C⋯N, 3.54 Å). In turn, these quartets are held together by different C–H⋯O and C–H⋯N hydrogen bonds with H⋯O, 2.54 Å, (C⋯O, 3.60 Å) and H⋯N, 2.67 Å, (C⋯N, 3.54 Å) respectively. However, in the crystal lattice, the sheets are not stacked translationally but are arranged in a crossed-sheet form. As a result, the juxtaposed molecules in the adjacent sheets interact with each other via the formation of helical chains among the molecules of CB (Fig. 1c). Further, the helical chains are bound together by the 110phen molecules (Fig. 1d).
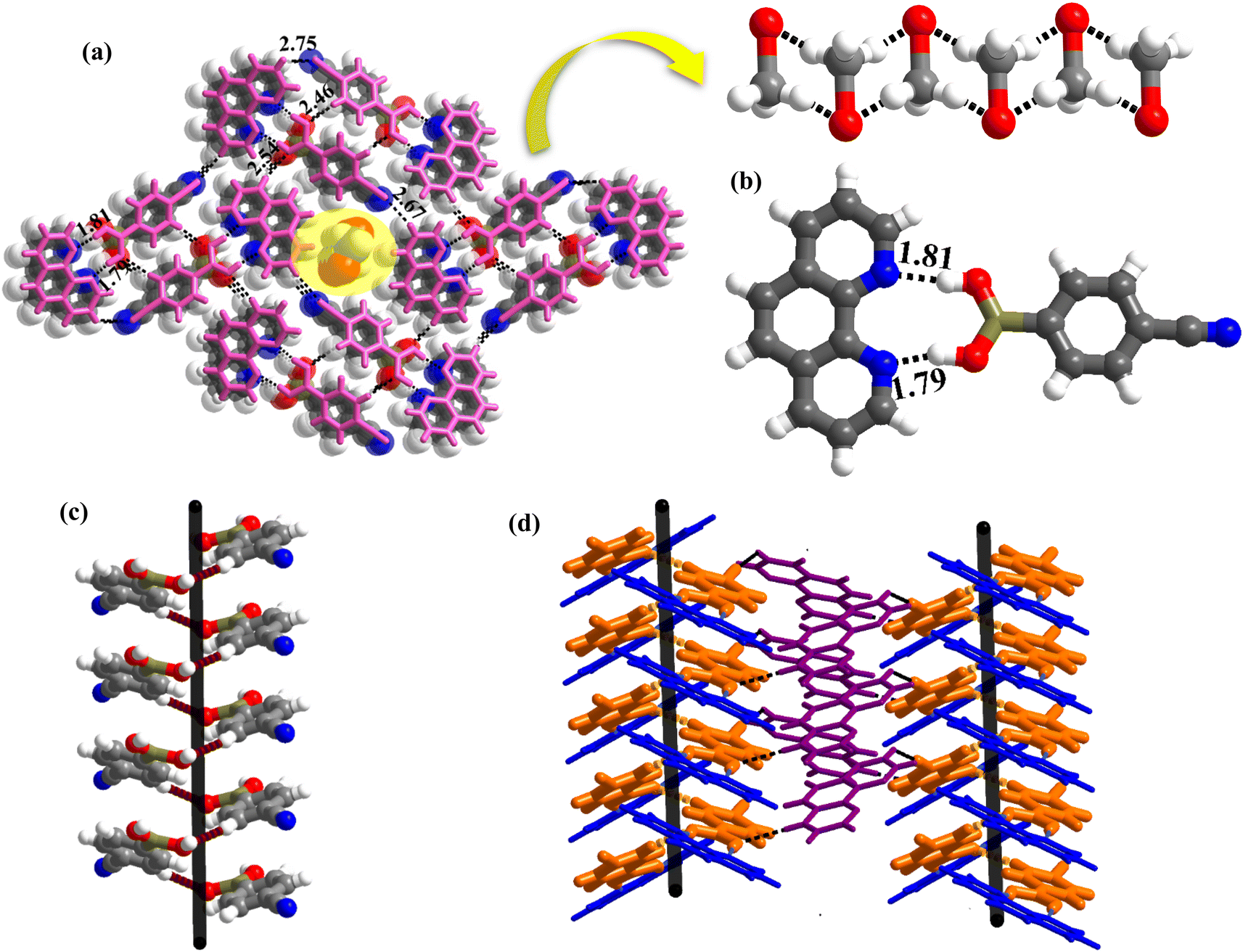 | ||
| Fig. 1 (a) Three-dimensional arrangement with voids, occupied by CH3OH molecules, in molecular complex, 1. (b) Basic recognition unit observed between complementary functional groups present in 1. (c) 1D helical chain and (d) left-handed and right-handed helical chains. | ||
:1 ratio, upon slow evaporation, forms needle-shaped crystals at ambient conditions. The molecules in the crystals are arranged in a monoclinic non-centrosymmetric space group P21, with an asymmetric unit consisting of co-formers in a 1:1 molecular ratio (Table S1†). Unlike in the crystals of 1, herein, –B(OH)2 moiety is found in a syn–anti conformation.Structural analysis shows that the building unit between CB and 47phen molecules in this complex is formed through a heteromeric pairwise O–H⋯N/C–H⋯O (H⋯N, 1.85 Å; O⋯N, 2.79 Å and H⋯O, 2.39 Å; C⋯O, 3.43 Å) hydrogen bonds in the form of Etter's notation, R22(8) (Fig. 2a), and yields a crisscrossed network in the crystal lattice (Fig. 2b). Further, the adjacent building units are bound together by an O–H⋯N (H⋯N, 1.98 Å; O⋯N, 2.88 Å) hydrogen bond formed between 47phen and CB molecules, invoking H-atom prevail on CB at the anti-position. Such an association, thereby, constitutes one-dimensional helical chains that are further held together by C–H⋯N (H⋯N, 2.54 Å; C⋯O, 3.59 Å) hydrogen bonds (Fig. 2c), leading to the construction of a double helix pattern.
 | ||
| Fig. 2 (a) Basic recognition unit observed in molecular complex, 2. (b) Criss-crossed network in the crystal lattice. (c) A double helix formed between CB and 47phen. | ||
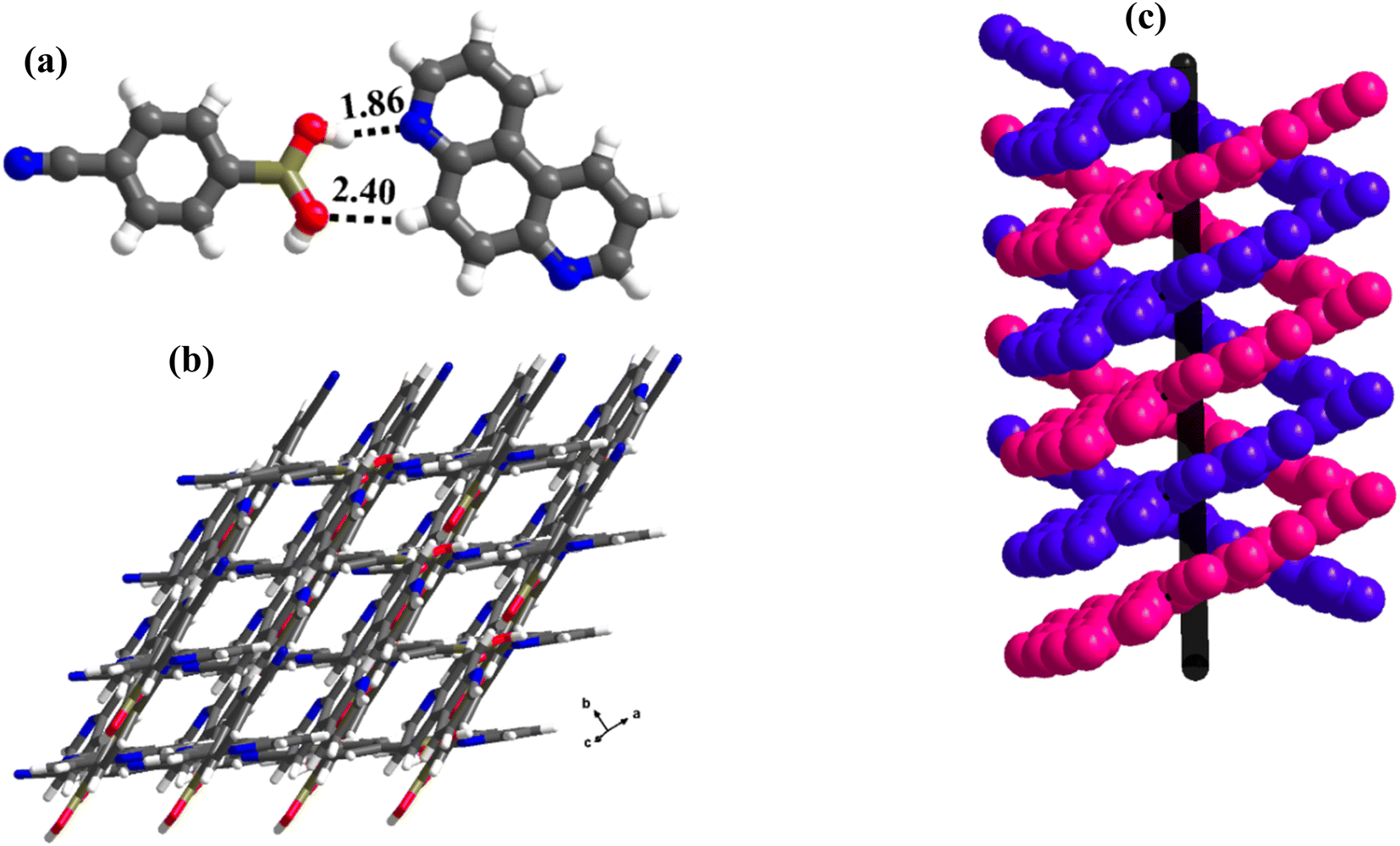
:1 ratio) forms crystals, 3, at ambient conditions, during slow-evaporation of a CH3OH solution of the co-formers. Structure determination by single-crystal X-ray diffraction method on a good-quality crystal reveals that the co-formers in the crystals of 3 crystallize in a triclinic space group, P, with an asymmetric unit consisting of molecules of CB and 17phen in a 1:1 ratio (Table S1†).Similar to 2, here also the –B(OH)2 moiety of CB espouses a syn–anti conformation, with unprecedented interactions with the co-former, 17phen. In general, –B(OH)2 has been addressed as a hybrid of –COOH and –CONH2 based on the homomeric interactions in the native structures of several boronic acids, but it mostly mimics molecular recognition features of –COOH in many complexes, though a few recognition patterns of amides are also realized.21 However, a significant pattern observed in amide-mediated co-crystals, for example, dimers of –CONH2 moieties holding the co-formers through the second –NH group, is not so observed in the complexes of boronic acids (see Scheme S1†).22
In complex, 3, interestingly, such an unusual amide feature is observed, for the first time, among boronic acid molecular complexes. Thus, molecules of CB form dimers through homomeric O–H⋯O (H⋯O, 1.81 Å; O⋯O, 2.78 Å) hydrogen bonds formed between the –B(OH)2, in the form of centrosymmetric R22(8) pattern. Additionally, other H-atoms on both paired molecules establish interaction with 17phen molecules via heteromeric O–H⋯N (H⋯N, 1.84 Å; O⋯N, 2.72 Å) hydrogen bonds, thus, creating tetrameric units (Fig. 3a). These units are further held together by C–H⋯O (H⋯O, 2.32 Å; O⋯N, 3.30 Å) hydrogen bonds, as shown in Fig. 3b. Nonetheless, the arrangement constitutes crossed tape networks in the crystal lattice, which are stabilized by homomeric C–H⋯N (H⋯N, 2.73 Å; C⋯N, 3.70 Å), hydrogen bonds (Fig. 3c).
