DOI:
10.1039/D3RA02913A
(Paper)
RSC Adv., 2023,
13, 17282-17296
Synthesis, structural studies and computational evaluation of cyclophanes incorporating imidazole-2-selones†
Received
2nd May 2023
, Accepted 17th May 2023
First published on 14th June 2023
Abstract
We report new cyclophanes containing imidazole-2-selone groups linked by xylylene rings. A set of imidazole-2-selone cyclophanes is synthesized by reaction corresponding to imidazolium cyclophanes with selenium in the presence of K2CO3. The structural behavior of the new imidazole-2-selone cyclophanes was determined by 1H and 13C NMR spectra and X-ray diffraction studies. Cyclophanes incorporating o-xylylene or mesitylene-m-cyclophane linked by selone groups were mutually syn in both the solid state and solution, and the cyclophanes showed a conformation similar to the cone conformation of calix[4]arenes. Cyclophanes incorporating p-xylylene or m-xylylene linked by selone groups showed two conformations in the solution: one mutually syn and the other mutually anti. There was no interconversion for both conformations observed on the NMR timescale. In the solid state, three conformations were detected for the p-xylylene-linked cyclophane: one is mutually syn and the other two are mutually anti and partial cone conformations. In the m-xylylene-linked case, only anti-conformation was characterized in the solid state. A density functional analysis was conducted to interpret the stability of the studied compounds and shed light on their origin. The energy preference analysis is in consistent agreement with the observed geometries and their co-existence.
Introduction
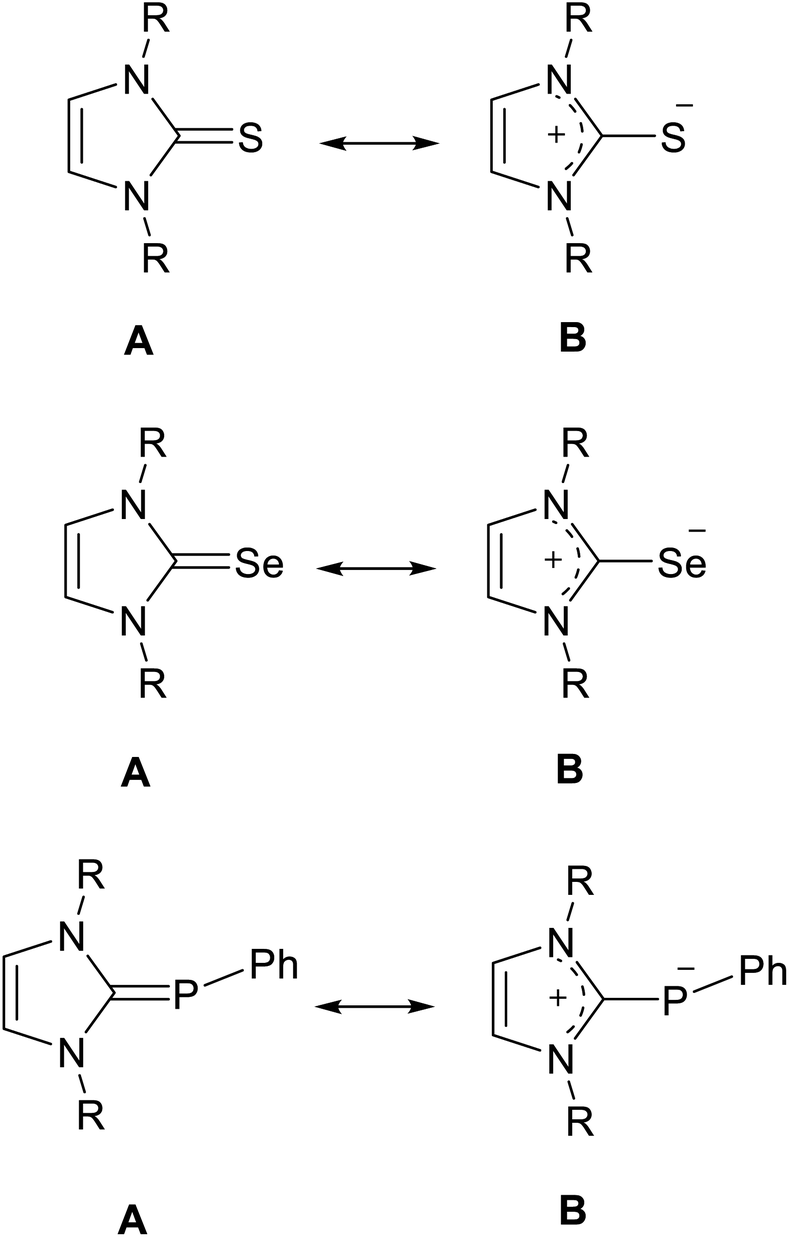
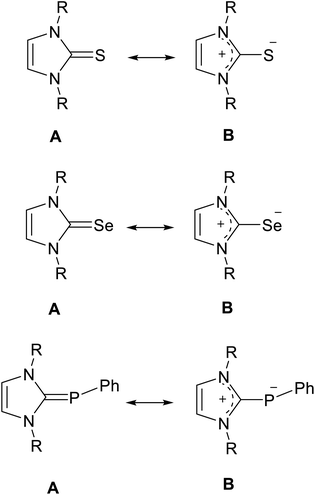
N-Heterocyclic carbenes (NHCs) can be used in various fields for different purposes, including organo transition-metal chemistry, catalysis, and the fields of biochemistry and medicine.1–7 Interestingly, NHCs can act as σ-donor and π-acceptor ligands, which made them interesting chemical structures in different fields of chemistry.1,8,9 Recently, NMR studies have been reported to determine the π-acceptor strength of NHCs by measuring the 31P or 77Se NMR chemical shifts of NHC–phosphinidene or NHC–Se adducts.10–13 In particular, X-ray studies of NHC–S adducts showed that the NHC–S moiety is planar and that the C–S bond distance (∼1.70 Å) is longer than a typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond distance (∼1.63 Å).14–19 Phosphinidene, selenium and sulfur adducts can be represented as the resonance hybrid by two structures (Fig. 1): the neutral hetero-alkene (A) with CP/Se/S character and a zwitterionic structure (B) with a C–P/Se/S character.12,14,15,20,21
S bond distance (∼1.63 Å).14–19 Phosphinidene, selenium and sulfur adducts can be represented as the resonance hybrid by two structures (Fig. 1): the neutral hetero-alkene (A) with CP/Se/S character and a zwitterionic structure (B) with a C–P/Se/S character.12,14,15,20,21
 |
| | Fig. 1 Resonance structures of the NHC sulfur, selenium and phosphinidene adducts. | |
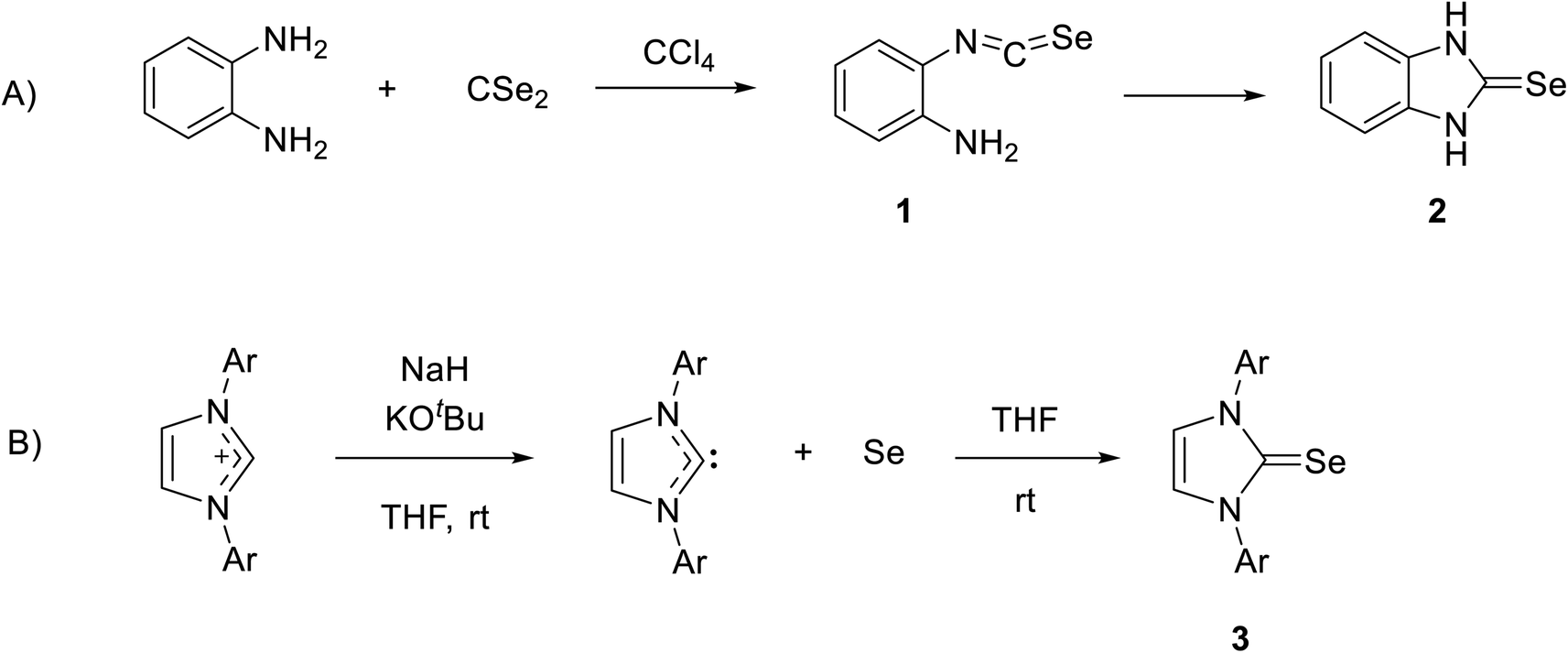
Recently, NHC selenium adducts (selones) have received significant attention owing to their promising applications, such as catalysis and their relevance in medicinal chemistry.22–32 The first NHC-based selone was synthesized by reacting o-phenylenediamine with carbon diselenide in CCl4 to form 2-isoselenocyanatoaniline 1, and then product 1 was cyclized by the intermolecular reaction of isoselenocyanate with an amino group to obtain selone 2 in high yield (Scheme 1A).33 The interesting procedure of synthesis of stable selone compounds is by the reaction of an imidazolium salt with base and then the addition of elemental selenium. For example, diarylimidazolium salt reacts with selenium to form 1,3-diarylimidazole-2-selenones 3 (Scheme 1B).34
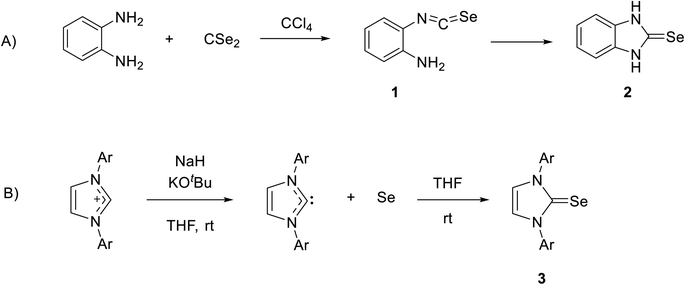 |
| | Scheme 1 (A) The first synthesis of selone by reaction of o-phenylenediamine with carbon diselenide. (B) Synthesis of stable selone by reaction of an imidazolium salt with Se. | |
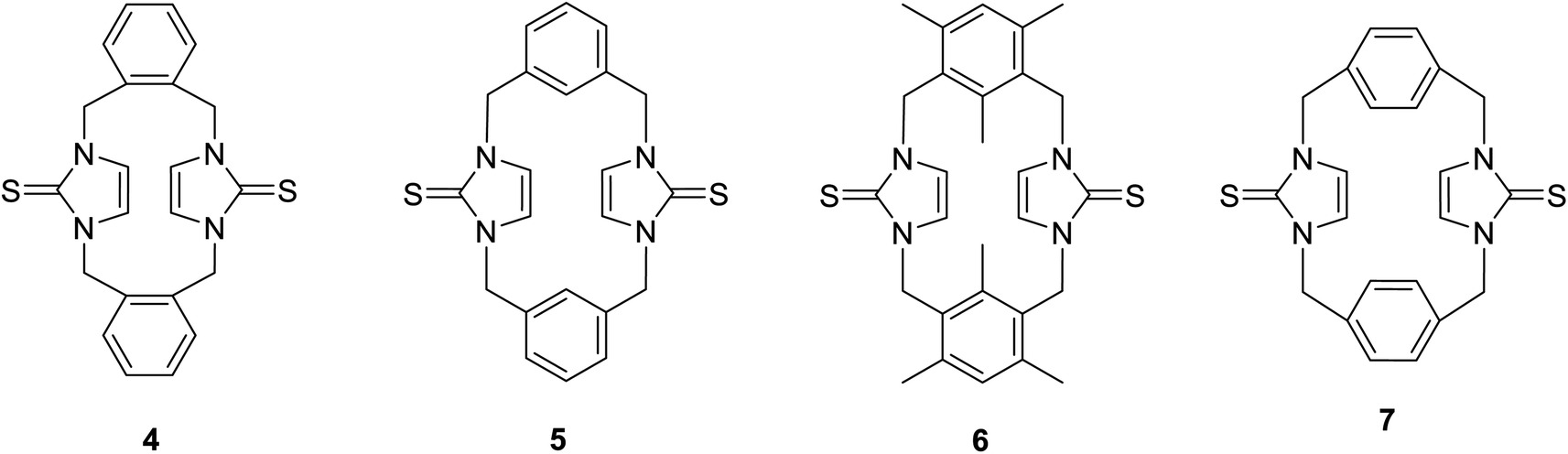
Previously, we reported the synthesis of some cyclophanes, including imidazole-2-thione units, such as 4–7, from corresponding imidazolium-linked cyclophanes (Fig. 2), and we studied their interesting conformational behaviour using X-ray diffraction and NMR studies.18 Many imidazole-2-selones are known.12,32,34–42 However, there are no reports of cyclophanes incorporating imidazole-2-selone moieties. As an extension of our study with imidazole-2-thiones, herein, we report the first synthesis of some cyclophanes, including imidazole-2-selone moieties, and study their conformational behaviour in solution and the solid state as well as use DFT calculations. Imidazole-2-selone cyclophanes may have great potential in various fields, including medicinal chemistry, catalysis, materials science, and supramolecular chemistry. Further research is needed to explore their full potential and to develop their practical applications.
