
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of composition changes on the structure and electronic properties of jamesonite: a DFT study
Xi Zhoua,
Cuihua Zhao *ab,
Jianhua Chen*ab and
Yao Fenga
*ab,
Jianhua Chen*ab and
Yao Fenga
aSchool of Resources, Environment and Materials, Guangxi University, Nanning 530004, China. E-mail: chzhao@gxu.edu.cn; jhchen20056@sina.com
bState Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, Guangxi University, Nanning 530004, China
First published on 5th September 2023
Abstract
Models of jamesonite with different compositions were built by different amounts of Sb or Pb substitution at Fe sites, and their structures and electronic properties were studied using the DFT method. The structure and properties of jamesonite significantly changed after Sb or Pb substitution. The lengths of the Sb–S and Pb–S bonds are larger than those of the corresponding Fe–S bonds of pure jamesonite, and the polarization of iron atoms adjacent to substitute atoms is weakened. After one Sb atom substitution, the S atoms bonded to Sb (substitution atom) gain more charges than those before Sb substitution. The Sb atom has more positive charges than the corresponding Fe atom before Sb substitution. For one Pb substitution system, the electrons transfer from the substituted Pb to adjacent S atoms, and the larger negative charge of the S atoms causes a slightly stronger Pb–S bond. With increasing Sb or Pb content, the electronic structural changes of the adjacent atoms are similar to those of one Sb or Pb substitution. However, the increase of 4-coordination Sb or 4-coordination Pb with the decrease of Fe atom changes the electronic structure of jamesonite, which will change its flotation performance.
1. Introduction
Owing to the complexity of the mineral formation process and influence of the ore-forming environment, the mineral composition in ore changes accordingly, resulting in differences in mineral flotation behavior.1 For example, sphalerite containing cadmium has good floatability, while sphalerite containing iron has poor flowability.2 Due to the difference in iron content (Fe(1−x)S, 0 < x ≤ 0.125), pyrrhotite forms monoclinic (Fe7S8) and hexagonal (Fe9S10) structures, leading to the difference of their magnetic and flotation behaviors.3–5 Abraitis et al. reported that the type and content of impurities in pyrite can affect its conductivity.6 Lan et al. found that galena containing Ag can improve its flotation recovery, and it is beneficial to the adsorption of collector ions on galena with the increase of silver content.7,8The flotation of a mineral is closely related to its surface chemical properties. Most studies have shown that the crystal structure and the ligand atoms reflect the basic nature of the minerals, and affect the interaction of the reagents with the mineral surface in the flotation process, which may be critical in the mineral flotation.9 Moreover, different mineral compositions often result in different coordination structures. Jamesonite (Pb4FeSb6S14) is a complex lead–zinc resource, which is rich in valuable metals, such as antimony (Sb), lead (Pb), zinc (Zn), bismuth (Bi) and silver (Ag).10 It is also a typical sulfide ore known as a “needle-ore” or “feather-ore” mineral as well, which has lead-gray hair that is up to one-centimeter-long and like crystals combined in bunches filling cavities in quartz veins.11,12 Zhao et al. compared the electronic structures and the flotation ability of jamesonite, stibnite, and galena. The flotation behavior of jamesonite is different from those of stibnite and galena due to its complex coordination structure, which contains not only three-coordinated Sb but also four-coordinated Sb, as well as four-coordinated Fe and six-coordinated Pb.13 They also found that 4-coordination Sb is more active than 3-coordination Sb in jamesonite.
Changes in the crystal composition of sulfide minerals can significantly change their structure and properties, thereby affecting the flotation of sulfide minerals.14 Yang et al. investigated the occurrence and relationship of In and Fe by substituting Zn in sphalerite with Fe and In, respectively, and found that Fe promotes the formation of the In–S bonds of sphalerite, and tends to form solid solution impurities in the sphalerite crystal.15 Chen et al. proposed the substitution of Sb atoms with Ga atoms to form Ga–O bonds in the perovskite oxide BaSb2O6, resulting in lattice oxygen activation.16 The main metal elements in jamesonite are lead and antimony, with a theoretical content of 41.2% for lead and 33.7% for antimony. However, the content of lead and antimony in actual ores is not fixed in jamesonite due to the influence of coexisting minerals, which would affect the flotation properties of jamesonite. As a result, it is necessary to study the effect of changes in composition on the performance of jamesonite.
The properties of minerals depend on their electronic structure. A large number of studies have shown that DFT (Density Functional Theory) is an effective and reliable method to study the electric structure and properties (microcosmic mechanism) of materials.17–19 Li et al. investigated the effect of ten organic depressants on jamesonite by DFT.20 The interaction mechanism between jamesonite and the organic depressant was revealed by the frontier orbital calculation. Zhao et al. studied the flotation properties of jamesonite, stibnite, and galena by DFT calculation, which was in agreement with the experimental results.13
