
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of pharmacologically important oxindoles via the asymmetric aldol reaction of isatin and the investigation of the organocatalytic activity of new alicyclic β-amino acid derivatives†
Tünde Faragó a,
Attila M. Remetea,
István Szatmáriab,
Rita Ambrusc and
Márta Palkó*a
a,
Attila M. Remetea,
István Szatmáriab,
Rita Ambrusc and
Márta Palkó*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös utca 6, Szeged H-6720, Hungary. E-mail: palko.marta@szte.hu
bStereochemistry Research Group, Eötvös Loránd Research Network, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
cInstitute of Pharmaceutical Technology and Regulatory Affairs Faculty of Pharmacy, University of Szeged, Eötvös utca 6, Szeged H-6720, Hungary
First published on 26th June 2023
Abstract
This work involves the synthesis and subsequent development of a number of novel organocatalysts generated from β-amino acids bearing diendo and diexo norbornene skeletons to improve their catalytic characteristics. The aldol reaction between isatin and acetone selected as the model reaction, was used to test and study enantioselectivities. The potential impact on enantioselectivity control regarding enantiomeric excess (ee%) was probed by varying the reaction parameters, such as additive, solvent, catalyst loading, temperature and substrate range. The corresponding 3-hydroxy-3-alkyl-2-oxindole derivetives were produced by organocatalyst 7 with good enantioselectivity up to 57% ee in the presence of LiOH. Substrate screening was used to investigate a number of substituted isatins with excellent findings up to 99% ee. Another aspect of this effort involved employing high-speed ball mill apparatus to conduct a mechanochemical study to make this model reaction more environmentally benign and sustainable.
Introduction
The synthesis of natural compounds poses a great challenge for synthetic organic chemists, and it is a subject that merits ongoing attention. Many different natural products have the oxindole core and these compounds also possess a great deal of scientific interest due to their extensive spectrum of biological activities, which raised the interest of chemists, pharmacists and biologists alike. For instance, arundaphine, convolutamydines, maremycins and CPC-1 possess biological properties, such as antioxidative, anti-HIV and neuroprotection activity.1–6 The study of pharmaceuticals incorporating the oxindole scaffold and the discovery of advantageous synthesis pathways will certainly be the focus of pharmaceutical research in the future. Due to the significance of a sustainable future in today's world, green chemistry is becoming desirable and more and more popular. Sustainable catalysis is the core of green chemistry. In comparison to metal and enzyme catalysis, organocatalysis has additional benefits, including chemical efficiency, availability, low cost, durability and non-toxicity.7,8 There is still a need for innovative, efficient catalytic systems for the aldol reaction of isatin and ketones. Furthermore, it is crucial to have a better understanding of the aldol reaction in order to develop and use effective catalysts. The aldol reaction of isatin and ketones can be influenced by a number of different factors. The outcome of aldol reactions depends significantly on solvents used and the amount of solvents.2,9–13 When the amount of catalysts was increased, it was usual to witness abrupt drops in ee% followed by sudden increases. As Wang et al. reported in 2018, it is likely that this phenomenon is attributed to the retro-aldol reaction.14Only a handful of research teams have studied organocatalysts in the aldol process with low catalyst loadings (0.5–1 mol%) reporting good15 or excellent results.16 These were superior to those obtained by employing 10 mol% of organocatalyst. When using substituted isatins in the aldol reaction between isatin and acetone in numerous studies typically better results were achieved in comparison to those found by the parent compound.9,10,17 Position and properties of the substituents of isatin have a significant impact on the yield and the enantioselectivity of the aldol reaction. Good results in ee from 77% up to 94% were obtained utilizing 4-chloroisatin, 4-bromoisatin, 4-methylisatin and 4,7-dichloroisatin as reagent.17,18 Good but slightly lower enantiomeric excesses were found when the same substituents were applied in C-5, C-6, or C-7 positions of isatin.17,18
Various other carbonyl compounds, for example, acetone, cyclohexanone and substituted cyclohexanone derivatives were also tested in the aldol reaction of isatin and ketones with fair to good results.19–23
Over the past 20 years, there has been a strong incentive to develop effective organocatalysts to enhance the asymmetric aldol reaction of isatin and ketones, which is controlled by secondary amine organocatalysts such as proline derivatives.8,11,24–26 In 2016, Singh et al. discovered that proline derivative (S)-N-((S)-1-phenylethyl)pyrrolidine-2-carboxamide has stronger organocatalytic effects than simple (S)-proline.27 In 2015, Chen et al. found that this model reaction was effectively catalysed by the Li+ salt of (S)-phenylalanine.10 However, only a small number of researchers have investigated the use of alicyclic amino acid derivatives as organocatalysts to synthesise the aforementioned oxindole derivatives.11 There are only a handful of cage-type amine organocatalysts with isoquinuclidine or 2-azanorbornane backbones described in the literature.28 The majority of such catalysts are composed of the following components: a larger or more rigid backbone for steric influence, amino groups as enamine-forming sites around the side chains, and one or two sterically bulky phenyl groups on the side chain. In order to achieve good results in enantioselectivity, the presence of another asymmetric site may also be required.11,28
β-Amino amides with a cyclohexane skeleton were also studied in this model reaction. Under neat conditions, the best result was 37% ee (20 mol% catalyst loading without additives).11 In the proposed reaction mechanism, the reaction of acetone with the NH2 group of the catalyst generates an imine, which is in equilibrium with its enamine tautomer. Then the enamine orients isatin through π–π interactions between the phenyl ring of isatin and the enamine. Finally, the enamine motif performs nucleophilic attack on isatin and then hydrolysis regenerates the catalyst.11 Regarding (S)-leucinol as organocatalyst in aldol reaction of isatin, a syn- and anti-enamine equilibration was proposed in the first steps of the catalytic cycle.29
In recent years, there has been a surge in the utilisation of energy-efficient methods, including mechanochemical activation, ultrasonic irradiation and microwave heating. The use of high-speed ball milling (HSBM), a technique for advancing synthetically valuable processes without the need of solvents, increases gradually in organic synthesis.30–34 To estimate the energy transfer onto the material, a number of variables must be taken into account, including the rotation frequency of the mill, milling time, vessel radius, size, weight and number of balls, density, etc. In general, ball milling raises the interior temperature.35–37 Utilising HSBM to study the interaction between isatin and acetone has been rather uncommon up until now, although it has several benefits.34,38
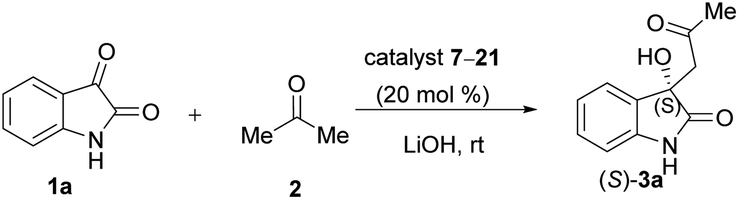
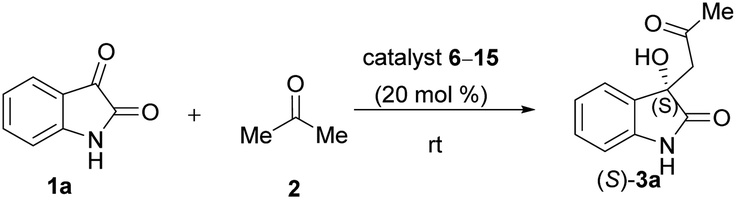
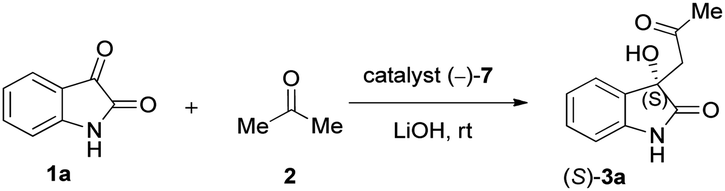
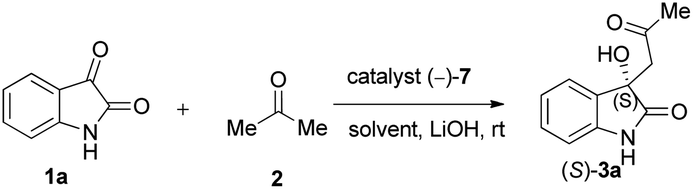
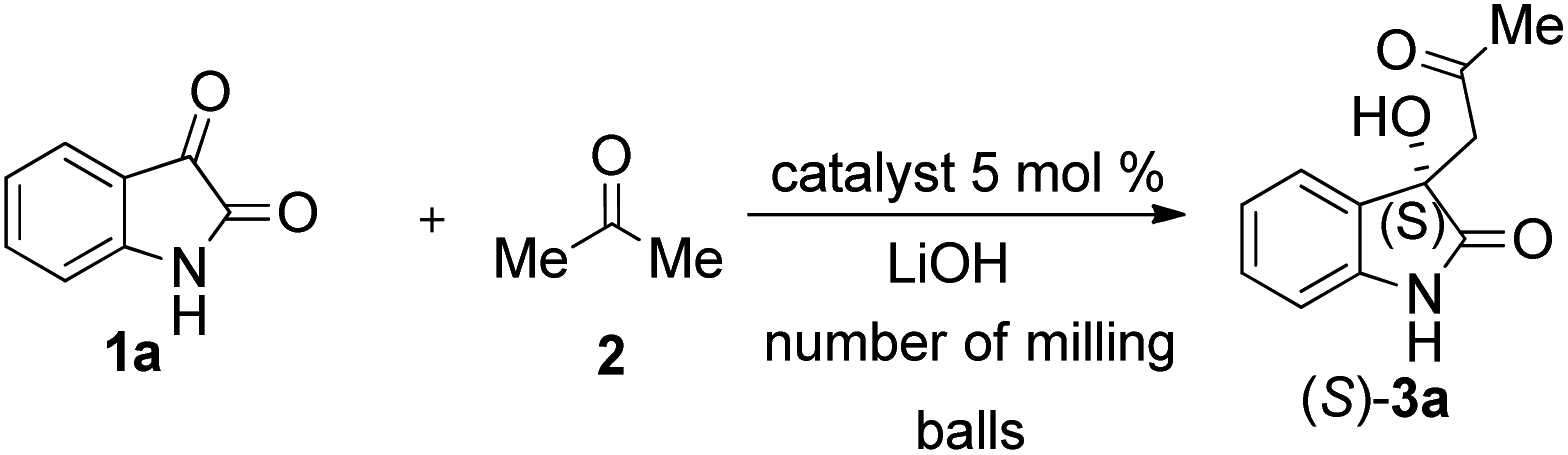
Due to these reasons, our aim was to synthesise novel diendo and diexo alicyclic amino acid derivatives with a norbornene skeleton. Furthermore, we also intended to investigate the catalytic activities of these compounds in the asymmetric aldol reaction between isatin (1a) and acetone (2) (Scheme 1). Additionally, we were interested in improving the catalytic efficacy of model reaction and identifying environmentally more benign and sustainable approaches to synthesise molecules containing the oxindole moiety.
