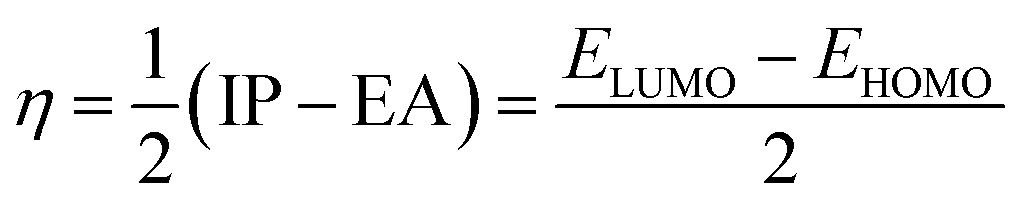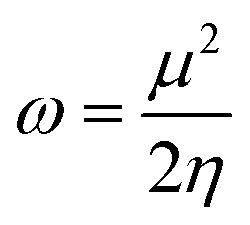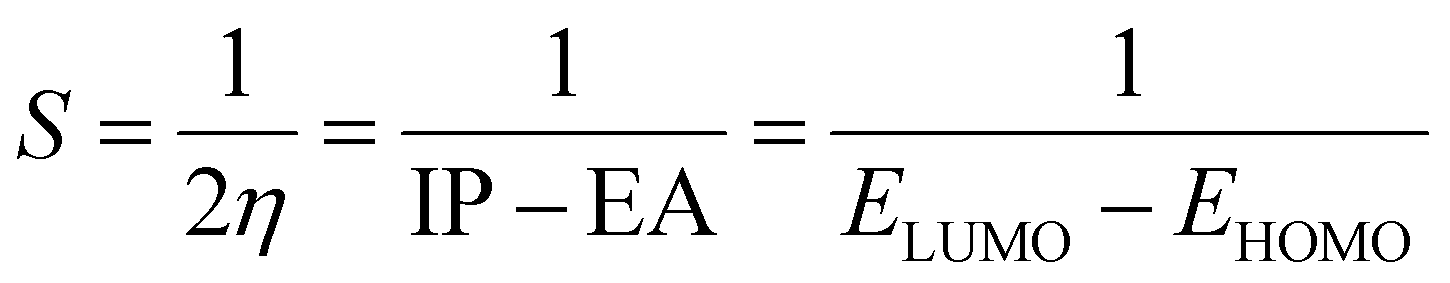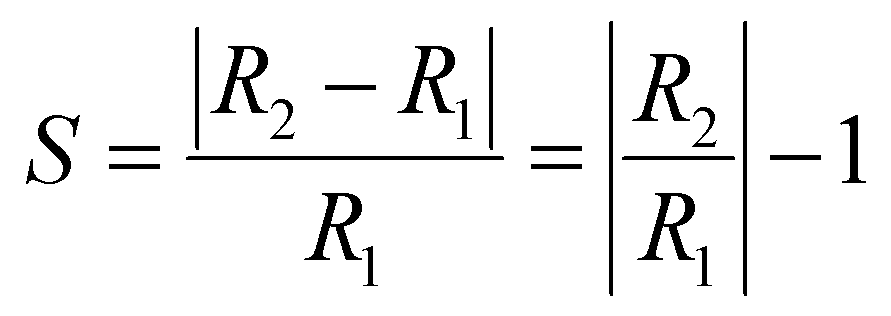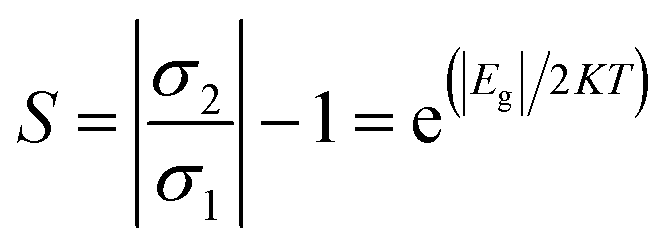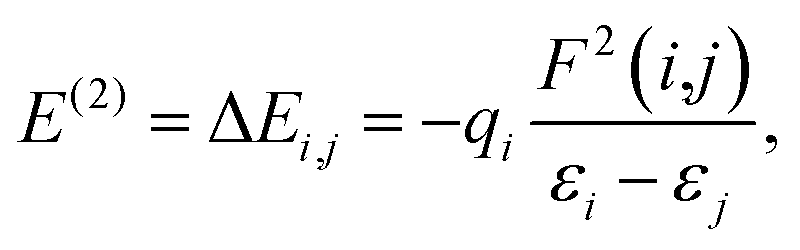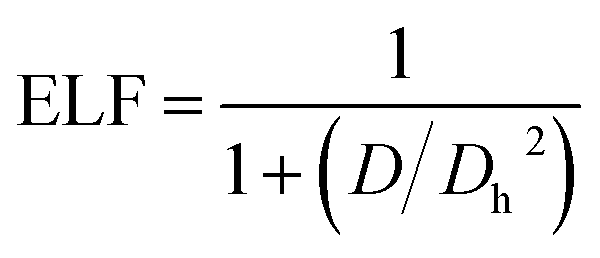DOI:
10.1039/D3RA02939E
(Paper)
RSC Adv., 2023,
13, 25391-25407
Retracted Article: Yttrium- and zirconium-decorated Mg12O12–X (X = Y, Zr) nanoclusters as sensors for diazomethane (CH2N2) gas†
Received
3rd May 2023
, Accepted 21st July 2023
First published on 24th August 2023
Abstract
Diazomethane (CH2N2) presents a notable hazard as a respiratory irritant, resulting in various adverse effects upon exposure. Consequently, there has been increasing concern in the field of environmental research to develop a sensor material that exhibits heightened sensitivity and conductivity for the detection and adsorption of this gas. Therefore, this study aims to provide a comprehensive analysis of the geometric structure of three systems: CH2N2@MgO (C1), CH2N2@YMgO (CY1), and CH2N2@ZrMgO (CZ1), in addition to pristine MgO nanocages. The investigation involves a theoretical analysis employing the DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory. Notably, the examination of bond lengths within the MgO cage yielded specific values, including Mg15–O4 (1.896 Å), Mg19–O4 (1.952 Å), and Mg23–O4 (1.952 Å), thereby offering valuable insights into the structural properties and interactions with CH2N2 gas. Intriguingly, after the interaction, bond length variations were observed, with CH2N2@MgO exhibiting shorter bonds and CH2N2@YMgO showcasing longer bonds. Meanwhile, CH2N2@ZrMgO displayed shorter bonds, except for a longer bond in Mg19–O4, suggesting increased stability due to shorter bond distances. The study further investigated the electronic properties, revealing changes in the energy gap that influenced electrical conductivity and sensitivity. The energy gap increased for Zr@MgO, CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO, indicating weak interactions on the MgO surface. Conversely, Y@MgO showed a decrease in energy, suggesting a strong interaction. The pure MgO surface exhibited the ability to donate and accept electrons, resulting in an energy gap of 4.799 eV. Surfaces decorated with yttrium and zirconium exhibited decreased energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), as well as decreased energy gap, indicating increased conductivity and sensitivity. Notably, Zr@MgO had the highest energy gap before CH2N2 adsorption, but C1 exhibited a significantly higher energy gap after adsorption, implying increased conductivity and sensitivity. The study also examined the density of states, demonstrating significant variations in the electronic properties of MgO and its decorated surfaces due to CH2N2 adsorption. Moreover, various analysis techniques were employed, including natural bond orbital (NBO), quantum theory of atoms in molecules (QTAIM), and noncovalent interaction (NCI) analysis, which provided insights into bonding, charge density, and intermolecular interactions. The findings contribute to a deeper understanding of the sensing mechanisms of CH2N2 gas on nanocage surfaces, shedding light on adsorption energy, conductivity, and recovery time. These results hold significance for gas-sensing applications and provide a basis for further exploration and development in this field.
1 Introduction
Toxic, carcinogenic, odorless, and extremely explosive gases like diazomethane (CH2N2) are potential threats to susceptible populations, leading to serious health concerns.1,2 Diazomethane is extremely explosive at room temperature, thereby becoming a subject of concern in the environment.3 In the production of methylene radical, CH2N2 is the main generator.4 The separation of diazomethane was accomplished in the year 1894; the processes of decomposition, isolation and purification are the essential aspects after successful separation.5 Moreover, diazomethane is dangerous during separation processes such as the injection of inert gas.6 In the thermal decomposition of diazomethane, a temperature range of 140–200 °C is required.7 Also, in the decomposition of this toxic gas, methylene and nitrogen are generated.8 Due to its toxic and hazardous nature, trapping or adsorption of diazomethane (CH2N2) gas is an increasing area of interest for researchers, especially those focused on the design and development of sensor devices with potency as efficient gas absorbers.
Due to their unique magnetic, mechanical, electrical, structural and optical properties, nanostructures are of vast interest to researchers.9 The wide range of applications of nanomaterials is not just limited to the chemical, medical, electronics and aviation industries.10 Over time, nanomaterials have become an object of interest to researchers in the area of gas sensing and drug development, such as drug design and drug delivery.11 As a result of their large porous surfaces and large surface–volume ratio, various reports are recorded on nanomaterials such as nanocages, nanotubes, nanoclusters, nanowires, and others. Various efforts have been made to evaluate and gain insights into the properties of fullerene and fullerene-like cages.12 Two-dimensional (2D) and three-dimensional (3D) nanomaterials have been given full attention due to different chemical agents.13 One of the most commonly and widely used two-dimensional nanostructures in the area of electronic sensors is graphene.14 Unfortunately, weak interaction has been observed and reported in pure graphene. Consequently, it is not suitable to be applied as a chemical sensor.15 Metal oxides, as inorganic nanomaterials, have drawn considerable attention due to their promising and unique physical and chemical properties.16 Fullerene-like cages such as MgO, CaO, and aluminum nitrides (AlNs) are of great use as a result of the closed electronic shells of the nanoclusters. The great stability and essential roles of the named nanomaterials in the development of proficient sensor devices.17 Due to their unique physical and chemical properties, low electron affinity, and large band gap, AlNs have made waves in this area of research.18 Ionic insulator nanostructures such as magnesium oxide (MgO), with a wide band gap and a variety of applications in catalysis, energy storage, sensors and ceramics, are used in this present study.19 Numerous approaches, such as functionalization by endohedral and exohedral encapsulation, have been suggested to tackle the problems arising from weak interactions and to enhance the electronic properties.20 Mohsen Doust et al. reported in their theoretical investigation of X12O12 (X = Be, Mg and Ca) in sensing CH2N2 (DFT study) that the energy calculations show a strong size-dependent adsorption, indicating that nanocages containing large atoms have the tendency to attract CH2N2 more strongly, hence showing effective binding. Significant changes to HOMO and LUMO energies have resulted from this adsorption.21 Also, in a DFT study to gain insight into the use of silicon carbide nanotubes (SiCNTs) in the catalytic decomposition of toxic diazomethane (DAZM) gas, Mohammad Solimannejad et al. reported that the DAZM molecule decomposed over the surface of (6,0) SiCNT with the activation energy of 0.523 eV. It was also concluded that adsorption over smaller diameters of SiCNT is thermodynamically more favorable than over larger diameters.22 Hitler Louis and team reported that the adsorption of phosgene gas follows an increasing pattern of adsorption energy: AlN < MgO < CaO, showing that CaO surface has the greatest negative adsorption energy of −4.79372 eV and −5.27066 eV using both levels of theory (PBE0 and ωB97XD), respectively. This is followed by MgO surface, with adsorption energy values of −1.2105 eV and −1.3279 eV for PBE0 and ωB97XD, respectively.23 In the adsorption of some small gas molecules such as N2O, NO2, NO, H2S, SO2, and SO3 on Mg12O12, Be12O12 and Ca12O12 surfaces, the adsorption was observed to follow an increasing trend of Ca12O12 > Mg12O12 > Be12O12, showing that these gases are fairly adsorbed on the Mg12O12 nanocage.24
In this present study, investigation was carried out on the adsorption of diazomethane (CH2N2) on pure magnesium oxide (MgO) and X-decorated (X = Y and Zr) magnesium oxide (MgO) surfaces. This study aimed at designing a potential nanosensor material for the efficient adsorption of diazomethane gas molecule in the atmosphere. Density functional theory (DFT) was employed throughout this study to carry out various geometric optimizations, calculations, and theoretical evaluations. All computational works have been accomplished within the framework of the ωB97XD range-separated DFT functional, assigning the Gen/SDD/6-311++G(d,p) basis set. Investigations on the nature of interactions, inter- and intramolecular interactions, and sensor mechanisms have been achieved by the computational parameters hailing from the frontier molecular orbital (FMO), HOMO–LUMO analysis and natural bond orbitals (NBO). Study of weak interactions was accomplished using the RDG and quantum theory of atoms in molecule (QTAIM) analysis. Topological electron localization function (ELF) was employed to study the chemical bonding features between the pure MgO and its decorated surface with the adsorbate (CH2N2) gas.