Although many studies have shown that the composition change of minerals would affect their flotation performance, the influence of the composition change of jamesonite on its flotation behavior has not been described in detail, especially the effect of composition changes on the electric structure and properties of jamesonite. In addition, iron is a magnetic substance, and the formation of iron oxyhydroxide on the surface of jamesonite would impact its flotation in an alkaline medium.21 Herein, we used the DFT method to study the structure and electronic property of jamesonite with different relative proportions of Fe, Sb, Pb, and S by Sb or Pb substitution at Fe sites. Our research focused on structural changes, density of states, Mulliken population analysis, and electron density. This study will provide a theoretical basis for the development of new reagents and processes for efficient flotation of jamesonite with different components.
2. Computational methods and models
2.1 Computational methods
All calculations were carried out by the DFT method based on first-principles. Geometry optimization of jamesonite was performed using GGA (Generalized Gradient Approximation).22,23 The k-point of the calculations was 1 × 1 × 4, and the cut-off energy was 350 eV. The convergence tolerances were set to the maximum energy change of 0.05 eV·per atom, the maximum stress of 0.1 GPa, the maximum force of 0.05 eV Å−1 and the maximum displacement of 0.002 Å. Valence electron configurations considered in this study included Fe 3d64s2, Pb 5d106s26p2, Sb 5s25p3 and S 3s23p4 states. In order to choose the calculation parameters suitable for the jamesonite system, three main parameters (correlation function, cut-off energy and k-point) for jamesonite were tested, which are shown in Tables 1, 2 and 3, respectively. The experimental values were also listed in the corresponding tables for comparison.24 By analyzing the calculation data of Tables 1, 2 and 3 (system energies and lattice parameters), GGA-PW91 was selected as the exchange–correlation functional, 350 eV was selected as the plane-wave cut off energy, and 1 × 1 × 4 was selected as the Monkhorst–Pack k-point sampling density. Under these parameters, the system energy is the lowest, and the lattice parameters calculated are a = 15.82 Å, b = 19.28 Å, and c = 4.05 Å, respectively, which are very close to the experimental ones (a = 15.75 Å, b = 19.13 Å, c = 4.03 Å),24 and the error of all parameters is less than 0.5%.| Methods | System energy (104 eV) | Lattice parameters (Å) | β | |||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| Computational functional | GGA-PW91 | −4.91 | 16.01 | 19.47 | 3.97 | 92.10 |
| GGA-PBE | −4.77 | 16.16 | 19.48 | 4.01 | 92.36 | |
| GGA-WC | −4.72 | 15.73 | 18.93 | 3.93 | 92.22 | |
| Experiment24 | — | — | 15.75 | 19.13 | 4.03 | 91.68 |
| Cut-off energy | System energy (104 eV) | Lattice parameters(Å) | β | ||
|---|---|---|---|---|---|
| a | b | c | |||
| 300 | −4.76 | 18.99 | 17.96 | 3.47 | 91.89 |
| 320 | −4.79 | 16.22 | 19.66 | 4.33 | 92.56 |
| 350 | −4.91 | 16.01 | 19.47 | 3.97 | 92.10 |
| 380 | −4.90 | 15.69 | 19.11 | 3.81 | 91.32 |
| 420 | −4.90 | 15.68 | 19.08 | 3.81 | 91.28 |
| Experiment24 | — | 15.75 | 19.13 | 4.03 | 91.68 |
| k-point | System energy (104 eV) | Lattice parameters(Å) | β | ||
|---|---|---|---|---|---|
| a | b | c | |||
| 1 × 1 × 1 | −4.91 | 16.01 | 19.47 | 3.97 | 92.10 |
| 1 × 1 × 4 | −4.94 | 15.82 | 19.28 | 4.05 | 91.79 |
| 2 × 2 × 1 | −4.89 | 15.43 | 18.71 | 3.99 | 91.42 |
| 2 × 2 × 4 | −4.88 | 15.13 | 19.54 | 4.13 | 92.05 |
| 3 × 3 × 1 | −4.88 | 16.27 | 18.61 | 3.89 | 92.11 |
| Experiment24 | 15.75 | 19.13 | 4.03 | 91.68 | |
2.2 Computational models
Jamesonite has a tetrahedral structure with monoclinic symmetry, and its space group is P21/a. The model of bulk jamesonite with 1 × 1 × 2 supercell is shown in Fig. 1, which includes 56 sulfur atoms, 4 iron atoms, 24 antimony atoms, and 16 lead atoms. The mole concentrations of Fe, Sb, Pb and S atoms are 4%, 24%, 16%, and 56% in jamesonite, respectively. It can be seen that the structure of jamesonite is axially symmetric along the diagonal. The four Fe atoms are marked as 1 to 4 (Fig. 1), including two iron atoms at the center (Fe1 and Fe2) and two iron atoms at the top angle (Fe3 and Fe4). The structure of jamesonite is quite complex. In jamesonite, the S atoms have four coordination modes of two, three, four, and five coordination, and the Sb atoms have two coordination modes of three and four coordination. In addition, the Fe and Pb atoms are four coordination and six coordination, respectively. The bond lengths of Fe–S in jamesonite are listed in Table 4, and the coordination and bonding information of all atoms are displayed in Table 5. The jamesonite models with different concentrations of Fe, Sb, and Pb were built by replacing the Fe atom with Sb or Pb.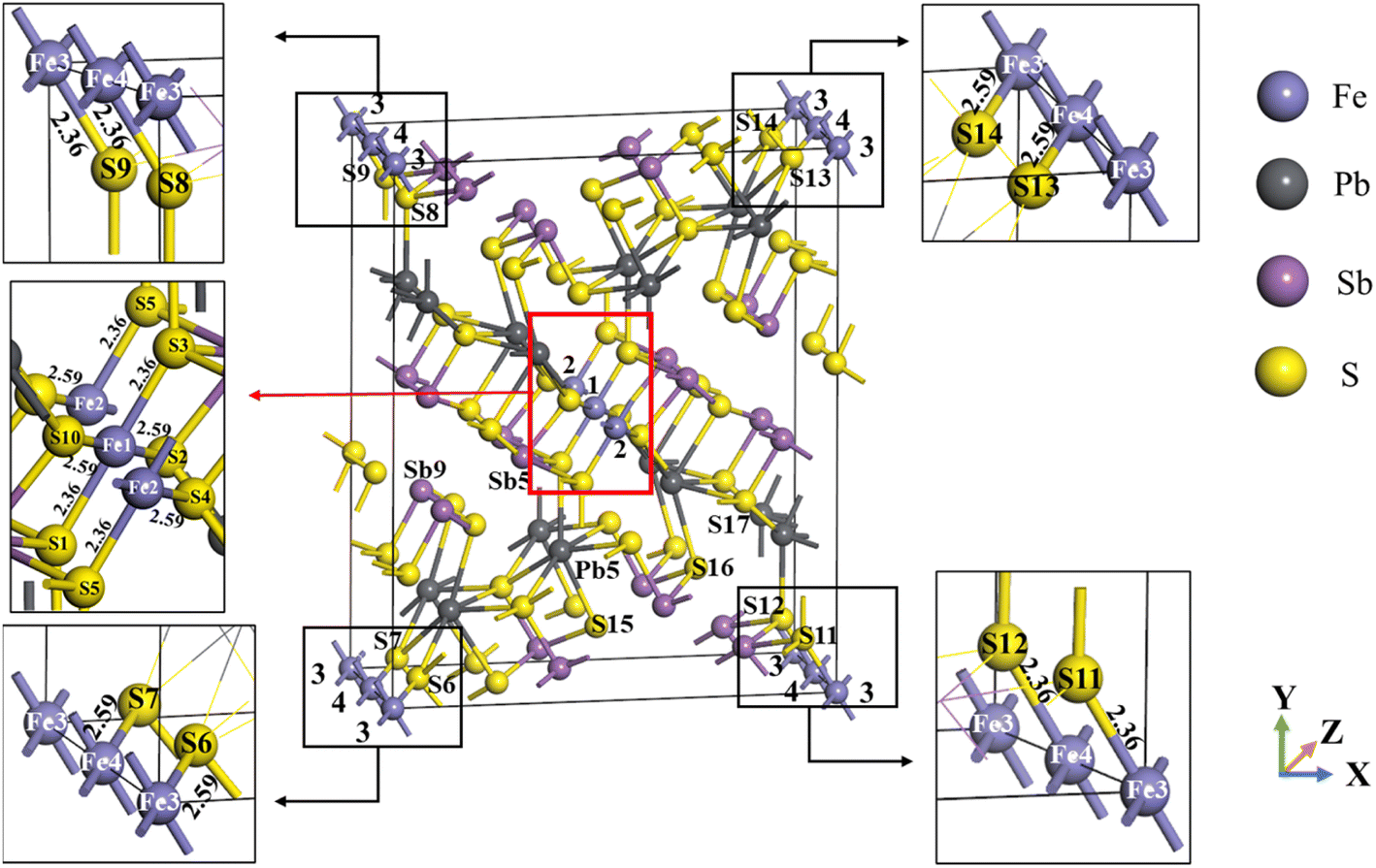 | ||