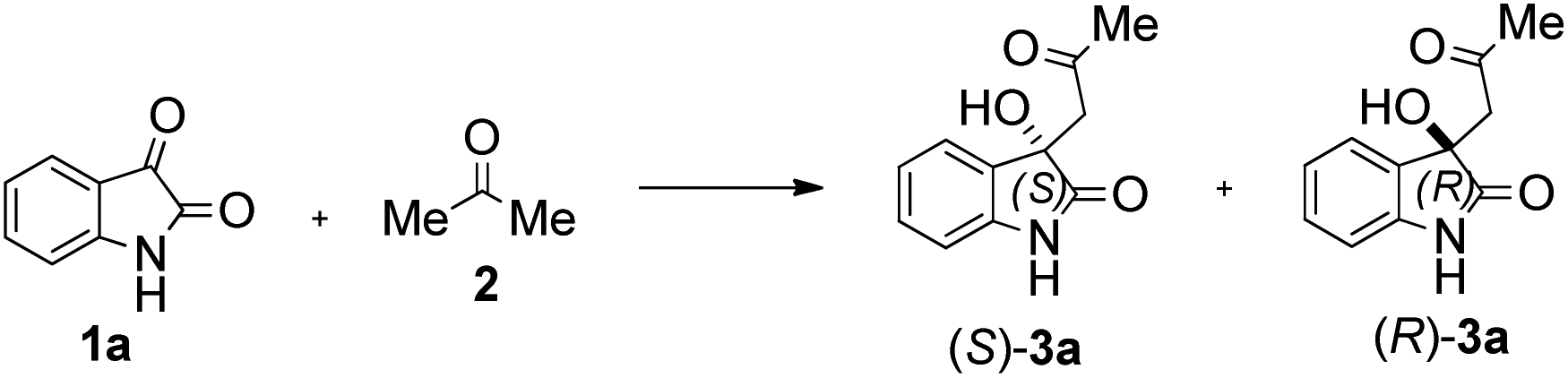 | ||
| Scheme 1 Aldol reaction between isatin (1a) and acetone (2). | ||
Results and discussion
According to the literature, 2-aminonorbornene ester enantiomer (−)-4 was prepared through diastereomer salt formation from racemic diendo 2-aminonorbornene ester (±)-4 utilising (+)-O,O′-dibenzoyltartaric acid (DBTA).39 Following the hydrolysis of the free ester (−)-4, the resulting amino acid was then reacted with di-tert-butyl dicarbonate to produce the diendo norbornene N-Boc-protected amino acid (−)-5.40 Urea (−)-6 was produced in a good yield when free ester (−)-4 and (S)-(−)-α-methylbenzyl isocyanate were reacted in diethyl ether at ambient temperature. N-Boc-protected diendo norbornene amino acid (−)-5 was transformed into amide (−)-7 in tetrahydrofuran (THF) at room temperature, using (S)-(−)-α-methylbenzyl amine in the presence of N,N′-diisopropylcarbodiimide (DIC) and hydroxybenzotriazole (HOBt) (Scheme 2).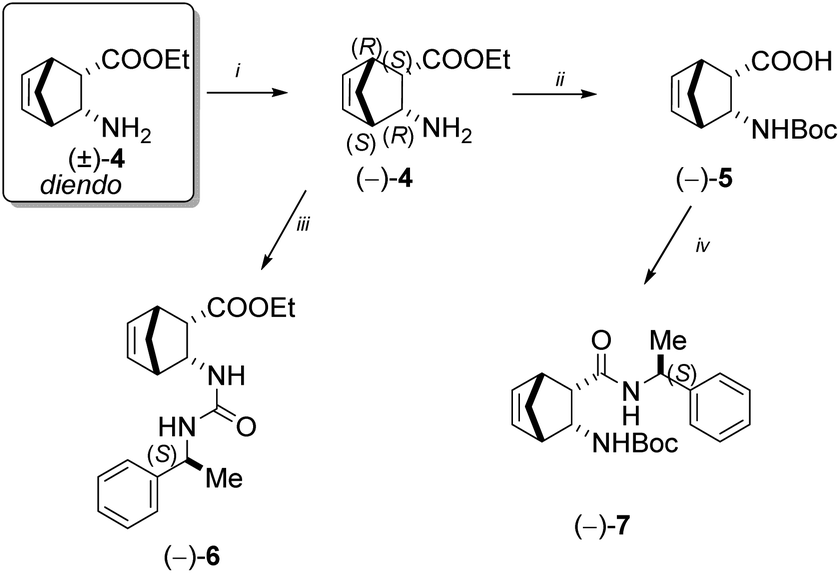 | ||
| Scheme 2 Reagents and conditions: (i) (1) (+)-DBTA, EtOH, 70 °C (2) NaOH, CHCl3/H2O; (ii) (1) MW, H2O, 100 °C; (2) Boc2O, dioxane/H2O, r.t.; (iii) (S)-(−)-α-methylbenzyl isocyanate, Et2O, r.t.; (iv) DIC, HOBt, (S)-(−)-α-methylbenzyl amine, THF, r.t., 24 h. | ||
Racemic diendo 2-aminonorbornene ester (±)-4 was also resolved with (−)-DBTA and the resulting enantiomeric amino ester (+)-4 was converted with (S)-(−)-α-methylbenzyl isocyanate into urea derivative (−)-8.41
Enantiomeric diendo norbornene β-amino acid was obtained by hydrolysis of ester (+)-4 under microwave heating. Afterwards, N-Boc protection yielded (+)-5, which was subjected to acid–amine coupling reactions with various amines in the presence of DIC and HOBt. Using this process, the following catalyst materials were prepared: (−)-9 with the use of (S)-(−)-1-(2-naphthyl)ethylamine, (−)-10 applying (S)-(−)-1-(1-naphthyl)ethylamine, (+)-11, (+)-12 using (R)-(+)-1-(2-naphthyl)ethylamine, (+)-13, (+)-14 utilizing (R)-(+)-1-(1-naphthyl)ethylamine and (−)-15 using propargylamine (Scheme 3).