2 Theoretical calculations
The studied systems were subjected to theoretical calculations using DFT/ωB97XD at the GenECP/6-311++G(d,p) level of theory.25,26 This computational method was utilized based on the findings from several literature reports.27 It can be said that this combination of computational methods offers a good balance between accuracy and computational cost, making it suitable for studying a wide range of molecular systems. Notably, the ωB97Xd functional incorporates long-range and dispersion corrections, enabling accurate treatment of noncovalent interactions and transition metal compounds. Additionally, the use of GenECP allows for efficient modeling of heavy atoms by replacing the inner-core electrons with effective core potentials, reducing computational complexity. The advantages of this theoretical algorithm over other algorithms lie in its accuracy, computational efficiency, and applicability. The combination of DFT with the ωB97Xd functional and appropriate basis sets provides accurate predictions of molecular properties. The GenECP method further enhances computational efficiency by reducing the computational burden associated with heavy atoms. Overall, this approach strikes a balance between accuracy and computational cost, making it versatile for studying various molecular systems and phenomena and offering valuable insights into their electronic structures and properties. Interestingly, geometric minimization was performed using GaussView 6.0.16 (ref. 28) and Gaussian16 (ref. 29) software, which aided in the optimization of the molecular structures. The output files generated from the calculation were used to obtain the HOMO–LUMO energies, which aided in understanding the conductivity and sensitivity of sensor materials and which were plotted with the help of Avogadro software.30 The quantum theory of atoms in molecule (QTAIM) analysis was done using the MultiWfn 3.7 program,31 and the pictorial view was obtained with the help of Visual Molecular Dynamics (VMD) software.32 The frontier molecular orbital and the global quantum descriptors, as presented in Table 3, were calculated using the following eqn (1)–(7).33| | |
Energy gap = ELUMO − EHOMO
| (1) |
| | |
−μ = 1/2(EHOMO + ELUMO) = χ
| (4) |
| |
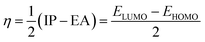 | (5) |
| |
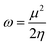 | (6) |
| |
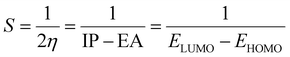 | (7) |
The adsorption energies for various complexes were calculated using eqn (8).
| | |
EAd(complex) = Ecomplex − (Eadsorbent + Eadsorbate)
| (8) |
Knowledge of inter- and intramolecular charge transfers is very important in the sensor community; hence, the second-order perturbation energy was considered while computing the natural bond orbital (NBO) using NBO version 3.9 in the Gaussian package. Weak interactions were studied using QTAIM and NCI analysis. As designed by Professor Richard Bader and his team, QTAIM analysis employs topological parameters such as density of all electrons ρ(r), electronic charge density V(r), energy density H(r), Laplacian of the electron density ∇2ρ(r), Hamiltonian kinetic energy K(r), and Lagrangian kinetic energy G(r) at critical points (CPs). The changes in the electronic state of the system were calculated via density of states analysis using Gaussium.34 Topological electron localization function was computed with the help of MultWfn software.
3 Results and discussion
3.1 Geometry and structural analysis
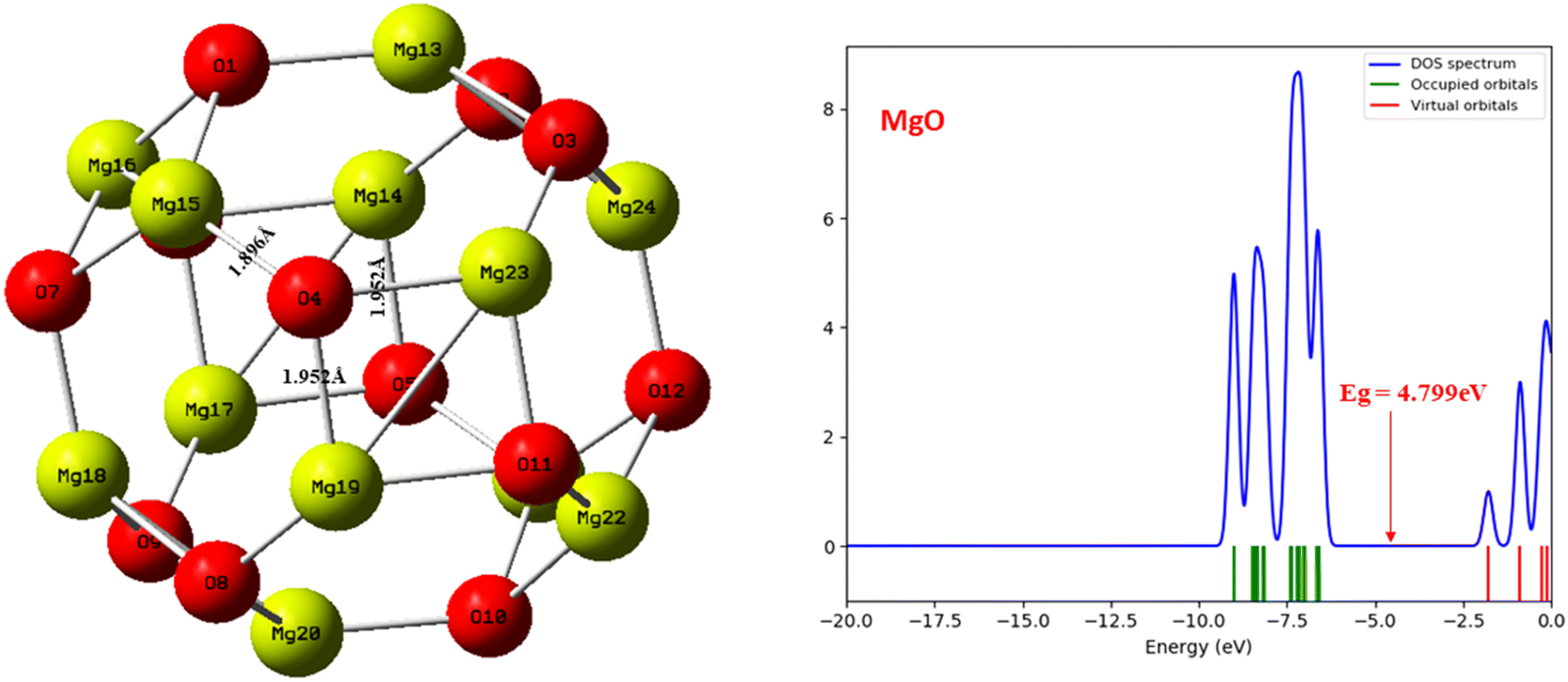
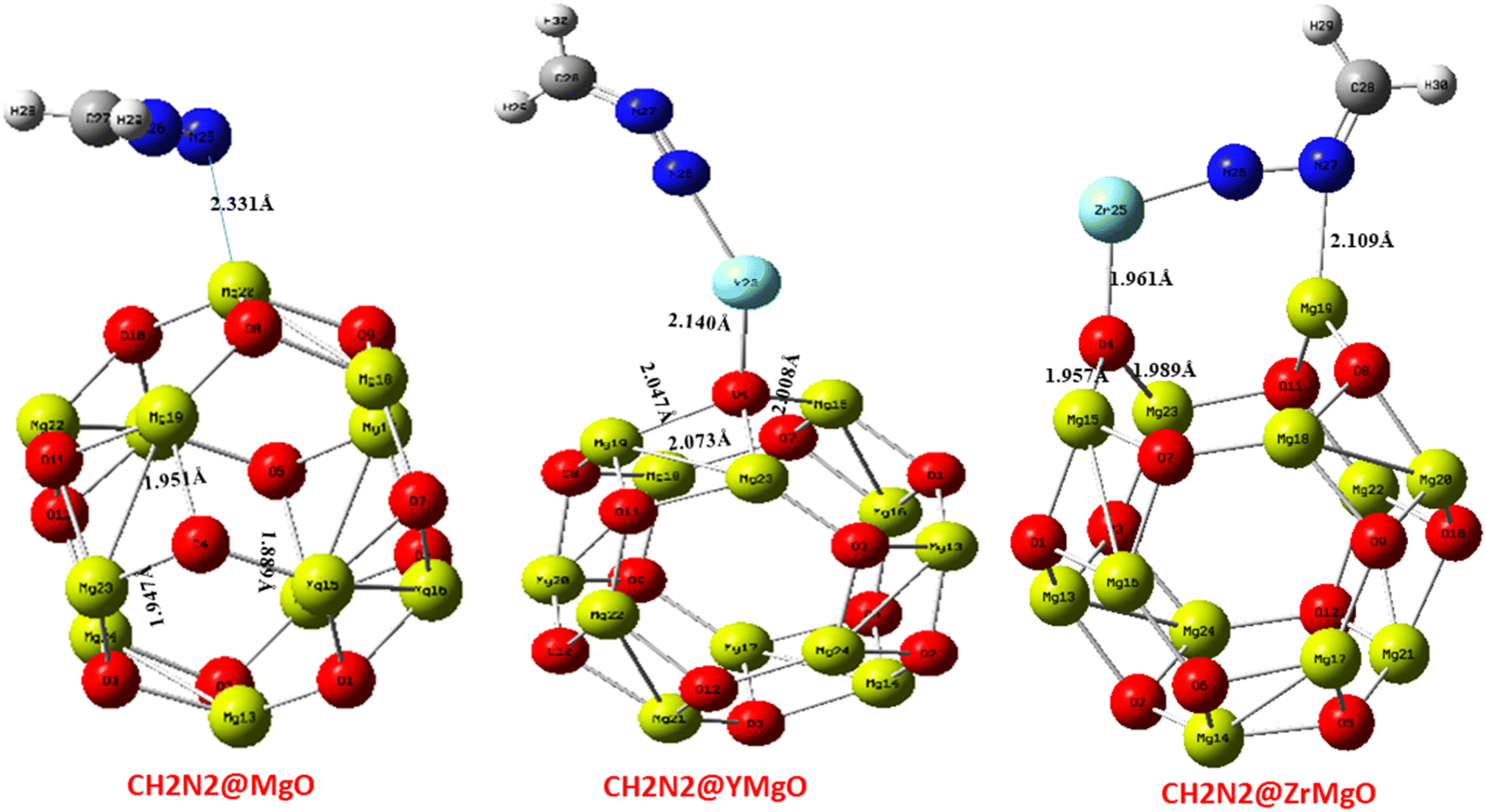
This study undertook a comprehensive evaluation of the geometrical structure and density of states (DOS) of the CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO systems, alongside the pristine MgO nanocage, with the aim of investigating the adsorption of CH2N2 on these nanomaterials. The geometry and structural parameters were optimized using the DFT/ωB97XD functional at the GenECP/6-311++G(d,p) level of theory, enabling the determination of minimum energies and configurations as well as the assessment of structural changes upon CH2N2 gas interaction. Fig. 1 provides a visual representation of the MgO cage, accompanied by a density of states plot that reveals a band gap value of 4.799 eV. Furthermore, Table 1 presents the calculated bond lengths for the MgO cage. Before the interaction, the selected bond lengths within the MgO cage were observed as Mg15–O4 (1.896 Å), Mg19–O4 (1.952 Å), and Mg23–O4 (1.952 Å), as shown in Fig. 1. Through these comprehensive evaluations, valuable insights were gained into the structural properties of the studied systems and their interaction with CH2N2 gas. These findings contribute to an enhanced understanding of the adsorption behavior and provide a solid foundation for future investigations into the CH2N2-sensing capabilities of the MgO nanocage and its decorated surfaces.
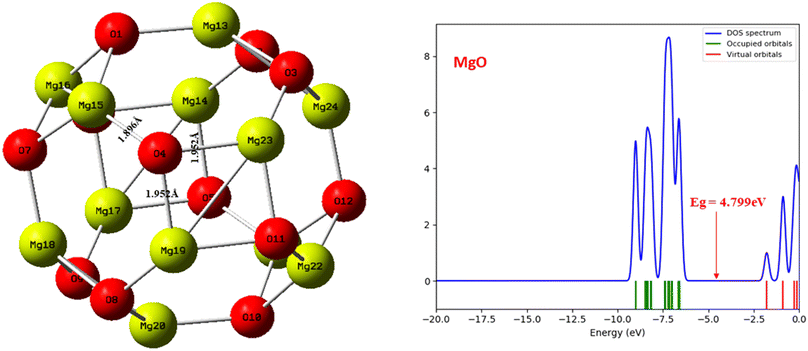 |
| | Fig. 1 Optimized structure of the MgO nanocage and density of states plot before interaction using DFT/ωB97XD at the GenECP/6-311++(d,p) level of theory. | |
Table 1 Selected bond lengths for the studied systems calculated at DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory
| System |
Bond |
Bond length (Å) |
| Before interaction |
After interaction |
| CH2N2@MgO (C1) |
Mg15–O4 |
1.896 |
1.889 |
| Mg19–O4 |
1.952 |
1.951 |
| Mg23–O4 |
1.952 |
1.947 |
| Mg20–N25 |
— |
2.331 |
| CH2N2@YMgO (CY1) |
O4–Y25 |
2.039 |
2.140 |
| Mg15–O4 |
1.896 |
2.008 |
| Mg19–O4 |
1.990 |
2.047 |
| Mg23–O4 |
3.066 |
2.073 |
| Y25–N26 |
— |
2.166 |
| CH2N2@ZrMgO (CZ1) |
O4–Zr25 |
1.954 |
1.961 |
| Mg15–O4 |
2.247 |
1.957 |
| Mg19–O4 |
2.028 |
3.119 |
| Mg23–O4 |
2.029 |
1.989 |
| Zr25–N26 |
— |
1.892 |
| Mg19–N27 |
— |
2.109 |
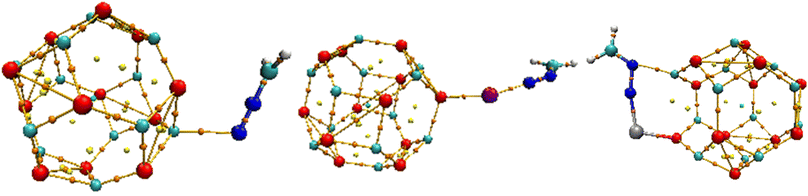
The selected bond lengths for the CH2N2@MgO interaction of Mg–O within the MgO cage before interaction were observed to be Mg15–O4 (1.896 Å), Mg19–O4 (1.952 Å), and Mg23–O4 (1.952 Å), respectively. The bond length appears to be shorter after interaction for the CH2N2@MgO interaction, with the bonds and bond lengths observed to be Mg15–O4 (1.889 Å), Mg19–O4 (1.951 Å), and Mg23–O4 (1.947 Å), respectively. The CH2N2@YMgO interactions for Mg–O within the MgO cage before interaction were observed to be Mg15–O4 (1.896 Å), Mg19–O4 (1.990 Å), and Mg23–O4 (3.066 Å), respectively. The bond length appears to be greater after interaction for the CH2N2@YMgO interaction, with the bonds and bond lengths observed to be Mg15–O4 (2.008 Å), Mg19–O4 (2.047 Å), and Mg23–O4 (2.073 Å), respectively. Lastly, for the CH2N2@ZrMgO interaction for Mg–O within the MgO cage before interaction, the bond lengths are observed to be Mg15–O4 (2.247 Å), Mg19–O4 (2.028 Å), and Mg23–O4 (2.029 Å), respectively. The bond length appears to be shorter after interaction for CH2N2@ZrMgO, observed to be Mg15–O4 (1.957 Å) and Mg23–O4 (1.989 Å), except for Mg19–O4 (3.119 Å), which appears greater after interaction. The axial bond distance of CH2N2@MgO with the bond and bond length Mg20–N25 (2.331 Å) appears to be higher than that of CH2N2@YMgO with bond and bond distance of Y25–N26 (2.166 Å) and that of CH2N2@ZrMgO with bonds and bond distances of Zr25–N26 (1.892 Å) and Mg19–N27 (2.109 Å). Notable results from Table 1 show that the compound CH2N2@ZrMgO had a shorter bond distance after interaction with the studied nanocage, and hence appears to be more stable as a result of its shorter bond distance.35 The optimized structure after interaction is depicted in Fig. 2.