| Fig. 1 Model of the jamesonite structure. | ||
| Bond name | Bond length (Å) |
|---|---|
| Fe1–S1 (Fe1–S3, Fe2–S5) | 2.36 |
| Fe1–S2 (Fe1–S10, Fe2–S4) | 2.59 |
| Fe3–S9 (Fe4–S8, Fe3–S11, Fe4–S12) | 2.36 |
| Fe3–S6 (Fe4–S7, Fe3–S14, Fe4–S13) | 2.59 |
| Atom species | Coordination number | Coordination atom | Type of bond |
|---|---|---|---|
| S | 2 | 6-Coordinated Pb | Pb–S |
| 3-Coordinated Sb | Sb–S | ||
| 3 | 6-Coordinated Pb | Pb–S | |
| Two 3-coordinated Sb | Sb–S | ||
| 3 | 6-Coordinated Pb | Pb–S | |
| 3-Coordinated Sb | Sb–S | ||
| 4-Coordinated Sb | Sb–S | ||
| 4 | 6-Coordinated Pb | Pb–S | |
| 4-Coordinated Fe | Fe–S | ||
| Two 4-coordinated Sb | Sb–S | ||
| 4 | Two 6-coordinated Pb | Pb–S | |
| 4-Coordinated Fe | Fe–S | ||
| 4-Coordinated Sb | Sb–S | ||
| 5 | Four 6-coordinated Pb | Pb–S | |
| 3-Coordinated Sb | Sb–S | ||
| Sb | 3 | Three 3-coordinated S | Sb–S |
| 3 | 2-Coordinated S | Sb–S | |
| 3-Coordinated S | Sb–S | ||
| 5-Coordinated S | Sb–S | ||
| 4 | 3-Coordinated S | Sb–S | |
| Three 4-coordinated S | Sb–S | ||
| Pb | 6 | 2-Coordinated S | Pb–S |
| Two 3-coordinated S | Pb–S | ||
| 4-Coordinated S | Pb–S | ||
| Two 5-coordinated S | Pb–S | ||
| 6 | Two 3-coordinated S | Pb–S | |
| Two 4-coordinated S | Pb–S | ||
| Two 5-coordinated S | Pb–S | ||
| Fe | 4 | Four 4-coordinated S | Fe–S |
2.3 Calculation of the binding energy
The binding energy with different doping atoms and concentrations were calculated as:| Eb = E(doped jamesonite) − E(pure jamesonite) − μ(A) + μ(B) | (1) |
3. Results and discussions
3.1 Structure of the Sb substitution system
Fig. 2 shows the structural models of jamesonite for Sb substitutions at the Fe sites, in which Fig. 2(a) is the model of one Sb atom substitution (Sb1 substitution at the Fe1 site) with the mole concentrations of Fe, Sb, and Pb atoms in jamesonite of 3%, 25% and 16%, respectively (Fe3Sb25Pb16S56); Fig. 2(b) is the model of two Sb atoms substitutions (Sb1 and Sb2 substitutions at Fe1 and Fe2 sites, respectively) with the mole concentrations of Fe, Sb and Pb of 2%, 26%, and 16%, respectively (Fe2Sb26Pb16S56); Fig. 2(c) is the model of three Sb atoms substitutions (Sb1, Sb2, and Sb3 substitutions at Fe1, Fe2, and Fe3 sites, respectively) with the mole concentrations of Fe, Sb, and Pb of 1%, 27%, and 16%, respectively (Fe1Sb27Pb16S56). The binding energies of the Sb-doped jamesonite with different Sb concentrations are displayed in Table 6. It was observed that the binding energies of all structures for the Sb-doped jamesonite system are negative, indicating that these structures are stable.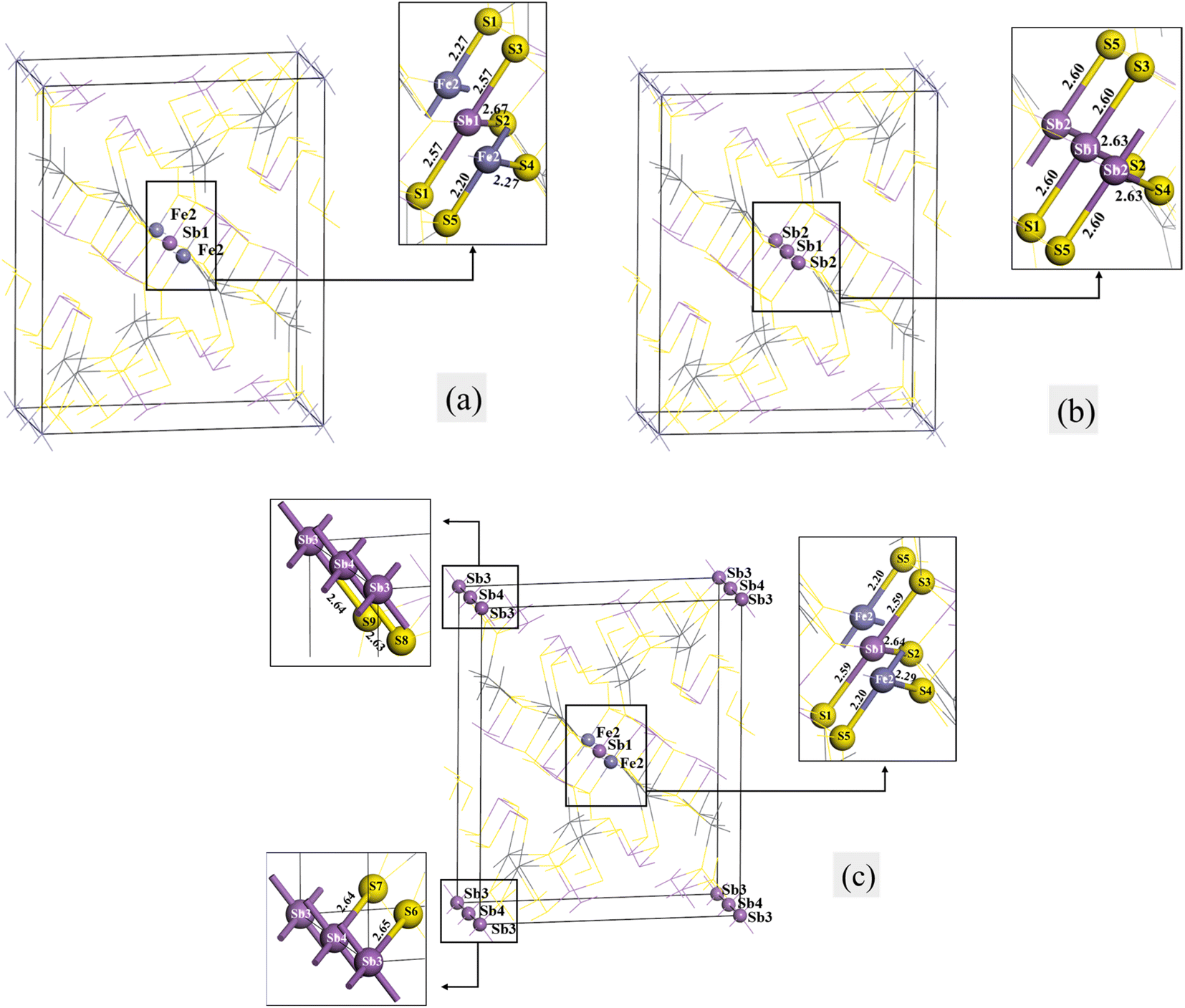 | ||
| Fig. 2 Models of single Sb substitution systems of jamesonite. (a) One Sb atom substitution; (b) two Sb atoms substitution; (c) three Sb atoms substitution. | ||
| Substitution type | Structure | Binding energy (eV) |
|---|---|---|
| Sb | S56Fe3Sb25Pb16 | −2.07 |
| S56Fe2Sb26Pb16 | −0.81 | |
| S56Fe1Sb27Pb16 | −0.24 |
The bond lengths of some bonds in the Sb substitution system of jamesonite are listed in Table 7. There are two types of S–Fe bonds for bulk jamesonite with the bond lengths of 2.18 Å (Fe1–S1 and Fe1–S3) and 2.35 Å (Fe1–S2) (Fig. 1). After Sb1 substitution of Fe1 (Fig. 2(a)), the bond lengths of the Sb1–S1 and Sb1–S2 bonds are 2.57 Å and 2.67 Å, respectively, which are larger than those of the corresponding Fe–S bonds before Sb1 substitution (Fig. 1). Furthermore, the lengths of the Fe2–S5 and Fe2–S4 bonds adjacent to the Sb1–S1 and Sb1–S2 bonds also change slightly compared to those of the corresponding Fe–S bonds. The bond length of Fe2–S5 increases from 2.18 Å to 2.20 Å, while that of Fe2–S4 decreases from 2.35 Å to 2.27 Å. However, the structure of jamesonite is still axially symmetric along the diagonal. After Sb1 and Sb2 replace Fe1 and Fe2, respectively, the bond lengths of the two types (Sb–S bonds) are 2.60 Å (Sb1–S1, Sb1–S3, and Sb2–S5) and 2.63 Å (Sb1–S2 and Sb2–S4), respectively (Fig. 2(b)). It was observed that the bond lengths of Sb1–S1 and Sb1–S3 are slightly larger than those of Sb1–S1 and Sb1–S3 for the one Sb substitution system, while the bond length of Sb1–S2 is slightly smaller than that of Sb1–S2 for the one Sb substitution system (Fig. 2(a)). However, the bond lengths of all Sb–S bonds are larger than those of the corresponding Fe–S bonds for pure jamesonite (Fig. 1). For Fig. 2(c), it can be seen that the length of the Sb1–S1 (2.59 Å) bond is close to that of Sb1–S1 (2.60 Å) of the two Sb substitution system (Fig. 2(b)). However, the bond lengths of S8–Sb4 (2.63 Å) and S9–Sb3 (2.64 Å) are slightly larger than that of the Sb1–S1 bond (Sb4–S8 and Sb3–S9 are the same type of bond with Sb1–S1). In addition, the length of the Pb1–S2 bond (0.64 Å) is close to that of Sb1–S2 (2.63 Å) of the two Sb substitution system (Fig. 2(b)), and the lengths of the Sb4–S7 (2.64 Å) and Sb3–S6 (2.65 Å) bonds are similar to that of the Pb1–S2 bond (Sb4–S7 and Sb3–S6 are the same type of bond with Sb1–S2). However, the structure of jamesonite changed greatly after Sb substitution, making its group space change from P21/a before Sb substitution to P1 after Sb substitution, and the structural change of jamesonite increases with the increase of Sb content.