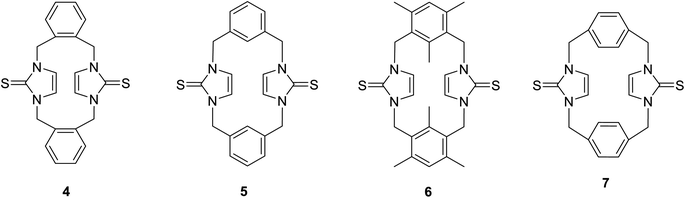 |
| | Fig. 2 Cyclophanes incorporating imidazole-2-thione moieties. | |
Results and discussion
Synthesis of the imidazole-2-selones
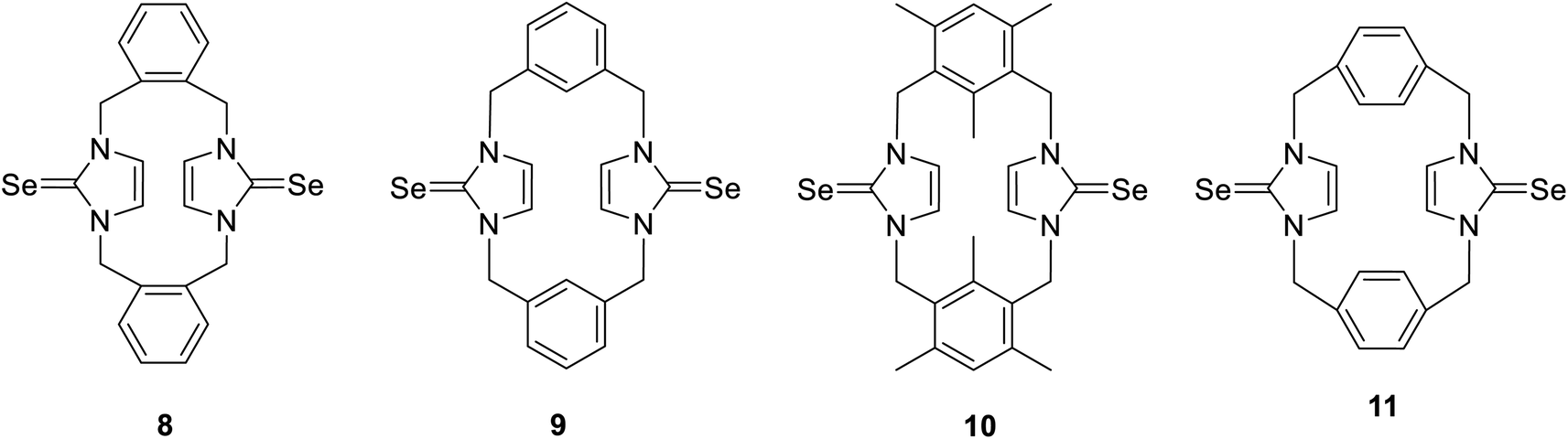
The cyclophanes incorporating imidazole-2-selone units 8–11 were synthesized by the reaction of the corresponding imidazolium-linked cyclophane salts with selenium and potassium carbonate in methanol at 60 °C overnight. The products were precipitated from the mixture and recrystallized to obtain a white powder with a moderate yield. The conformations of the cyclophanes incorporating imidazole-2-selone units 8–11 were characterized using X-ray diffraction and NMR methods.
Structures and conformations of imidazole-2-selones cyclophanes in solid state
Crystals 8, 10 and 11 were grown by the slow evaporation of a solution of the relevant compound in CH2Cl2, while 9·(CHCl3)2 was grown by the slow evaporation of a solution of the compound in CHCl3. The results of crystallographic studies are summarized in Tables 1 and 2 and Fig. 3–8.
Table 1 Crystal data of 8, 9, 10 and 11
| Complex |
8 |
9·(CHCl3)2 |
10 |
11 |
11′ |
11′′ |
| Empirical formula |
C22H20N4Se2 |
C24H22Cl6N4Se2 |
C28H32N4Se2 |
C22H20N4Se2 |
C22H20N4Se2 |
C22H20N4Se2 |
| Formula weight |
498.34 |
737.07 |
582.49 |
498.34 |
498.34 |
498.34 |
| Wavelength/Å |
1.54178 |
0.71073 |
1.54178 |
1.54178 |
1.54178 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
Orthorhombic |
Orthorhombic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/c |
Pbcm |
Pnma |
P21/c |
P21/c |
| a/Å |
17.8415 (4) |
7.2960 (2) |
11.533 (2) |
12.3930 (8) |
7.1187 (2) |
9.1890 (4) |
| b/Å |
10.5984 (3) |
19.9813 (5) |
12.4756 (13) |
18.8028 (13) |
14.2693 (6) |
12.1051 (4) |
| c/Å |
20.8127 (5) |
9.9038 (2) |
17.2625 (14) |
8.6492 (4) |
19.7393 (8) |
9.9941 (4) |
| β/° |
100.348 (2) |
103.675 (2) |
— |
— |
98.384 (4) |
114.539 (5) |
| V/Å3 |
3871.49 (17) |
1402.88 (6) |
2483.9 (5) |
2015.5 (2) |
1983.67 (13) |
1011.27 (8) |
| Z |
8 |
2 |
4 |
4 |
4 |
2 |
| ρ(calc)/Mg m−3 |
1.710 |
1.745 |
1.558 |
1.642 |
1.669 |
1.637 |
| μ/mm−1 |
4.894 |
3.230 |
3.902 |
4.700 |
4.776 |
3.671 |
| Crystal size/mm3 |
0.09 × 0.06 × 0.01 |
0.17 × 0.07 × 0.04 |
0.170 × 0.060 × 0.010 |
0.275 × 0.040 × 0.024 |
0.315 × 0.030 × 0.020 |
0.200 × 0.136 × 0.034 |
| θ range for data collection/° |
4.3 to 66.8 |
2.3 to 30.4 |
3.8 to 67.4 |
5.6 to 67.1 |
3.8 to 66.7 |
2.5 to 30.9 |
| Reflections collected |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 568 568 |
29526 |
11654 |
17634 |
17396 |
10805 |
| Independent reflections |
6875 |
4884 |
2302 |
1856 |
3530 |
3329 |
| R(int) |
0.0717 |
0.060 |
0.1747 |
0.1109 |
0.1204 |
0.0555 |
| Max./min. transmission |
1.00/0.918 |
1.00/0.923 |
1.00/0.658 |
1.00/0.695 |
1.00/0.875 |
1.00/0.756 |
| Restraints/parameters |
0/505 |
0/163 |
0/166 |
0/127 |
0/253 |
0/127 |
| Goodness-of-fit on F2 |
1.001 |
1.000 |
1.001 |
1.003 |
1.000 |
1.000 |
| R1[I > 2σ(I)] |
0.0487 |
0.0383 |
0.0738 |
0.0721 |
0.0588 |
0.0469 |
| wR2[I > 2σ(I)] |
0.1011 |
0.0838 |
0.1780 |
0.1832 |
0.1342 |
0.1053 |
| R1 (all data) |
0.049 |
0.038 |
0.074 |
0.072 |
0.059 |
0.047 |
| wR2 (all data) |
0.123 |
0.094 |
0.241 |
0.216 |
0.165 |
0.126 |
| Δρ(max/min)/e Å−3 |
1.51/−0.51 |
0.56/−0.48 |
1.02/−1.01 |
1.50/−0.46 |
1.17/−0.66 |
0.95/−1.25 |
| CCDC number |
2247962 |
2247957 |
2247960 |
2247959 |
2247961 |
2247958 |
Table 2 Bond lengths (Å) and angles (°) in imidazole-2-selones
| Compound |
Se–C |
N–C |
N–C–Se |
| 8 |
1.850 (6), 1.827 (6) |
1.475 (7), 1.480 (7) |
126.4 (4), 127.3 (4) |
| 1.843 (6), 1.847 (6) |
127.8 (4), 127.2 (4) |
| 9 |
1.841 (2) |
1.357 (3), 1.359 (3) |
126.98 (16), 127.39 (17) |
| 10 |
1.861 (10) |
1.382 (13), 1.344 (14) |
124.8 (9), 127.7 (7) |
| 11 |
1.867 (8) |
1.337 (10), 1.353 (9) |
126.3 (6), 125.6 (6) |
| 11′ |
1.849 (3) |
1.359 (4), 1.366 (4) |
126.8 (2), 127.8 (2) |
| 11′′ |
1.855 (7), 1.842 (7) |
1.353 (9), 1.362 (9) |
126.8 (5), 127.6 (5) |
| 1.380 (9), 1.380 (9) |
127.8 (5), 125.7 (5) |
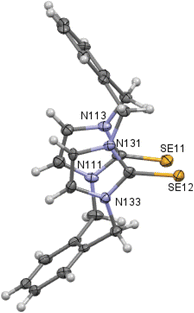 |
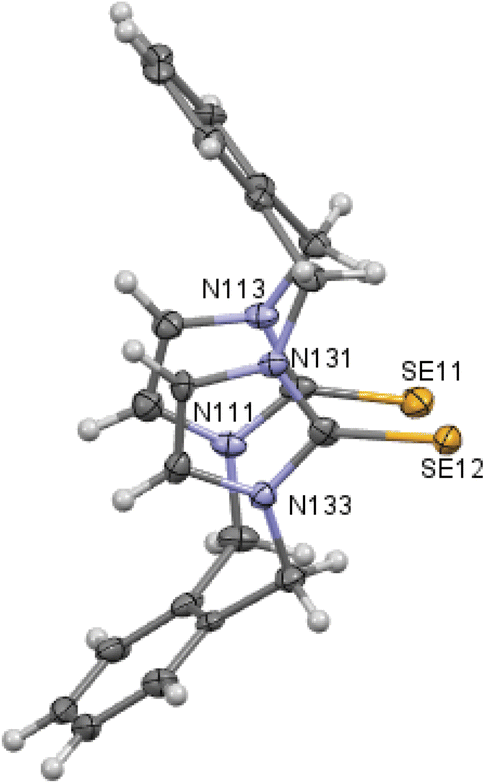
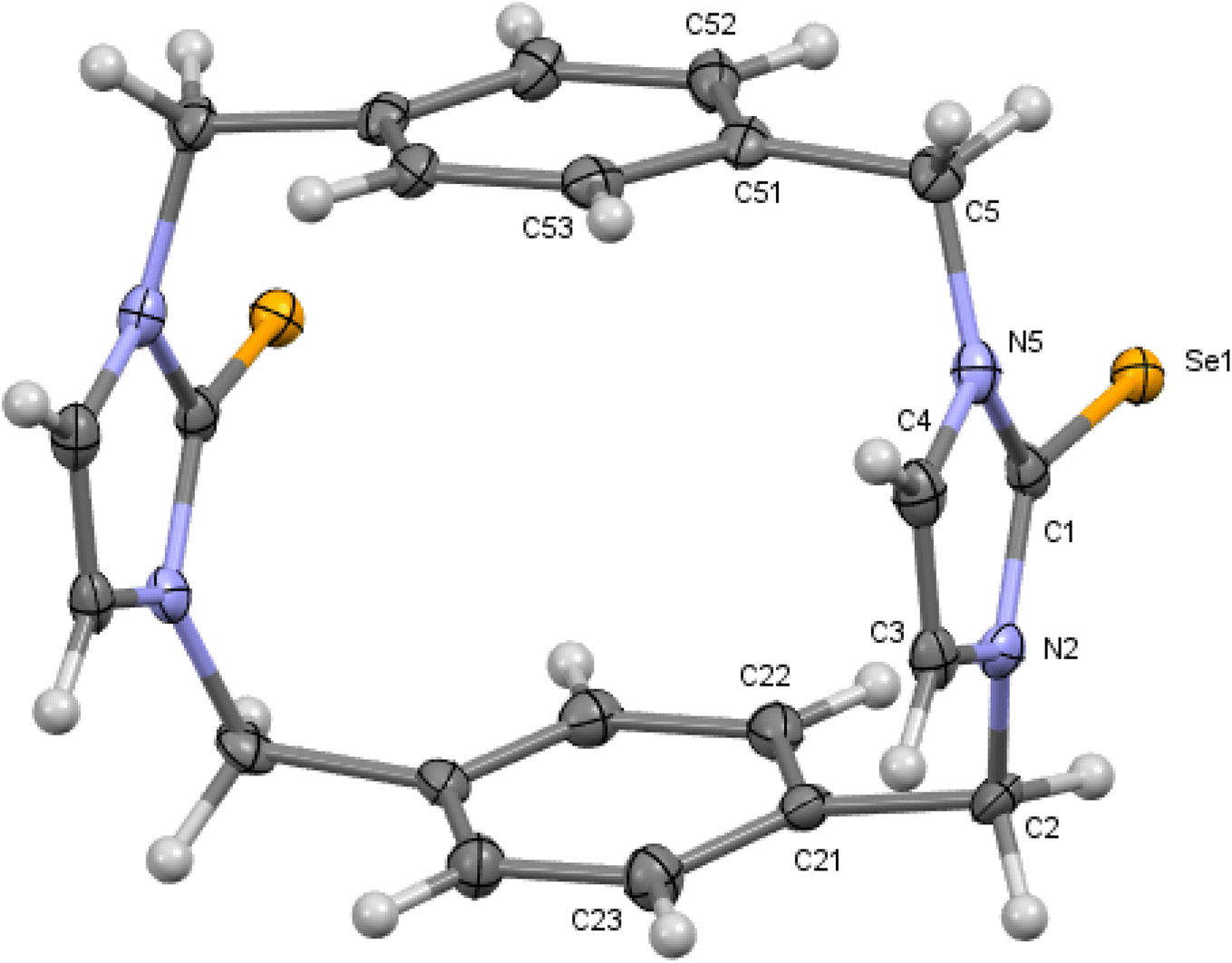