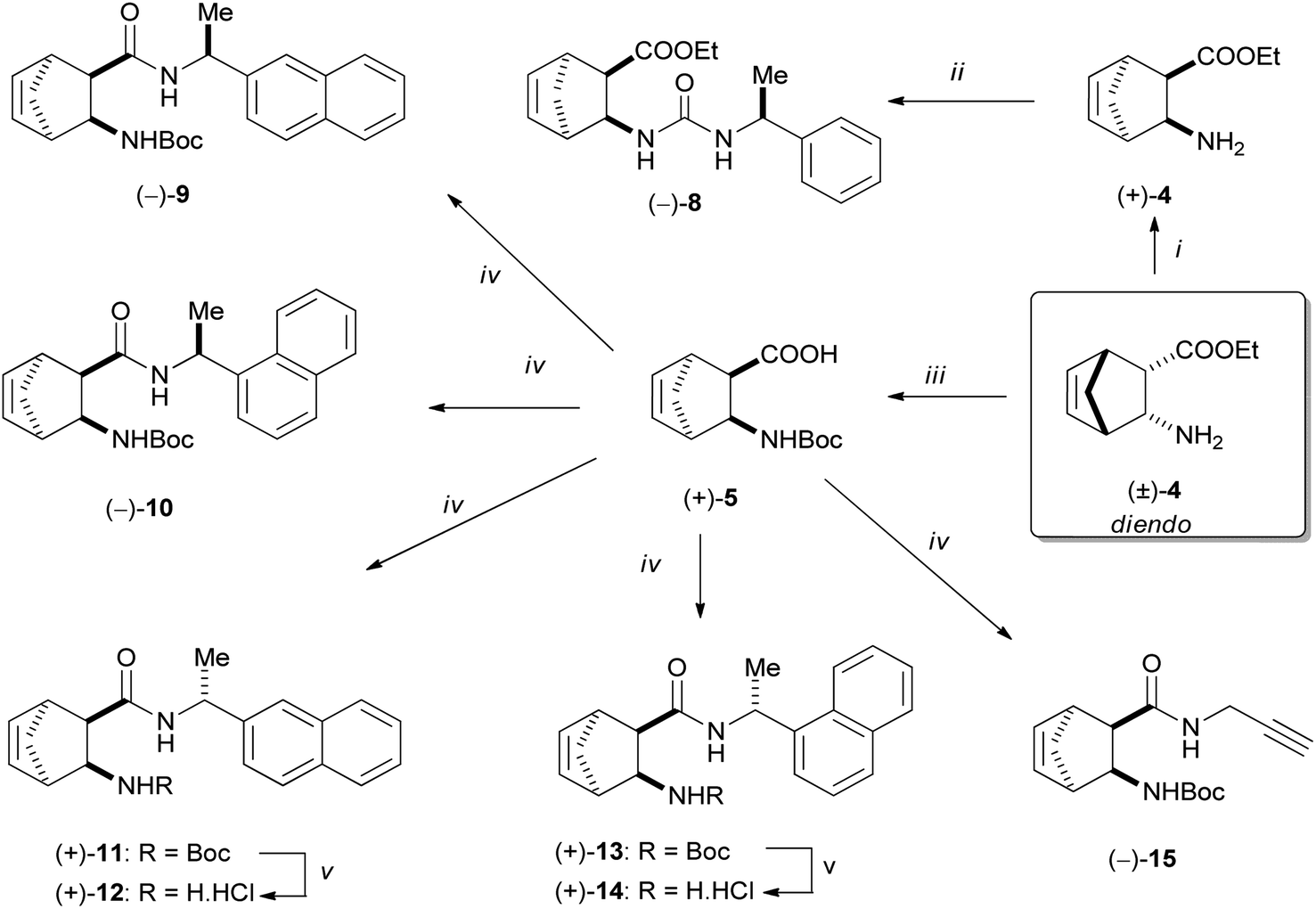 | ||
| Scheme 3 Reagents and conditions: (i) (1) (−)-DBTA, EtOH, 70 °C, (2) NaOH, CHCl3/H2O; (ii) (S)-(−)-α-methylbenzyl isocyanate, Et2O, r.t. (iii) (1) (−)-DBTA, EtOH, 70 °C; (2) NaOH, CHCl3/H2O, (3) MW, H2O, 100 °C; 4. Boc2O, dioxane/H2O, r.t.; (iv) DIC, HOBt, corresponding amine, THF, r.t., 24 h. (v) HCl/EtOH, 100 °C, 1 h. | ||
Resolution of racemic diexo aminonorbornene ester (±)-16 with (+)-O,O′-di-p-toluoyltartaric acid ((+)-DPTTA) in ethanol delivered enantiopure diexo aminonorbornene ester (+)-16. Free ester base (+)-16 was hydrolysed into the corresponding amino acid in a microwave reactor and then it was protected with Fmoc-OSu to produce diexo N-Fmoc-protected protected norbornene β-amino acid (+)-17.42 From enantiopure amino acid (+)-17, the following catalyst materials were prepared via acid–amine coupling processes: (+)-18 using (R)-(+)-1-(2-naphthyl)-ethylamine, (−)-19 utilizing (S)-(−)-1-phenylethylamine and (+)-20 using (R)-(+)-1-phenylethylamine (Scheme 4).
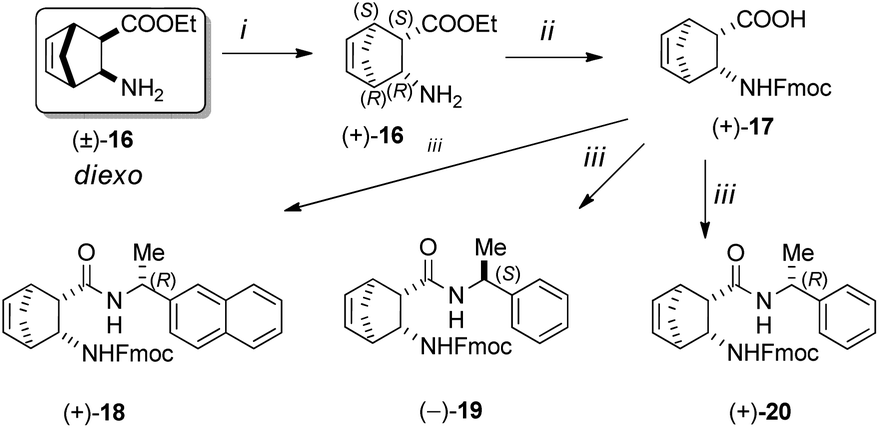 | ||
| Scheme 4 Reagents and conditions: (i) (1) (+)-DPTTA, EtOH, 70 °C, (2) NaOH, CHCl3/H2O; (ii) (1) MW, H2O, 100 °C; (2) Fmoc-OSu, acetone/H2O, NaHCO3, r.t.; (iii) DIC, HOBt, chiral amine, THF, r.t., 24 h. | ||
In the aldol reaction of isatin (1a) with acetone (2), a wide range of catalysts shown in Fig. 1 were investigated.