| Substitution systems | Bond type | Length (Å) |
|---|---|---|
| One Sb atom substitution | Sb1–S1 (Sb1–S3) | 2.57 |
| Sb1–S2 | 2.67 | |
| Fe2–S4 | 2.27 | |
| Fe2–S5 | 2.20 | |
| Two Sb atoms substitution | Sb1–S1 (Sb1–S3) | 2.60 |
| Sb1–S2 | 2.63 | |
| Sb2–S4 | 2.63 | |
| Sb2–S5 | 2.60 | |
| Three Sb atoms substitution | Sb1–S1 (Sb1–S3) | 2.59 |
| Sb1–S2 | 2.64 | |
| Fe2–S4 | 2.29 | |
| Fe2–S5 | 2.20 | |
| Sb3–S6 | 2.65 | |
| Sb4–S7 (Sb3–S9) | 2.64 | |
| Sb4–S8 | 2.63 |
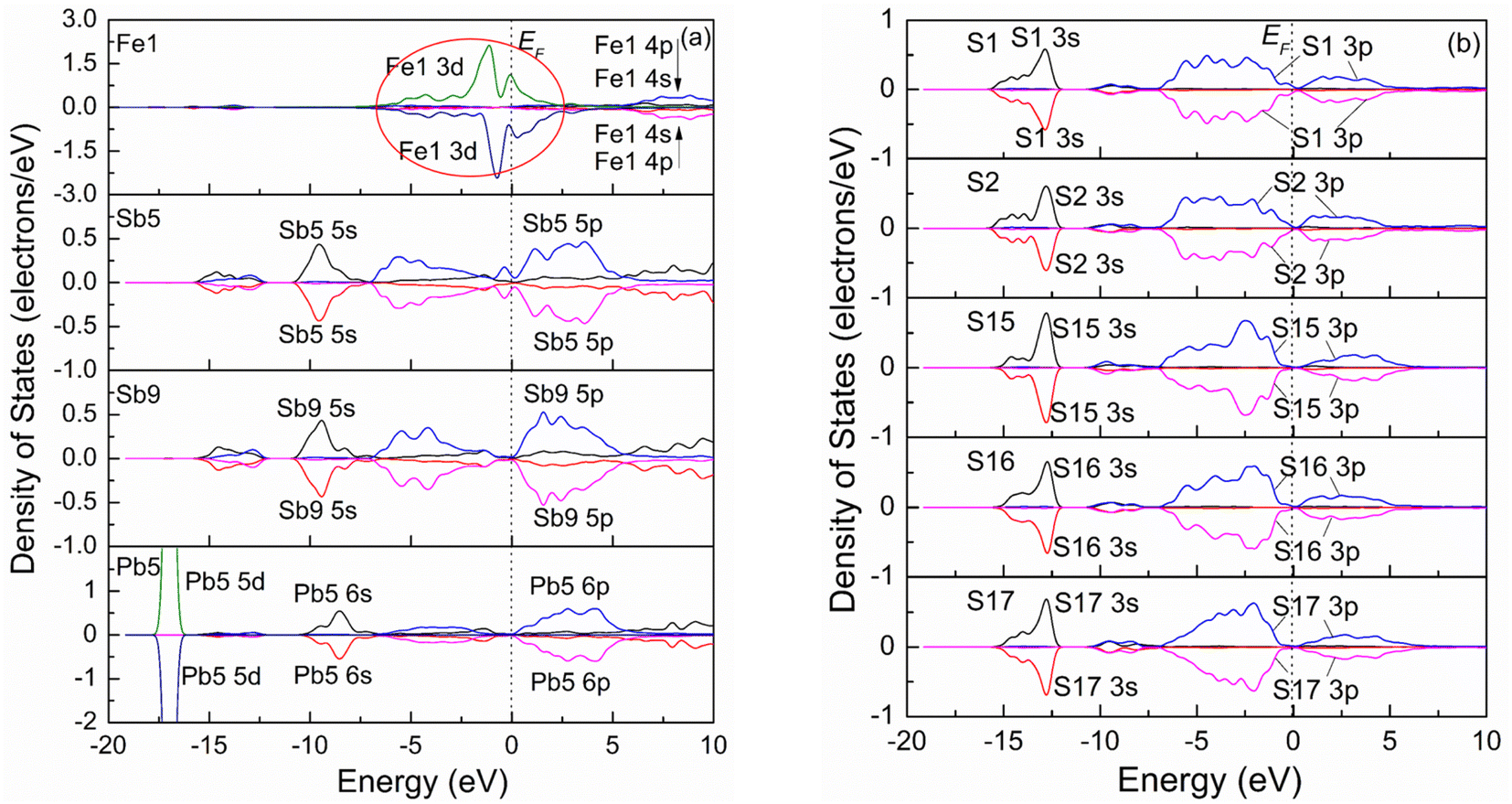 | ||
| Fig. 3 Spin polarizing partial density of states of pure jamesonite: (a) metal atoms; (b) S atoms. | ||
Fig. 4 shows the spin-polarization PDOSs curves of one Sb atom substitution (a), two Sb atoms substitution (b), and three Sb atoms substitution (c) at the Fe site. It is seen from Fig. 4(a) that the PDOS curve of the Fe2 atom for one Sb atom substitution (Fe3Sb25Pb16S56) changes compared to that of the pristine. Before Sb substitution at the Fe1 site, the DOS curve of Fe1 is the same as that of the Fe2 atom. However, the Fe atom is low spin state after Sb1 substitution at the Fe1 site. For S atoms (S1 and S2), it was found that the PDOSs shift to low energy compared to those of pristine jamesonite (Fig. 3(b)), and the DOSs in the conduction band extend towards high energy, while those in the valence band extend towards low energy. In addition, it is observed that the DOS curves of the S1 and S2 atoms are slightly different. These results suggest that interaction between S and Sb1 after Sb1 replaces Fe1 is different from that between S and Fe1 before Sb1 replaces Fe1, and the interaction between S1 and Sb1 is also different from that between S2 and Sb2. For two Sb atoms substitution (Fe2Sb26Pb16S56), it is found from Fig. 4(b) that the PDOSs of Sb1 and Sb2 are similar, and become greatly changed when compared with the DOS curve of Sb1 (Fig. 4(a)). Near the Fermi energy, the DOSs of Sb 5p in the conduction band and valence band increase significantly, and a small peak of Sb 5s appears at about 1.25 eV. The DOS curves of the S atoms (S1 and S2) bonded to Sb1 are also different from the pristine and one Sb substitution jamesonite. For one thing, the DOS curves of S1 and S1 for the two Sb substitution system shift to high energy compared to those of the one Sb substitution system, and they have low energy compared to those of pure jamesonite. For another thing, the DOS of S1 and S2 for the two Sb substitution system in the range of −7.5 to 0 eV is larger than those of the corresponding one Sb substitution system and pure jamesonite. As for the three Sb atoms substitution (Fe1Sb27Pb16S56), it is observed from Fig. 4(c) that the PDOS curve of Fe2 after three Sb substitution is similar to that of the Fe atom for pure jamesonite (Fig. 4(a)). The PDOSs of Sb3 and Sb4 are slightly similar to those of Sb1 and Sb2 for the two Sb atoms substitution jamesonite (Fig. 4(b)), which is similar to that of Sb5 (3-coordinated) of pure jamesonite. However, the DOS curve of Sb1 is similar to that of Sb9 (4-coordinated) of pure jamesonite (Fig. 3(a)), but different from that of Sb1 for the one Sb (Sb1) substitution system (Fig. 4(a)). The DOS of S1 and S2 for the three Sb substitution system is slightly different from those of S1 and S2 of the one Sb substitution system. The DOS curves of S1 and S2 shift to higher energy compared to those of S1 and S2 of the one Sb substitution system (Fig. 4(a)), but shift to low energy compared to those of S1 and S2 of pure jamesonite (Fig. 3(b)). The DOS curves of S1 and S2 in the conduction band show a parabola shape, and the DOSs are almost 0 when the energy exceeds 5 eV, which is different from those of the one Sb substitution system. These results show that the substitution of Sb with different mole concentrations at Fe sites can affect the electronic properties of jamesonite to a certain extent.
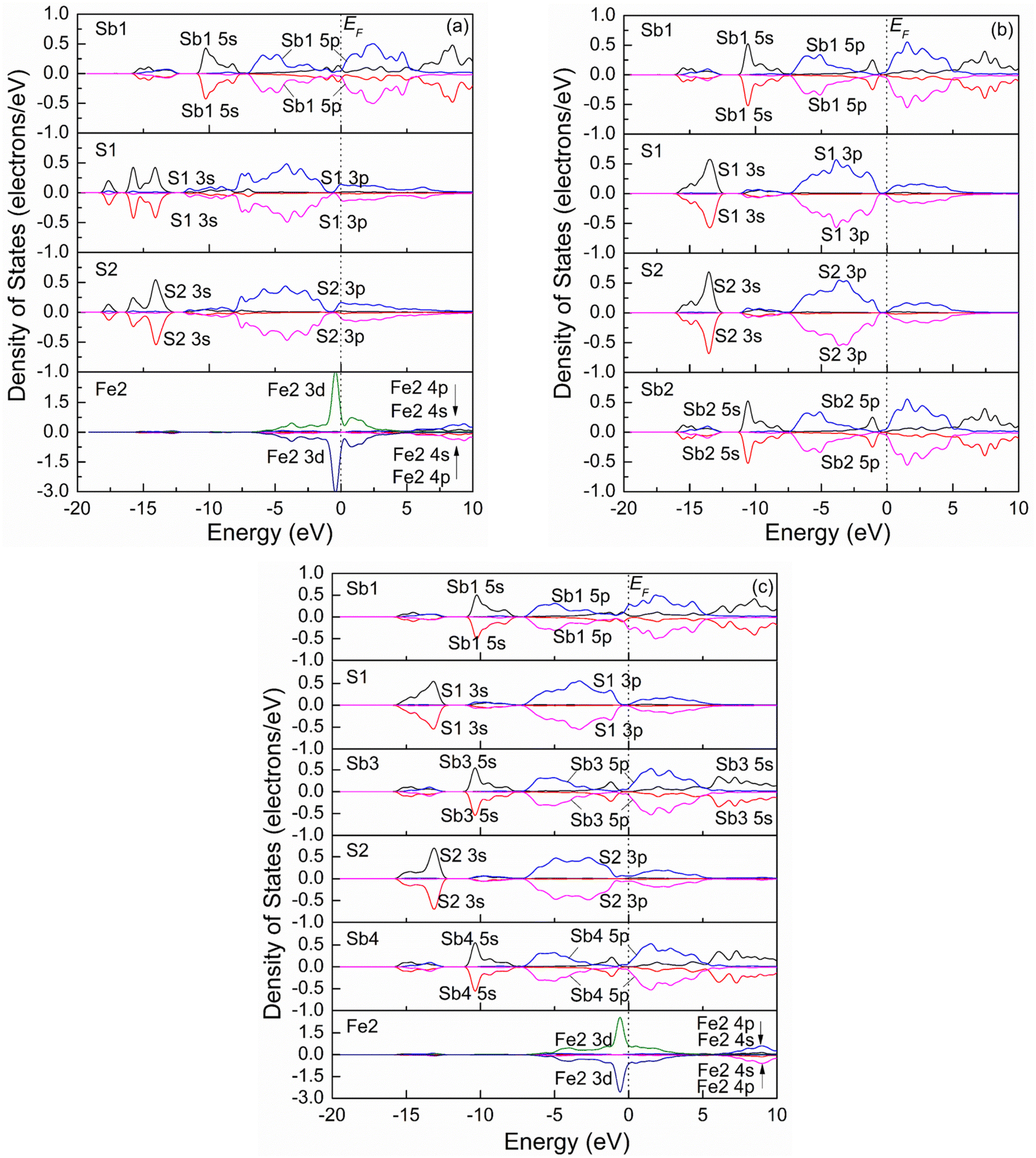 | ||
| Fig. 4 Spin partial density of jamesonite for Sb substitution at the Fe site: (a) one Sb atom substitution; (b) two Sb atoms substitution; (c) three Sb atoms substitution. | ||
Our previous experimental study showed that the presence of antimony leads to poor floatability of jamesonite in alkaline media, and is particularly sensitive to lime, which suggested that the change of the antimony content in jamesonite can have a significant impact on its flotation performance.13 In this study, as the content of Sb increases, the DOS curve of the Sb 3p orbital in the valence band moves towards the Fermi level, and the activity of the Sb atoms increases. It can be inferred that increasing the Sb content will change the electronic properties of jamesonite, thereby changing its flotation performance, which is consistent with the conclusions of the experimental study.13
The coordination structure of all Fe atoms in jamesonite is the same. We take the Fe1 atom as an example to analyze the spatial structure of the atoms around Fe (Fig. 5(a)). The Fe1 atom is coordinated with four S atoms. Among these four sulfur atoms, S1 and S3 are coordinated with one lead and two antimony atoms, and S2 and S10 are coordinated with one antimony and two lead atoms. It is observed from the bond length and coordination structure that S3 with its coordination atoms and S1 with its coordination atoms are symmetrical about Fe1. Similarly, S2 with its coordination atoms and S10 with its coordination atoms are also symmetrical about Fe1. Therefore, we only discuss the Mulliken charge populations and electron density difference of the lower half of the dotted line in Fig. 5(a).