| | Fig. 3 Crystal structure (50% probability level for the displacement ellipsoids) of a single molecule of 8. Selected bond lengths (Å) and angles (°): Se11–C112 1.850 (6), Se12–C132 1.827 (6), C11–N111 1.475 (7), C11–C142 1.520 (9), N111–C112–Se11 126.4 (4), N113–C112–Se11 127.3 (4), N131–C132–Se12 127.8 (4), N133–C132–Se12 127.2 (4). | |
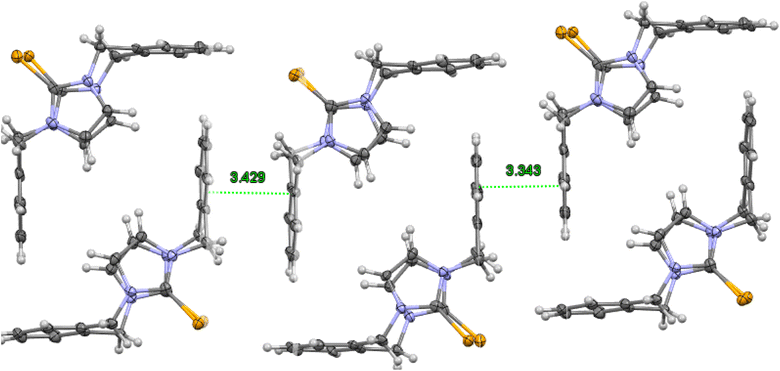 |
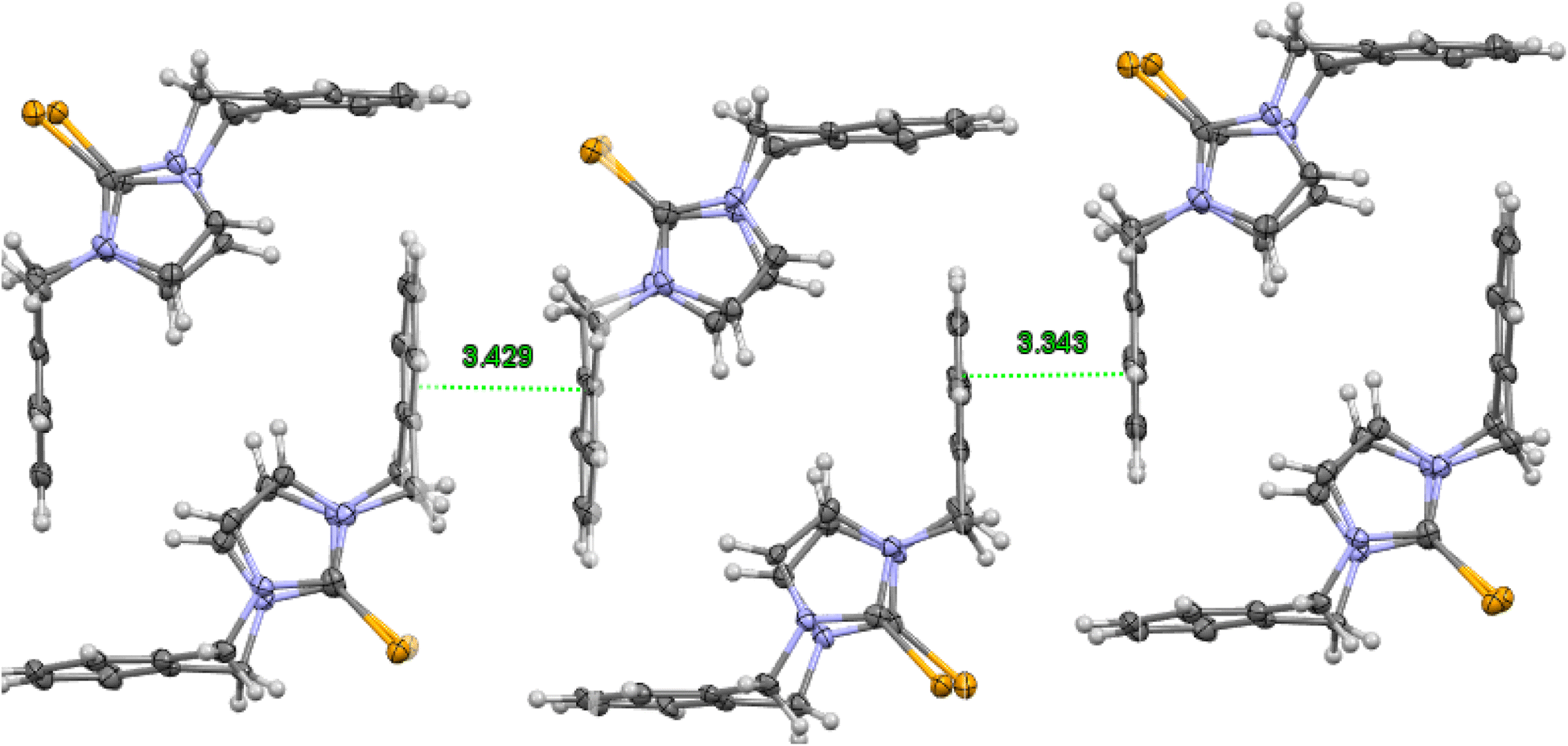
| | Fig. 4 Crystal structure (50% probability level for the displacement ellipsoids) of a column of interlocked pairs of 8. | |
 |
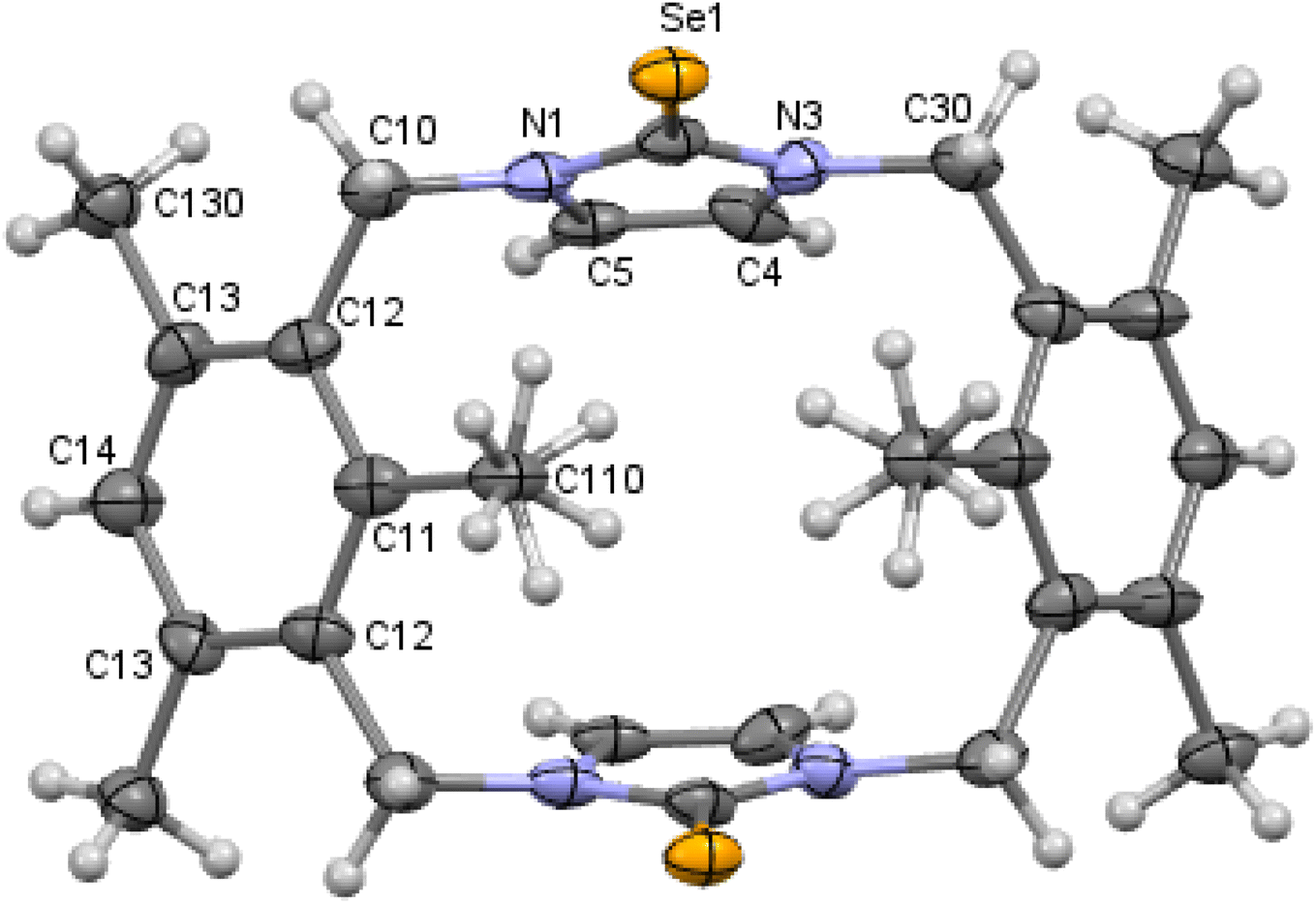
| | Fig. 5 Crystal structure (30% probability level for the displacement ellipsoids) of a single molecule of 10. Selected bond lengths (Å) and angles (°): Se1–C2 1.861 (10), N1–C2 1.382 (13), C2–N3 1.344 (14), C4–C5 1.340 (15), N3–C2–Se1 127.7 (7), and N1–C2–Se1 124.8 (9). | |
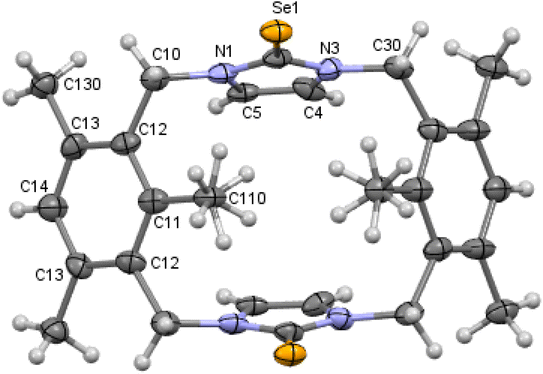
 |
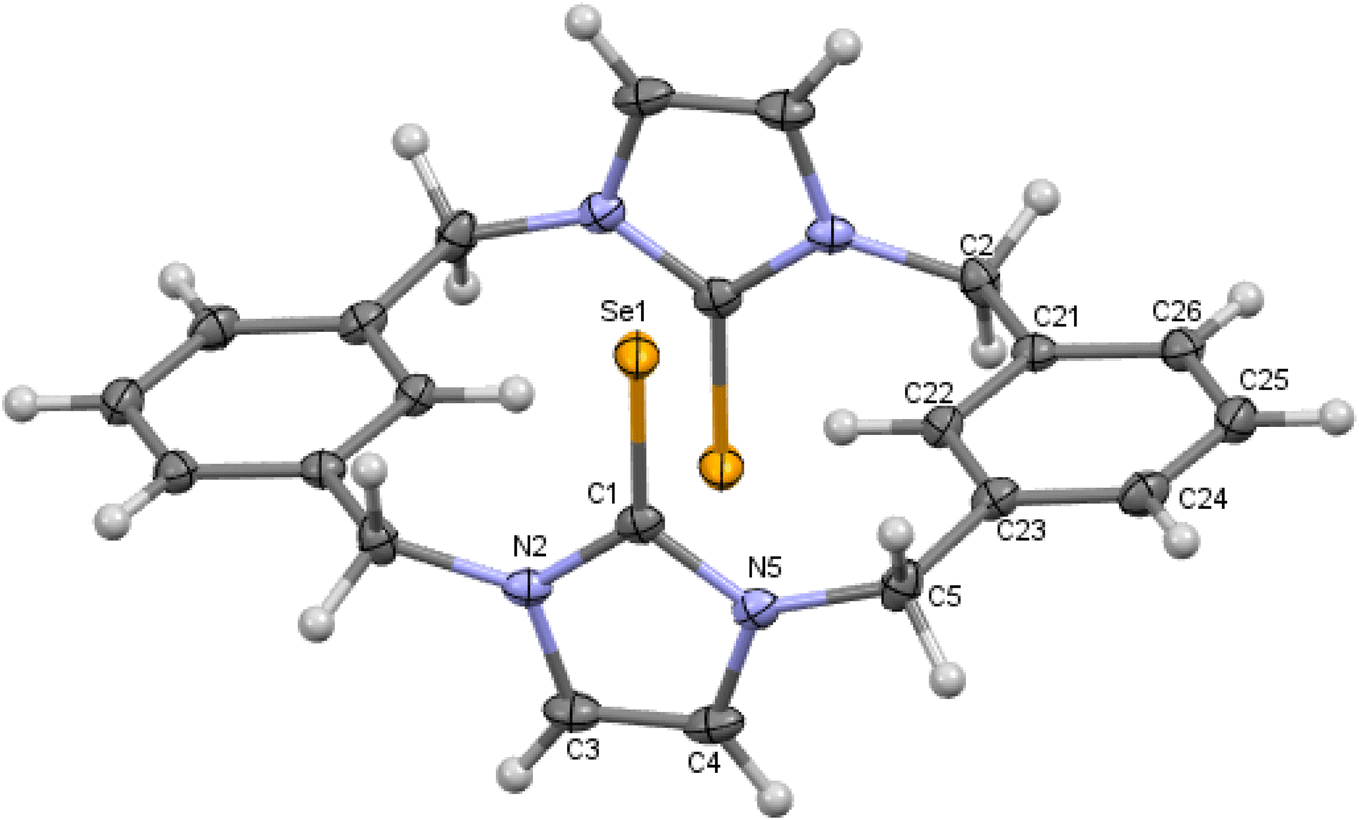
| | Fig. 6 Crystal structure (50% probability level for the displacement ellipsoids) of a single molecule of 9. Selected bond lengths (Å) and angles (°): Se1–C1 1.841 (2), N2–C1 1.357 (3), C1–N5 1.359 (3), C4–C5 1.334 (3), N2–C1–Se1 126.98 (16), N5–C1–Se1 127.39 (17). | |
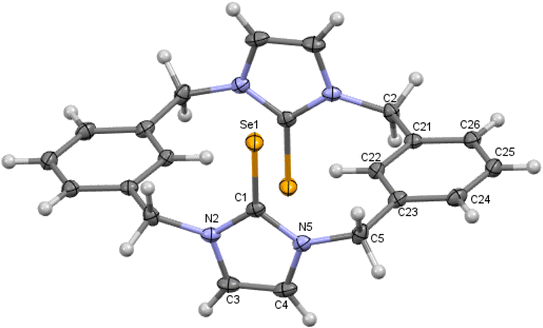
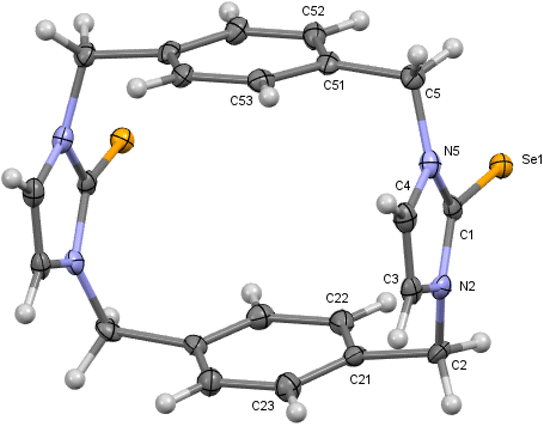 |
| | Fig. 7 Crystal structure (30% probability level for the displacement ellipsoids) of a single molecule of 11. Selected bond lengths (Å) and angles (°): Se1–C1 1.867 (8), N2–C1 1.337 (10), C1–N5 1.353 (9), C4–C3 1.282 (11), N2–C1–Se1 126.3 (6), N5–C1–Se1 125.6 (6). | |
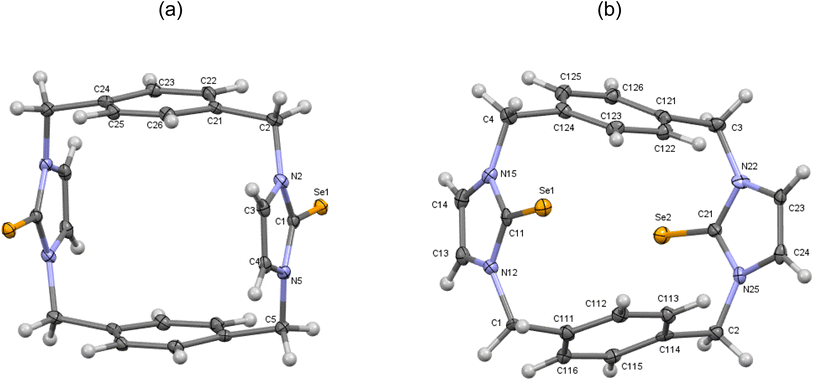 |
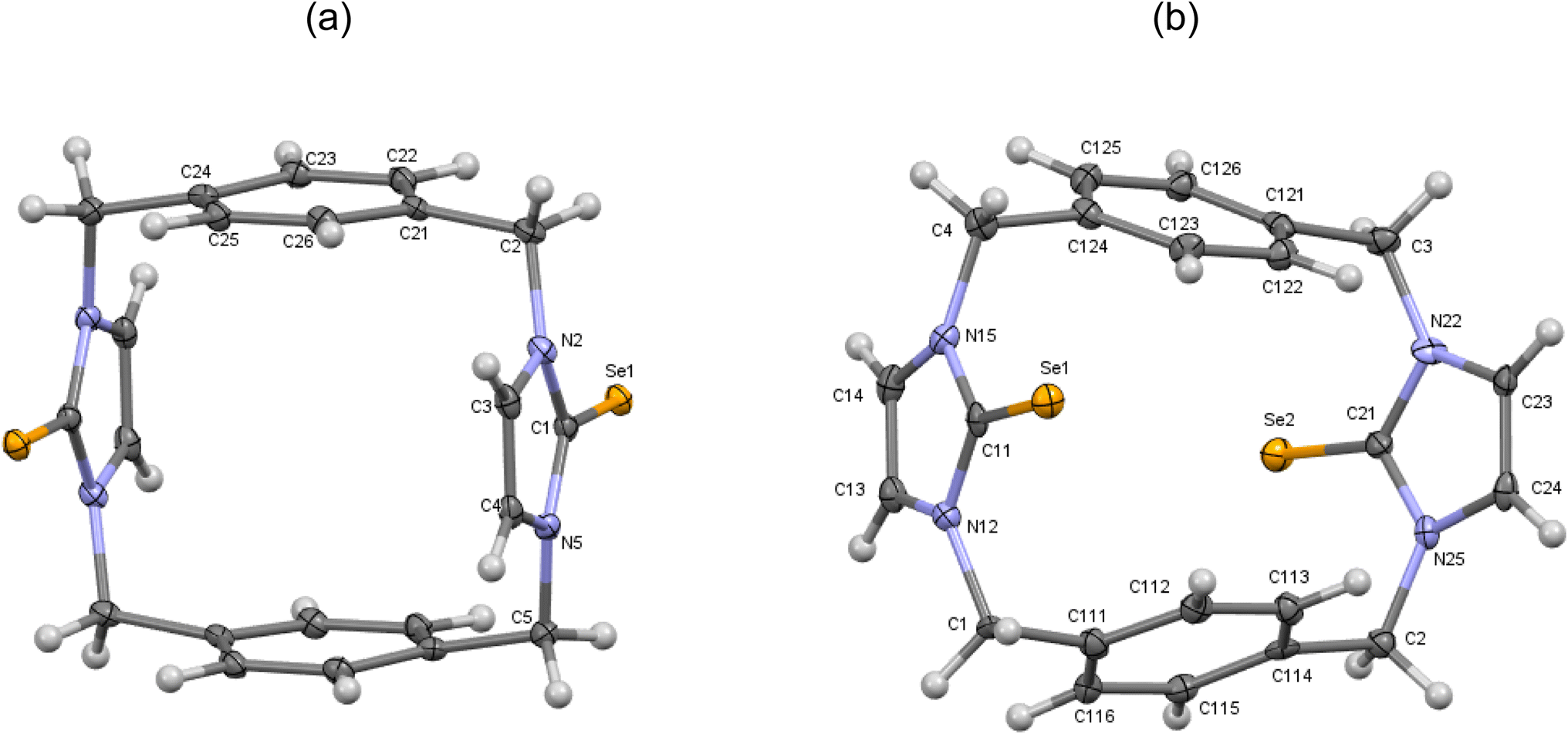
| | Fig. 8 (a) Crystal structure (30% probability level for the displacement ellipsoids) of a single molecule of 11′. Selected bond lengths (Å) and angles (°): Se1–C1 1.849 (3), N2–C1 1.359 (4), C1–N5 1.366 (4), C4–C3 1.347 (5), N2–C1–Se1 126.8 (2), N5–C1–Se1 127.8 (2). (b) Crystal structure (30% probability level for the displacement ellipsoids) of a single molecule of 11′′. Selected bond lengths (Å) and angles (°): Se1–C11 1.855 (7), Se2–C21 1.842 (7), N15–C11 1.353 (9), C11–N12 1.362 (9), C14–C13 1.338 (11), N22–C21 1.380 (9), C21–N25 1.380 (9), C24–C23 1.331 (11), N15–C11–Se1 126.8 (5), N12–C11–Se1 127.6 (5), N22–C21–Se2 127.8 (5), N25–C21–Se2 125.7 (5). | |
X-ray studies demonstrate that the conformation of cyclophanes incorporating imidazole-2-selone units 8–11 is broadly similar to that observed in the cyclophanes incorporating imidazole-2-thione units 4–7. For o-cyclophane 8, the benzene and imidazole-2-selone rings making a ‘cup shape’ and the orientation of CSe groups are exo for the macrocyclic ring to yield conformation similar to the cone conformation of calix[4]arenes. The imidazole-2-selone rings are nearly parallel; therefore, the cup is slightly flattened. Obviously, the bending of the CSe bonds out of the N2C2 planes of the imidazole-2-selones is a result of steric repulsion between the Se atoms (Fig. 3).
The molecule structures form pairs to obtain interlocking cup conformation. Then, the pairs accumulate into columns because of interactions between the benzene rings (intermolecular distance between C6 planes is ∼3.343 Å and 3.429 Å) (Fig. 4).
Mesitylene m-cyclophane 10 also has a conformation similar to the cone conformation of calix[4]arenes. However, in this case, the imidazole-2-selone rings are separated. Therefore, the unfavorable steric interactions between the Se atoms and the methyl substituents in each of the benzene rings are nonexistent, and the “cup conformation” is now completely pinched. Obviously, the structure of the m-cyclophane allows for the separation of the selone rings such that the CSe bond is not bent outside the C3N2 plane (cf. the case of o-cyclophane 8). The observed conformation of 10 is similar to the conformation of the corresponding imidazole-2-thione 6 and contradicts the conformation of the parent imidazolium-linked cyclophane, in which the C2–H in the imidazolium moieties is oriented into the cavity formed between the mesitylene rings. The corresponding orientation of the imidazole thione or selone groups in 6 or 10 might be unfavorable because of the electron repulsion between the electron-rich CS/Se groups.
