
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of NO3−, Cl−, and CH3COO− anions and diethylene glycol on the morphological, structural, antidiabetic, and cell viability properties of CeO2 nanoparticles†
Shaidatul Najihah Matussin a,
Fazlurrahman Khanbc,
Pathum Chandikabc,
Mohammad Hilni Harunsania,
Norhayati Ahmadd,
Young-Mog Kimbce,
Won-Kyo Jungbcf and
Mohammad Mansoob Khan*a
a,
Fazlurrahman Khanbc,
Pathum Chandikabc,
Mohammad Hilni Harunsania,
Norhayati Ahmadd,
Young-Mog Kimbce,
Won-Kyo Jungbcf and
Mohammad Mansoob Khan*a
aChemical Sciences, Faculty of Science, Universiti Brunei Darussalam, Jalan Tungku Link, Gadong, BE 1410, Brunei Darussalam. E-mail: mmansoobkhan@yahoo.com; mansoob.khan@ubd.edu.bn
bMarine Integrated Biomedical Technology Center, The National Key Research Institutes in Universities, Pukyong National University, Busan, 48513, Republic of Korea
cResearch Center for Marine Integrated Bionics Technology, Pukyong National University, Busan, 48513, Republic of Korea
dEnvironmental and Life Sciences, Faculty of Science, Universiti Brunei Darussalam, Jalan Tungku Link, Gadong, BE 1410, Brunei Darussalam
eDepartment of Food Science and Technology, Pukyong National University, Busan 48513, Republic of Korea
fMajor of Biomedical Engineering, Division of Smart Healthcare and New-Senior Healthcare Innovation Center (BK21 Plus), Pukyong National University, Busan 48513, Republic of Korea
First published on 22nd May 2023
Abstract
Cerium oxide (CeO2) nanoparticles (NPs) were synthesized using a modified conventional polyol method. The ratio of diethylene glycol (DEG) and water in the synthesis was varied, and three different cerium precursor salts (Ce(NO3)3, CeCl3, and Ce(CH3COO)3) were used. The structure, size, and morphology of the synthesized CeO2 NPs were studied. An average crystallite size of 13 to 33 nm was obtained from the XRD analysis. Spherical and elongated morphologies of the synthesized CeO2 NPs were acquired. Average particle sizes in the range of 16–36 nm were obtained by varying different ratios of DEG and water. The presence of DEG molecules on the surface of CeO2 NPs was confirmed using FTIR. Synthesized CeO2 NPs were used to study the antidiabetic and cell viability (cell cytotoxicity) properties. Antidiabetic studies were carried out using α-glucosidase enzymes inhibition activity. CeO2 synthesized using Ce(NO3)3 and CeCl3 precursors showed approximately 40.0% α-glucosidase enzyme inhibition activity, while CeO2 synthesized using Ce(CH3COO)3 showed the lowest α-glucosidase enzyme inhibition activity. Cell viability properties of CeO2 NPs were investigated using an in vitro cytotoxicity test. CeO2 NPs prepared using Ce(NO3)3 and CeCl3 were non-toxic at lower concentrations, while CeO2 NPs prepared using Ce(CH3COO)3 were non-toxic at all concentrations. Therefore, polyol-mediated synthesized CeO2 NPs showed quite good α-glucosidase inhibition activity and biocompatibility.
1. Introduction
Cerium oxide (CeO2) is commonly explored for various applications due to its unique nature and high oxygen storage capacity (OSC). It can easily switch its oxidation state between Ce3+ and Ce4+ in the stable fluorite structure.1,2 CeO2 has attracted intensive attention due to its size, morphology, and orientation-dependent properties. Various methods have been used to synthesize CeO2 nanoparticles (NPs), nanowires or nanotubes, such as precipitation, spray pyrolysis, hydrothermal, solvothermal, and sonochemical reactions.3–6 Furthermore, CeO2 has remarkable properties such as high ionic conductivity, relatively high mechanical strength, strong adsorption, and photoluminescence in the UV-vis range. These properties are interesting for applications in catalysis, fuel cells, solar cells, sensors, oxygen storage, polishing, luminescent materials, promoters of three-way catalysts in automotive pollution control, UV blockers, and energy storage.7–14Postprandial hyperglycemia is a major complication of type II diabetes patients.15 The increasing trend of type II diabetes mellitus (DM) has become a serious medical concern. Therefore, it is crucial to control postprandial blood glucose levels by exploring and developing new therapeutic agents. α-Glucosidase converts complex carbohydrates into glucose, which is then absorbed into the small intestine, increasing blood glucose levels.16,17 In type II DM, inhibition of α-glucosidase (digestive enzyme) is favorable to delaying glucose absorption to the small intestine and allowing for a more controlled absorption.18 Administration of drugs to maintain the blood glucose level can be through conventional oral route, subcutaneous route, transdermal, pulmonary and inhalation.19 Subcutaneous injections are considered effective compared to oral route due to peptides are unstable in the gastrointestinal environment.20,21 However, till date, no reports have been found on the injectable CeO2 NPs for inhibition of α-glucosidase enzyme. Nevertheless, Jan et al. reported on the inhibition of α-glucosidase using green synthesized CeO2.22 It was found that the CeO2 inhibited 31.28% of α-glucosidase. To the best of the authors' knowledge, there has been no other report on the inhibition of α-glucosidase activity using CeO2 that was synthesized using the polyol method.
The polyol synthesis method, which involves ethylene glycol, diethylene glycol, and glycerol, among others, was first introduced in 1989 by Fievet, Lagier, and Figlarz.23 The polyol synthesis started with Co, Ni, Cu, and Pt particles and was extended to metal oxide NPs.23 The polyol process offers multiple advantages, such as similar solubilities of compounds to that of water.24 Therefore, simple and low-cost metal salts can be used as starting materials. The chelating effect of polyols helps in controlling particle nucleation, particle growth, and aggregation of NPs.23,24 Apart from that, the high boiling points of polyols allow for high reaction temperatures without using autoclaves or high pressure. Further benefits of the polyols are the reductive properties for direct preparation of elemental metals, the easy removal of the polyols from the particle surface after synthesis, and the scalability and implementation in continuous-flow synthesis. Polyols are considered to have low to moderate toxicity and are highly biodegradable, which can be considered as green solvents.23 In order to produce smaller particles of CeO2 with defined morphologies, the polyol method was employed in this study.
Furthermore, the role of counter-anion in the metal salt precursors on the shape-selective growth of nanomaterials has not been widely studied.25 It is said that the inorganic anions themselves might be selectively adsorbed on particular facets and thus greatly affecting the structure, size, and morphology of the nanomaterials.26 Therefore, in this study, the effect of different precursors such as Ce(NO3)3·6H2O, CeCl3·7H2O, and Ce(CH3COO)3·H2O were used in the syntheses of CeO2 via polyol method were investigated. Apart from that, the ratio of diethylene glycol (DEG) and H2O used in the synthesis was also varied to study its effect on the structural and morphological properties of the synthesized CeO2 NPs. Finally, the different sets of the synthesized CeO2 NPs were tested for their biological activities, such as α-glucosidase inhibition and its cytotoxicity (cell viability) activities.
2. Experimental method
2.1. Chemicals used
For the synthesis, cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, 99%), cerium(III) chloride heptahydrate (CeCl3·7H2O, 99%), and cerium acetate hydrate (Ce(CH3COO)3, 99%) and cerium(IV) oxide were all obtained from Sigma-Aldrich. Diethylene glycol (C4H10O3, 99%) was obtained from Alfa Aesar. Throughout the reaction, double distilled water (purified using Aquatron, England) was used. For α-glucosidase inhibition activity of CeO2, α-glucosidase (≥50 units per mg protein), (PNPG, C12H15NO8, 99%), acarbose (C25H43NO18, ≥95%), sodium carbonate (Na2CO3, ≥99.5%), sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O) and sodium phosphate monobasic monohydrate (NaHPO4·H2O, ≥99%) were obtained from Sigma-Aldrich. The cytotoxicity tests were carried out using MTT assay (3,4,5-dimethylthiazol-2-yl)-2-5-diphenyl tetrazolium bromide and RAW 264.7 cell lines (mouse macrophage) obtained from American Type of Culture Collection (ATCC, Rockville, MD, USA); penicillin/streptomycin, and fetal bovine serum (FBS) from GIBCO™, Invitrogen Corporation, USA; and dimethyl sulfoxide were obtained from Sigma-Aldrich.2.2. Synthesis of CeO2 NPs
For the synthesis of CeO2 NPs, two variables were chosen, which are (i) the DEG/H2O ratio and (ii) different anions precursors i.e., Ce(NO3)3·6H2O, CeCl3·7H2O, and Ce(CH3COO)3. For the synthesis, a typical reflux setup was utilized in which paraffin oil was used to heat the solution in the round bottom flask. Different DEG/H2O ratio were varied in the synthesis, i.e., 0/25, 5/20, 10/15, 15/10, 20/5, and 25/0 mL of DEG and double distilled H2O and based on the ratio, the solvents were mixed together to make up to 25 mL of the total volume (Table S1†). The amount of cerium precursor was weighed in order to prepare 0.05 M in a 25 mL solvent (DEG + water). The mixture was stirred at room temperature for 5 min and heated slowly to 80 °C. Subsequently, 4 mL of 1 M of NaOH solution was added dropwise using a burette. The solution was then stirred and heated at 100 °C for 4 h. Subsequently, gel-like precipitate was obtained. The product was centrifuged at 3500 rpm for 5 min and washed for three times using water. The obtained product was finally calcined at 600 °C for 2 h before it was ground to yield powder CeO2 NPs. The schematic diagram for the synthesis method can be found in Fig. S1.†2.3. Instrumentations
The crystal phase identification of NPs was investigated using a Shimadzu XRD-7000 X-ray diffractometer (XRD) with Cu Kα radiation (λ = 1.5418 Å). To investigate the different functional groups related to DEG and NP formation, Fourier-transform infrared spectroscopy (FTIR) of finely dried and ground CeO2 and KBr was carried out using FT-IR (Shimadzu IRPrestige-21, Japan) in the range of 400–4500 cm−1 via KBr method at room temperature. The morphologies of the synthesized CeO2 NPs were studied using field emission transmission electron microscopy (FE-TEM) and selected area electron diffraction (SAED) conducted with JEM-F200 (JEOL Ltd, Tokyo, Japan). X-ray photoelectron spectroscopy (XPS) was performed on Kratos Analytical, AXIS Nova. For α-glucosidase activities, the absorbance of the enzyme-substrate solution was measured using a UV-vis spectrophotometer (Shimadzu UV 1900, Japan). For the cell cytotoxicity study, the absorbance was measured at 540 nm using a microplate reader (Gen 5™ ELISA Bio Tek, Winooski, VT, USA).2.4. Inhibition of α-glucosidase activity
The inhibition of α-glucosidase activities using CeO2 NPs synthesized from different precursors, and varied DEG/H2O ratio was tested in triplicates. A standard procedure of the α-glucosidase assay was followed with slight modification.27 Different concentrations of CeO2 NPs (0.5, 1.5, and 2.5 mg) were used in the reaction. Phosphate buffer solution (PBS, 1 M, pH 6.8) was mixed before 250 μL of 1.5 U mL−1 of the enzyme was put in. The reaction solution was incubated at 37 °C for 10 min. After the incubation period, 250 μL of 0.5 mM of p-nitrophenyl α-D-glucopyranosidase (PNPG) was added to the reaction solution and incubated further at 37 °C for 30 min. The reaction was stopped by adding 1 mL of 0.2 M sodium carbonate solution. The overall solution was centrifuged before the absorbance of the solution was measured at 405 nm. The percentage inhibition of α-glucosidase activities was estimated using the following eqn (1):28
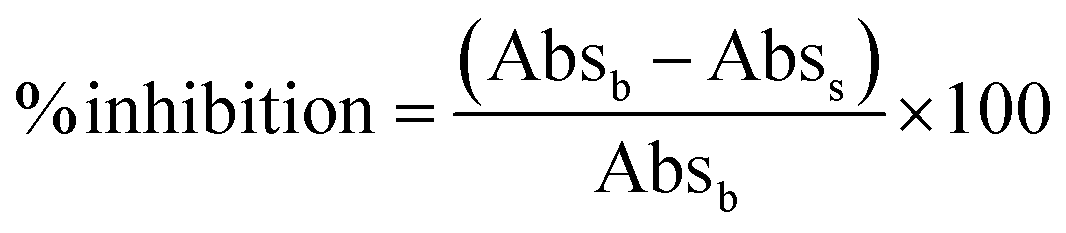 | (1) |
2.5. Cytoxicity test/cell viability test
MTT was used to perform the in vitro cytotoxicity of CN, CC, and CA against the mouse macrophage cell line RAW 264.7. Cells were cultured in Dulbecco's minimum Eagle's medium (DMEM) supplemented with 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, and 10% FBS and maintained in 5% CO2 at 37 °C. RAW 264.7 macrophage cells pre-incubated for 24 hours were seeded into a 96-well microtiter plate, treated with different concentrations of CN, CC, and CA, and re-incubated for 24 hours. The cytotoxicity test was carried out by adding a fresh culture medium containing MTT reagent (1 mg mL−1) and incubating for 4 h. Subsequently, formazan crystals in each well were dissolved in 100 μL of dimethyl sulfoxide (DMSO). The absorbance was measured at 540 nm using a microplate reader (Gen 5 ™ELISA Bio Tek, Winooski, VT, USA). All experiments were conducted in triplicate.3. Results and discussion
Three different sets of CeO2 NPs were synthesized using the polyol synthesis method, and the products are labeled as CN, CC, and CA NPs, which stand for CeO2 synthesized using Ce(NO3)3·6H2O, CeCl3·7H2O, and Ce(CH3COO)3·H2O respectively. In this article, the codes CN1 to CN6, CC1 to CC6, and CA1 to CA6 indicate the respective precursor salts used and the DEG/H2O ratio used in the synthesis (0/25, 5/20, 10/15, 15/10, 20/5, and 25/0 mL, respectively).3.1. X-ray diffraction
X-ray diffraction analysis of the synthesized CeO2 NPs was carried out as depicted in Fig. 1. By comparing with the JCPDS 00-004-0593 standard (Fig. S2†),29 cubic phases CeO2 have been successfully prepared (Fig. 1) using three different cerium precursors i.e. Ce(NO3)3·6H2O, CeCl3·7H2O, and Ce(CH3COO)3·H2O. No extra peaks have been detected, indicating that the synthesized CeO2 NPs using Ce(NO3)3·6H2O are in the pure form (Fig. 1(a)). In order to see the effect of DEG/H2O ratio on the structural properties of the synthesized CeO2 NPs, average crystallite sizes were estimated using the Debye–Scherrer's formula (eqn (2)):30
D = kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (2) |
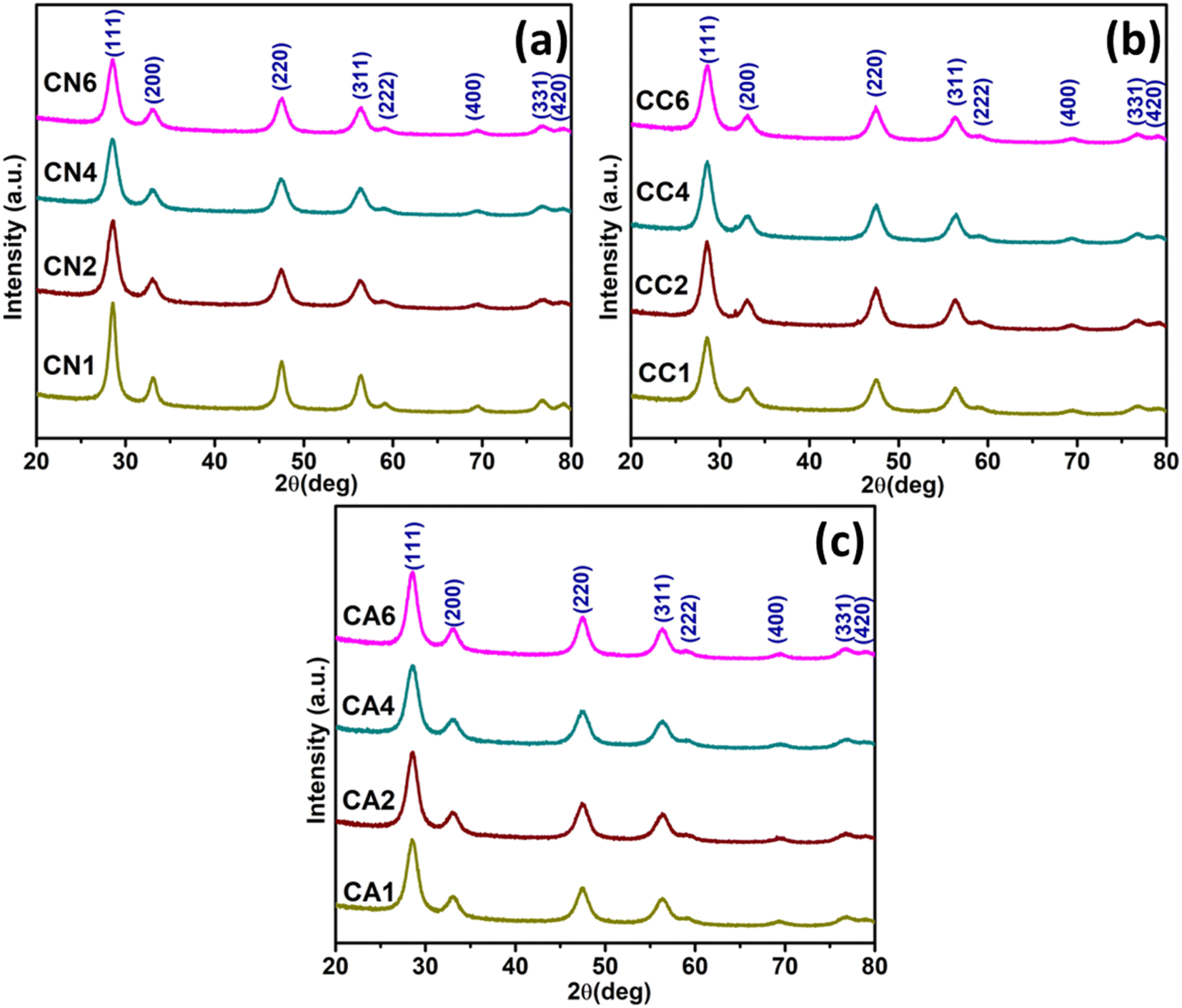 | ||
| Fig. 1 XRD patterns of the synthesized CeO2 NPs using (a) Ce(NO3)3·6H2O, (b) CeCl3·7H2O, and, (c) Ce(CH3COO)3·H2O precursors. | ||
| Sample | Average crystallite size (nm) | Lattice parameter, α (Å) | Cell volume (Å3) | Peak ratio between 1st and 2nd | Average particle size (nm) from TEM |
|---|---|---|---|---|---|
| CN1 | 21.92 | 5.411 | 158.39 | 4.13 | 28 |
| CN2 | 18.92 | 5.410 | 158.35 | 4.32 | 26 |
| CN4 | 23.24 | 5.427 | 159.81 | 4.62 | 21 |
| CN6 | 20.21 | 5.419 | 159.20 | 4.08 | 16 |
| CC1 | 15.31 | 5.415 | 158.78 | 4.46 | 34 |
| CC2 | 19.59 | 5.392 | 156.76 | 3.98 | 36 |
| CC4 | 33.87 | 5.426 | 159.73 | 3.73 | 25 |
| CC6 | 13.14 | 5.395 | 157.02 | 4.53 | 28 |
| CA1 | 15.98 | 5.435 | 160.52 | 4.08 | 29 |
| CA2 | 14.52 | 5.412 | 158.50 | 4.30 | 21 |
| CA4 | 20.07 | 5.365 | 154.39 | 3.39 | 21 |
| CA6 | 29.10 | 5.406 | 157.98 | 4.08 | 20 |
Similarly, cubic CeO2 has been successfully synthesized using CeCl3·7H2O according to JCPDS 00-004-0593 (Fig. 1(b)).29 The average crystallite size was increased from 15.31 nm (CC1) to 33.87 nm (CC4) in which 0, 5, and 15 mL of DEG were used, and the average crystallite size was reduced back to 13.14 when 25 mL DEG was used in the synthesis. Nevertheless, comparable lattice parameters were observed (Table 1). There is an insignificant change between the peak height of CC1, CC2, CC4, and CC6 positioned at about 28°. The peak height ratios between the first and second peaks differ slightly from one another in that the ratios are 4.46, 3.98, 3.73, and 4.53 for CC1 to CC6, respectively. The cell volume was observed to be comparable.
Accordingly, cubic CeO2 has also been successfully synthesized using Ce(CH3COO)3·H2O which is similar to JCPDS 00-004-0593 (Fig. 1(c)).29 No extra peak was observed in the XRD spectra, indicating no impurities were detected. The average crystallite size among CA1 and CA2 was comparable, which were 15.98 and 14.52 nm, respectively. This means there was no change in crystallite size when 0 and 5 mL of DEG were used. However, the crystallite size became larger when 15 mL and 25 mL of DEG was used which showed about 20.07 and 29.10 nm, respectively. Despite the difference in crystallite size, comparable lattice parameters were also observed. The cell volume was reduced from 160.52 to 154.39 nm when more DEG was used. There is an insignificant change between the peak intensity at 29° for CA1, CA2, CA4, and CA6. The peak height ratio between the first and second peaks are 4.08, 4.30, 3.39, and 4.08 for CA1 to CA6, respectively. Overall, this might suggest that the addition of DEG influences the average crystallite size of CeO2. All the estimated average crystallite sizes, lattice parameters, cell volumes, and peak ratios can be found in Table 1.
In order to see the effects of anions on the structural properties of the synthesized CeO2, the average crystallite sizes of CN1, CC1, and CA1 (all were synthesized without DEG) were compared. The crystallite size became smaller when it changed from NO3− to Cl− anion precursor. The average crystallize size was retained when CH3COO− anion precursor was used. However, for CN6, CC6, and CA6 (all were synthesized using 100% DEG), only the crystallite size of CeO2 NPs synthesized using Ce(NO3)3 (CN6) and CeCl3 (CC6) shows similar values as CN1 and CC1, respectively. CA6 shows the largest crystallite size amongst the NPs, demonstrating that the CH3COO− anion could not produce a smaller crystallite size as expected. However, DEG has some effects on the average crystallite size of the synthesized CeO2.31
Therefore, in order to investigate the effects of DEG/H2O ratio, the structural properties of the three groups of synthesized CeO2 NPs (CN, CC, and CA NPs) were also compared. For CN, CN1 and CN2 show remarkable findings. The crystallite size was obviously reduced when 5 mL of DEG was introduced, showing DEG has minimized the average crystallite size. However, when adding more DEG, the crystallite sizes of CN4 and CN6 were similar to CN1. Thus, adding more DEG has no effect on reducing the crystallite size of CN NPs.
On the other hand, in CC NPs, adding 5 mL and 15 mL DEG certainly increased the average crystallite sizes, as can be seen for CC1 to CC4 (Table 1). However, when 100% DEG was used, the crystallite size was reduced to even lower than the crystallite size of CC1. The interaction between DEG and Cl− might have been the reason for the crystallite size reduction.32 Interestingly, for CA NPs, adding 5 mL of DEG has no significant effect on the crystallite size, as can be observed in the case of CA1 and CA2. However, introducing more DEG has increased the crystallite size of the CeO2 NPs. Therefore, the interaction between CH3COO− ions and DEG might lead to the increase in the crystallite size of CeO2 NPs.
3.2. X-ray photoelectron spectroscopy
The chemical state and the electronic structure of the elements formed in the selected CN, CC, and CA NPs were investigated using XPS. Fig. 2(a) shows the complete survey scan spectra of the NPs, which confirmed the presence of Ce 3d, O 1s, and C 1s. Typical characteristics peaks of Ce 3d are shown in Fig. 2(b). CN1 and CN6 show a significant difference in the peak intensity despite showing similar peak positions. CN1 shows peaks at 879.73, 886.50, 895.77, 898.38, 905.04, and 914.10 eV, while CN6 shows peaks at 879.84, 886.07, 895.56, 898.17, 904.83, and 914.10 eV, respectively. Similarly, CC1 shows higher peak intensity than CC6. Shift was observed in the case of CC1 and CC6. The position of the Ce 3d peaks of CC1 are shown at 880.39, 886.94, 896.43, 898.93, 905.59, and 914.86 eV. Whereas, Ce 3d in the case of CC6 are formed at 879.96, 886.39, 895.89, 898.61, 905.59, and 914.42 eV. The shift in the peak position might be due to the presence of oxygen vacancies and Ce3+ and other lattice defects.33,34 However, CA1 shows a lower intensity than CA6. The position of the Ce 3d peaks of CA1 are 880.16, 886.39, 896.21, 898.72, 904.95, and 914.54 eV, and the position of the Ce 3d peaks of CA6 are 879.96, 886.50, 895.77, 898.61, 904.95, and 914.33 eV. It can be seen that peak position and height were influenced by DEG/H2O ratio and different cerium anions.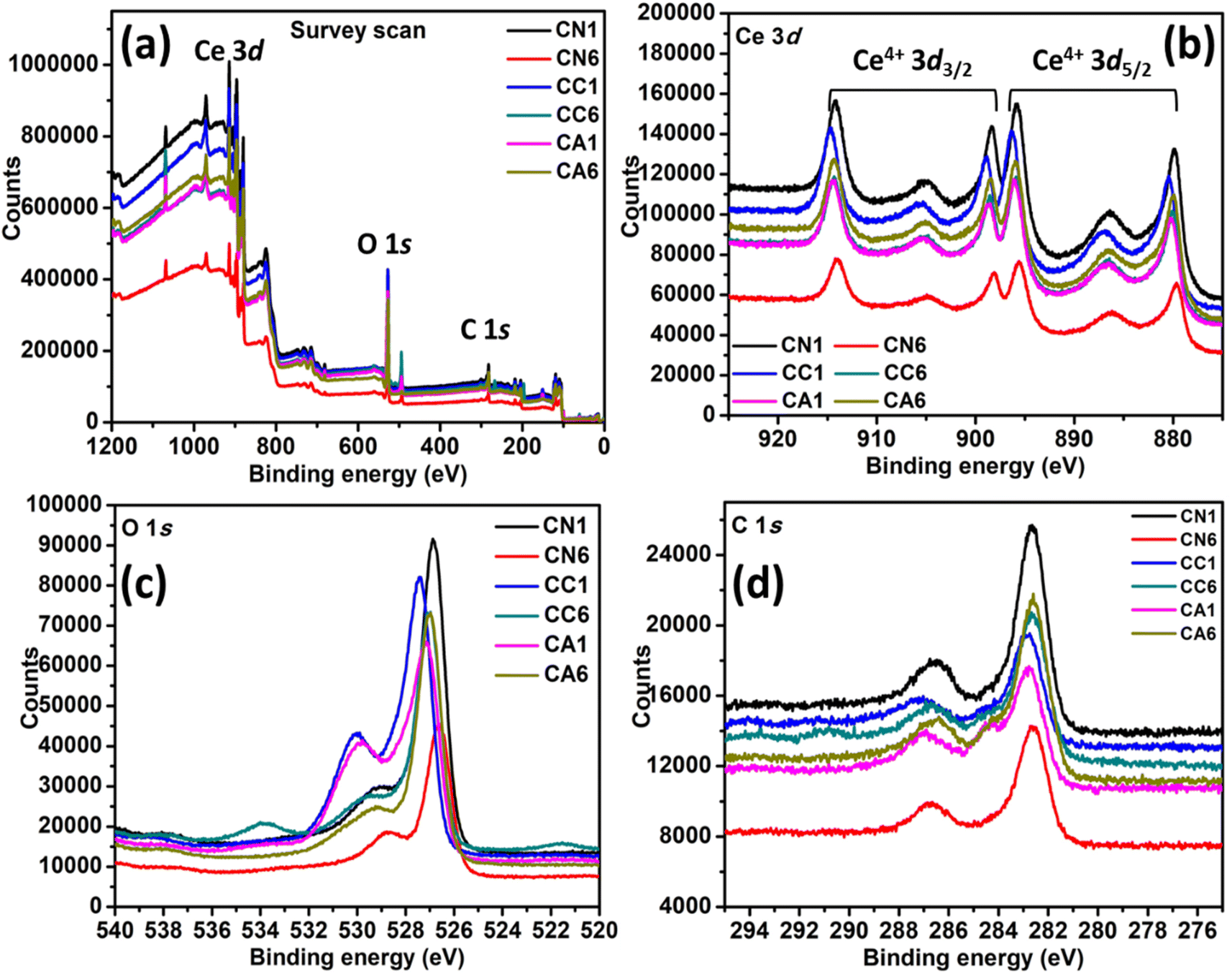 | ||
| Fig. 2 XPS spectra of selected CN, CC, and CA NPs: (a) Survey scan, (b) Ce 3d, (c) O 1s, and (d) C 1s. | ||
The XPS spectrum of O 1s can be seen in Fig. 2(c), in which all peaks exhibit two asymmetrical peaks, indicating the presence of O2−, OH−, and O− at the surface of the nanostructures. The peak at higher binding energy i.e. 531–532 eV is attributed to O2− vacancies and adsorbed –OH, while the peak at lower binding energy value of 529 eV is attributed to the metal–oxygen binding.35 CN1 and CN6 also show a significant difference in the XPS peak height. CN1 exhibits two asymmetrical peaks at 526.83 and 529.22 eV. On the other hand, CN6 has shifted to lower binding energies i.e., 526.61 and 528.79 eV. CC6 also shifted to lower energy compared to CC1. The O 1s peaks of CC1 are at 527.48 and 530.01 eV, while the O 1s peaks of CC6 are at 529.57 and 527.00 eV. Interestingly, CC6 shows an additional peak at 533.94 eV, which can be attributed to H2O adsorption.36 CA1 shows the O 1s peaks at 527.17 and 529.84 eV, while CA6 shows peaks at 527.90 and 529.32 eV. The typical C 1s peaks were also observed in the spectra (Fig. 2(d)), which were derived from the carbon coating used in the analysis. The atomic percentage of C 1s, O 1s, and Ce 3d are shown in Table 2.
| Samples | Atomic percentage (%) | ||
|---|---|---|---|
| C 1s | O 1s | Ce 3d | |
| CN1 | 26.2 | 51.0 | 22.8 |
| CN6 | 29.1 | 51.5 | 19.4 |
| CC1 | 17.7 | 61.8 | 20.5 |
| CC6 | 27.3 | 52.8 | 20.0 |
| CA1 | 21.5 | 61.7 | 16.8 |
| CA6 | 28.7 | 48.9 | 22.4 |
3.3. Transmission electron microscopy
In general, DEG acts as a solvent and a capping agent which limits particle growth, prevents aggregation of NPs, and retains a uniform dispersion without adding additives.37 However, the relative amount of water added, defined by the hydrolysis ratio, can affect the nature of the final compound i.e., the morphology of the synthesized CeO2.38 Therefore, in this study, different amounts of DEG and water were varied, and the morphologies of the synthesized CeO2 were investigated using TEM.Fig. 3 shows the TEM images of the CeO2 NPs synthesized using Ce(NO3)3·6H2O. The effect of various amounts of DEG was observed with respect to their size and morphology. Fig. 3(a) displays CN1 NPs synthesized with 0 mL of DEG. The synthesized CN1 NPs show a mix of spherical and elongated particles with an average size of about 28 nm. When 5 mL of DEG (CN2) was used in the synthesis, the particle size was decreased to an average size of about 26 nm (Fig. 3(d)).
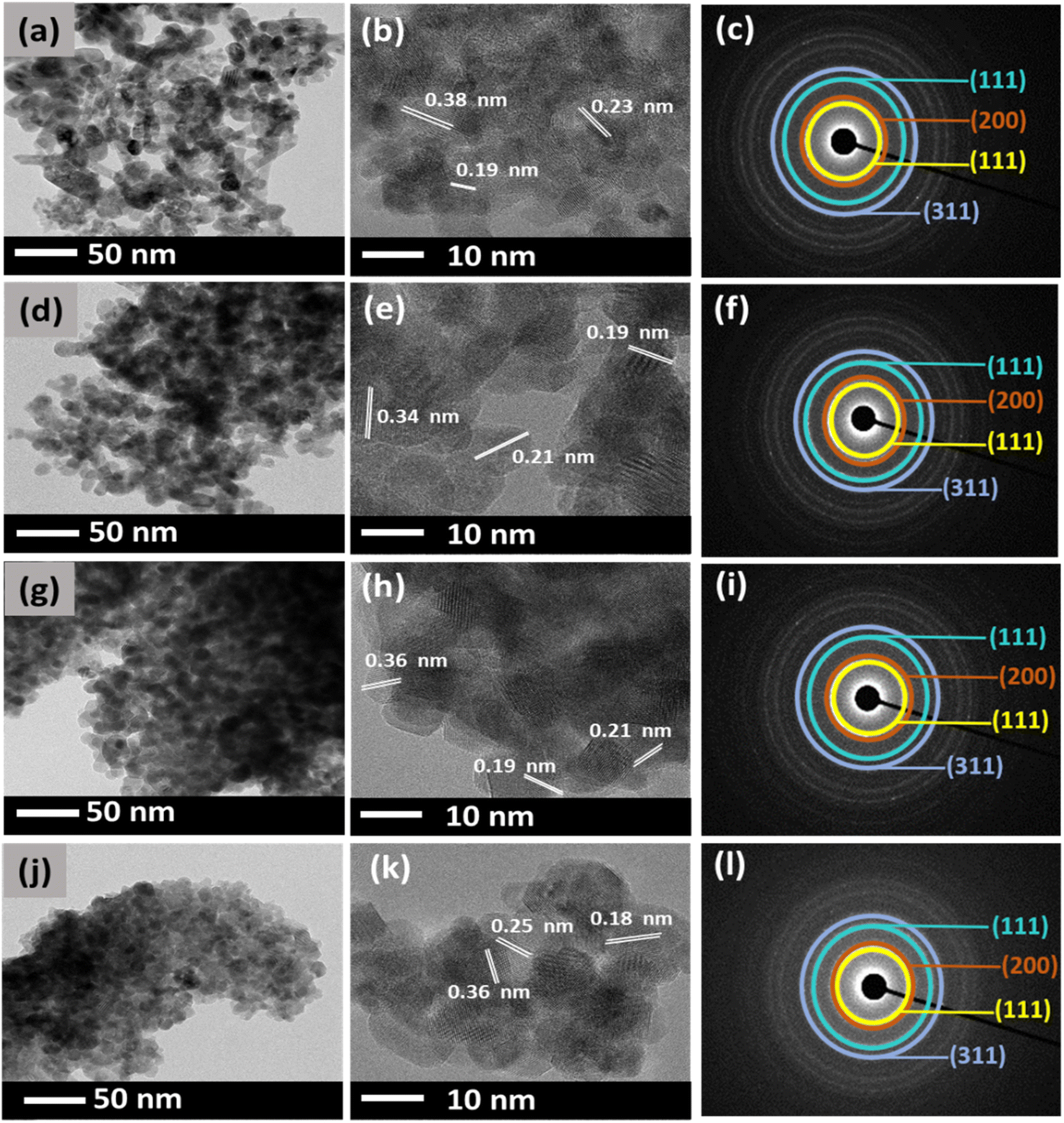 | ||
| Fig. 3 TEM, HRTEM, and SAED of: (a)–(c) CN1, (d)–(f) CN2, (g)–(i) CN4, and (j)–(l) CN6. | ||
Moreover, the morphology was clearly retained. This suggests that a small amount of DEG might affect the size of CeO2 NPs. Moreover, spherical particles were found to be more profound in Fig. 3(g) when 15 mL DEG was used. The synthesized CN4 showed an average particle size of ∼21 nm. Interestingly, as shown in Fig. 3(j), CN6 shows a further reduction of the particle size to about 16 nm when 25 mL of DEG was used, despite its highly agglomerated spherical morphology of CeO2 NP. This finding shows that further addition of DEG might lead to smaller particles and significant spherical morphology particles. On the other hand, it can be said that water is responsible for the enlargement of particle size. The d-spacing values of the lattice planes were also determined from the HR-TEM images (Fig. 3(b), (e), (h), and (k)). The d-spacing values were estimated to be around 0.38, 0.23, and 0.19 nm, which corresponds to the (111), (200), and (220) planes of the fluorite structure of cubic CeO2.39 Moreover, the diffraction pattern of CN NPs was also examined by selected electron diffraction (SAED) analysis as shown in Fig. 3(c), (f), (i), and (l). It was observed that they exhibit four broad rings, which are attributed to the (111), (200), (220), and (311) reflections of the fluorite cubic CeO2 structure.40
Different effects have taken place on the synthesis of CeO2 using CeCl3·7H2O. Fig. 4 shows the TEM images of the synthesized CeO2 NPs at the varied amount of DEG. Fig. 4(a) shows the morphology of the synthesized CeO2 NPs without DEG at all. Non-uniform elongated morphology was observed in CC1 NPs with an average particle size of 34 nm, indicating that no DEG was involved in controlling the growth of particles when CeCl3·7H2O was used. However, for CC2 NPs, when 5 mL of DEG was used, the particles started to display a mixture of spherical and fiber-like morphologies, as shown in Fig. 4(d) with an average particle size of 36 nm.
 | ||
| Fig. 4 TEM, HRTEM, and SAED of: (a)–(c) CC1, (d)–(f) CC2, (g)–(i) CC4, and (j)–(l) CC6. | ||
Fig. 4(g) shows CC4 NPs with similar particle morphology with a reduced average particle size of about 25 nm when 15 mL DEG was used. Finally, in the case of CC6 NPs, 25 mL of DEG (Fig. 4(j)) was used to produce spherical particles exhibiting an average particle size of around 28 nm. Similar to CN NPs, the d-spacing values of CC NPs, as shown in Fig. 4(b), (e), (h), and (k) were estimated to be around 0.35, 0.21, and 0.19 nm, which corresponds to the (111), (200), and (220) planes of the fluorite structure of cubic CeO2.39 SAED of CC1, CC2, CC4, and CC6 that are depicted in Fig. 4(c), (f), (i), and (l) show four distinct diffraction rings which can be attributed to the (111), (200), (220), and (311) reflections of the fluorite cubic CeO2 structure.40 This further supports the XRD and confirms the synthesis of CeO2 NPs.
Different observation was seen when CeO2 NPs was synthesized using Ce(CH3COO)3·H2O. Firstly, for CA1 NPs, with 0 mL of DEG, smaller spherical particle sizes with an average value of 29 nm are shown in Fig. 5(a). CA2 NPs showed similar morphology to CA1 NPs with 21 nm (Fig. 5(d)) when 5 mL of DEG was used. Highly agglomerated spherical particles were observed in CA4 NPs when 15 mL DEG showed about 21 nm of its average particle size. Similar spherical particles was seen agglomerated, resulting in an average particle size of 20 nm (Fig. 5(g)) when 25 mL of DEG was used in the synthesis of CA6 NPs. It indicates that the DEG was not involved in restricting particle growth (Fig. 5(j)). Therefore, in the case of CA NPs, the DEG has a minor effect on the size and particle morphology of CeO2. CA NPs showed d-spacing values of around 0.36, 0.25, and 0.19 nm, as shown in Fig. 5(b), (e), (h), and (k), which corresponds to the (111), (200), and (220) planes of the fluorite structure of cubic CeO2.39 SAED for CA1, CA2, CA4, and CA6 shows four distinct diffraction rings were observed in Fig. 5(c), (f), (i), and (l) show which is attributed to the (111), (200), (220), and (311) reflections of the fluorite cubic CeO2 structure.40
 | ||
| Fig. 5 TEM, HRTEM, and SAED of: (a)–(c) CA1, (d)–(f) CA2, (g)–(i) CA4, and (j)–(l) CA6. | ||
To investigate the effect of anions (NO3−, Cl− and CH3COO−) on the morphology of CN1, CC1, and CA1 (CeO2 synthesized using 0 mL DEG) as well as CN6, CC6, and CA6 (CeO2 synthesized using 25 mL DEG) were compared and discussed. By changing the anion precursor without the addition of DEG, the morphology of CeO2 was greatly influenced. The spherical morphology obtained in CN1 could not be retained when Cl− anion precursor was used. However, when CH3COO− anion precursor was employed, the morphology was similar to CN1. As reported earlier, the morphology of Fe2O3 using NO3− anion precursor resulted in micrometer particles.41 This suggests that NO3− ions are known to have different adsorption strengths on different crystal facets, which might cause faster growth of nanocrystals.41 Hence, large particles were seen in CN NPs. Furthermore, in another study, porous ZnO platelets were obtained using Zn(CH3COO)2 precursor.42 The study found that microplatelets were produced due to the high stability of the coordination complex of Zn2+ and deprotonated glycerol. The temperature of the liquid phase synthesis is not high enough to thermally decompose the complex. Hence, larger particles were mostly seen in CA NPs in which 100 °C might not be high enough to decompose the Ce3+-DEG complex. On the other hand, the Cl− anion precursor led to well-defined shape of CeO2 compared to CeO2 in CN NPs. This might be due to only a moderate reaction temperature is needed for a metal halide and polyol reaction to occur.32 This makes the system useful and more versatile.
The interaction between anion precursors and DEG also produced a variety of morphologies, as can be seen in Fig. 3–5. In brief, Ce(NO3)3 precursor produced densely populated spherical particles (CN6) in which the direction of particle growth changed from a mixture of spherical and elongated CeO2 particles (CN1) to a spherical morphology when 25 mL DEG was used. On the other hand, CC6 produced a significant spherical morphology compared to CC1. This suggests that the amount of DEG significantly impacts the morphology of CeO2 NPs synthesized using Cl− anion precursor. On the other hand, the spherical morphology of CA1 was retained when 25 mL DEG was introduced in CA6. In addition, the addition of DEG did not significantly affect the particle size of CA.
There are other factors that could influence the morphology of the synthesized CeO2, namely: reflux temperature, duration of a synthesis reaction, the relative ratio of DEG and H2O, as well as the basicity of the polyol solution.43 In this study, a reflux temperature of 100 °C was used for 4 h to synthesize different sets of CeO2 NPs, as mentioned in the experimental method section. Although in most reports, a reflux temperature of 180 °C for a 4 h reaction was commonly used.44–46 However, the addition of water in this study limits the rise of temperature beyond 100 °C. In general, an increase in temperature would lead to larger particles. However, based on the literature, CeO2 synthesized at 180 °C was mostly in the range of 10 to 50 nm.37,47 This might be due to the best performance of polyol synthesis taking place at about 50 °C lower than its boiling point (244 °C).48 This suggests that 180 °C is the optimum reaction temperature for the polyol synthesis method.
Furthermore, a defined amount of water is needed to transform the precursors into metal oxides.32 The production of metal oxides depends mainly on the amount of water present in the solution. In one study, a reaction between zinc acetate dihydrate and DEG led to the formation of ZnO.49 However, no ZnO was formed when the synthesis was performed using dehydrated Zn(CH3COO)2 and anhydrous DEG.32 However, one should note that the more water is added, it could lead to larger particles.32 Hence, agglomerated particles were mostly observed in this study. Ideally, the more DEG is added, the smaller the particle size. However, various morphologies were obtained due to different anions and other factors mentioned above. The estimated average particle size of CN, CC and CA NPs can be found in Table 1.
3.4. Fourier transform infrared spectroscopy
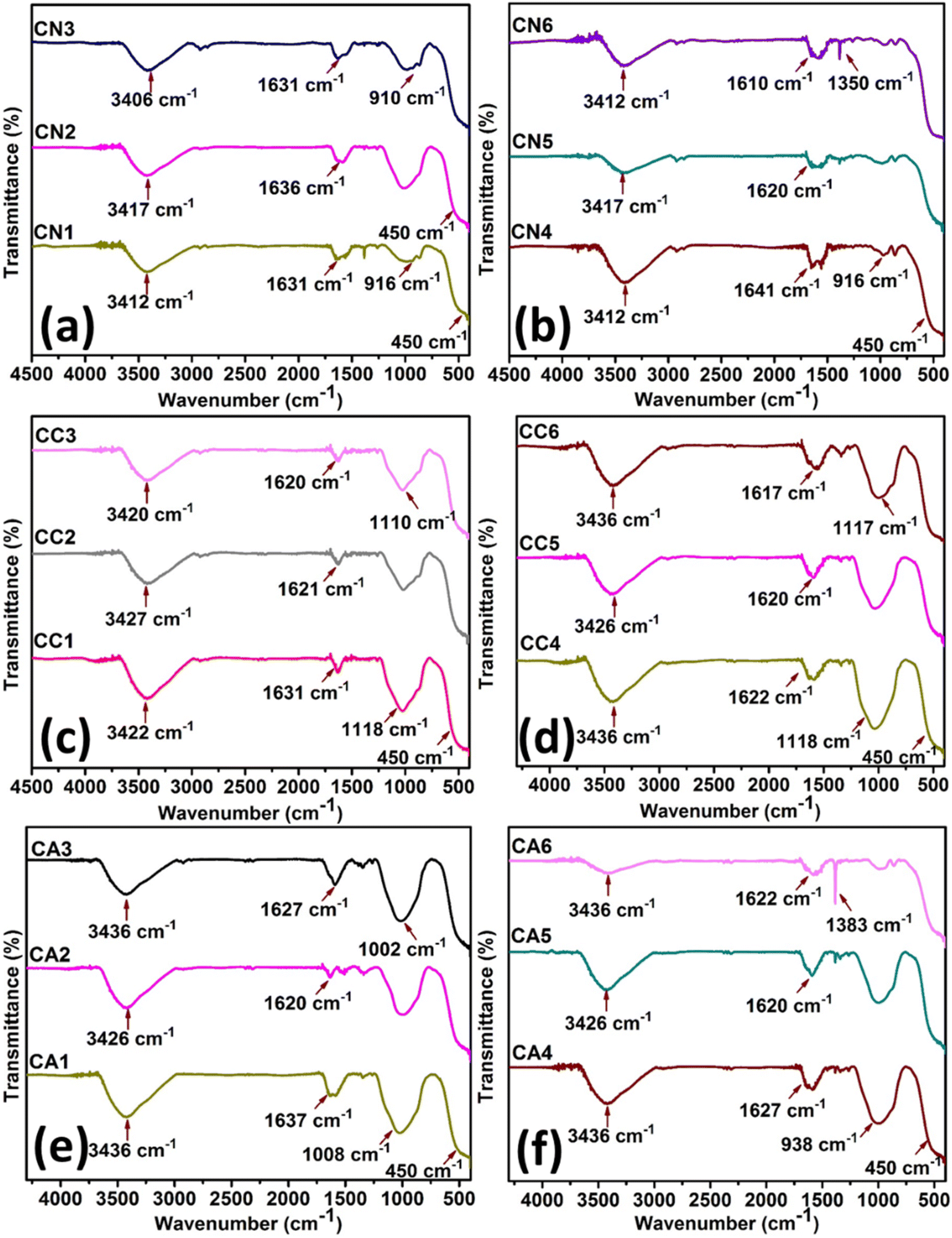
Fig. 6 shows the FT-IR spectra of CeO2 NPs of CN (Fig. 6(a) and (b)), CC (Fig. 6(c) and (d)), CA (Fig. 6(e) and (f)). CN NPs (Fig. 6(a) and (b)) showed a number of characteristic bands as shown by: stretching mode of O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O occurring at ∼1600 cm−1, the C–O stretching at 1100–1000 cm−1, O–H of moisture at ∼3400 cm−1 as well as NO3− residues due to the precursor salts at ∼900 cm−1. The band of Ce–O at 450 cm−1 confirmed the successful synthesis of CeO2.50 The minor difference between the intensities of CN1 to CN6 might be due to the amount of DEG as well as the nitrate residues in the NPs.
O occurring at ∼1600 cm−1, the C–O stretching at 1100–1000 cm−1, O–H of moisture at ∼3400 cm−1 as well as NO3− residues due to the precursor salts at ∼900 cm−1. The band of Ce–O at 450 cm−1 confirmed the successful synthesis of CeO2.50 The minor difference between the intensities of CN1 to CN6 might be due to the amount of DEG as well as the nitrate residues in the NPs.
 | ||
| Fig. 6 FTIR spectra of CeO2 NPs: (a) and (b) CN NPs, and, (c) and (d) CC NPs, and (e) and (f) CA NPs. | ||
Similar observations can be seen for CeO2 in CC NPs (Fig. 6(c) and (d)). The C–O stretching and stretching mode of O–CO at about 1100–1000 cm−1 and ∼1600 cm−1, respectively, were also observed in the region. The O–H stretching vibrations at ∼3390 cm−1 were seen to be at similar intensity despite the difference in the amount of H2O. The band of Ce–O positioned at 450 cm−1 confirmed the successful synthesis of CeO2.50 Interestingly, the peak at ∼938–1100 cm−1 (C–O stretching) in Fig. 6(e) and (f) was reduced with more DEG in the CeO2 NPs synthesized using Ce(CH3COO)3·H2O. On the other hand, C–H bending (∼1310 cm−1) became more apparent due to the presence of acetate ions.50 Similar to other synthesized CeO2 NPs, The O–H stretching vibrations at ∼3390 cm−1 were seen to be at similar intensity despite the difference in the amount of H2O. The successful synthesis of CeO2 was confirmed by the presence of Ce–O band at about 450 cm−1.50
4. Applications
4.1. α-Glucosidase inhibition activity
The α-glucosidase inhibition activity was carried out to determine if the synthesized NPs were able to inhibit the activity of the α-glucosidase enzyme. An α-glucosidase enzyme is involved in carbohydrate metabolism via its action on catalyzing the cleavage of oligosaccharides and disaccharides into monosaccharides.51 The ability to inhibit the α-glucosidase activity can prolong the digestion and absorption of carbohydrate, reducing blood glucose levels.52 A range of dosages of the synthesized CeO2 (CN, CC, and CA) was tested to check and confirm its ability to inhibit α-glucosidase activity. Acarbose was used as a positive control, and the inhibition of α-glucosidase using commercially available CeO2 was also tested as a positive control to compare with the synthesized CeO2 (Fig. 7 and Table S2†).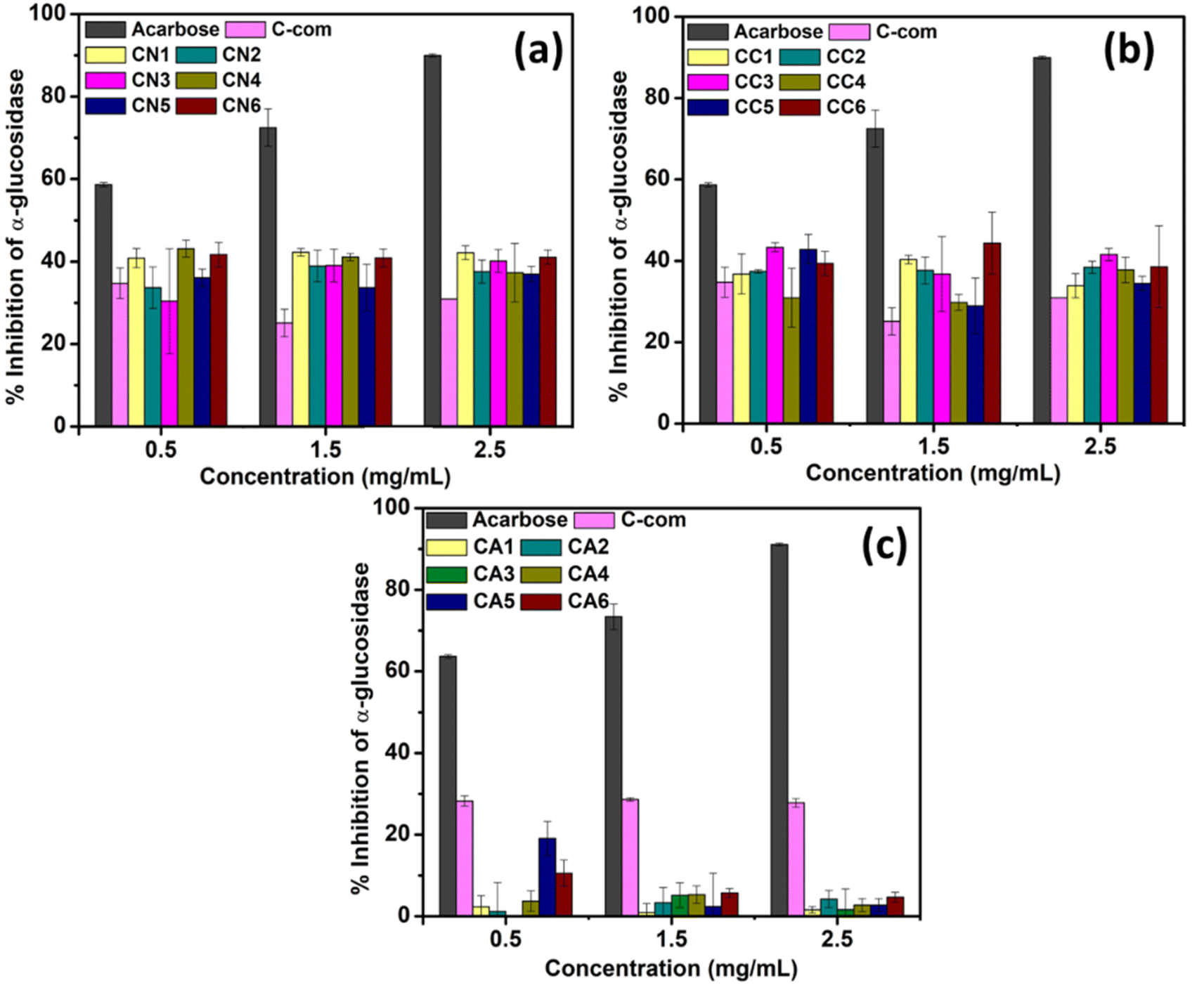 | ||
| Fig. 7 Inhibition of α-glucosidase activity using CeO2 NPs from (a) CN, (b) CC, and (c) CA NPs. | ||
Fig. 7(a) shows the inhibition activity of α-glucosidase using CeO2 synthesized using Ce(NO3)3·6H2O (CN1–CN6). Amongst CN1–CN6, CN4 showed better inhibition (43.15 ± 2.05%) at 0.5 mg mL−1 concentration. However, by increasing the dosage, the inhibition efficiency dropped by 4.65% and 21.92% at 1.5 and 2.5 mg mL−1, respectively (Fig. 7(a)). CN1 and CN6 also showed comparable efficiency at the lowest concentrations, which are 40.86 ± 3.70% and 41.70 ± 2.91, respectively. At 1.5 mg mL−1, CN1, CN4, and CN6 also showed similar behavior, although CN1 showed the highest inhibition efficiency percentage (42.29 ± 0.93%). Similarly, CN1 showed the highest inhibition activity at 2.5 mg mL−1. CN2 showed a lower response (33–37%) compared to CN1, CN4, and CN6 despite having similar morphology. However, as the dosage increases, the response increased which could be attributed to the smaller particle size. From this study, CeO2 NPs that was synthesized using 0, 50, and 100% DEG showed better responses in inhibiting α-glucosidase activity. Based on the TEM images, all of them possess a spherical morphology. The performance consistency of these materials from the doses suggested that the morphology might not directly influence inhibition activity. On the other hand, the obtained crystallite sizes for CN1, CN4, and CN6 are comparable (∼20–23 nm). This finding proposes that CeO2 NPs with crystallite size lower or higher than 20–23 nm might show lower inhibition activity.
The α-glucosidase inhibition activity using CeO2 NPs prepared using CeCl3·7H2O is shown in Fig. 7(b). In this case, CC3 showed the highest α-glucosidase inhibition activity at both 0.5 and 2.5 mg mL−1 which are about 43.33 ± 1.11 and 41.57 ± 1.54, respectively (Table S2†). CC5 also showed a comparable response at 0.5 mg mL−1 which is about 42.83 ± 3.63. Higher α-glucosidase inhibition percentage of CC3 and CC5 might be due to their morphology. However, at 1.5 mg mL−1, CC1 and CC6 showed higher α-glucosidase inhibition activities. This might be due to the sheet-like structure of CeO2 NPs as well as the lower crystallite size (15.31 and 13.14 nm for CC1 and CC6).
On the otherhand, the α-glucosidase activity shown by CeO2 NPs synthesized using Ce(CH3COO)3·H2O showed different response (Fig. 7(c) compared to Fig. 7(a) and (b)). Apart from the commercially available CeO2, none of the materials (CA1–CA6) showed remarkable responses, although CA5 showed a slight response at 0.5 mg mL−1. This trend, however, could not be observed at a higher dosage. The activity of enzymes can be regulated by interactions with regulatory molecules that can activate or inhibit enzyme activity. In summary, among the tested CeO2 (CN, CC, and CA NPs), CN and CC NPs showed α-glucosidase enzyme inhibitory with the increase in concentration.
4.2. General mechanism of α-glucosidase inhibition activity
Mammalian α-glucosidase in the mucosal brush border of the small intestine catalyzes the end step of digestion of complex carbohydrates.16 Inhibitor of α-glucosidase enzyme functions to delay the breakdown of carbohydrates in the small intestine, slowing down carbohydrate absorption in the gut and reducing glucose in the blood. In Fig. 8, the enzymatic reaction mechanisms between 4-nitrophenyl α-D-gluocipyranoside (PNPG) and α-glucosidase (Fig. 8(a)) and reaction between different α-glucosidase inhibitors with α-glucosidase (Fig. 8(b)–(d)) are shown. The substrate (PNPG) used in the reaction binds to the enzyme's active sites, forming an enzyme-substrate complex. The substrate is broken down into smaller compounds (Fig. 8(a)).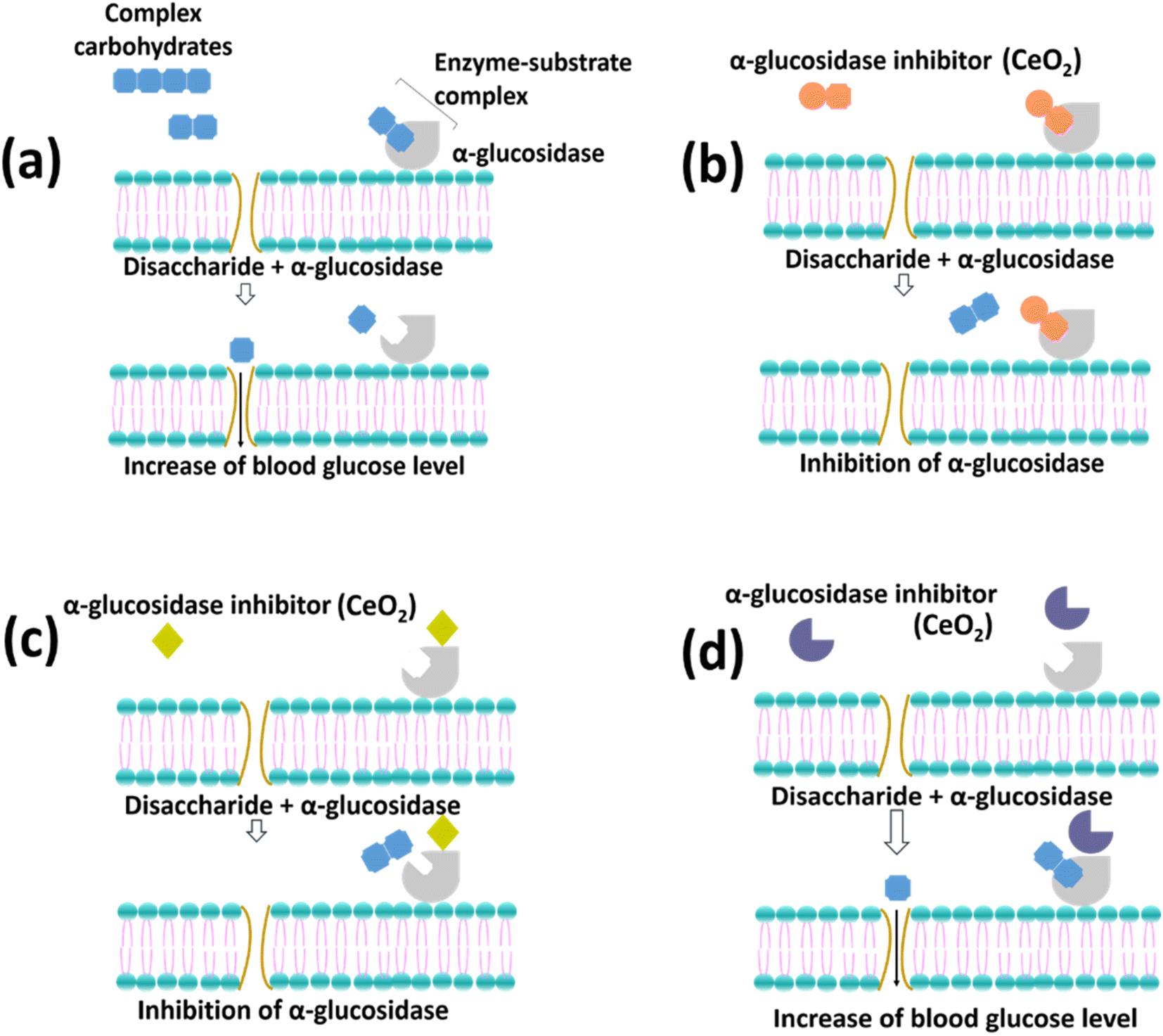 | ||
| Fig. 8 The enzymatic reaction with (a) PNPG and (b)–(d) reaction in the presence of different types of inhibitors (adapted from Assefa et al. with modification).16 | ||
Fig. 8(b) shows that the morphology of the inhibitors can fit in the active site of the enzyme, which therefore competes with the PNPG. On the other hand, Fig. 8(c) illustrates the attachment of the inhibitors on the enzyme where the active sites of the enzyme change resulting in the difficulties of the PNPG to bind to the enzyme. Finally, Fig. 8(d) describes the inability of the inhibitors to alter the enzyme, which results in the successful formation of the enzyme-substrate complex. Overall, Fig. 8(a) and (d) show the unsuccessful inhibition of the enzymes, while Fig. 8(b) and (c) show the successful inhibition of the enzymes.
The inhibition using CeO2 NPs could not be determined based on the percentage inhibition alone. However, it can be estimated by comparing the activity between CeO2 NPs and the positive control, acarbose which was used in this study. The percentage inhibition of acarbose increased with increased dosage, indicating the possibility of either mechanism, as shown in Fig. 8(b) and (c) to inhibit α-glucosidase enzyme. On the other hand, CN and CC NPs showed a moderately high percentage inhibition of α-glucosidase (>40.0%). Thus, the enzymatic reaction is closely illustrated by Fig. 8(b). The CN and CC materials might behave as a competitive inhibitor with PNPG compounds. Hence, the percentage inhibition of α-glucosidase activity achieved by CN and CC materials was not exceed 50%. Furthermore, the inhibition activity using CN and CC can also be illustrated by Fig. 8(c). According to studies using carbon nanoparticles (CNPs), the CNPs might combine with α-glucosidase by noncovalent bonding to alter the structure of the enzyme.53,54 Moreover, Cha et al. reported that morphology play an important role in penetrating the grooves where the active center of the enzyme is located.55 Hence, interfering with its reconfiguration is needed for the catalytic activity. Therefore, this hinders the combination of the substrate with the enzyme. In the case of CA NPs, the enzymatic reaction might be due to either the materials being competitive with PNPG (Fig. 8(b)) or the materials not binding to the enzymes (Fig. 8(d)). Therefore, less percentage inhibition of α-glucosidase was observed.
4.3. Cytotoxicity test/cell viability test
The cytotoxicity of CN, CC, and CA NPs on RAW 264.7 macrophage cells was evaluated by MTT assay. All the synthesized NPs did not exhibit cytotoxic effects at concentrations of 62.5, 125, 250, and 500 μg mL−1 following the 24 h incubation after the treatment (Fig. 9). As shown in Fig. 9(a), CN1 showed some cytotoxicity at 2000 μg mL−1, exhibiting about 80% cell viability. However, at lower concentrations, CN1 showed lower cell cytotoxicity. CN4 showed the highest cytotoxicity against RAW 246.7 macrophages, showing only about 60% cell viability.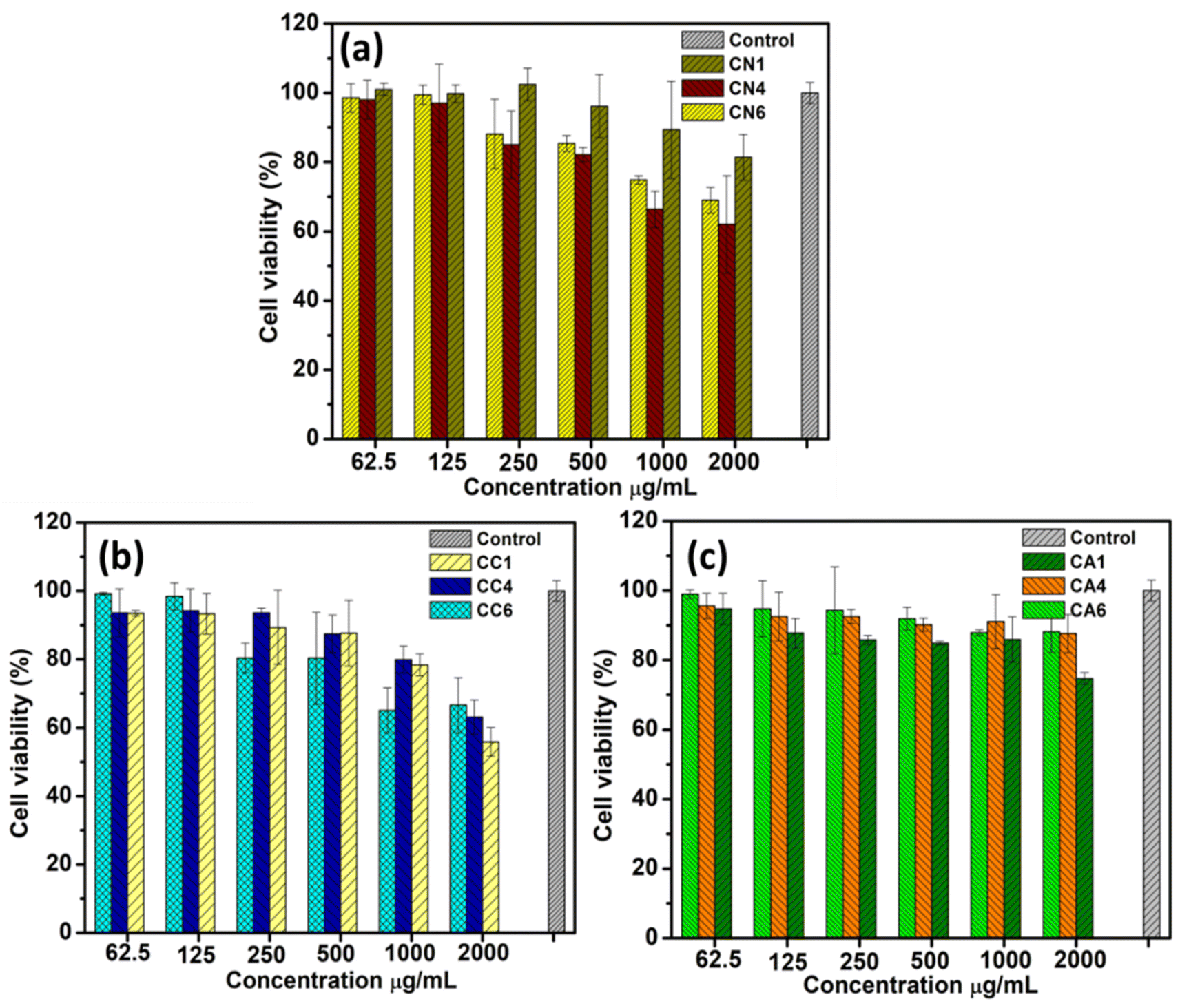 | ||
| Fig. 9 Cell viability (%) of selected (a) CN, (b) CC, and (c) CA NPs. | ||
On the other hand, CC1 showed the most cytotoxic at 2000 μg mL−1 (Fig. 9(b)). However, when lowering the concentration, CC1 showed lower cytotoxic and was comparable to CC4 (75–95% cell viability). CC6 was observed to show no effect at lower concentrations i.e., 62.5 and 125 μg mL−1. However, at a concentration ranging from 250–1000 μg mL−1, the cell viability of CC6 was the lowest compared to CC1 and CC4. Therefore, within this range of concentration, CC6 showed a certain degree of cytotoxicity. Interestingly, at 2000 μg mL−1, the cell viability of CC6 was the highest, i.e., about 65% for CC1 and CC4. Both CN and CC NPs showed some cytotoxicity towards the RAW 264.7 macrophages. Based on the previous section, both CN and CC NPs showed α-glucosidase inhibition activity. This might show that the medium cytotoxicity of CN and CC NPs might cause some inhibition of α-glucosidase inhibition activity. Similar results were shown by Saravanakumar et al. in which they reported biocompatible CeO2 NPs that showed α-glucosidase inhibition activity.56
CA NPs (Fig. 9(c)) showed low cytotoxicity at concentrations ranging from 62.5 to 2000 μg mL−1, yet only CA1 showed slight cytotoxicity at the concentration of 2000 μg mL−1 compared with the CA4 and CA6. This can also be seen that the low α-glucosidase inhibition activity of CA NPs was seen in Fig. 7(c). Overall, the present result indicates that CA1–CA6 and CN1 at all concentrations and CC and other CN NPs between 62.5 to 125 μg mL−1 have high potential in pharmacological applications since they were not toxic to human cells at various concentrations which is in agreement with previous reports.57,58
5. Conclusion
Cerium oxide nanoparticles (CeO2 NPs) was synthesized using a modified conventional polyol method. In this study, the diethylene glycoland water (DEG)/H2O ratio was varied from 0/25, 5/20, 10/15, 15/10, 20/5, and 25/0 mL, respectively. Apart from that, three different cerium precursor salts (Ce(NO3)3, CeCl3, and Ce(CH3COO)3) were also used to synthesize CeO2 NPs. The structures, size, and morphologies of the synthesized CeO2 were studied based on the variations of factors in the synthesis. An average crystallite size of 13 to 33 nm was obtained from the XRD analysis for all the synthesized CeO2 NPs using NO3−, Cl−, and CH3COO− cerium anions precursors, as well as the different DEG/H2O ratio. The synthesized CeO2 showed spherical, sheet-like, and fiber-like morphologies. Average particle sizes in the range of 16–36 nm were obtained by varying the DEG/H2O ratio and using different cerium anion precursors. The presence of DEG molecules on the surface of CeO2 NPs was confirmed using FTIR. Inhibition of α-glucosidase enzymes using the synthesized CeO2 NPs was carried out. CeO2 synthesized using Ce(NO3)3 and CeCl3 precursors showed approximately 40.0% inhibition activity, while CeO2 synthesized using Ce(CH3COO)3 showed the lowest α-glucosidase enzyme inhibition activity. The probable mechanism of α-glucosidase enzyme inhibition activity using CN, CC, and CA NPs was also discussed. Cell cytotoxicity was also conducted, and it was found that CN and CC NPs were not cytotoxic to RAW 264.7 macrophage cells at lower concentrations, while CA NPs were found to be non-toxic at all concentrations. Therefore, CeO2 NPs synthesized using different DEG/H2O ratio and cerium anions precursors are biocompatible and have shown potential in α-glucosidase inhibition property.Author contributions
Shaidatul Najihah Matussin: methodology; investigation, data curation; writing – original draft. Fazlurrahman Khan: methodology, investigation, data curation, review & editing. Pathum Chandika: methodology, investigation, data curation; writing – original. Mohammad Hilni Harunsani: supervision, writing – review & editing. Norhayati Ahmad: supervision, writing – review & editing. Young-Mog Kim: resources, formal analysis. Won-Kyo Jung: resources, formal analysis. Mohammad Mansoob Khan: supervision, conceptualization, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to acknowledge the FRC grant (UBD/RSCH/1.4/FICBF(b)/2022/046) received from Universiti Brunei Darussalam, Brunei Darussalam. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (2021R1A6A1A03039211 and 2022R1A2B5B01001998).References
- A. Dhall and W. Self, Cerium Oxide Nanoparticles: A Brief Review of Their Synthesis Methods and Biomedical Applications, Antioxidants, 2018, 7(8), 1–13, DOI:10.3390/antiox7080097.
- S. N. Naidi, M. H. Harunsani, A. L. Tan and M. M. Khan, Green-Synthesized CeO2 Nanoparticles for Photocatalytic, Antimicrobial, Antioxidant and Cytotoxicity Activities, J. Mater. Chem. B, 2021, 9, 5599–5620, 10.1039/D1TB00248A.
- G. Spezzati, K. Fant, A. Ahniyaz, M. Lundin-Johnson, E. J. M. Hensen, H. Langermans and J. P. Hofmann, Synthesis, Physicochemical Characterization, and Cytotoxicity Assessment of CeO2 Nanoparticles with Different Morphologies, Eur. J. Inorg. Chem., 2017, 2017(25), 3184–3190, DOI:10.1002/ejic.201700248.
- C. M. Magdalane, K. Kaviyarasu, J. J. Vijaya, B. Siddhardha and B. Jeyaraj, Photocatalytic Activity of Binary Metal Oxide Nanocomposites of CeO2/CdO Nanospheres: Investigation of Optical and Antimicrobial Activity, J. Photochem. Photobiol., B, 2016, 163, 77–86, DOI:10.1016/j.jphotobiol.2016.08.013.
- D. Liu, J. Tian, Y. Tang, J. Li, S. Wu, S. Yi, X. Huang, D. Sun and H. Wang, High-Power Double-Face Flow Al-Air Battery Enabled by CeO2 Decorated MnOOH Nanorods Catalyst, Chem. Eng. J., 2021, 406(2020), 126772, DOI:10.1016/j.cej.2020.126772.
- S. N. Matussin, M. H. Harunsani and M. M. Khan, CeO2 and CeO2-Based Nanomaterials for Photocatalytic, Antioxidant and Antimicrobial Activities, J. Rare Earths, 2023, 41(2), 167–181, DOI:10.1016/j.jre.2022.09.003.
- A. J. Khan, M. Hanif, M. S. Javed, S. Hussain, W. Zhong, M. Saleem and Z. Liu, Energy Storage Properties of Hydrothermally Processed, Nanostructured, Porous CeO2 Nanoparticles, J. Electroanal. Chem., 2020, 865, 114158, DOI:10.1016/j.jelechem.2020.114158.
- J. Kullgren, K. Hermansson and P. Broqvist, Supercharged Low-Temperature Oxygen Storage Capacity of Ceria at the Nanoscale, J. Phys. Chem. Lett., 2013, 4(4), 604–608, DOI:10.1021/jz3020524.
- Y. Chen, Y. Zhang, J. Baker, P. Majumdar, Z. Yang, M. Han and F. Chen, Hierarchically Oriented Macroporous Anode-Supported Solid Oxide Fuel Cell with Thin Ceria Electrolyte Film, ACS Appl. Mater. Interfaces, 2014, 6(7), 5130–5136, DOI:10.1021/am5003662.
- M. M. Khan, Metal Oxide Powder Photocatalysts, Elsevier Ltd, 2018, DOI:10.1016/B978-0-08-101977-1.00002-8.
- M. E. Khan, M. M. Khan and M. H. Cho, Ce3+-Ion, Surface Oxygen Vacancy, and Visible Light-Induced Photocatalytic Dye Degradation and Photocapacitive Performance of CeO2-Graphene Nanostructures, Sci. Rep., 2017, 7(1), 1–17, DOI:10.1038/s41598-017-06139-6.
- M. M. Khan, S. A. Ansari, D. Pradhan, D. H. Han, J. Lee and M. H. Cho, Defect-Induced Band Gap Narrowed CeO2 Nanostructures for Visible Light Activities, Ind. Eng. Chem. Res., 2014, 53(23), 9754–9763, DOI:10.1021/ie500986n.
- M. M. Khan, S. F. Adil and A. Al-Mayouf, Metal Oxides as Photocatalysts, J. Saudi Chem. Soc., 2015, 19(5), 462–464, DOI:10.1016/j.jscs.2015.04.003.
- S. N. Matussin, F. Khan, M. H. Harunsani, Y.-M. Kim and M. M. Khan, Visible-Light-Induced Photocatalytic and Photoantibacterial Activities of Co-Doped CeO2, ACS Omega, 2023, 8(13), 11868–11879, DOI:10.1021/acsomega.2c07058.
- P. Senthilkumar, L. Surendran, B. Sudhagar and D. S. Ranjith Santhosh Kumar, Facile Green Synthesis of Gold Nanoparticles from Marine Algae Gelidiella Acerosa and Evaluation of Its Biological Potential, SN Appl. Sci., 2019, 1(4), 284, DOI:10.1007/s42452-019-0284-z.
- S. T. Assefa, E. Y. Yang, S. Y. Chae, M. Song, J. Lee, M. C. Cho and S. Jang, Alpha Glucosidase Inhibitory Activities of Plants with Focus on Common Vegetables, Plants, 2020, 9(1), 1–16, DOI:10.3390/plants9010002.
- A. Asok, S. Ghosh, P. A. More, B. A. Chopade, M. N. Gandhi and A. R. Kulkarni, Surface Defect Rich ZnO Quantum Dots as Antioxidants Inhibiting α-Amylase and α-Glucosidase: A Potential Anti-Diabetic Nanomedicine, J. Mater. Chem. B, 2015, 3(22), 4597–4606, 10.1039/c5tb00407a.
- A. R. Prasad, S. M. Basheer, L. Williams and A. Joseph, Highly Selective Inhibition of α-Glucosidase by Green Synthesised ZnO Nanoparticles–In Vitro Screening and In Silico Docking Studies, Int. J. Biol. Macromol., 2019, 139, 712–718, DOI:10.1016/j.ijbiomac.2019.08.033.
- J. Dholakia, B. Prabhakar and P. Shende, Strategies for the Delivery of Antidiabetic Drugs via Intranasal Route, Int. J. Pharm., 2021, 608, 121068, DOI:10.1016/j.ijpharm.2021.121068.
- C. Y. Wong, J. Martinez and C. R. Dass, Oral Delivery of Insulin for Treatment of Diabetes: Status Quo, Challenges and Opportunities, J. Pharm. Pharmacol., 2016, 68(9), 1093–1108, DOI:10.1111/jphp.12607.
- R. Khair, P. Shende and Y. A. Kulkarni, Nanostructured Polymer-Based Cochleates for Effective Transportation of Insulin, J. Mol. Liq., 2020, 311, 113352, DOI:10.1016/j.molliq.2020.113352.
- H. Jan, M. A. Khan, H. Usman, M. Shah, R. Ansir, S. Faisal, N. Ullah and L. Rahman, TheAquilegia Pubiflora(Himalayan Columbine) Mediated Synthesis of Nanoceria for Diverse Biomedical Applications, RSC Adv., 2020, 10(33), 19219–19231, 10.1039/d0ra01971b.
- H. Dong, Y. C. Chen and C. Feldmann, Polyol Synthesis of Nanoparticles: Status and Options Regarding Metals, Oxides, Chalcogenides, and Non-Metal Elements, Green Chem., 2015, 17(8), 4107–4132, 10.1039/c5gc00943j.
- F. Fievet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J. Y. Piquemal, L. Sicard and G. Viau, The Polyol Process: A Unique Method for Easy Access to Metal Nanoparticles with Tailored Sizes, Shapes and Compositions, Chem. Soc. Rev., 2018, 47(14), 5187–5233, 10.1039/c7cs00777a.
- S. N. Matussin, A. Rahman and M. M. Khan, Role of Anions in the Synthesis and Crystal Growth of Selected Semiconductors, Front. Chem., 2022, 10, 1–13, DOI:10.3389/fchem.2022.881518.
- T. Herricks, J. Chen and Y. Xia, Polyol Synthesis of Platinum Nanoparticles: Control of Morphology with Sodium Nitrate, Nano Lett., 2004, 4(12), 2367–2371, DOI:10.1021/nl048570a.
- U. M. Badeggi, J. A. Badmus, S. S. Botha, E. Ismail, J. L. Marnewick, C. W. J. Africa and A. A. Hussein, Biosynthesis, Characterization, and Biological Activities of Procyanidin Capped Silver Nanoparticles, J. Funct. Biomater., 2020, 11(3), 1–20, DOI:10.3390/JFB11030066.
- A. S. Alqahtani, S. Hidayathulla, T. Rehman, A. A. Elgamal, R. A. El Dib and M. F. Alajmi, Biomolecules, 2019, 10(1), 61 CrossRef.
- G. Seong, M. Dejhosseini and T. Adschiri, A Kinetic Study of Catalytic Hydrothermal Reactions of Acetaldehyde with Cubic CeO2 Nanoparticles, Appl. Catal., A, 2018, 550(2017), 284–296, DOI:10.1016/j.apcata.2017.11.023.
- M. A. M. Khan, W. Khan, M. Ahamed and A. N. Alhazaa, Microstructural Properties and Enhanced Photocatalytic Performance of Zn Doped CeO2 Nanocrystals, Sci. Rep., 2017, 7(1), 1–11, DOI:10.1038/s41598-017-11074-7.
- H. Nosrati, R. Sarraf-Mamoory, M. Canillas Perez, D. Q. S. Le, R. Zolfaghari Emameh and C. E. Bünger, Characteristics of Hydroxyapatite-Reduced Graphene Oxide Composite Powders Synthesized via Hydrothermal Method in the Absence and Presence of Diethylene Glycol, Open Ceram., 2021, 5, 100067, DOI:10.1016/j.oceram.2021.100067.
- N. Markus and N. Pinna, Solvent-Controlled Synthesis, in Engineering Materials and Processes, Springer, London, 2009, pp. 53–95, DOI:10.1007/978-1-84882-671-7_4.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Ce 3d XPS Investigation of Cerium Oxides and Mixed Cerium Oxide (CexTiyOz), Surf. Interface Anal., 2008, 40(3–4), 264–267, DOI:10.1002/sia.2686.
- S. N. Matussin, F. Khan, M. H. Harunsani, Y.-M. Kim and M. M. Khan, Effect of Pd-Doping Concentrations on the Photocatalytic, Photoelectrochemical, and Photoantibacterial Properties of CeO2, Catalysts, 2023, 13(1), 96, DOI:10.3390/catal13010096.
- M. Tou, R. Michalsky and A. Steinfeld, Solar-Driven Thermochemical Splitting of CO2 and In Situ Separation of CO and O2 across a Ceria Redox Membrane Reactor, Joule, 2017, 1(1), 146–154, DOI:10.1016/j.joule.2017.07.015.
- J.-H. Jhang, J. A. Boscoboinik and E. I. Altman, Ambient Pressure X-Ray Photoelectron Spectroscopy Study of Water Formation and Adsorption under Two-Dimensional Silica and Aluminosilicate Layers on Pd(111), J. Chem. Phys., 2020, 152(8), 084705, DOI:10.1063/1.5142621.
- A. Guleria, P. Pranjali, M. K. Meher, A. Chaturvedi, S. Chakraborti, R. Raj, K. M. Poluri and D. Kumar, Effect of Polyol Chain Length on Proton Relativity of Gadolinium Oxide Nanoparticles for Enhanced Magnetic Resonance Imaging Contrast, J. Phys. Chem. C, 2019, 123(29), 18061–18070, DOI:10.1021/acs.jpcc.9b04089.
- C. Feldmann and H.-O. Jungk, Polyol-Mediated Preparation of Nanoscale Oxide Particles, Angew. Chem., Int. Ed., 2001, 40(2), 359–362, DOI:10.1002/1521-3773(20010119)40:2<359::AID-ANIE359>3.0.CO;2-B.
- L. He, Y. Ren, Y. Fu, B. Yue, S. C. Edman Tsang and H. He, Morphology-Dependent Catalytic Activity of Ru/CeO2 in Dry Reforming of Methane, Molecules, 2019, 24(3), 526, DOI:10.3390/molecules24030526.
- S. N. Naidi, M. H. Harunsani, A. L. Tan and M. M. Khan, Structural, Morphological and Optical Studies of CeO2 Nanoparticles Synthesized Using Aqueous Leaf Extract of Pometia Pinnata, Bionanoscience, 2022, 12(2), 393–404, DOI:10.1007/s12668-022-00956-4.
- S. Jain, A. Panigrahi and T. K. Sarma, Counter Anion-Directed Growth of Iron Oxide Nanorods in a Polyol Medium with Efficient Peroxidase-Mimicking Activity for Degradation of Dyes in Contaminated Water, ACS Omega, 2019, 4(8), 13153–13164, DOI:10.1021/acsomega.9b01201.
- H. Dong and C. Feldmann, Porous ZnO Platelets via Controlled Thermal Decomposition of Zinc Glycerolate, J. Alloys Compd., 2012, 513, 125–129, DOI:10.1016/j.jallcom.2011.10.004.
- R. Brayner, T. Coradin and F. Fiévet, Nanomaterials: A Danger or a Promise?, ed. R. Brayner, F. Fiévet and T. Coradin, Springer, London, 2013, DOI:10.1007/978-1-4471-4213-3.
- Y. Kuang, Y. Cao, M. Liu, G. Zu, Y. Zhang, Y. Zhang and R. Pei, Geometrical Confinement of Gadolinium Oxide Nanoparticles in Poly(Ethylene Glycol)/Arginylglycylaspartic Acid-Modified Mesoporous Carbon Nanospheres as an Enhanced T1 Magnetic Resonance Imaging Contrast Agent, ACS Appl. Mater. Interfaces, 2018, 10(31), 26099–26107, DOI:10.1021/acsami.8b09709.
- R. Di Corato, F. Gazeau, C. Le Visage, D. Fayol, P. Levitz, F. Lux, D. Letourneur, N. Luciani, O. Tillement and C. Wilhelm, High-Resolution Cellular MRI: Gadolinium and Iron Oxide Nanoparticles for in-Depth Dual-Cell Imaging of Engineered Tissue Constructs, ACS Nano, 2013, 7(9), 7500–7512, DOI:10.1021/nn401095p.
- A. Hedlund, M. Ahrén, H. Gustafsson, N. Abrikossova, M. Warntjes, J. I. Jönsson, K. Uvdal and M. Engström, Gd2O3 Nanoparticles in Hematopoietic Cells for MRI Contrast Enhancement, Int. J. Nanomed., 2011, 6, 3233–3240, DOI:10.2147/ijn.s23940.
- X. Miao, S. L. Ho, T. Tegafaw, H. Cha, Y. Chang, I. T. Oh, A. M. Yaseen, S. Marasini, A. Ghazanfari, H. Yue, K. S. Chae and G. H. Lee, Stable and Non-Toxic Ultrasmall Gadolinium Oxide Nanoparticle Colloids (Coating Material = Polyacrylic Acid) as High-Performance: T 1 Magnetic Resonance Imaging Contrast Agents, RSC Adv., 2018, 8(6), 3189–3197, 10.1039/c7ra11830a.
- H. Dong, Y. C. Chen and C. Feldmann, Polyol Synthesis of Nanoparticles: Status and Options Regarding Metals, Oxides, Chalcogenides, and Non-Metal Elements, Green Chem., 2015, 17(8), 4107–4132, 10.1039/c5gc00943j.
- D. Jézéquel, J. Guenot, N. Jouini and F. Fiévet, Submicrometer Zinc Oxide Particles: Elaboration in Polyol Medium and Morphological Characteristics, J. Mater. Res., 1995, 10(1), 77–83, DOI:10.1557/JMR.1995.0077.
- A. Gondolini, E. Mercadelli, A. Sanson, S. Albonetti, L. Doubova and S. Boldrini, Effects of the Microwave Heating on the Properties of Gadolinium-Doped Cerium Oxide Prepared by Polyol Method, J. Eur. Ceram. Soc., 2013, 33(1), 67–77, DOI:10.1016/j.jeurceramsoc.2012.08.008.
- G. Chen and M. Guo, Rapid Screening for α-Glucosidase Inhibitors from Gymnema Sylvestre by Affinity Ultrafiltration–HPLC-MS, Front. Pharmacol., 2017, 8, 228, DOI:10.3389/fphar.2017.00228.
- K. M. Khan, F. Rahim, A. Wadood, N. Kosar, M. Taha, S. Lalani, A. Khan, M. I. Fakhri, M. Junaid, W. Rehman, M. Khan, S. Perveen, M. Sajid and M. I. Choudhary, Synthesis and Molecular Docking Studies of Potent α-Glucosidase Inhibitors Based on Biscoumarin Skeleton, Eur. J. Med. Chem., 2014, 81, 245–252, DOI:10.1016/j.ejmech.2014.05.010.
- T. Shao, P. Yuan, L. Zhu, H. Xu, X. Li, S. He, P. Li, G. Wang and K. Chen, Carbon Nanoparticles Inhibit α-Glucosidase Activity and Induce a Hypoglycemic Effect in Diabetic Mice, Molecules, 2019, 24(18), 3257, DOI:10.3390/molecules24183257.
- M. Zhang, H. Wang, B. Wang, Y. Ma, H. Huang, Y. Liu, M. Shao, B. Yao and Z. Kang, Maltase Decorated by Chiral Carbon Dots with Inhibited Enzyme Activity for Glucose Level Control, Small, 2019, 15(48), 1–7, DOI:10.1002/smll.201901512.
- S. H. Cha, J. Hong, M. McGuffie, B. Yeom, J. S. Vanepps and N. A. Kotov, Shape-Dependent Biomimetic Inhibition of Enzyme by Nanoparticles and Their Antibacterial Activity, ACS Nano, 2015, 9(9), 9097–9105, DOI:10.1021/acsnano.5b03247.
- K. Saravanakumar, A. Sathiyaseelan, A. V. A. Mariadoss and M.-H. Wang, Antioxidant and Antidiabetic Properties of Biocompatible Ceria Oxide (CeO2) Nanoparticles in Mouse Fibroblast NIH3T3 and Insulin Resistant HepG2 Cells, Ceram. Int., 2021, 47(6), 8618–8626, DOI:10.1016/j.ceramint.2020.11.230.
- A. Sathiyaseelan, K. Saravanakumar, A. V. A. Mariadoss and M.-H. Wang, Biocompatible Fungal Chitosan Encapsulated Phytogenic Silver Nanoparticles Enhanced Antidiabetic, Antioxidant and Antibacterial Activity, Int. J. Biol. Macromol., 2020, 153, 63–71, DOI:10.1016/j.ijbiomac.2020.02.291.
- G. Chinnasamy, S. Chandrasekharan and S. Bhatnagar, Biosynthesis of Silver Nanoparticles from Melia Azedarach: Enhancement of Antibacterial, Wound Healing, Antidiabetic and Antioxidant Activities, Int. J. Nanomed., 2019, 14, 9823–9836, DOI:10.2147/IJN.S231340.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02474a |
| This journal is © The Royal Society of Chemistry 2023 |