DOI:
10.1039/D3RA02656F
(Paper)
RSC Adv., 2023,
13, 23050-23060
Surface-decorated porphyrinic zirconium-based metal–organic frameworks (MOFs) using post-synthetic self-assembly for photodegradation of methyl orange dye†
Received
21st April 2023
, Accepted 22nd July 2023
First published on 31st July 2023
Abstract
We report herein the surface decoration of a water-soluble free-base porphyrin, namely, 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin-tetra(p-toluenesulfonate) (H2TMPyP), over three different zirconium-based metal–organic frameworks of different linker structure and functionality; namely UiO66, UiO66-NH2, and MIP-202, via self-assembly. The synthesized MOFs along with the resulting complexes have been characterized via spectroscopic and analytical techniques (XRD, FT-IR, TEM, N2 adsorption/desorption, and laser scanning confocal microscopy). The self-assembly of H2TMPyP with the examined three MOFs was observed by using the steady-state absorption and fluorescence, as well as the fluorescence lifetime studies. It was evident that the highest complex interaction was recorded between porphyrin and UiO-66-NH2 compared with the lowest interactions between porphyrin and MIP-202. This is in good agreement with the high surface area and pore volume of UiO-66 (1100 m2 g−1 and 0.68 cm3 g−1) and compared to that of MIP-202 (94 m2 g−1 and 0.26 cm3 g−1). The photocatalytic activities of the three porphyrin entities immobilized zirconium-based MOFs were compared toward methyl orange dye degradation from aqueous solution under visible light irradiation (λex = 430 nm). The photocatalytic studies render the fabrication of the self-assembled H2TMPyP@UiO-66-NH2 composite as a promising material for dye degradation from polluted wastewater.
1. Introduction
During the last few decades, heterogeneous photocatalysis has emerged as one of the most interesting research topics. It has been used for generating solar fuels, water treatment, and nitrogen gas reduction.1 The typical photocatalytic process can be subdivided into three main stages: light absorption, charge separation forming an exciton, and interaction of the charge with a substrate.2
Metal–organic frameworks (MOFs) are among the most promising candidates as heterogeneous photocatalysts.3,4 MOFs used in photocatalysis are classified into three main types. In type I MOFs, the metallic clusters (SBUs) are regarded as 0D semiconductors, while the organic linker molecules are considered spacers that separate them throughout the MOF crystal. Type II MOFs, on the other hand, have functional dyes as organic linkers linked together in ordered and organized arrays in the MOF crystal using metallic nodes. In type III MOFs, the photocatalytic species (the organic dye in our case) of suitable size is encapsulated inside the MOF pores via in situ preparation5,6 Another way to form type III MOF photocatalysts is by soaking the MOF of suitable binding sites in a dye solution.7 This method does not give the maximum loading capacity unless the pore size, volume, and binding sites of the MOF are suitable for the guest dye molecule, but it is still much more straightforward than the in situ method. Although this method is much less complex than the previous method, it gives much fewer loading percentages than the in situ preparation.
Porphyrins are well-known for their outstanding optical properties; thus, they are used as photocatalysts8,9 solar energy conversion10,11 and biomedical applications.12,13 The synthesis of porphyrin-based metal–organic framework complexes and their utilization as light-harvesting materials was extensively investigated in the literature.14 One way to load porphyrins on metal–organic frameworks is by in situ preparation. Eddaoudi et al. managed for the first time to encapsulate H2TMPyP in the pores of the strongly anionic rho-ZMOF by in situ preparation.15 The resulting “ship in a bottle” structure of H2TMPyP inside the MOF cavities could be achieved due to the meticulous design of MOF of large pore size along with small pores around the MOF cavities to prevent leaching. Although this technique can produce significantly higher porphyrin loading percentages in the resulting complex, it is unsuitable for all MOFs.
Another way to add porphyrin to the MOF structure is via post-synthetic self-assembly. Rabiee et al. introduced porphyrins post-synthetically to the surface of NH2-MIL-53 by mixing it with leaf extracts to enhance its sensitivity for the detection of ultra-trace concentrations of ssDNA, sgRNA, anti-cas9 proteins, and recombinant SARS-CoV-2 spike antigen.16 Moreover, Kan et al. used a similar technique by mixing UiO-66 with TPP-SH in CH2Cl2 for several hours at room temperature to obtain a surface-decorated porphyrinic metal–organic framework, which proved its efficiency as a PDT agent.17 This method produces lower yields than the in situ method. However, this method is easier to apply and provides more accessibility to the porphyrin molecules.
Using post-synthetic self-assembly in DMF, Wang et al. examined the DOPA functionalization of three isoreticular, unfunctionalized Zr-based MOFs with various linker lengths (UiO-66, UiO-67, and BUT-30).18 This technique was employed to alter the nanoMOF surface chemistry while preserving crystallinity and permanent porosity by coordinating to exposed metal-containing units.
Herein, we investigate the post-synthetic surface decoration of a free-base tetra cationic porphyrin (H2TMPyP) with three synthesized zirconium-based metal–organic frameworks possessing different linkers; terephthalic acid for UiO-66, 2-aminoterepthalic acid for the amine-functionalized UiO-66, and aspartic acid for MIP-202. The selection of the three linkers is based on targeting different potential modes of interaction with the tetra cationic porphyrin, and thus varying the loading of the porphyrin on the selected MOFs (Scheme 1). The photocatalytic activity of the three complexes was also evaluated for Methylene orange (MO), and Methyl orange (MO) degradation in aqueous media using a continuous-flow photoreactor under visible light.
 |
| | Scheme 1 Schematic representation of the building blocks of the three MOFs and their targeted non-covalent interactions with H2TMPyP. | |
2. Materials and methods
2.1. Chemicals and materials
All reagents and chemicals were of the best available analytical reagent and were used as received without purification. Deionized H2O was obtained from a US Filter Corporation deionization system. 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin-tetra(p-toluenesulfonate) (H2TMPyP) was purchased from (TCI-Ace, JAPAN). Zirconium tetrachloride (ZrCl4) was purchased from Acros Organics, 1,4-benzenedicarboxylic acid (BDC), and 2-aminobenzene-1,4-dicarboxylic acid (BDC-NH2) were purchased from Sigma-Aldrich. All the measurements were performed at room temperature and under atmospheric pressure. Quartz cuvettes with a 1.00 cm path length were used for all measurements.