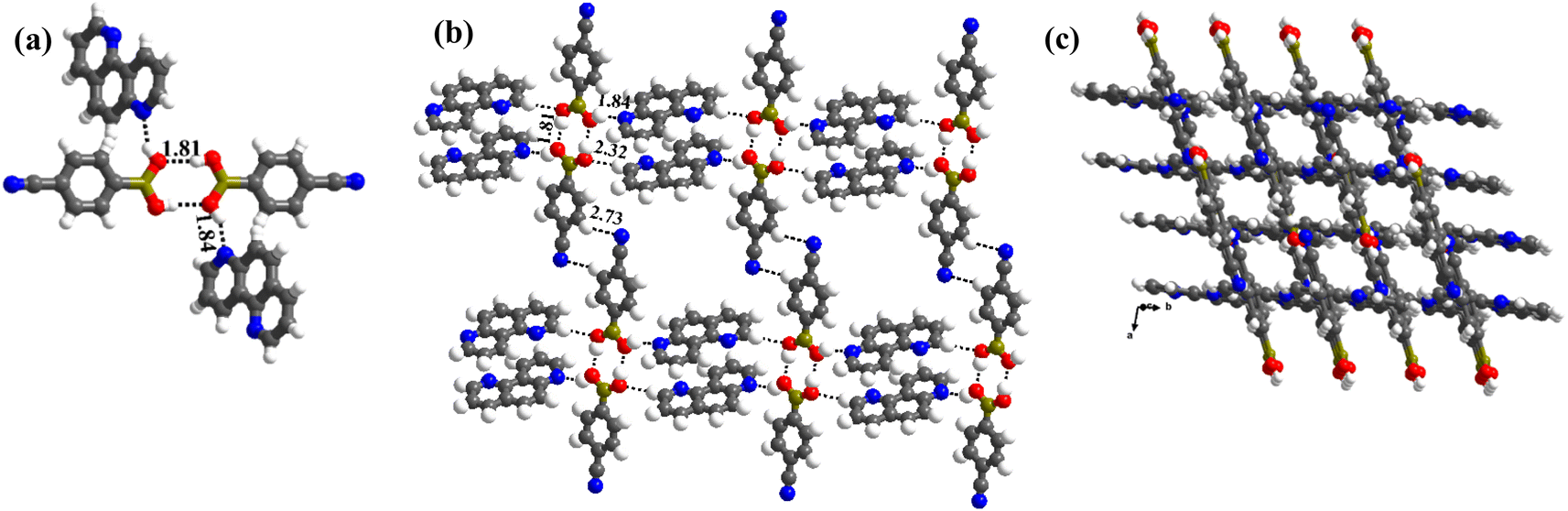 | ||
| Fig. 3 (a) A typical tetrameric unit in the crystal structure of 3. (b) Interaction between the tetramers in the crystal lattice. (c) Crossed tape networks in molecular complex, 3. | ||
:1 molecular ratio. The constituents of an asymmetric unit are displayed in the form of ORTEP (Table S1†).Conformation of –B(OH)2 in this complex is also in the form of syn–anti as observed in 2 and 3. Furthermore, the basic recognition patterns with the co-former reflect the amide pattern, the same as found in complex 3. Thus, CB molecules interact with each other, yielding homomeric dimers in a centrosymmetric O–H⋯O (H⋯O, 1.78 Å; O⋯O, 2.75 Å) hydrogen bonding pattern, as shown in Fig. 4a. These dimers further form a tetrameric unit by interacting with acr molecules through heteromeric O–H⋯N (H⋯N, 1.80 Å; O⋯N, 2.69 Å) hydrogen bond, as shown in Fig. 4a. In such a pattern, though acr molecules appear to be like hanging pendants connected to a chain of CB molecules, in further self-assembly, acr molecules are connected to adjacent tetramers by C–H⋯O (H⋯O, 2.42 Å; C⋯O, 3.45 Å) hydrogen bonds formed between aromatic –CH groups on acr and –O atom of –B(OH)2 moiety (Fig. 4a). Such aggregates are held together as crinkle chains in the crystal lattice by C–H⋯N (H⋯N, 2.47 Å; C⋯N, 3.46 Å) hydrogen bond, as depicted in Fig. 4 (acr molecules are not shown for brevity purposes). Complete details of all the pertinent hydrogen bond distances are given in Table 2.
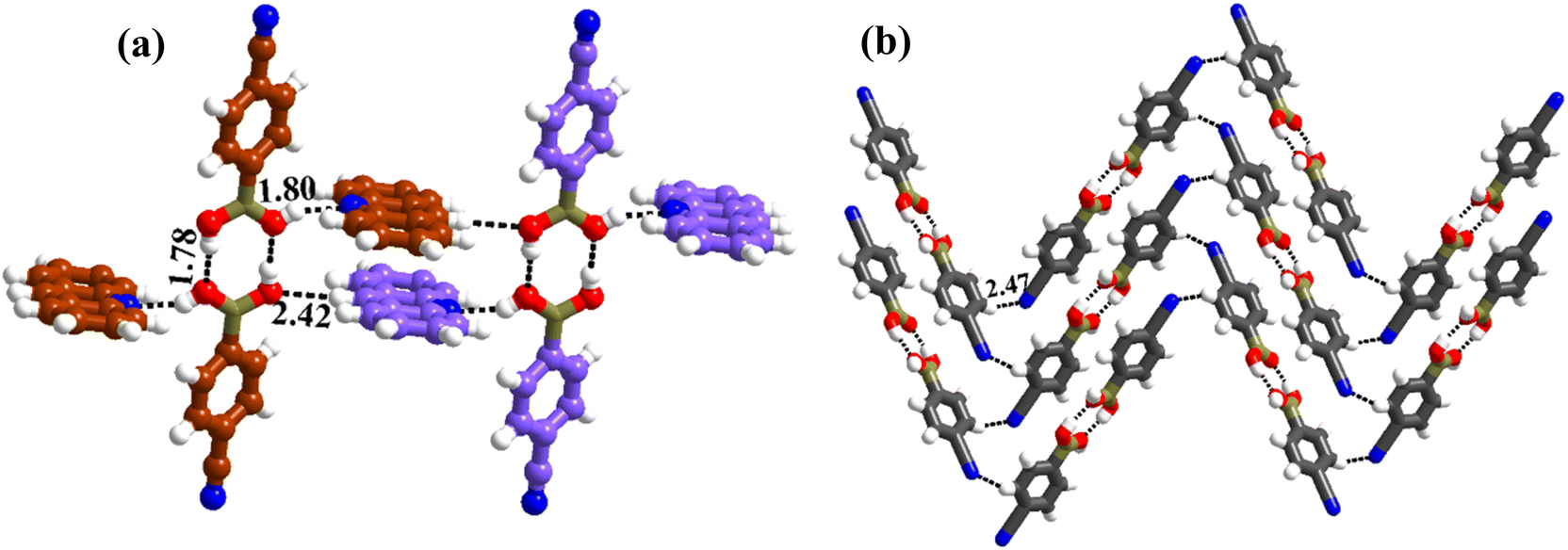 | ||
| Fig. 4 (a) Basic interaction in the form of a tetramer and between the adjacent tetramers in complex, 4. (b) Crinkle arrangement of CB molecules through C–H⋯N hydrogen bonds. | ||
The structural features of complexes 1–4, show clear variations due to the diverse conformations of –B(OH)2 influenced by N-atom locations on aza-donors. For example, 110phen, having juxtaposed N-atoms directs to have a syn–syn conformation in CB molecules, while widely separated N-atoms mediated 17phen, induces syn–anti conformations in CB molecules, as also observed in the literature even with linear ligands like 4,4′-bipyridine (bpy) and bpyee.