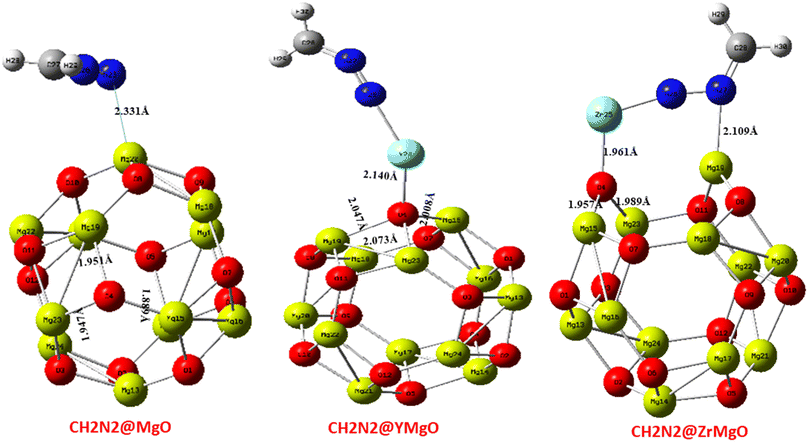 |
| | Fig. 2 Optimized structure of the MgO nanocage after interaction, obtained via DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory. | |
3.2 Electronic state analysis
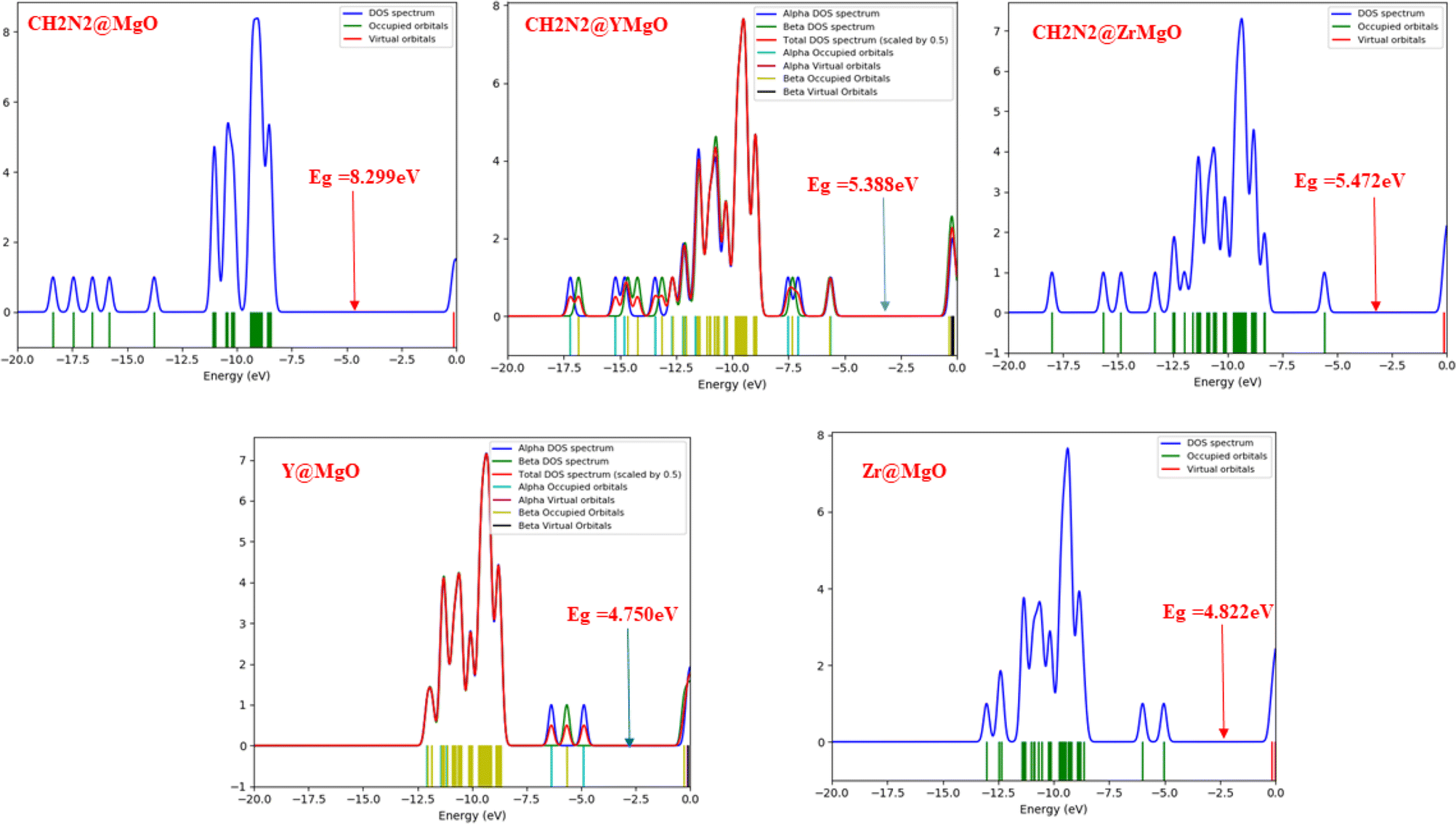
The density of state plots for this study are depicted in Fig. 3, which reveal more information about the electronic pattern of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the MgO-decorated nanocage before and after adsorption with CH2N2 gas. According to ref. 36, the conductivity of a surface can be investigated via the density of states. The density of state plots also make it possible to visualize slight changes as a result of weak interactions between the adsorbent and the adsorbate. As displayed in Fig. 3, it is noticeable that a change in the energy state of the HOMO and the LUMO of the studied systems appears near the Fermi level. A change in the energy gap can bring about a change in the electrical conductivity, hence bringing a rise in sensitivity, and an observable change in the energy gap as was noticed after decoration, which indicates an increase in the adsorption strength of Y@MgO, Zr@MgO, CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO interactions, respectively. With respect to the MgO cage, an increment is observed in the energy gap values of Zr@MgO, CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO with energy values of 4.822 eV, 8.299 eV, 5.388 eV, and 5.472 eV, respectively, indicating weak interaction in the MgO surface, while a decrement in energy value is observed in Y@MgO, with the value of 4.750 eV, indicating a strong interaction in the MgO surface, which implies strong sensitivity along the bond in MgO. The intensity of the HOMO with the virtual orbital LUMO is depicted in Fig. 3, and the detailed information on the geometry structure is presented in Tables S1–S3.†
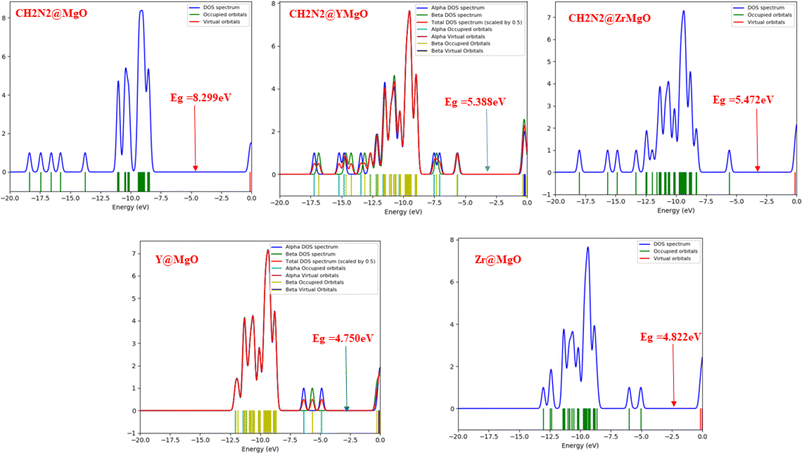 |
| | Fig. 3 Density of states plots for the interactions between MgO, its decorated cages, and the complexes. | |
3.3 Bader topological analysis
In an attempt to gain complete insight into the intermolecular interactions of the investigated complexes, quantum theory of atoms in molecule (QTAIM) analysis, by R. F. Bader et al., was invoked.36 Structural investigations may not provide complete insight into the intermolecular interactions due to the fact that atoms are characterized by different sets of traits that have differences with relatively small limits.37,38 The information on the properties of the molecule at bond critical points (BCPs) is provided by QTAIM.39 The point at which the first derivative of the charge density (density of all electrons) vanishes is called the critical point.40 One of the most valuable methods in gaining more insights into hydrogen and non-hydrogen-bonding interactions is the atoms in molecule (AIM) analysis. In this study, topological parameters such as the bond critical point (BCP), electron density ρ(r), Laplacian of the electron density ∇2(r), Lagrangian kinetic energy G(r), Hamiltonian kinetic energy K(r), potential electron energy density V(r), total electron energy density H(r), G(r)/V(r) ratio, electron localization function (ELF), ellipticity of electron density (ε), eigenvalues (λ1, λ2 and λ3) and the λ1/λ3 ratio were utilized. Table 2 shows the aforementioned computed and calculated topological parameters, and Fig. 4 shows the pictorial view of the analysis of atoms in molecules. It can be observed from Table 2 that electron density values are all less than one (ranging from 0.128 to 0.677), indicating the accumulation of electron density between two atoms that are bonded.41 Classification into partially covalent, noncovalent, and strongly covalent was achieved using the ∇2ρ(r) and H(r) simultaneously. This follows that: ∇2ρ(r) > 0 and H(r) < 0; ∇2ρ(r) > 0 and H(r) > 0; and ∇2ρ(r) < 0 and H(r) < 0 denoting partially covalent, noncovalent, and strongly covalent, respectively. The partial covalent bond observed in all complexes is due to the resulting condition (H(r) < 0; ∇2ρ(r) > 0). For example, in CH2N2@MgO, the ∇2ρ(r) and H(r) values of 0.128 and −0.440 indicate partial covalence. Similarly, in CH2N2@YMgO (∇2ρ(r) = 0.422 and H(r) = −0.372) and CH2N2@ZrMgO (∇2ρ(r) = 0.677 and H(r) = −0.391) indicate partial covalence. Also, the nature of intermolecular interactions of the studied systems can be accounted for using the λ1/λ3 values. The λ1/λ3 was observed to be less than one in the case of CH2N2@YMgO and CH2N2@ZrMgO, depicting the presence of strong intermolecular interactions.42 Insights into the regions of charge density accumulation and bond stability were gained via the ellipticity of electron density (ε).43 Lower ellipticity often signifies greater stability, and vice versa.44 From Table 2, the lower ε values of 0.023 a.u., 0.015 a.u., and (0.109, 0.037 a.u.) for CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO, respectively, imply the stability of the following bonds: N25–Mg20, O4–Y25, N26–Zr25, and N27–Mg19. The least and greatest ellipticity indexes are found in CH2N2@YMgO (ε = 0.015 a.u.) and CH2N2@ZrMgO (ε = 0.109 a.u.), showing that CH2N2@YMgO forms the most stable complexes, with CH2N2@ZrMgO having the least stability.