2.2. Synthesis of zirconium-based MOFs
2.2.1. Synthesis of UiO-66. UiO-66 was synthesized via the conventional solvothermal method shown in Scheme 2. Briefly, in a 100 mL screw-top bottle, 1,4-benzenedicarboxylic acid (BDC) (0.83 g), zirconium tetrachloride ZrCl4 (1.17 g), and 37% HCl (0.8 mL) were mixed and sonicated in 30 mL of DMF for 20 minutes, then heated in the oven for 24 h at 120 °C. The resulting MOF was cooled to room temperature, centrifuged for 15 minutes at 6000 rpm, and washed several times using (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixed reagent of DMF and anhydrous methanol. The resulting UiO-66 was activated in a vacuum oven at 100 °C overnight.
:1) mixed reagent of DMF and anhydrous methanol. The resulting UiO-66 was activated in a vacuum oven at 100 °C overnight.
 |
| | Scheme 2 Schematic representation of the general procedure of synthesizing UiO-66 using a conventional solvothermal technique. | |
2.2.2. Synthesis of UiO-66-NH2. Similarly, the amine-functionalized UiO-66 was synthesized UiO-66 was synthesized. Briefly, in a 100 mL screw-top bottle, 2-aminobenzene-1,4-dicarboxylic acid (BDC-NH2) (0.27 g), zirconium tetrachloride ZrCl4 (0.25 g), and 37% HCl (0.8 mL) were mixed and sonicated in 30 mL of DMF for 20 minutes, then heated in the oven for 24 h at 120 °C. The resulting MOF was cooled to room temperature, centrifuged for 15 minutes at 6000 rpm, and washed several times using (1:1) mixed reagent of DMF and anhydrous methanol. The resulting UiO-66 was activated in a vacuum oven at 100 °C overnight.
2.2.3. Synthesis of MIP-202. MIP-202 was synthesized according to the green hydrothermal procedure made by Diab et al.19 In a 100 mL beaker, Aspartic acid (2.8 g) was dispersed in 10 mL of deionized water using ultrasonication for 10 minutes. On the other hand, zirconium tetrachloride ZrCl4 (2.3 g) was mixed and sonicated in 10 mL of deionized water for 30 minutes. The ZrCl4 solution was then added to the aspartic acid solution dropwise under sonication. The mixture was then transferred to a reflux system and heated for 24 hours at 100 °C under magnetic stirring at 500 rpm. The resulting MOF was cooled to room temperature, centrifuged for 15–20 minutes at 6000 rpm, washed several times with water and ethanol, and dried in a vacuum oven at 60 °C overnight.
2.2.4. Synthesis of MOF@H2TMPyP composites. All the MOF@dye composites were synthesized using the same procedure followed by Xie et al.7 The MOFs were soaked in a concentrated aqueous solution of H2TMPyP and stirred overnight at 1000 rpm to achieve maximum H2TMPyP loading. The resulting complex was washed with deionized water multiple times, centrifuged at 9000 rpm for 3 minutes, and dried in a vacuum oven at 70 °C overnight.
2.3. Characterization techniques
In order to confirm the crystallographic structures of the three prepared zirconium-based MOFs, X-ray diffraction (Shimadzu-X-lab 600, Japan) was carried out at 40 kV with Cu Kα radiation. Steady-state absorption and fluorescence spectra were recorded using a UV-vis spectrophotometer (Shimadzu UV-2600) and spectrofluorometer (Shimadzu RF-6000), respectively. Laser microscopy measurements were performed on MOF films prepared using drop casting on a glass substrate using a Keyence 3D laser microscope. Transmission electron microscopy (TEM) images were captured using a JEOL 2100 microscope operating at an accelerating voltage of 200 kV. The information on functional groups of the prepared materials was determined using FTIR (Shimadzu 8400s, Japan).
2.4. Photocatalytic activity of porphyrin@MOF complexes toward methyl orange dye degradation in aqueous solution
The three fabricated MOF composites were compared against their potential photocatalytic degradation of methyl orange dye under visible light irradiation (λex = 430 nm). The photocatalytic experiment was conducted using a continuous-flow reactor (Vapour tech. UV-150) at room temperature (25 °C), keeping a constant flow rate of (5 mL min−1), and a constant catalyst dose of 10 mg per 100 mL of the dye solution. The working procedure is similar to the procedure previously discussed by Safo et al.20 Each catalyst sample was added to a 100 mL dye solution (20 ppm) under stirring without adding any buffers. The samples were kept in the dark for 60 minutes in order to reach the adsorption–desorption equilibrium. After that, the samples were subjected to the LED lamp irradiation (λex = 430 nm) in the (UV-150) reactor for additional 60 minutes, at which the product was collected every 10 minutes, centrifuged for 5 minutes at 7000 rpm to remove any dispersed catalyst, and decanted. The absorbance of the supernatant at 464 nm was determined using a spectrophotometer to detect the photodegradation of the dye. The photocatalytic degradation efficiency was evaluated according to the following equation:
where (ηdeg) stands for the photocatalytic degradation efficiency, (A0) indicates the initial absorbance, and (ΔA) stands for the difference between initial absorbance and absorbance at a given time.
3. Results and discussion
3.1. Characterization of MOFs and MOF complexes
All the MOF@dye composites were synthesized using the same procedure followed by Xie et al.21 The MOFs were soaked in a concentrated aqueous solution of H2TMPyP and stirred overnight at 1000 rpm to achieve maximum H2TMPyP loading. The resulting complex was washed with deionized water multiple times, centrifuged at 9000 rpm for 3 minutes, and dried in a vacuum oven at 70 °C overnight. The loading efficiency (η) of the H2TMPyP was calculated by the following equation:22
where m0 is the mass of the dye in the initial dye solution, ms is the mass of H2TMPyP in the supernatant after loading, and mMOF is the mass of the MOF. The ms value was determined by fitting the absorbance value of the supernatant on the calibration curve of H2TMPyP at λ = 520 nm (Fig. S1†). According to this method, the porphyrin loading efficiency on the surface of the MOFs was found to be the lowest in the case of MIP-202 (η = 0.09%), the loading efficiency increased to 0.15% in the case of UiO-66, while UiO-66-NH2 achieved the highest loading efficiency with (η = 0.21%), which is more than the double of MIP-202. The stability of the resulting complexes using UV-vis spectroscopy in aqueous media has been tested for 18 hours and they were found to be stable under ambient conditions of temperature and pH. Also, the three normalized absorption spectra of the three complexes shown in Fig. S10† show the presence of the porphyrin peak at 425 nm, which confirms the loading of the porphyrin on the MOF.
3.1.1. XRD. As previously mentioned, three zirconium-based metal–organic frameworks were synthesized namely, UiO-66, UiO-66-NH2, and MIP 202. As shown in Fig. 1, the XRD data exhibited three significant peaks in the case of UiO-66 at 7.24°, 8.38°, and 25.56°. UiO-66-NH2 also has three identified peaks at 7.19°, 8.31°, and 25.55° with a sharp shoulder at 25.18° corresponding to the (111), (002), and (006) planes respectively. These results are in good agreement with the values in the literature.16 On the other hand, MIP-202 has distinguishable peaks at 8.84°, 9.84°, 19.78° and 21.56° corresponding to the (111), (200), (420), and (440) planes as previously investigated in the literature. Accordingly, XRD patterns confirm the successful preparation of the three zirconium-based metal–organic frameworks. Although the XRD data shown in Fig. 1 did not exhibit a significant change in the PXRD patterns after immobilization of the three MOF types with H2TMPyP, it may prove that the MOF's crystallinity was preserved before and after adding the porphyrin.
 |
| | Fig. 1 XRD of the synthesized UiO-66, UiO-66-NH2, and MIP-202. | |
3.1.2. HR-TEM. High-resolution transmission electron microscopy (HR-TEM) was used to confirm the self-assembly between the porphyrin and synthesized zirconium-based MOFs. The TEM images in Fig. 2a–c reveal the aggregation of MOF particles as a result of the complex formation between H2TMPyP and the three MOF types. The highest aggregation is observed in the UiO-66-NH2 complex, and the lowest level of aggregation is observed in the MIP-202 complex. These results suggested that the interactions between the porphyrin dye and UiO-66-NH2 are higher than that between the porphyrin dye and MIP-202.
 |
| | Fig. 2 HR-TEM images of (a) H2TMPyP@UiO-66-NH2 complex, (b) H2TMPyP@UiO-66 complex, and (c) H2TMPyP@MIP-202 complex. | |
In order to confirm the loading of H2TMPyP on the MOFs, TEM-EDS elemental analysis was performed for the pristine MOFs as well as the three MOF complexes (Fig. S2–S7†). The nitrogen peaks could not be detected in the TEM-EDS spectra due to their overlapping with carbon and oxygen peaks. However, the presence of nitrogen in the sample could be detected from elemental mapping. The presence of sulfur, which is only present in H2TMPyP in the complex samples in very small amounts as well as the increasing carbon ratio in H2TMPyP@UiO-66-NH2 and H2TMPyP@UiO-66 samples can confirm the porphyrin loading on the MOFs. The carbon ratio in the H2TMPyP@MIP-202 slight decrease compared to the MIP-202 sample along with the unexpected increase in the sulfur ratio may suggest that the linker exchange mechanism can be favorable in the case of MIP-202. Overall, the EDS mapping reveals uniform and homogenous distributions of the elements in the three MOF complexes, including C, O, N, S, and Zr along the surfaces.
3.1.3. BET. The nitrogen adsorption–desorption isotherms of UiO-66 and MIP-202 were studied to gain insight into their surface area and pore volume. This important information helps to understand the physical characteristics of the material, which are crucial for determining its suitability for various applications. The results of the nitrogen adsorption–desorption isotherms showed the amount of nitrogen adsorbed by the UiO-66 and MIP-202 samples as a function of pressure. From these measurements, the BET surface area and pore volume of UiO-66 were determined to be 1100 m2 g−1 and 0.6834 cm3 g−1, respectively. It is noteworthy to mention that this surface area is comparable and matches the surface area reported for UiO-66 MOF.23 As a result, one favorable probability for the H2TMPyP molecules to assemble on the surface of the UiO-66 as it offers a large space for adsorption, as both the high surface area and pore volume provide ample space for the H2TMPyP molecules to be adsorbed inside the pores of H2TMPyP.16,21 Also, there is a probability for the diffusion of the porphyrin molecules by linker exchange as reported in the literature.24,25 However, the nitrogen adsorption–desorption isotherms of the complexes (Fig. 3) show surface area values of 835 and 996 m2 g−1 and pore volume values of 0.4317 and 0.5230 cm3 g−1 for the H2TMPyP@UiO-66-NH2 and H2TMPyP@UiO-66 complexes, respectively. This slight decrease in the surface area can be explained in terms of the successful porphyrin loading on the MOF. The surface area and pore volume of MIP-202 bio-MOF were also determined to be around 94 m2 g−1 and 0.26 cm3 g−1, respectively. It is noteworthy to mention that the N2 isotherm and surface area values are consistent with previous reports of the MIP-202 bio-MOF and that the resulting porosity is attributed to the shrinking of the MOF micropores towards the nitrogen.16
 |
| | Fig. 3 N2 adsorption–desorption isotherms of (a) H2TMPyP@UiO-66-NH2, and (b) H2TMPyP@UiO-66. | |
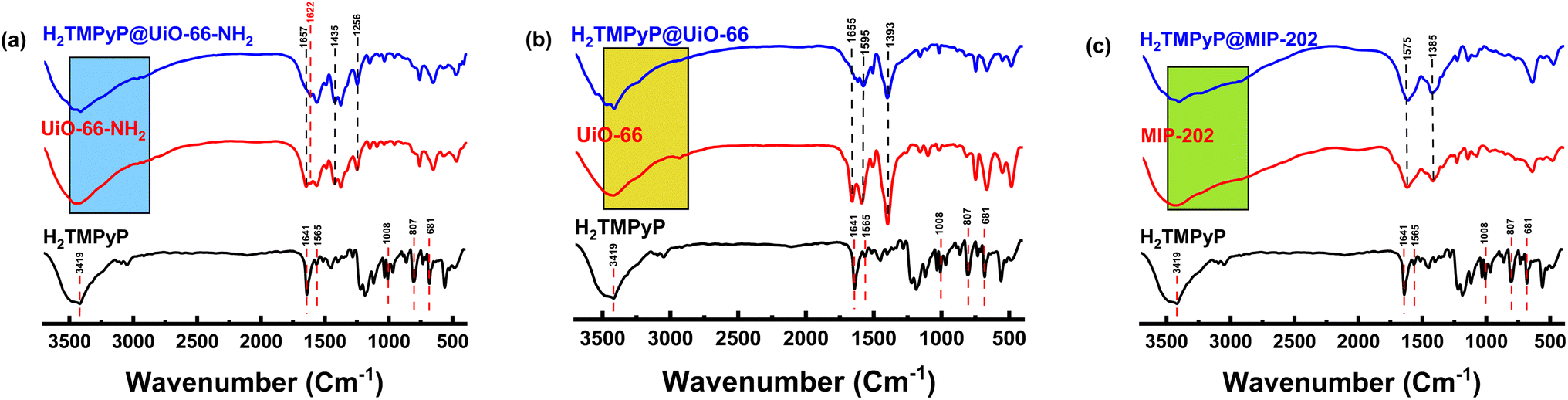
3.1.4. FT-IR. The FTIR spectra obtained from H2TMPyP, Zr-MOFs, and their complexes are shown in Fig. 5. Revealing spectral changes before and after the surface decoration with H2TMPyP. The spectrum of H2TMPyP exhibits fingerprint bands attributed to the amino groups present in the structure of the macrocycle and the pyridinium groups at 681, 733, 807, 1008, 1033, 1565, and 1641 cm−1, corresponding to in-plane vibration deformation porphyrin, out-of-plane N–H bending, out-of-plane C–H bending, C–H rocking vibrations, in-plane N–H bending and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch vibrations, respectively.26,27 On the other hand, the FT-IR spectrum of UiO-66-NH2 (Fig. 4a) exhibited a strong peak at 1657 cm−1, which can be related to the ν(CO) stretching of N,N-dimethylformamide (DMF). The absence of this band in the complex indicates the complete exchange of DMF with water.28,29 The 3000–2800, and 3600–3100 cm−1 bands are assigned to the stretching vibration of CH2 and unreacted amino groups, respectively. However, the fact that these signals became slightly stronger after the addition of H2TMPyP can be attributed to the successful incorporation of the H2TMPyP on the MOF surface.28 The characteristic stretching vibration peaks of NH2 on the MOF appeared at 1435 and 1256 cm−1. Another band at 1622 cm−1 can be attributed to the NC bond (stretching vibration).28,30
C stretch vibrations, respectively.26,27 On the other hand, the FT-IR spectrum of UiO-66-NH2 (Fig. 4a) exhibited a strong peak at 1657 cm−1, which can be related to the ν(CO) stretching of N,N-dimethylformamide (DMF). The absence of this band in the complex indicates the complete exchange of DMF with water.28,29 The 3000–2800, and 3600–3100 cm−1 bands are assigned to the stretching vibration of CH2 and unreacted amino groups, respectively. However, the fact that these signals became slightly stronger after the addition of H2TMPyP can be attributed to the successful incorporation of the H2TMPyP on the MOF surface.28 The characteristic stretching vibration peaks of NH2 on the MOF appeared at 1435 and 1256 cm−1. Another band at 1622 cm−1 can be attributed to the NC bond (stretching vibration).28,30
 |
| | Fig. 4 FT-IR spectra of H2TMPyP (black), Zr-based MOFs (red), and MOF@H2TMPyP complexes (blue). | |
Likewise, UiO-66 exhibited a (CO) stretching peak arising from DMF residues at 1655 cm−1, which diminishes clearly in the complex (Fig. 4b). The MIP-202 (Fig. 4c) has a characteristic peak at 3447 cm−1 corresponding to the symmetric and asymmetric vibrations of the NH2 group of the aspartate linker. This peak became sharper in the case of H2TMPyP@MIP-202 due to the interference with the sharp porphyrinic peak at 3419 cm−1 indicating successful loading of the porphyrins. Moreover, the presence of two other peaks can be noticed at 2924 and 2851 cm−1 corresponding to the symmetrical and asymmetrical stretching vibration of methylene groups (CH2) in the aspartate linker. The characteristic double peak of MIP-202 in the range between 1400–1600 cm−1 can be attributed to the symmetric stretching vibration of the carboxylate group in the amino acid.
3.2. Spectroscopic studies for the self-assembly complexes
3.2.1. UV-vis spectroscopy. UV-vis absorption spectra of H2TMPyP in aqueous media (Fig. S8†) show a conventional sharp Soret band resulting from the (S0 → S2) transition at around 422 nm. This band undergoes a slight blue shift upon increasing the concentration of H2TMPyP from 0.25 to 2.91 μM to become 423.5 nm. Also, Q-band resulting from the (S0 → S1) transition can be observed at around 520 nm. Photometric titrations have been conducted on H2TMPyP using multiple additions of three Zr-based MOFs, namely UiO-66-NH2, UiO-66, and MIP-202. Before the addition of UiO-66-NH2, H2TMPyP has shown a main sharp absorption band at 422 nm and another smaller absorption band at 519 nm. Upon titration with UiO-66-NH2 (Fig. 5a), we could notice a significant decrease in the absorbance intensity accompanied by an 8 nm red shift of the main band to reach 430 nm with two clear isosbestic points at 411 and 431 nm. Similar behavior was recorded upon the addition of UiO-66 and MIP-202 to the aqueous solutions of H2TMPyP (Fig. 5b and c). This decrease of the absorbance along with the red shift suggests the self-assembly of H2TMPyP over the surface of the examined types of MOFs (UiO-66-NH2, UiO-66, and MIP-202) through different binding modes. Based on the Benesi–Hildebrand method shown in Fig. 5d,31 the binding constants (Kb) for the Zr-based MOF complexes with H2TMPyP were calculated in the following order: 1.13 × 105 M−1 (for UiO-66-NH2), 4.89 × 104 M−1 (for non-functionalized UiO-66), while the MIP-202 possessed the lowest binding constant (Kb = 3.19 × 104 M−1). Implying that the dominant mode of interaction during the formation of the H2TMPyP@UiO66 complex is π–π stacking, while in the case of H2TMPyP@UiO66-NH2 hydrogen bonding between the pyridinium ring of the porphyrin and the amine group of the MOF may lead to added stability to the formed complex along with π–π stacking.
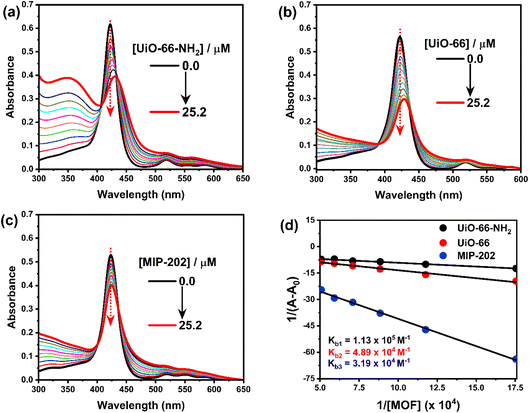 |
| | Fig. 5 UV-vis absorption spectra of photometric titration of H2TMPyP (3.0 μM) in the presence of: (a) UiO-66, (b) UiO-66-NH2, (c) MIP-202, (d) Benesi–Hildebrand plot for the three titrations to determine the binding constant Kb. | |
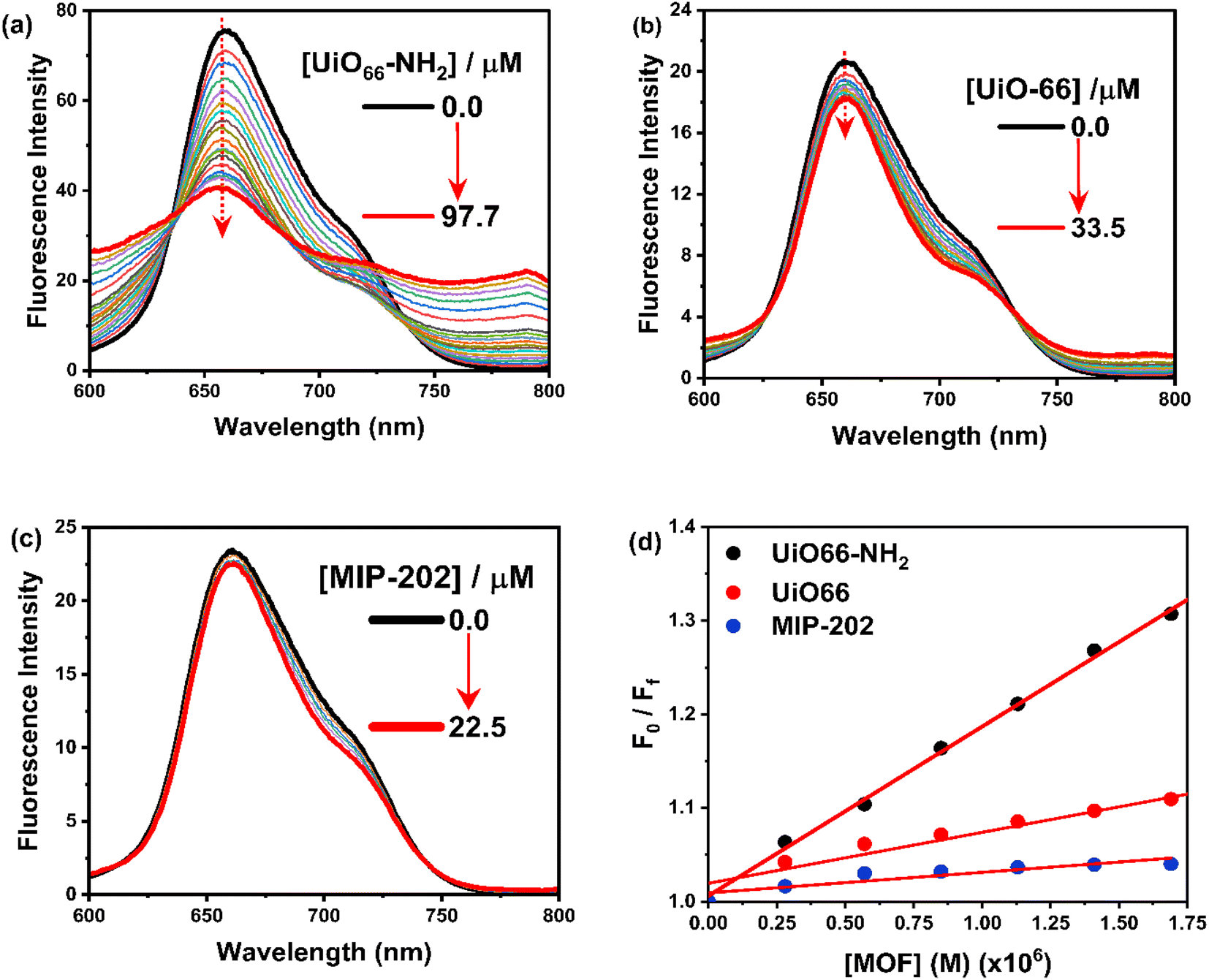
3.2.2. Steady-state and time-resolved fluorescence measurements. The fluorescence spectrum of H2TMPyP in an aqueous medium exhibited a broad emission band at 658.5 nm, from which the energy of the singlet excited state was calculated to be 1.88 eV (Fig. 6a).32 By adding further various amounts of UiO-66-NH2 to a solution of H2TMPyP, a significant fluorescence quenching of the emission band of H2TMPyP was observed accompanied by a considerable red-shift (≈3 nm), and the appearance of a new weak emission band at 790 nm. Similarly, the fluorescence quenching of H2TMPyP was observed in the presence of UiO-66 and MIP-202 (Fig. 6b and c). Similarly, upon titration using UiO-66, we could notice a 6.5 nm red shift in the main band with two isosbestic points at 410, and 429 nm (Fig. 6b). The emission spectra (Fig. 6b) show noticeable fluorescence quenching. Also, the vibrational resolution of the shoulder increased. After adding MIP-202 (Fig. 6c), we could notice the formation of two isosbestic points at 613, and 744 nm along with slight emission quenching of H2TMPyP with a non-significant red shift (≈1 nm). Similar behavior concerning the bathochromic shifting of the H2TMPyP Soret band was explained in the literature in terms of the flattening of the porphyrin rings on the surface due to π–π stacking during its self-assembly with the negatively charged chemically converted graphene (CCG)33 and LAPONITE®34 which suggests that the red-shifting can arise from the porphyrin ring flattening. Moreover, the formation of a stable isosbestic point after several additions of the three MOFs eliminates the possibility of any contribution of J-aggregates formation to this bathochromic shift.33,34 Based on the Stern–Volmer plots shown in Fig. 6d, it is apparent that the rate constants of the self-assembly process decrease were found to be in the order of UiO-66-NH2 (1.8 × 104 L M−1) > UiO-66 (5.4 × 103 L M−1) > MIP-202 (2.2 × 103 L M−1).35
 |
| | Fig. 6 Fluorescence spectra for titration of H2TMPyP (3.0 μM) of H2TMPyP (3.0 μM) in the presence of (a) UiO-66, (b) UiO-66-NH2, (c) MIP-202. The concentrations of MOFs were kept at (0–25.2 μM). (d) Stern–Volmer plots. | |
The fluorescence lifetime measurements were also performed to track the steady-state fluorescence measurements in a quantitative way using 439 nm excitation light (Fig. 7 and S9†). The decay profile of H2TMPyP could be fitted as a single-exponential decay, where the fluorescence lifetime (τ0) of H2TMPyP was found to be 2.75 ns. Upon adding UiO-66-NH2, we could observe a gradual formation of a bi-exponential process with gradual quenching in fluorescence lifetime. At the end of the titration, the addition of 97.7 μM of UiO66-NH2 resulted in a fast decay component (τ1) with a lifetime of 0.3 ns and a slow decay component (τ2) with a lifetime of 1.9 ns. On the other hand, upon adding 87.3 μM of UiO-66 a bi-exponential decay profile becomes clear with a relatively faster decay component (τ1) with a lifetime of 0.5 ns, and a slower decay component (τ2) with a lifetime of 4.3 ns. Upon adding 49.9 μM of UiO-66, a bi-exponential decay profile becomes clear with a relatively faster decay component (τ1) with a lifetime of 1.6 ns.
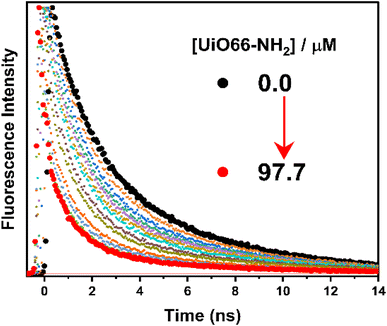 |
| | Fig. 7 TCSPC decay profiles for titration of H2TMPyP (3.0 μM) with UiO-66 (0–97.7 μM). The excitation light was fixed at 439 nm. | |
3.2.3. Laser scanning confocal microscopy. The use of laser scanning confocal microscopy provided additional evidence for the H2TMPyP's successful self-assembly with the three Zr-MOFs under examination. Fig. 8a depicts the image of the UiO-66-NH2 under examination, while Fig. 8b depicts the image of the H2TMPyP@UiO-66-NH2 that was self-assembled. As can be seen, the brightness exhibited in the case of H2TMPyP@UiO-66-NH2 would indicate that H2TMPyP units were successfully loaded onto the surface of UiO-66-NH2. Similar behavior was seen when H2TMPyP was loaded onto the surfaces of UiO-66 (Fig. 9c and d) and MIP-202 (Fig. 9e and f). The matching of the Q-band of the loaded photo-excited H2TMPyP units across the surface of MOFs with the wavelength of the laser source utilized in the microscope (658 nm) may be the cause of the brightness of the self-assembled composites (Fig. S9†). A further indicator of the increasing porphyrin loading in the samples is the observed pattern of gradual brightness increase in the three composites (H2TMPyP@UiO-66-NH2 > H2TMPyP@UiO-66 > H2TMPyP@MIP-202), which is consistent with the aforementioned absorption and fluorescence measurements (Fig. 5–7). When porphyrin units were loaded onto the surface of GO, a similar pattern of behavior was seen.36
 |
| | Fig. 8 Laser scanning confocal microscope images of: (a) UiO-66-NH2, (b) H2TMPyP@UiO-66-NH2 complex, (c) UiO-66, (d) H2TMPyP@UiO-66 complex, (e) MIP-202, and (f) H2TMPyP@MIP-202 complex. | |
 |
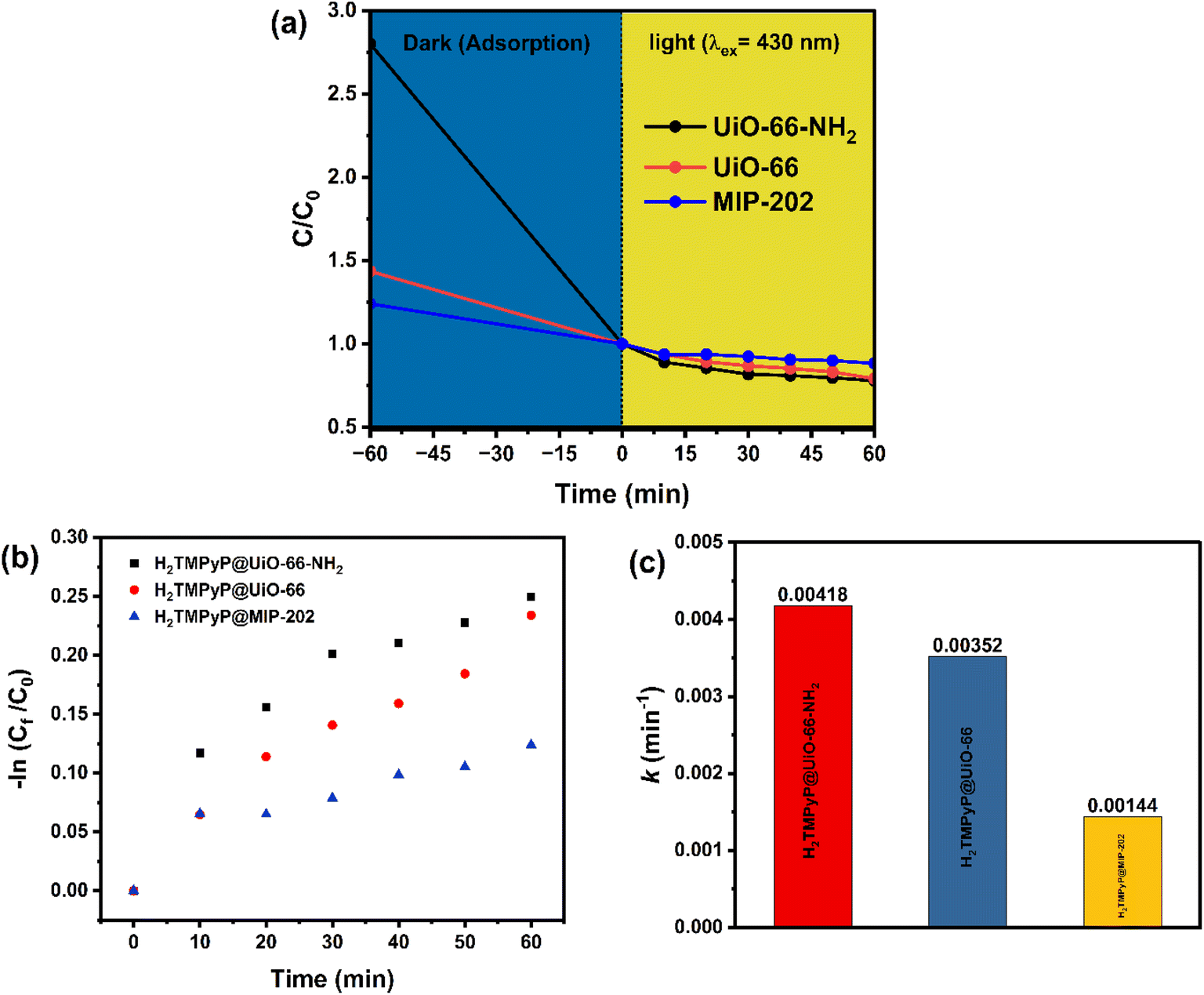
| | Fig. 9 (a) Rate of photodegradation of MO dye under (λex = 430 nm) light irradiation [conditions: catalyst dosage of 10 mg, MB concentration of 20 mg L−1], (b) first-order reaction kinetics of the photocatalytic degradation of MO using H2TMPyP@MOF composites, and (c) rate constant values (k) of the photocatalytic degradation reaction of MO using H2TMPyP@MOF composites. | |
3.3. Evaluation of the photocatalytic degradation of methyl orange dye
A significant variation in the absorption capacity was indicated from the results illustrated in Fig. 9 for the three fabricated MOF composites with the UiO-66-NH2 having the highest absorption capacity followed by UiO-66 and MIP-202 respectively. Moreover, the three MOF composites showed photocatalytic degradation efficiencies of 21.5%, 20.6%, and 11.5% for the UiO-66-NH2, UiO-66, and MIP-202 samples, respectively, which can be elaborated in terms of the varying porphyrinic loading on the surface. In order to understand the reaction kinetics, the rate constant of the photodegradation reaction was calculated using a pseudo-first-order model (Fig. 9a) using the following equation: kt = −ln(C0/Ct), where (kt) is the rate constant in min−1, (t) is time in minutes, (C0) represents the dye concentration before degradation, and (Ct) represents the dye concentration after a given amount of time (t). As seen in Fig. 9b, the rate constants for the photocatalytic degradation reactions of MO using the three MOF composites were calculated and found to be 0.0042 min−1 in the case of H2TMPyP@UiO-66-NH2, 0.0035 min−1 for H2TMPyP@UiO-66, and 0.0014 min−1 for H2TMPyP@MIP-202. The values of the regression coefficients (R2) were found to be 0.9233, 0.9406, and 0.98514 respectively. The previous findings prove that the H2TMPyP@UiO-66-NH2 composite had the highest reaction rate value with a slight advantage over the H2TMPyP@UiO-66 composite, which is in good agreement with the previously reported data.
4. Conclusion
The surface decoration of UiO-66-NH2, UiO-66, and MIP-202 using a water-soluble porphyrin H2TMPyP via post-synthetic self-assembly was investigated via various spectroscopic and microscopic techniques. The loading of H2TMPyP and the stability of the three composites were found to be significantly dependent on the type of targeted non-covalent interactions with the linker as observed from the laser fluorescence images, as well as the steady-state absorption and fluorescence measurements. The BET results showed a high surface area and pore volume of UiO-66 (1100 m2 g−1 and 0.6834 cm3 g−1) compared to that of MIP-202 (surface area = 94 m2 g−1 and pore volume = 94 cm3 g−1). Keeping into consideration the large diameter of the porphyrin molecule (approximately 0.26 nm) along with the low pore diameter of the three MOFs confirms the presence of the H2TMPyP on the surface. Also, a comparative study on the photocatalytic degradation of Methyl orange dye from aqueous solution using the three composites was conducted under visible light using a continuous-flow reactor at room temperature. The photocatalytic degradation efficiencies were found to be directly proportional to the porphyrinic loading on the surface of the three MOFs.
Conflicts of interest
The authors declare no conflict of interest.
References
- X. Yang and D. Wang, Photocatalysis: From Fundamental Principles to Materials and Applications, ACS Appl. Energy Mater., 2018, 1(12), 6657–6693 CrossRef CAS.
- S. Fukuzumi, Electron Transfer: Mechanisms and Applications, Wiley-VCH, 2020 Search PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Metal–Organic Frameworks as Photocatalysts for Solar-Driven Overall Water Splitting, Chem. Rev., 2022, 123(1), 445–490 CrossRef.
- C. Lin, C. Han, H. Zhang, L. Gong, Y. Gao, H. Wang, Y. Bian, R. Li and J. Jiang, Porphyrin-Based Metal–Organic Frameworks for Efficient Photocatalytic H2 Production under Visible-Light Irradiation, Inorg. Chem., 2021, 60(6), 3988–3995 CrossRef CAS PubMed.
- C. Wu and M. Zhao, Incorporation of Molecular Catalysts in Metal–Organic Frameworks for Highly Efficient Heterogeneous Catalysis, Adv. Mater., 2017, 29, 1605446 CrossRef PubMed.
- L. Zeng, X. Guo, C. He and C. Duan, Metal–Organic Frameworks: Versatile Materials for Heterogeneous Photocatalysis, ACS Catal., 2016, 6(11), 7935–7947 CrossRef CAS.
- W. Chen, Y. Zhuang, L. Wang, Y. Lv, J. Liu, T. L. Zhou and R. J. Xie, Color-Tunable and High-Efficiency Dye-Encapsulated Metal-Organic Framework Composites Used for Smart White-Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2018, 10(22), 18910–18917 CrossRef CAS.
- L. Wang, H. Fan and F. Bai, Porphyrin-based photocatalysts for hydrogen production, MRS Bull., 2020, 45, 49–56 CrossRef.
- K. Rybicka-Jasińska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, Porphyrins as Promising Photocatalysts for Red-Light-Induced Functionalizations of Biomolecules, ACS Org. Inorg. Au, 2022, 2, 422–426 CrossRef PubMed.
- W. Zhu, N. Sharma, Y.-M. Lee, M. E. El-Khouly, S. Fukuzumi and W. Nam, Use of singlet oxygen in the generation of a mononuclear nonheme iron(IV)-Oxo complex, Inorg. Chem., 2023, 62(10), 4116–4123 CrossRef CAS PubMed.
- N. Sharma, J. jieun, K. Ohkubo, L. Ying-Min, M. E. El-Khouly, W. Nam and S. Fukuzumi, Long-lived photoexcited state of a Mn(IV)-Oxo complex binding scandium ions that is capable of hydroxylation benzene, J. Am. Chem. Soc., 2018, 140(27), 8405–8409 CrossRef CAS PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy, Photochem. Photobiol., 2009, 85(5), 1053–1074 CrossRef PubMed.
- R. R. Cheruku, J. Cacaccio, F. A. Durrani, W. A. Tabaczynski, R. Watson, A. Marko, R. Kumar, M. E. El-Khouly, S. Fukuzumi, J. R. Missert, R. Yao, M. Sajjad, D. Chandra, K. Guru and R. K. Pandey, Epidermal growth factor receptor-targeted multifunctional photosensitizers for bladder cancer imaging and photodynamic therapy, J. Med. Chem., 2019, 62(5), 2598–2617 CrossRef CAS.
- J. Chen, Y. Zhu and S. Kaskel, Porphyrin-Based Metal–Organic Frameworks for Biomedical Applications, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, Zeolite-like Metal−Organic Frameworks as Platforms for Applications: On Metalloporphyrin-Based Catalysts, J. Am. Chem. Soc., 2008, 130(38), 12639–12641 CrossRef CAS PubMed.
- N. Rabiee, M. Rabiee, S. Sojdeh, Y. Fatahi, R. Dinarvand, M. Safarkhani, S. Ahmadi, H. Daneshgar, F. Radmanesh, S. Maghsoudi, M. Bagherzadeh, R. S. Varma and E. Mostafavi, Porphyrin Molecules Decorated on Metal–Organic Frameworks for Multi-Functional Biomedical Applications, Biomolecules, 2021, 11(11), 1714 CrossRef CAS.
- J. L. Kan, Y. Jiang, A. Xue, Y. H. Yu, Q. Wang, Y. Zhou and Y. B. Dong, Surface Decorated Porphyrinic Nanoscale Metal-Organic Framework for Photodynamic Therapy, Inorg. Chem., 2018, 57(9), 5420–5428 CrossRef CAS PubMed.
- S. Wang, W. Morris, Y. Liu, C. M. McGuik, Y. Zhou, J. T. Hupp, O. K. Farha and C. A. Mirkin, Surface specific functionalization of nanoscale metal-organic frameworks, Angew. Chem., Int. Ed., 2015, 54, 14738–14762 CrossRef CAS PubMed.
- K. E. Diab, E. Salama, H. S. Hassan, A. Abd El-Moneim and M. F. Elkady, Biocompatible MIP-202 Zr-MOF Tunable Sorbent for Cost-Effective Decontamination of Anionic and Cationic Pollutants from Waste Solutions, Sci. Rep., 2021, 11, 6619 CrossRef CAS PubMed.
- K. Safo, H. Noby, M. Matatoshi, H. Naragino and A. H. Shazly, Statistical optimization modeling of organic dye photodegradation process using slag nanocomposite, Res. Chem. Intermed., 2022, 48(10), 4183–4208 CrossRef CAS.
- W. Chen, Y. Zhuang, L. Wang, Y. Lv, J. Liu, T. L. Zhou and R. J. Xie, Color-Tunable and High-Efficiency Dye-Encapsulated Metal-Organic Framework Composites Used for Smart White-Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2018, 10(22), 18910–18917 CrossRef CAS.
- M. He, J. Zhou, J. Chen, F. Zheng, D. Wang, R. Shi, Z. Guo, H. Wang and Q. Chen, Fe3O4@carbon@zeolitic Imidazolate Framework-8 Nanoparticles as Multifunctional pH-Responsive Drug Delivery Vehicles for Tumor Therapy in vivo, J. Mater. Chem. B, 2015, 3(46), 9033–9042 RSC.
- Y. Cao, H. Zhang, F. Song, T. Hunag, J. Ji, Q. Zhong, W. Chu and Q. Xu, UiO-66-NH2/GO composite: Synthesis, characterization and CO2 adsorption performance, Materials, 2018, 11(4), 589 CrossRef PubMed.
- Z. Li, T. M. Rayder, L. Luo, J. Byers and C.-K. Tsung, Aperture-Opening Encapsulation of a Transition Metal Catalyst in a Metal–Organic Framework for CO2 Hydrogenation, J. Am. Chem. Soc., 2018, 140(26), 8082–8085 CrossRef CAS.
- J. V. Morabito, L.-Y. Chou, Z. Li, C. M. Manna, C. A. Petroff, R. J. Kyada, J. M. Palomba, J. A. Byers and C.-K. Tsung, Molecular Encapsulation beyond the Aperture Size Limit through Dissociative Linker Exchange in Metal–Organic Framework Crystals, J. Am. Chem. Soc., 2014, 136(36), 12540–12543 CrossRef CAS.
- R. X. Wang, J. J. Fan, Y. J. Fan, J. P. Zhong, L. Wang, S. G. Sun and X. C. Shen, Platinum Nanoparticles on porphyrin functionalized graphene nanosheets as superior catalysts for methanol electrooxidation, Nanoscale, 2014, 6(24), 14999–15007 RSC.
- D. Lozano-López, M. Galván-Valencia, I. Rojas-de Soto, R. A. Escalona-Villalpando, J. Ledesma-García and S. Durón-Torres, Immobilization of Glucose Oxidase on Glutathione Capped CdTe Quantum Dots for Bioenergy Generation, Catalysts, 2022, 12(12), 1659 CrossRef.
- J. Zhu, L. Wu, Z. Bu, S. Jie and B. G. Li, Polyethyleneimine-Modified UiO-66-NH2(Zr) Metal-Organic Frameworks: Preparation and Enhanced CO2 Selective Adsorption, ACS Omega, 2019, 4(2), 3188–3197 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory, Chem. Mater., 2011, 23(7), 1700–1718 CrossRef CAS.
- Y. Fang, L. Zhang, Q. Zhao, X. Wang and X. Jia, Highly Selective Visible-Light Photocatalytic Benzene Hydroxylation to Phenol Using a New Heterogeneous Photocatalyst UiO-66-NH2-SA-V, Catal. Lett., 2019, 149(9), 2408–2414 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons, J. Am. Chem. Soc., 2002, 71, 2703–2707 CrossRef.
- R. Teixeira, V. V. Serra, P. M. R. Paulo, S. M. Andrade and S. M. B. Costa, Encapsulation of Photoactive Porphyrinoids in Polyelectrolyte Hollow Microcapsules Viewed by Fluorescence Lifetime Imaging Microscopy (FLIM), RSC Adv., 2015, 5(96), 79050–79060 RSC.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, Chemically Converted Graphene Induced Molecular Flattening of 5,10,15,20-Tetrakis(1-Methyl-4-Pyridinio)Porphyrin and Its Application for Optical Detection of Cadmium(II) Ions, J. Am. Chem. Soc., 2009, 131(37), 13490–13497 CrossRef CAS.
- Z. Chernia and D. Gill, Flattening of TMPyP Adsorbed on Laponite. Evidence in Observed and Calculated UV-vis Spectra, Langmuir, 1999, 15(5), 1625–1633 CrossRef CAS.
- The quenching rate, as well as the mechanism of quenching, can be derived using the Stern–Volmer equation: F0/F = 1 + Kqτ0[Q] = 1 + KSV[Q], where F0 is the fluorescence before quenching, F is the fluorescence after adding the quencher, [Q] represents the concentration of the quencher, KSV is the Stern–Volmer constant, Kq is the bimolecular quenching rate and τ0 is the fluorescence lifetime before quenching. By plotting F0/F against [Q] we could obtain three linear Stern–Volmer plots with different slopes for the three complexes (Fig. 7d). The linear plots suggest that the main quenching mechanism is complex formation.
- N. El-Shafai, M. E. El-Khouly, M. El-Kemary, M. S. Ramadan and M. S. Masoud, Self-Assembly of Porphyrin on Graphene Oxide in Aqueous Medium: Fabrication, Characterization, and Photocatalytic Studies, Photochem. Photobiol. Sci., 2019, 18(8), 2071–2079 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a