3.2. Molecular complexes of CB with linear N-donor ligands
In the literature, CB molecular complexes with linear ligands (bpy and bpyee), are noted to be hydrated only. Also, co-crystals of CB with bpyea and azopy (higher homologous of bpy and analogous to bpyee) are not yet reported in the literature. Hence, co-crystallization of CB with bpyea and azopy has been carried out and the derived structural features are illustrated below.:1 ratio CB and bpyea co-formers in CH3OH and subsequently evaporating the solution slowly at ambient conditions. Though water is not used in the crystallization process, the asymmetric unit is noted to be associated with a water molecule also, which is in accordance with the nature of acid CB as is known to form hydrated complexes.15 In fact, the structure analysis by single-crystal X-ray diffraction method, further discloses that complex 5 is isomorphous to the molecular complex formed by its analog, bpyee, which also gives a monohydrated molecular complex while crystallizing from a CH3OH solution. The contents of the asymmetric unit are shown in Table S1,† representing water molecules also, in the form of ORTEP.The analysis further discloses that the –B(OH)2 moiety is present in syn–anti conformation as observed in 2–4. In addition, the co-formers are arranged in a stacked sheets form in the crystal lattice as shown in Fig. 5a, with each sheet comprising both co-formers. The co-formers within each sheet are held together by O–H⋯N (H⋯N, 1.78 Å; O⋯N, 2.72 Å) hydrogen bonds between –N atom of bpyea and –B(OH)2 moiety, as also observed in the other structures discussed above, and are further connected through water molecules (see Fig. 5b). Thus, linear ribbons are formed in a one-dimensional arrangement with the adjacent primary blocks connected by two O–H⋯N (H⋯N, 1.88 and 2.03 Å; O⋯N, 2.79 and O⋯N, 2.94 Å) hydrogen bonds formed between water and –N atoms of bpyea and nitrile (–CN) groups. Such linear ribbons are further held together by O–H⋯O (H⋯O, 1.77 Å; O⋯O, 2.68 Å) hydrogen bonds formed by water molecules with –B(OH)2 moiety as well as by some C–H⋯O hydrogen bonds with aza-donor molecules, as vividly shown in Fig. 5c.
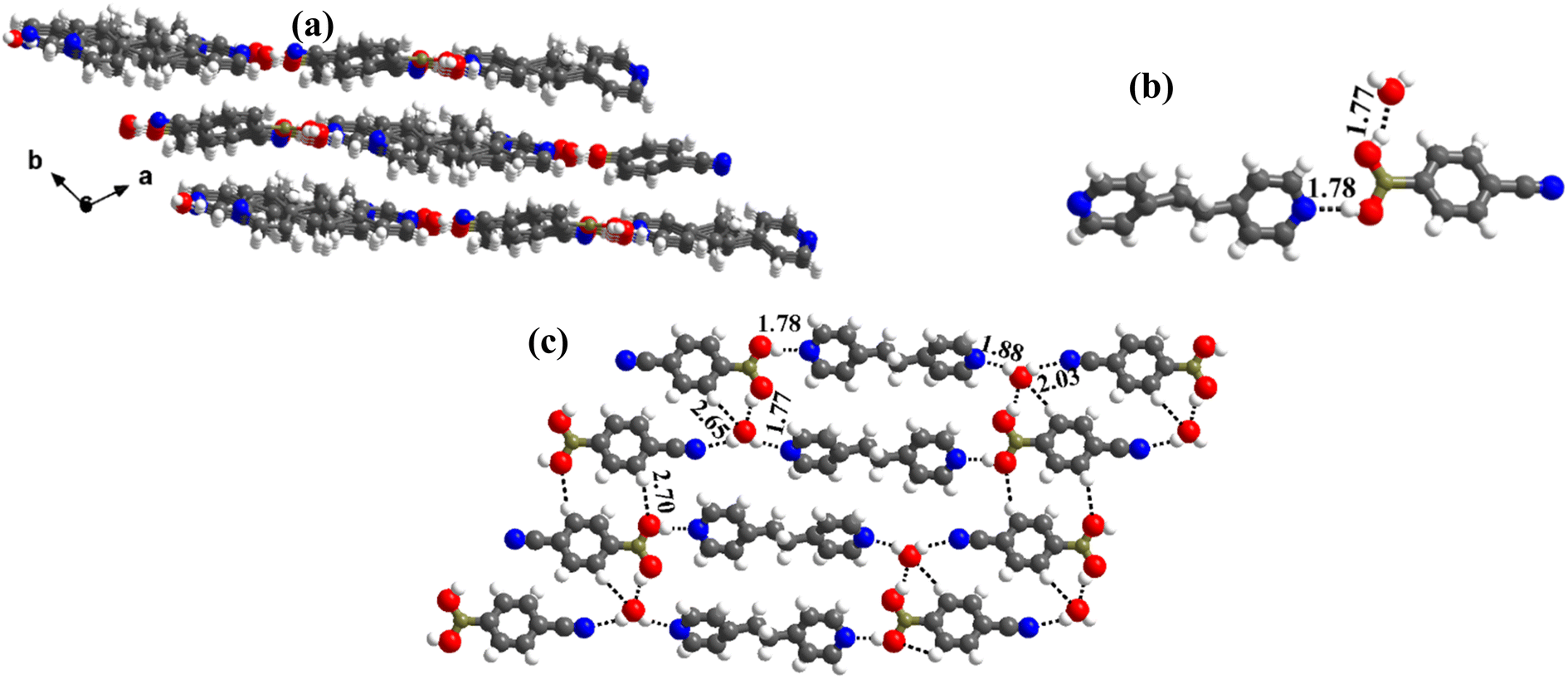 | ||
| Fig. 5 (a) Sheet arrangement of the molecules in the crystal lattice. (b) Basic recognition of CB with bpyea in hydrated complex 5. (c) Two-dimensional linear ribbon arrangement. | ||
The structure analysis reveals that the co-crystals of azopy and CB, obtained from CH3NO2 (labeled as 6), crystallize in a monoclinic space group, P21/c, with unit cell dimensions, as presented in Table 1. The contents of the asymmetric unit as depicted in Table S1,† reveals that structure 6 is an anhydrous complex of CB and azopy in a 2:1 ratio.
The analysis of molecular features of co-formers in 6 exposes that –B(OH)2 moiety is in unusual syn–syn conformation, as observed in complex 1, despite the absence of juxtaposed –N atoms on aza-donor molecules, expressing the two –OH groups of –B(OH)2 moiety as vicinal diols. In fact, the observed conformation is quite intriguing and certainly accounted for the molecular recognition process between the co-former molecules CB and azopy. Further, the azo-bridge (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) in azopy molecules is noted to be disordered in a 2:1 ratio. Thus, the co-formers are held together by heteromeric O–H⋯N (H⋯N, 1.87 Å; O⋯N, 2.76 Å) hydrogen bonds between molecules of CB and azopy; however, an unusual homomeric O–H⋯N (H⋯N, 1.98 Å; O⋯N, 2.91 Å) hydrogen bond is also observed, which is formed between CB molecules (formed between –B(OH)2 and nitrile group), a hitherto unknown pattern in any of the CB complexes. The pattern (both homomeric and heteromeric), along with its propagation is shown in Fig. 6a. Such an association further leads to the formation of crossed ribbon structures, as shown in Fig. 6b.
N) in azopy molecules is noted to be disordered in a 2:1 ratio. Thus, the co-formers are held together by heteromeric O–H⋯N (H⋯N, 1.87 Å; O⋯N, 2.76 Å) hydrogen bonds between molecules of CB and azopy; however, an unusual homomeric O–H⋯N (H⋯N, 1.98 Å; O⋯N, 2.91 Å) hydrogen bond is also observed, which is formed between CB molecules (formed between –B(OH)2 and nitrile group), a hitherto unknown pattern in any of the CB complexes. The pattern (both homomeric and heteromeric), along with its propagation is shown in Fig. 6a. Such an association further leads to the formation of crossed ribbon structures, as shown in Fig. 6b.
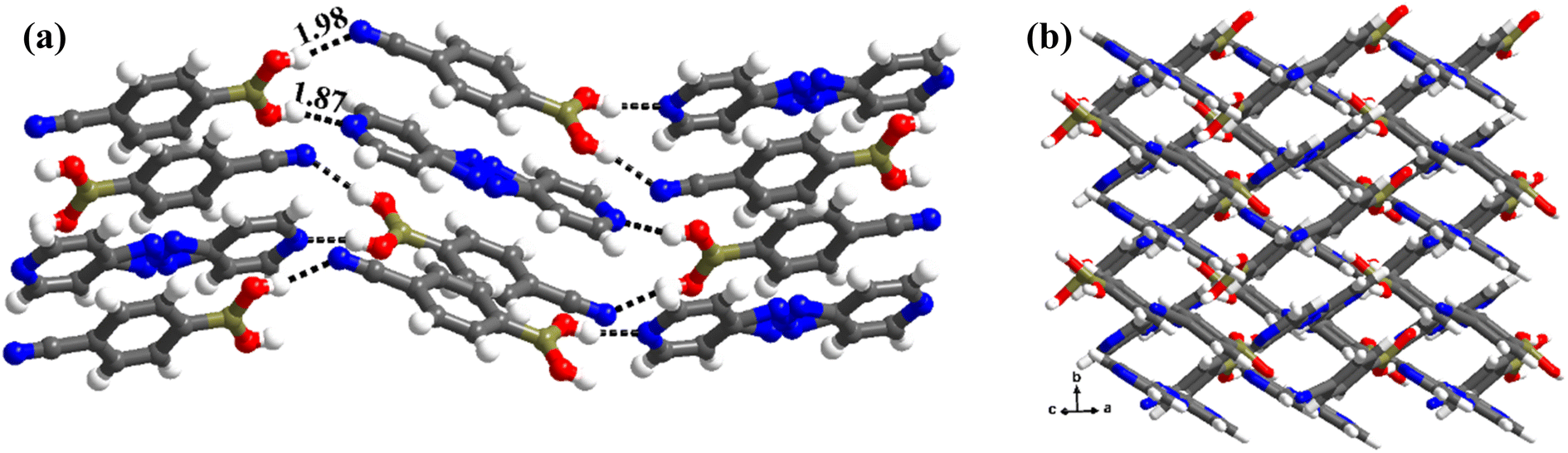 | ||
| Fig. 6 (a) Propagation of chain arrangement in the molecular complex, 6. (b) Crossed ribbon arrangement present in the crystal lattice. | ||
space group, with an asymmetric unit having 1:1 molecules of co-formers and a water molecule. The constituents of the asymmetric unit are shown in Table S1.† Crystals of 6a (see Table 1) are found to be isomorphous with PELMUR (a = 8.918(2), b = 9.508(2), c = 12.009(3) Å and α = 71.75(1), β = 70.21(1), γ = 89.23(1)°). Also, 6a and PELMUR show an isostructural relationship, despite the –B(OH)2 moiety present in different conformations (syn–syn, 6a; syn–anti, PELMUR). The structural features of 6a are described below.The needle-shaped crystals, however, on the other hand interestingly have shown entirely a new feature with the formation of co-crystals generated between in situ converted monoester of CB and azopy. These crystals are labeled 6b and the structural features are discussed in detail in later sections.