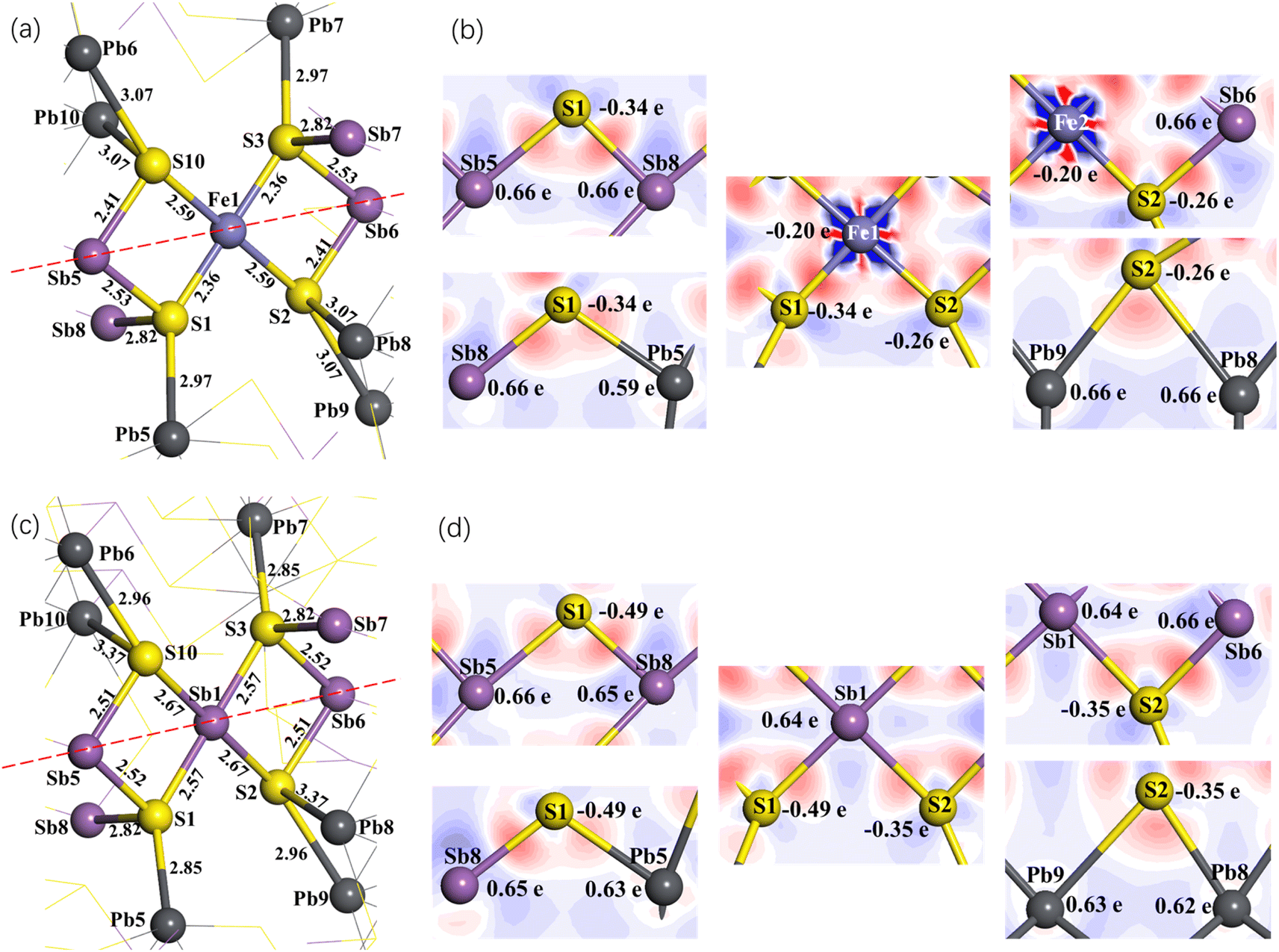 | ||
| Fig. 5 Spatial structure of the atoms around Fe1 (a), electron density difference and Mulliken charge populations of jamesonite of pure (b); spatial structure of the atoms around Sb1 (c) and electron density difference and Mulliken charge populations (d) of one Sb atom substitution on the Fe1 site for jamesonite. Numbers in (a) and (c) represent the bond length in Å, and that in (b) and (d) represent the Mulliken charge population. | ||
Fig. 5(b) shows the electron density difference and Mulliken charge populations of pure jamesonite (lower half of Fig. 5(a)). It is found that the red area around S is larger than that around the Fe1 atom, indicating that the electrons in the Fe–S bonds (Fe1–S1 and Fe1–S2) gravitate toward the S atoms. According to the Mulliken charge populations, the Fe1, S1 and S2 atoms are negatively charged (Fe1: −0.20e; S1: −0.34e; S2: −0.26e). However, the S atoms have more negative charges than the Fe1 atom, and the S1 atom has more negative charges than S2, suggesting that the covalence of the Fe1–S1 bond is stronger than that of the Fe1–S2 bond. This result is consistent with that of the bond length. The bond length of Fe1–S1 (2.36 Å) is less than that of Fe1–S2 (2.56 Å). For the Sb5–S1, Sb8–S1 and Pb5–S1 bonds, it is observed that the electrons are basically distributed around the S1 atom, and there are almost no electrons around the Pb5, Sb5 and Sb8 atoms. Therefore, the S1 atom is negatively charged (−0.34e), while te Sb5, Sb8 and Pb5 atoms are positively charged (Sb5: 0.66e; Sb8: 0.66e; Pb5: 0.59e). However, the Pb5 atom has less positive charge than the Sb atoms, which is confirmed by the distribution of electrons around the Sb5, Sb8 and Pb5 atoms. Similarly, for the Sb6–S2, Pb8–S2 and Pb9–S2 bonds, it is found that the electrons are basically distributed around the S2 atom, and there are almost no electrons around the Sb6, Pb8 and Pb9 atoms. The S2 atom is negatively charged (−0.26e), while the Sb6, Pb8 and Pb9 atoms are positively charged (Sb6: 0.66e; Pb8: 0.66e; Pb9: 0.66e). Pb8 and Pb9 have the same charges, so the strengths of the Pb8–S2 and Pb9–S2 bonds are the same, which is confirmed by the bond length. The lengths of the Pb8–S2 and Pb9–S2 bonds are the same (3.07 Å). The length of the Sb6–S2 bond is smaller than those of the Sb5–S1 and Sb8–S1 bonds, suggesting that the Sb6–S2 bond is stronger than the Sb5–S1 and Sb8–S1 bonds.