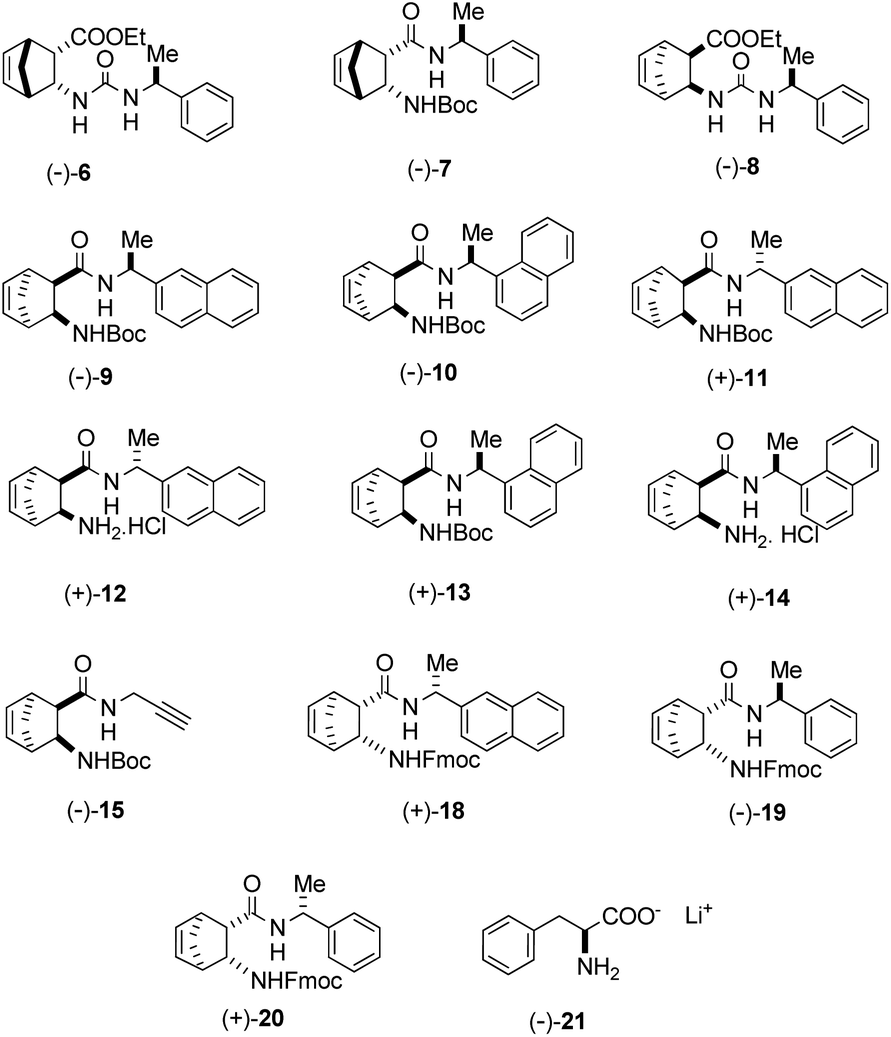 | ||
| Fig. 1 Structure of catalysts 6–21. Derivatives (−)-15 and (−)-21 were prepared according to literature procedure.10,40 | ||
The ratio of catalysts to be evaluated in the aldol reaction of isatin (1a) and acetone (2) was often studied in the literature. At first, catalysts 7–21 were screened at a 20 mol% concentration at room temperature. In these reactions, a very small amount (1 mmol) of LiOH as co-catalyst was also used (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb | eec (%) (S) config.d |
| a Organocatalysts 6–21 (20 mol%), isatin (0.28 mmol), acetone (5 mL, 325 equiv.), LiOH (1 mmol) as additive, stirred at room temperature for specified time.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44 | ||||
| 1 | (−)-6 | 2 | 95 | 15 |
| 2 | (−)-7 | 0.5 | 94 | 43 |
| 3 | (−)-8 | 5 | 90 | 5 |
| 4 | (−)-9 | 4 | 89 | Rac. |
| 5 | (−)-10 | 3 | 88 | Rac. |
| 6 | (+)-11 | 2 | 90 | 48 |
| 7 | (+)-12 | 2 | 92 | 51 |
| 8 | (+)-13 | 3 | 90 | 7 |
| 9 | (+)-14 | 2 | 96 | 49 |
| 10 | (−)-15 | 1 | 94 | 3 |
| 11 | (+)-18 | 3 | 89 | 23 |
| 12 | (−)-19 | 2 | 80 | 3 |
| 13 | (+)-20 | 3 | 85 | 23 |
| 14 | (−)-21 | 6 | 96 | 53 |
Derivatives (−)-7, (−)-8, (+)-11, (+)-12 and (+)-14 gave quite good results presented in Table 1 (entries 2, 3, 6, 7 and 9). In all cases, high conversion values were observed shown already by TLC monitoring. A comparison of the results with respect to the enantiomeric excess of (+)-11 and (+)-13 showed that the use of β-amino acid derivative (+)-11, bearing the ((R)-1-(naphthalen-2-yl)ethyl)carbamoyl substituted diendo norbornene scaffold as catalyst, gave a remarkably better result, than ((R)-1-(naphthalen-1-yl)ethyl)carbamoyl-substituted compound (+)-13 (Table 1 entry 6 and 8). One plausible explanation is that when the catalysts contain protecting groups, the transition state of the aldol reaction between isatin and acetone prefers the substituent (R)-(+)-1-(2-naphthyl)ethyl rather than the substituent of (R)-(+)-1-(1-naphthyl)ethyl. However, when the protective groups from (+)-11 and (+)-13 were removed, (+)-12 and (+)-14 produced HCl salts that performed better than their protected analogue counterparts (Table 1, entries 6 and 8 versus entries 7 and 9). When the Fmoc-protected diexo norbornene β-amino amides (+)-18 and (−)-19 were used instead of Boc-protected diendo norbornene β-amino amides (−)-7 and (+)-11, large drops were registered in enantiomeric excesses (in Table 1, compare entries 2 and 6 with entries 11 and 12). In the case of Fmoc as the protecting group, ee values in the aldol reaction between isatin and acetone dropped significantly. Supposedly, it is related to a structural reason, namely the significant steric demand of Fmoc in comparison to Boc. Obviously, this could affect the corresponding transition state.
When our catalysts were tested without LiOH as co-catalyst, only the hydrochloride salts (+)-12 (derived from (R)-(+)-1-(2-naphthyl)ethylamine) and (+)-14 (derived from (R)-(+)-1-(1-naphthyl)ethylamine) afforded good results (Table 2, entries 6 and 7). Furthermore, when only catalysts without LiOH were applied in the aldol reaction, treatment times had to be increased significantly.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb | ee%c (S) config.d |
| a Reactions were carried out (20 mol% catalyst loading) with isatin derivatives (0.28 mmol) and acetone (5 mL, 325 equiv.).b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44 | ||||
| 1 | (−)-6 | 72 | 89 | 7 |
| 2 | (−)-7 | 96 | 70 | 1 |
| 3 | (−)-8 | 90 | 75 | 3 |
| 4 | (−)-9 | 72 | 80 | Rac. |
| 5 | (+)-11 | 72 | 85 | 3 |
| 6 | (+)-12 | 48 | 90 | 49 |
| 7 | (+)-14 | 48 | 95 | 57 |
| 8 | (−)-15 | 48 | 91 | 3 |
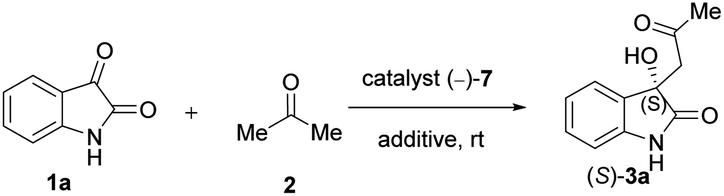
Based on the results summarised in Tables 1 and 2, our best catalyst, compound (−)-7 was chosen for further tests. In the next steps the model reaction was optimised to increase the enantiomeric excess. Thus, the possible effects of additives, catalyst loading, solvent and temperature on the model reaction were studied.
First, various additives were examined in order to enhance the enantiomeric excess (ee%) (Table 3). We found that the LiOH originally applied is the best additive: in its presence, the reaction is both fast and moderately enantioselective (Table 3, entry 7). Other bases also accelerated the reaction, but failed to provide reasonable ee values (Table 3, entries 5 and 6). When LiOH was replaced with acids, the most commonly applied additives in this reaction according to the literature, the reaction became sluggish and barely enantioselective (Table 7, entries 1–3). Water as an additive failed as well (Table 7, entry 4). Therefore, we decided to use a very small amount of LiOH (1 mmol) together with our catalysts during further reactions.
|
||||
|---|---|---|---|---|
| Entrya | Additive | Time (h) | Yieldb | eec (%) (S) config.d |
| a Reactions were carried out with (−)-7 (20 mol%), isatin (0.28 mmol), acetone (5 mL, 325 equiv.) and additive (1 mmol) stirred at room temperature for specified time.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44 | ||||
| 1 | TFA | 12 | 75 | 15 |
| 2 | HCOOH | 12 | 70 | 13 |
| 3 | Benzoic acid | 12 | 80 | Rac. |
| 4 | H2O | 12 | 84 | 3 |
| 5 | NaOH | 4 | 72 | 9 |
| 6 | KOH | 4 | 83 | 13 |
| 7 | LiOH | 0.5 | 94 | 43 |
Subsequently, by changing the catalyst loading from 5 mol% to 20 mol%, the impact of various catalyst loadings of (−)-7 was examined on the model reaction (Table 4). As shown in entry 1 of Table 4, the use of 5 mol% catalyst loading applied under the most environmentally benign condition, turned out to be the most enantioselective. Note, that in this case, a short treatment was satisfactory. When the catalyst loading was increased, a sharp reduction in ee values was observed followed by a rapid surge. Possible background processes, like the retro-aldol reaction, may explain this phenomenon.14
|
||||
|---|---|---|---|---|
| Entrya | Catalyst mol% | Time (min) | Yieldb | eec (%) (S) config.d |
| a Organocatalyst (−)-7 in specified mol%, isatin (0.28 mmol) and acetone (5 mL, 325 equiv.) stirred at room temperature for specified times in the presence of LiOH (1 mmol).b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44 | ||||
| 1 | 5 | 30 | 96 | 57 |
| 2 | 10 | 60 | 89 | 13 |
| 3 | 15 | 60 | 90 | 11 |
| 4 | 20 | 30 | 94 | 43 |
Then, the partially optimised model reaction (5 mol% (−)-7 catalyst, 1 mmol LiOH additive, room temperature) was performed in various bicomponent solvents (prepared by mixing 1–163 equivalents of acetone with a polar protic or a polar aprotic solvent) in order to study the possible solvent effects. In the case of ACN/acetone mixtures, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture produced an almost racemic product, but decreasing the amount of acetone somewhat improved ee% (Table 5, entries 17–20). In all other cases, the 1:1 mixture provided the highest enantioselectivity, whereas decreasing the amount of acetone greatly decreased ee% (Table 5, entries 1–16). This was especially true for acetone/2-methyl-2-butanol (2M2B) and acetone/THF mixtures, whereas the presence of MeOH and EtOH was better tolerated. However, because all the above mixtures provided inferior yield and ee% compared to pure acetone (Table 4, entry 1), acetone was utilised as a solvent in further studies.