In contrast to 8 and 10, m-cyclophane 9 has an anti conformation (Fig. 6), which is similar to the 1,2-alternate conformation of calix[4]arene. The imidazole-2-selone rings are parallel, with their CSe groups located in opposite directions c.
X-ray diffraction shows three crystal structures of p-cyclophane. First, in the crystal structure of p-cyclophane 11, the CSe groups in the imidazole-2-selone rings are oriented in the same direction to obtain a syn conformation (Fig. 7). The macrocycle of p-cyclophane 11 is in the form of approximately parallelepiped. The two imidazole-2-selone rings are almost parallel (dihedral angle 1.68°) and occupy opposite faces separated by a distance of about 6.517 Å, and the aryl groups are approximately parallel (dihedral angle 8.55°) and occupy two opposite faces separated by a distance of about 5.198 Å. The dihedral angles between the imidazole-2-selone and aryl group planes within the macrocycle are 89.84° and 89.72°. Obviously, there is some strain within the macrocycle. The bond between carbon methylene and carbon arene ring slightly deviates from the plane of the aryl group by 7.6 (4)°, and the bond between carbon methylene and nitrogen atom in the selone ring deviates from the plane of the imidazole-2-selone moiety by 12.3 (6)°.
Second, in the crystal structures of p-cyclophane (11′ and 11′′), the CSe groups in the imidazole-2-selone rings are oriented in opposite directions to obtain an anti conformation (Fig. 8). The macrocycle of p-cyclophane 11′ is also in the form of a parallelepiped (Fig. 8a). The two imidazole-2-selone rings are parallel (dihedral angle 0°) and occupy opposite faces separated by 6.283 Å; the aryl groups are also parallel (dihedral angle 0°), and the distance between the two opposite faces is about 5.464 Å. The dihedral angles between the imidazole-2-selone and aryl group planes within the macrocycle are 89.71 (10)°. As observed in 11, there is also some strain within the macrocycle in 11′. The bond between carbon methylene and carbon arene ring slightly deviates from the plane of the aryl group by 6.4 (3)°, and the bond between carbon methylene and nitrogen atom in the selone ring deviates from the plane of the imidazole-2-selone moiety by 10.9 (3)°. In contrast to 11 and 11′, the macrocycle of p-cyclophane 11′′ adopts a conformation similar to the partial cone conformation of calix[4]arenes (Fig. 8b). The imidazole-2-selone rings are approximately parallel (dihedral angle 15.76°), and the CSe bond in one of the selone rings is directed slightly into the cavity formed between the arene rings. Evidently, the aryl groups are now unparallel (dihedral angle 44.32°).
Conformation of imidazole-2-selone cyclophanes in a solution state
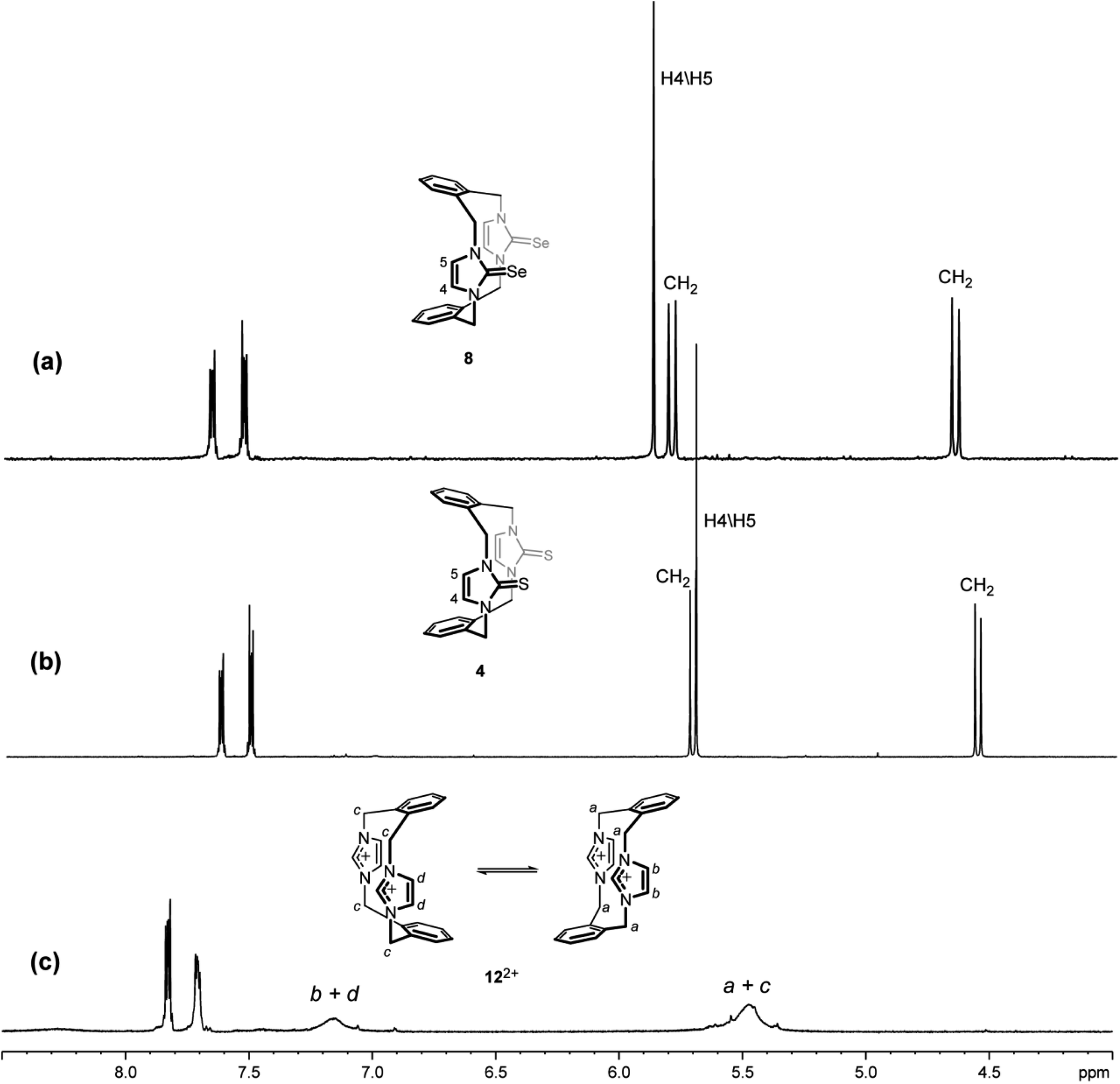
The NMR spectra of the imidazole-2-selone ligands in DMSO-d6 solution at room temperature showed signals predicted for the suggested conformations (Table 3). The 1H NMR spectrum of 8 is consistent with the structure shown in the solid state, in which the cyclophane macrocycle adopts a cone conformation. H4/H5 protons in the imidazole-2-selone ring show a remarkably upfield chemical shift (∼5.88 ppm), which is an indication that the H4/H5 hydrogen atoms are shielded by the anisotropy of the cyclophane's benzene rings. Notably, the benzylic protons (exo and endo environments) show the pair of sharp doublets (∼5.81 and ∼4.65 ppm), which implies that the cone conformation of 8 is rigid on the NMR timescale. Obviously, the 1H NMR spectrum of the 8 shows signals consistent with those seen in the corresponding imidazole-2-thione 4,18 while the 1H NMR spectrum of the parent imidazolium cyclophane 122+ appears broad signals because the imidazolium cyclophane is rapidly interconverted to several conformations in solution43 (Fig. 9). Presumably, the conformational ability of the imidazole-2-thione and imidazole-2-selone cyclophane structure decreased owing to an increase in bulkiness of the thione and selone groups in 4 and 8, respectively, compared to the imidazolium groups in 122+.