:1 ratio along with a water molecule also, form a stacked layers structure in a three-dimensional arrangement (along a-axis), as shown in Fig. 7a. In such an arrangement, the –B(OH)2 moiety, in a syn–syn confirmation, establishes interaction through O–H⋯N (H⋯N, 1.81 and 1.87 Å; O⋯N, 2.77 and 2.79 Å) and O–H⋯O (H⋯O, 1.97 Å; O⋯O, 2.72 Å) hydrogen bonds, directly with N-atom of azopy and with the aid of water molecules, respectively. Thus, a cyclic six molecular unit (two of each CB, azopy and H2O) is realized, as pictorially depicted in Fig. 7b. The cyclic units (highlighted in Fig. 7c) are further held together in a two-dimensional arrangement by establishing a centrosymmetric homomeric C–H⋯N (H⋯N, 2.71 Å; C⋯N, 3.67 Å) hydrogen bonding mediated R22(10) pattern between the adjacent CB molecules (see Fig. 7c).
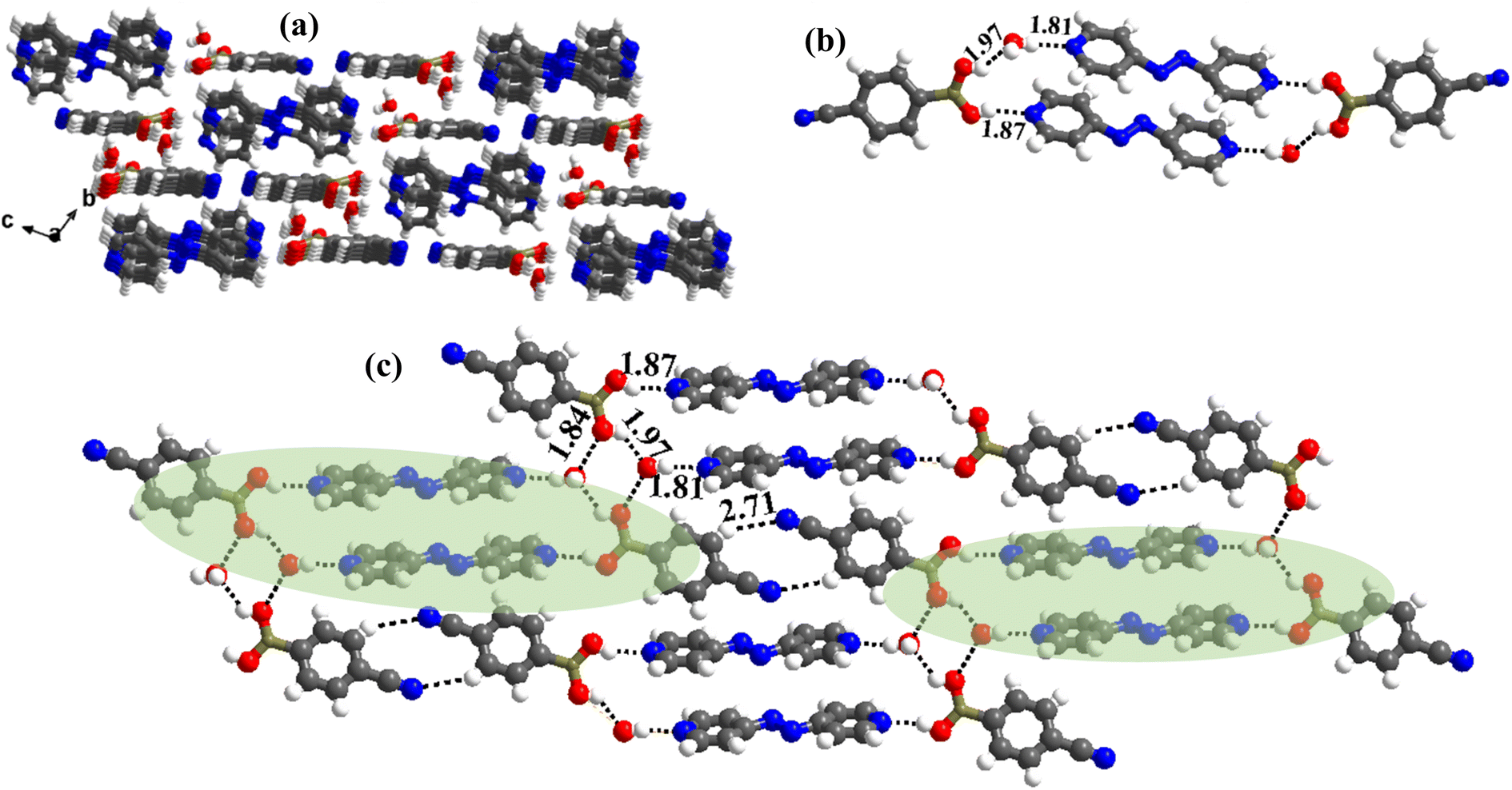 | ||
| Fig. 7 (a) Three-dimensional stacked layer arrangement in the molecular complex, 6a. (b) The six-membered cyclic network formed in molecular recognition. (c) Interactions of the hexameric unit in the crystal lattice. | ||
It is well documented in the literature that hydrates and anhydrous forms of the same co-formers are generally prepared by varying the crystallization conditions, such as solvents, temperature, pH, etc.23 Incidentally, by altering the solvent of crystallization, azopy has been noted to form both anhydrous and hydrated complexes 6 and 6a, with a noteworthy feature that the hydrated form, 6a, obtained from CH3OH is isomorphous with the reported structure (PELMUR) of its analog, bpyee. Since, the anhydrous form of PELMUR is not known so far, crystallization of CB and bpyee has been further carried out from different solvents to explore the possibly unnoticed anhydrous form. Indeed, such experiments gave crystals of different geometry 7 and 7a from CH3CN and C2H5OH solutions, respectively, as discussed below.
X-ray diffraction analysis of crystal 7 reveals that it is adorably an anhydrous complex of CB and bpyee and also shows isomorphism features with that of complex 6, crystallizing in the same monoclinic space group P21/c and similar unit cell dimensions. Apart from that 6 and 7 are also isostructural in terms of –B(OH)2 moiety conformation (syn–syn), molecular packing in 2D and 3D arrangements, with the exception of hydrogen bond parameters (distances and angles) as displayed in Fig. S1.† Thus, in 7 also, homomeric (O–H⋯N with H⋯N, 1.85 Å; O⋯N, 2.74 Å) and heteromeric (O–H⋯N with H⋯N, 2.01 Å; O⋯N, 2.96 Å, Table 2) hydrogen bonds mediated crisscross ribbon structure is observed.
The crystals have an asymmetric unit in a 2:1 ratio of the co-formers along with two water molecules, as shown in Table S1,† in the form of an ORTEP drawing. The molecules are arranged in the crystal lattice through the stacking of planar sheets (Fig. 8b), as also observed in the structure of 5, which has a co-former (bpyea), one of the close analogs of bpyee; but totally with a different recognition pattern between the co-formers. Furthermore, the structure of 7a is completely distinct from the other structures (1–6) discussed above. In 7a, the co-formers (CB and bpyee) do not interact with each other directly through any significant recognition pattern. Thus, the molecules form a host–guest network with the host being CB and water molecules with bpyee as guest species. In the host network, CB molecules form homomeric O–H⋯O hydrogen bonds (H⋯O, 1.81 Å; O⋯O, 2.77 Å) in the form of dimers and further interact with water molecules also by O–H⋯O hydrogen bonds (H⋯O, 1.84 Å; O⋯O, 2.71 Å) as depicted in Fig. 8a. The guest moieties (bpyee) interact with the host lattice through a series of hydrogen bonds; O–H⋯N (H⋯N, 1.79 Å; O⋯N, 2.71 Å) with water molecules and with CB molecules by C–H⋯O as well as C–H⋯N hydrogen bonds, as shown in Fig. 8c.
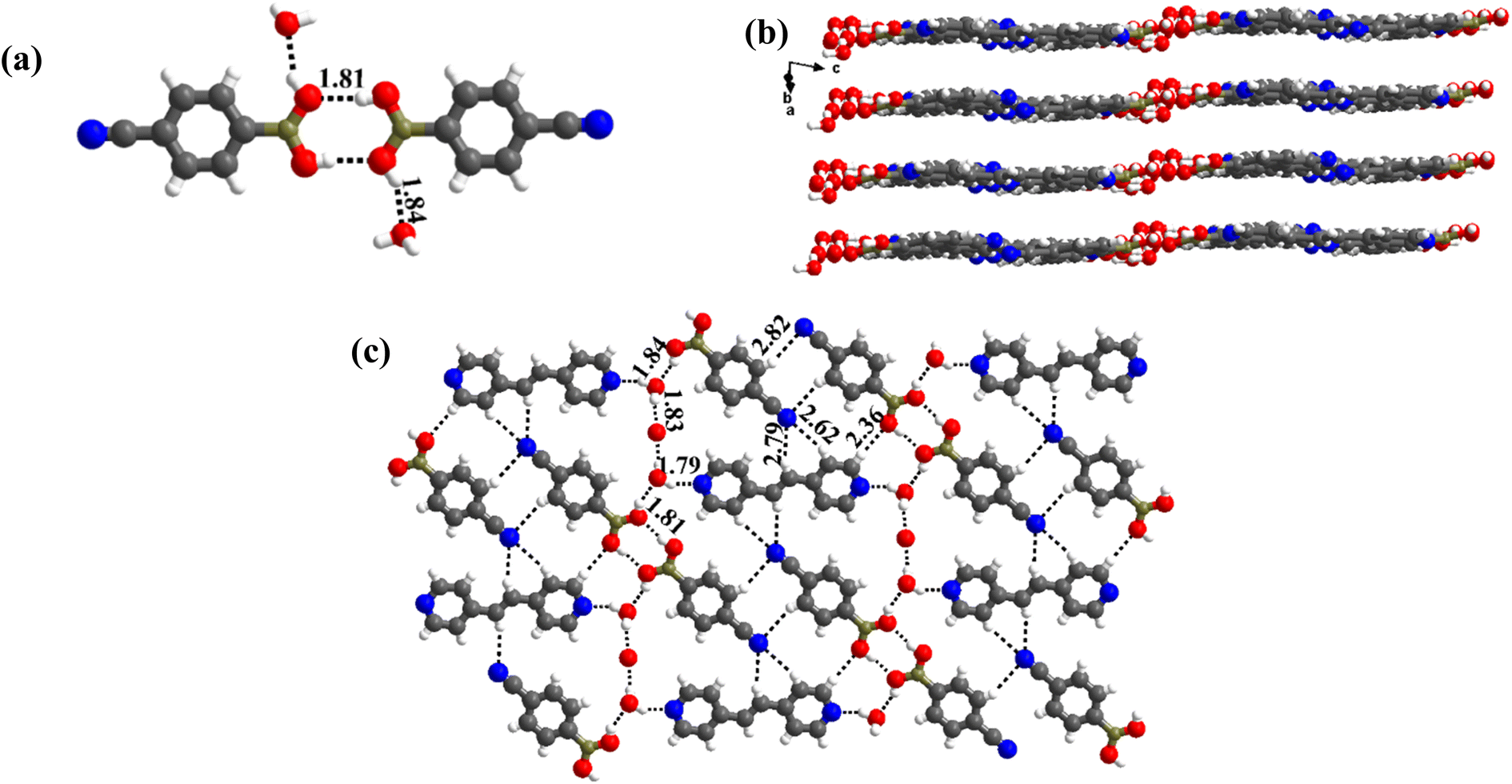 | ||
| Fig. 8 (a) Basic recognition of CB with bpyee in hydrated complex 7a. (b) Planar-sheet arrangement of the molecules in the crystal lattice. (c) Two-dimensional host–guest network. | ||
The crystals of 6b crystallize in a monoclinic P21/c space group (Table 1), with the asymmetric units having the co-formers (monoester of CB and azopy) in a 2:1 ratio. The constituents are shown in Table S1,† in the form of ORTEP.
The crystal structure analyses of 6b further illustrate that the recognition moiety between the constituents is a three-component unit, with two O–H⋯N (H⋯N, 1.87 Å; O⋯N, 2.75 Å) hydrogen bonds with each azopy molecule glued to two molecules of monoester of CB. Because methylation destroys the symmetry of –B(OH)2, the observed conformation of B(OH)(OCH3) may be regarded as anti with respect to the position of –OCH3 and –OH groups. Thus, interaction is established through the participation of anti-H-atom and the recognition pattern is shown in Fig. 9a.
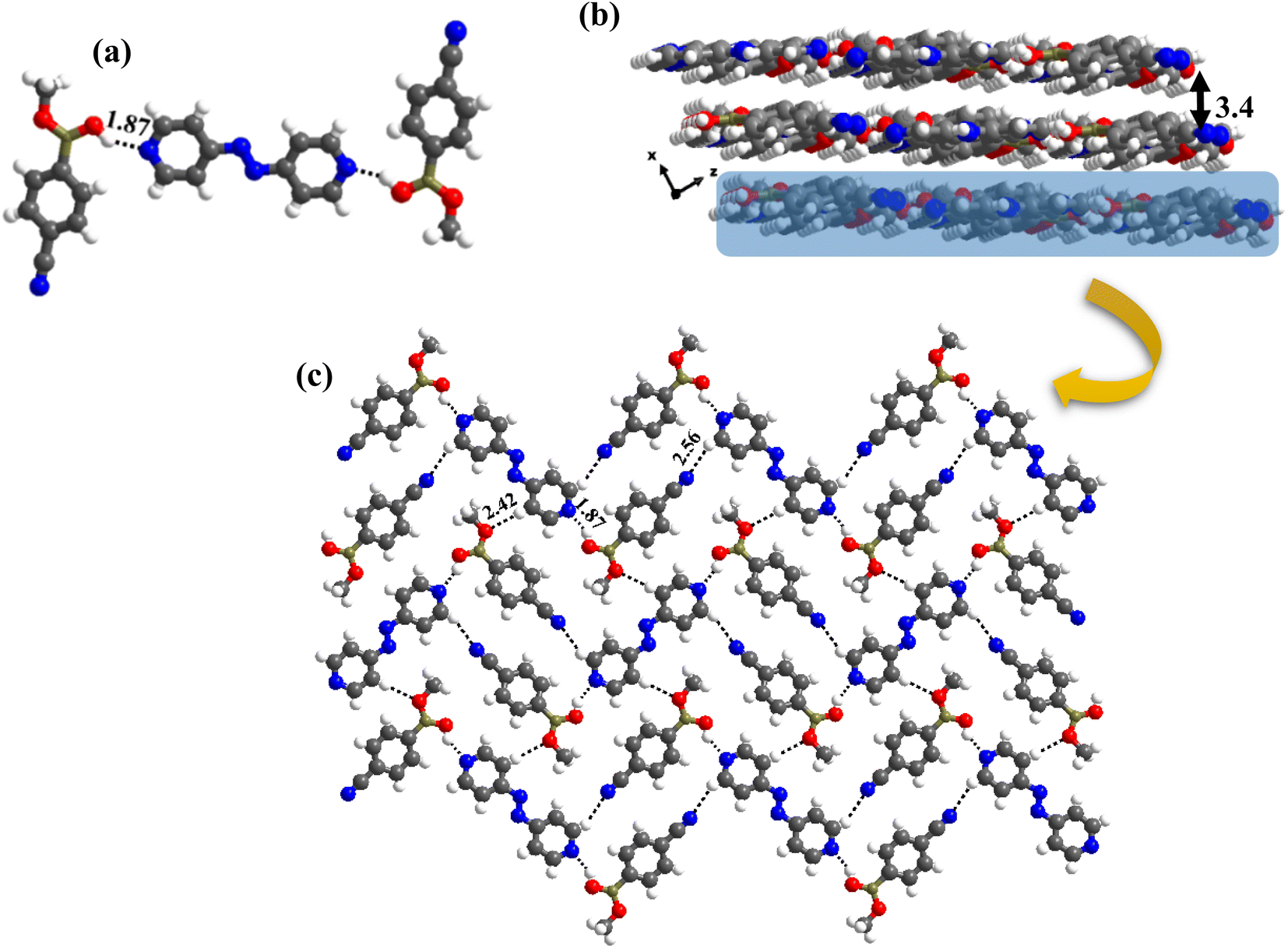 | ||
| Fig. 9 (a) Molecular recognition observed in the monoester with azopy. (b) Stacked sheet arrangement found in the molecular complex 6b, separated by 3.4 Å. (c) Two-dimensional arrangement of the molecules in the crystal lattice, 6b. | ||
Further, from the packing analysis, the arrangement of the molecules in the crystal lattice is in the form of stacked sheets, being separated at a distance of 3.4 Å. A typical arrangement of such a stacked sheet structure is shown in Fig. 9b. Within a typical sheet, the tri-components described above are held together through C–H⋯O (H⋯O, 2.42 Å; C⋯O, 3.47 Å) and C–H⋯N (H⋯N, 2.56 Å; C⋯N, 3.47 Å) hydrogen bonds formed by –OCH3 and –CN groups with hydrogen atoms of azopy molecules (Fig. 9c).
Since 6b was obtained as a concomitant form together with a hydrated form 6a, co-crystallization of CB with bpyee has also been repeated from a CH3OH solution to evaluate if any concomitant crystals were missed out earlier, as only the hydrated form was reported in the literature from such crystallization experiment. The experiment reveals that this is indeed, the case with the observation of concomitant crystals of different morphologies.
Upon characterization by single-crystal X-ray diffraction, one of the forms was found to be reported hydrated structure (PELMUR), while, the other one being with an asymmetric unit comprising of in situ transformed monoester of CB and bpyee (labeled as 7b), as observed in 6b. Further, 7b is also noted to be isomorphous and isostructural 6b, as both crystallize in the same space group P21/c and with similar unit cell parameters (Table 1), as well as similar structural arrangement in the crystal lattices except for hydrogen bond distances and angles. The arrangement of molecules in 7b is placed in Fig. S2.†
It should be noted that crystals of 1–5, were also obtained from CH3OH; but no methylation of CB was observed. Such a transformation may be described, as shown in Scheme 2, in terms of the loss of a water molecule from CB molecules, in the vicinity of either azopy/bpyee, which is generally a rare transformation in the co-crystallization process, but otherwise mostly found in the covalent synthesis of base-catalyzed methanolysis reactions.24
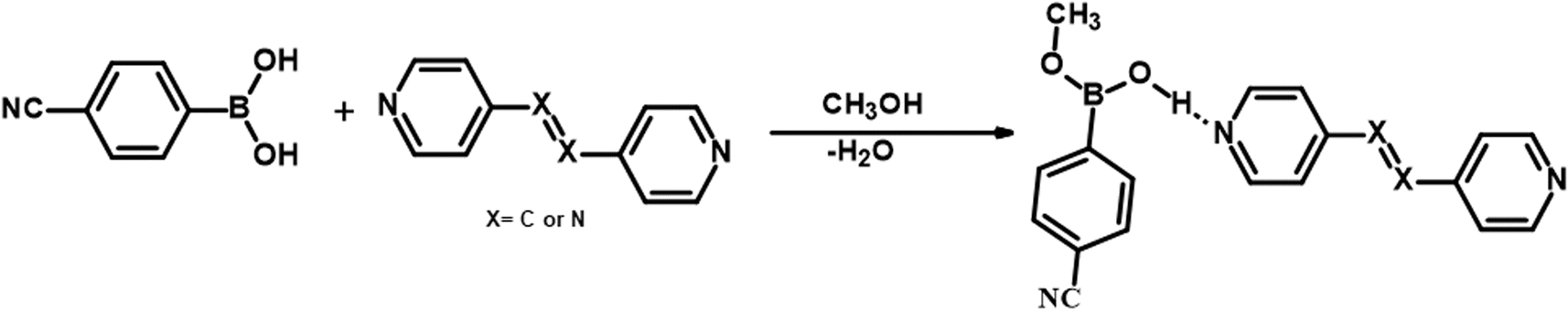 | ||
| Scheme 2 | ||
Based on the single-crystal X-ray diffraction data obtained from a single-crystal of the samples, it is evident that the CB molecule forms various exotic supramolecular assemblies with all N-donor molecules. So, in order to ensure the homogeneity of the bulk product, Powder X-Ray Diffraction (PXRD) analysis was conducted on the ground samples of the crystals obtained through solution growth. The result of the analysis demonstrates that the simulated PXRD patterns are corroborated with the experimental patterns, providing confirmation of the exclusive formation of the desired products, as depicted in Section S3.†
4. Correlation of structural features and intermolecular interactions energy
4.1. Structural features
The calculated Is, Π, RMSD, PXRD similarity and χ for all co-crystal pairs are listed in Table S2.† Among all structures, co-crystal pairs 6/7 and 6b/7b show the highest Is values 97.5 and 97.0%, respectively, as both the pairs are isomorphous as listed in Table 3. However, the hydrated structures of azopy (6a) and bpyea (5) as well as each of them along with the reported hydrated structure of bpyee, show similar Is values (∼65–70%) as observed in 6/7 and 6b/7b, but the Π values diverge more (Table 3), which may be due the difference in packing arrangement observed in these crystal lattices.
The Xpac analysis, computing dissimilarity index (χ), reinforces the observed isostructural features. Herein the points (δa, δp) are closer to the origin in the structures of 6/7, 6b/7b, and 6a/PELMUR pairs, suggesting a stronger correlation. In each of these examples, the observed Xpac dissimilarity index (χ = 3.2, 2.2 and 2.6), is shown in the right corner of Fig. 10.
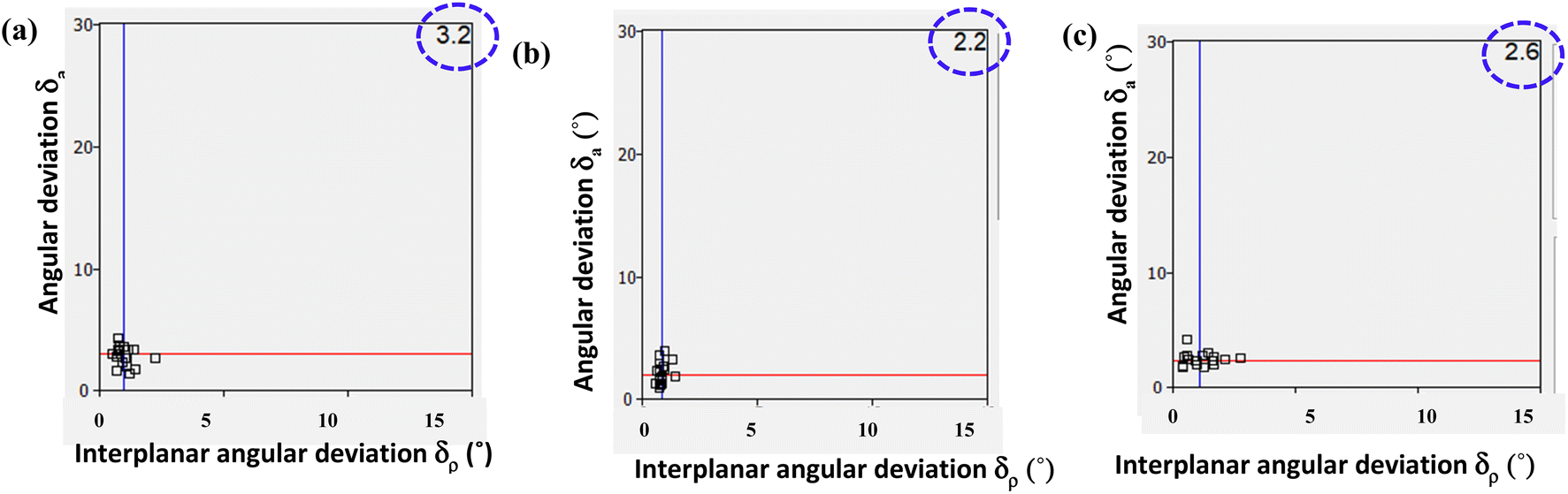 | ||
| Fig. 10 XPac plot of interplanar angular deviation vs. angular deviation (°) for the pair of (a) 6/7, (b) 6b/7b and (c) 6a/PELMUR illustrating the degree of similarity (top right corner value indicates dissimilarity index). | ||
 | ||
| Fig. 11 Presentation of experimental shapes (top) and computed BFDH morphology (bottom) for each structure. | ||
Shapes of crystals, referred to as morphology, in general, indeed will be the reflection of the packing (arrangement) of molecules in crystal lattices. But, depending upon the growth conditions, often the experimental shapes of the crystals do not always truly corroborate with the observed arrangement of molecules deduced from the structural evaluation carried out by single-crystal X-ray diffraction. Thus, several computational evaluation methods are developed to understand the true morphology of the crystals and thereby compare them with the experimental observations to authenticate the real shapes of the crystals.
Among the available different methodologies and procedures, morphology prediction modules available within Mercury software,28 Bravais–Friedel–Donnay–Harker (BFDH) morphologies, are widely populated to establish a correlation between the experimental and predicted shapes, mainly based on the evaluation of the packing of the molecules.29
To assess the observed morphology correlating with the packing patterns, BFDH morphologies are generated considering attachment energy (AE), as listed in Table S3,† and BFDH models. The evolved geometries for all crystals are compiled in Fig. 11, which also shows the experimental geometry of the crystals. It is apparent that experimental and computed morphologies show a high degree of overlap, grouping into four types – blocks, needles, plates, and a blade, as observed experimentally also.
4.2. Energy of intermolecular interactions
Intermolecular interactions with gradient energy direct the course of the packing of molecules in crystal lattices. X-ray diffraction data not only assess the determination of positions of the atoms in the lattices but also facilitates the estimation of different characteristics of intermolecular interactions quantitatively in terms of bond distances, angles, etc., that enable further estimate the stability factors like packing coefficients, lattice energy, intermolecular interactions energy, etc. In general, as numerous interactions are present in any crystal structure and assessment to establish the correlation between structures is tedious following truly numerical parameters, which leads to the evolution of different types of pictorial representation of strength and energy patterns. Among, Hirshfeld analysis gives a presentation of quantification of the strength of each type of intermolecular interaction through dnorm and 2D-finger plots, while ‘Energy Frameworks’ project total energy evaluating contributions from each interaction as represented by gradient cylinders. Thus, energy calculations have been carried out on all structures 1–7, 6a, 6b, 7a and 7b using Crystal Explorer which is embedded with routines/algorithms to carry out Hirshfeld and energy frameworks.In the following section the details of dnorm and corresponding 2D fingerprint plots of each co-crystal, 1–7, 6a, 6b, 7a and 7b, are shown in Fig. 12, wherein each structure shows a unique Hirshfeld surface and 2D fingerprint plots (details of analysis given in Tables S4 and S5†). The intensity of the surface is determined based on the strength of intermolecular interactions in different color codes and intensities. Thus, deep bright red on the surface at respective donor and acceptor atoms indicates the presence of strong O–H⋯O or O–H⋯N interactions; C–H⋯O appears as a faint red spot and furthermore, white and blue regions on the surface determine the moderate to very weak interactions. Hence, herein Hirshfeld surface of CB has been generated to investigate the influence of different N-donors with varied backbones in overall packing in the crystals (Fig. 12).
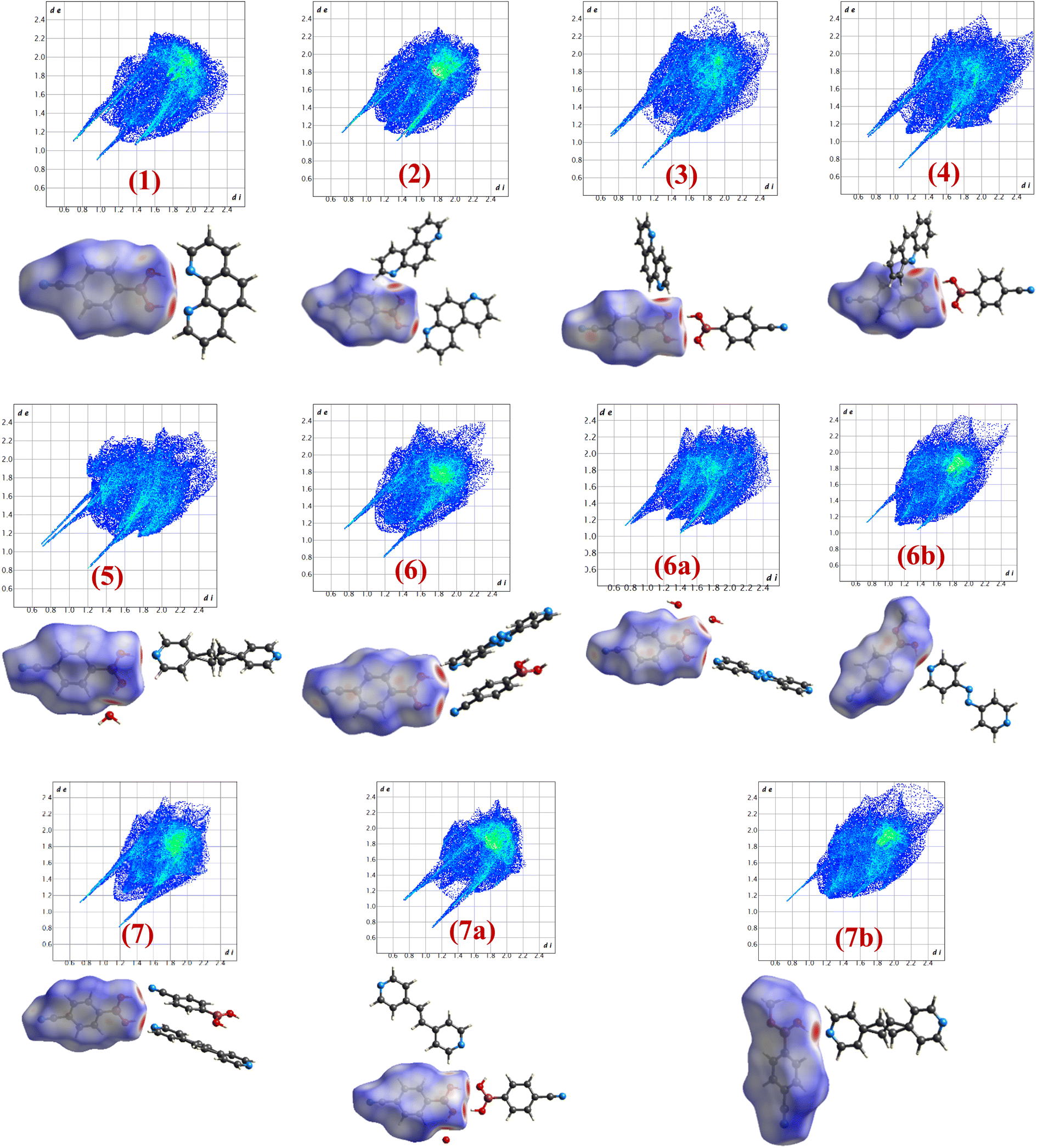 | ||
| Fig. 12 Hirshfeld surfaces and 2D fingerprint plots of 1–7, 6a, 6b, 7a and 7b. | ||
Analysis of dnorm plots shows differently shaded regions in the proximity of N-donor molecules reflecting hydrogen-bonded molecular recognition patterns, either homomeric/heteromeric or both together. For example, structures 1, 2, 5, 6a, 6b and 7b show only heteromeric while in structures 3 and 4 both homomeric as well as heteromeric could be visualized; however, in 7a only homomeric interaction is apparent as the heteromeric interaction appears faintly shaded, thus confirming the standalone nature of 7a. Also, the isostructural pairs (6/7 and 6b/7b) show similar Hirshfeld surface and 2D fingerprint plots.
From 2D fingerprint analysis it is accentuated that in almost all the cases except 6b and 7b (crystals with monoester form of CB), two prominent long spikes at de + di ≈ 1.8 Å, which represent the H⋯O/O⋯H or H⋯N/N⋯H interactions, with the close contacts range for H⋯O (10–24%) and H⋯N (18–25%), as details are shown in Fig. 13. However, 6b and 7b show only one spike which is due to the –OH group of monoesters CB.
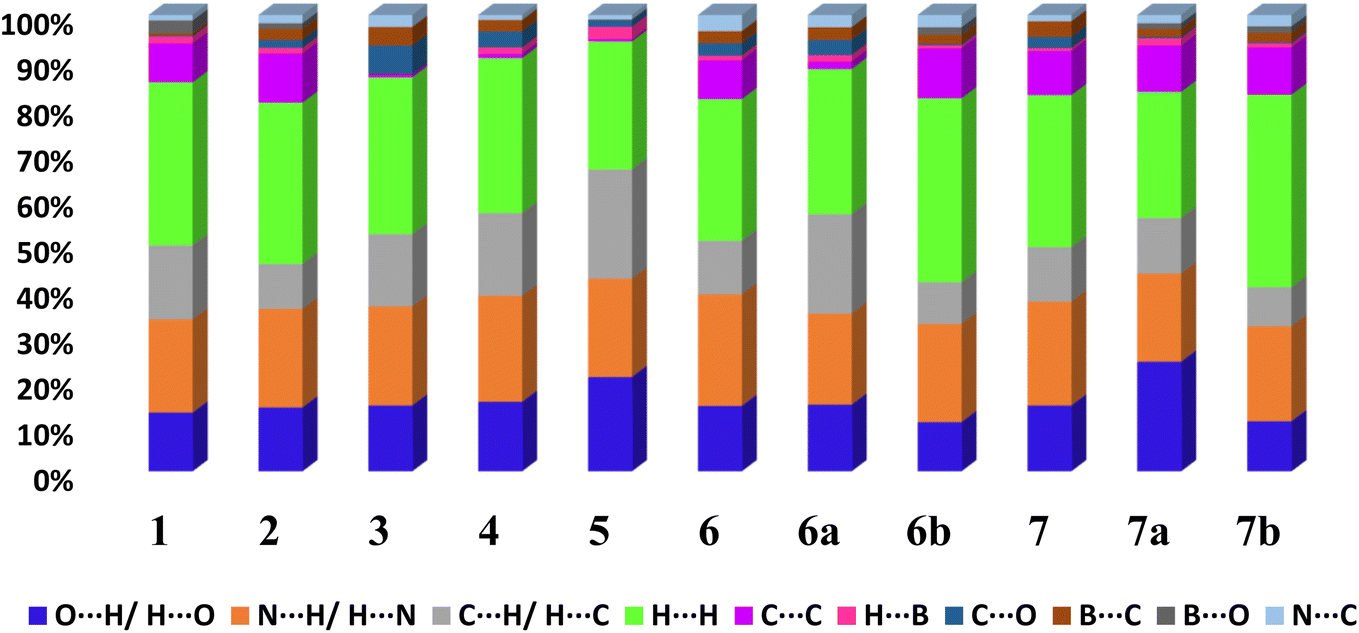 | ||
| Fig. 13 The relative contribution of different types of intermolecular interactions in crystal lattices considering CB as a dominant surface 1–7, 6a, 6b, 7a and 7b. | ||
Further, the finger plots disclose that all crystal structures herein follow the general packing features observed in other hydrogen-bonded structures with variable contributions from different intermolecular interactions. Thus, it is apparent from Fig. 13 that all structures show the approximately equal contribution of strong hydrogen bonds (∼30%) and secondary hydrogen bonds about 20% with an overall van der Waals of ∼30%. In each fingerprint plot, the van der Waals interactions are represented by light to intense green spots at around 3.4 Å, which is due to the C–C interaction or often referred to as π–π interaction.
This approach has proven to be particularly useful in the study of molecular crystals, where the nature and strength of intermolecular interactions can have a significant impact on the properties and behaviour of the crystals. For example, the utilization of energy frameworks has proven to be valuable in establishing structure–property relationships in molecular crystals, including the study of mechanical behaviour, such as bending, shearing and brittleness of molecular crystals.31 The basic frameworks are represented as electrostatic energy (Eele, red cylinders), dispersion energy (Edis, green cylinders) and total energy (Etot, blue cylinders).
Therefore, energy frameworks are developed to analyze and comprehend the diverse crystals packing of co-crystals 1–7, 6a, 6b, 7a and 7b, by generating overlapped energy frameworks onto the packing plots, as represented in Fig. 14, for selected structures (1, 5, 6, 7 and 7a) while the same was shown for all the structures in Table S5.†
 | ||
| Fig. 14 Pictorial representation of the energy frameworks for some selected co-crystals. | ||
Analysis of all the plots reveals that electrostatic interactions play a pivotal role in the crystal packing compared to dispersive forces as the radius of green cylinders is so low compared to red ones, except in structure 7a. A close look at the electrostatic interactions reveals that in structure 1, the high thickness of the cylinders compared to other structures with high stabilization energy (−93.1 kJ mol−1) indeed reflects the significance of distinct dual heteromeric O–H⋯N hydrogen bonds, among all structures in this study, being facilitated by juxtaposed N-atoms on 110phen. Similar features are also observed in structure 5 (hydrated CB and bpyea).
Further, a striking feature is that in structure 7a, dispersion energy is prominent over electrostatic (Fig. 14), as clearly visible with thick green cylinders, which is indeed in agreement with the structural features in 7a as the heteromeric interactions are obsolete in the form of only weak interactions like C–H⋯N and C–H⋯O hydrogen bonds. In fact, the representation authenticates the observation of standalone structure (host–guest type) features of 7a, in this series as discussed in the corresponding structural description above. A noteworthy observation that could be deduced is that homomeric interactions between CB molecules despite being in the category of strong hydrogen bonds (O–H⋯O) could contribute only a little towards the total energy of the crystal lattice.
In general, heteromeric interactions are known to be stronger than homomeric interactions. In fact, the same is reinforced with the energy terms observed in structures 3 and 4 (Table S5†), wherein both form homomeric and heteromeric interactions simultaneously, as heteromeric O–H⋯N hydrogen bonds show higher energy −46.3 and −48.9 kJ mol−1 compared to the homomeric O–H⋯O hydrogen bonds, which are at −43.6 and −42.8 kJ mol−1, respectively for 3 and 4.
In addition, the isostructural features are well reflected with the energy frameworks showing similar energy gradients in the structures 6/7 and 6b/7b (see Fig. 14). However, in all other structures, the contributions towards total energy are through the equal competence of electrostatic and dispersive energy, as represented in ESI (Table S5†).
5. Conclusions
Some supramolecular assemblies of CB with different N-donor compounds, distinguished by geometrical flexibility have been discussed highlighting the significance of –B(OH)2 moiety for its ability to form a variety of complexes, particularly due to the conformational flexibility that leads to the formation of hydrogen bonds in topologically different patterns. Though a majority of the complexes –B(OH)2 show a syn–anti conformation, the formation of syn–syn noted in some complexes indeed strengthens its prevalence, especially in the presence of N-donors with juxtaposed N-atoms, for instance, 110phen compared to analogous 14- and 17phen. Also, observation of different complexes with the same co-formers particularly in the form of hydrates and anhydrous further signifies the importance of multiple crystallization experiments by varying solvents of crystallization, specifically with linear N-donor ligands. Furthermore, the study highlights the in situ covalent bond formation with ease, being influenced by selective co-formers that possess reactive functionality. Thus, CB undergoes monoester transformation in the co-crystallization studies with N-donors bpyee and azopy that have CC and NN, respectively, while it remains intact in the presence of bpyea (an analog of bpyee and azopy), which has saturated (–CH2–)2 moiety only. The observed structural features are well corroborated through computational methods and also by calculating isostructurality, evaluation of the strength of intermolecular interactions (Hirshfeld surface analysis) and energy frameworks using Crystal Explorer. Furthermore, experimentally observed morphology of the crystals in the form of needles, blocks, plates and a blade are validated by performing BFDH morphology calculations using Mercury.