:1 mixture produced an almost racemic product, but decreasing the amount of acetone somewhat improved ee% (Table 5, entries 17–20). In all other cases, the 1:1 mixture provided the highest enantioselectivity, whereas decreasing the amount of acetone greatly decreased ee% (Table 5, entries 1–16). This was especially true for acetone/2-methyl-2-butanol (2M2B) and acetone/THF mixtures, whereas the presence of MeOH and EtOH was better tolerated. However, because all the above mixtures provided inferior yield and ee% compared to pure acetone (Table 4, entry 1), acetone was utilised as a solvent in further studies.
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Acetone equiv. | Time (h) | Yieldb | eec (%) (S) config.d |
| a Reactions were carried out with (−)-7 (5 mol%), isatin (0.28 mmol), acetone (amount given in the table) and solvent (overall volume: 5 mL), in the presence of LiOH (1 mmol), stirred at room temperature for the specified time.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44e 2-Methyl-2-butanol. | |||||
| 1 | MeOH | 163 | 12 | 56 | 39 |
| 2 | MeOH | 65 | 12 | 50 | 7 |
| 3 | MeOH | 6.5 | 12 | 67 | 1 |
| 4 | MeOH | 1 | 12 | 48 | 7 |
| 5 | EtOH | 163 | 12 | 73 | 28 |
| 6 | EtOH | 65 | 12 | 75 | 25 |
| 7 | EtOH | 6.5 | 12 | 50 | 1 |
| 8 | EtOH | 1 | 12 | 69 | 3 |
| 9 | 2M2Be | 163 | 12 | 60 | 7 |
| 10 | 2M2Be | 65 | 12 | 70 | 7 |
| 11 | 2M2Be | 6.5 | 12 | 55 | 1 |
| 12 | 2M2Be | 1 | 12 | 46 | 1 |
| 13 | THF | 163 | 12 | 78 | 15 |
| 14 | THF | 65 | 12 | 81 | 1 |
| 15 | THF | 6.5 | 12 | 86 | 5 |
| 16 | THF | 1 | 12 | 74 | 1 |
| 17 | ACN | 163 | 12 | 83 | 3 |
| 18 | ACN | 65 | 12 | 86 | 1 |
| 19 | ACN | 6.5 | 12 | 80 | 9 |
| 20 | ACN | 1 | 12 | 89 | 11 |
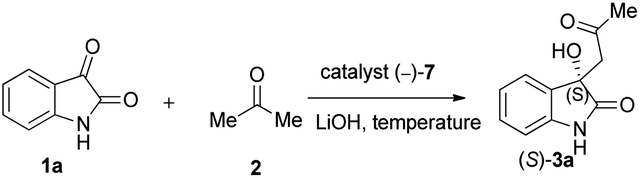
Finally, the effect of temperature was investigated ranging from −20 °C to 75 °C, under the already optimised conditions (Table 6). As shown, both too high and too low temperatures gave unsatisfactory results (Table 6, entries 1, 2, 4 and 5). On the other hand, when the reaction was carried out at room temperature, it worked really effectively (Table 6, entry 3). Notably, performing reactions at room temperature is quite convenient from a practical viewpoint.
|
||||
|---|---|---|---|---|
| Entrya | Temperatureb (°C) | Time (h) | Yieldc | eed (%) (S) config.e |
| a Isatin (0.28 mmol), organocatalyst (−)-7 (5 mol%) and LiOH (1 mmol) were dissolved in acetone (5 mL, 325 equiv.) and stirred at the specified temperature for the specified time.b Microwave reactor: 50 °C, 75 °C.c Isolated yield after purification by column chromatography.d The ee was determined by HPLC using Chiralpak IA column.e The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,29,43,44 | ||||
| 1 | −20 | 48 | 89 | 3 |
| 2 | 4 | 48 | 90 | 1 |
| 3 | 25 | 0.5 | 96 | 57 |
| 4 | 50b | 0.5 | 92 | Rac. |
| 5 | 75b | 0.5 | 87 | Rac. |
We were interested in exploring the issue of substrates after adjusting the reaction conditions; therefore, various substituted isatin derivatives were examined in model reactions. The purpose of the study was to determine the effect of substitution of the aromatic ring on isatin. Hence, 5-iodoisatin (1b), 7-chloroisatin (1c), 5-fluoroisatin (1d), 5-nitroisatin (1e), 5-methylisatin (1f), 4,7-dichloroisatin (1g) and 5-bromoisatin (1h) were examined. As described in Table 7, catalysts (−)-7 (entries 2, 6, 10, 13, 16, 19 and 22), (−)-8 (entries 3, 7, 11, 14, 17, 20 and 23), (+)-14 (entries 4 and 8) and (−)-21 (entries 1, 5, 9, 12, 15, 18, 21 and 24) were studied under the optimised reaction conditions (acetone as solvent, 5–20 mol% catalyst, room temperature, LiOH additive (1 mmol) in the cases of catalysts (−)-7 and (−)-8, no additives in the cases of catalysts (+)-14 and (−)-21).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Catalyst | R1 | R2 | Product | Time (h) | Yieldb | eec (%) (S) config.d |
| a The isatin derivative (0.28 mmol) and the catalyst system [5 mol% (−)-7 and 1 mmol LiOH; 5 mol% (−)-8 and 1 mmol LiOH; 20 mol% (+)-14; or 20 mol% (−)-21] were dissolved in acetone (5 mL, 325 equiv.), and stirred at room temperature for the specified time.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.10,18,43,45,46 | |||||||
| 1 | (−)-21 | H | H | 3a | 48 | 92 | 53 |
| 2 | (−)-7 | 5I | H | 3b | 72 | 67 | 27 |
| 3 | (−)-8 | 5I | H | 3b | 72 | 82 | 35 |
| 4 | (+)-14 | 5I | H | 3b | 50 | 84 | 45 |
| 5 | (−)-21 | 5I | H | 3b | 48 | 78 | 65 |
| 6 | (−)-7 | H | Cl | 3c | 72 | 75 | 81 |
| 7 | (−)-8 | H | Cl | 3c | 72 | 90 | 93 |
| 8 | (+)-14 | H | Cl | 3c | 70 | 93 | 39 |
| 9 | (−)-21 | H | Cl | 3c | 48 | 81 | 98 |
| 10 | (−)-7 | 5F | H | 3d | 72 | 63 | 30 |
| 11 | (−)-8 | 5F | H | 3d | 72 | 67 | 47 |
| 12 | (−)-21 | 5F | H | 3d | 72 | 72 | 37 |
| 13 | (−)-7 | 5NO2 | H | 3e | 72 | 76 | 45 |
| 14 | (−)-8 | 5NO2 | H | 3e | 72 | 80 | 9 |
| 15 | (−)-21 | 5NO2 | H | 3e | 72 | 73 | 41 |
| 16 | (−)-7 | 5Me | H | 3f | 72 | 74 | 5 |
| 17 | (−)-8 | 5Me | H | 3f | 72 | 69 | 15 |
| 18 | (−)-21 | 5Me | H | 3f | 72 | 65 | 39 |
| 19 | (−)-7 | 4Cl | Cl | 3g | 72 | 70 | 99 |
| 20 | (−)-8 | 4Cl | Cl | 3g | 72 | 77 | 83 |
| 21 | (−)-21 | 4Cl | Cl | 3g | 72 | 82 | 97 |
| 22 | (−)-7 | 5Br | H | 3h | 72 | 64 | 79 |
| 23 | (−)-8 | 5Br | H | 3h | 72 | 68 | 75 |
| 24 | (−)-21 | 5Br | H | 3h | 72 | 70 | 49 |
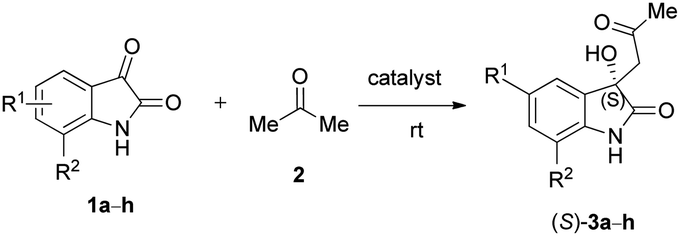
First, results with our norbornene catalyst derivatives are discussed. Aldol reaction of 5-iodoisatin (1b) and acetone yielded moderate to good (27–45%) ees (Table 7, entries 2–4). When 7-chloroisatin (1c) and acetone were applied as substrates, catalyst (−)-7 gave the (S) product with 81% enantiomeric excess (Table 7, entry 6). Then we studied catalyst (−)-8 (ref. 41) with 7-chloroisatin and the enantiomeric excess reached an excellent value of 93% of the (S) product (Table 7, entry 7). Experiments with 5-fluoroisatin (1d), 5-nitroisatin (1e) and 5-methylisatin (1f) gave moderate results regarding ee% (Table 7, entries 10–11, 13–14 and 16–17). In contrast, 4,7-dichloroisatin (1g) provided the (S) product with excellent enantiomeric excesses of 99% and 83% (Table 7, entries 19 and 20). Testing 5-bromoisatin (1h) gave good results (Table 7, entries 22–23), especially with catalyst (−)-7 (Table 7, entry 22, 79% enantiomeric excess). In general, organocatalysts have been found to give better results in ee% values with chlorinated and brominated isatins compared to isatin itself. Guo et al. made similar observations when a chiral diamine catalyst was investigated in the asymmetric cross aldol reaction of isatin and acetone.17
Different substituted isatins were also screened with acetone and catalyst (−)-21 (Table 7, entries 1, 5, 9, 12, 15, 18, 21 and 24).10 Note that this catalyst was already known in the literature, but it was not examined with these substituted isatin derivatives. In each case, in particular, when 7-chloroisatin (1c) and 4,7-dichloroisatin (1g) were used, very good enantiomeric excesses were observed (Table 7, entries 9 and 21, 98% and 97% ees). These results are in agreement with the behaviour of our catalysts (−)-7 and (−)-8 derived from β-amino acids as well as the experiences of Guo et al. To sum up, moderately electron-withdrawing chlorine or bromine substituents at the C-4, C-5 and/or C-7 positions of the isatin ring are beneficial to the enantiomeric excess in the case of a wide variety of catalysts.17
One of our goals was to study the model reaction using cyclohexanone as well. However, these reactions could not be successfully monitored because, in our measurement setup, the syn and anti isomers and enantiomers of the products could not be separated on the chiral IA column.