Table 3 1H NMR dataa for imidazole-2-selones
| Compound |
CH3 |
CH2 |
H4/H5 |
Aromatics |
| Recorded at 500.10 MHz and ambient temperature from solutions in DMSO-d6. Normal type indicates anti conformation, italics type indicates syn conformation, and bold type indicates overlapping signals of both conformations. Normal type indicates anti conformation; italics type indicates syn conformation. |
| 8 |
— |
4.65 (4H, d, 2JH,H 15 Hz) |
5.88 (4H, s) |
7.67–7.65, 7.54–7.52 |
| 5.80 (4H, d, 2JH,H 15 Hz) |
(8H, AX pattern) |
| 9b |
— |
5.73 (4H, d, 2JH,H 15 Hz) |
7.31 (4H, s) |
7.43–7.39 (4H, m) |
| 4.96 (4H, d, 2JH,H 15 Hz) |
7.35–7.31 (8H, m) |
| 5.89 (4H, d, 2JH,H 15 Hz) |
7.11 (4H, s) |
6.25 (2H, s) |
| 5.02 (4H, d, 2JH,H 15 Hz) |
6.18 (2H, s) |
| 10 |
2.40 (12H, s) |
5.62 (4H, d, 2JH,H 15 Hz) |
5.79 (4H, s) |
7.01 (2H, s) |
| 1.55 (6H, s) |
4.80 (4H, d, 2JH,H 15 Hz) |
| 11c |
— |
5.54 (4H, d, 2JH,H 15 Hz) |
7.42 (4H, s) |
7.18 (8H, s) |
| 4.66 (4H, d, 2JH,H 15 Hz) |
| 5.22 (4H, d, 2JH,H 15 Hz) |
7.15 (4H, s) |
7.48 (8H, s) |
| 4.81 (4H, d, 2JH,H 15 Hz) |
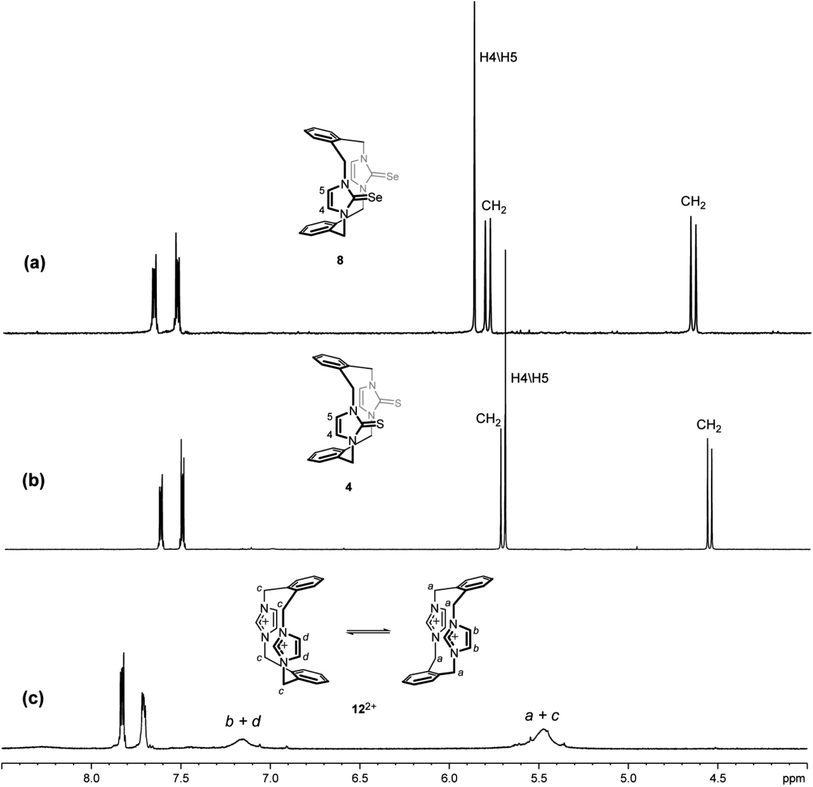 |
| | Fig. 9 Downfield region of the 1H NMR spectra (500 MHz, DMSO-d6) for solutions of (a) the ligand of imidazole-2-selone 8, (b) the ligand of imidazole-2-thione 4 and (c) its precursor imidazolium-linked cyclophane salt 12·2Br. | |
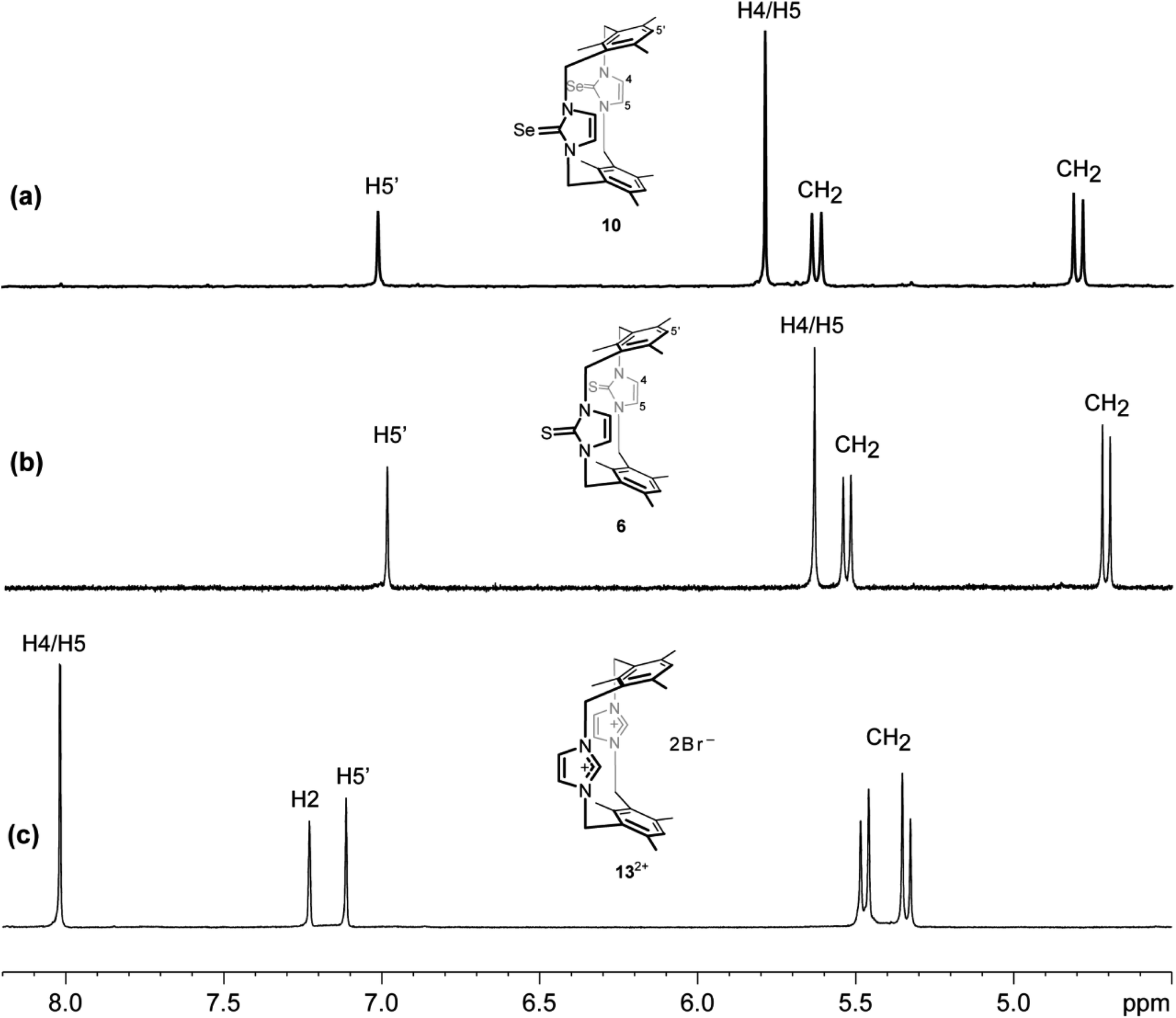
The 1H NMR spectrum (DMSO-d6 solution at room temperature) of 10 is also consistent with the cone-type conformation in the solution, as observed in the solid state. Again, the 1H NMR chemical shift of the H4/H5 protons in the imidazole-2-selone ring is markedly upfield chemical shift (∼5.79 ppm, compared with ∼8.02 ppm seen for H4/H5 protons in the parent imidazolium cyclophane 132+). Notably, the corresponding protons in imidazole-2-thione 6 are significantly upfield (∼5.63 ppm). This upfield shift observed in 10 and 6 also indicates that the H4/H5 protons are shielded by the ring resonance of the benzene groups and, therefore, shows that the conformation in the solution is consistent with that seen in the solid state. H4/H5 protons in 132+ show a far downfield (8.02 ppm), and this indicates that the H4/H5 protons are deshielded by the effects of the benzene groups because of cyclophane formed in the 1,3-alternate type conformation and also due to the positive charge effects in the imidazolium rings. Interestingly, the pair of sharp doublets (i.e., an AX pattern) for the benzylic protons (exo and endo environments) indicates that rapid interconversion between the two equivalent forms of this conformation does not occur in the solution. This provides additional evidence that the cone conformation is rigid in solution, as observed in the solid state. Rigidity of conformation has been shown similarly for the precursor imidazole-2-thione 6 and imidazolium-linked cyclophane 132+ (Fig. 10).18,43
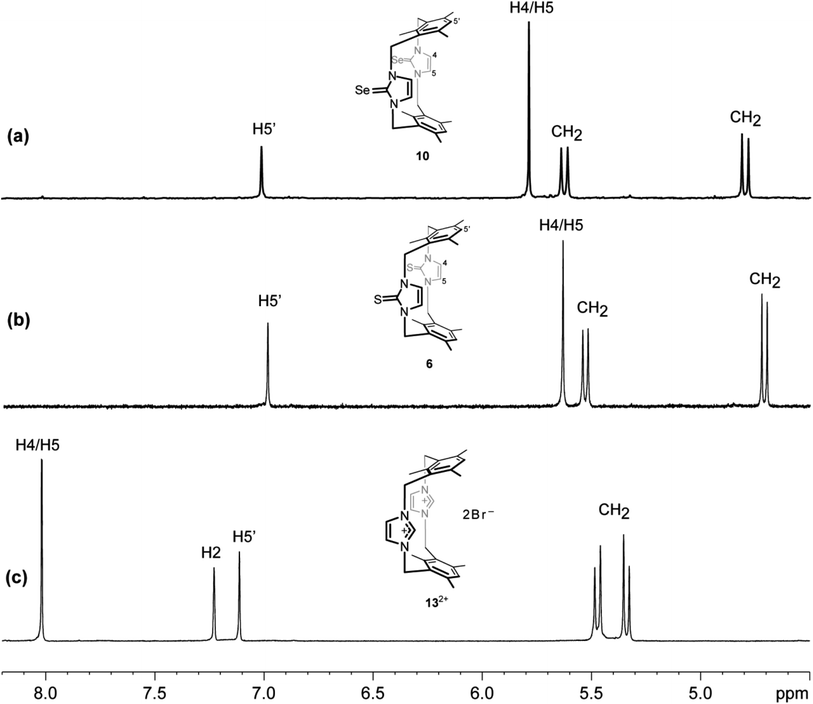 |
| | Fig. 10 Downfield region of the 1H NMR spectra (500 MHz, DMSO-d6) for solutions of (a) the ligand of imidazole-2-selone 10, (b) the ligand of imidazole-2-thione 6 and (c) its precursor imidazolium-linked cyclophane salt 13·2Br. | |
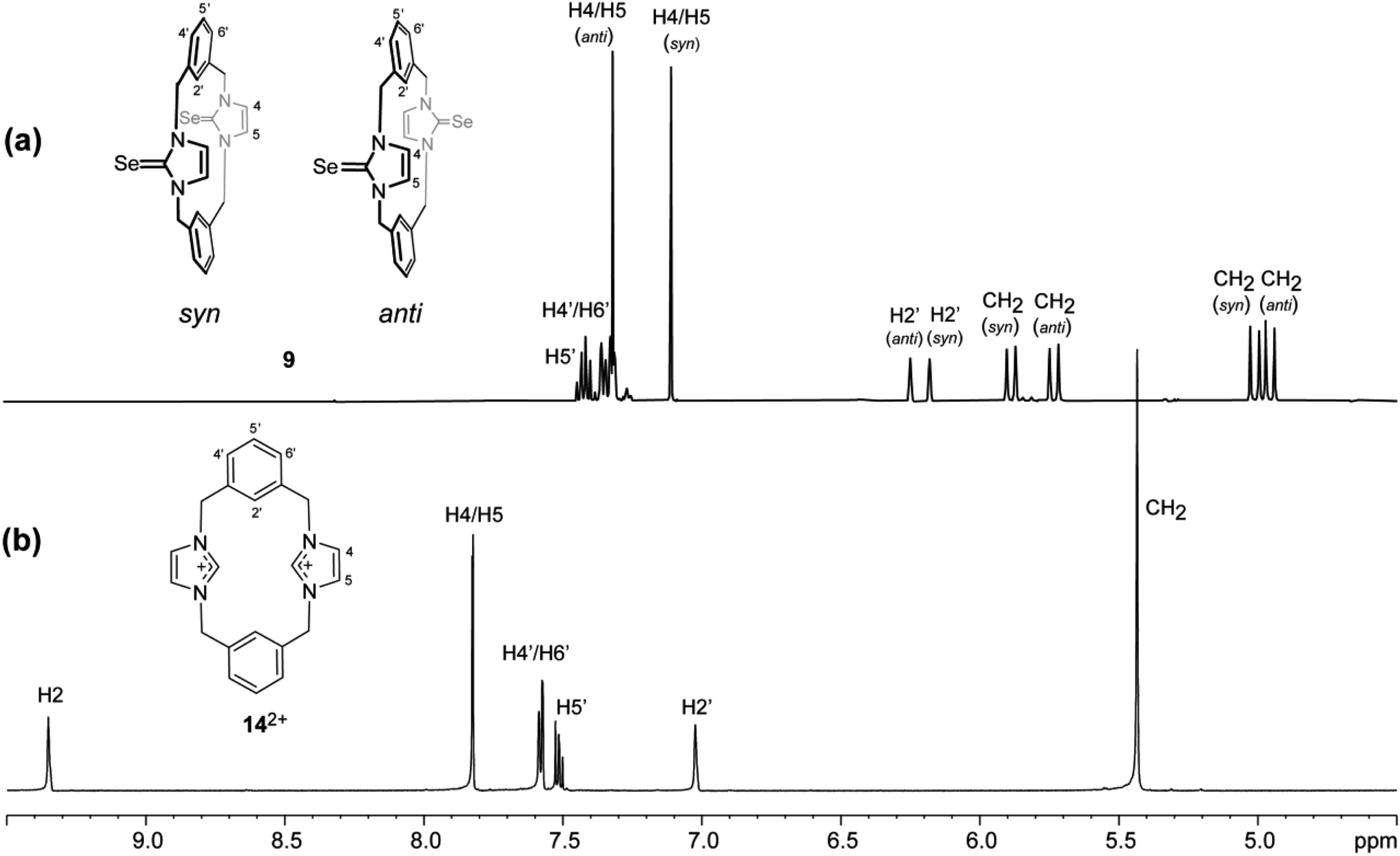
The 1H NMR spectrum of 9 shows two sets of signals with integrals in equal ratios (Fig. 11). These signals were assigned to two conformations based on different orientations of the two imidazole-2-selone moieties: one of them has syn conformation, and the other has anti conformation. In each conformation, the benzylic protons show one pair of doublets, indicating that both conformations of 9 are rigid on the NMR timescale (i.e. the rotation of imidazole-2-selone rings about their N–N axes do not occur in the solution). This result is consistent with the case for imidazole-2-thione 5 (ref. 18) and contradicts the parent imidazolium cyclophane 142+, which has conformational lability.43 The two conformations are present in nearly equal amounts in the NMR timescale, and it is not able to assign the set signals in each conformation based on splitting patterns and chemical shifts. However, the crystals formed of 9 show only anti conformation based on the X-ray study (see above), while the powder formed of 9 presents as a syn conformation.