Abbreviations
| CB | Cyanophenylboronic acid |
| 110phen | 1,10-Phenanthroline |
| 47phen | 4,7-Phenanthroline |
| 17phen | 1,7-Phenanthroline |
| acr | Acridine |
| bpyea | 1,2-Bis(4-pyridyl)ethane |
| bpyee | 1,2-Bis(4-pyridyl)ethene |
| azopy | 4,4′-Azopyridine |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge IIT Bhubaneswar for infrastructure and DST-SERB, New Delhi, for the financial support.References
- (a) J. C. MacDonald and G. M. Whitesides, Chem. Rev., 1994, 94, 2383–2420 CrossRef CAS; (b) D. Braga and F. Grepioni, Crystal Engineering: from Molecules and Crystals to Materials, Springer, 1999 Search PubMed; (c) P. Alivisatos, P. F. Barbara, A. W. Castleman, J. Chang, D. A. Dixon, M. L. Klein, G. L. McLendon, J. S. Miller, M. A. Ratner and P. J. Rossky, Adv. Mater., 1998, 10, 1297–1336 CrossRef; (d) J. L. Atwood, Comprehensive Supramolecular Chemistry II, Elsevier, 2017 Search PubMed.
- (a) G. R. Desiraju, Nature, 2001, 412, 397–400 CrossRef CAS PubMed; (b) L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243–19256 CrossRef CAS PubMed; (c) D. Braga and F. Grepioni, Acc. Chem. Res., 2000, 33, 601–608 CrossRef CAS PubMed; (d) J. Bernstein, J. J. Novoa, R. Boese and S. A. Cirkel, Chem.–Eur. J., 2010, 16, 9047–9055 CrossRef CAS PubMed; (e) S. Ahn, J. PrakashaReddy, B. M. Kariuki, S. Chatterjee, A. Ranganathan, V. R. Pedireddi, C. N. R. Rao and K. D. M. Harris, Chem.–Eur. J., 2005, 11, 2433–2439 CrossRef CAS PubMed.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed; (b) S. Subramanian and M. J. Zaworotko, Coord. Chem. Rev., 1994, 137, 357–401 CrossRef CAS; (c) Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao and Y. Zhang, Nat. Commun., 2019, 10, 3074 CrossRef PubMed.
- (a) P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS; (b) K. S. Eccles, R. E. Morrison, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2014, 14, 2753–2762 CrossRef CAS; (c) S. Easmin and V. R. Pedireddi, Cryst. Growth Des., 2023, 23, 2802–2811 CrossRef CAS.
- (a) P. S. Pereira Silva, R. A. E. Castro, E. Melro, M. R. Silva, T. M. R. Maria, J. Canotilho and M. E. S. Eusébio, J. Therm. Anal. Calorim., 2015, 120, 667–677 CrossRef CAS; (b) M. Nowak, A. J. Dyba, J. Janczak, A. Morritt, L. Fábián, B. Karolewicz, Y. Z. Khimyak, D. E. Braun and K. P. Nartowski, Mol. Pharm., 2022, 19, 456–471 CrossRef CAS PubMed.
- (a) I. Georgiou, S. Kervyn, A. Rossignon, F. De Leo, J. Wouters, G. Bruylants and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 2710–2727 CrossRef CAS PubMed; (b) M. R. Shimpi, N. SeethaLekshmi and V. R. Pedireddi, Cryst. Growth Des., 2007, 7, 1958–1963 CrossRef CAS; (c) S. Forensi, A. Stopin, F. de Leo, J. Wouters and D. Bonifazi, Tetrahedron, 2020, 76, 131299 CrossRef CAS; (d) S. Varughese, S. B. Sinha and G. R. Desiraju, Sci. China: Chem., 2011, 54, 1909–1919 CrossRef CAS.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) D. J. Wallace and C.-y. Chen, Tetrahedron Lett., 2002, 43, 6987–6990 CrossRef CAS; (c) A. N. Cammidge and K. V. Crépy, Chem. Commun., 2000, 1723–1724 RSC.
- (a) J. N. Cambre and B. S. Sumerlin, Polymer, 2011, 52, 4631–4643 CrossRef CAS; (b) B. Wang, K. Yoshida, K. Sato and J.-i. Anzai, Polymers, 2017, 9, 202 CrossRef PubMed.
- (a) G. T. Williams, J. L. Kedge and J. S. Fossey, ACS Sens., 2021, 6, 1508–1528 CrossRef CAS; (b) T. D. James, K. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed., 1996, 35, 1910–1922 CrossRef.
- (a) W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346–368 CrossRef CAS PubMed; (b) B. Muz, A. K. Azab, L. Confalonieri, E. Del Grosso, S. Fallarini, D. Imperio and L. Panza, Bioorg. Med. Chem., 2022, 59, 116659 CrossRef CAS PubMed.
- (a) S.-T. Yang, J. Kim, H.-Y. Cho, S. Kim and W.-S. Ahn, RSC Adv., 2012, 2, 10179–10181 RSC; (b) Y. Kubo, R. Nishiyabu and T. D. James, Chem. Commun., 2015, 51, 2005–2020 RSC; (c) S. D. Brucks, D. N. Bunck and W. R. Dichtel, Polymer, 2014, 55, 330–334 CrossRef CAS.
- (a) V. R. Pedireddi and N. SeethaLekshmi, Tetrahedron Lett., 2004, 45, 1903–1906 CrossRef CAS; (b) M. Talwelkar and V. R. Pedireddi, Tetrahedron Lett., 2010, 51, 6901–6905 CrossRef CAS; (c) J. J. Campos-Gaxiola, B. A. García-Grajeda, I. F. Hernández-Ahuactzi, J. A. Guerrero-Álvarez, H. Höpfl and A. Cruz-Enríquez, CrystEngComm, 2017, 19, 3760–3775 RSC; (d) P. Tomaszewski, M. Wiszniewski, J. Serwatowski, K. Woźniak, K. Durka and S. Luliński, Dalton Trans., 2018, 47, 16627–16637 RSC.
- (a) S. Weinstein, L. Leiserowitz and E. Gil-Av, J. Am. Chem. Soc., 1980, 102, 2768–2772 CrossRef CAS; (b) A. D. Burrows, Supramolecular Assembly via Hydrogen Bonds I, 2004, pp. 55–96 Search PubMed; (c) N. Shan, A. Bond and W. Jones, Cryst. Eng., 2002, 5, 9–24 CrossRef CAS.
- (a) D. G. Hall, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, John Wiley & Sons, 2006 Search PubMed; (b) D. G. Hall, Boronic acids: Preparation and Applications in Organic Synthesis and Medicine 2005, pp. 1–99 CrossRef; (c) N. Fujita, S. Shinkai and T. D. James, Chem.–Asian J., 2008, 3, 1076–1091 CrossRef CAS PubMed; (d) T. D. James, Beilstein J. Org. Chem., 2016, 12, 391–405 CrossRef CAS; (e) R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- M. TalwelkarShimpi, S. Öberg, L. Giri and V. R. Pedireddi, Cryst. Growth Des., 2017, 17, 6247–6254 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2014, 70, C1437 Search PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- G. Bergerhoff, M. Berndt and K. Brandenburg, J. Res. Natl. Inst. Stand. Technol., 1996, 101, 221 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- (a) M. Talwelkar and V. Pedireddi, Tetrahedron Lett., 2010, 51, 6901–6905 CrossRef CAS; (b) S. SeethaLekshmi, S. Varughese, L. Giri and V. Pedireddi, Cryst. Growth Des., 2014, 14, 4143–4154 CrossRef CAS.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed.
- J. PrakashaReddy and V. R. Pedireddi, Tetrahedron, 2004, 60, 8817–8827 CrossRef CAS.
- (a) V. Nagarajan, M. R. Shimpi and V. R. Pedireddi, J. Mol. Struct., 2013, 1050, 216–221 CrossRef CAS; (b) K. Fucke, S. A. Myz, T. P. Shakhtshneider, E. V. Boldyreva and U. J. Griesser, New J. Chem., 2012, 36, 1969–1977 RSC.
- (a) I. B. Banković-Ilić, Z. B. Todorović, J. M. Avramović, A. V. Veličković and V. B. Veljković, Fuel Process. Technol., 2015, 137, 339–350 CrossRef; (b) H. Liu, J. Chen, L. Chen, Y. Xu, X. Guo and D. Fang, ACS Sustainable Chem. Eng., 2016, 4, 3140–3150 CrossRef CAS.
- S. Ranjan, R. Devarapalli, S. Kundu, S. Saha, S. Deolka, V. R. Vangala and C. M. Reddy, IUCrJ, 2020, 7, 173–183 CrossRef CAS PubMed.
- (a) R. Thakuria and A. Nangia, Cryst. Growth Des., 2013, 13, 3672–3680 CrossRef CAS; (b) D. Cinčić, T. Friščić and W. Jones, New J. Chem., 2008, 32, 1776–1781 RSC; (c) A. J. Cruz Cabeza, G. M. Day, W. S. Motherwell and W. Jones, J. Am. Chem. Soc., 2006, 128, 14466–14467 CrossRef.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448–460 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- J. Prywer, J. Cryst. Growth, 2004, 270, 699–710 CrossRef CAS.
- (a) D. Dey, S. Bhandary, S. P. Thomas, M. A. Spackman and D. Chopra, Phys. Chem. Chem. Phys., 2016, 18, 31811–31820 RSC; (b) C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- (a) S. P. Thomas, M. W. Shi, G. A. Koutsantonis, D. Jayatilaka, A. J. Edwards and M. A. Spackman, Angew. Chem., Int. Ed., 2017, 56, 8468–8472 CrossRef CAS PubMed; (b) M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC; (c) M. Ghora, P. Majumdar, M. Anas and S. Varghese, Chem.–Eur. J., 2020, 26, 14488–14495 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP plots, powder X-ray diffraction patterns, packing analysis, isostructurality, Hirshfeld surface analysis and interaction energy. CCDC 2260224, 2260225, 2260226, 2260227, 2260231, 2260232, 2260233, 2260234, 2260235, 2260236 and 2260237. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra03936f |
| This journal is © The Royal Society of Chemistry 2023 |