The norbornene skeleton is a rigid and sterically bulky structure, which has a significant impact on the outcome of reactions.
In order to demonstrate the necessity of norbornene skeleton of our organocatalysts, its potential impacts were investigated. Therefore, a few β-amino acid derivatives containing other skeletons [(+)-22, (+)-23, (+)-24 and (+)-25] were synthesised (Fig. 2). These compounds have the same chiral side chain moieties, which were effective with our best catalysts. In the model reaction, they all gave racemic aldol products. On the basis of these results, we propose that the norbornene skeleton has a significant effect on the organocatalytic activity.
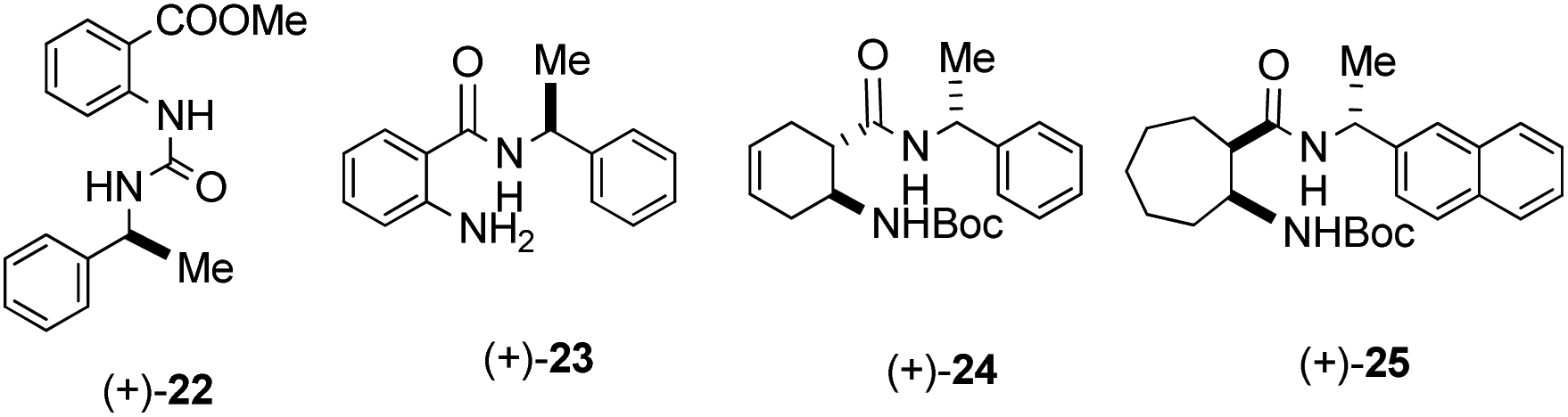 | ||
| Fig. 2 Structure of catalysts 22–25. | ||
In order to interpret our results correctly, it is important to notice that our catalysts, based on their functional groups, can be divided into three distinct groups.
The first group, β-carbamido esters, has only two representatives [(−)-6 and (−)-8]. In both cases, the presence of LiOH greatly accelerated reactions and somewhat increased enantioselectivity, but with isatin, ee of the product never increased above 15% (see Table 1, entries 1 and 3 and Table 2, entries 1 and 3). Therefore, detailed discussion of these catalysts is omitted.
The second group, β-amino amide hydrochloride salts, has 2 representatives – (+)-12 and (+)-14. Their enantioselectivity is moderate to good (49–57%) and it is only weakly affected by the presence or absence of LiOH (see Table 1, entries 7 and 9 and Table 2, entries 6 and 7). Interestingly, the side chain chiral centre also has low impact on their enantioselectivity. Reactions utilizing these catalysts are greatly accelerated by LiOH. We propose that these catalysts follow a mechanism that is very similar to that described by Gavendova et al.11 First, condensation of acetone with the free amine form of the catalyst yields an imine. Then, LiOH deprotonates this imine to form a rather nucleophilic aza-enolate. Reprotonation of this aza-enolate produces a nucleophilic enamine. The side chain aryl group of these active species interacts with isatin via π–π stacking, orienting isatin in such a way that Re-face attack of the aza-enolate or enamine is preferred over Si-face attack (Scheme 5). This mechanism explains that enantioselectivity is an inherent property of the catalyst (Li+ is not involved in orientation of the reactants). It also accounts for the effect of LiOH on the rate of the reaction. First, OH− accelerates tautomerisation of imines to enamines. Second, the aza-enolate intermediates of this base-promoted tautomerisation are more nucleophilic and more reactive than enamines. Finally, LiOH deprotonates the hydrochloride derivative of the catalyst, transforming it into the catalytically active free amine form.
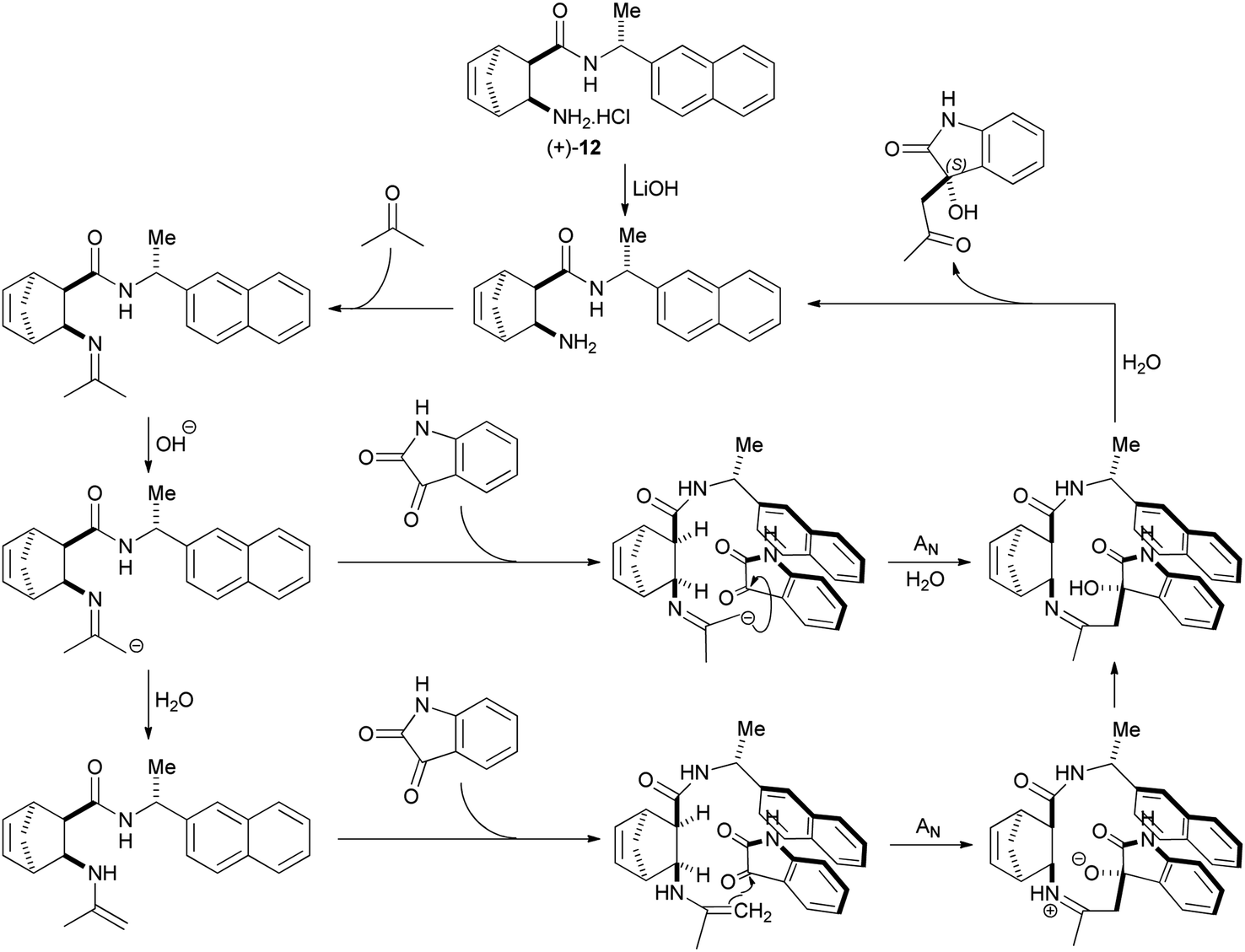 | ||
| Scheme 5 Mechanism of asymmetric cross aldol reaction with β-amino amide hydrochloride salt catalyst (+)-12. | ||
The third group, N-Boc-protected β-amino amides, was the focus of our investigation. As a consequence, this group has the highest number of representatives (9 compounds). In this group, the effect of LiOH was investigated only in the case of four-membered derivatives.
It was found that LiOH accelerates aldol reactions catalysed by (−)-7, (−)-9, (+)-11 and (−)-15, and it also significantly increases enantioselectivity of catalysts (−)-7 and (+)-11 (compare Table 1, entries 2, 4, 6 and 10 with Table 2, entries 2, 4, 5 and 8). In the case of catalyst (−)-7, other bases were investigated as well, but they mostly increased reaction rates (see Table 3, entries 4–7).
Because nucleophilicity of the Boc-protected nitrogen is greatly reduced, enamine formation between N-Boc-protected β-amino amide catalysts and acetone is unlikely. We propose that these catalysts utilise a different mechanism. First, the reaction of LiOH with acetone generates a lithium enolate, which exists as a close ion pair in the reaction media of moderate polarity. It is known that Li+ has much stronger coordinative ability than other alkali metal ions (because the small size of Li+ results in high surface charge density), and Li+ preferentially interacts with hard Lewis bases (e.g. oxygen-donor ligands). Consequently, the Li+ counterion of the enolate can interact with the hard carbonyl oxygen donor atoms of the catalyst, too. Moreover, the catalyst also interacts with isatin via π–π stacking. As a result of these interactions, the enolate and isatin are oriented in such a way that the Re-face attack is favoured (Scheme 6).
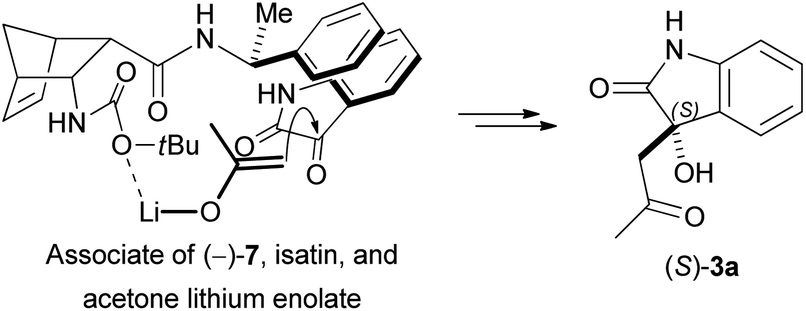 | ||
| Scheme 6 The key step of the mechanism of asymmetric cross aldol reaction with catalyst (−)-7. | ||
This mechanism is in accordance with our observations. In the absence of OH−, the nucleophile would be the enol form of acetone, which is less reactive than the enolate of acetone. Therefore, OH− is responsible for accelerating the reaction. Note that any base with sufficient solubility can perform this role (see Table 3, entries 4–7). In contrast, enantioselectivity requires specifically Li+, because its unique coordinative ability is necessary for orientation of the enolate.
As part of our work, mechanochemical investigations were undertaken to make the model reaction more sustainable and environmentally benign. We studied the model reaction under HSBM conditions, testing catalyst loading, milling frequency and reaction time. All high-speed ball milling measurements were initially performed using a Retsch 400 Mixer Mill equipped with a 10 mL agate jar and 5 mm agate balls. In order to prevent temperature rise of the mill, we applied pauses during the milling process. However, when using higher frequencies, this approach turned out to be futile. Neither catalyst loading higher than 5 mol% nor using too low and too high frequencies yielded successful results. At too low frequency, the reaction time was very long. In contrast, when using a too high frequency, the product was racemic. The most effective milling frequencies, which produced satisfactory results, were found to be 15 and 20 Hz (Table 8). It may be surmised that increasing the milling frequency causes an increase of the temperature, which is known to decrease enantioselectivity in solution-phase experiments (see Table 6, entries 3–5).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Frequency (Hz) | Time (min) | Yieldb | ee%c (S) config.d |
| a A mixture of isatin (0.28 mmol), LiOH (1 mmol), organocatalyst (5 mol%) and acetone (30 μL, 1.95 equiv.) was milled with 3 balls at the specified frequency for the specified time.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,43–45 | |||||
| 1 | (−)-8 | 15 | 30 | 50 | N.D. |
| 2 | (−)-8 | 15 | 60 | 56 | 31 |
| 3 | (−)-8 | 15 | 120 | 48 | 33 |
| 4 | (−)-8 | 20 | 30 | 61 | 19 |
| 5 | (−)-8 | 20 | 60 | 59 | 35 |
| 6 | (−)-8 | 20 | 120 | 43 | 21 |
| 7 | (−)-21 | 15 | 30 | 67 | 57 |
| 8 | (−)-21 | 15 | 60 | 65 | 39 |
| 9 | (−)-21 | 15 | 120 | 61 | 23 |
| 10 | (−)-21 | 20 | 30 | 70 | 47 |
| 11 | (−)-21 | 20 | 60 | 69 | 37 |
| 12 | (−)-21 | 20 | 120 | 61 | 31 |
We also screened the possible effect of the number of milling balls. The results demonstrated that in the case of urea derivative (−)-8, applying 4 balls gave the best results (Table 9, entries 1–3), while in the case of catalyst (−)-21, increasing the number of balls to 4 or 5 provided inferior results in ee% (Table 9, entries 4–6).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Frequency (Hz) | Number of milling balls | Yieldb | ee%c (S) config.d |
| a A mixture of isatin (0.28 mmol), LiOH (1 mmol), organocatalyst (5 mol%) and acetone (30 μL, 1.95 equiv.) was milled with the specified number of balls for the already optimised time. In the case of catalyst (−)-8, milling time was 60 min, while regarding catalyst (−)-21 the milling time was 30 min.b Isolated yield after purification by column chromatography.c The ee was determined by HPLC using Chiralpak IA column.d The absolute configuration was determined by comparison of the specific optical rotation with literature data.2,43–45 | |||||
| 1 | (−)-8 | 20 | 3 | 68 | 35 |
| 2 | (−)-8 | 20 | 4 | 70 | 96 |
| 3 | (−)-8 | 20 | 5 | 61 | 5 |
| 4 | (−)-21 | 15 | 3 | 69 | 57 |
| 5 | (−)-21 | 15 | 4 | 73 | 45 |
| 6 | (−)-21 | 15 | 5 | 64 | 33 |
Conclusions
In conclusion, organocatalysts 6–25 were synthesised, characterised and tested in the aldol reaction between isatin and acetone as a model reaction. The application of these organocatalysts significantly shortened the reaction time. By employing alicyclic amino acid derivatives as organocatalysts, it was successfully established that a norbornene skeleton, a substituted aromatic chiral amine and an amino group attached to the norbornene backbone are required to provide good control of enantioselectivity. It was observed that reactions took place more rapidly when LiOH was present. In the cases of a few catalysts, the presence of LiOH was also beneficial to enantioselectivity.Our best working organocatalyst (−)-7 was used to optimise the model reaction. LiOH proved to be the most efficient additive. 5 mol% catalyst loading provided better results than higher catalyst loadings. Possible solvent effects were studied by performing the reaction in acetone/cosolvent mixtures. Numerous polar cosolvents were used, and the acetone/cosolvent ratio was varied within a wide range. With our catalyst (−)-7, pure acetone as solvent provided the best results. Room temperature, the most environmentally benign condition, proved to be the best choice concerning the use of our catalyst. In the end, under the fully optimised conditions (5 mol% catalyst loading, 3.57 equiv. LiOH additive, acetone as both the solvent and the substrate, room temperature), the aldol adduct formed in 96% yield and 57% ee (S) in a mere 30 minutes.