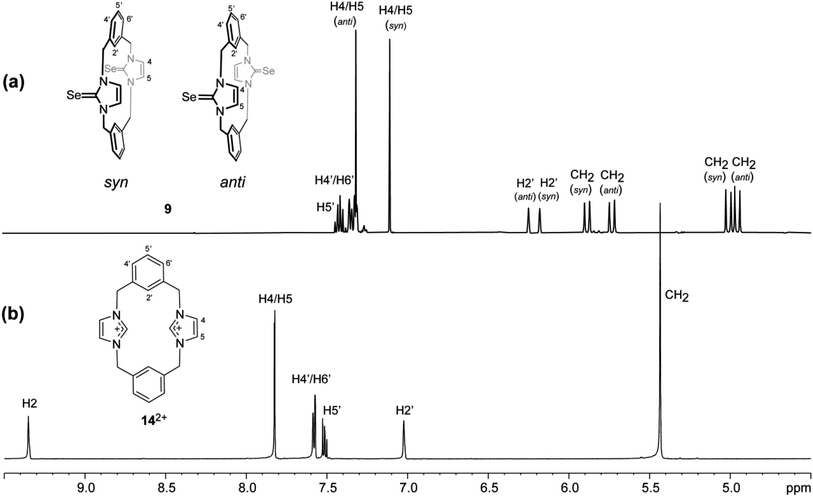 |
| | Fig. 11 Downfield region of the 1H NMR spectra (500 MHz, DMSO-d6) for solutions of (a) the ligand of imidazole-2-selone 9 and (b) its precursor imidazolium-linked cyclophane salt 14·2Br. | |
In both conformations of 9, the splitting patterns and chemical shifts of the different proton environments are similar. Therefore, we are not able to assign a special conformation to a special set of signals in NMR. Notably, the similarity of signals in NMR shows that there is no interesting difference in shielding because of, for example, the resonance in arene rings. This interpretation is consistent with that seen in the solid state in which the arene rings are directed with their planes approximately parallel to the N–N axes of the imidazole-2-selone rings, so the environments of the CH2 and H4/H5 protons are away from the regions of shielding. These considerations apply to the anti and syn conformations.
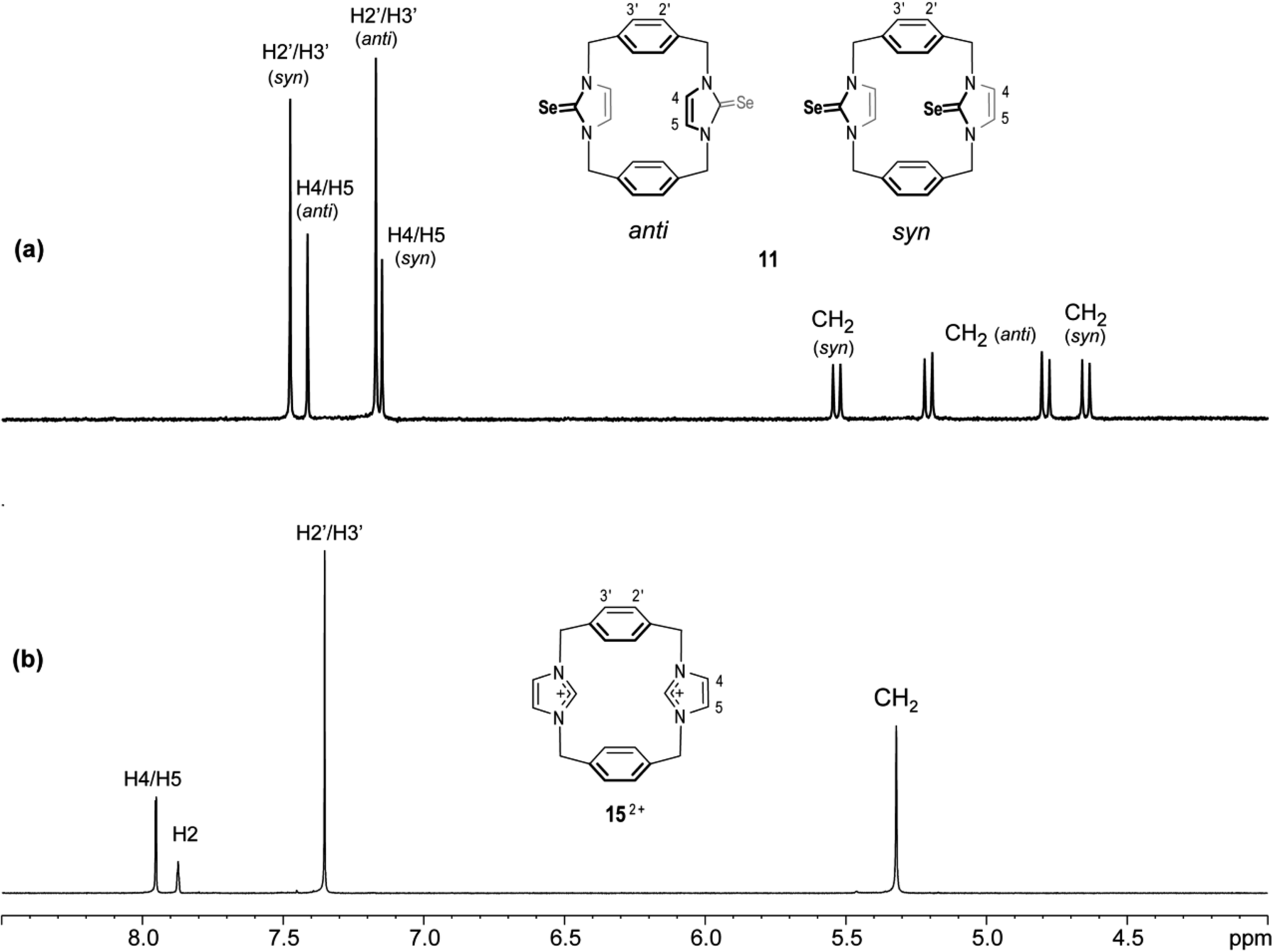
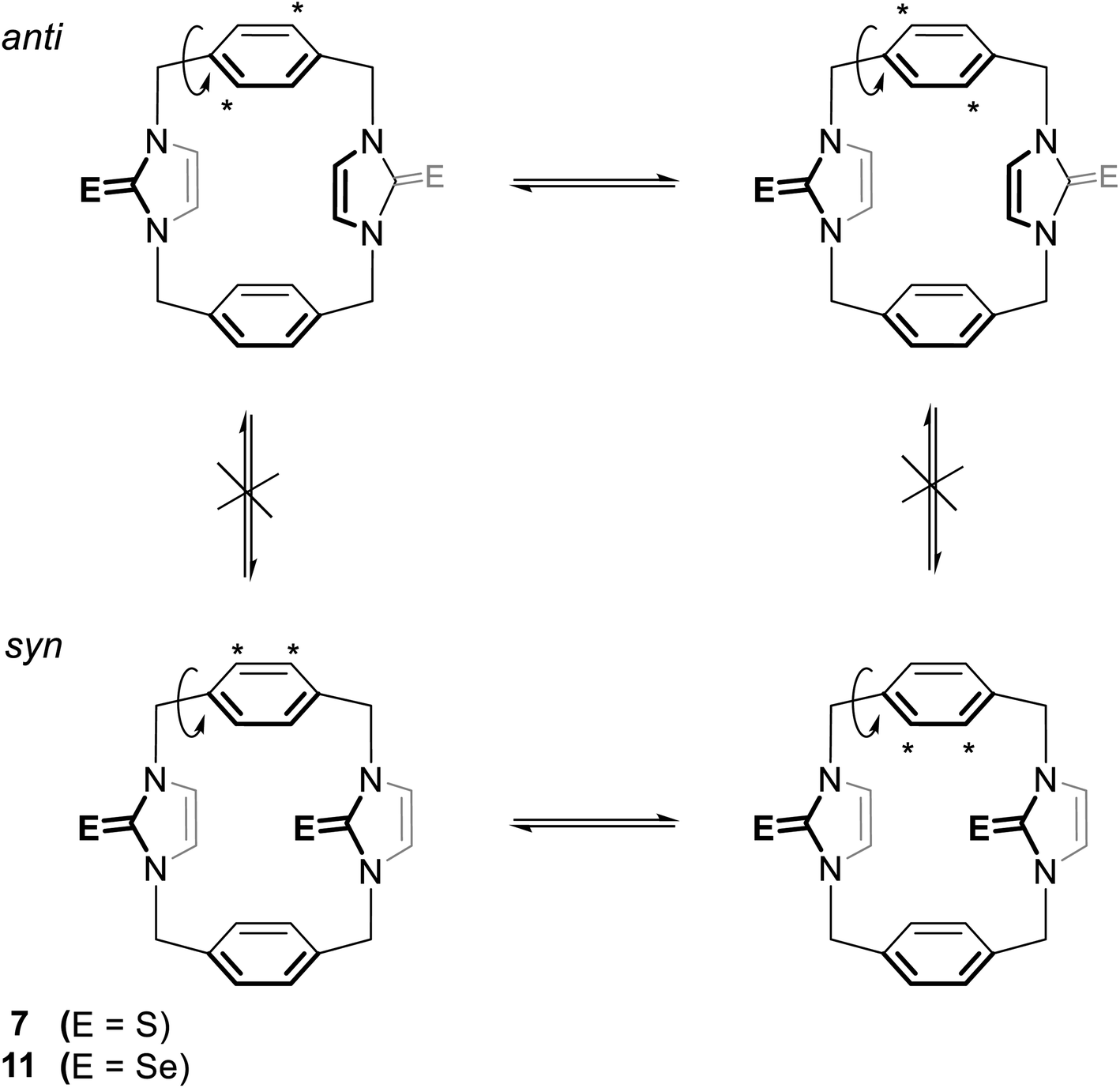
The 1H NMR spectra of 11 again show two sets of signals that are perhaps assigned to syn and anti conformations (Fig. 12). Obviously, both conformations are present in nearly equal amounts in the NMR timescale. Again, we are not able to assign the set signals to exact conformation based on chemical shifts and splitting patterns. Interestingly, the recrystallisation of 11 provided three types of crystals (as observed in the X-ray diffraction). The yellow needle crystals can be attributed to the syn conformation, and the colorless plate crystals can be attributed to the anti conformation based on the X-ray study, while the colorless needle crystals formed the other conformation attributed to the partial cone conformation. Notably, for anti and syn conformations of solutions of 11 in DMSO-d6 and in acetone-d6, arene ring protons H2′/H3′ should be shown as two environments. Therefore, in both cases, H2′/H3′ protons show one sharp single signal (even in acetone-d6 at −25 °C). These considerations suggest that the arene rings are rapidly rotated about their C1′–C4′ axis on the NMR timescale (Fig. 13); therefore, a partial cone conformation does not appear in the solution, as seen in the solid state. The observed that two sets of NMR signals for syn and anti conformations indicate that the selone moieties are not rotated about their N–N axes on the NMR timescale.