Fig. 5(c) shows the spatial structure around Sb1 after the replacement of the Fe1 atom with Sb1. It is found that similar to the symmetry about Fe1 before Sb1 substitution, S3 with its coordination atoms and S1 with its coordination atoms are symmetrical about Sb1, and S2 with its coordination and S10 with its coordination are also symmetrical about Sb1 atom. Therefore, we also discuss the Mulliken charge populations and electron density difference of the lower half of the dotted line in Fig. 5(c).
Fig. 5(d) shows the electron density difference and Mulliken charge populations of one Sb (Sb1) substitution on Fe site for jamesonite (lower half of Fig. 5(c)). It is found that the blue area around Sb1 is larger than that around the Fe1 atom before substitution, indicating that the electrons are transferred to the S atoms (S1 and S2) after Sb1 substitution. So, Sb1 is positively charged (0.64e), and the negative charges of the S1 and S2 atoms increase. S1 has more negative charges than S2, leading to the Sb1–S1 bond being stronger than the Sb1–S2 bond, which is confirmed by the bond lengths (Sb1–S1: 2.57 Å; Sb1–S2: 2.67 Å). The Sb5–S1 and Sb8–S1 bonds after Sb1 substitution exhibit little change compared to those of pure jamesonite, including the electronic distribution between S1 and Sb5, and S1 and Sb8, and the bond lengths of Sb5–S1 and Sb8–S1, and charges of Sb5 and Sb8. However, the Pb5 charge increases slightly (from 0.59e to 0.63e), leading to the increase of the Pb5–S1 bond strength. The bond length of Pb5–S1 changes from 2.97 Å before Sb1 substitution to 2.85 Å after Sb1 substitution. For the Sb6–S2, Pb8–S2 and Pb9–S2 bonds, it is found that the charge of Sb6 has not changed compared to that before Sb1 substitution. However, the bond length of Sb6–S2 increases from 2.41 Å to 2.51 Å. The charges of Pb8 and Pb9 decrease slightly from 0.66e to 0.62e and 0.63e, respectively. However, the strengths of the Pb8–S2 and Pb9–S2 bonds increase slightly, which may be due to the increase of the negative charge of the S2 atom (from −0.26 to −0.35e).
The structure change and charge distribution around the substitution atom (Sb) caused by two or three antimony atoms substitution on Fe sites are similar to those caused by one antimony substitution, so it is not described here.
3.2 Structure of the Pb substitution system
The models of jamesonite with different mole concentrations of Pb substitution on Fe sites were built. These include the following models: one lead atom (Pb1) substitution at the Fe1 site with the mole concentrations of Fe, Sb and Pb atoms in jamesonite of 3%, 24% and 17%, respectively (Fe3Sb24Pb17S56) (Fig. 6(a)); two lead atoms (Pb1 and Pb2) substitution at the Fe1 and Fe2 sites with the mole concentrations of Fe, Sb and Pb atoms in jamesonite of 2%, 24% and 18%, respectively (Fe2Sb24Pb18S56) (Fig. 6(b)); and three lead atoms (Pb1, Pb2 and Pb3) substitution at the Fe1, Fe2 and Fe3 sites with the mole concentrations of Fe, Sb and Pb atoms in jamesonite of 1%, 24% and 19%, respectively (Fe1Sb24Pb19S56) (Fig. 6(c)). It is found that the structure of jamesonite changes greatly after the Pb atoms substitution with different mole concentrations. The group space of jamesonite changes from P21/a to P1, as in the Sb substitution system. The binding energies of the Pb-doped jamesonite with different Pb concentrations are displayed in Table 8. It is also observed that the binding energies of all structures for the Pb-doped jamesonite system are negative, indicating that these structures are stable.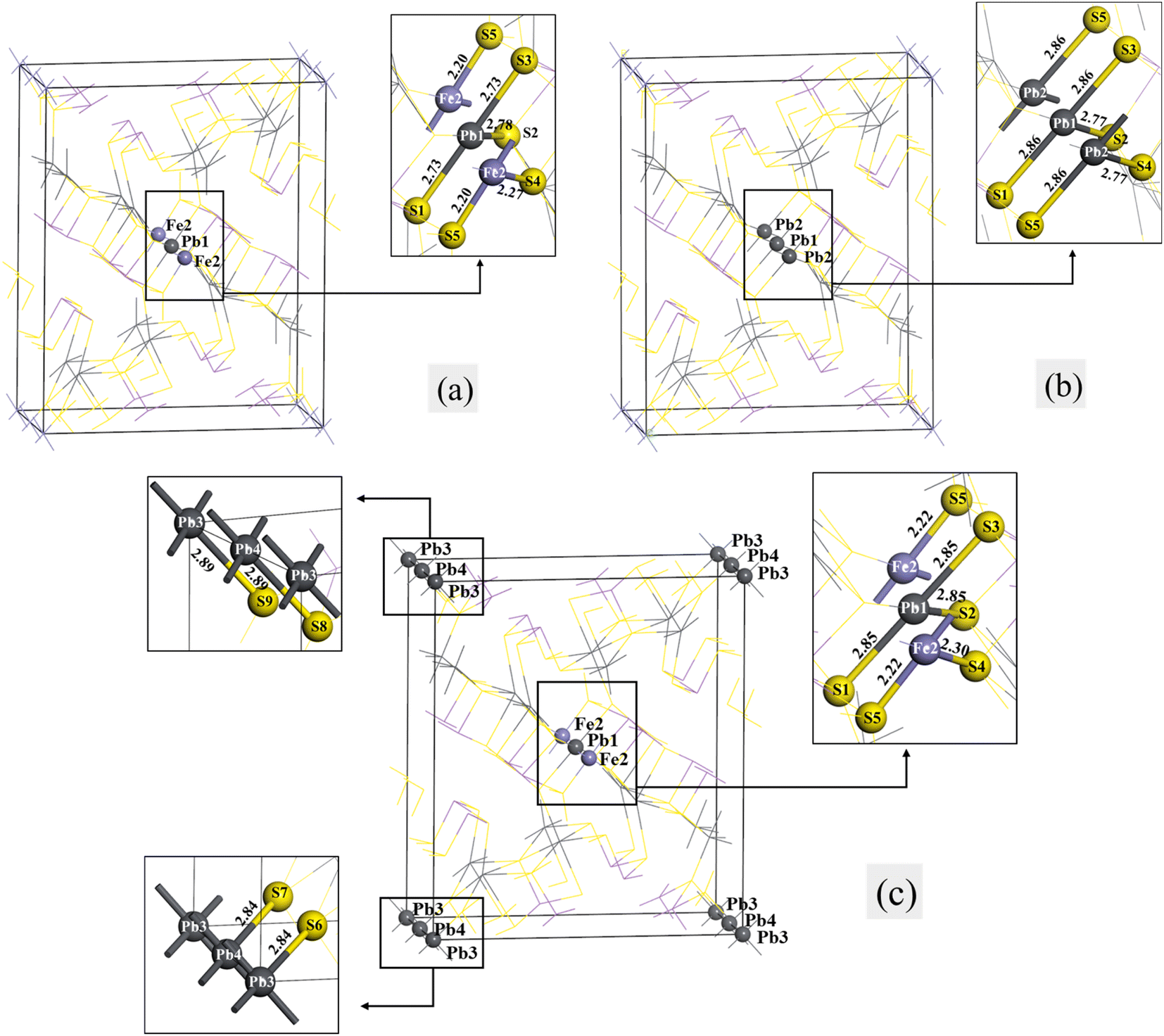 | ||
| Fig. 6 Models of the single Pb substitution systems of jamesonite: (a) one Pb atom substitution; (b) two Pb atoms substitution; (c) three Pb atoms substitution. | ||
| Substitution type | Structure | Binding energy (eV) |
|---|---|---|
| Pb | S56Fe3Sb24Pb17 | −2.95 |
| S56Fe2Sb24Pb18 | −1.93 | |
| S56Fe1Sb24Pb19 | −0.90 |
The bond lengths of some bonds in the Pb substitution systems of jamesonite are listed in Table 9. It can be seen from Fig. 6(a) that the lengths of the Pb–S bonds (Pb1–S1:2.73 Å; Pb1–S2:2.78 Å) after one Pb atom substitution at the Fe1 site are much larger than those of the corresponding Fe–S bond of pure jamesonite (Fe1–S1:2.18 Å; Fe1–S2: 2.35 Å). The bond lengths retain diagonal symmetry along with the central atoms (Pb1 and Fe2 atoms), like the one Sb atom substitution system (Fig. 2(a)). Interestingly, the bond lengths of Fe2–S4 and Fe2–S5 are the same as the one Sb substitution system. For two Pb atoms substitution system (Fig. 6(b)), the lengths of all Pb–S bonds increase (2.86 Å and 2.77 Å) compared with those of the corresponding Fe–S bonds of pure jamesonite (2.18 Å and 2.35 Å). As for the three Pb atoms substitution system (Fig. 6(c)), the structural change is larger than that for the one and two Pb atoms substitution systems. The lengths of all Pb–S bonds increase compared with those of the corresponding S–Fe bonds before Pb substitution. The lengths of all Pb1–S bonds (Pb1–S1, Pb1–S2 and Pb1–S3) are 2.85 Å, and the lengths of the Pb3–S6 and Pb3–S9 bonds are 2.84 Å and 2.89 Å, respectively, and those of Pb4–S7 and Pb4–S8 bonds are also 2.84 Å and 2.89 Å, respectively. In addition, the lengths of the Fe2–S bonds change after the three Pb atoms substitution. The bond length of Fe2–S5 increases from 2.18 Å to 2.22 Å, while that of Fe2–S4 decreases from 2.35 Å to 2.30 Å.