Table 2 Topological parameters: bond critical point (BCP), electron density ρ(r), Laplacian of the electron density ∇2(r), Lagrangian kinetic energy G(r), Hamiltonian kinetic energy K(r), potential electron energy density V(r), total electron energy density H(r), G(r)/V(r) ratio, electron localization function (ELF), ellipticity of electron density (ε), eigenvalues (λ1, λ2 and λ3) and the λ1/λ3 ratio
| System |
Bond |
CP |
ρ(r) |
∇2ρ(r) |
G(r) |
V(r) |
H(r) |
−G(r)/V(r) |
ELF |
ε |
λ1 |
λ2 |
λ3 |
λ1/λ3 |
| CH2N2@MgO (C1) |
N25–Mg20 |
48 |
0.187 |
0.128 |
0.759 |
−0.760 |
−0.440 |
0.999 |
0.678 |
0.023 |
0.176 |
−0.245 |
0.163 |
1.080 |
| CH2N2@YMgO (CY1) |
O4–Y25 |
55 |
0.750 |
0.422 |
0.109 |
−0.113 |
−0.372 |
0.965 |
0.109 |
0.015 |
0.626 |
−0.101 |
−0.103 |
−6.078 |
| CH2N2@ZrMgO (CZ1) |
N26–Zr25 |
48 |
0.138 |
0.677 |
0.207 |
−0.246 |
−0.391 |
0.842 |
0.206 |
0.109 |
−0.180 |
−0.106 |
−0.200 |
0.900 |
| N27–Mg19 |
71 |
0.335 |
0.259 |
0.181 |
−0.195 |
−0.145 |
0.928 |
0.670 |
0.037 |
0.360 |
−0.498 |
−0.516 |
−0.698 |
 |
| | Fig. 4 Pictorial view of the analysis of quantum theory of atoms in molecules. | |
3.4 HOMO–LUMO analysis
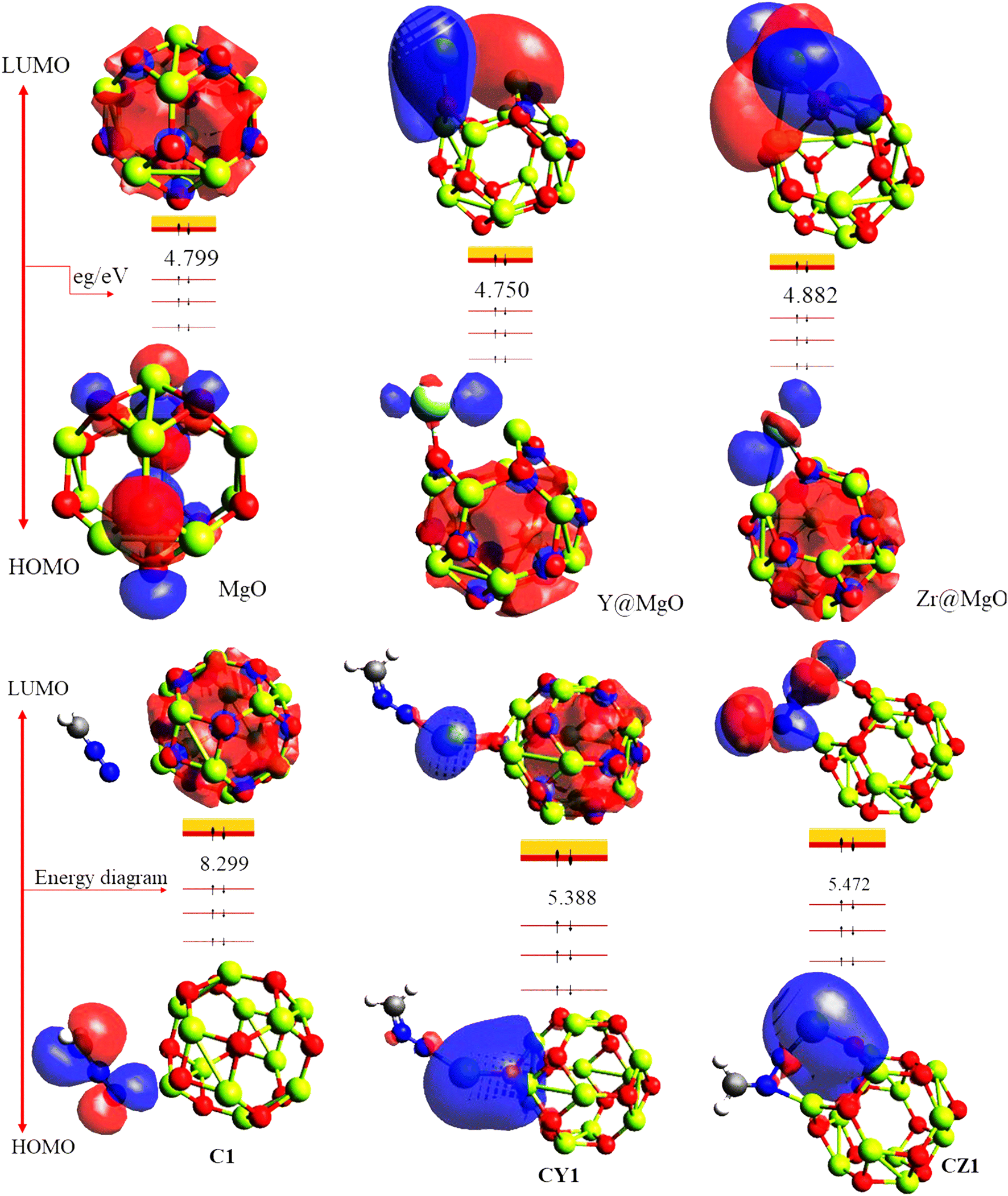
In the research of sensor materials, the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are particularly important.45–47 This is because the relevant information acquired through these properties can be used to predict the characteristics of a sensor material. The difference in the energy between the HOMO and LUMO is termed the energy gap.48 Energy gap is very crucial in affirming the sensitivity, conductivity, and selectivity of a sensor material, in that a small energy gap indicates an increase in the conductivity and reactivity of the studied materials. The HOMO, LUMO, and the corresponding energy gap of the studied systems are presented in Table 3, and the pictorial view of the charge distribution within the studied systems is presented in Fig. 5 for better understanding of their electronic distribution. The DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory was employed in this study to obtain the information presented herein. From Table 3, the pure-surface MgO nanocage was observed to have −6.579 eV as its ability to donate electrons from the outer shell; meanwhile, −1.780 eV was seen to be its capability to accept the loosely bonded electron, giving rise to an energy gap of 4.799 eV. This pure surface was further decorated to check the influence of the metal-decorated surface on gas adsorption. The decorated surface brought about a decrease in both the energies of the HOMO and LUMO, and consequently the energy gap of the system, which indicates an increase in the conductivity and sensitivity of the studied systems according to literature.49 Yttrium-decorated MgO (Y@MgO) was observed to have the HOMO values of −4.865 eV and a total decrease in the LUMO value of −0.116 eV, which give rise to an energy gap of 4.750 eV. Also, zirconium-decorated MgO (Zr@MgO) was observed to have a decreased HOMO value of −5.036 eV, with a corresponding LUMO value of −0.154 eV, giving rise to an energy gap of 4.882 eV. It can be seen that before adsorption of diazomethane (CH2N2), which is an extremely sensitive explosive yellow gas, Zr@MgO was observed to have the higher energy gap, which makes it more stable compared to the studied surface. Upon adsorption of the CH2N2 gas on the studied surface, C1 was observed to have a much higher energy gap of 8.299 eV, compared to the decorated surfaces CY1 and CZ1, with the energy gaps of 5.388 and 5.472, respectively. The drastic decrease in the energy gap of the decorated surface implies that decoration of the MgO nanocage increased the conductivity and sensitivity of the pure surface. This is in agreement with previously reported works that the doping and decoration of pure surfaces increases the conductivity and sensitivity of the studied systems.50 From Fig. 5, it can be observed that before adsorption of CH2N2 gas, the LUMO was highly delocalized at the decorated metals, while the HOMO was evenly distributed on both the surface and the adsorbate. It can be seen that the HOMO is highly distributed at the nanocage. After interaction of the surface with CH2N2, C1 was observed to have its HOMO highly distributed at the adsorbate, while the LUMO is on the nanocage. CY1 was observed to have its HOMO distributed through the decorated metal and the adsorbate, while the LUMO is highly distributed on the nanocage and the decorated metal. This was similarly observed on the CZ1 complex. In order to fully understand the behavior of diazomethane based on the electronic properties of the modelled systems, the global quantum descriptors were calculated by employing the popular Koopmans approximation.51 The quantum descriptors presented in Table 3 were calculated using equations presented in the literature.52 The famous Koopmans approximation53 was used to further understand and assess the sensitivity and conductivity of the examined sensor material in terms of chemical hardness (η), described as the resistance of atoms or atom groups to charge transfer;54 electronegativity (χ), the tendency of an atom or atom group to attract electrons;55 chemical potential (μ) or electronic potential, the measure of an atom or group of atoms' likelihood of escaping its ground/non-excited state;56 electrophilicity index (ω), a measure of the electron acceptor affinity to gain an additional electronic charge from the surrounding systems;57 and chemical softness (σ), which accurately depicts an atom's proclivity to accept electrons/electrons.58 As a result, these characteristics provide additional light on the reactivity, stability, and intermolecular interactions of chemical systems, as well as the electric and optical properties of the same systems, which were approximated using Koopmans' approximation and conceptual density theory. These characteristics, as shown in Table 3, were found to be in excellent agreement with other researched objectives and provide additional insight into the changes in the electrical properties of the studied system before and after adsorption.
Table 3 Energy of the highest occupied molecular orbital (EHOMO), lowest unoccupied molecular orbital (ELUMO), band/energy gap (Eg), electron potential (IP), electron affinity (EA), chemical hardness (η), chemical softness (σ), chemical potential (μ), electrophilicity (ω), and Fermi level energy (EFL), all in electron volt (eV)
| System |
HOMO |
LUMO |
Band gap |
IP |
EA |
σ |
η |
μ |
ω |
EFL |
| MgO |
−6.579 |
−1.780 |
4.799 |
6.579 |
1.780 |
0.417 |
2.399 |
−4.180 |
20.957 |
4.180 |
| Y@MgO |
−4.865 |
−0.116 |
4.750 |
4.865 |
0.116 |
0.421 |
2.374 |
−2.490 |
7.366 |
2.490 |
| Zr@MgO |
−5.036 |
−0.154 |
4.882 |
5.036 |
0.154 |
0.410 |
2.441 |
−2.595 |
8.220 |
2.595 |
| CH2N2@MgO (C1) |
−8.429 |
−0.130 |
8.299 |
8.429 |
0.130 |
0.121 |
4.149 |
−4.280 |
2.207 |
4.280 |
| CH2N2@YMgO (CY1) |
−5.614 |
−0.227 |
5.388 |
5.614 |
0.227 |
0.186 |
2.694 |
−2.921 |
1.583 |
2.921 |
| CH2N2@ZrMgO (CZ1) |
−5.578 |
−0.106 |
5.472 |
5.578 |
0.106 |
0.183 |
2.736 |
−2.842 |
1.476 |
2.842 |
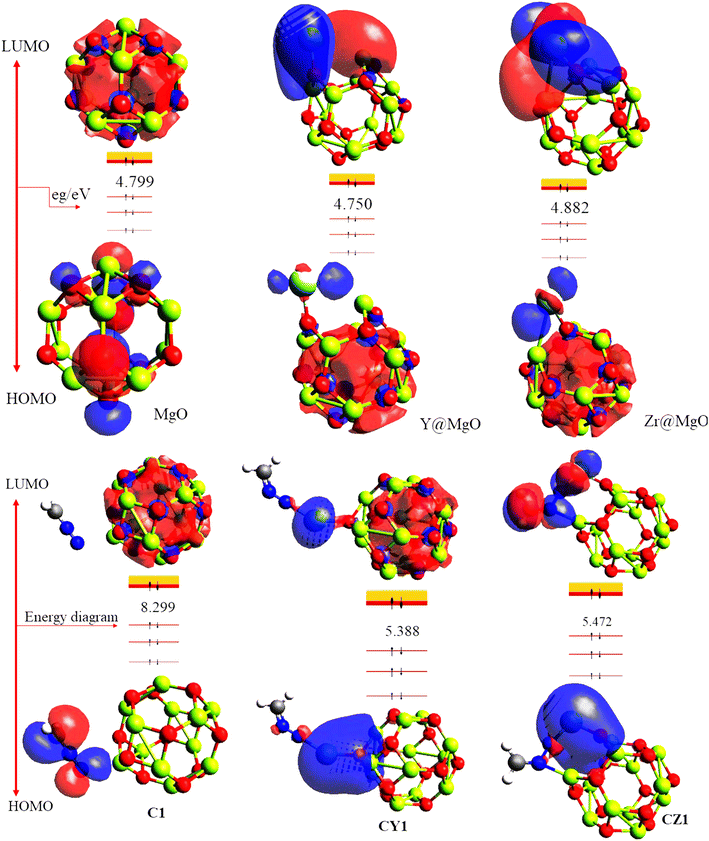 |
| | Fig. 5 Pictorial projection of the HOMO–LUMO isosurfaces of the studied compounds. | |
3.5 Sensor mechanisms
The characteristics and performance of a sensor device are determined by the sensor mechanism parameters.59 These parameters determine the adsorption potency, sensitivity, and conductivity of materials to be used as potential sensor device.60 The mechanisms of sensing include, but are not limited to, adsorption energy, conductivity, work function (φ), fraction of electron transfer, Fermi energy level, and recovery time.
3.5.1 Adsorption energy. Initial structural equilibration was carried out using the DFT/ωB97XD/SDD/GenECP/6-311++G(d,p) level of theory. To ascertain stable adsorption configuration, the studied complexes, surfaces, and gas molecule were optimized separately. Minimum energy configurations were carried out via optimization.61–66 Table 4 presents the calculated adsorption energy values for the investigated system. The adsorption energy values of −4.817 eV and −7.422 eV obtained for CH2N2@YMgO (CY1) and CH2N2@ZrMgO (CZ1), respectively, depict negative adsorption enthalpies. The adsorption mechanism is best described as chemisorption, owing to the negative adsorption enthalpy. For CH2N2@MgO (C1), the adsorption value of 12.520 eV was obtained. Due to the positive adsorption energy, the adsorption mechanism is best described as physisorption. The greater the negative value of the adsorption energy, the better the adsorption performance.67,68 Among the studied complexes, CZ1 complex exhibits the greatest negative adsorption energy of −7.422 eV, indicating that CZ1 complex shows the best adsorption performance among its studied counterparts.
Table 4 The adsorption energy and selected sensor parameter values for the studied complexes
| Complex |
Ecomplex |
Esurface |
Egas |
Eadsorb |
EFL |
φ |
| CH2N2@MgO (C1) |
−3453.861587 |
−3305.703928 |
−148.617753 |
12.520 |
4.280 |
−4.280 |
| CH2N2@YMgO (CY1) |
−3492.219653 |
−3343.424884 |
−148.617753 |
−4.817 |
2.921 |
−2.921 |
| CH2N2@ZrMgO (CZ1) |
−3500.974304 |
−3352.083800 |
−148.617753 |
−7.422 |
2.842 |
−2.842 |
3.5.2 Conductivity. Variability in the electronic behavior of the studied complexes could lead to changes in the conductivity and resistivity of that system. The ability to move electrons from the valence band to the conduction band is affected by electrical conductivity.69 The relation between conductivity and energy gap is shown in eqn (9):From eqn (9), the electrical conductivity, constant, temperature, and Boltzmann constant are represented by σ, A, T, and K, respectively. The sensing response was analyzed based on eqn (10),
| |
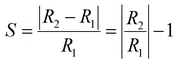 | (10) |
R1 and
R2 denote the resistivity of the MgO, YMgO and ZrMgO nanocages and their complexes, respectively. The inverse relationship between electrical conductivity and resistivity is shown in
eqn (11).
| |
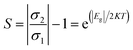 | (11) |
σ1 and
σ2 are the electrical conductivities of the MgO, YMgO, and ZrMgO nanocages and complexes, respectively.
Table 3 lists the changes in HOMO value, LUMO value, and energy gap of the studied nanocages after interaction with CH
2N
2 molecules. The least energy gap values of 5.388 eV and 5.578 eV are observed for
CY1 and
CZ1 complexes, respectively, indicating better sensitivity and electrical conductivity as compared to the
C1 complex.
3.5.3 Recovery time. The strength of interaction is among the essential parameters in designing sensor devices. The strength of an adsorption process can be determined by the interaction strength.70 The magnitude of adsorption energy can be related to the recovery time, which in turn depicts the interaction strength.71 The direct relation between adsorption energy and recovery time depicts that the greater the adsorption energy, the greater the recovery time.72 This relation is presented in eqn (12). From Table 4, CZ1 complex with the greatest negative adsorption energy (EAd = −7.422) has the greatest recovery time (eqn (12)).V0 is the attempt frequency, K is the Boltzmann constant (approx. ∼2.0 × 10−3 kcal mol−1 K−1), and T is the temperature.