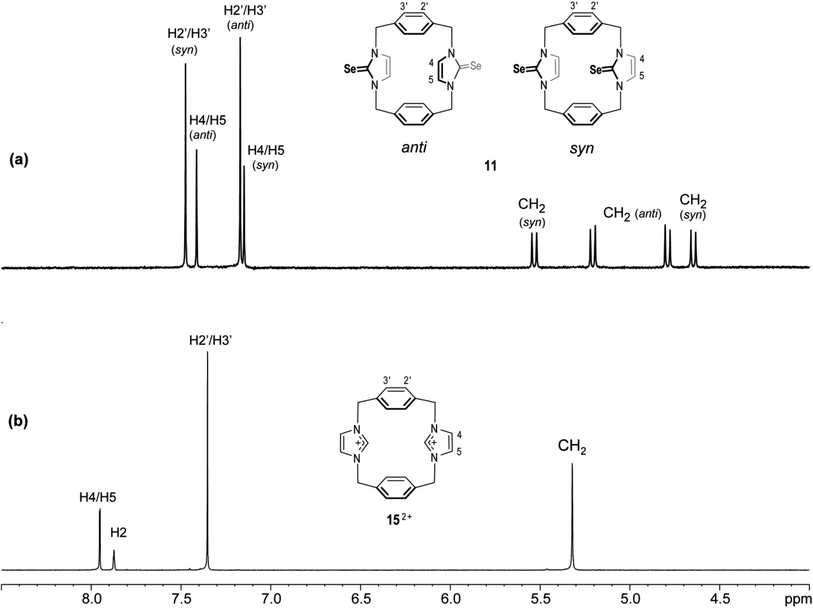 |
| | Fig. 12 Downfield region of the 1H NMR spectra (500 MHz, DMSO-d6) for solutions of (a) the ligand of imidazole-2-selone 11 and (b) its precursor imidazolium-linked cyclophane salt 15·2Br. | |
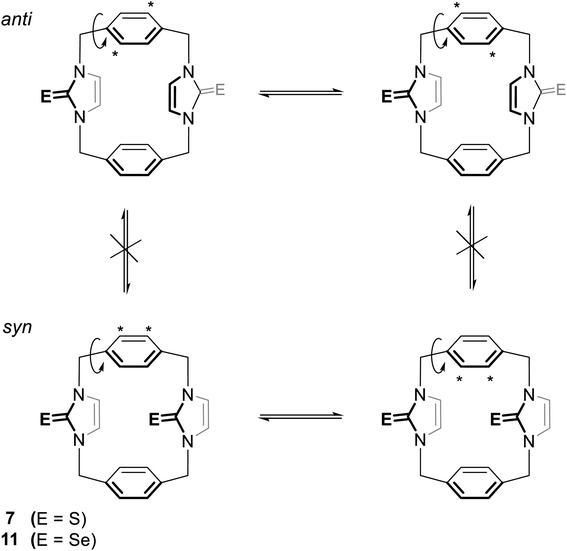 |
| | Fig. 13 The syn and anti conformations of 7 (ref. 18) and 11. In each conformation, the asterisk refers to two equivalent protons on the arene rings. The rotation of this ring around its C1′–C4′ axis leads to the labeled protons replacing places with the unlabeled protons so that all the protons in the arene rings are equivalent. | |
This result shows that both conformations are rigid in solution, as seen in the solid state. Obviously, the rigidity of conformation is similar to the precursor imidazole-2-thione 7 and imidazolium-linked cyclophane 152+ (Fig. 12).18,43
In the 13C NMR spectra of the imidazole-2-selone ligands, the signal for the CSe occurs in the range of 157 ppm, which is close to that reported for the related imidazole-2-selone,13,34,44 but slightly upfield compared to that of the CS (∼163 ppm) in imidazole-2-thione ligands.18
DFT analysis
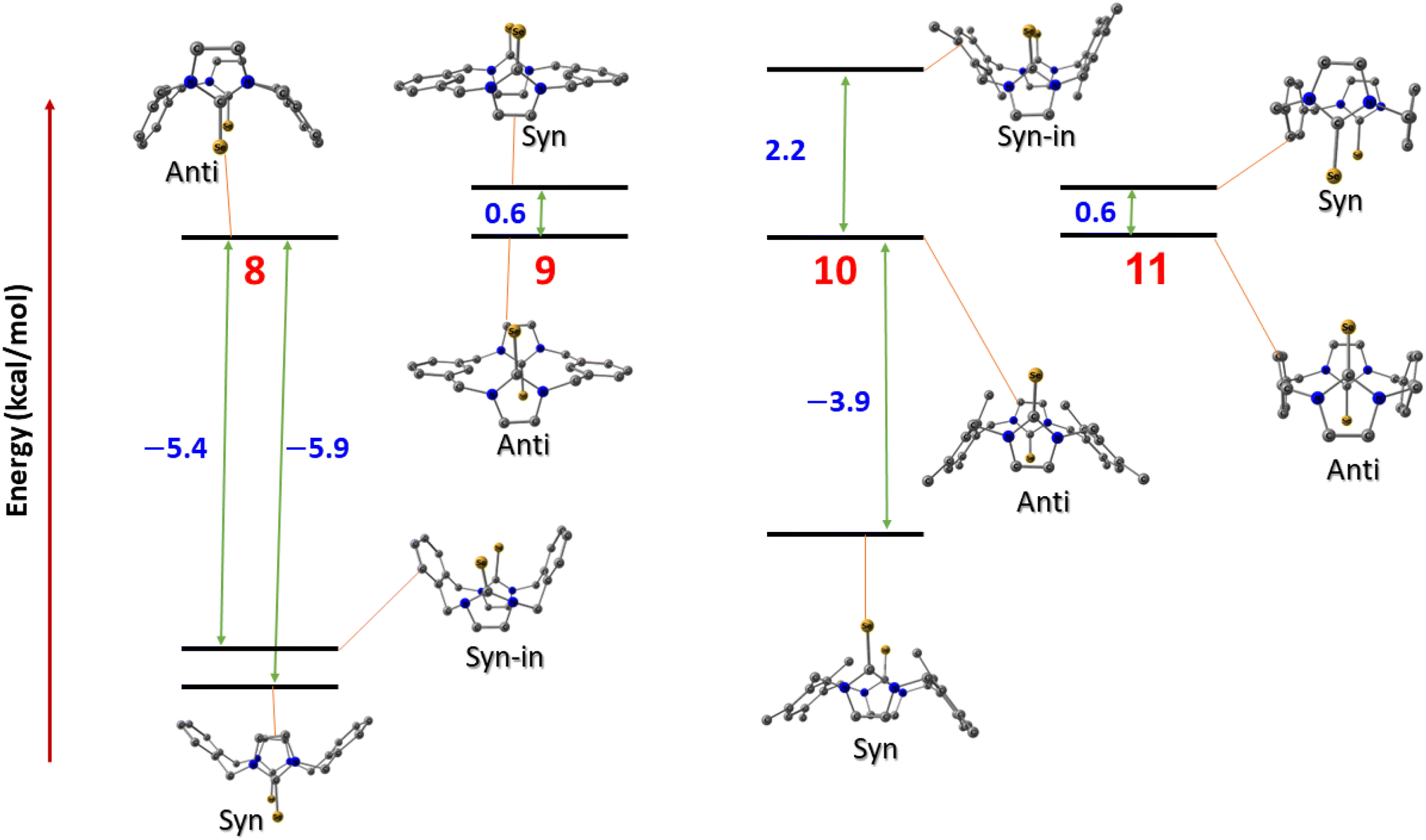
To investigate the relative stability of the different conformations of imidazole-2-selone cyclophanes, we conducted a series of calculations employing density functional theory (DFT) to interpret the origin of the stability of the crystal structures that we isolated experimentally. Four compounds were investigated with their possible spatial conformations 8 and 10 with three possible conformations: syn, where the two selone rings are oriented opposite to the xylylene rings; anti, where the CSe group in selone rings is located on the opposite side of each other; and syn_in, where the two selone rings point toward the direction of the two aromatic rings. The other two conformations (9 and 11) have only two conformations (syn and anti) owing to their semi planar nature.
Fig. 14 shows the relative energies of the different conformations calculated using the Beck functional (BP86). The negative values indicate the energy preference of the syn geometries over the anti ones. From first glance, one can assume that pairs 8 and 10, and 9 and 11 have the same energy trend in their conformational preferences. This assumption seems to be true for the latter geometries (9 and 11), where anti conformers tend to be marginally more stable over the syn structures in the semi planar molecules, which can explain the crystal co-existence of the two conformers. However, the above assumption is not true for the 8 and 10 compounds. Although both the anti symmetric structures are not energetically preferred in these systems, the energy trends are contrasted when it comes to syn_in conformers. In 8 compounds, we see syn < syn_in < anti trend, and the 10 compound stability order is syn < anti < syn_in, where the syn_in is the least stable conformer. These computed figures agree with the experimental geometries for syn geometry that we isolated and reported in the experiments.
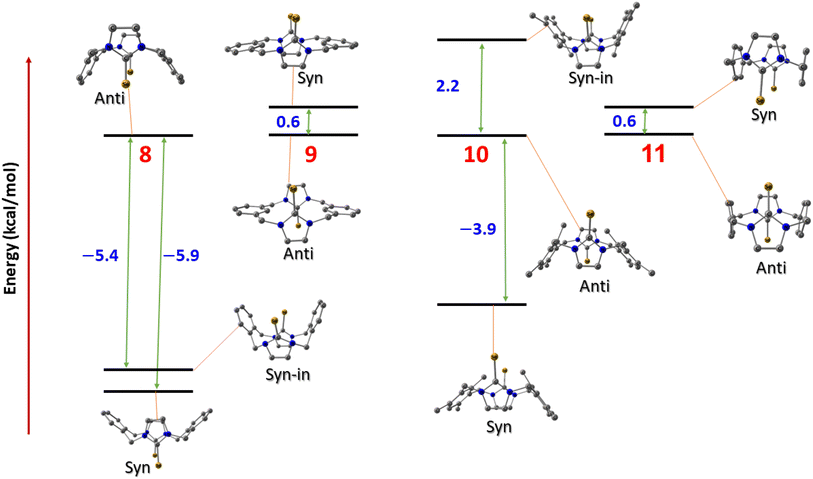 |
| | Fig. 14 The conformation relative energies in kcal mol−1 for imidazole-2-selones. | |
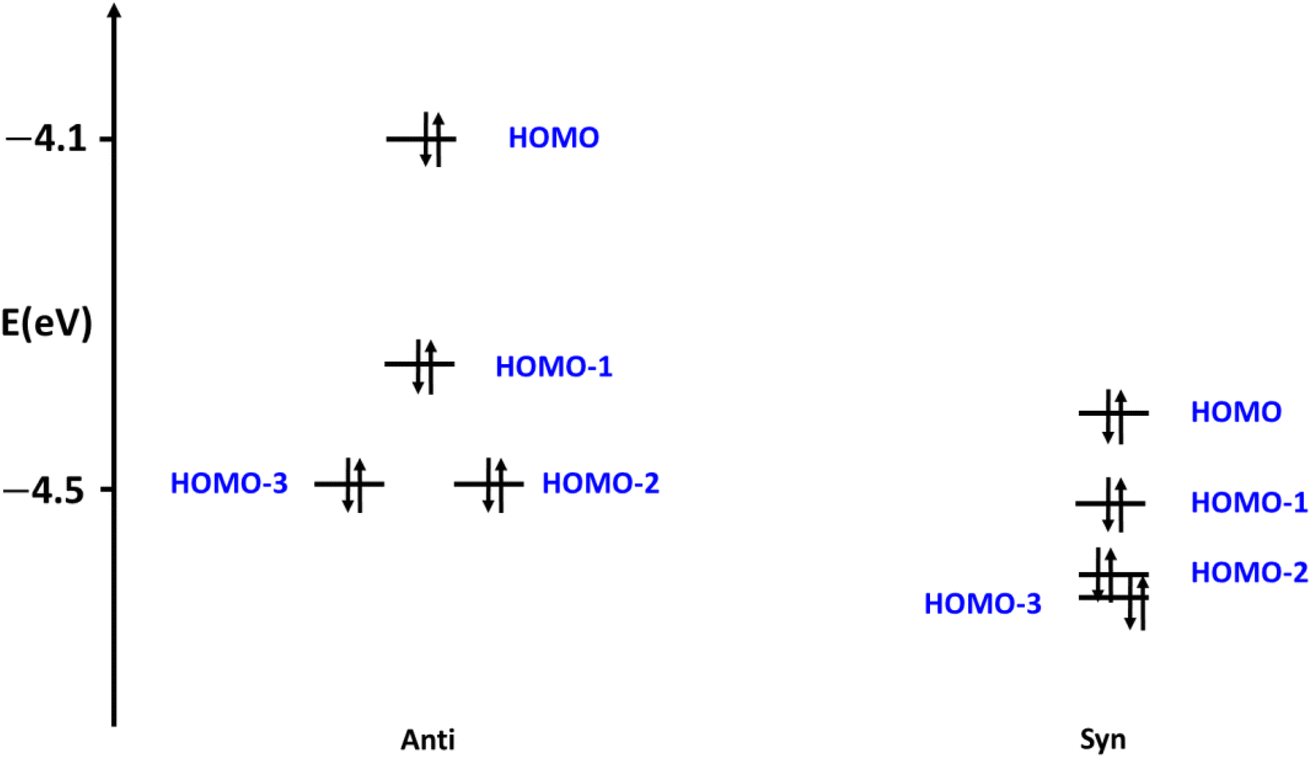
To shed light on the origin of the stability of the different conformers, we were inspired by Walsh's prominent orbital–structure correlation diagram.45 For that, we illustrated the four of the highest occupied molecular orbitals (HOMOs) of both syn and anti conformers of 10 (Fig. 15). These energies contribute to a significant share influencing the final total energy of the system. The Kohn–Sham relative molecular orbitals were represented (eV) to facilitate the obvious contrast energy levels of the two conformers. The eigenvalues of the molecular states of the syn conformers recorded obvious stability over all the orbital energies of the anti conformers. The HOMO of the anti c destabilized the HOMO orbitals by 0.27 eV, while the HOMO-2 and HOMO-3 increase by only 0.10 eV and 0.11 eV, respectively. Similar justification can be drawn for the other conformational stabilities of the other of the structures shown in Fig. 14.
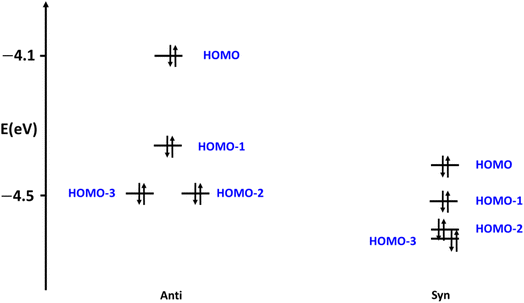 |
| | Fig. 15 The occupied frontier orbitals of 10 conformers. | |
Conclusion
Imidazole-2-selone cyclophanes can be synthesized by reacting imidazolium cyclophanes with selenium in the presence of a mild base. These cyclophanes are stable white powdered compounds and were thoroughly characterized using X-ray diffraction, NMR spectroscopy, and mass spectrometry. Similar to the imidazole-2-thione cyclophanes, the imidazole-2-selone cyclophanes with selone groups connected by o-xylylene or mesitylene-m-xylylene rings exhibit only the syn conformation, while those with selone groups connected by m-xylylene or p-xylylene rings exhibit both syn and anti conformations. Unlike the parent imidazolium cyclophanes, which are conformationally labile, the synthesized thione and selone cyclophanes are significantly more conformationally rigid. Therefore, no interconversion between the syn and anti conformations can be detected on the NMR timescale. The geometrical analysis of various compounds was carried out by DFT calculation, and the calculations included the different possible conformations that resulted from orienting the selone groups in different directions. These calculations agree with the experimental data, which confirmed the stability of both anti and syn conformers in 9 and 11 compounds and the unique stability of syn geometries in 8 and 10.
Experimental section
General procedures
Nuclear magnetic resonance spectra were recorded using a Bruker ARX500 (500.13 MHz for 1H and 125.77 MHz for 13C) or Bruker ARX 600 (600.13 MHz for 1H, 150.90 MHz for 13C) spectrometer at ambient temperature. 1H and 13C NMR chemical shifts were referenced to solvent resonance of the solvent (DMSO). When necessary, assignments were determined by 1H–13C HSQC (heteronuclear single quantum correlation) and 1H–13C HMBC (heteronuclear multiple bond coherence) spectra. Microanalyses were performed by The School of Chemistry and Molecular Bioscience, University of Queensland, Australia. High-resolution mass spectra were measured using Agilent LCMS 6510 Q-TOF and Waters LCT Premier XE spectrometers using the APCI method with MeCN:H2O (9:1) as the solvent.
Synthesis of imidazole-2-selones
1. Synthesis of compound 8. A solution of o-cyclophane salt 12·2Br (306 mg, 060 mmol) in CH3OH (5 mL) was added to a mixture of selenium (99 mg, 1.25 mmol) and K2CO3 (165 mg, 1.25 mmol) in CH3OH (15 mL). The mixture was refluxed for 48 h. The mixture was filtrated, and the solution was extracted with CHCl3. The solvent was evaporated to obtain product 8 as a white powder (198 mg, 67%). Found: C, 51.92; H, 3.99; N, 10.92% C22H20N4Se2(H2O)0.5 requires C, 52.08; H, 4.17; N, 11.04%. 1H NMR (500 MHz, DMSO-d6): 7.67–7.65 (m, 4H, H4′/H5′), 7.54–7.52 (m, 4H, H3′/H6′), δ 5.88 (s, 4H, H4/H5), 5.81–5.79 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.67–4.64 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). 13C NMR (125.75 MHz, DMSO-d6): δ 155.75 (C2), 134.94 (C1′/C2′), 133.72 (C3′/C6′), 129.63 (C4′/C5′), 118.49 (C4/C5), 50.91 (CH2). HRMS (APCI+): calcd for C22H20N4Se2 [M + H]+, m/z 501.0097 found, m/z 501.0052.
2. Synthesis of compound 9. A solution of m-cyclophane salt 14·2Br (209 mg, 0.40 mmol) in CH3OH (5 mL) was added to a mixture of selenium (67 mg, 0.85 mmol) and K2CO3 (118 mg, 0.85 mmol) in CH3OH (10 mL). The mixture was refluxed for 48 h. The mixture was filtrated, and the solution was extracted with CHCl3. The solvent was evaporated to obtain product 9 as a white powder (120 mg, 60%). Found: C, 52.11; H, 3.97; N, 10.91% C22H20N4Se2(H2O)0.5 requires C, 52.08; H, 4.17; N, 11.04%. 1H NMR (500 MHz, DMSO-d6): syn; δ 7.43–7.39 (m, 4H, H5′), 7.35–7.31 (m, 8, H4′/H6′), 6.18 (s, 2H, H2′), 7.11 (s, 4H, H4/H5), 5.89 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 5.02 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). anti; δ 7.43–7.39 (m, 4H, H5′), 7.35–7.31 (m, 8, H4′/H6′), 6.25 (s, 2H, H2′), 7.31 (s, 4H, H4/H5), 5.73 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.96 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). 13C NMR (125.75 MHz, DMSO-d6): syn; δ 157.80 (C2), 137.52 (C1′/C3′), 128.48 (C5′), 126.20 (C4′/C6′), 122.12 (C2′), 120.42 (C4/C5), 51.29 (CH2). anti; δ 156.95 (C2), 137.29 (C1′/C3′), 128.37 (C5′), 125.82 (C4′/C6′), 121.89 (C2′), 119.96 (C4/C5), 51.22 (CH2). HRMS (APCI+): calcd for C22H20N4Se2 [M + H]+, m/z 501.0097 found, m/z 501.0067.
3. Synthesis of compound 10. A solution of mesityl-cyclophane salt 13·2Br (235 mg, 0.40 mmol) in CH3OH (5 mL) was added to a mixture of selenium (68 mg, 0.85 mmol) and K2CO3 (120 mg, 0.85 mmol) in CH3OH (10 mL). The mixture was refluxed for 48 h. The mixture was filtrated, and the solution was extracted with CHCl3. The solvent was evaporated to obtain product 10 as a white powder (134 mg, 58%). Found: C, 53.88; H, 5.23; N, 8.86% C28H32N4Se2(CHCl3)0.4 requires C, 54.12; H, 5.18; N, 8.89%. 1H NMR (DMSO-d6, 500 MHz): δ 7.01 (s, 2H, H5′), 5.79 (s, 4H, H4/H5), 5.64–5.61 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.81–4.78 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 2.40 (s, 12H, CH3), 1.55 (s, 6H, CH3). 13C NMR (DMSO-d6, 125.75 MHz): δ 156.45 (C2), 138.33 (C2′), 136.92 (C4′/C6′), 133.39 (C1′/C3′), 129.86 (C5′), 118.53 (C4/C5), 46.96 (Cbenzylic), 19.57 (CH3), 17.66 (CH3). HRMS (APCI+): calcd for C28H33N4Se2 [M + H]+, m/z 585.1036. Found, m/z 585.1043.
4. Synthesis of compound 11. A solution of p-cyclophane salt 15·2Br (152 mg, 0.30 mmol) in CH3OH (5 mL) was added to a mixture of selenium (50 mg, 0.65 mmol) and K2CO3 (90 mg, 0.65 mmol) in CH3OH (10 mL). The mixture was refluxed for 48 h. The mixture was filtrated, and the solution was extracted with CHCl3. The solvent was evaporated to obtain product 11 as a white powder (105 mg, 52%). Found: C, 52.32; H, 3.93; N, 10.79% C22H20N4Se2(H2O)0.5 requires C, 52.08; H, 4.17; N, 11.04%. 1H NMR (500 MHz, DMSO-d6): syn; δ 7.48 (s, 8H, H2′/H3′), 7.15 (s, 4H, H4/H5), 5.23–5.20 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.82–4.79 (d, 2JH,H = 13 Hz, 4H, benzylic HCH). anti; δ 7.18 (s, 8H, H2′/H3’), 7.42 (s, 4H, H4/H5), 5.56–5.53 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.68–4.65 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). 1H NMR (500 MHz, acetone-d6): syn; δ 7.66 (s, 8H, H2′/H3′), 6.99 (s, 4H, H4/H5), 5.88–5.85 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.64–4.61 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). anti; δ 7.28 (s, 8H, H2′/H3′), 7.29 (s, 4H, H4/H5), 5.47–5.44 (d, 2JH,H = 15 Hz, 4H, benzylic HCH), 4.84–4.81 (d, 2JH,H = 15 Hz, 4H, benzylic HCH). HRMS (APCI+): calcd for C22H20N4Se2 [M + H]+, m/z 501.0097 found, m/z 501.0089.
Computational details
In this study, Kohn–Sham DFT was utilized to calculate the compounds under investigation. To optimize the geometry of the compounds, we utilized the general gradient approximation (GGA) functional BP86,46,47 which was composed of the Becke 1988 exchange functional and the Perdew 86 correlation functional. For more accurate energies on the optimized geometries, Ahlrichs' triple zeta valence polarized (def2-TZVP)48 basis set was employed, as well as the resolution of the identity (RI) approach and corresponding auxiliary basis sets,49 along with Grimme's D3 dispersion correction.50 All calculations were carried out using the Turbomole 7.3 package.
X-ray crystal structure determination
Crystals 8, 10 and 11 were grown by the slow evaporation of a solution of the relevant compound in CH2Cl2, and crystals of 9·(CHCl3)2 were grown by the slow evaporation of a solution of the compound in CHCl3. Crystallographic data were collected at 100(2) K using either an Oxford Diffraction Gemini or an Oxford Diffraction Xcalibur diffractometer with Mo Kα or Cu Kα radiation. Following multi-scan or analytical absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the program SHELXL-2015.51 Water molecule hydrogen atoms were refined with geometries restrained to ideal values. All remaining hydrogen atoms were added at calculated positions and refined using riding models with isotropic displacement parameters based on those of the parent atoms. Anisotropic displacement parameters were employed throughout the non-hydrogen atoms.
Author contributions
Ahmed Hassoon Mageed: conceptualization, investigation, methodology, writing – original draft. Karrar Al-Ameed: software, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia.
References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef PubMed.
- S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS PubMed.
- M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed.
- H. Valdés, D. Canseco-González, J. M. Germán-Acacio and D. Morales-Morales, J. Organomet. Chem., 2018, 867, 51–54 CrossRef.
- A. H. Mageed, J. Organomet. Chem., 2019, 902, 120965 CrossRef CAS.
- A. H. Mageed, B. W. Skelton, A. N. Sobolev and M. V. Baker, Eur. J. Inorg. Chem., 2018, 2018, 109–120 CrossRef CAS.
- K. Al-Ameed and A. H. Mageed, Polyhedron, 2021, 205, 115323 CrossRef CAS.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed.
- R. R. Rodrigues, C. L. Dorsey, C. A. Arceneaux and T. W. Hudnall, Chem. Commun., 2014, 50, 162–164 RSC.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- V. Rani, H. B. Singh and R. J. Butcher, Eur. J. Inorg. Chem., 2017, 2017, 3720–3728 CrossRef CAS.
- D. Sathyanarayana, S. K. Raja and R. Shunmugam, Spectrochim. Acta, Part A, 1987, 43, 501–506 CrossRef.
- E. Raper, J. Creighton, R. Oughtred and I. Nowell, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 355–360 CrossRef.
- S. Sauerbrey, P. K. Majhi, G. Schnakenburg, A. J. Arduengo III and R. Streubel, Dalton Trans., 2012, 41, 5368–5376 RSC.
- K. B. Wiberg and Y. Wang, Arkivoc, 2011, 45, 56 Search PubMed.
- A. H. Mageed, B. W. Skelton, A. N. Sobolev and M. V. Baker, Tetrahedron, 2018, 74, 2956–2966 CrossRef CAS.
- A. H. Mageed and K. Al-Ameed, New J. Chem., 2021, 45(39), 18433–18442 RSC.
- A. Decken, C. J. Carmalt, J. A. Clyburne and A. H. Cowley, Inorg. Chem., 1997, 36, 3741–3744 CrossRef CAS PubMed.
- G. Kjellin and J. S. Strom, Acta Chem. Scand., 1969, 23, 2888–2899 CrossRef CAS.
- W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612–5620 RSC.
- N. Ghavale, S. T. Manjare, H. B. Singh and R. J. Butcher, Dalton Trans., 2015, 44, 11893–11900 RSC.
- C. N. Babu, K. Srinivas and G. Prabusankar, Dalton Trans., 2016, 45, 6456–6465 RSC.
- H.-N. Zhang, W.-G. Jia, Q.-T. Xu and C.-C. Ji, Inorg. Chim. Acta, 2016, 450, 315–320 CrossRef CAS.
- A. K. Sharma, H. Joshi, R. Bhaskar and A. K. Singh, Dalton Trans., 2017, 46, 2228–2237 RSC.
- G. Roy, M. Nethaji and G. Mugesh, J. Am. Chem. Soc., 2004, 126, 2712–2713 CrossRef CAS PubMed.
- Y. Yamashita and M. Yamashita, J. Biol. Chem., 2010, 285, 18134–18138 CrossRef CAS PubMed.
- A. Kamal, M. A. Iqbal and H. N. Bhatti, Rev. Inorg. Chem., 2018, 38, 49–76 CrossRef CAS.
- K. N. Sharma, N. Satrawala, A. K. Srivastava, M. Ali and R. K. Joshi, Org. Biomol. Chem., 2019, 17, 8969–8976 RSC.
- W. Cao, L. Wang and H. Xu, Nano Today, 2015, 10, 717–736 CrossRef CAS.
- A. Kamal, M. Nazari, M. Yaseen, M. A. Iqbal, M. B. K. Ahamed, A. S. A. Majid and H. N. Bhatti, Bioorg. Chem., 2019, 90, 103042 CrossRef CAS PubMed.
- J. Warner, J. Org. Chem., 1963, 28, 1642–1644 CrossRef CAS.
- D. J. Nelson, A. Collado, S. Manzini, S. Meiries, A. M. Slawin, D. B. Cordes and S. P. Nolan, Organometallics, 2014, 33, 2048–2058 CrossRef CAS.
- M. Yaqoob, S. Gul, N. F. Zubair, J. Iqbal and M. A. Iqbal, J. Mol. Struct., 2020, 1204, 127462 CrossRef CAS.
- N. Iqbal, M. Yaqoob, M. Javed, M. Abbasi, J. Iqbal and M. A. Iqbal, Comput. Theor. Chem., 2021, 1197, 113135 CrossRef CAS.
- R. Ashraf, M. A. Iqbal, H. N. Bhatti, M. R. S. A. Janjua and M. El-Naggar, ChemistrySelect, 2020, 5, 10970–10981 CrossRef CAS.
- H. Buhl, K. Verlinden, C. Ganter, S. B. Novaković and G. A. Bogdanović, Eur. J. Inorg. Chem., 2016, 2016, 3389–3395 CrossRef CAS.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2015, 2416–2425 CrossRef CAS.
- S. Yadav, R. Deka and H. B. Singh, Chem. Lett., 2019, 48, 65–79 CrossRef CAS.
- M. S. S. Jamil and N. A. Endot, Molecules, 2020, 25, 5161 CrossRef CAS PubMed.
- S. V. Vummaleti, D. J. Nelson, A. Poater, A. Gómez-Suárez, D. B. Cordes, A. M. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC.
- M. V. Baker, M. J. Bosnich, D. H. Brown, L. T. Byrne, V. J. Hesler, B. W. Skelton, A. H. White and C. C. Williams, J. Org. Chem., 2004, 69, 7640–7652 CrossRef CAS PubMed.
- M. T. Aroz, M. C. Gimeno, M. Kulcsar, A. Laguna and V. Lippolis, Group 11 Complexes with Imidazoline-2-thione or Selone Derivatives, Wiley Online Library, 2011 Search PubMed.
- T. A. Walsh, Adv. Chem. Phys., 1953, 2, 171–290 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- K. Eichkorn and O. Treutler, Chem. Phys. Lett., 1995, 240, 283 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Karrar Al-Ameed
* and
Karrar Al-Ameed