We suggest that β-amino amide catalysts, which have a free primary amino group, promote the investigated cross aldol reaction via enamine catalysis. In these cases, the main role of LiOH is to accelerate the reaction. In contrast, N-protected β-amino amides follow a different mechanism, which involves coordination of both the catalyst and the enolate of acetone to the same Li+ ion. In these cases, LiOH has a double role: it accelerates the reaction and provides enantioselectivity.
Substituent effects were studied by transforming various substituted isatins. When 7-chloroisatin were used as substrate, catalyst (−)-7 gave the (S) product with high ee (81%). With urea derivative (−)-8 in the case of 7-chloroisatin, enantioselectivity reached an outstanding value of 93% providing the (S) product. Reaction of 4,7-dichloroisatin in the presence of catalysts (−)-7 and (−)-8 provided the (S) product in excellent ees (99% and 83%, respectively). The analogous reaction of 5-bromoisatin in the presence of catalysts (−)-7 and (−)-8 provided the (S) product in good ees (79% and 75%, respectively). When there is an electron-withdrawing group in the C-4, C-5 or C-7 position, organocatalysts, in general, provided better ee values. Oxindole derivatives 3a–h were isolated and characterized.
HSBM was employed to perform the model reaction in a more environmentally and ecologically benign way. Most importantly, solvent usage was greatly decreased. We thoroughly examined the effects of catalyst loading, milling frequency and reaction time. The transformation of isatin in the presence of catalyst (−)-8 was improved in HSBM conditions up to an ee of 96%. Catalyst (−)-21 also showed superior performance under HSBM conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors' thanks are due to the Hungarian Research Foundation (OTKA No. K-138871) and the Ministry of Human Capacities, Hungary grant, TKP-2021-EGA-32. T. F: was supported by the ÚNKP-22-3-SZTE-150 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund. The high-resolution mass spectrometric (HRMS) analysis was performed by Robert Berkecz. Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.Notes and references
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20–38 CrossRef CAS.
- M. Kinsella, P. G. Duggan and C. M. Lennon, Tetrahedron: Asymmetry, 2011, 22, 1423–1433 CrossRef CAS.
- S. Nakamura, N. Hara, H. Nakashima, K. Kubo, N. Shibata and T. Toru, Chem.–Eur. J., 2008, 14, 8079–8081 CrossRef CAS PubMed.
- T. B. S. Giorno, Y. L. L. Ballard, M. S. Cordeiro, B. V. Silva, A. C. Pinto and P. D. Fernandes, Pharmacol., Biochem. Behav., 2015, 135, 13–19 CrossRef CAS PubMed.
- K. Furuta, Y. Mizuno, M. Maeda, H. Koyama and Y. Hirata, Chem. Pharm. Bull., 2017, 65, 1093–1097 CrossRef CAS PubMed.
- P. Sharma, K. R. Senwar, M. K. Jeengar, T. S. Reddy, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Eur. J. Med. Chem., 2015, 104, 11–24 CrossRef CAS PubMed.
- M. E. Abbasov and D. Romo, Nat. Prod. Rep., 2014, 31, 1318–1327 RSC.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- J.-R. Chen, X.-P. Liu, X.-Y. Zhu, L. Li, Y.-F. Qiao, J.-M. Zhang and W.-J. Xiao, Tetrahedron, 2007, 63, 10437–10444 CrossRef CAS.
- G. Chen, Y. Ju, T. Yang, Z. Li, W. Ang, Z. Sang, J. Liu and Y. Luo, Tetrahedron: Asymmetry, 2015, 26, 943–947 CrossRef CAS.
- M. Gavendova, C. M. Lennon, L. Coffey, P. Manesiotis and M. Kinsella, ChemistrySelect, 2019, 4, 8246–8254 CrossRef CAS.
- Y. Zou, C.-Y. Li, M. Xiang, W.-S. Li, J. Zhang, W.-J. Wan and L.-X. Wang, Tetrahedron Lett., 2022, 97, 153780 CrossRef CAS.
- A. M. Al-Majid, A. S. Alammari, S. Alshahrani, M. Haukka, M. S. Islam and A. Barakat, RSC Adv., 2022, 12, 6149–6165 RSC.
- J. Wang, Z.-X. Deng, C.-M. Wang, P.-J. Xia, J.-A. Xiao, H.-Y. Xiang, X.-Q. Chen and H. Yang, Org. Lett., 2018, 20, 7535–7538 CrossRef CAS PubMed.
- H. Liao, C. Shen, F. Shen, P. Zhang and W. Su, Curr. Organocatal., 2015, 2, 71–76 CrossRef CAS.
- A. Kumar and S. S. Chimni, Tetrahedron, 2013, 69, 5197–5204 CrossRef CAS.
- J.-T. Guo, B.-Q. Zhang, Y. Luo, Z. Guan and Y.-H. He, Asian J. Org. Chem., 2017, 6, 605–608 CrossRef CAS.
- H. Lu, J. Bai, J. Xu, T. Yang, X. Lin, J. Li and F. Ren, Tetrahedron, 2015, 71, 2610–2615 CrossRef CAS.
- C. Cruz-Hernández, P. Hernández-González and E. Juaristi, Synthesis, 2018, 50, 3445–3459 CrossRef.
- C. Cruz-Hernández, P. Hernández-González and E. Juaristi, Synthesis, 2018, 50, 1827–1840 CrossRef.
- Y. Liu, P. Gao, J. Wang, Q. Sun, Z. Ge and R. Li, Synlett, 2012, 23, 1031–1034 CrossRef CAS.
- J. Wang, Q. Liu, Q. Hao, Y. Sun, Y. Luo and H. Yang, Chirality, 2015, 27, 314–319 CrossRef CAS PubMed.
- M. Raj, N. Veerasamy and V. K. Singh, Tetrahedron Lett., 2010, 51, 2157–2159 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- Y. Zheng and M. A. Avery, Tetrahedron, 2004, 60, 2091–2095 CrossRef CAS.
- G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418–7421 CrossRef CAS PubMed.
- G. D. Yadav and S. Singh, Tetrahedron: Asymmetry, 2016, 27, 463–466 CrossRef CAS.
- A. Ogasawara, U. V. Subba Reddy, C. Seki, Y. Okuyama, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Tetrahedron: Asymmetry, 2016, 27, 1062–1068 CrossRef CAS.
- A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Org. Lett., 2007, 9, 5473–5476 CrossRef CAS PubMed.
- B. Rodríguez, T. Rantanen and C. Bolm, Angew. Chem., 2006, 118, 7078–7080 CrossRef.
- I. N. Egorov, S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, B. C. Ranu, V. L. Rusinov and O. N. Chupakhin, RSC Green Chem. Ser., 2020, 22, 302–315 RSC.
- J. G. Hernández and E. Juaristi, J. Org. Chem., 2011, 76, 1464–1467 CrossRef PubMed.
- E. Machuca, Y. Rojas and E. Juaristi, Asian J. Org. Chem., 2015, 4, 46–53 CrossRef CAS.
- B. Rodríguez, A. Bruckmann and C. Bolm, Chem.–Eur. J., 2007, 13, 4710–4722 CrossRef PubMed.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- B. Rodríguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213–2233 CrossRef.
- J. G. Hernández, V. García-López and E. Juaristi, Tetrahedron, 2012, 68, 92–97 CrossRef.
- Y. Cheng, Z.-T. Huang and M.-X. Wang, Curr. Org. Chem., 2004, 8, 325–351 CrossRef CAS.
- M. Palkó, M. El Haimer, Z. Kormányos and F. Fülöp, Molecules, 2019, 24, 772 CrossRef PubMed.
- B. Fekete, M. Palkó, I. Mándity, M. Haukka and F. Fülöp, Eur. J. Org. Chem., 2016, 3519–3527 CrossRef CAS.
- I. Nekkaa, D. Bogdán, T. Gáti, S. Béni, T. Juhász, M. Palkó, G. Paragi, G. K. Tóth, F. Fülöp and I. M. Mándity, Chem. Commun., 2019, 55, 3061–3064 RSC.
- C. Shen, F. Shen, H. Xia, P. Zhang and X. Chen, Tetrahedron: Asymmetry, 2011, 22, 708–712 CrossRef CAS.
- S. Allu, N. Molleti, R. Panem and V. K. Singh, Tetrahedron Lett., 2011, 52, 4080–4083 CrossRef CAS.
- Y. Lu, Y. Ma, S. Yang, M. Ma, H. Chu and C. Song, Tetrahedron: Asymmetry, 2013, 24, 1082–1088 CrossRef CAS.
- K. Tseke, C. Lennon, J. O'Mahony and M. Kinsella, Eur. J. Org. Chem., 2021, 5767–5774 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03528j |
| This journal is © The Royal Society of Chemistry 2023 |