3.6 Second-order perturbation theory analysis
Knowledge of inter- and intramolecular charge transfer, as well as system conjugation, is critical in adsorption investigations. This is because information concerning inter-charge transfer aids in recognizing a sensor material's capacity to easily interact with the adsorbate.73,74 Natural bond orbital analysis is one of the available computational tools that have recently been used to study inter- and intramolecular charge transfer, resulting in a detailed understanding of the bonding concept. It was used in this study to obtain more information about the electronic charge transfer, conductivity, and sensitivity of the pure MgO nanocage and its decorated surface towards the adsorption of CH2N2 gas. The inter- and intramolecular charge transfer in this inquiry was carried out using NBO analysis utilizing the DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory, and the results are shown in Table 5. The second-order perturbation energy measures the type and strength of the interaction between the donor and acceptor orbitals in the adsorbate–adsorbent interaction. According to the literature, the larger the second-order perturbation energy E(2), which is measured in kcal mol−1, the stronger the interaction and the adsorption of such sensor material. Using eqn (13), the interaction of pure MgO and its decorated metals (Y and Zr) on the adsorption of CH2N2 gas was evaluated.| |
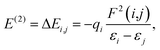 | (13) |
where qi is the donor occupancy, εi, and εj stand for the diagonal elements in the Fock matrix, and F(i,j) represents the off-diagonal elements of the Fock matrix, respectively.
Table 5 Donor (i), acceptor (j), transitions, stabilization energies, diagonal and off-diagonal elements of the studied systems
| System |
Transition |
Donor (i) |
Acceptor (j) |
E(2)/kcal mol−1 |
E(j−) − E(i) |
F(i,j) |
| MgO |
LP → LP* |
LP(3)O1 |
LP*Mg13 |
6.08 |
0.54 |
0.052 |
| LP(4)O12 |
LP*Mg22 |
10.95 |
1.03 |
0.097 |
| Y@MgO |
LP → LP* |
LP(4)O1 |
LP*Mg13 |
10.00 |
1.29 |
0.146 |
| LP(3)O4 |
LP*Mg15 |
5.46 |
0.58 |
0.074 |
| Zr@MgO |
LP → LP* |
LP(4)O1 |
LP*Mg13 |
21.19 |
1.27 |
0.149 |
| LP → σ* |
LP(2)O1 |
σ*Mg15–Zr25 |
11.11 |
0.61 |
0.075 |
| CH2N2@MgO (C1) |
LP → LP* |
LP(3)O1 |
LP*Mg13 |
11.52 |
0.72 |
0.083 |
| LP(4)O1 |
LP*Mg13 |
21.91 |
1.25 |
0.151 |
| CH2N2@YMgO (CY1) |
LP → LP* |
LP(4)O2 |
LP*Mg13 |
6.54 |
1.28 |
0.118 |
| σ → σ* |
σN26–N27 |
σ*Mg23–Y25 |
0.04 |
0.96 |
0.008 |
| LPN26 |
LP*Y25 |
22.58 |
0.83 |
0.177 |
| CH2N2@ZrMgO (CZ1) |
LP → LP* |
LP(3)O1 |
LP*Mg13 |
10.90 |
0.74 |
0.082 |
| LP(4)O1 |
LP*Mg13 |
21.08 |
1.29 |
0.150 |
| LP(4)O2 |
LP*Mg24 |
15.82 |
1.26 |
0.129 |
| LP(2)O3 |
σ*O4 – Zr25 |
0.08 |
0.63 |
0.006 |
As presented in Table 5, three major transitions were observed between the donor and acceptor orbitals. The transitions were LP → LP*, LP → σ*, and σ → σ*, and it could be seen that the major contribution in the electronic transition was from LP → LP*, which could be explained based on the lone pair of electrons from the nitrogen atom in the adsorbate and the decorated metals. The result presented herein shows that there was an increase in the stabilization energy of the studied system upon decoration of the pure surface of MgO. This is in agreement with the frontier molecular orbital analysis due to the same observation upon decoration of the surface. Before adsorption, the perturbation energy of the studied surface was observed to have the range MgO < Y@MgO < Zr@MgO, with the higher values of 10.95 kcal mol−1, 10.00 kcal mol−1, and 21.19 kcal mol−1. After adsorption, an increase in the values of the perturbation energies was observed, which implies an increase in the strength of gas adsorption on the studied decorated surface. As mentioned earlier, higher values of stabilization energy indicate stronger interaction and increase the conductivity of the studied systems. From Table 5, after adsorption, (C1) was observed to have the higher perturbation energy at the following donor acceptor orbitals: LP(3)O1 → LP*Mg13 and LP(4)O1 → LP*Mg13, with the energy values of 11.52 kcal mol−1 and 21.91 kcal mol−1. Similarly, yttrium-decorated MgO (CY1) was observed to have an increase in perturbation energy from LP(4)O2 → LP*Mg13, σN26–N27 → σ*Mg23–Y25 and LPN26 → LP*Y25, with the perturbation energies of 6.54 kcal mol−1, 0.04 kcal mol−1, and 22.58 kcal mol−1, respectively. On the other hand, decoration of the pure MgO nanocage with Zr metal was observed to increase the adsorption strength of the surface; this phenomenon was also observed in the HOMO–LUMO analysis as the decoration brought about a decrease in energy gap of the system, indicating the increased electrical conductivity and sensitivity of the system. The CZ1 system was observed to have only one major contribution of LP → LP* in the orbital interactions. The higher values of the stabilization energies were from LP(3)O1 → LP*Mg13, LP(4)O1 → LP*Mg13, and LP(4)O2 → LP*Mg24, corresponding to the perturbation values of 10.90 kcal mol−1, 21.08 kcal mol−1, and 15.82 kcal mol−1. From this analysis, it can be inferred that decoration of the pure nanocage with selected metals increases the strength of the interaction between the donor and acceptor orbitals. It is very important to point out here that an inverse relationship exists between the natural bond orbital (NBO) analysis and the frontier molecular orbitals, such that the system with the least energy gap of 5.388 eV had the higher perturbation energy of 22.58 kcal mol−1.
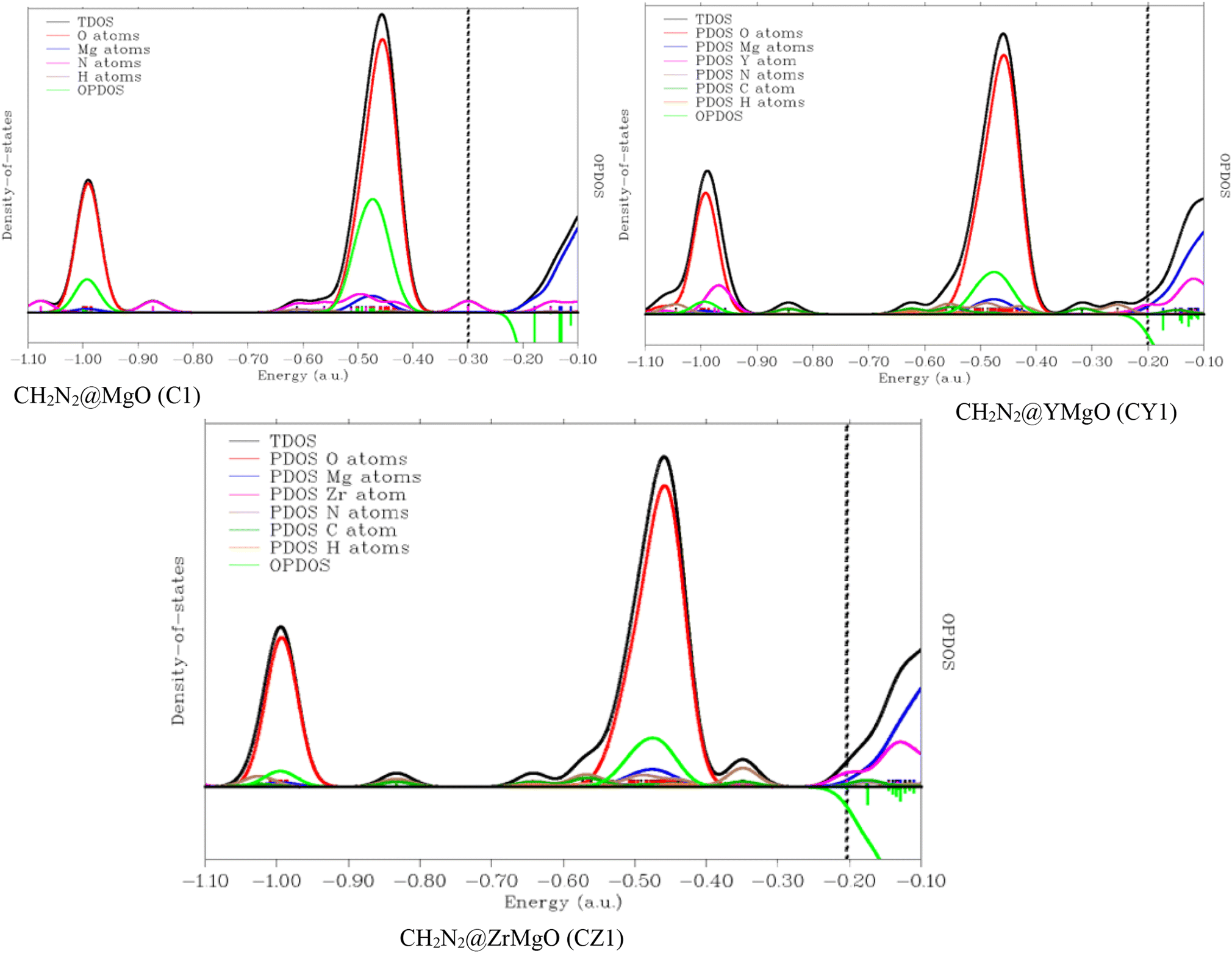
3.7 Density of states (DOS) analysis
Understanding the changes in the electrical characteristics of pure MgO surfaces and their Y- and Zr-decorated counterparts following CH2N2 adsorption is critical, since it aids in anticipating the sensing applications of these surfaces.75 In order to adequately compare the electrical characteristics of MgO and its decorated surface, the geometry energy of the modelled systems was reduced and brought to a minimum using system geometry optimization. Fig. 1 and 2 show the optimized structures, and the detailed electrical parameters of the simulated systems were determined at DFT/ωB97XD/6-311++G (d,p) level of theory. We computed the electronic density of states (DOS), partial density of states (PDOS), and total density of states (TDOS) for the complexes, i.e., the interaction between the absorbate and surface, and the results are shown in Fig. 6. Fig. 6 shows the DOS plotted in the range of −5.0 a.u. to 25.0 a.u. and −2.0 a.u. to 27.0 a.u. for the adsorbent–adsorbate interaction. This DOS was carried out to analyze the mechanism and sensing ability of the studied systems and the changes in the electronic states of the modelled senor material upon adsorption of the detected gas. This analysis helps in understanding the changes in the electronic states, which helps in determining the surface that best detects CH2N2 gas. A discrete DOS across the Fermi level for all the modelled systems (C1, CY1, and CZ1) in detecting CH2N2 gas molecules was observed, demonstrating their metallic nature most especially in the decorated surface.76 DOS of the investigated systems was obtained by employing Multiwfn software. Furthermore, in the neighborhood of the Fermi level, CY1 has the maximum availability of electronic states, although the difference in DOS for all the modelled systems is not significant. As seen in Fig. 6, a number of peaks are also observed in the valence and conduction bands; as a result, the observed CH2N2 aided variations in the electronic properties of the DOS of MgO, and its decorated surfaces are significant enough to be used for sensing applications. The antibonding behavior of the OPDOS found here might be better explained by the unfavorable overlapping in the orbital phase. It is obvious that the covalently bound atoms are the sole contributors to the occupied border MOs. The left axis correlates to TDOS and PDOS, whereas the right axis refers to OPDOS. The distinct lines in red, blue, pink, and green show the PDOS of MgO, N, Y, and Zr and the overlapping partial density of states, respectively.
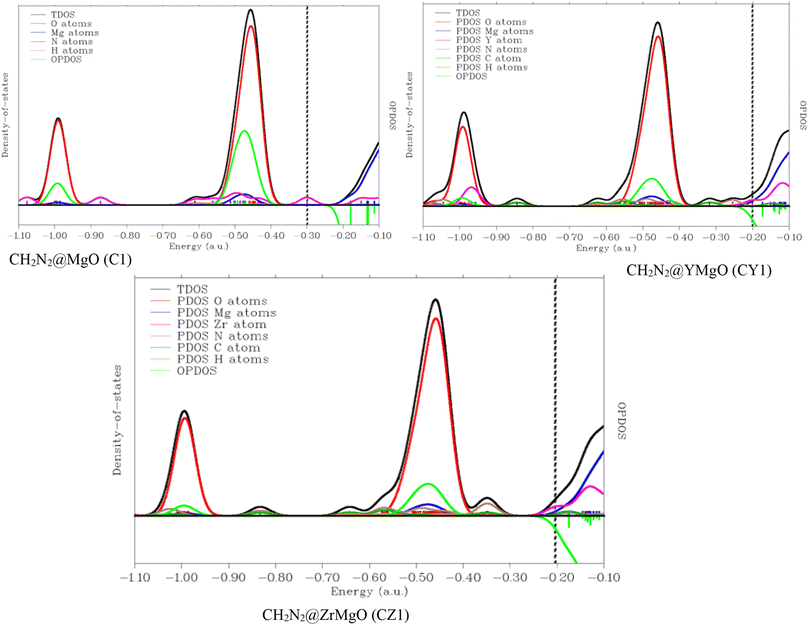 |
| | Fig. 6 Density of states (DOS) plots for CH2N2@MgO (C1), CH2N2@MgO (CY1), and CH2N2@ZrMgO (CZ1) obtained from multifunctional wave function analyzer. | |
3.8 Electron localization function
Becke and Edgecombe76 developed a valuable topological analysis called the electron localization function, which was first constructed in the framework of the Hartree–Fock approximation and shows the chance of locating an electron in the vicinity of another electron with the same spin. The Pauli exclusion principle has been extensively researched using ELF.77 This approximation is technically stated as follows:| |
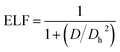 | (14) |
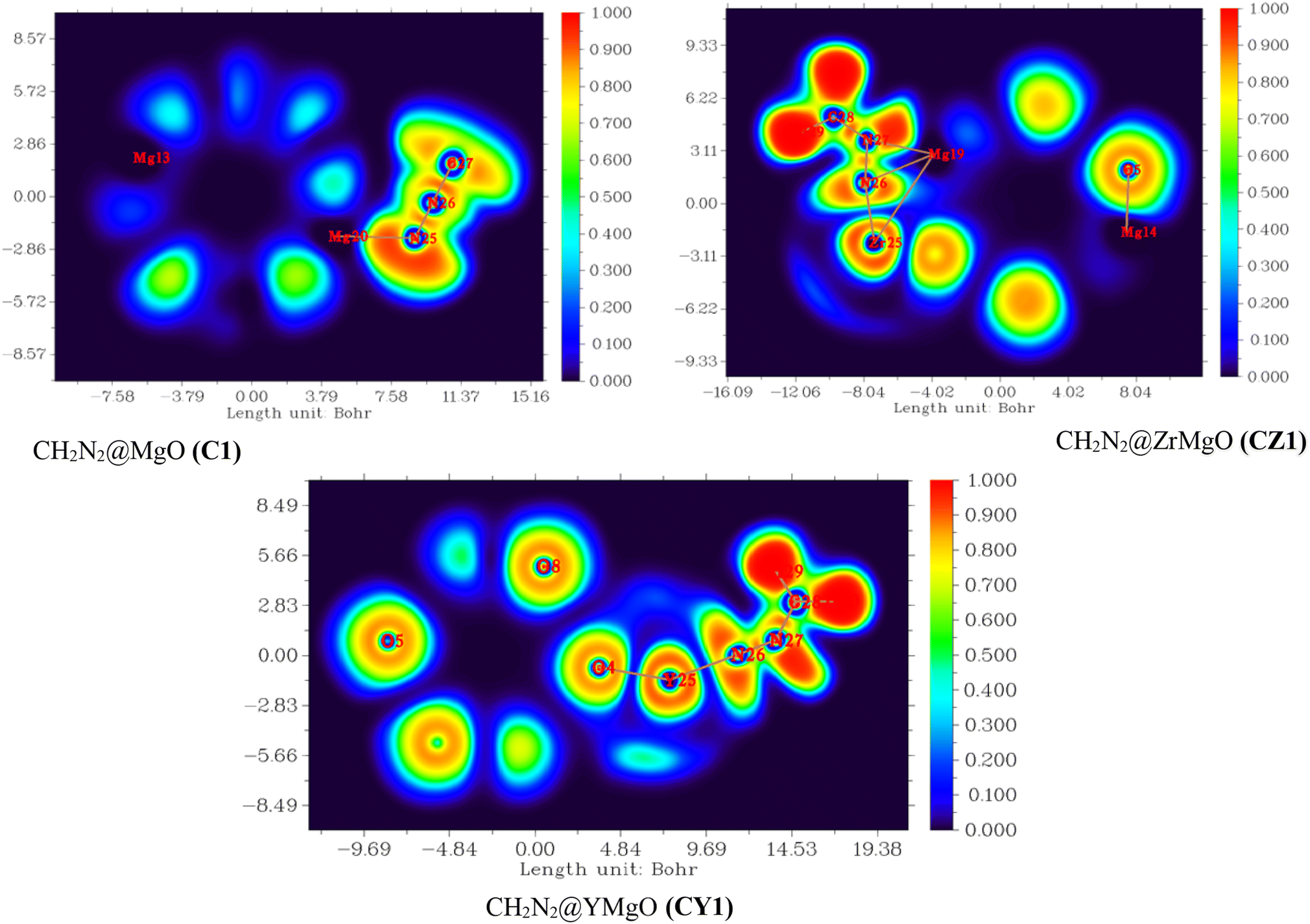
From this equation, D and Dh are the curvature of the electron-pair density for an identical-spin electron system. Literature review shows that the region occupied by paired electrons and kinetic energy usually takes a lower value reading than the values of ELF close to unity, indicating a localized domain, although low values reflect a metallic nature.78 From the QTAIM, which is one of the topological analyses, the following ELF values were observed for the studied systems (C1): 0.679 a.u., (CY1) 0.109 a.u., and (CZ1) with 0.206 a.u, and 0.670 a.u., respectively. The values obtained showed the interaction of CH2N2@YMgO (CY1) to be closer to zero, while its counterparts are close to unity. To further analyze the chemical bonding concept and to support the QTAIM analysis, topological analysis of the electron localization function ELF was employed in this study. ELF is reported to be the local measure of the effect of Pauli repulsion on the kinetic energy density. Weaker repulsion in the region of space is observed to have an ELF closer to one; hence, as previously reported in Table 2, C1 and CZI were observed to have weaker repulsion. However, CY1 had parallel pairing, since the ELF value was observed to be closer to zero. Different maps of the electron localization function, as presented in Fig. 7, were calculated with the help of MultiWfn as previously reported in the computational details. In Fig. 7, the dark-blue region is an indication of a decrease in the electron delocalization function. This implies that electrons tend to be localized in the outer side of the region. As we can see from the pictorial view of the HOMO–LUMO analysis, for C1, the electron exhibits a localization domain at the adsorbate; for the decorated surface CY1, there was an ionic bond formation that exhibits a localization domain between Y23–N25, and the localized region extends towards Y23 due to the charge transfer from the surface to the absorbate CH2N2. Similarly, for CZ1 complex, the electron delocalization function was mostly localized at Zr25–N25 and Mg10–N27. As a result, the HOMO–LUMO analysis, QTAIM, and ELF all indicate the same pattern. Thus, ELF topological analysis offers a thorough tool for evaluating the electrophilic substitution site and, more broadly, for localizing reactive sites in clusters. This also gives qualitative visual information via the graphical depiction of the regions of localization. The ELF descriptive approach augments the reactive sites of the cluster afforded by the molecules' frontier orbitals.
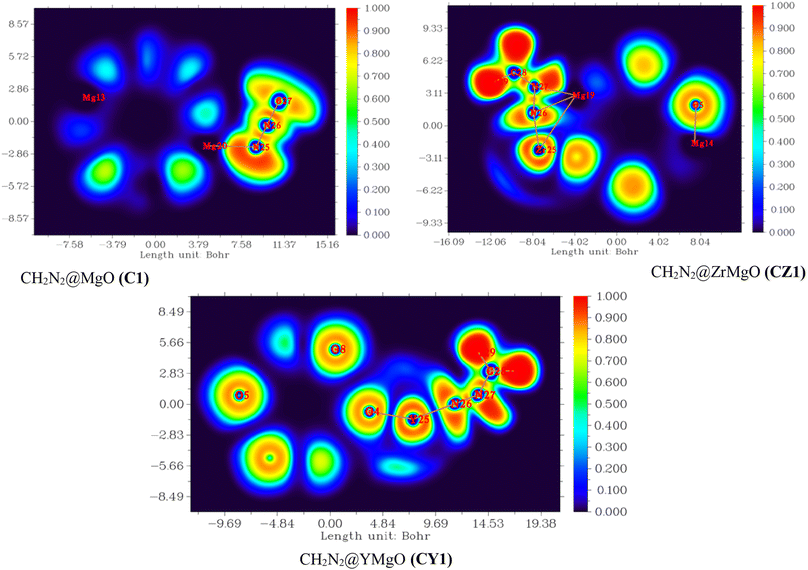 |
| | Fig. 7 Electron localization function (ELF) map for CH2N2@MgO (C1), CH2N2@YMgO (CY1), and CH2N2@ZrMgO (CZ1) obtained from the multifunctional wave function analyzer. | |
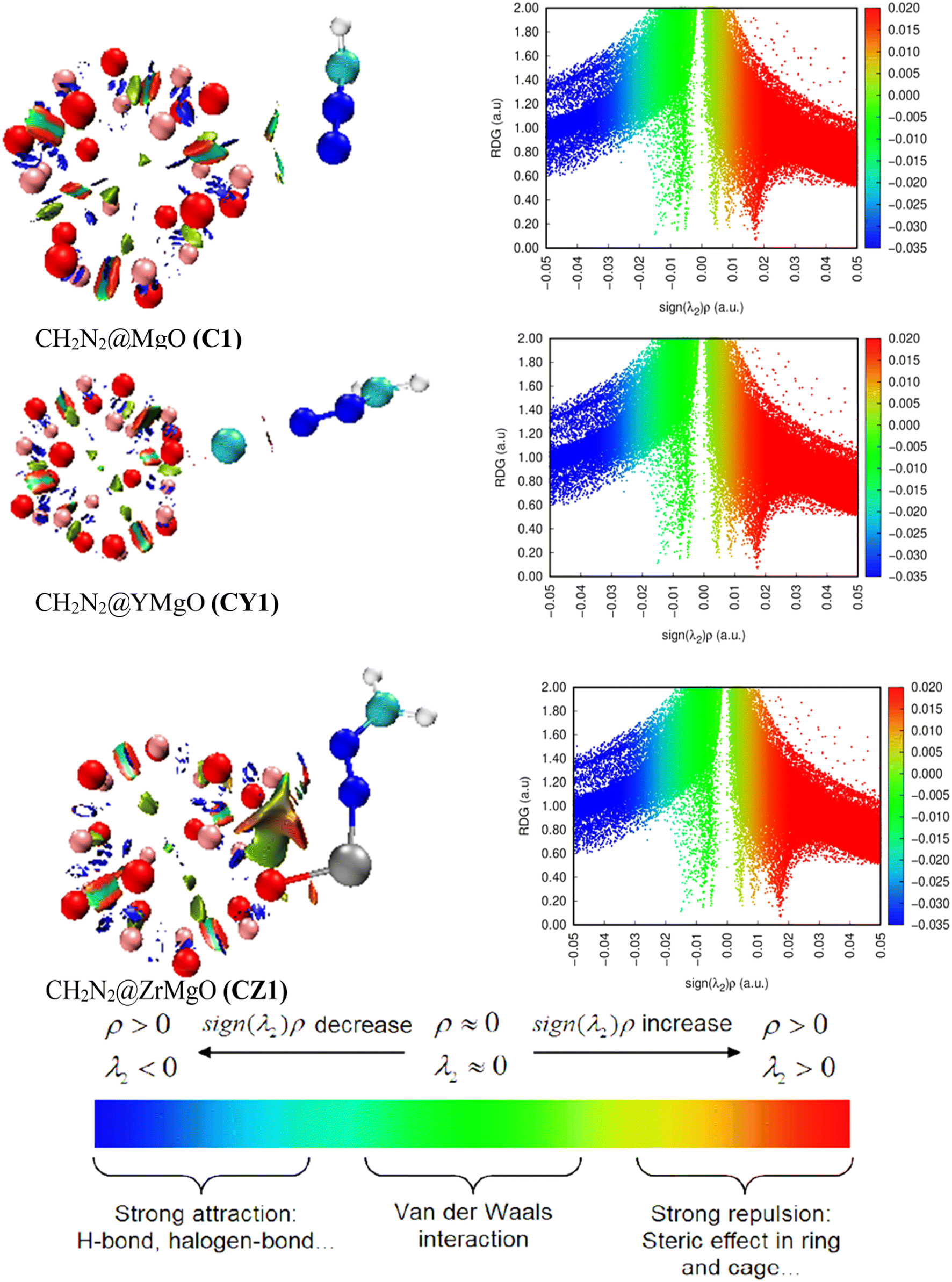
3.9 Noncovalent interaction
The DFT/ωB97XD method at the GenECP/6-311++G(d,p)/SDD level of theory was used for the energy minimization of the pure MgO nanocage and its decorated nanomaterial alongside the adsorbate CH2N2. MultiWfn and VMD were used to obtain the pictorial view of the noncovalent interaction, as presented in Fig. 8. Noncovalent interaction (NCI) analysis was performed to illuminate and vividly quantify the inter- and intramolecular interactions between the surfaces and the adsorbate. As a result, NCI varies from covalent bond interaction in that it does not necessitate electron exchange but rather supports widespread variations in electromagnetic interactions between molecules or within a molecule.79 Electrostatic interaction effect, van der Waals forces, and hydrophobic effect are several dimensions or kinds of noncovalent contact that are usually stated to exist. Without a doubt, the formation of NCI is studied in terms of the release of chemical energy, which is typically on the scale of 1–5 kcal mol−1 (1000–5000 calories per 6.02 × 1023 molecules). Computational imaging of intramolecular and intermolecular interactions inside and between the complexed molecules showed numerous remarkable zones enabled by the isosurfaces, including van der Waals contacts, steric repulsion, and strong attractive interactions. From the NCI analysis, the green isosurfaces show weak interactions caused by van der Waals dispersion forces, which are used to explain charge fluctuation, resulting in a nonspecific and nondirectional attraction. The red isosurfaces, on the other hand, show strong interactions caused mostly by steric repulsion, but they also impact the conformation and reactivity of ions and molecules. The isosurfaces are depicted with colors ranging from blue to red depending on the values of λ2. Hydrogen bonding is represented by deep blue color, while red indicates repulsive forces. According to literature review, small changes in the electronic density result in a large change in the reduced density gradient value, and the interaction in NCI is characterized by the Laplacian of the electron density (∇ρ(r)), along with three principal axes with three eigenvalues I of the Hessian matrix and the Laplacian, and is expressed in eqn (15).| | |
(∇ρ(r)) = λ1 + λ2 + λ3,
| (15) |
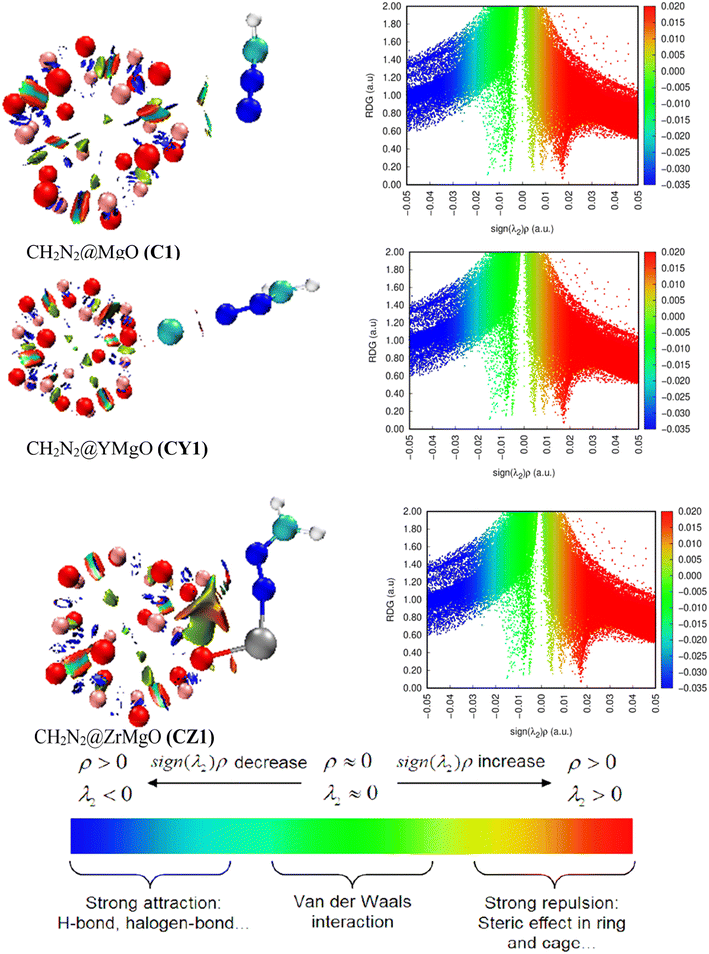 |
| | Fig. 8 Visualization of noncovalent interactions for CH2N2@MgO (C1), CH2N2@YMgO (CY1), and CH2N2@ZrMgO (CZ1) obtained from the VMD package. | |
From eqn (15), much information about the type of bond formation is examined by λ2 in such a way that if λ2 has negative values, the contact is considered hydrogen bonding, whereas positive values of λ2 represent repulsive force. From the result of the QTAIM analysis, C1 had −0.245 a.u., CY1 had −0.101 a.u., and CZ1 had −0.200 a.u. and −0.516 a.u., respectively, indicating that all the studied systems had strong interaction between the surface and the adsorbate. For better understanding, Fig. 8 presents the NCI 2D-RGD graph. From the plot, a wide blue peak was observed from the C1 complex, confirming the higher positive values of the adsorption energy, which is an indication of the physically adsorbed system. On the other hand, the two decorated complexes were observed to have higher spike peaks spread around the decorated atoms. The appearance of the green isosurface in the 3D plot of the CY1 complex reveals intramolecular and intermolecular interactions. Furthermore, among the three complexes, the depth of the blue color bonded by the RDG isosurfaces and the accompanying spike peaks are very similar, indicating strong attraction between the modelled nanosurfaces and the interacted gas (CH2N2), as the spike peaks are approximately ranged from 0.00 to 1.40 a.u., respectively.
4 Conclusions
This study extensively analyzed yttrium- and zirconium-decorated Mg12O12–X (X = Y, Zr) nanoclusters as sensors for diazomethane (CH2N2) gas utilizing the power of DFT at the ωB97XD/GenECP/6-311++G(d,p)/SDD level of theory. These modelled systems CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO, along with the pristine MgO nanocages, were investigated with the goal of constructing a suitable nanosensor material for effective adsorption of CH2N2 gas molecules. Bond lengths within the MgO cage were observed as Mg15–O4 (1.896 Å), Mg19–O4 (1.952 Å), and Mg23–O4 (1.952 Å). These insights shed light on the structural properties and interactions with CH2N2 gas. Post-interaction, CH2N2@MgO exhibited shorter bond lengths; CH2N2@YMgO had longer bond lengths; and CH2N2@ZrMgO showed shorter bond lengths except for Mg19–O4. CH2N2@ZrMgO achieved greater stability due to a shorter bond distance. Electronic properties were investigated, revealing changes in the energy gap, which influenced electrical conductivity and sensitivity. The energy gap increased for Zr@MgO, CH2N2@MgO, CH2N2@YMgO, and CH2N2@ZrMgO, indicating stable interactions on the MgO surface. Y@MgO exhibited a decreased energy value, suggesting an increased conductivity and reactivity. The pure MgO surface donated and accepted electrons, resulting in an energy gap of 4.799 eV. Decoration with yttrium and zirconium decreased the energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), as well as the energy gap, indicating increased conductivity and sensitivity. Zr@MgO had the highest energy gap before CH2N2 adsorption, but after adsorption, C1 exhibited a significantly higher energy gap, indicating increased conductivity and sensitivity. The quantum theory of atoms in molecules (QTAIM) revealed partial covalent bonding in all complexes. Intermolecular interactions were indicated by λ1/λ3 values, with CH2N2@YMgO and CH2N2@ZrMgO showing strong interactions. Delocalization of the LUMO and distribution of the HOMO were observed before and after CH2N2 adsorption, consistent with previous studies on enhanced conductivity and sensitivity through surface decoration. Regarding adsorption energy, CH2N2@YMgO (CY1) showed an energy of −4.817 eV, while CH2N2@ZrMgO (CZ1) had an even more negative value of −7.422 eV, indicating chemisorption with negative enthalpy. In contrast, CH2N2@MgO (C1) had a positive adsorption energy of 12.52 eV, suggesting physisorption. CZ1 exhibited the highest negative adsorption energy of −7.422 eV, indicating superior adsorption performance. Adsorption energy was correlated with recovery time, with higher energy leading to longer recovery times. CZ1, with the most negative energy of −7.422 eV, had the longest recovery time. This study provides insights into CH2N2 gas sensing mechanisms on nanocage surfaces, including adsorption energy, conductivity changes, and recovery times, with implications for gas-sensing applications.
Data availability
All data are contained within the manuscript and ESI.†
Authors contributions
Hitler Louis: project conceptualization, design, and supervision. Terkumbur E. Gber: writing, results extraction, analysis, and manuscript first draft. Obinna C. Ngana and Ismail O. Amodu: manuscript revision, review, and proofreading. Ernest E. Ekereke and Innocent Benjamin: manuscript proofreading. Adedabo Adeyinka: resources, review, and editing.
Conflicts of interest
All authors declare zero financial or interpersonal conflict of interest that could have influenced the research work or results reported in this research paper.
Acknowledgements
This research was not funded by any governmental or nongovernmental agency. The authors would like to acknowledge the Centre for High Performance Computing (CHPC) at the University of Johannesburg, South Africa, for providing computational resources for this research project.
References
- A. S. Perry, I. Yamamoto, I. Ishaaya and R. Y. Perry, Insecticides in agriculture and environment: retrospects and prospects, Springer Science & Business Media, 2013, vol. 146, p. 111180 Search PubMed.
- R. A. Maurya, C. P. Park, J. H. Lee and D. P. Kim, Continuous in situ generation, separation, and reaction of diazomethane in a dual-channel microreactor, Angew. Chem., Int. Ed. Engl., 2011, 50, 5952–5955 CrossRef CAS PubMed.
- D. Dallinger, B. Gutmann and C. O. Kappe, The concept of chemical generators: on-site on-demand production of hazardous reagents in continuous flow, Acc. Chem. Res., 2020, 53, 1330–1341 CrossRef CAS PubMed.
- W. Braun, A. M. Bass and M. Pilling, Flash photolysis of ketene and diazomethane: The production and reaction kinetics of triplet and singlet methylene, J. Chem. Phys., 1970, 52, 5131–5143 CrossRef CAS.
- H. Yang, B. Martin and B. Schenkel, On-demand generation and consumption of diazomethane in multistep continuous flow systems, Org. Process Res. Dev., 2018, 22, 446–456 CrossRef CAS.
- R. Kipfer, W. Aeschbach-Hertig, F. Peeters and M. Stute, Noble gases in lakes and ground waters, Rev. Mineral. Geochem., 2002, 47, 615–700 CrossRef CAS.
- E. W. R. Steacie, The thermal decomposition of diazomethane, J. Phys. Chem., 2002, 35, 1493–1495 CrossRef.
- U. Abd Rani, L. Y. Ng, C. Y. Ng, E. Mahmoudi, Y. S. Ng and A. W. Mohammad, Sustainable production of nitrogen-doped carbon quantum dots for photocatalytic degradation of methylene blue and malachite green, J. Water Process Eng., 2021, 40, 101816 CrossRef.
- A. R. Ingole, S. R. Thakare, N. T. Khati, A. V. Wankhade and D. K. Burghate, Green synthesis of selenium nanoparticles under ambient condition, Chalcogenide Lett., 2010, 7, 485–489 CAS.
- Carbon nanotechnology: recent developments in chemistry, physics, materials science and device applications, ed. L. Dai, 2006. vol. 26, pp. 274–286 Search PubMed.
- S. Mallakpour and V. Behranvand, Polymeric nanoparticles: Recent development in synthesis and application, eXPRESS Polym. Lett., 2016, 10, 895 CrossRef CAS.
- A. S. Sethulekshmi, J. S. Jayan, A. Saritha and K. Joseph, Insights into the reinforcibility and multifarious role of WS2 in polymer matrix, J. Alloys Compd., 2021, 876, 160107 CrossRef CAS.
- M. D. Mohammadi, H. Y. Abdullah, S. Bhowmick and G. Biskos, A comprehensive investigation of the intermolecular interactions between CH2N2 and X12Y12 (X= B, Al, Ga; Y= N, P, As) nanocages, Can. J. Chem., 2021, 99, 733–741 CrossRef CAS.
- M. Derakhshi, S. Daemi, P. Shahini, A. Habibzadeh, E. Mostafavi and A. A. Ashkarran, Two-dimensional nanomaterials beyond graphene for biomedical applications, J. Funct. Biomater., 2022, 13, 27 CrossRef CAS PubMed.
- M. D. Mohammadi, H. Y. Abdullah, S. Bhowmick and G. Biskos, Theoretical investigation of X12O12 (X= Be, Mg, and Ca) in sensing CH2N2: A DFT study, Comput. Theor. Chem., 2021, 1198, 113–168 Search PubMed.
- C. Gao, Z. Guo, J. H. Liu and X. J. Huang, The new age of carbon nanotubes: An updated review of functionalized carbon nanotubes in electrochemical sensors, Nanoscale, 2012, 4, 1948–1963 RSC.
- O. K. Andersen, S. T. John, P. Jena, C. T. Chan, S. Q. Shen, Z. Liu, and N. H. Quang, Day 3: Thursday, 8 September 2011. In Welcome Message from Conference Chairmen Info-4 Acknowledgement Info-5 Committees Info-6 Conference General Information Info-8 Conference Location Info-11, 2011, vol. 5, pp. 1070–1081 Search PubMed.
- C. Liu, Z. Hu, Q. Wu, X. Wang, Y. Chen, H. Sang and N. Xu, Vapor− solid growth and characterization of aluminum nitride nanocones, J. Am. Chem. Soc., 2005, 127, 1318–1322 CrossRef CAS PubMed.
- J. Gangwar, B. K. Gupta and A. K. Srivastava, Prospects of Emerging
Engineered Oxide Nanomaterials and their Applications, Def. Sci. J., 2016, 66, 34–40 Search PubMed.
- I. Y. Jeon, D. Chang, N. A. Kumar and J. B. Baek, Functionalization of carbon nanotubes, Carbon Nanotubes: Polym. Nanocompos., 2011, 6, 91–110 Search PubMed.
- M. D. Mohammadi, H. Y. Abdullah, S. Bhowmick and G. Biskos, Theoretical investigation of X12O12 (X= Be, Mg, and Ca) in sensing CH2N2: A DFT study, Comput. Theor. Chem., 2021, 1198, 113168 CrossRef CAS.
- M. Solimannejad, S. Kamalinahad and E. Shakerzadeh, Silicon carbide nanotubes (SiCNTs) serving for catalytic decomposition of toxic diazomethane (DAZM) gas: a DFT study, Mol. Phys., 2018, 116, 414–422 CrossRef CAS.
- H. Louis, I. O. Amodu, T. O. Unimuke, T. E. Gber, B. B. Isang and A. S. Adeyinka, Modeling of Ca12O12, Mg12O12, and Al12N12 nanostructured materials as sensors for phosgene (Cl2CO), Mater. Today Commun., 2022, 32, 103946 CrossRef CAS.
- H. M. Badran, K. M. Eid, S. Baskoutas and H. Y. Ammar, Mg12O12 and Be12O12 Nanocages as Sorbents and Sensors for H2S and SO2 Gases: A Theoretical Approach, Nanomaterials, 2022, 12, 1757 CrossRef CAS PubMed.
- W. Emori, H. Louis, S. A. Adalikwu, R. A. Timothy, C. R. Cheng, T. E. Gber and A. S. Adeyinka, Molecular modeling of the spectroscopic, structural, and bioactive potential of tetrahydropalmatine: insight from experimental and theoretical approach, Polycyclic Aromat. Compd., 2022, 1–18 Search PubMed.
- B. Shi, L. Yuan, T. Tang, Y. Yuan and Y. Tang, Study on electronic structure and excitation characteristics of cyclo [18] carbon, Chem. Phys. Lett., 2020, 741, 136975 CrossRef CAS.
- H. Louis, O. J. Ikenyirimba, T. O. Unimuke, G. E. Mathias, T. E. Gber and A. S. Adeyinka, Electrocatalytic activity of metal encapsulated, doped, and engineered fullerene-based nanostructured materials towards hydrogen evolution reaction, Sci. Rep., 2022, 12, 15608 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith, and J. M. Millam, GaussView, version 6.0. 16, Semichem Inc, Shawnee Mission KS, 2016, vol. 27 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- Z. Liu, T. Lu and Q. Chen, An sp-hybridized all-carboatomic ring, cyclo [18] carbon: Electronic structure, electronic spectrum, and optical nonlinearity, Carbon, 2020, 165, 461–467 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. D. Mohammadi, F. Abbas, H. Louis, L. E. Afahanam and T. E. Gber, Intermolecular interactions between nitrosourea and polyoxometalate compounds, ChemistrySelect, 2022, 7, e202202535 CrossRef CAS.
- H. Louis, D. Etiese, T. O. Unimuke, A. E. Owen, A. O. Rajee, T. E. Gber and E. N. Nfor, Computational design and molecular modeling of the interaction of nicotinic acid hydrazide nickel-based complexes with H 2 S gas, RSC Adv., 2022, 12, 30365–30380 RSC.
- F. Weinhold, The Path to Natural Bond Orbitals, Isr. J. Chem., 2022, 62, e202100026 CrossRef CAS.
- R. F. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- H. Louis, C. M. Chima, I. O. Amodu, T. E. Gber, T. O. Unimuke and A. S. Adeyinka, Organochlorine detection on transition metals (X= Zn, Ti, Ni, Fe, and Cr) anchored fullerenes (C23X), ChemistrySelect, 2023, 8, e202203843 CrossRef CAS.
- K. Gkionis, H. Kruse, J. A. Platts, A. Mladek, J. Koca and J. Sponer, Ion binding to quadruplex DNA stems. Comparison of MM and QM descriptions reveals sizable polarization effects not included in contemporary simulations, J. Chem. Theory Comput., 2014, 10(3), 1326–1340 CrossRef CAS PubMed.
- P. M. Utsu, C. A. Anyama, T. E. Gber, A. A. Ayi and H. Louis, Modeling of transition metals coordination polymers of benzene tricarboxylate and pyridyl oxime-based ligands for application as antibacterial agents, J. Indian Chem. Soc., 2023, 100993 CrossRef CAS.
- C. A. Anyama, H. Louis, B. E. Inah, T. E. Gber, J. O. Ogar and A. A. Ayi, Hydrothermal Synthesis, crystal structure, DFT studies, and molecular docking of Zn-BTC MOF as potential antiprotozoal agents, J. Mol. Struct., 2023, 1277, 134825 CrossRef CAS.
- M. I. Ofem, H. Louis, J. A. Agwupuye, U. S. Ameuru, G. C. Apebende, T. E. Gber, J. O. Odey, M. Neksumi and A. A. Ayi Ayi, Synthesis, spectral characterization, and theoretical investigation of the photovoltaic properties of (E)-6-(4-(dimethylamino) phenyl) diazenyl)-2-octyl-benzoisoquinoline-1, 3-dione, BMC Chem., 2022, 16, 109 CrossRef CAS PubMed.
- E. A. Eno, J. I. Mbonu, H. Louis, F. S. Patrick-Inezi, T. E. Gber, T. O. Unimuke and O. E. Offiong, Antimicrobial activities of 1-phenyl-3-methyl-4-trichloroacetyl-pyrazolone: Experimental, DFT studies, and molecular docking investigation, J. Indian Chem. Soc., 2022, 99, 100524 CrossRef CAS.
- C. G. Apebende, T. O. Magu, F. C. Asogwa, I. B. Onyebuenyi, T. O. Unimuke and T. E. Gber, Density functional theory study of the influence of activating and deactivating groups on Naphthalene, Results Chem., 2022, 4, 100669 CrossRef CAS.
- Y. Eken, N. Almeida, C. Wang and A. K. Wilson, SAMPL7: Host–guest binding prediction by molecular dynamics and quantum mechanics, J. Comput.-Aided Mol. Des., 2021, 35, 63–77 CrossRef CAS PubMed.
- M. I. Ofem, C. A. Ayi, H. Louis, T. E. Gber and A. A. Ayi, Influence of anionic species on the molecular structure, nature of bonding, reactivity, and stability of ionic
liquids-based on 1-butyl-3-methylimidazolium, J. Mol. Liq., 2023, 25, 122657 CrossRef.
- D. O. Odey, H. Louis, D. K. Ita, H. O. Edet, P. B. Ashishie, T. E. Gber and A. G. Effa, Intermolecular interactions of cytosine DNA nucleoside base with Gallic acid and its Methylgallate and Ethylgallate derivatives, ChemistrySelect, 2023, 8, e202203832 CrossRef CAS.
- F. Liu, N. Wang, C. Shi, J. Sha, L. Ma, E. Liu and N. Zhao, Phosphorus doping of 3D structural MoS2 to promote catalytic activity for lithium-sulfur batteries, J. Chem. Eng., 2022, 431, 133923 CrossRef CAS.
- H. Louis, T. E. Gber, F. C. Asogwa, E. A. Eno, T. O. Unimuke, V. M. Bassey and B. I. Ita, Understanding the lithiation mechanisms of pyrenetetrone-based carbonyl compound as cathode material for lithium-ion battery: Insight from first principle density functional theory, Mater. Chem. Phys., 2022, 278, 125518 CrossRef CAS.
- H. Liu, W. C. Silva, L. S. G. de Souza, A. Veiga, L. Seixas, K. Fujisawa and D. Grasseschi, 3d Transition Metal Coordination on Monolayer MoS2: A facile doping method to functionalize surfaces, Nanoscale, 2022, 38, 14595–14617 Search PubMed.
- L. E. Afahanam, H. Louis, I. Benjamin, T. E. Gber, I. J. Ikot and A. L. E. Manicum, Heteroatom (B, N, P, and S)-doped cyclodextrin as a hydroxyurea (HU) drug nanocarrier: a computational approach, ACS Omega, 2023, 8, 9861–9872 CrossRef CAS PubMed.
- M. A. Akpe, H. Louis, T. E. Gber, C. M. Chima, O. I. Brown and A. S. Adeyinka, Modeling of Cu, Ag, and Au-decorated Al12Se12 nanostructured as sensor materials for trapping of chlorpyrifos insecticide, Comput. Theor. Chem., 2023, 1226, 114218 CrossRef CAS.
- A. Farhan, E. U. Rashid, M. Waqas, H. Ahmad, S. Nawaz, J. Munawar and M. Bilal, Multifunctional graphene-based nanocomposites and nanohybrids for the abatement of agro-industrial pollutants in aqueous environments-A review, Environ. Pollut., 2022, 57, 119557 CrossRef PubMed.
- M. D. Mohammadi, H. Y. Abdullah, H. Louis and G. E. Mathias, 2D Boron Nitride Material as a sensor for H2SiCl2, Comput. Theor. Chem., 2022, 11, 37–42 Search PubMed.
- H. Louis, T. E. Gber, D. E. Charlie, T. C. Egemonye and M. M. Orosun, Detection of hydroxymethanesulfonate (HMS) by transition metal-anchored fullerene nanoclusters, J. Iran. Chem. Soc., 2023, 20, 713–729 CrossRef CAS.
- B. E. Inah, J. F. Eze, H. Louis, H. O. Edet, T. E. Gber, E. A. Eno and A. S. Adeyinka, Adsorption and gas-sensing investigation of oil dissolved gases onto nitrogen and sulfur doped graphene quantum dots, Chem. Phys., 2023, 10, 02–65 Search PubMed.
- J. A. Agwupuye, T. E. Gber, H. E. Edet, M. Zeeshan, S. Batool, O. E. Duke and G. E. Egbung, Molecular modeling, DFT studies and biological evaluation of methyl 2, 8-dichloro-1, 2-dihydroquinoline-3-carboxylate, Chem. Phys., 2023, 6, 100–146 Search PubMed.
- U. J. Undiandeye, H. Louis, T. E. Gber, T. C. Egemonye, E. C. Agwamba, I. A. Undiandeye and B. I. Ita, Spectroscopic, conformational analysis, structural benchmarking, excited state dynamics, and the photovoltaic properties of Enalapril and Lisinopril, J. Indian Chem. Soc., 2022, 99, 100500 CrossRef CAS.
- S. Sarfaraz, M. Yar, A. A. Khan, R. Ahmad and K. Ayub, DFT investigation of adsorption of nitro-explosives over C2N surface: Highly selective towards trinitro benzene, J. Mol. Liq., 2022, 352, 118–652 CrossRef.
- C. G. Apebende, H. Louis, A. E. Owen, I. Benjamin, I. O. Amodu, T. E. Gber and F. C. Asogwa, Adsorption properties of metal functionalized fullerene (C59Au, C59Hf, C59Ag, and C59Ir) nanoclusters for application as a biosensor for hydroxyurea (HXU): insight from theoretical computation, Z. Phys. Chem., 2022, 236, 1515–1546 CrossRef CAS.
- H. Louis, I. O. Amodu, E. A. Eno, I. Benjamin, T. E. Gber, T. O. Unimuke and A. S. Adeyinka, Modeling the Interaction of F-gases on Ruthenium-Doped Boron Nitridenanotube, Chem. Afr., 2023, 9, 1–19 Search PubMed.
- W. Yao, H. Guan, K. Zhang, G. Wang, X. Wu and Z. Jia, Nb-doped PtS2 monolayer for detection of C2H2 and C2H4 in on-load tap-changer of the oil-immersed transformers: A first-principles study, Chem. Phys. Lett., 2022, 802, 139755 CrossRef CAS.
- T. T. Uzah, I. J. Mbonu, T. E. Gber and H. Louis, Synergistic effect of KI and urea on the corrosion protection of mild steel in 0.5 M H2SO4: Experimental and computational insights, Results Chem., 2023, 10, 09–81 Search PubMed.
- G. Li, J. Li, W. Tan, M. Yang, H. Wang and X. Wang, Effectiveness and mechanisms of the adsorption of carbendazim from wastewater onto commercial activated carbon, Chemosphere, 2022, 13, 5231 Search PubMed.
- H. Louis, B. B. Isang, T. O. Unimuke, T. E. Gber, I. O. Amodu, A. I. Ikeuba and A. S. Adeyinka, Modeling of Al12N12, Mg12O12, Ca12O12, and C23N nanostructured as potential anode materials for sodium-ion battery, J. Solid State Electrochem., 2023, 27, 47–59 CrossRef CAS.
- E. I. I. Emmanuel, O. E. Duke, H. Louis, T. E. Gber, U. J. Undiandeye, A. Imojara and I. J. Ikot, Molecular Modeling of Cu (II), Zn (II), and Hg (II) Metal Complexes of (E)-Benzo [d][1, 3-dioxol-6-ylimino) methyl-4-bromophenol Schiffbase as Potential Antibacterial Agent, Chem. Afr., 2023, 28, 1–18 Search PubMed.
- K. Koperwas, F. Kaśkosz, F. Affouard, A. Grzybowski and M. Paluch, The role of the diffusion in the predictions of the classical nucleation theory for quasi-real systems differ in dipole moment value, Sci. Rep., 2022, 12, 1–10 CrossRef PubMed.
- E. C. Agwamba, H. Louis, P. O. Olagoke, T. E. Gber, G. A. Okon, C. F. Fidelis and A. S. Adeyinka, Modeling of magnesium-decorated graphene quantum dot nanostructure for trapping AsH 3, PH 3 and NH 3 gases, RSC Adv., 2023, 13, 13624–13641 RSC.
- M. Doust Mohammadi and H. Y. Abdullah, The adsorption of chlorofluoromethane on pristine, Al-, Ga-, P-, and As-doped boron nitride nanotubes: a PBC-DFT, NBO, and QTAIM study, ChemistrySelect, 2020, 5, 12115–12124 CrossRef CAS.
- H. O. Edet, H. Louis, T. E. Gber, P. S. Idante, T. C. Egemonye, P. S. Ashishie and A. S. Adeyinka, Heteroatoms (B, N, S) doped quantum dots as potential drug delivery system for isoniazid: insight from DFT, NCI, and QTAIM, Heliyon, 2023, 9, 131019 Search PubMed.
- S. J. Burden, B. D. Weedon, A. Turner, L. Whaymand, A. Meaney, H. Dawes and A. Jones, Intensity and duration of physical activity and cardiorespiratory fitness, Pediatrics, 2022, 26, 101930 Search PubMed.
- G. J. Ogunwale, H. Louis, T. E. Gber and A. S. Adeyinka, Modeling of pristine, Ir-and Au-decorated C60 fullerenes as sensors for detection of Hydroxyurea and Nitrosourea drugs, J. Environ. Chem. Eng., 2022, 10, 108802 CrossRef CAS.
- H. Louis, E. E. Ekereke, B. B. Isang, A. I. Ikeuba, I. O. Amodu, T. E. Gber and E. C. Agwamba, Assessing the Performance of Al12N12 and Al12P12 Nanostructured Materials for Alkali Metal Ion (Li, Na, K) Batteries, ACS Omega, 2022, 7, 46183–46202 CrossRef CAS PubMed.
- E. E. Ekereke, O. C. Ikechukwu, H. Louis, T. E. Gber, D. E. Charlie, A. I. Ikeuba and A. S. Adeyinka, Quantum capacitances of alkaline-earth metals: Be, Ca, and Mg integrated on Al12N12 and Al12P12 nanostructured—insight from DFT approach, Monatsh. Chem., 2023, 154, 355–365 CrossRef CAS.
- I. Oyo-Ita, H. Louis, V. C. Nsofor, H.
O. Edet, T. E. Gber, F. O. Ogungbemiro and A. S. Adeyinka, Studies on transition metals (Rh, Ir, Co) doped silicon carbide nanotubes (SiCNT) for the detection and adsorption of acrolein: Insight from DFT approach, Mater. Sci. Eng., B, 2023, 296, 116668 CrossRef CAS.
- E. A. Eno, H. Louis, T. O. Unimuke, T. E. Gber, J. A. Akpanke, I. O. Amodu and O. E. Offiong, Modeling of Re (I) tricarbonyl complexes against SARS-CoV-2 receptor via DFT, in-silico molecular docking, and QSAR, Chem. Phys., 2022, 5, 100105 Search PubMed.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397, DOI:10.1063/1.458517.
- M. Doust Mohammadi and H. Y. Abdullah, The adsorption of chlorofluoromethane on pristine, and Al-and Ga-doped boron nitride nanosheets: a DFT, NBO, and QTAIM study, J. Mol. Model., 2020, 26, 1–15 CrossRef PubMed.
- H. Louis, D. E. Charlie, I. O. Amodu, I. Benjamin, T. E. Gber, E. C. Agwamba and A. S. Adeyinka, Probing the reactions of thiourea (CH4N2S) with metals (X= Au, Hf, Hg, Ir, Os, W, Pt, and Re) anchored on fullerene surfaces (C59X), ACS Omega, 2022, 7, 35118–35135 CrossRef CAS PubMed.
- S. Bibi, S. Ur-Rehman, L. Khalid, I. A. Bhatti, H. N. Bhatti, J. Iqbal and H. X. Zhang, Investigation of the adsorption properties of gemcitabine anticancer drug with metal-doped boron nitride fullerenes as a drug-delivery carrier: a DFT study, RSC Adv., 2022, 12, 2873–2887 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Hitler Louis
ab,
Hitler Louis