| Substitution systems | Bond type | Length (Å) |
|---|---|---|
| One Pb atom substitution | Pb1–S1 (Pb1–S3) | 2.73 |
| Pb1–S2 | 2.78 | |
| Fe2–S4 | 2.27 | |
| Fe2–S5 | 2.20 | |
| Two Pb atoms substitution | Pb1–S1 (Pb1–S3) | 2.86 |
| Pb1–S2 | 2.77 | |
| Pb2–S4 | 2.77 | |
| Pb2–S5 | 2.86 | |
| Three Pb atoms substitution | Pb1–S1 (Pb1–S3) | 2.85 |
| Pb1–S2 | 2.85 | |
| Fe2–S4 | 2.30 | |
| Fe2–S5 | 2.22 | |
| Pb3–S6 (Pb4–S7) | 2.84 | |
| Pb3–S9 | 2.89 | |
| Pb4–S8 | 2.89 |
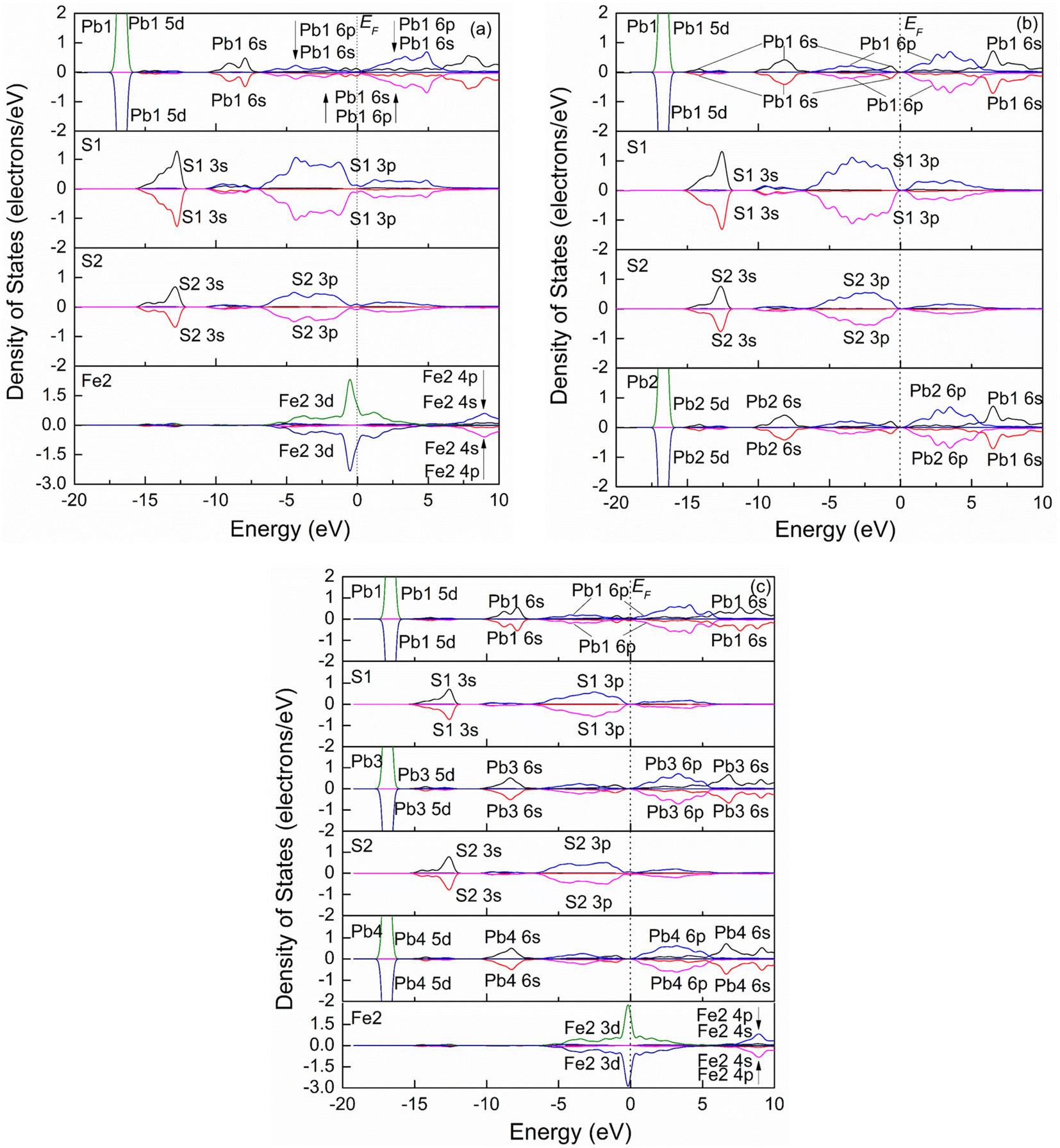 | ||
| Fig. 7 Spin partial density of states of jamesonite of the Pb substitution systems. (a) One Pb atom substitution; (b) two Pb atoms substitution; (c) three Pb atoms substitution. | ||
Fig. 8(a) shows the spatial structure of the atoms around Pb1 for jamesonite with one Pb atom substitution (Fe3Sb24Pb17S56). Similar to the Sb substitution system, S3 and S1 with their coordination atoms are symmetrical about Pb1, and S2 and S10 atoms with their coordination are also symmetrical about the Pb1 atom. Here, we also discuss the Mulliken charge populations and electron density difference of the lower half of the dotted line in Fig. 8(a).
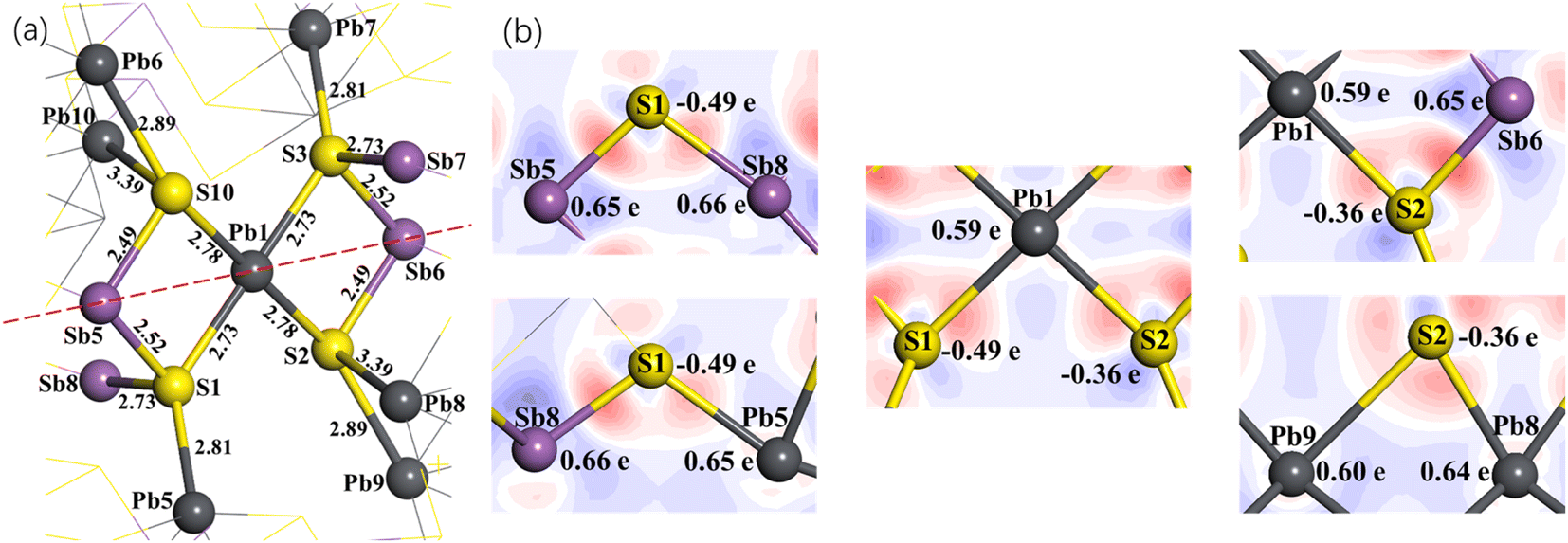 | ||
| Fig. 8 Spatial structure of the atoms around Pb1 (a), and electron density difference and Mulliken charge populations (b) of the one Pb atom substitution on the Fe1 site for jamesonite. Numbers in (a) and (b) represent the bond length in Å and Mulliken charge population. | ||
Fig. 8(b) shows the electron density difference and Mulliken charge populations for the lower half of Fig. 8(a). It can be seen that the blue area around Pb1 is larger than that around the Fe1 atom of pure jamesonite, and is close to that around Sb1 of one Sb substitution, suggesting that the electrons transfer from Pb1 to the S1 and S2 atoms after Pb substitution. The charge of Pb1 is 0.59e, which is close to that of Sb1 (0.64e). The charges of the S1 and S2 atoms are −0.49e and −0.36e, respectively, which are similar to those of S1 and S2 for the one Sb substitution system (S1: −0.49e; S2: −0.35e). The larger negative charge of S1 than S2 atom causes a slightly stronger Sb1–S1 bond (Pb1–S1: 2.73 Å) than the Sb1–S2 bond (Sb1–S2: 2.78 Å). Similar to the Sb substitution system, the Sb5–S1 and Sb8–S1 bonds of Fe3Sb24Pb17S56 also change a little compared to those of pure jamesonite (Fig. 5(a) and (b)) and the one Sb substitution system (Fig. 5(c) and (d)), including the electronic distribution between the Sb5–S1 and Sb8–S1 bonds, and charges of the Sb5 and Sb8 atoms. However, the Pb5–S1 bond (Pb5–S1: 2.81 Å) is much stronger than that of pure jamesonite (Pb5–S1: 2.97 Å), and slightly stronger than that of the one Sb substitution system (Pb5–S1: 2.85 Å). The charge of Pb5 for the one Pb substitution system is 0.65e, which is more than that of pure jamesonite, and close to that of the one Sb substitution system. In addition, it is found that the lengths of the Pb8–S2 and Sb6–S2 bonds after Pb substitution (Pb8–S2: 3.39 Å; Sb6–S2: 2.49 Å) increase, while that of the Pb9–S2 bond (2.89 Å) decreases compared to those of pure jamesonite (Pb8–S2: 3.07 Å; Sb6–S2: 2.41 Å; Pb9–S2: 3.07 Å), which is similar to those of the Sb substitution system (Pb8–S2: 3.37 Å; Sb6–S2: 2.51 Å; Pb9–S2: 2.96 Å). It is also observed that the change of these bond lengths after Sb and Pb substitution is attributed mainly to the change of the charge of S1 and S2 bonded with the substitution atoms. Similarly, the structure change, Mulliken charge population, and electron density difference around the substitution atom (Pb) caused by two or three lead atoms substitution on Fe sites are similar to those caused by the one lead substitution, which is not described here.
The flotation property of jamesonite is closely related to its composition. The jamesonite models by Sb or Pb substitution at Fe sites decrease the Fe content, which is more conducive to the flotation of jamesonite.20 On the other hand, Sb or Pb substitution at Fe sites changes the chemical composition of Sb or Pb in jamesonite, thus changing the flotation behavior of jamesonite. Sb substitution at the Fe site increases 4-coordinated Sb, which is more active than 3-coordinated Sb.13 Similarly, Pb substitution at the Fe site is four coordinated, which is more active than the six-coordinated Pb (the PDOS curve of the 4-coordinated Pb shifts to higher energy compared to that of 6-coordinated Pb). These substitutions have greatly changed the electronic structure and properties of jamesonite, especially polyatomic substitutions. With the increase in the concentration of substituted elements (Sb or Pb), the electronic structure of jamesonite further changes, so the influence on its flotation property may be greater.
4. Conclusions
The models of jamesonite with different Fe, Sb, and Pb contents were built by Sb or Pb substitution at Fe sites, and their electronic structures were studied by DFT method. The Fe atom of pure jamesonite is a high spin state, while the low spin state occurs for Fe atoms in Sb and Pb substitution systems. After Sb or Pb substitution at the Fe sites, the structure of jamesonite significantly changes, but remains axially symmetric along the diagonal. The bond lengths of the Sb–S and Pb–S bonds are larger than those of the corresponding Fe–S bonds before Sb or Pb substitution. The S1 and S2 atoms that are bonded to Sb1 gain more charges (S1: −0.49e; S2: −0.35e) after one Sb substitution (Sb1) than those before Sb1 substitution (S1: −0.34e; S2: −0.26e). The Sb1 atom has more positive charges (0.64e) than the Fe1 atom before Sb1 substitution (−0.20e). For the one Pb (Pb1) substitution system, the electrons transfer from Pb1 (0.59e) to adjacent S1 and S2 atoms (S1: −0.49e; S2: −0.36e), and the larger negative charge of S1 than S2 atom cause a slightly stronger Pb1–S1 bond than the corresponding Fe1–S1 of pure jamesonite. For multi-Sb and Pb atoms substitution systems, the interaction between the substitution atoms and surrounding atoms is a little similar to that of the one Sb and Pb atom substitution system. However, the increases of the 4-coordination Sb or 4-coordination Pb with the decrease of the Fe atom greatly change the property of jamesonite.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 51964004) and the high-performance computing platform of Guangxi University.References
- C. S. Chehreh, M. Rudolph, T. Leistner, J. Gutzmer and U. A. Peuker, Int. J. Min. Sci. Technol., 2015, 25, 877–883 CrossRef.
- H. Tang, Z. B. Deng, Y. Tang, X. Tong and Z. Q. Wei, Sep. Purif. Technol., 2023, 312, 123316 CrossRef CAS.
- R. G. Arnold, Am. Mineral., 1966, 51, 1221–1227 CAS.
- C. H. Zhao, B. Z. Wu and J. H. Chen, J. Cent. South Univ., 2015, 22, 466–471 CrossRef CAS.
- C. S. Horng and A. P. Roberts, Geochem., Geophys., Geosyst., 2018, 19, 3364–3375 CrossRef.
- P. K. Abraitis, R. A. D. Pattrick and D. J. Vaughan, Int. J. Miner. Process., 2004, 74, 41–59 CrossRef CAS.
- L. H. Lan, J. H. Chen, Y. Q. Li, P. Lan, Z. Yang and G. Y. Ai, Trans. Nonferrous Met. Soc. China, 2016, 26, 272–281 CrossRef CAS.
- B. X. Song, X. R. Dong, X. Y. Qiu, Z. Hu and Y. Wang, J. Alloys Compd., 2021, 868, 159105 CrossRef CAS.
- J. H. Chen, Miner. Eng., 2021, 171, 107067 CrossRef CAS.
- J. W. Anthony, R. A. Bideaux, K. W. Bladh and M. C. Nichols, Handbook of Mineralogy, Mineral Data Publ., 2001 Search PubMed.
- M. S. Sakharova, Trudy Mineral. Muz. Muzeya, 1955, 7, 112–126 CAS.
- E. V. Shannon, Am. Mineral., 1925, 10, 194–197 CAS.
- C. H. Zhao, J. H. Chen, Y. Q. Li, Q. He and B. Z. Wu, Trans. Nonferrous Met. Soc. China, 2015, 25, 590–596 CrossRef CAS.
- Y. H. Hu, M. R. Wu, R. Q. Liu and W. Sun, Miner. Eng., 2020, 150, 106272 CrossRef CAS.
- X. Yang, Y. Q. Li and J. H. Chen, Miner. Eng., 2022, 183, 107596 CrossRef CAS.
- L. Chen, X. Zhao, F. Dong and Y. Sun, J. Hazard. Mater., 2022, 436, 29089 Search PubMed.
- K. Nakada and A. Ishii, Solid State Commun., 2011, 151, 13–16 CrossRef CAS.
- M. G. Medvedev, I. S. Bushmarinov, J. W. Sun, J. P. Perdew and K. A. Lyssenko, Science, 2017, 355, 49–52 CrossRef CAS PubMed.
- J. H. Chen and Y. Q. Li, Miner. Eng., 2022, 179, 107469 CrossRef CAS.
- J. H. Chen, Y. Q. Li, Q. R. Long, Z. W. Wei and Y. Chen, Int. J. Miner. Process., 2011, 100, 54–56 CrossRef CAS.
- J. T. Medina, M. R. Perez, E. P. Beas, I. A. Dominguez, M. U. F. Guerrero, A. M. T. Ruiz, M. P. Labra, J. C. J. Tapia and F. R. B. Hernandez, Charact. Miner., Met., Mater., 2021, 543–550 Search PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- Y. Matsushita and Y. Ueda, Inorg. Chem., 2003, 42, 7830–7838 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |