
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the process of intrinsic point defects in PuO2
Huilong Yu†
,
Shuaipeng Wang†,
Ruizhi Qiu ,
Gan Li,
Haibo Li,
Xin Xiang and
Wenhua Luo*
,
Gan Li,
Haibo Li,
Xin Xiang and
Wenhua Luo*
Institute of Materials, China Academy of Engineering Physics, Mianyang, 621907, China. E-mail: luowenhua@caep.cn
First published on 31st July 2023
Abstract
Intrinsic point defects are known to play a crucial role in determining the physical properties of solid-state materials. In this study, we systematically investigate the intrinsic point defects, including vacancies (VPu and VO), interstitials (Pui and Oi), and antisite atoms (PuO and OPu) in PuO2 using the first-principles plane wave pseudopotential method. Our calculations consider the whole charge state of these point defects, as well as the effect of oxygen partial pressure. This leads to a new perspective on the process of intrinsic point defects in PuO2. We find that the antisite atoms OPu and PuO are more likely to appear in O-rich and O-deficient environments, respectively. Interestingly, the most energetically favorable type of Schottky defect is {2VPu3−: 3VO2+} in an O-rich environment, while {4VO1+: VPu4−} is preferred in an O-deficient environment. These results differ from the commonly known {VPu4−: 2VO2+} type of Schottky defect. Moreover, under O-deficient conditions, we predict that the stable cation Frenkel defect is {VPu4+: Pui4+}, while the most stable anion Frenkel defect is {VO2+: Oi2−} under O-rich conditions. Lastly, we find that the only two types of antisite pairs that can appear are {OPu5−: PuO5+} and {OPu6−: PuO6+}, with the latter being the more stable configuration. These unconventional defect configurations provide a new viewpoint on the process of intrinsic point defects in PuO2 and lay theoretical foundations for future experiments.
1 Introduction
The study of plutonium oxides has attracted significant attention from scientists due to their critical role in the permanent storage of Pu-based radioactive waste and the nuclear fuel cycle.1–6 Their complex chemical reactions and impressive physical properties have also contributed to their importance. Many researchers have investigated various physical and chemical properties of PuO2 because it is considered the standard in plutonium reuse, separation, and permanent storage.7–11 The redox properties of Pu-oxides are crucial to plutonium science and reactor safety because different oxidation states lead to different solubility levels, which can impact the chemical corrosion of the plutonium surface.3–6 This topic has been explored extensively by researchers, as highlighted in several studies.1–11There has been considerable research interest in stoichiometric PuO2; however, studies on non-stoichiometric PuO2 remain inadequate.1,8 In reality, under various external environments, PuO2 with a fluorite structure is non-stoichiometric.10,12,13 The stoichiometry of PuO2 is known to have a significant impact on important physical properties like lattice parameters, thermodynamical stability, and melting temperature.14 Therefore, it is essential to investigate the characteristics of defects in non-stoichiometric PuO2 composition to understand the variation in its physical properties.
Direct measurement of formation energies and defect concentrations for non-stoichiometric PuO2 is challenging and intricate.8 As a result, theoretical models have been developed in the defect properties of PuO2. However, conventional DFT that applies the local density approximation (LDA) or generalized gradient approximation (GGA), proves failure in accounting for the strong correlation of 5f electrons, describing PuO2 as an incorrect ferromagnetic (FM) conductor.8,15,16 Nonetheless, correction schemes such as self-interaction correction (SIC), DFT+U, and hybrid functional have been implemented to mitigate the inefficiencies associated with conventional DFT in PuO2.16,17
Nowadays research on point defects in PuO2 primarily focus on only four types of isolated point defects interstitial (Pui, Oi) and vacancies (VPu, VO) in PuO2, which are described with their full formal charges state: Pui4+, Oi2−, VPu4−, and VO2+.1 Antisite defects such as OPu and PuO rarely come into play due to their high formation energy in metallic oxide, although conclusive evidence regarding their role in PuO2 is lacking.18,19 M. Freyss has suggested that oxygen Frenkel pair formation is more difficult than Schottky defect formation, and that the plutonium Frenkel pair defect have lower formation energy compared to Schottky defects.7 However, charge-neutral combinations of anion and cation with full formal charge states (e.g., VPu4− + 2VO2+ for Schottky defects, VO2+ + Oi2− for oxygen Frenkel pairs, and VPu4− + Oi4+ for plutonium Frenkel pairs) are artificially assumed to be sources of intrinsic point defects during the defect formation process.21,22 In reality, the charge state of intrinsic point defects changes with oxygen potential and Fermi energy, and the equilibrium charge state for VO is neutral.
The insufficient understanding of the defect process in PuO2 has encouraged us to conduct a comprehensive analysis of its innate point defects. We have employed the first-principles plane-wave pseudopotential method to investigate the charge states and formation energies of these defects, along with their variation with oxygen partial pressure in PuO2. Therefore, this work will constitute a benchmark for future experimental or theoretical studies.
2 Computational method and model
Total energy calculations are carried out with VASP code, which employs the project augmented-wave (PAW) method and generalized gradient approximation (GGA) functional.23 To account for strong correlations, the Hubbard model was used with the DFT+U method, incorporating values of U = 4.75 eV and J = 0.75 eV for 5f electrons of Pu.1 These values are tested and the qualitative conclusion in this work is not affected by the choice of U and J. A 2 × 2 × 2 supercell, consisting of 32 plutonium atoms and 64 oxygen atoms, was constructed to create the defect model. Brillouin zone integrations were performed using a 3 × 3 × 3 k-point mesh. The electron wave function was expanded in plane waves up to a cutoff energy of 500 eV, which is sufficient to ensure that errors in intrinsic point defect formation energies are less than 1 meV.In this study, shown in Fig. 1, six types of point defects are considered. These include interstitial (Oi, Pui), vacancy (VO, VPu), and antisite atom (OPu, PuO) defects that are created by introducing, removing, or substituting a corresponding atom in the PuO2 supercell. Four typical intrinsic point defect processes of forming plutonium Frenkel, oxygen Frenkel, Schottky defects, and antisite pairs defect are examined. Charge-neutral conditions are imposed for defects in pure PuO2 bulk. A Frenkel pair is composed of a vacancy and a corresponding interstitial atom (Oi + VO, Pui + VPu), while a Schottky defect comprises several vacancy defects (mVPu + nVO). On the other hand, the antisite pair defect consists of two types of antisite atoms (OPu and PuO) separated by different distances.
 | ||
| Fig. 1 (a) The 2 × 2 × 2 supercell of PuO2. (b) The description of six types isolated intrinsic point defect in the PuO2 unit cell. | ||
The formation energy ΔEfdef(X, q) of intrinsic point defect X in charge state q is given by the following equation: 23
 | (1) |
According to the thermodynamic equilibrium condition, the heat of formation must be equal to the sum of the chemical potentials of atoms in PuO2 to ensure the stability of the compound PuO2. The equation condition can be expressed as
| μPuO2 = μPu + 2μO | (2) |
 | (3) |
Thus, two extreme conditions of O-rich and O-deficient are taken into account here, as the condition of PuO2 is relative temperature and pressure fixed. Under O-rich condition, μO and μPu are determined by the total energy of an oxygen molecule, i.e., μO = μO2/2, μPu = μPuO2 − 2 μO. While in O-deficient environment, μO and μPu are determined by the total energy of the α-Pu unit cell, i.e., μPu = μmetalPu, μO = (μPuO2 − μPu)/2.
3 Results and discussion
3.1 Isolated intrinsic point defect
The formation energies of six types of isolated intrinsic point defects (PuO, OPu, Oi, Pui, VPu, and VO) as a function of the Fermi level EF according to eqn (1) are presented in Fig. 2, under O-rich and O-deficient conditions. Only the energetically favourable charge states are labelled in Fig. 2 for clarity. It is important to note that this study represents the first investigation of two types of antisite point defects (OPu and PuO). The formation energies of PuO under O-deficient conditions and OPu under O-rich conditions are significantly negative and the smallest within some range of EF. This indicates that OPu and PuO would likely be formed under O-rich and O-deficient conditions, respectively. Therefore, when investigating defect properties in PuO2, it is critical to consider the antisite atom PuO and OPu.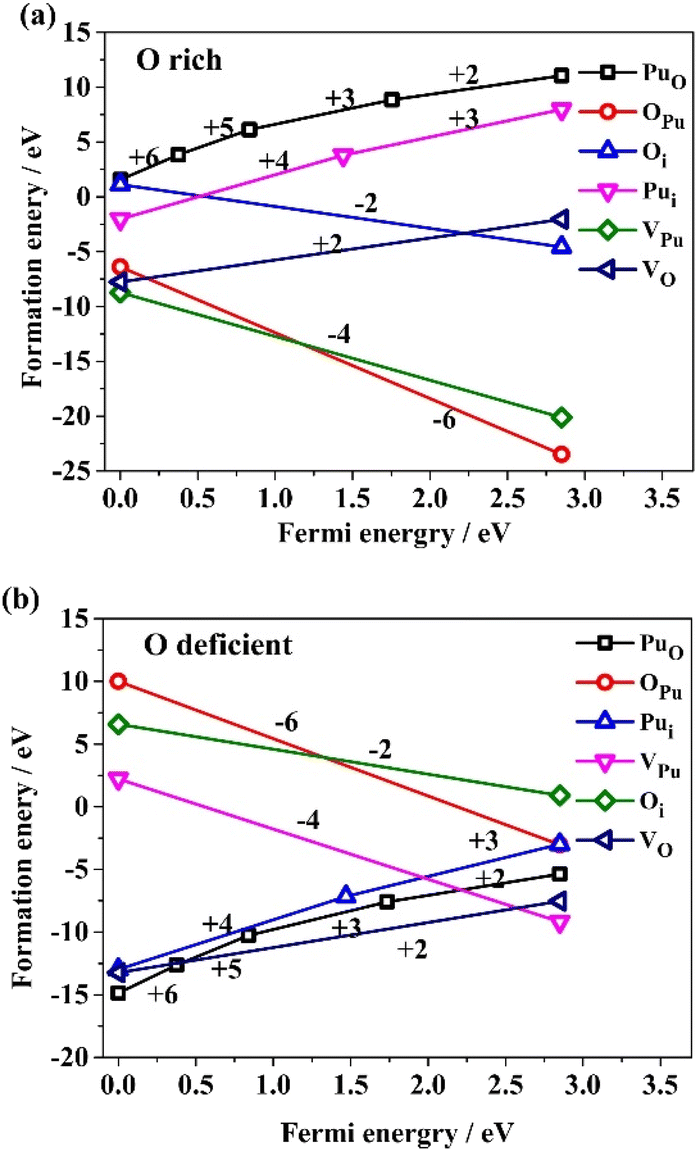 | ||
| Fig. 2 The formation energies correspond to the most favourable charge state of the isolated intrinsic point defects (PuO, OPu, Oi, Pui, VPu and VO) under conditions of O-rich (a) and O-deficient (b), versus Fermi energy EF. | ||
From Fig. 2a, it can be concluded that plutonium-related defects tend to have higher formation energies than oxygen-related defects under O-rich condition. Conversely, as shown in Fig. 2b, oxygen-related defects have higher formation energies than plutonium-related defects under O-deficient condition. Additionally, in both O-rich and O-deficient environments, the most stable state charge for each defect species varies with the Fermi level. Specifically, when EF is located near the top of the valence band under O-rich condition, intrinsic point defect species are ranked in order of rising sequence of formation energies: VPu4−, VO2+, OPu6−, Pui4+, Oi2−, and PuO6+. When EF is positioned near the bottom of the conduction band under O-rich conditions, the ranking changes to OPu6−,VPu4−, Oi2−, VO2+, Pui3+, and PuO2+. Under the O-deficient condition, when EF locate near the top of valence band, the ranking changes to PuO6+, VO2+, Pui4+, VPu4−, Oi2−, and OPu3−. When EF position is near the bottom of the conduction band under O-deficient condition, the ranking changes to VPu4−, VO2+, PuO2+, Pui3+, OPu5−, and Oi2−.
The presence of VPu4− and OPu6− are strong indications for the existence of PuO2±x under certain environmental condition.8 Previous research has ignored the intrinsic point defects of antisite atoms OPu and PuO, failing to provide convincible evidence. However, this study shows that VPu4− and OPu6− are the dominant defect configurations under O-rich conditions, while VO2+ and PuO6+ dominate under O-deficient conditions. These results suggest that antisite atoms are the dominant intrinsic defects in both scenarios. Specifically, under O-rich conditions, adding oxygen atoms to existing Pu vacancies is more favourable than adding them interstitially, as indicated by the lower formation energy of OPu6− compared to Oi2−. Conversely, under O-deficient conditions, VO2+ accounts for the lowest formation energy at mostly Fermi level. The most favourable structure for interstitial atom Pui varies from +4 to +3, thus explaining why PuO commonly has a charge of +6 or +5 near the valence band, as it forms via adding a Pu atom to the existing O vacancy. It is also noteworthy that the formation energies of PuO and Pui under O-rich conditions, and Oi and OPu under deficient conditions are too high to form in PuO2 at the equilibrium state, hence qualifying as minor defects.
3.2 Schottky defect
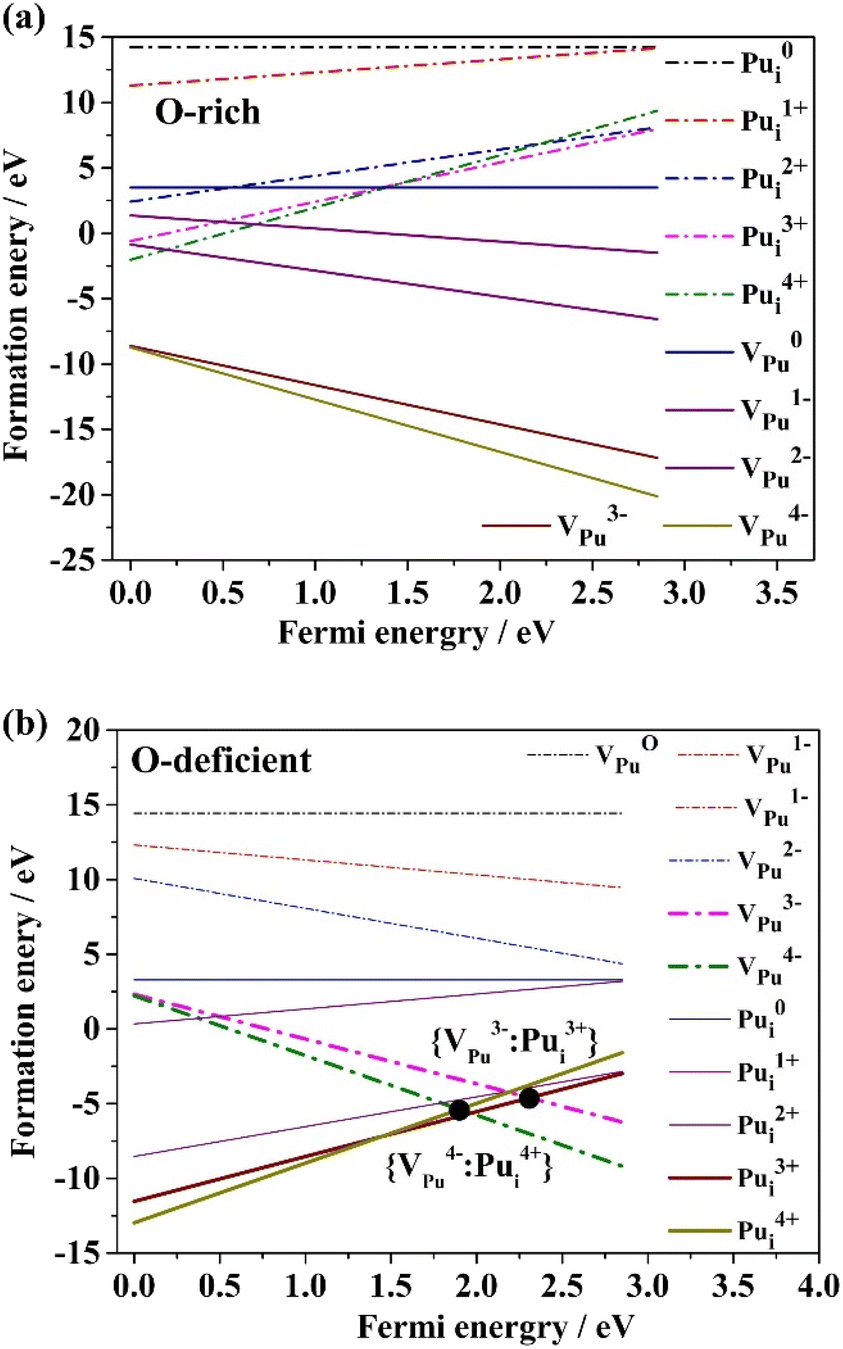
The Schottky defect in PuO2 is a combination of cation and anion defects, often artificially considered to be of triplet (VPu4+ + 2VO2−), given that PuO2 is an ionic compound of Pu4+: O2−. In this study, we present the formation energies of various Schottky defects with respect to the Fermi energy, as shown in Fig. 3. Our results indicate that the main stable charge states for VPu are +3 and +4 throughout the range of the Fermi level of PuO2. While the most stable state for VO is not +1, the formation energy of VO+ is lower than zero under O-deficient conditions, suggesting that it is stable. Although Schottky defects in PuO2 could consist of several ion compounds besides (VPu4+ + 2VO2−), we calculated the formation energies of other combinations, including (2VPu3+ + 3VO2−) and (4VO+ + VPu4−). The results indicate that the formation energies of these combinations are much too high, implying that the vacancy defects should be spaced far enough apart from each other to form a Schottky defect. In fact, under extreme conditions, the space between vacancies could be so great that their interactions become infinitely small. Furthermore, the average formation energy of each component must have an equal value for a Schottky defect to form. | ||
| Fig. 3 Formation energies of oxygen vacancies and plutonium vacancies in various charge states within whole EF level, and possible Schottky defect combination formed in PuO2 under the conditions of (a) O-rich, (b) O-deficient. | ||
The point of intersection Fig. 3 represents a possible Schottky defect in PuO2. Two cases of Schottky defects are shown in Fig. 3a and b. All cases are obtained by enumeration. When exposed to O-rich conditions, as seen in Fig. 3a, the average formation energies of three possible Schottky defects {2VPu3−: 3VO2+}, {VPu4−: 2VO2+}, and {2VPu1−: VO2+} are −20.26 eV, −12.10 eV, and −2.44 eV, respectively. According to the thermodynamic theory, the primary Schottky defect is {2VPu3−: 3VO2+} due to its lowest formation energy, rather than {VPu4−: 2VO2+}, which was customarily considered as the primary defect under O-rich conditions. This finding provides a fresh perspective on Schottky defects in PuO2. Further, {2VPu1−: VO2+} acts as a minor Schottky defect; its formation energy is significantly higher than that of the other defects.
Under O-deficient condition, the average formation energies of {4VO1+: VPu4+} and {3VO1+: VPu3−} are −5.75 eV and −4.01 eV, respectively, as shown in Fig. 3b. Interestingly, the VO charge state of +1 is not the most stable for VO. The main Schottky defect is {4VO1+: VPu4−}, which has the lowest formation energy under O-deficient conditions, despite not being consistent with the commonly considered {VPu4−: 2VO1+}. However, the small formation energy difference between these two types of Schottky defects indicates that neither will dominate in PuO2.
When comparing an O-deficient condition and an O-rich condition, the average formation energies of the prominent Schottky defect species ({2VPu3−: 3VO2+} and {VPu4−: 2VO2+}) are significantly lower in the O-rich environment. This suggests that it is much easier to form dominant Schottky defect species in an O-rich condition. Notably, the Schottky defect species differ entirely between O-rich and O-deficient conditions, indicating that the formation of Schottky defects is dependent on oxygen partial pressure. These results agree with earlier findings.8
3.3 Frenkel defect
The Frenkel defect in PuO2 comprises of an anion Frenkel {Oi: VO} and a cation Frenkel {Pui: VPu}, whose charge states are commonly considered as {Oi2−: VO2+} and {Pui4+: VPu4−}. However, just like described in Section 3.2 for the charge states of VPu (+3 and +4), the custom standpoint for the cation Frenkel defect may differ. To further investigate this, the relative energy of the PuO2 supercell was calculated, which includes Frenkel defects ({Oi: VO} and {Pui: VPu}) at different separation distances between a vacancy and corresponding interstitial atom. Fig. 4 presents the structural model of the Frenkel defect in the 2 × 2 × 2 supercell of PuO2, where the separation distance between the interstitial atom and vacancy is 1NN (the nearest neighbour). In addition, Fig. 5 illustrates the relative energies of the Frenkel defect for varying separation distances between the vacancy and interstitial atom. The outcome suggests that the relative energies of the Frenkel defect decrease as the distance increases, indicating that the most stable configuration happens when the cation Frenkel defect has an interstitial atom Pui closest to the vacancy VPu (1 NN). The stability behaviour of the anion Frenkel defect is consistent with that of the cation Frenkel defect. It's noteworthy that Frenkel defects differ significantly from Schottky defects, whose stability increases as the VPu vacancy moves away from the VO vacancy. | ||
| Fig. 4 The structural models of (a) anion and (b) cation Frenkel defects in the 2 × 2 × 2 supercell of PuO2, when the space distance between vacancy atoms is the nearest neighbour. | ||
 | ||
| Fig. 5 Relative energies of the (2 × 2 × 2) PuO2 supercell including (a) oxygen and (b) plutonium Frenkel defect as a function of separate distance between corresponding vacancy and interstitial atom. | ||
The formation energies of possible plutonium Frenkel defects have been calculated under both O-rich and O-deficient conditions, as shown in Fig. 6. It is worth noting that under O-rich conditions, no plutonium Frenkel defect can form. Conversely, under O-deficient conditions, two types of plutonium Frenkel defects, i.e., {VPu3−: Pui3+} and {VPu4+: Pui4+}, are formed with formation energies of −4.52 eV and −5.35 eV, respectively. Intriguingly, the difference in formation energy between the two types of defects is minimal (<1 eV), suggesting that a significant proportion of {VPu3−: Pui3+} may also form in PuO2, besides {VPu4+: Pui4+}. This provides a new perspective on oxygen Frenkel defects under O-deficient condition.
 | ||
| Fig. 6 Formation energies of plutonium vacancies in various charge states within whole EF level, and possible plutonium Frenkel defect combination formed in PuO2 under the condition of O-rich and O-deficient. | ||
The possible oxygen Frenkel defects' formation energies under both O-rich and O-deficient conditions are presented in Fig. 7. Notably, no plutonium Frenkel defect is formed under O-rich conditions, which deviates from typical expectations. However, two types of oxygen Frenkel defects, i.e., {VO+: Oi−} and {VO2+: Oi2−}, do form under O-deficient condition, and their respective formation energies are 2.17 eV and −3.30 eV. Of these two defects, the {VO2+: Oi2−} variant is the more prevalent oxygen Frenkel defect due to its lower average formation energy. Conversely, the formation energy of 2.17 eV for the Frenkel defect {VO+: Oi−} is too large to emerge under O-deficient condition.
 | ||
| Fig. 7 Formation energies of oxygen vacancies in various charge states within whole EF level, and possible oxygen Frenkel defect combination formed in PuO2 under the condition of O-rich and O-deficient. | ||
The results suggest that the presence of an oxygen environment significantly affects the formation of both oxygen Frenkel defects and plutonium Frenkel defects. While charge states do affect the formation energy of plutonium Frenkel defects, this effect is relatively minor compared to that of oxygen Frenkel defects. These findings are consistent with earlier research on PuO2.21,26
3.4 Antisite pairs
The formation energies of the antisite pair were computed under both O-rich and O-deficient conditions, as illustrated in Fig. 8. Conventionally, an antisite pair cannot be formed. In Fig. 8, it is observed that the antisite atom OPu is unable to form under O-rich conditions, whereas PuO readily forms under O-deficient conditions. The formation energies for the two antisite pairs {OPu5+: PuO5−} and {OPu6+: PuO6−} under O-deficient conditions are −1.58 eV and −2.26 eV, respectively. The negligible difference (<1 eV) between the formation energy of these two types of antisite pairs implies that both of them dominate as defects, providing a new insight into antisite pairs under O-deficient conditions. This outcome indicates that the oxygen environment exerts a significant influence on antisite pairs' formation. However, the charged states' effect on the antisite pair's formation energy is comparatively minor. | ||
| Fig. 8 ForFimation energies of antisite atoms in various charge states, and possible antisite defect combinations formed in PuO2 under the condition of both O-rich and O-deficient. | ||
4 Conclusions
In summary, we have systematically investigated the process of intrinsic point defects including vacancies, interstitials, and particularly antisite atoms in PuO2 by the first-principles plane-wave pseudopotential calculations. The whole charge state of these point defects and the effect of oxygen partial pressure are considered.For the individual point defects, the most stable charge state varies with the Fermi energy EF and oxygen environment. Under O-rich condition, VPu4− is the most stable near the top of valence band, while OPu6− is the most stable near the bottom of conduction band. In O-deficient environment, the most stable charge states in both cases change to PuO6+ and VPu4−, respectively. It is surprising to find that the antisite atoms OPu and PuO dominate the behaviour of point defect in PuO2.
Schottky defects can take three possible forms, i.e., {2VPu3−: 3VO2+}, {VPu4−: 2VO2+}, and {2VPu1−: VO2+}, under O-rich condition with {2VPu3−: 3VO2+} being the most stable. Under O-deficient condition, Schottky defects can take two forms, {4VO1+: VPu4+} and {3VO1+: VPu3−} with the latter being more stable.
Cation Frenkel defects can occur in two forms {VPu3−: Pui3+} and {VPu4+: Pui4+} under the O-deficient condition, with the latter being more stable. Anion Frenkel defects can occur in two forms {VO+: Oi−} and {VO2+: Oi2−} under O-rich condition, with the latter being more stable.
Two types of antisite pairs were found to be possible under O-deficient conditions, {OPu5−: PuO5+} and {OPu6−: PuO6+}, with the latter being more stable.
Overall, our results demonstrate that the charge states and existing forms of the intrinsic point defect processes strongly depend on the oxygen condition. From the energetics of the defects, the relative stability ranked in order of rising sequence of formation energies: Schottky defects > cation Frenkel > anion Frenkel > antisite pair. This indicates that the dominant defects in PuO2 are Schottky defects, being consistent with the experiment.8,10
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21601167 and 22176181) and the Foundation of President of China Academy of Engineering Physics (Grant No. YZJJZQ2022011).References
- B. Sun, H. Liu, H. Song, G. Zhang, H. Zheng, X. Zhao and P. Zhang, Phys. Lett. A, 2012, 376, 2672–2676 CrossRef CAS.
- H. Yu, D. Meng, H. Huang and G. Li, J. Nucl. Mater., 2014, 452, 6–9 CrossRef CAS.
- K. Moore and G. van der Laan, Rev. Mod. Phys., 2009, 81, 235–298 CrossRef CAS.
- M. Freyss, N. Vergnet and T. Petit, J. Nucl. Mater., 2006, 352, 144–150 CrossRef CAS.
- M. Cooper, M. Rushton and R. Grimes, J. Phys.: Condens. Matter, 2014, 26, 105401 CrossRef CAS PubMed.
- J. Haschke and T. Ricketts. Los Alamos Tech. Rep., 1995, LA-12999-MS Search PubMed.
- X. Tian, T. Gao, C. Lu, J. Shang and H. Xiao, Eur. Phys. J. B, 2013, 86, 1–7 CrossRef.
- Y. Lu, Y. Yang and P. Zhang, J. Alloys Compd., 2015, 649, 544–552 CrossRef CAS.
- C. Guéneau, N. Dupin, B. Sundman, C. Martial, J. Dumas, S. Gossé, S. Chatain, F. De Bruycker, D. Manara and R. Konigs, J. Nucl. Mater., 2011, 419, 145–167 CrossRef.
- C. Guéneau, A. Chartier, P. Fossati, L. Brutzel and P. Martin, Compr. Nucl. Mater., 2012, 2, 21–59 Search PubMed.
- B. Dorado and P. Garcia, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195139 CrossRef.
- M. Freyss and T. Petit, International Conference of Computational Methods in Sciences and Engineering 2004 (ICCMSE 2004) Search PubMed.
- L. Zhang, B. Sun, Q. Zhang, H. Liu, K. Liu and H. Song, Appl. Surf. Sci., 2020, 516, 146164 CrossRef CAS.
- M. Kato, T. Uchida and T. Sunaoshi, Phys. Status Solidi C, 2013, 10, 189–192 CrossRef CAS.
- B. Ao, R. Qiu, H. Lu, X. Ye, P. Shi, P. Chen and X. Wang, J. Phys. Chem. C, 2015, 119, 101–108 CrossRef CAS.
- X. Wen, R. Martin, T. Henderson and G. Scuseria, Chem. Rev., 2012, 113, 1063–1096 CrossRef PubMed.
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- X. Xiang, G. Zhang, X. Wang, T. Tang and Y. Shi, Phys. Chem. Chem. Phys., 2015, 17, 29134–29141 RSC.
- S. Zhang, S. Wei and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 075205 CrossRef.
- T. Mccleskey, E. Bauer, Q. Jia, A. Burrell, B. Scott, S. Condradson, A. Mueller, L. Roy, X. Wen, G. Scuseria and R. Martin, J. Appl. Phys., 2013, 113, 013515 CrossRef.
- P. Tiwary, A. van de Walle, B. Jeon and N. Grønbech-Jensen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 1126–1129 CrossRef.
- L. Petit L, A. Svane, Z. Szotek, W. Temmerman and G. Stocks, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 045108 CrossRef.
- K. Matsunaga, T. Tanaka, T. Yamamoto and Y. Ikuhara, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 085110 CrossRef.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 035406 CrossRef.
- M. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, 1998, 4th edn Search PubMed.
- M. Qin, M. Cooper, E. Kuo, M. Rushton, R. Grimes, G. Lumpkin and S. Middleburgh, J. Phys.: Condens. Matter, 2014, 26, 495401 CrossRef CAS PubMed.
Footnote |
| † These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |