
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into fourth generation selective inhibitors of (C797S) EGFR mutation combating non-small cell lung cancer resistance: a critical review
Mostafa A. Mansour a,
Asmaa M. AboulMagda,
Samar H. Abbasb,
Hamdy M. Abdel-Rahman*cd and
Mohamed Abdel-Azizb
a,
Asmaa M. AboulMagda,
Samar H. Abbasb,
Hamdy M. Abdel-Rahman*cd and
Mohamed Abdel-Azizb
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University in Beni-Suef (NUB), Beni-Suef 62513, Egypt
bMedicinal Chemistry Department, Faculty of Pharmacy, Minia University, Minia 61519, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt. E-mail: hamdym@aun.edu.eg
dPharmaceutical Chemistry Department, Faculty of Pharmacy, Badr University in Assiut (BUA), Assiut 2014101, Egypt
First published on 21st June 2023
Abstract
Lung cancer is the second most common cause of morbidity and mortality among cancer types worldwide, with non-small cell lung cancer (NSCLC) representing the majority of most cases. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) are among the most commonly used targeted therapy to treat NSCLC. Recent years have seen the evaluation of many synthetic EGFR TKIs, most of which showed therapeutic activity in pertinent models and were classified as first, second, and third-generation. The latest studies have concluded that their efficacy was also compromised by additional acquired mutations, including C797S. Because second- and third-generation EGFR TKIs are irreversible inhibitors, they are ineffective against C797S containing EGFR triple mutations (Del19/T790M/C797S and L858R/T790M/C797S). Therefore, there is an urgent unmet medical need to develop next-generation EGFR TKIs that selectively inhibit EGFR triple mutations via a non-irreversible mechanism. This review covers the fourth-generation EGFR-TKIs' most recent design with their essential binding interactions, the clinical difficulties, and the potential outcomes of treating patients with EGFR mutation C797S resistant to third-generation EGFR-TKIs was also discussed. Moreover, the utilization of various therapeutic strategies, including multi-targeting drugs and combination therapies, has also been reviewed.
1. Introduction
1.1 Overview
A broad spectrum of malignancies are included under the umbrella term “cancer”.1 Unrestricted cell growth and proliferation, as well as uncontrollable cell migration and organ/tissue spreading, are the major hallmark characteristics of these malignancies.2,3 A normal cell may become a tumor-affected cell due to the disruption of multiple regulatory, signal transduction, and apoptotic pathways by a number of different variables.4–6 In 2020, with more than 1.38 million deaths worldwide, lung cancer was the most frequent type of cancer that causes death, with non-small cell lung cancers (NSCLC) representing about 80% of lung cancer cases.7–9 Evenly NSCLC is reported to be less responsive to chemotherapy and radiation treatment compared to small cell lung cancers (SCLC).10,11 Chemotherapy has not significantly enhanced survival for the majority of lung cancer patients, and it is clear that chemotherapy's effectiveness in the treatment of NSCLC has reached a plateau.12 Targeted small molecules block signaling pathways that influence the development and advancement of cancer. As these pathways are preferentially activated in cancer cells compared to normal cells, targeted therapies are likely more tolerable than conventional cytotoxic agents.131.2 Epidermal growth factor receptor (EGFR) family and their inhibitors
Targeted therapy, a unique type of chemotherapy, is a helpful mechanism to stop cancer cells from growing and spreading by targeting specific genes or proteins.14,15 Advancing understanding of the cellular and molecular biology will improve chemists' abilities to target specific function in cancer cells. The trans-membrane glycoprotein known as the epidermal growth factor receptor (EGFR) is a cytoplasmic tyrosine kinase; its physiological function is to regulate the development and homeostasis of epithelial tissue.16,17 The cytoplasmic region of the kinase includes the kinase domain, which mediates the transfer of a phosphate group to the kinases with an ATP binding pocket found between the N- and C-lobes of the EGFR.18,19Epidermal growth factor (EGF) and transforming growth factor (TGF), the two ligands for the EGFR, cause the activation of dormant receptors through homo- or hetero-dimerization. Numerous downstream signaling cascades, including RAS/RAF/ERK, STAT, and PI3K/AKT/mTOR, are triggered by activation and auto-phosphorylation of the intracellular EGFR tyrosine kinase domain of receptors.20,21 This is followed by a cascade of processes in the cytoplasm, including cell growth and apoptosis inhibition, represented in (Fig. 1).
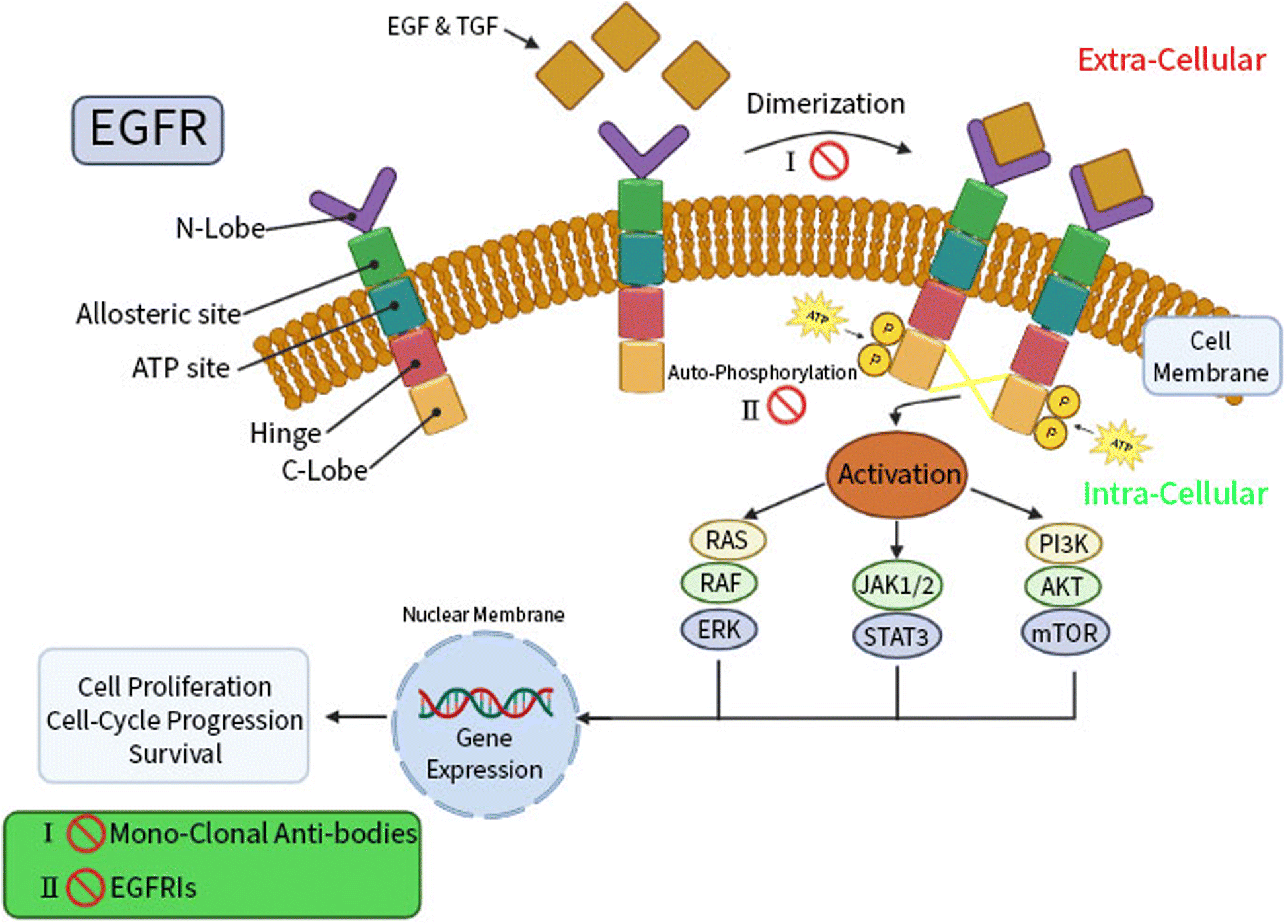 | ||
| Fig. 1 The EGFR structure, signaling pathways, and functions. | ||
EGFR is a crucial player in carcinogenesis and is highly expressed in breast and lung cancers.22,23 Accordingly, EGFR inhibitors are recommended by international guidelines as the first-line treatment in patients with advanced EGFR mutations in breast and lung cancers, regarding the higher efficacy and safety of these agents rather than standard chemotherapy.24 Two therapeutically effective pharmacological strategies for EGFR inhibition therapy are reported; monoclonal antibodies and small-molecule tyrosine kinase inhibitors (TKIs).25 In order to prevent ligand-induced EGFR tyrosine kinase activation, EGFR monoclonal antibodies can compete for binding to the inactive extracellular domain of EGFR receptors.26 In the same context, small-molecule EGFR TKIs exerted their mechanism of action via decreasing EGFR auto-phosphorylation and downstream signaling by reversibly competing with adenosine 5′ triphosphate (ATP) for binding to the intracellular tyrosine kinase domain of EGFR.27
It was reported that adenylylimidodiphophate (AMPPNP) is a form of ATP that cannot be hydrolyzed and linked to the wild-type EGFR (EGFRWT) according to the later crystal structure (PDB ID: 3VJO); thus, AMPPNP has to typical hydrogen bonds with Met793 and Gln791, this activates the downstream signaling and cascade of process in the cytoplasm include cell growth, and apoptosis inhibition28–31 (Fig. 2).
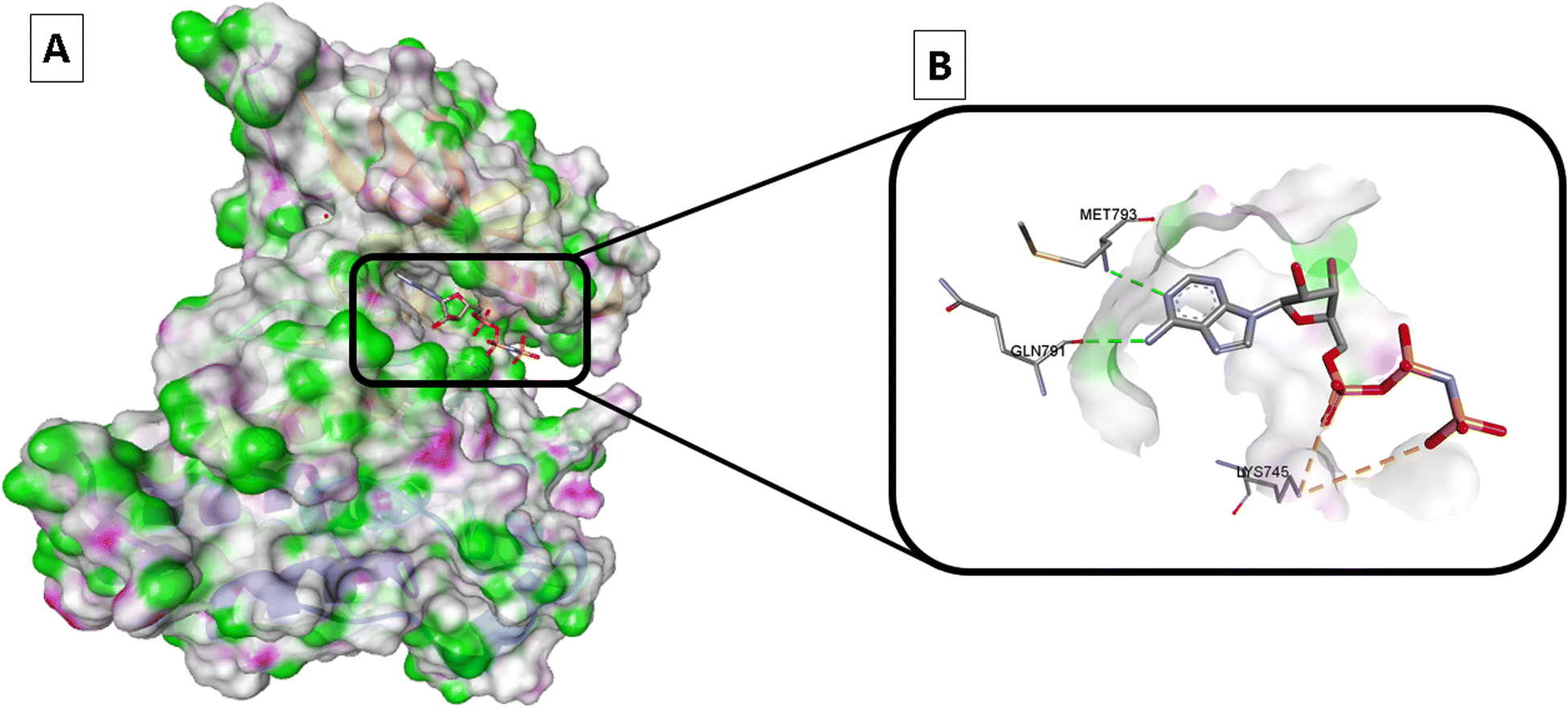 | ||
| Fig. 2 Wild-type EGFR bound to AMPPNP (PDB: 3VJO): (A) 3D binding mode of AMPPNP, receptor shown as hydrogen bond surface; (B) magnified 3D binding mode showing interactions with key residues; the figure was generated using BIOVIA Discovery Studio 2021. | ||
Since the development of EGFR TKIs for the treatment of NSCLC, hundreds of small molecules have been designed and evaluated as EGFR TKIs; these compounds are divided from first-to fourth-generation EGFR TKIs32,33 (Fig. 3).
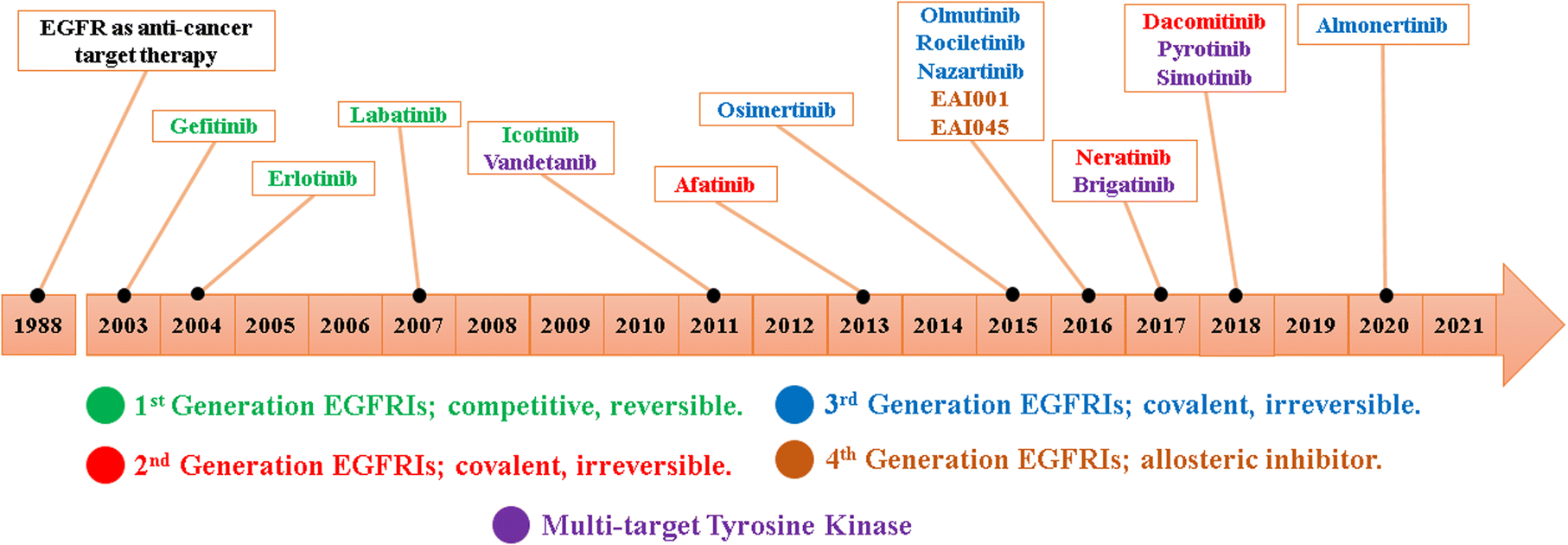 | ||
| Fig. 3 EGFR TKIs' development timeline. | ||
1.3 Role of EGFR-TKIs in NSCLC and their resistance mechanisms
EGFR plays an important role in the development and progression of lung cancer.34 The progression of lung cancer begins with abnormal cellular multiplication, which prompts normal cells to transition into malignant forms.35 In both NSCLC and SCLC, EGFR plays a significant role in the initiation and activation of signaling events. There are three different types of sub-categories incorporated within NSCLC: (i) adenocarcinomas, (ii) squamous cell carcinomas, and (iii) large cell carcinomas. Overexpression of EGFR has been found in all of these subgroups of NSCLC, with the greatest rates seen in squamous tumors (90%) and the lowest rates seen in adenocarcinomas (40%), and very little rates seen in large cell adenocarcinoma.36 According to the literature, EGFR has been shown to be a potential therapeutic target in the treatment of NSCLC. Targeting the suppression of EGFR function can be accomplished using of two primary methods: (i) the deactivation of intracellular kinase signaling through TKIs; and (ii) the neutralization of EGFR and ligands through the use of antibodies.37Nowadays, mutations in the EGFR gene and various resistance mechanisms have been extensively studied in patients with lung cancer.38 Both intrinsic (primary) and acquired (secondary) chemo-resistance reduce the response rate to EGFR TKIs therapy.39 Inappropriate EGFR activation in cancer is mainly caused by genomic locus amplification, and that leads to full mutation.40 The primary resistance mechanisms include point mutations in exon 18, deletions or insertions in exon 19, insertions, duplications and point mutations in exon 20 and exon 21 of EGFR gene, that mutation affected 4–10% of NSCLC patients.41 As a result of this primary mutation, NSCLC may develop primary innate resistance to reversible and irreversible EGFR-TKIs.42
Since the secondary mutation or amplification of the EGFR has been observed to increase in tumors under therapeutic pressure, it is also widely recognized as a biomarker of drug resistance.43 The secondary mutation is characterized by the ATP-binding cleft of EGFR containing the gatekeeper mutant T790M, which is associated with acquired resistance to first- and second-generation EGFR TKIs in 50% to 60% of NSCLC patients.44 The T790M mutation reduces the affinity of EGFR TKIs while it increases the affinity of ATP for the EGFR, even though it causes steric hindrance in the interaction of TKIs with EGFR.45 This enhances the ability of mutant NSCLC cells to survive by preventing EGFR inhibition by first- and second-generation EGFR-TKIs.46 The T790M mutation-mediated resistance to first- and second-generation EGFR-TKIs in NSCLC has been overcome by third-generation EGFR-TKIs, which have been developed and produced.46,47
The 797 location of the ATP binding pocket of the EGFR-tyrosine kinase domain is where the third-generation EGFR-TKIs can covalently engage with a cysteine residue.48 After some time, individuals using third-generation EGFR-TKIs begin to acquire heterogeneous resistance to these medications.49 Third-generation EGFR-TKI resistance is primarily caused by the appearance of a tertiary point mutation (C797S) in the ATP binding cleft. Deleting the secondary T790M mutation is another known factor in this secondary resistance.50,51
More recently, the fourth-generation of EGFR TK inhibitors has been introduced to the clinical evaluation to fight against EGFR tertiary mutation (C797S).52 The studies are devoted to the use of multi-target agents and combination therapy to overcome epigenetic mutation and boost the effectiveness of EGFR TK inhibitors.53 As well as the design and discovery of specific inhibitors targeting the mutant EGFR. In this regard, small molecule EGFR TKIs degraders have been evaluated to deal with these mutations in the tyrosine kinase domain of EGFR for combating resistance NSCLC.54
This review article aims to categorize the current EGFR TKIs and provides a summary based on their structures. Additionally, it highlighted how the most significant protein–drug interactions influenced selectivity that may encourage the development of more potent selective fourth-generation EGFR TKIs that can fight against EGFR tertiary mutation (C797S).
1.4 EGFR modulation pipeline to overcome EGFR genetic code mutations in NSCLC
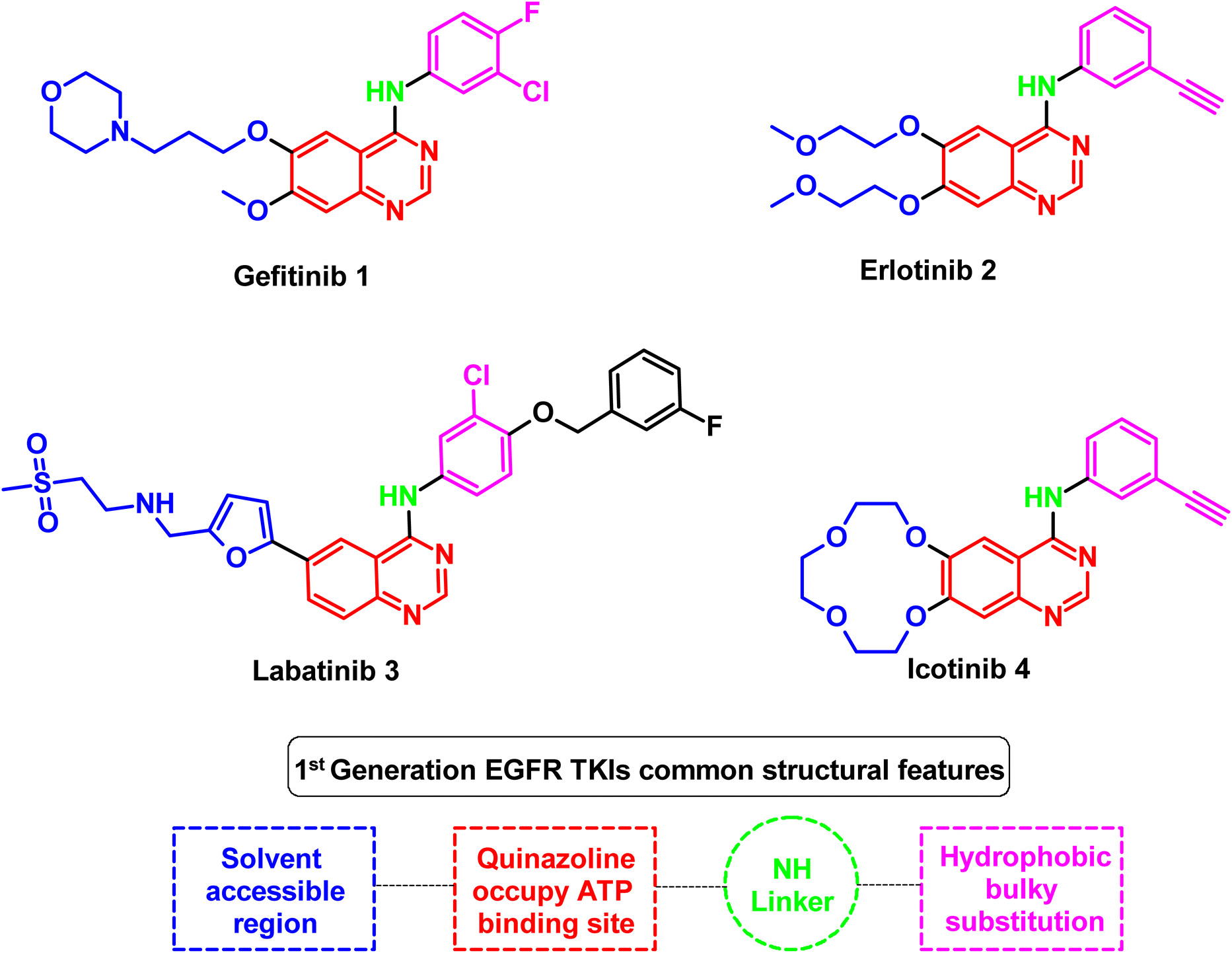 | ||
| Fig. 4 Representative molecules of 1st generation EGFR TKIs and their structural common features. | ||
As an example for the first-generation EGFR TKIs, gefitinib (1) formed strong hydrogen bond interaction with Met 769 located in the ATP binding site with N quinazoline moiety, which serves as hydrogen bond acceptor, as well as the substituted bulky hydrophobic group, occupied the hydrophobic pocket and the dialkyloxy moiety pointed forward the solvent accessible region, as illustrated in Fig. 5.60
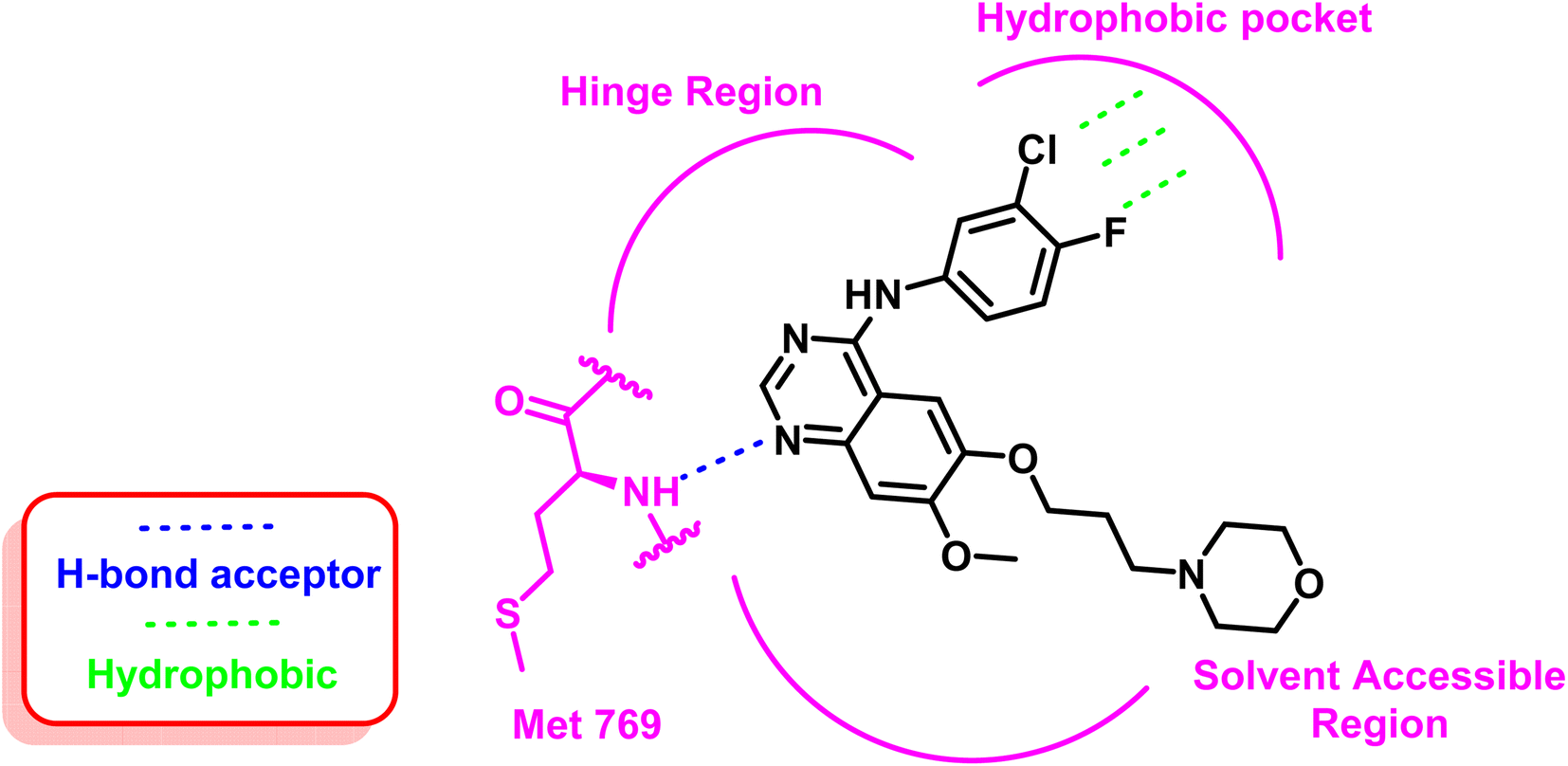 | ||
| Fig. 5 2D interaction of gefitinib (1) at the EGFR TK binding site. | ||
EGFR's kinase domain encoded by exons 18 to 24, with the ATP-binding pocket, is the site of more than 90% of kinase-activating mutations.61 Thus, NSCLC patients are thought to be the most likely to have tumor genome activation mutations, such as a single point replacement in exon 21 by the L858R and a deletion of exon 19 (delE746-A750) of the EGFR gene. EGFR TKIs require at least one single mutation (L858R and Del19) to be active against NSCLC.62
For treating patients with these mutations (Del19 and L858R), the first-generation EGFR TKIs (gefitinib (1) and erlotinib (2)) showed great sensitivity, with positive clinical responses ranging from 50%–80%.63 These mutations serve as prognostic indicators for treating NSCLC patients since they provide antitumor activity and more prolonged progression-free survival (PFS) in patients who received EGFR TKI therapy.64 After almost a year of treatment, patients responding to these molecules acquired secondary drug resistance, dramatically reducing the efficacy.
Different resistance mechanisms have been identified; in most cases, the gatekeeper T790M mutation, also known as the replacement of secondary threonine 790 with the bulkier methionine residue (T790M) in exon 20, is the most relevant one.65 Since the threonine 790 gatekeeper residue controlled the inhibitor's affinity to the ATP binding pocket, this mutation improved ATP's affinity for the EGFR active site, preventing EGFR inhibition by first-generation EGFR inhibitors.33 Subsequently, most clinical reports indicated that T790M accounted for half of the acquired resistant NSCLC cases to first-generation EGFR TKI.66
 | ||
| Fig. 6 Representative molecules of 2nd generation EGFR TKIs and their common structural features. | ||
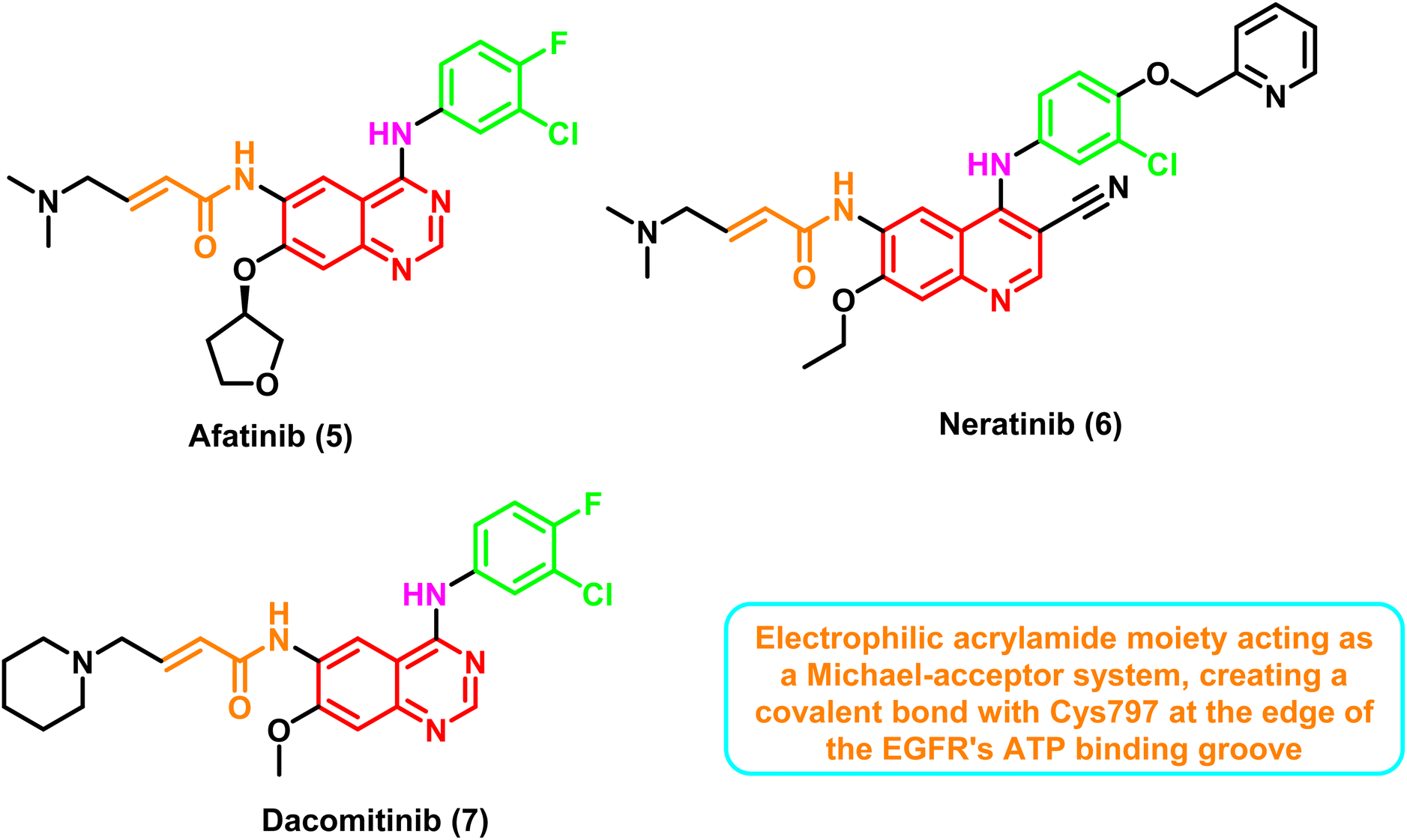
Inhibitors in this class are characterized by having an electrophilic acrylamide moiety acting as a Michael-acceptor system, creating a covalent bond with Cys797 at the edge of the EGFR's ATP binding groove.70 The binding of an aniline moiety to the back pocket of the ATP-binding domain raised the possibility of an interaction with the gatekeeper Met790 residue.
These inhibitors formed two strong hydrogen bond interactions at the ATP binding site towards Met769 and the mutated residue Met790 with the typical hydrophobic interaction to the hydrophobic pocket. Additionally, the important covalent interaction to Cys797 contains the active thiol group through the electrophilic acrylamide moiety, Fig. 7.71
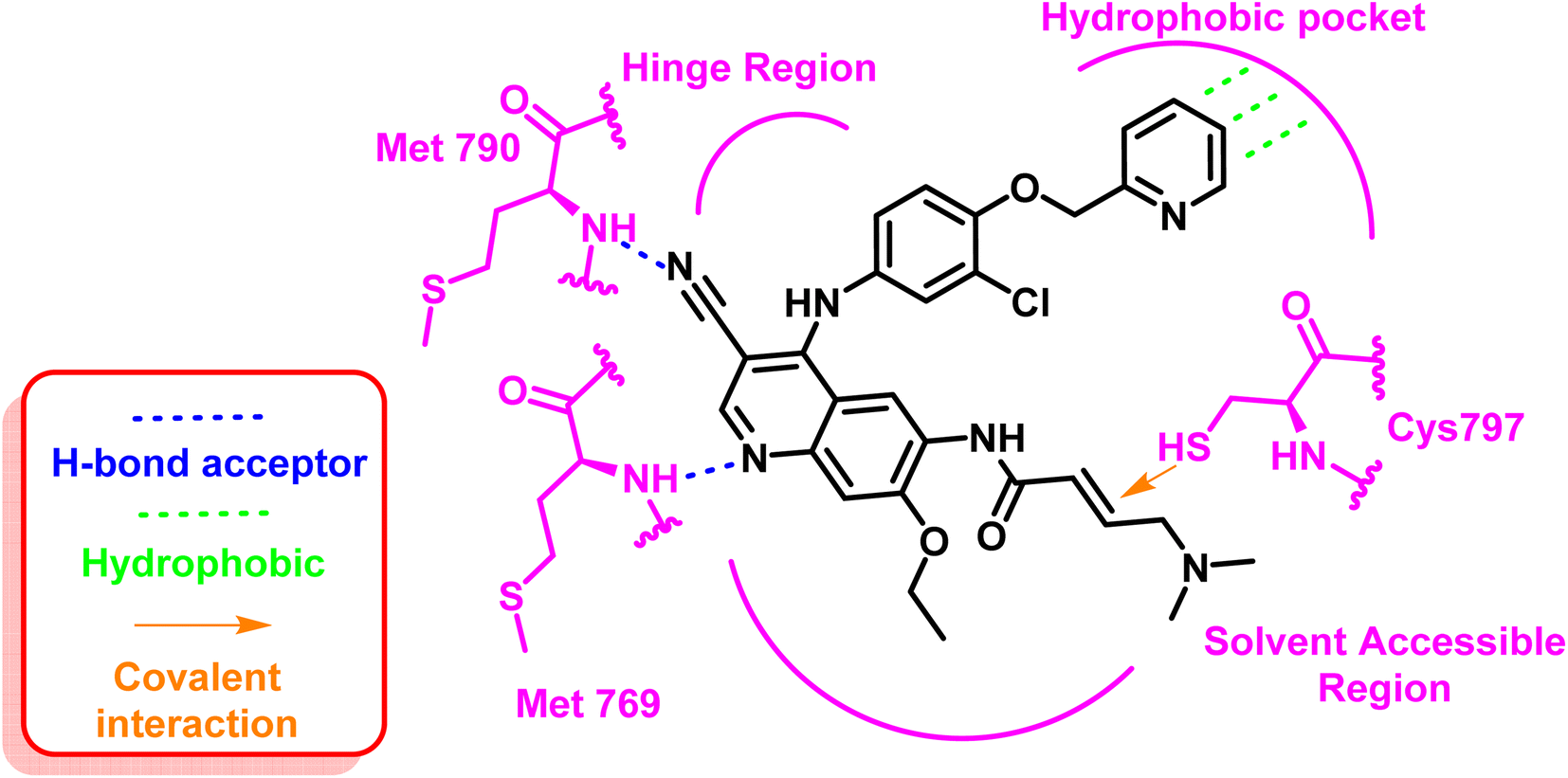 | ||
| Fig. 7 2D diagram of neratinib (6) at the binding site of mutant EGFRT790M TK. | ||
Patients with metastatic NSCLC whose tumors contain EGFR exon 19 deletion or exon 21 (L858R) mutation have just one first-line therapy approved to date: afatinib (5).72 Unfortunately, the majority of second-generation inhibitors had dose-limiting toxicity (skin rash and gastrointestinal) and an unacceptably low maximum tolerated dosage (MTD), which significantly restricted their therapeutic application.73 Targeting additional human epidermal growth factor receptors (HER-family members such as HER2) and the dose-limiting toxicity that results from attacking both mutant T790M and wild-type EGFR appear to be these drugs' two main clinical development obstacles.74
 | ||
| Fig. 8 Representative molecules of 3rd generation EGFR TKIs and their structural common features. | ||
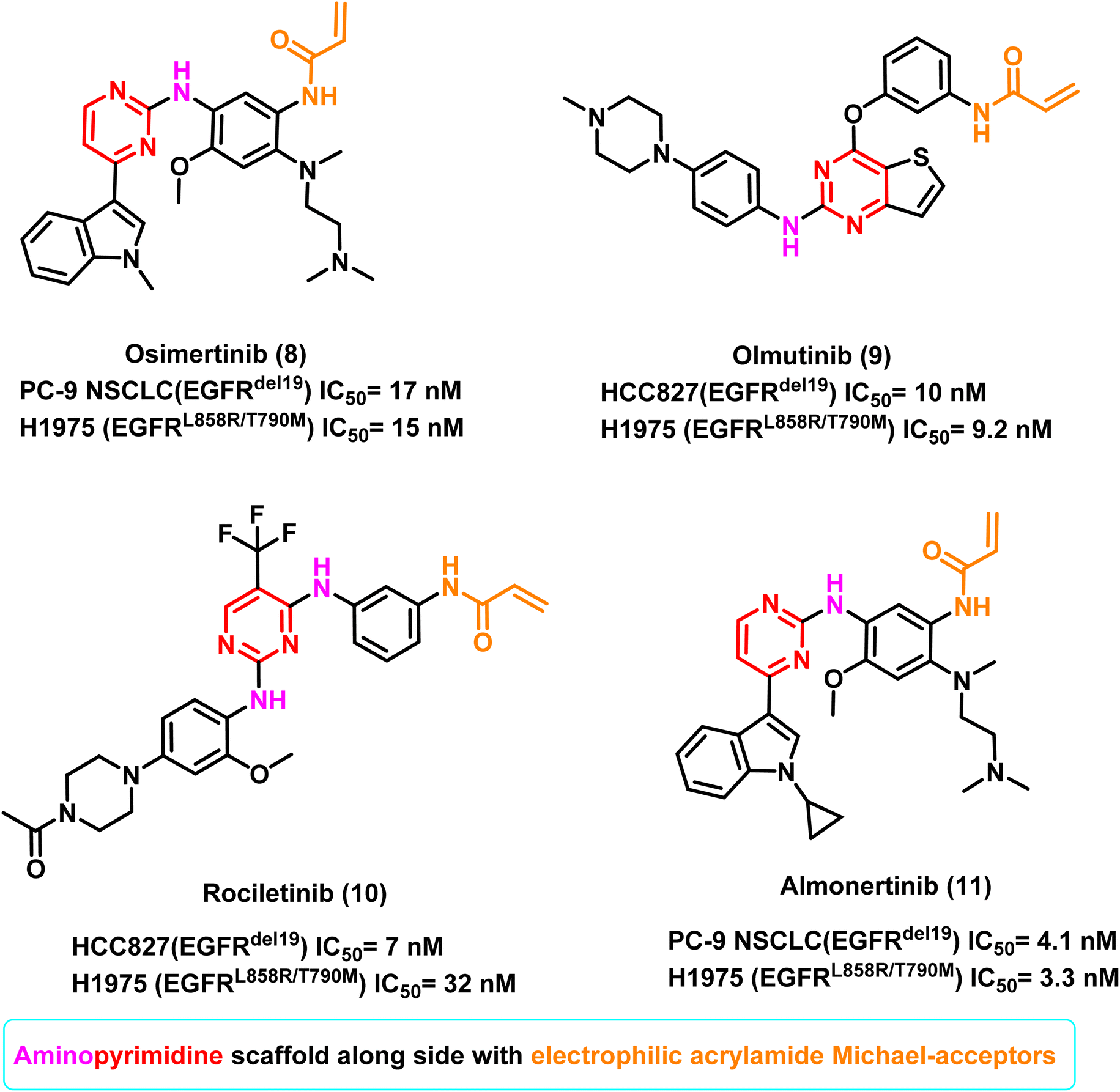
Osimertinib (8) is the only third-generation EGFR TKI with great effectiveness and few side effects, and it is the only drug approved by FDA for treating NSCLC patients with the T790M mutation.78,79 It inhibited the proliferation of PC-9 NSCLC (EGFRdel19)cells and H1975 (EGFRL858R/T790M) cells selectively, with IC50 of 17 nM and 15 nM, respectively. It also substantially inhibited tumour regressions in both PC9 and H1975 tumour xenograft models, resulting in FDA approval in 2015 for treating NSCLC patients with an EGFRT790M positive mutation.78,79
Moreover, olmutinib (9) is a thieno[3,2-d]pyrimidine-based compound which demonstrated potent inhibitory activity against H1975 (EGFRL858R/T790M) and HCC827 (EGFRdel19) cells, with IC50 values of 9.2 nM and 10 nM, respectively. It was introduced in 2016 and was used to treat patients with locally progressed or metastatic EGFRT790M positive NSCLC.79
Furthermore, in an enzymatic assay, rociletinib (10), a selective mutant EGFRL858R/T790M inhibitor, showed little activity against EGFRwt. It inhibited the proliferation of H1975 (EGFRL858R/T790M) and HCC827 (EGFRdel19) cells with IC50 values of 32 and 7 nM, respectively. It exhibited a 17-fold greater selectivity against H1975 (carrying EGFRL858R/T790M) than A431 cells (EGFRwt, IC50 value of 547 nM against A431).80 Almonertinib (11) bears a cyclopropyl group in place of the methyl group of osimertinib (8), which improves its in vitro activity. It inhibits EGFR phosphorylation with IC50 values of 3.3 nM in the HCC827 (EGFRdel19) cell line, 4.1 nM in the PC9 (EGFRdel19) cell line, 3.3 nM in the NCI-H1975 (EGFRL858R/T790M) cell line, and 596.6 nM in A431 (EGFRWT) cell line.81
Multiple mechanisms for the occurrence of resistance in third-generation EGFR TKIs have been identified through biomarker studies and mutation analysis techniques; the C797S mutation is regarded as the primary mechanism of resistance in all third-generation EGFR TKIs.82 Changing the nucleophilic cysteine C797 to the less reactive serine S797 in exon 20 of the EGFR tyrosine kinase domain is regarded as the most significant mechanism of mutation in biopsy samples. This mutation precluded the formation of a covalent bond between the inhibitors and the cysteine residue located at position 797 in the ATP-binding pocket's edge.83 Two additional mutations, EGFRL718Q and EGFRL844V, led to resistance to the third-generation of EGFR TKIs.84
Because of these limitations, there is an immediate need for new EGFR TKIs that can successfully suppress mutated EGFR and target acquired resistance using alternative binding mechanisms that are located away from the ATP binding site.
The fourth-generation EGFR-TKIs were required due to the frequently acquired C797S mutation. According to reports, EAI001 (12) and EAI045 (13) are fourth-generation EGFR allosteric kinase inhibitors that have therapeutic effects despite the C797S mutation.86 Due to their unique binding locations with the target, allosteric kinase inhibitors often function as a potential complementary therapeutic method to ATP-competitive kinase inhibitors. It doesn't bind to Cys797 because its residue is outside the allosteric binding pocket87,88 (Fig. 9).
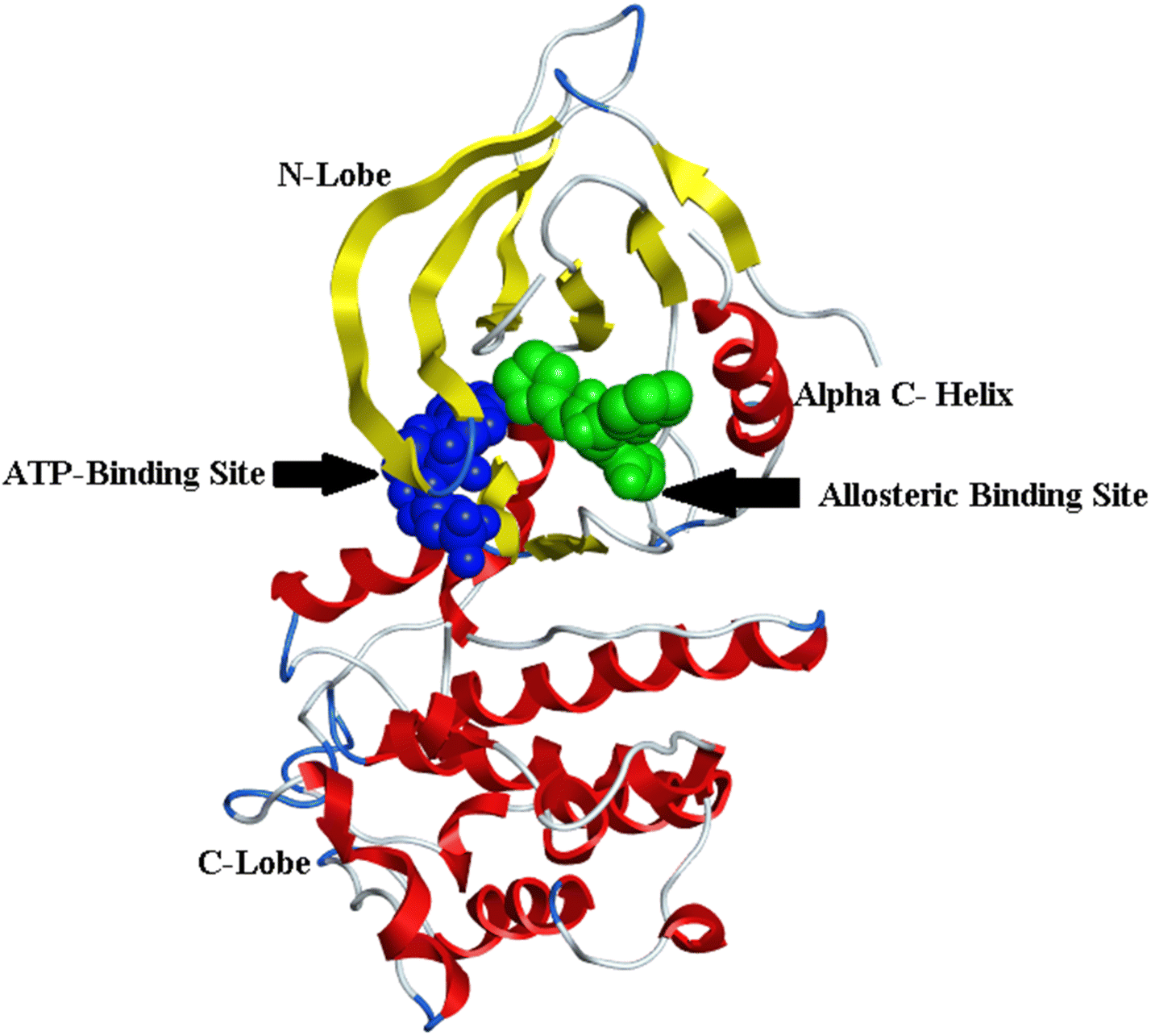 | ||
| Fig. 9 Overall view of the structure of EGFRL858R/T790M/C797S showed the ATP binding site in blue color and the allosteric binding site in green, the figure was generated using the crystal structure of mutant receptor (PDB ID: 6LUB) and BIOVIA Discovery Studio 2021. | ||
2. Development of EGFR TKIs against C797S mutation
Fourth-generation EGFR TKIs are allosteric kinase inhibitors that frequently act as a potential complementary therapeutic approach to ATP-competitive kinase inhibitors because of their distinct binding sites with the target. High-throughput screening (HTS) technique is used to select the prototype fourth-generation EGFR TKI oxoisoindoline phenyl-acetamide derivative EAI001 (12) from a library of 2.5 million compounds at a concentration of 1 μM ATP with EGFRL858R/T790M IC50 of 24 nM. EAI0045 (13) is also an allosteric, non-ATP competitive inhibitor of mutant EGFR (EGFRL858R/T790M IC50 = 3 nM) developed through further phenyl ring modification of EAI001 (12)87 (Fig. 10).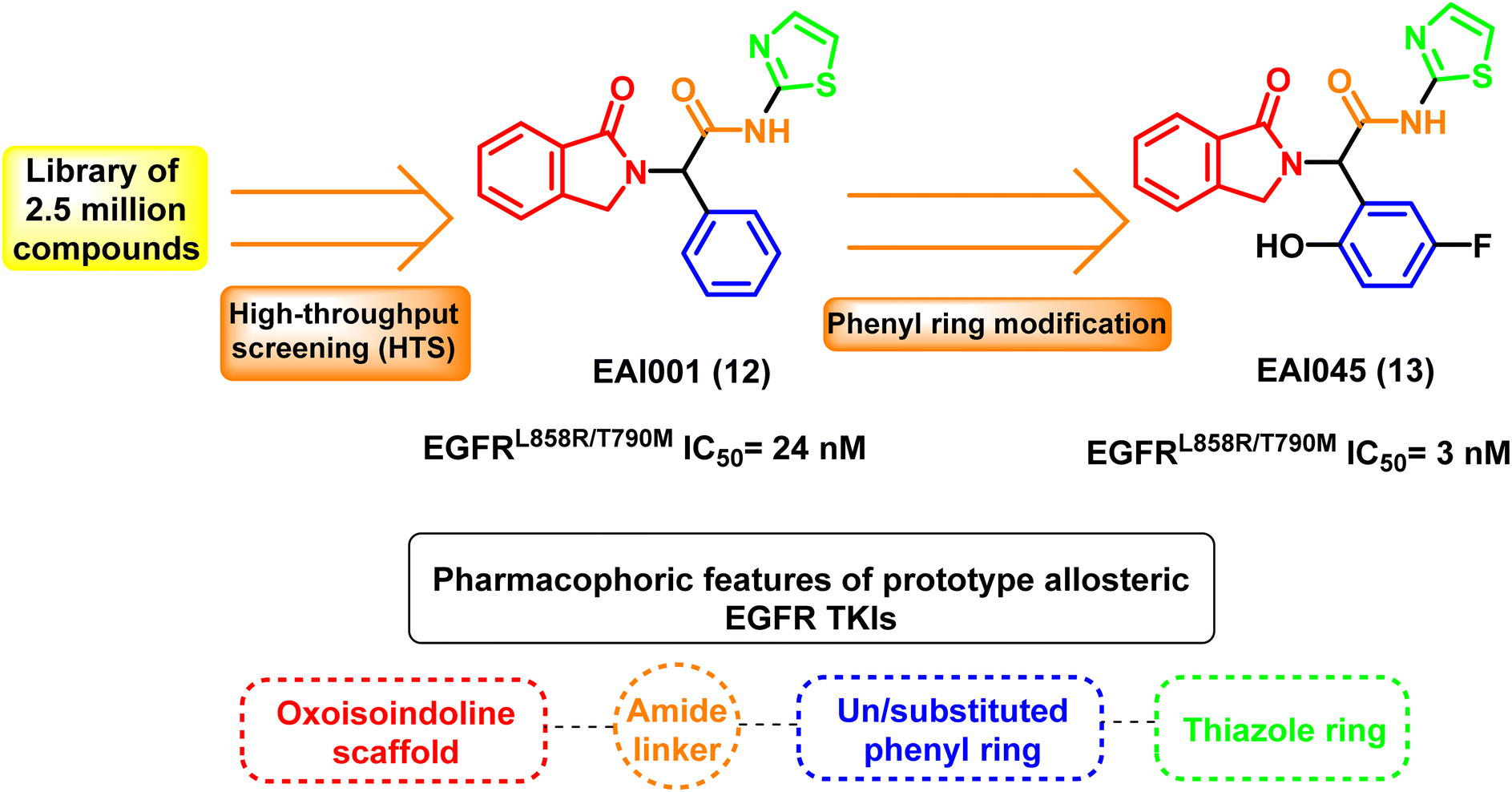 | ||
| Fig. 10 Prototype allosteric EGFR TKIs and their common pharmcophoric features. | ||
The X-ray crystal structure between the molecule and the mutant EGFRT790M reveals that EAI001 (12) is linked to the allosteric area adjacent to the conventional ATP binding site, resulting in an inactive conformation of the alpha C-helix. Additionally, the selectivity of the mutant EGFRT790M/C797S, compared to the EGFRWT, was supported by direct contact in a “Y-shaped” arrangement between the aminothiazole group of EAI001 (12) and the gatekeeper Met790 residue.87
Early investigations into the cellular function of EAI045 (13) revealed that it potently reduced, but did not entirely eradicate, EGFR auto-phosphorylation in H1975 cells, an NSCLC cell line with the mutation L858R/T790M. In addition, stably transfected NIH-3T3 cells with mutant EGFRL858R/T790M showed a better outcome.89 Additionally, EAI045 (13) succeed in suppressing EGFR Y1173 phosphorylation in H1975 cells with half maximal effective concentration (EC50) equal to 2 nM, which is specific for mutant EGFR.90
The modified analog EAI045 (13) is attached to the kinase in its “C-helix out” state. The asymmetric dimer interaction prevents the C-helix from moving outside of the receiver component but not the activator.91 Therefore, it is suggested that EAI045 (13) was a powerful inhibitor of the mutant receptor's activator subunit but a much weaker inhibitor of the receiver subunit, which contains the confined C-helix.91 This result could explain both the insufficient suppression of EGFR auto-phosphorylation and the apparent discrepancy between the biochemical and cellular potencies of the allosteric inhibitor because the mutant receptor favors dimer formation.
Further research verified that the inability of EAI045 (13) to access the two subunits of the asymmetric dimer of EGFR kinase is the cause of its low clinical efficacy.92 However, its success in the design modification strategy provides a new avenue for allosteric inhibitor research in the future. The urgent need for new small molecules capable of inhibiting EGFR mutations through non-reversible inhibition of double mutant EGFRT790M/C797S has prompted researchers to seek out new EGFR allosteric inhibitors that will be discussed in the following sections:
2.1 Purines based derivatives
A group of 8-phenylamino-9H-purine compounds had been reported as potent and irreversible EGFR inhibitors by Y. Hei and his coworkers. Compound (14) was the first one of these chemotype derivatives to inhibit EGFRL858R/T790M/C797S, exhibiting moderate inhibitory activity (IC50 = 114 nM) against this mutant enzyme.93 In 2020, H. Lei and his colleagues created a series of 9-heterocyclyl substituted 9H-purine derivatives, which serve as EGFRL858R/T790M/C797S TKIs with a fruitful modification that can be summarized in the following points: (i) maintaining the purine as a main scaffold, (ii) replacing the sulphide linker by –NH– linker to afford a hydrogen bond donor moiety, (iii) adding an extra hydrogen bond acceptor moiety and finally, (iv) cyclization of terminal acyclic aliphatic nitrogen containing moiety to piperazine one. Among these compounds, compounds (15) and (16) exhibited greatly anti-proliferative activities and mutant EGFRL858R/T790M/C797S inhibitory activities with IC50 equal to 22 nM and 18 nM, respectively94 (Fig. 11).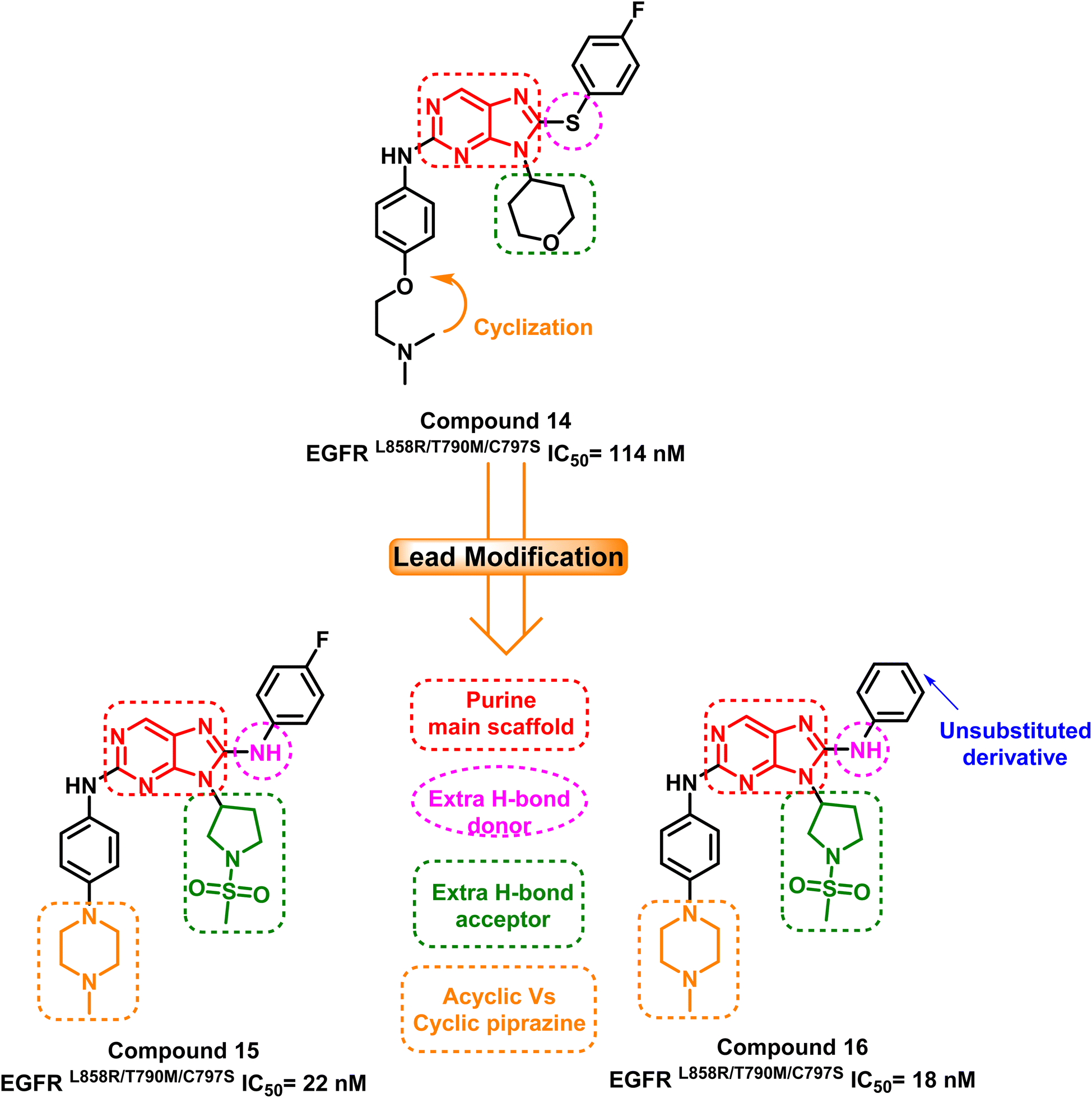 | ||
| Fig. 11 The optimization process of purine-based inhibitors 15, 16 using compound (14) as lead compound. | ||
Compound (16) showed potent anti-proliferative effects against both the HCC827 and H1975 cell lines, significantly suppress the mutant EGFR phosphorylation and induced cell apoptosis.94 The authors believed that two hydrogen bond interactions were formed with Met793 in the hinge region between the nitrogen atom at position C-1 and the hydrogen atom from amino at position C-2 of the 9H-purine scaffold which are responsible for its activity. Also an extra–hydrogen bond interaction was observed with Thr854 when the –NH– linker replaced the –S– linker in the previously reported compound (14).
Additionally, adding a methylsulfonyl pyrrolidine group adds increases the number of hydrogen bond acceptor moieties to the mutant Ser797 residue. These findings could explain why compound (16) exhibited potent enzyme inhibitory activity with mutant EGFRL858R/T790M/C797S. Therefore, adding an additional hydrogen bond interaction with mutant Ser797 can be considered an effective design strategy for fourth-generation EGFR-TKIs (Fig. 12).
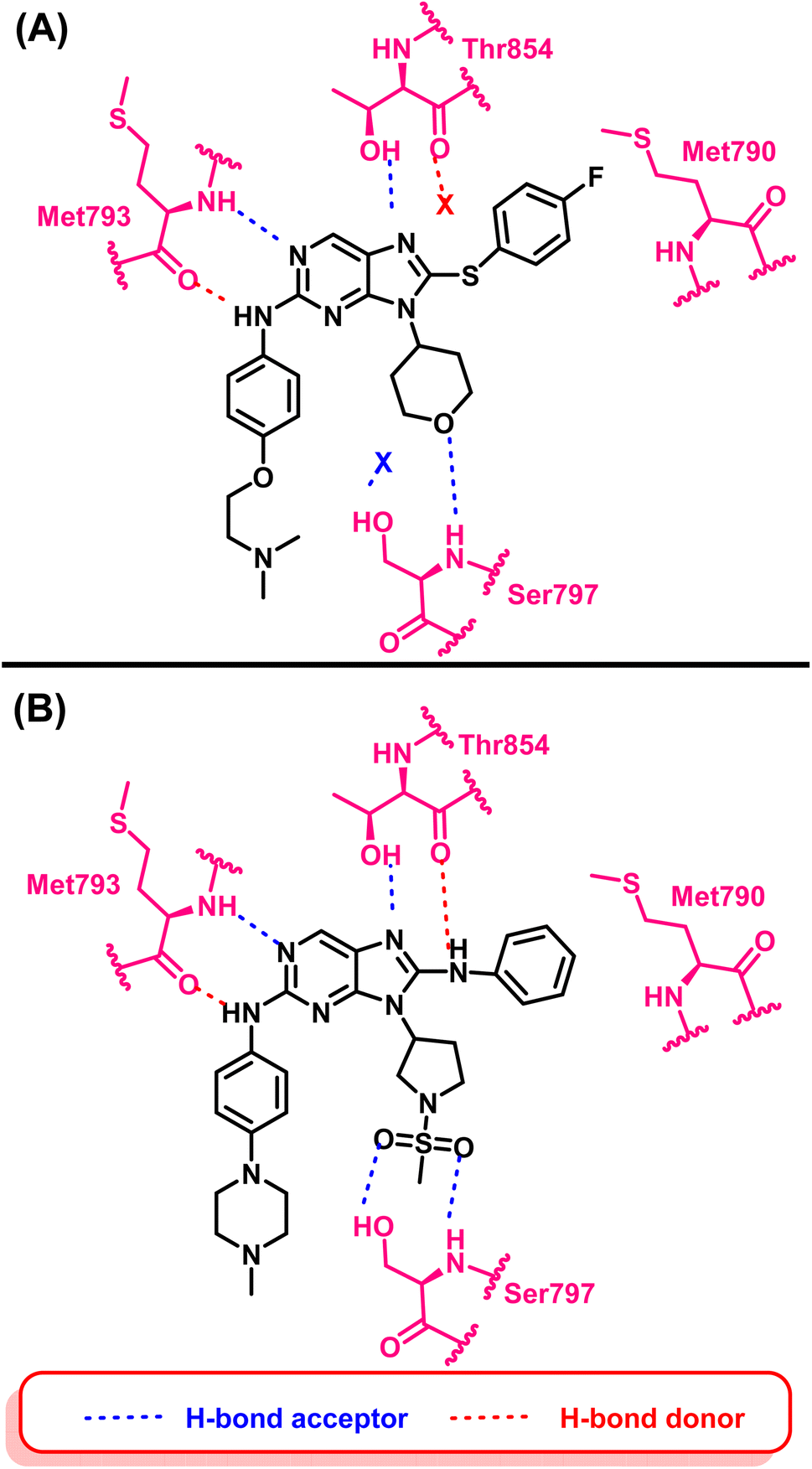 | ||
| Fig. 12 (A) 2D interactions of compound (14) with the active site of mutant EGFRL858R/T790M/C797S TK, (B) 2D interactions of compound (16) with active site of mutant EGFRL858R/T790M/C797S TK. | ||
2.2 Pyrimidine based derivatives
Z. Su and his research associates designed and synthesized a variety of pyrimidine-indole derivatives with the terminal hydroxyl of the alkyl chain to expand an extra binding interaction with the Asp855 in the conservative DFG site to overcome the resistance mutation 19D/T790M/C797S. Compound (17) stood out among them due to its notable inhibitory effect against EGFR19D/T790M/C797S (IC50 = 15.3 nM) and high selectivity over EGFRWT (IC50 > 1000 nM).95The 2D interaction diagram explained the good inhibitory effect for compound (17); it binds to Lys745 and Asp855 located in the allosteric binding site of the EGFR TK as well as aminopyrimidine scaffold forms a bidentate formation with Met793 residue and the indole moiety occupied the hydrophobic back pocket95 (Fig. 13).
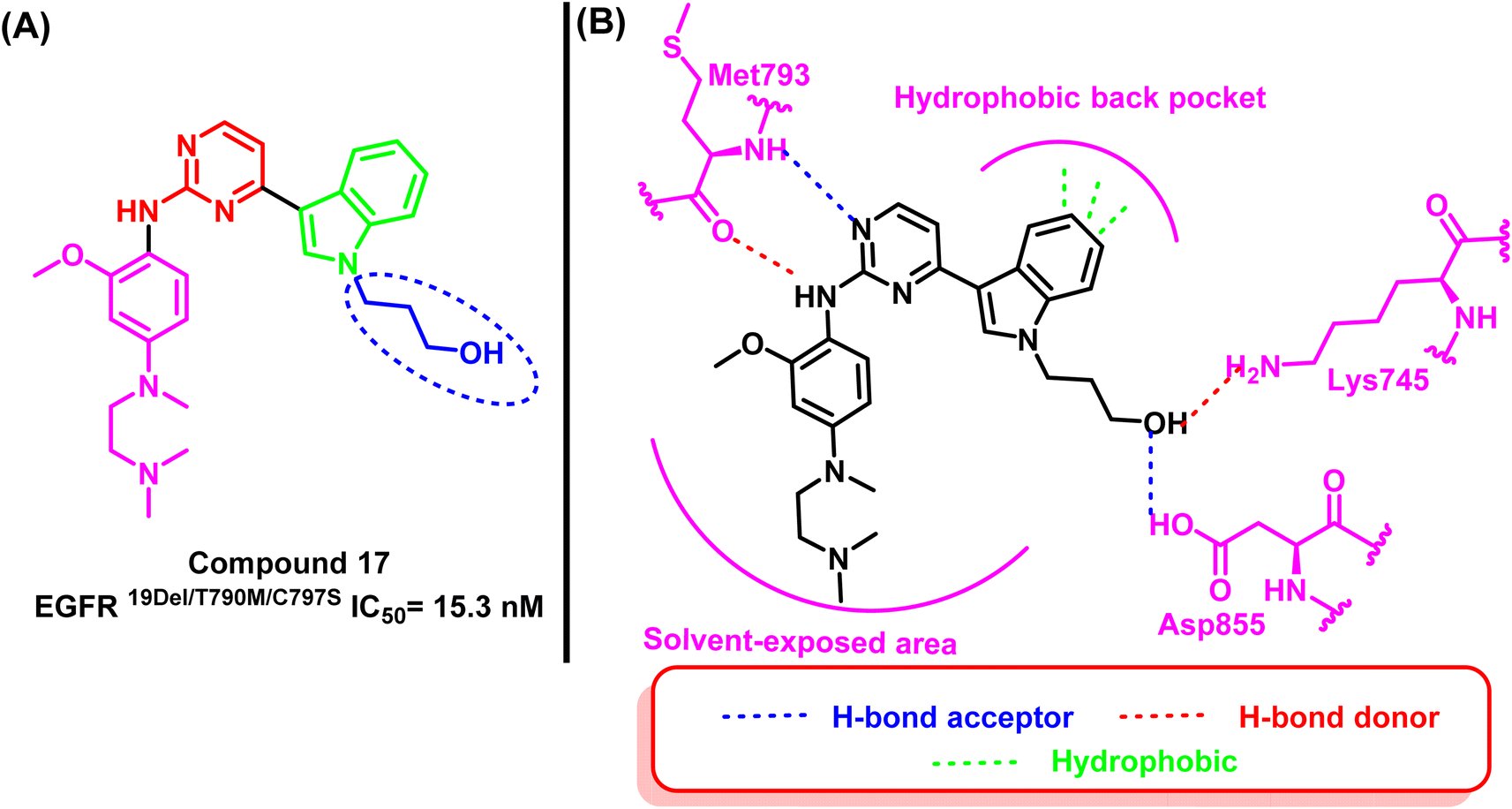 | ||
| Fig. 13 (A) Compound (17), (B) 2D interactions of compound (17) with active site of mutant EGFRL858R/T790M/C797S TK. | ||
Additionally, compounds (18) and (19) were developed as representative of a novel category of N2,N4-diphenylpyrimidine-2,4-diamine derivatives bearing amino/methyl sulfonyl moieties (Fig. 14). Both compounds exhibited a potent inhibitory concentration (IC50) of 0.2 nM for the mutant EGFRdel19/T790M/C797S 96. These compounds retain the aminopyrimidine scaffold to form a bidentate interaction with Met793 in addition to amino/methyl sulphonyl moiety as a strong hydrogen bond acceptor that bound to Lys745 and Asp855 residues (Fig. 15). So these findings offered interesting prospects for further research and development for new candidates served as fourth-generation EGFR inhibitors.
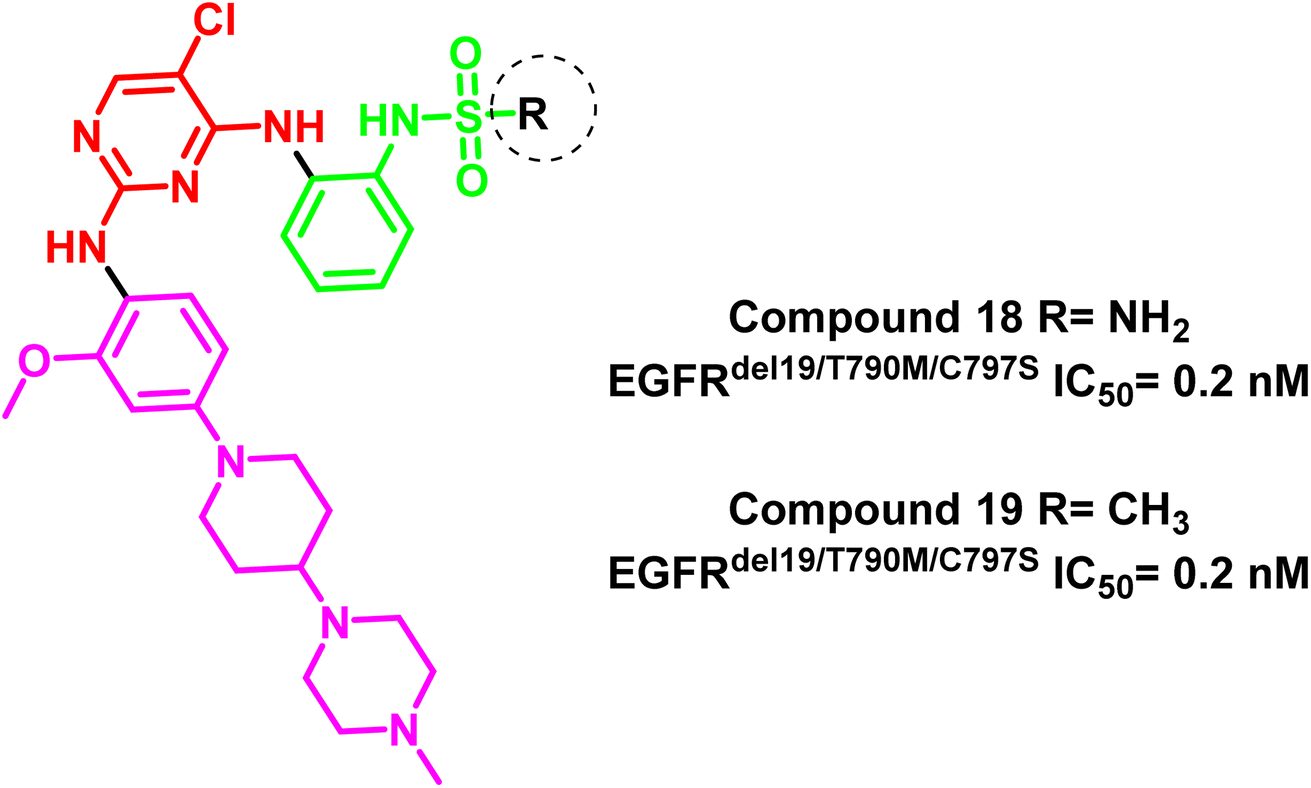 | ||
| Fig. 14 The amino pyrimidine compounds (18) and (19). | ||
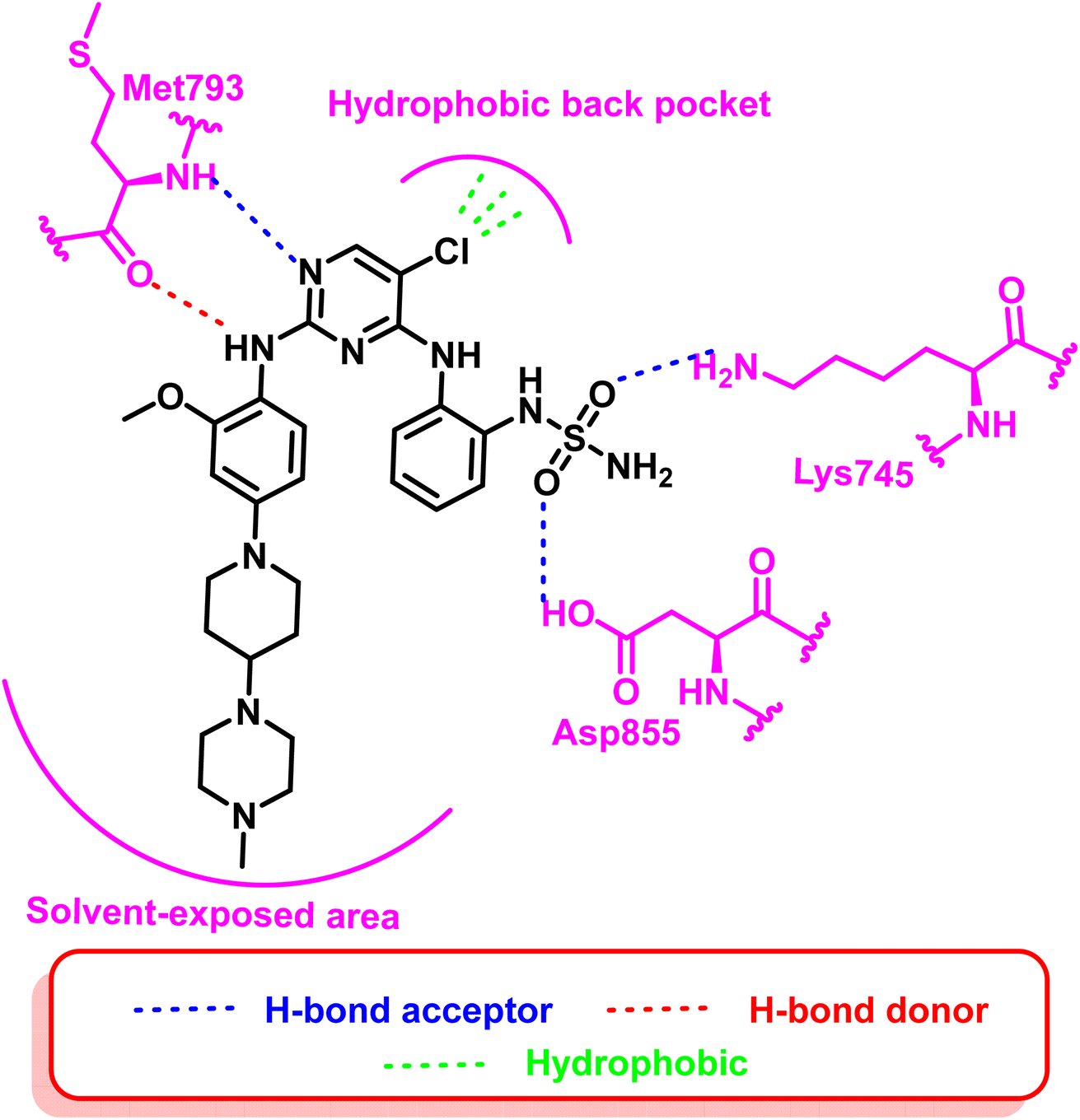 | ||
| Fig. 15 2D diagram of compound (18) at the active site of mutated EGFR TK. | ||
Furthermore, a series of WZ4002 (20) (a third-generation EGFR TKI) optimized analogs have been developed, and their antiproliferative effects on various cell lines and their EGFR kinase inhibitory efficacy against six different EGFR kinases have been evaluated. At a concentration of 10 μM, the ortho-hydroxyacetamide derivative (21) completely inhibited all six kinases, including the mutant EGFRL858R/T790M/C797S, with 99% inhibition97 (Fig. 16).
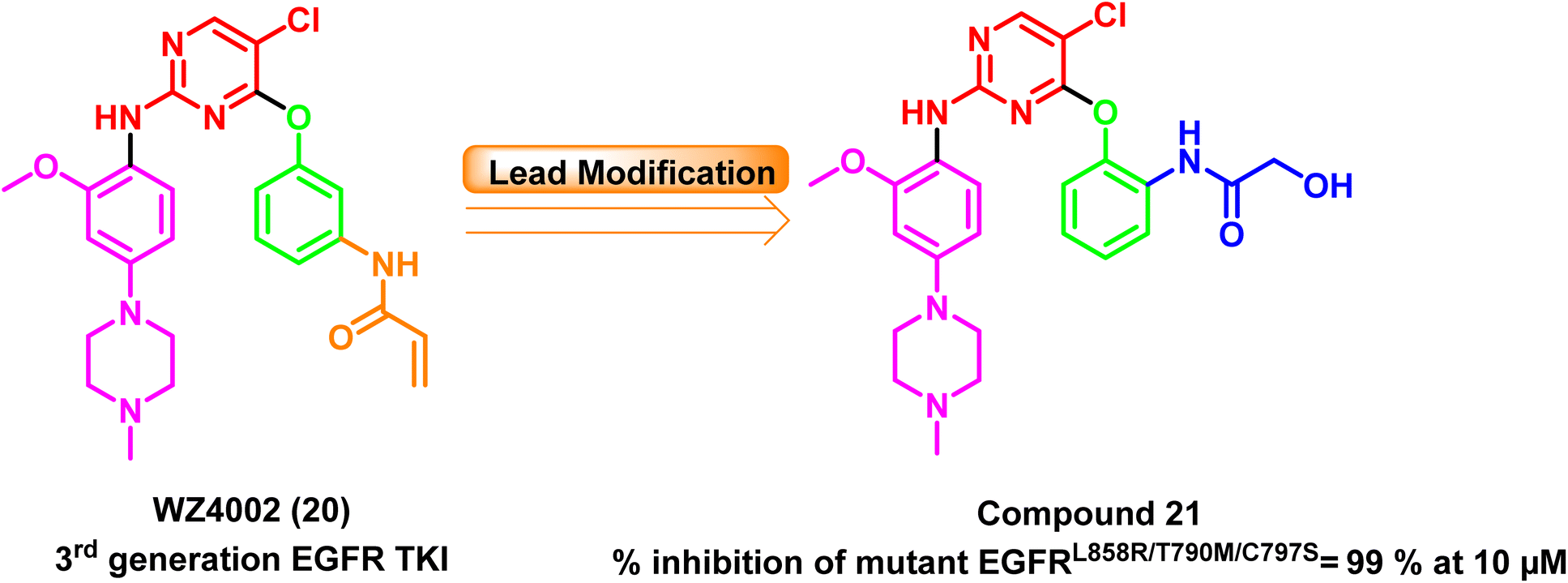 | ||
| Fig. 16 Optimization process for compound (21) from WZ4002 (20). | ||
2.3 Pyrrolopyrimidine/pyrimidopyrimidine/pyridopyrmidine derivatives
Merging the pyrimidine ring with another nitrogen-containing five or six-membered ring showed a good progression in designing the fourth-generation EGFR TKIs. Jonas' research team analyzed the X-ray structure of the T790M mutant EGFR TK to develop the lead compound (22), which inhibits mutant EGFRL858R/T790M with an IC50 of 176 nM.98 Lead optimization of compound (22) via O-alkyl substitution variation and addition of methyl piperazine moiety resulted in the synthesis of pyrrolopyrimidines (23–25) as potent EGFRL858R/T790M/C797S inhibitors (Fig. 17).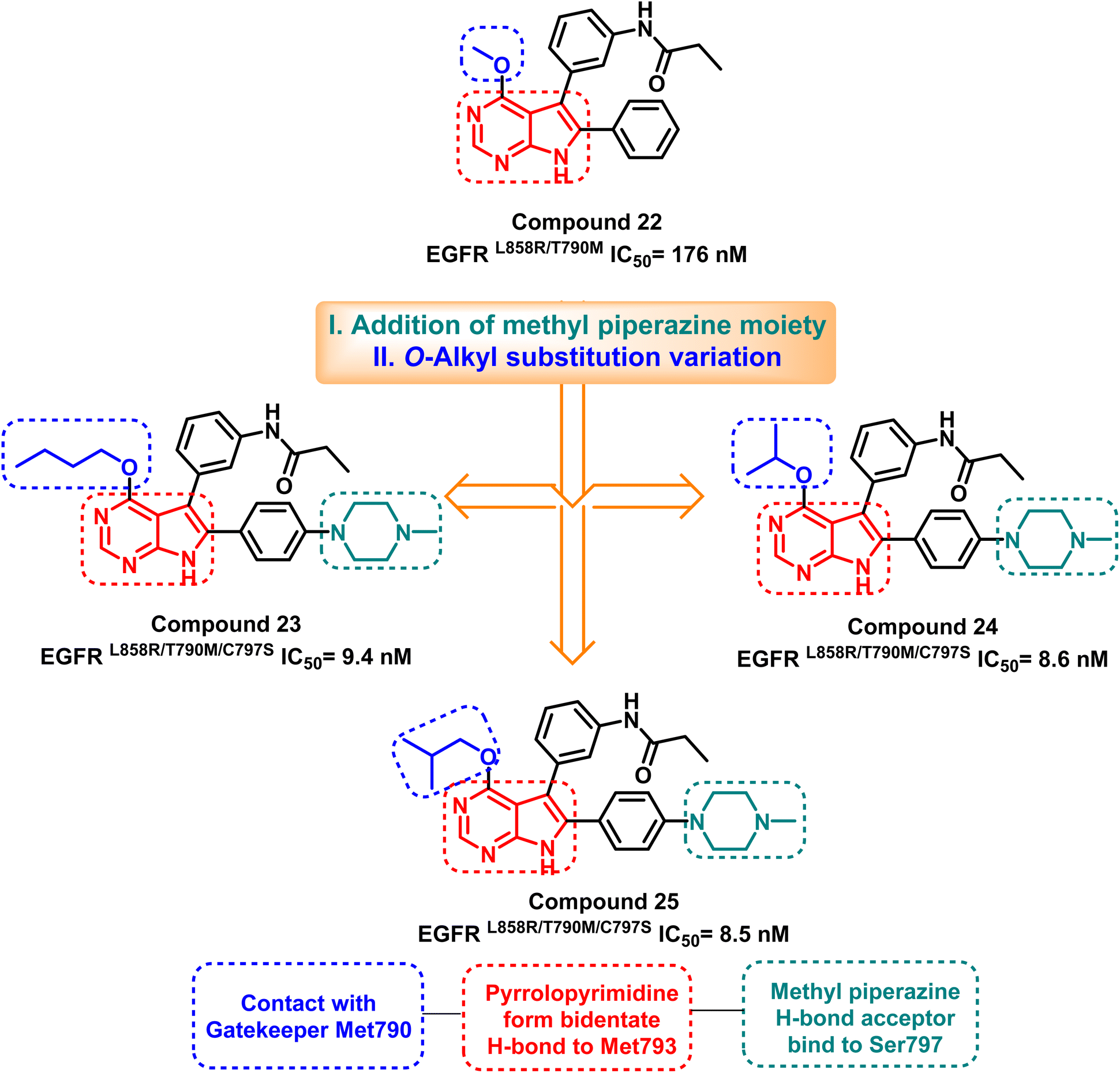 | ||
| Fig. 17 The optimization process of compounds (23–25) from compound (22) and their basic pharmacophore and binding residues. | ||
These compounds exhibited a high level of efficacy, around 9 nM against the resistant EGFRL858R/T790M/C797S. Molecular docking analysis showed the core pyrrolopyrimidine of these compounds could form a bidentate hydrogen bond with Met793, while the addition of piperazine moiety provides interaction with mutant Ser797. As well as, O-alkyl substitution with a bigger contact surface with the gatekeeper Met790 as in compound (25) having the isobutyl moiety showed relatively potent activity, with an IC50 of 8.5 nM (ref. 98) (Fig. 18).
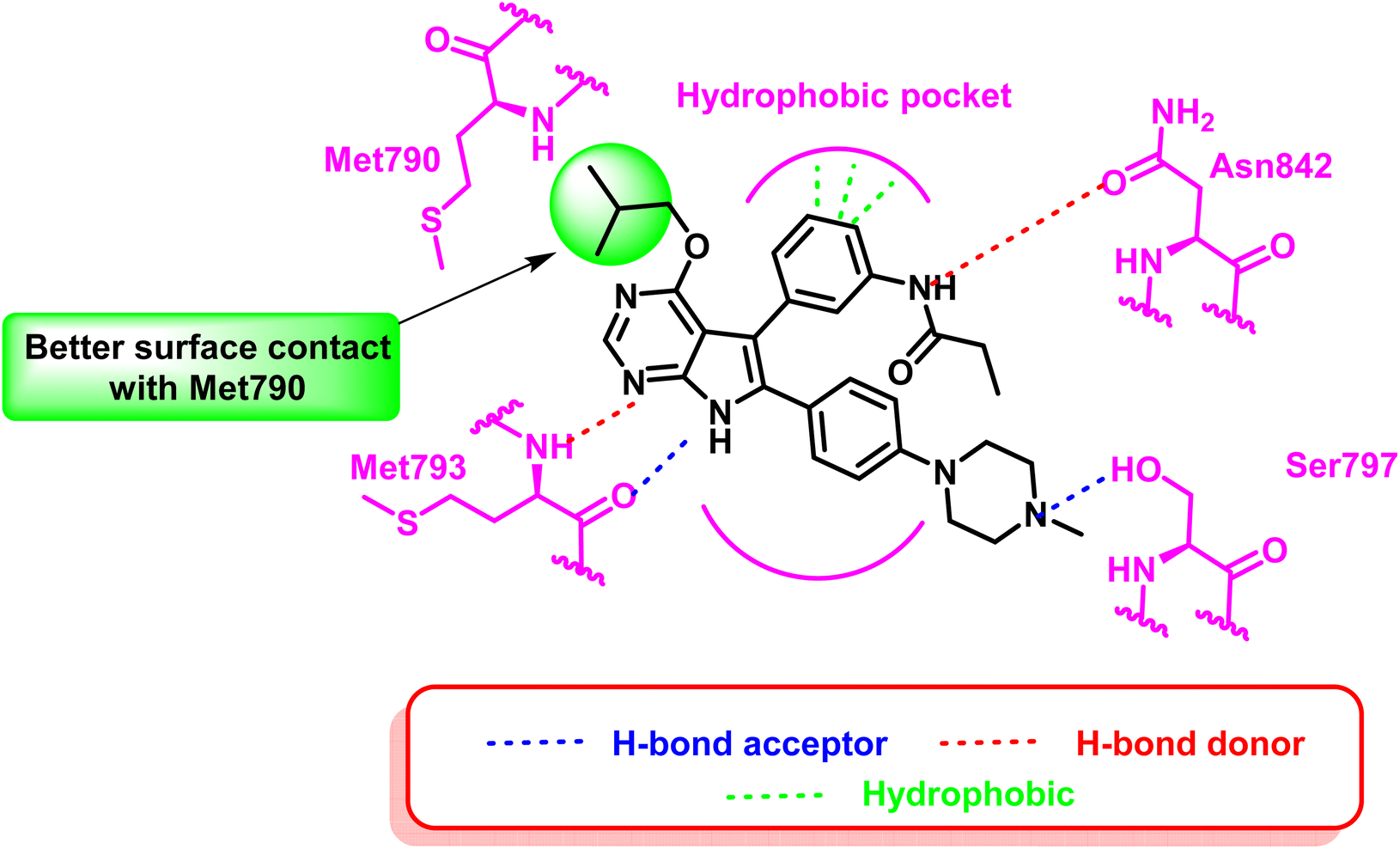 | ||
| Fig. 18 2D diagram of compound (25) at the binding site of mutant EGFRL858R/T790M/C797S TK. | ||
On the other hand, pyrimidopyrimidinone derivatives (26) and (27) were reported by Lu et al. to be effective as EGFRC797S inhibitors. These compounds were found by a random screening of a kinase inhibitor library that contained about 3000 different compounds.99–101 Compound (26) showed significant activity of EGFRL858R/T790M/C797S with an IC50 value of 5.8 nM. As well as compound (27) showed a potent inhibitory effect in the enzyme-linked immunosorbent assay (ELISA) against the kinase activity of EGFRL858R/T790M/C797S with an IC50 value of 3.1 nM. The “hinge” residue Met793 of the protein and the dihydropyrimido[4,5-d]pyrimidinone nucleus interact via a bidentate hydrogen bond. The 2-chlorophenyl group is located in a hydrophobic back pocket that contains numerous significant amino acid residues. Meanwhile, a hydrogen bond is formed between the nitrogen of Lys745 and the carbonyl of the main scaffold. Also, the propionamide group extended in the solvent-exposed region, and the NH moiety formed a hydrogen bond with Leu718 (ref. 101) (Fig. 19).
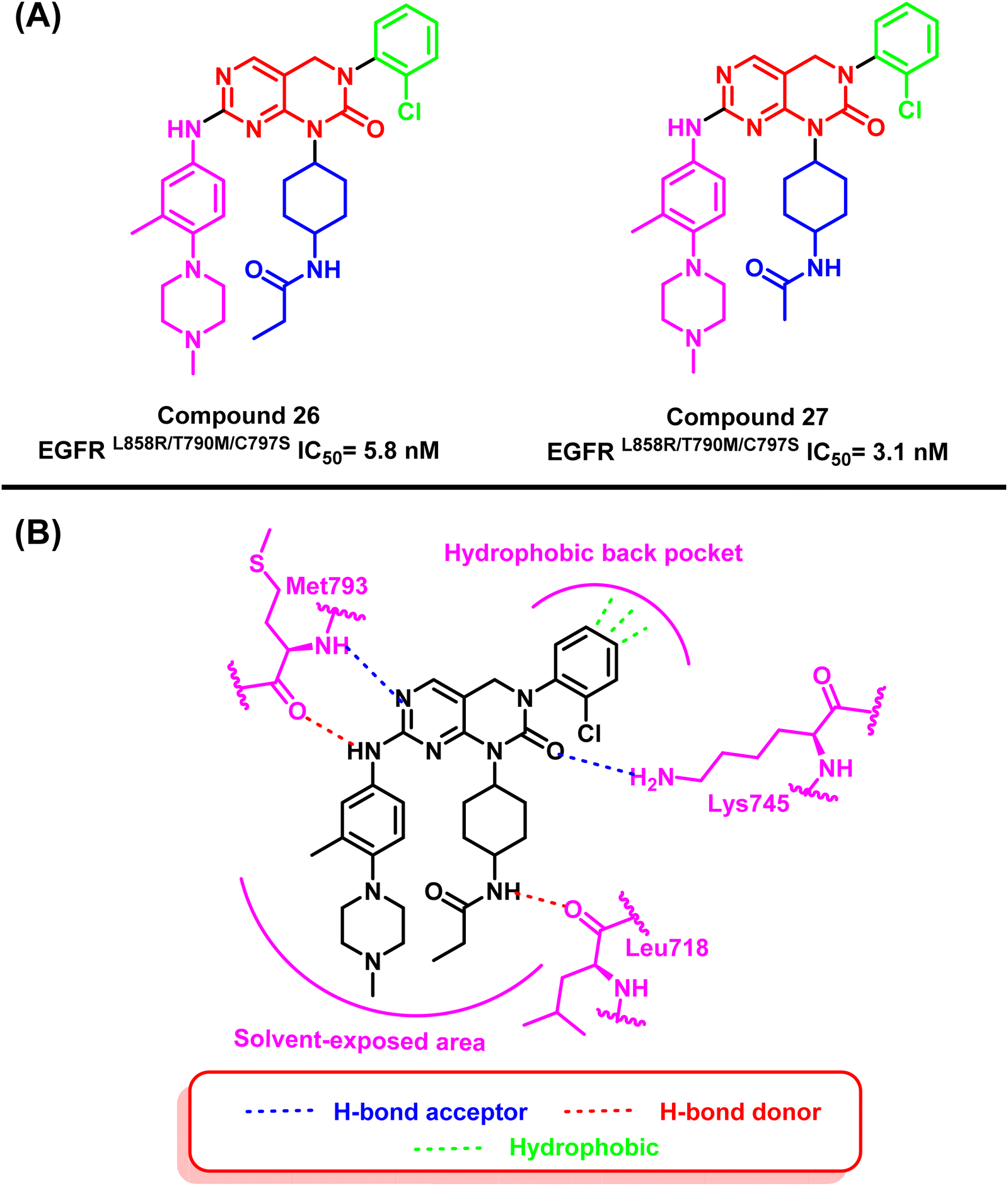 | ||
| Fig. 19 (A) Compounds (26) and (27), (B) 2D interactions of compound (26) with active site of mutant EGFR TK. | ||
Also, compound (28) is a representative compound of a series of 5-methylpyrido[2,3-d]pyrimidine derivatives that were deployed as selective EGFRL858R/T790M/C797S inhibitors. This compound's IC50 for the mutant EGFRL858R/T790M/C797S was 27.5 nM, compared to EGFRWT (IC50 > 1.0 μM), which was clearly less potent on EGFRwt 102 (Fig. 20). Additionally, based on the Ding group research,101 Zhang et al. developed a series of 2,4,6-trisubstituted pyrido[3,4-d]pyrimidine derivatives as potential fourth-generation EGFR-TKIs. Compound (29) was the most effective one in this study, with IC50 values that suppressed the proliferation of HCC827 and H1975 cell lines by 0.044 μM and 0.40 μM, respectively. Also, it significantly inhibited EGFRL858R/T790M/C797S with IC50 = 7.2 nM.103 It was noticed that methyl substitution of the main scaffold reduces the potency of this family with a negative impact on mutation selectivity (Fig. 20).
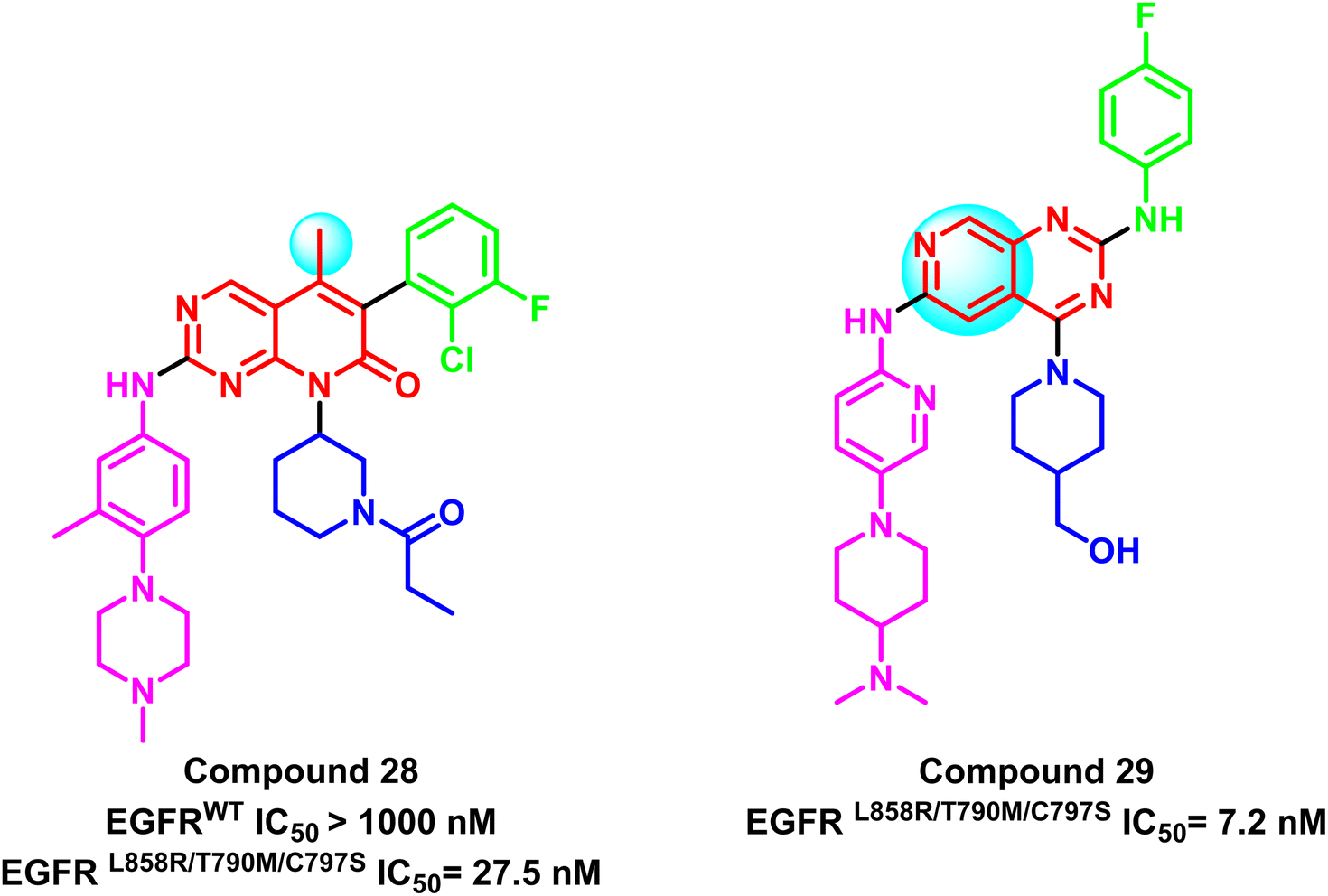 | ||
| Fig. 20 Examples of pyrido[2,3-d]pyrimidine derivatives compounds (28) and (29). | ||
2.4 Quinazoline/quinoline based derivatives
The structure of EAI045 (13) inspires the design and the synthesis of many quinazoline-4-one derivatives, which were synthesized by replacing the 2-aminothiazole amide group with a more rigid, non-hydrolyzed amidic group embedded in the core of the quinazoline scaffold. The designed candidates were considered constrained analogs of EAI045 (13) with enhanced pharmacokinetic, pharmacodynamic, and safety features. According to the findings of the biochemical testing, compound (30) had an inhibitory effect against EGFRL858R/T790M/C797S, with an IC50 value = 5.3 μM (ref. 104) (Fig. 21).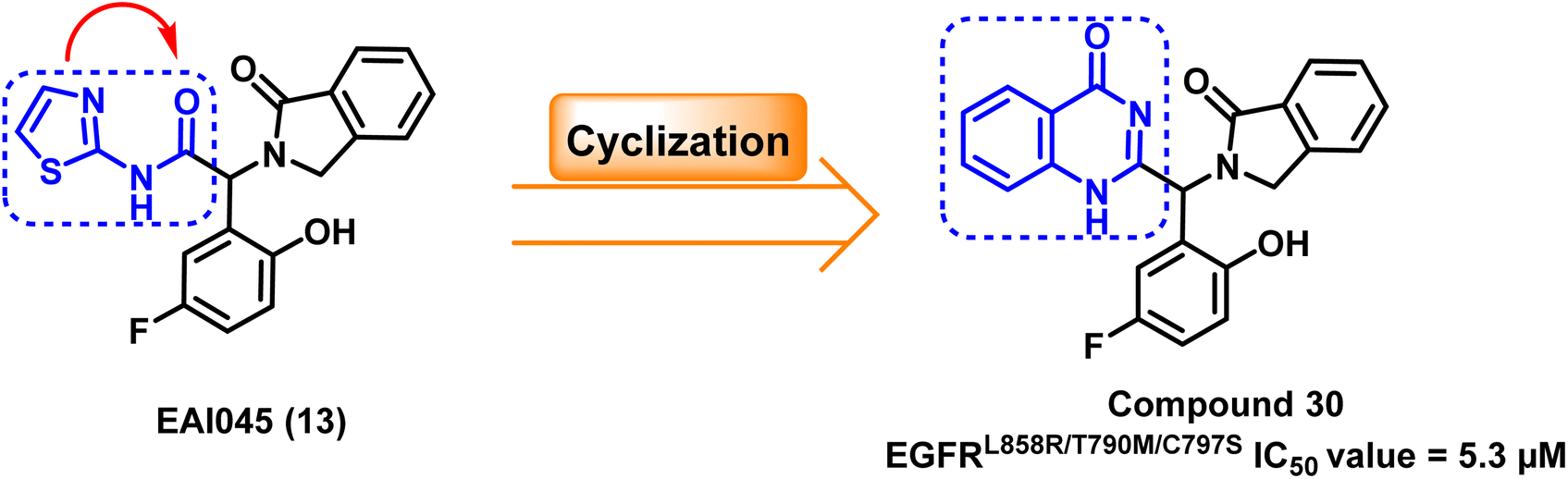 | ||
| Fig. 21 The optimization process of compound (30) from the prototype lead compound EAI045 (13). | ||
Furthermore, a molecular hybridization between vandetanib (31), a quinazoline-based member of the first-generation of EGFR TKIs, and EAI0045 (13) were combined through chemical modification to produce compound (32), which strongly inhibited the activity of the EGFRL858R/T790M/C797S (IC50 = 2.2 nM). The hybridized molecule was found to bind to the mutated EGFRT790M, and it occupied both the ATP binding site and the allosteric binding site, according to the X-ray crystal structure, as it carried both the required pharmacophores.105 Unfortunately, fast plasma clearance, low oral bioavailability, and oral exposure precluded further research on this derivative (Fig. 22).
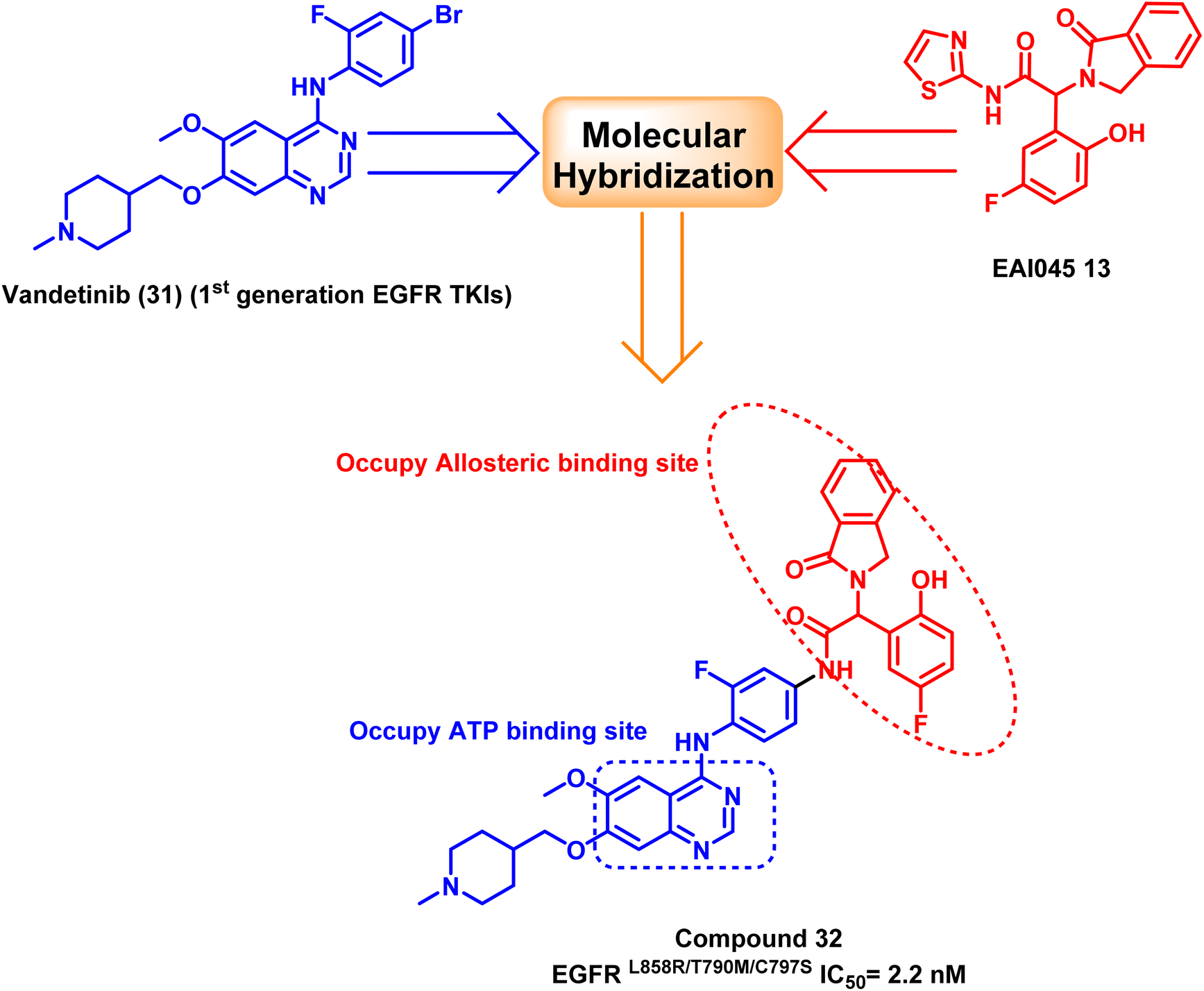 | ||
| Fig. 22 The optimization process of compound (32) via molecular hybridization. | ||
Additionally, compound (33) was identified as the fourth-generation triple mutant EGFRdel746-750/T790M/C797S inhibitor based on the virtual screening strategy, with potent kinase inhibitory enzymatic activity (IC50 = 149 nM) and 163-fold selectivity over EGFRWT 106. Substitution variation of the 2-phenolic moiety resulted in the development of compounds (34) and (35).
With EGFRdel19/T790M/C797S IC50 = 46 nM, compound (34) bearing the methyl ester may provide a hydrogen bond acceptor bind to Ser797; however, it may suffer from hydrolysis to corresponding acid analog, which may affect its pharmacodynamics proprieties. The 5-cyano derivative (35) exhibited about 1000 times more selectivity against the triple mutant EGFRdel19/T790M/C797S (IC50 = 17.9 nM) than EGFRWT. This could be explained by the forming of strong interactions with the mutant residue Ser797 by cyano moiety as hydrogen bond acceptor106 (Fig. 23).
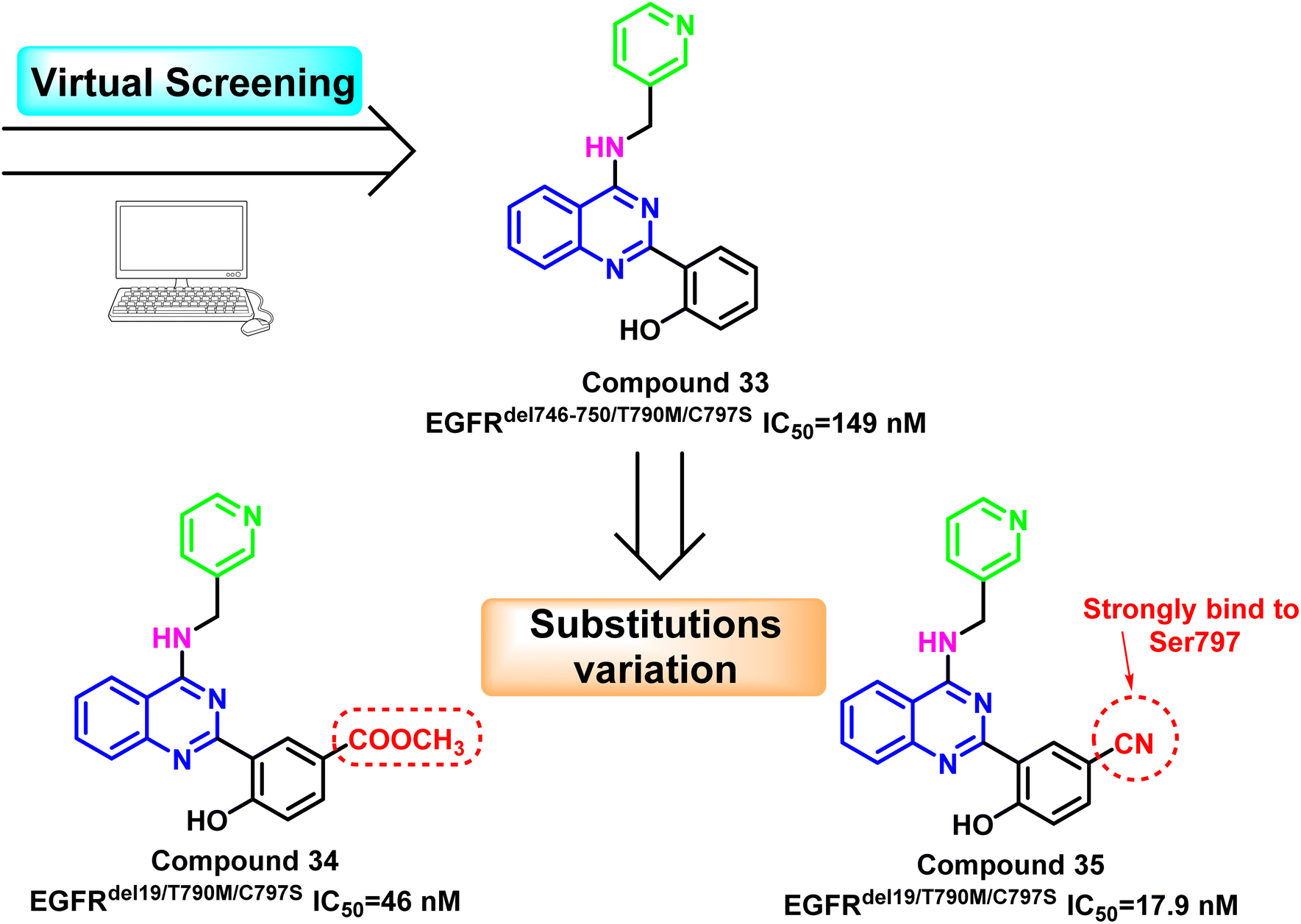 | ||
| Fig. 23 The optimization process of quinazoline compounds (33–35). | ||
Also, a series of substituted quinoline compounds were designed and synthesized, according to K. S. Karnik and his coworkers, as mutant L858R/T790M/C797S EGFR-TKIs.107 Significantly anti-proliferative and enzyme-based inhibitory efficiency of EGFR L858R/T790M/C797S was demonstrated by the substituted quinolone compounds (36–38). With an IC50 value of 124 nM for compound (37), it exhibited a higher EGFRL858R/T790M/C797S inhibitory effect (Fig. 24).
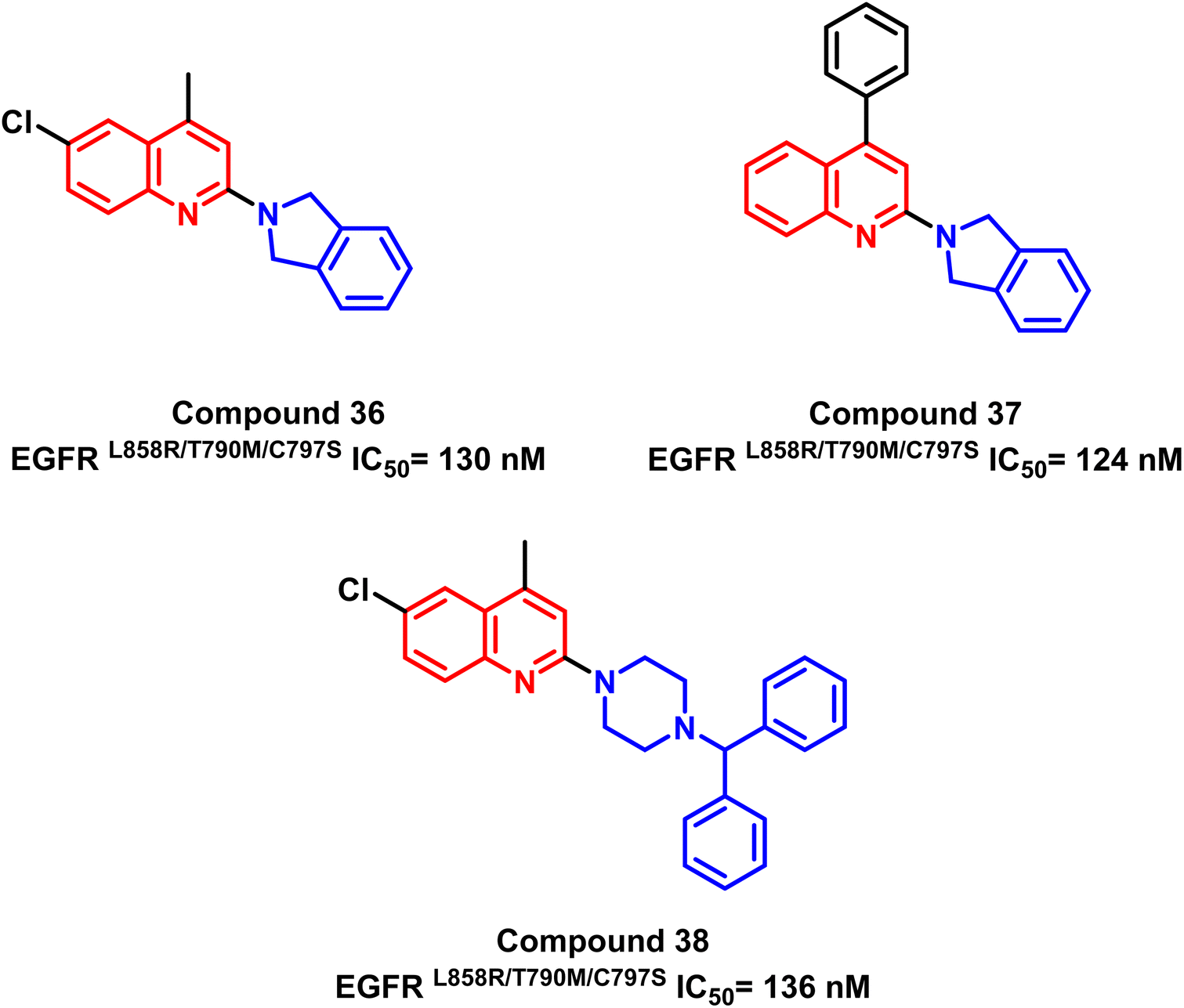 | ||
| Fig. 24 Quinoline derivatives (36–38) as EGFRL858R/T790M/C797S TK inhibitors. | ||
2.5 Oxoisoindoline phenyl-acetamide-piperazine based derivatives
A ligand extension strategy was applied to the lead compound EAI045 (13) by adding a phenyl-piperazine moiety to the para substitution of isoindolinone ring. With an IC50 value of 0.1 μM, the newly developed drug (39) was relatively the most potent against the mutant EGFRL858R/T790M/C797S.108 The phenyl-piperazine extension could provide better exposure to the allosteric pocket to have better binding (Fig. 25).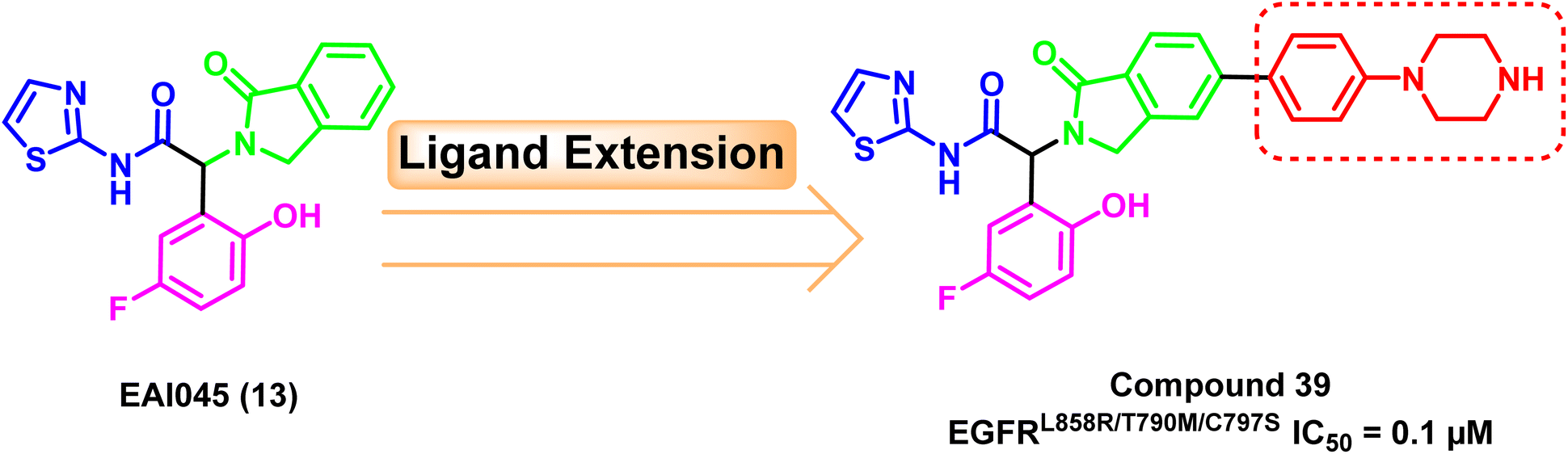 | ||
| Fig. 25 The optimization process of compound (39). | ||
Furthermore, it exhibited good in vivo efficacy after monotherapy and excellent results if it is administrated in combination therapy with osimertinib (8) in mice leading to a much less amount of residual tumor.108
2.6 Trisubstituted imidazoles/benzoimidazole derivatives
Tri-substituted imidazole derivatives containing an aliphatic alcohol chain moiety were evaluated as EGFRL858R/T790M/C797S mutant reversible inhibitors via selective p38 MAP kinase inhibition.109 Among these derivatives, compounds (40) and (41) showed significant inhibitory activities against the mutant EGFRL858R/T790M/C797S with IC50 values of 7.64 nM and 8.83 nM respectively, unfortunately without selectivity over EGFRWT (IC50 less than 0.5 nM).110 In addition, the synthesized compounds shared the following standard pharmacophoric features: (i) para-fluoro-phenyl ring pointed towards mutant Met790 residue, (ii) central imidazole ring, (iii) pyrrolopyridine moiety provides H-bond acceptor and H-bond donor moiety to form bidentate H-bond to Met793, and (iv) aliphatic chain with H-bond donor targeted towards Asn842 residue (Fig. 26).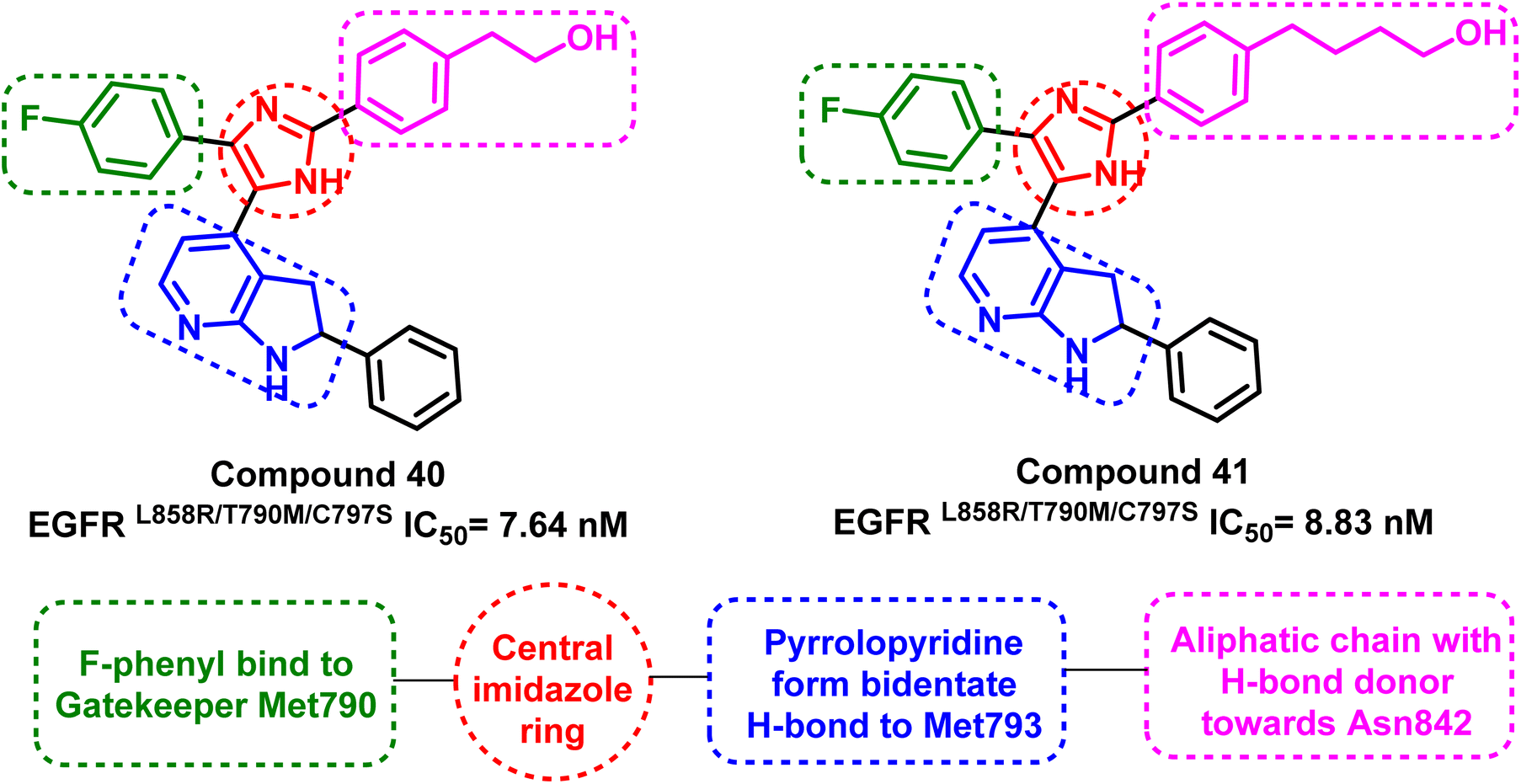 | ||
| Fig. 26 Common structural features of compounds (40) and (41). | ||
Moreover, another series of compounds (43) and (44) were designed via optimization compound (42). (Fig. 27) Both compounds successfully inhibit the mutant EGFRL858R/T790M/C797S with IC50 values 8 nM and 7 nM, respectively, if compared to the parent molecule compound (42), which inhibits the mutant EGFRL858R/T790M/C797S with IC50 more than 149 μM.111 These compounds are modified with three carbons aliphatic alcohol chain, which was long enough to form a strong hydrogen bond with Asn842 residue. As well as, N-(4-methoxyphenyl)acrylamide or N-(4-methoxyphenyl)propionamide may afford interaction with mutant Ser797 residue of EGFR TK, as illustrated in the 2D diagram in Fig. 28.
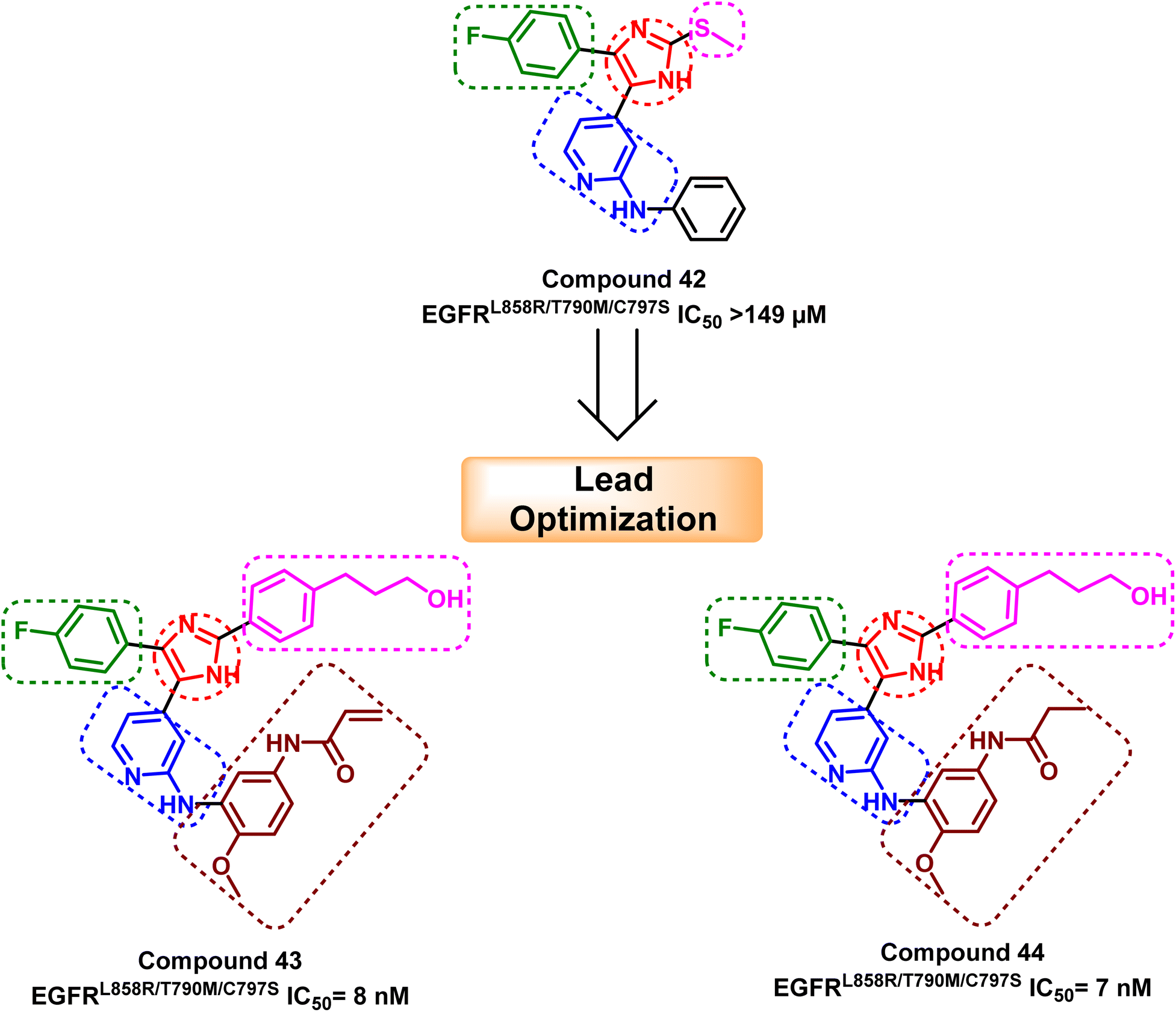 | ||
| Fig. 27 The optimization process of compounds (43) and (44) from the lead compound (42). | ||
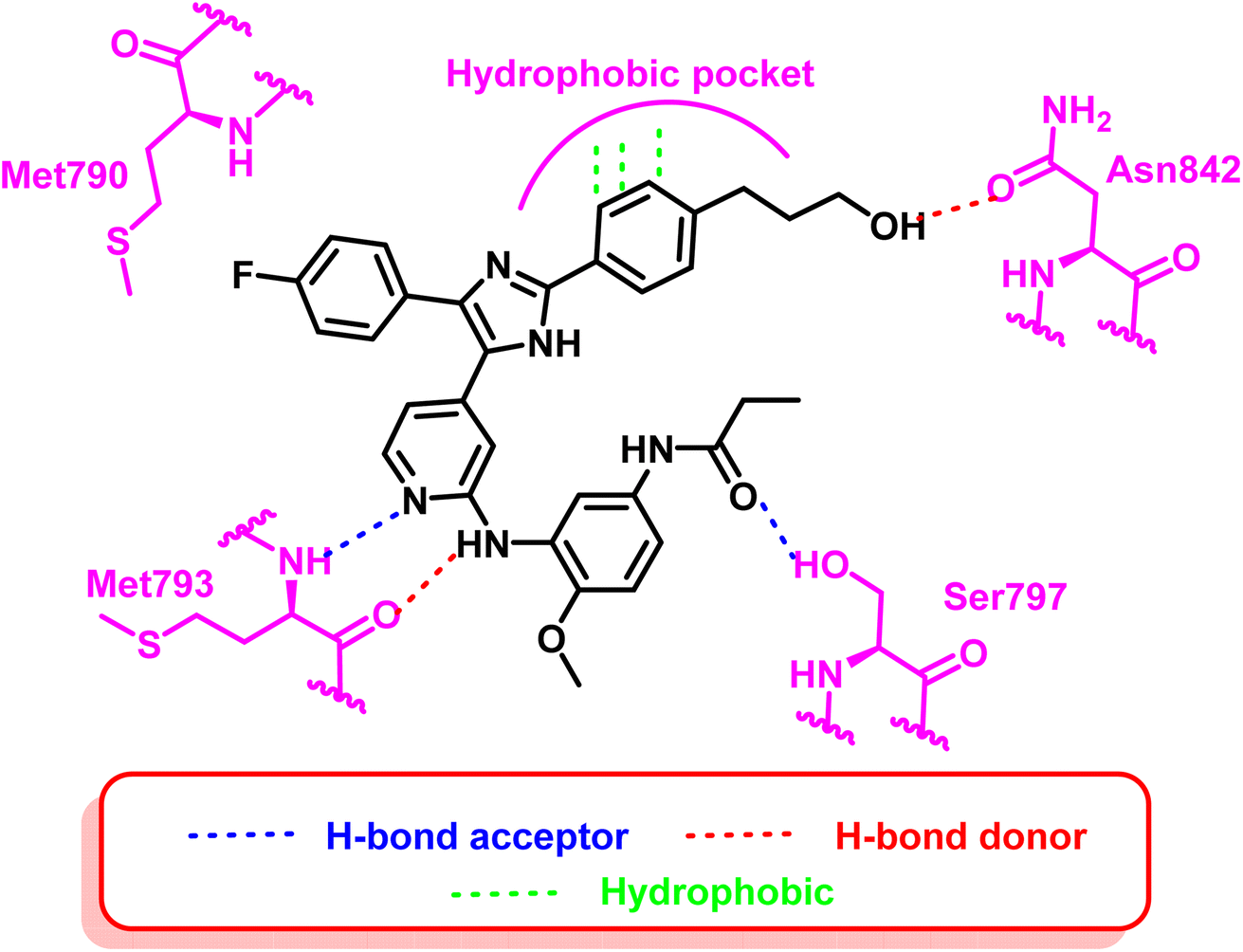 | ||
| Fig. 28 2D diagram of compound (44) at the binding site of the mutant EGFRL858R/T790M/C797S. | ||
Also, a survey through the literature demonstrated that the benzimidazole skeleton has widespread use in synthesizing EGFR inhibitors. H. Engelhardt et al. discovered a potent benzimidazole derivative (45) with highly selective but modest action that inhibits EGFRdel19/T790M/C797S at IC50 = 790 nM through a library of selective kinase inhibitors.112
After that, compound (45) was macro cyclized to rigidize the molecule, yielding the target compound (46) with potent inhibitory activity against EGFRdel19/T790M/C797S with IC50 = 0.6 nM. Compound (46) suppressed triple cis mutations in the presence of EGFRdel19 and complicated EGFR mutations containing T790M and C797S mutations. Simultaneously, compound (46) caused tumor regression in anti-osimertinib EGFRdel19/T790M/C797S mice xenograft models. The macrocyclization of the target compounds generally results the compounds having good in vivo and in vitro effectiveness112 (Fig. 29).
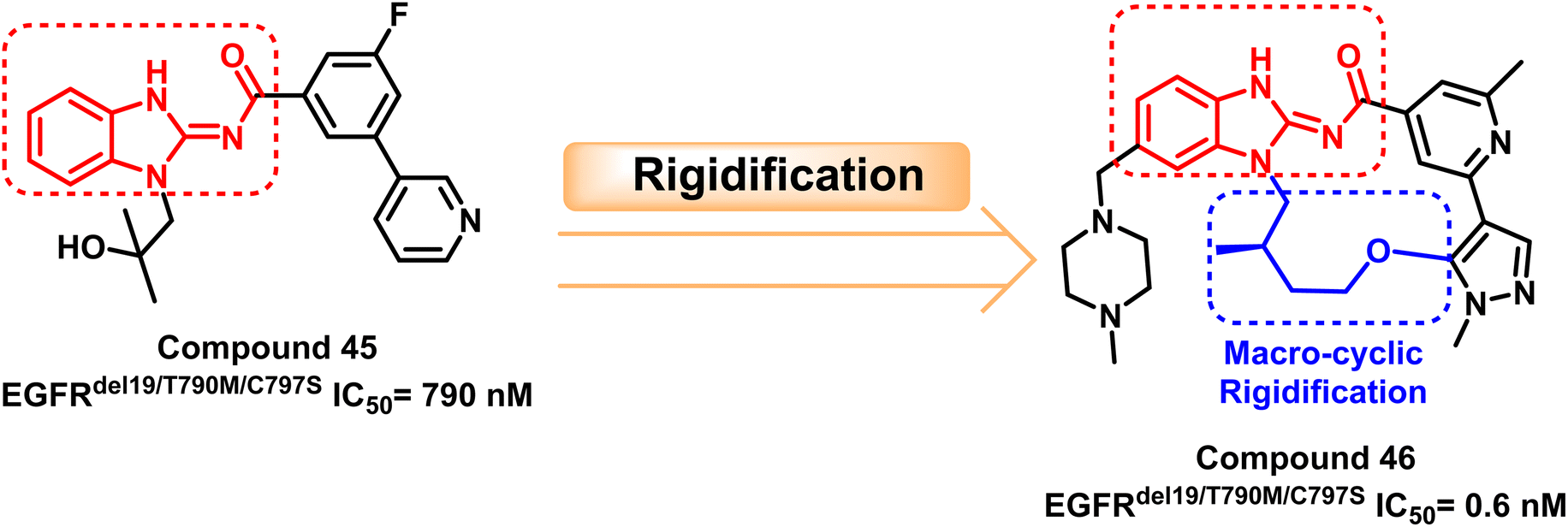 | ||
| Fig. 29 The optimization process of compounds (46) through rigidification of compound (45). | ||
2.7 Spiro/aryl/phosphorous/oxygen based derivatives
A series of spiro/aryl/phosphorous/oxygen derivatives were reported as fourth-generation kinase inhibitors of the mutant EGFRdel19/T790M/C797S. The IC50 values for compounds (47–49) were less than 100 nM, demonstrating remarkable anti-proliferative activity against all tested cell lines (Ba/F3, A431, and NCI-H1975). Notably, this group of spiro/aryl/phosphorous/oxygen derivatives also exhibited potent anticancer activity in xenograft mouse models carrying the C797S mutation. Moreover, the experiments' results indicated that these molecules appointed a new scaffold for the inhibition of EGFR TK with C797S mutation96 (Fig. 30). All these analogs shared an amino pyrimidine scaffold alongside phosphorus oxygen moiety, which acts as a strong H-bond acceptor pharmacophore, as well as a spiro aryl tail.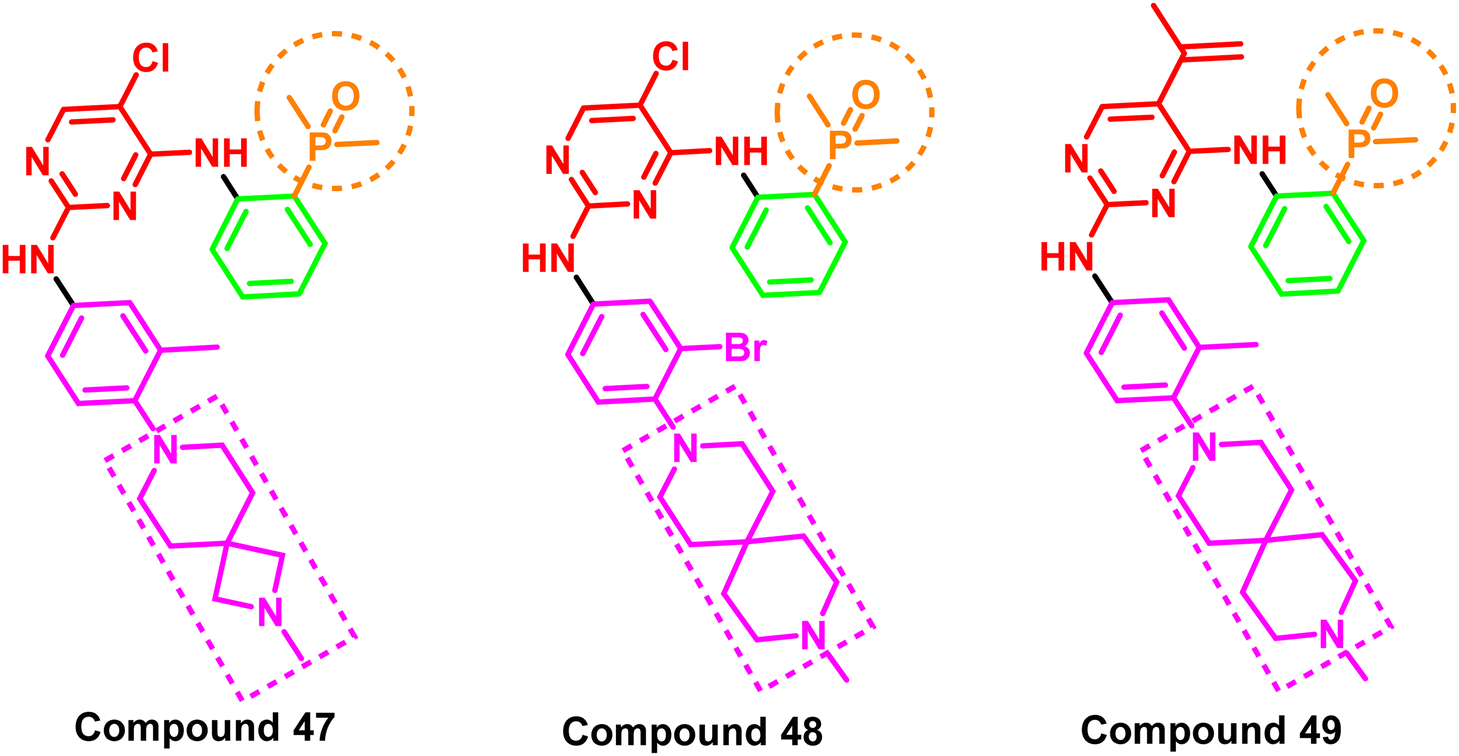 | ||
| Fig. 30 Spiro/aryl/phosphorous/oxygen compounds (47–49). | ||
2.8 Natural products as EGFR inhibitors
One of the most plentiful sources for current drug discovery hits or leads is natural products.113 In 2003, Facompré et al. discovered the marine alkaloid lamellarin N (50) as a topoisomerase I inhibitor.114 Nishiya et al. developed Lamellarin derivative (51) via modification of ring C of the original natural product's scaffold with two tails attached to its phenolic group and methoxy substituent. This compound was tested against the EGFRC797S and showed effective inhibition.115The double or triple mutations of EGFRC797S resistant to osimertinib (8) were inhibited by the bis(N,N-dimethylethanamine)ether derivative compound (51) in a specific manner. It also reduced downstream signal transduction and EGFR phosphorylation in triplet mutant EGFR PC-9 cells. Therefore, compound (51) can serve as a new EGFR-TKI structural skeleton to stop the development of cross-resistance to well-known medications like osimertinib (8)115 (Fig. 31).
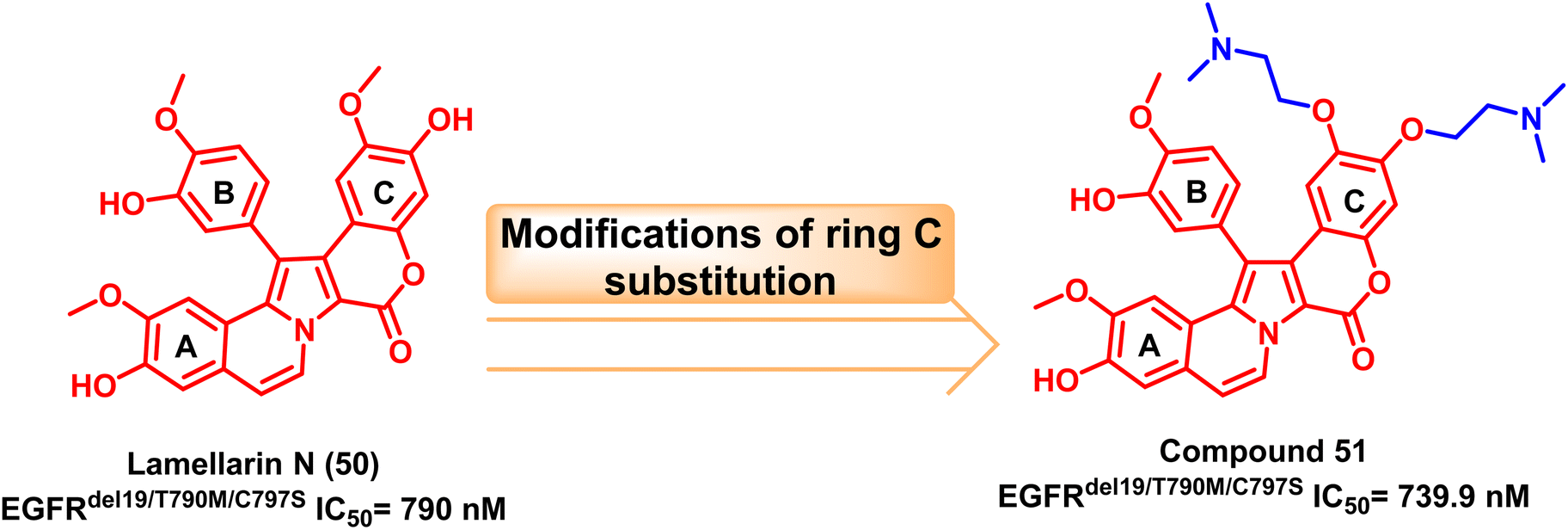 | ||
| Fig. 31 Marine alkaloid lamellarin N (50) and its modified analogue compound (51). | ||
2.9 Drug design based on PROTAC
Proteolytic targeting chimeras (PROTACs), which cause targeted protein degradation, have been a popular drug discovery approach in the last ten years.116 PROTAC is a heterobifunctional small molecule that may remove particular undesirable proteins. It consists of a linker and two active domains; the first is a ligand for the E3 ubiquitin ligase, and the second is a ligand bound to the protein of interest. Target proteins are induced to become ubiquitinated by PROTAC and then selectively destroyed by 26S proteolytic enzymes. Increased selectivity for protein degradation and escape mechanisms, such as acquired resistance mutations and target protein overexpression, could result from PROTAC.117,118Based on brigatinib (52), an ATP-competitive EGFR inhibitor with high affinity to the ATP binding site and multi-kinase activities. Zhang et al. developed and synthesized powerful and specific EGFRDel19/T790M/C797S triple-mutated PROTAC degraders. According to mechanistic research, molecular linking of brigatinib (52) with VHL (E3 ubiquitin ligase) (53) provide compound (54) that exclusively mediated the degradation of EGFR protein via proteasome pathway with a DC50 value of 8 nM; it decreased EGFR protein levels in Ba/F3 cells containing the triple mutation EGFRdel19/T790M/C797S in a dose- and time-dependent manner119 (Fig. 32).
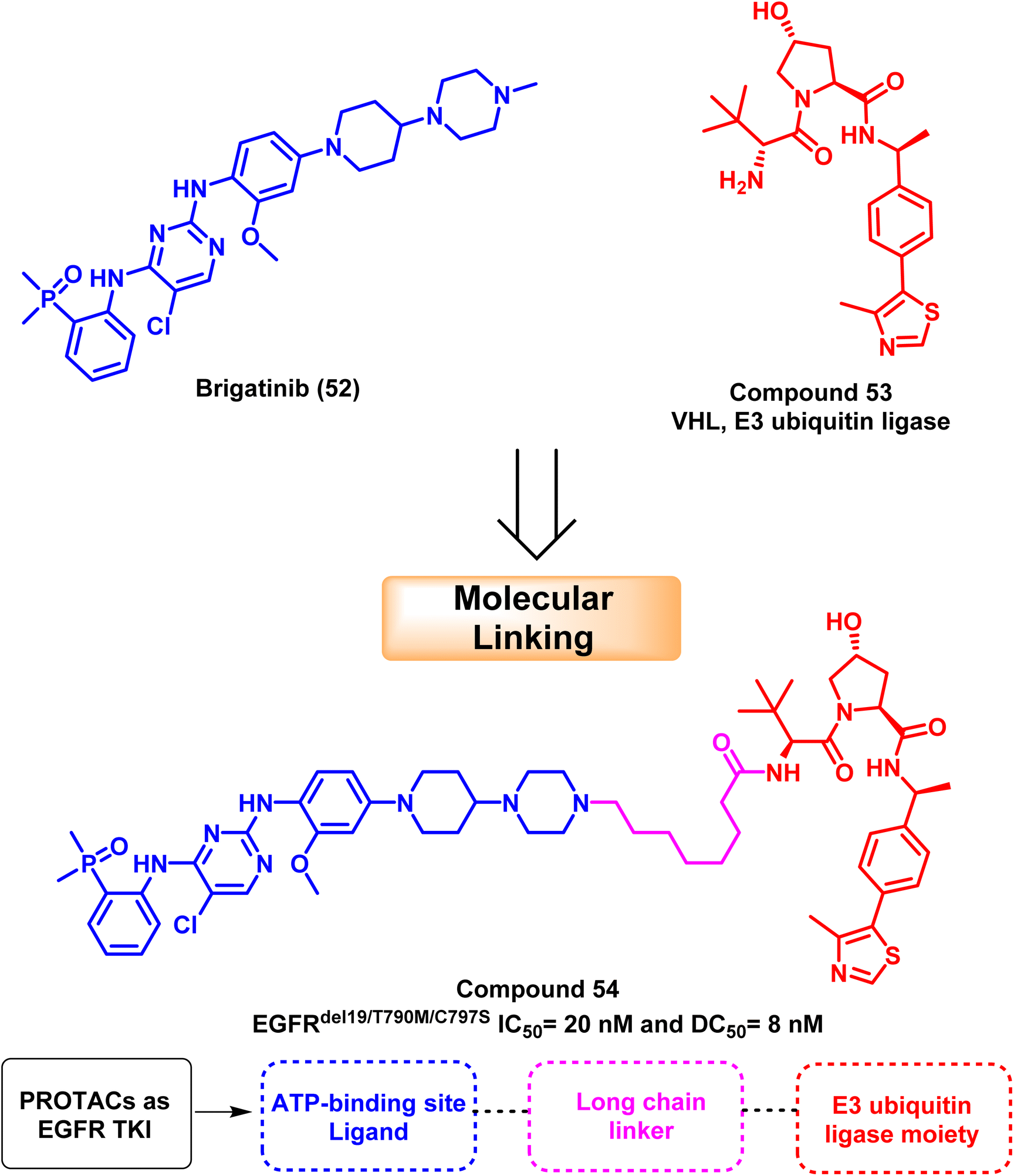 | ||
| Fig. 32 The optimization process of PROTAC compound (54). | ||
Compound (54) has an IC50 of 20 nM for suppressing Ba/F3 EGFRdel19/T790M/C797S cells, and it can successfully prevent the activation of intracellular EGFR downstream, signaling ERK and AKT. The resistance caused by the EGFRC797S mutation can be overcome by developing new medications in the clinic using compound (54) as a lead candidate.119
3. Allosteric inhibitors with a Y-shaped structure for the C797S EGFR mutation
The use of an allosteric approach was successful in overcoming the C797S EGFR resistance. Studies on the binding interactions of allosteric inhibitors, such as EAI045 (13) and JBJ-04-125-02 1 (55) (non-ATP competitive inhibitors of mutant EGFRT790M), indicated that the inhibitor has a Y-shaped structure.87 The binding model of these allosteric inhibitors, which have a shape of a “Three-blade propeller,” is made up of asymmetric carbon and three-ring attachments; two are hydrophobic groups that have aromatic ring structures, and an amido isothiazole moiety is the third attachment, which participates in an electron donor/acceptor bonding with the receptor87 (Fig. 33).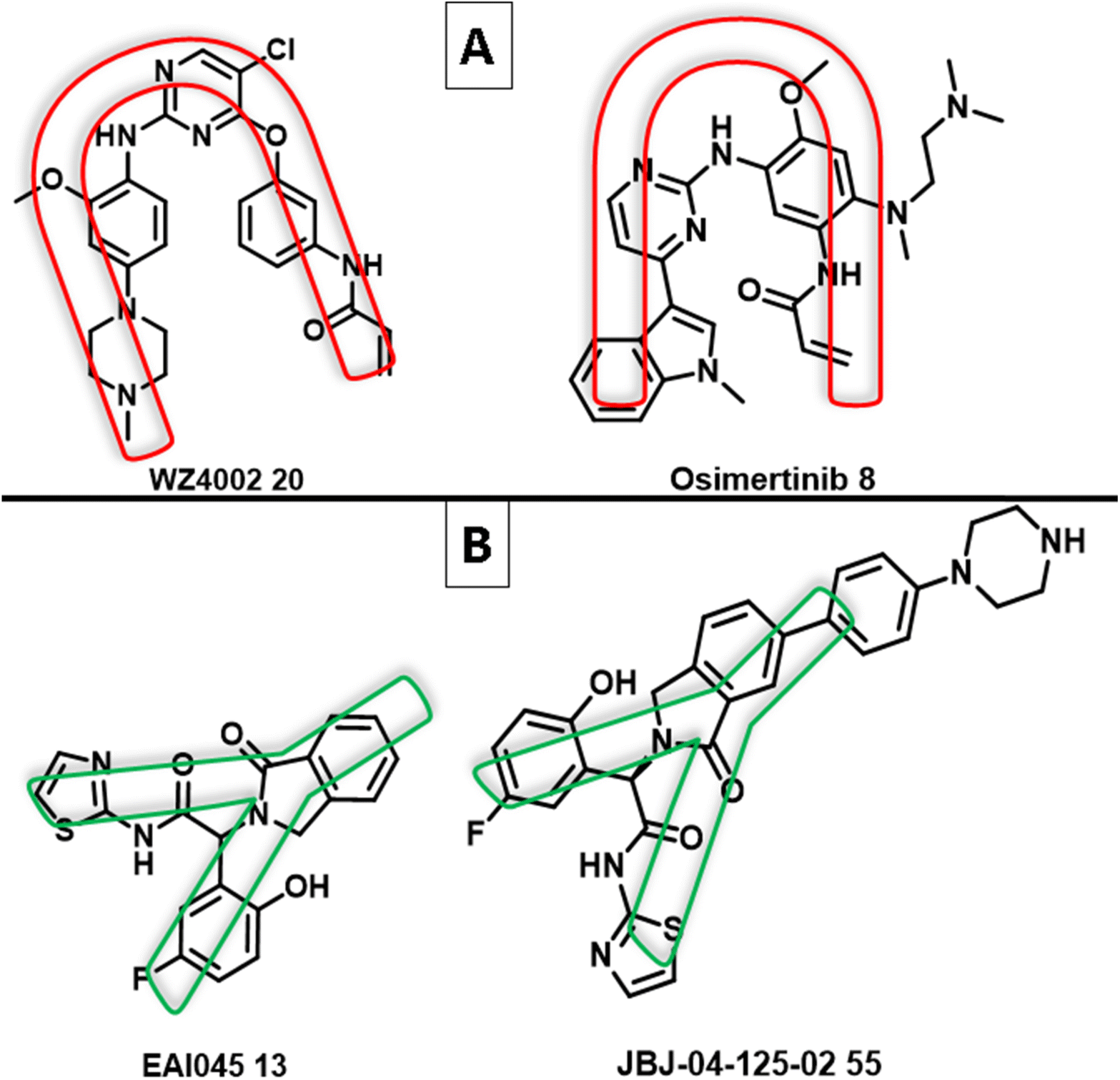 | ||
| Fig. 33 (A) U-shape of 3rd generation EGFR TKIs WZ4002 (20) and osimertinib (8); (B) Y-shape of the non-ATP competitive inhibitors of mutant EGFRT790M EAI045 (13) and JBJ-04-125-02 (55). | ||
While the mutant EGFRT790M kinase domain's X-ray crystal structure with the third-generation TKI inhibitors (WZ4002 (20) and osimertinib (8)) showed that the pyrimidine core was arranged in a U-shape with an aniline ring harboring a hydrophilic group and an acrylamide moiety120 (Fig. 33). Most small compounds were developed as selective mutant EGFR inhibitors based on altering U- to Y-shaped structures. Glucokinase activators (GKAs) were discovered using structure-based virtual screening techniques as mutant EGFRC797S inhibitors from the ZINC database, containing all necessary pharmacophores and a Y-shaped conformation similar to EAI045 (13).121
The study is based on the interactions between the ligand and the EGFR enzyme binding pocket that contains various mutations, such as the L858R EGFR, T790M mutant EGFR, and C797S mutant EGFR. The findings show that all types of EGFR mutations (L858R, T790M, C797S) can be inhibited by GKAs via mutant-selective allosteric inhibition. Using the final filter of Jargan's Rule of Three, Lipinski's Rule of Five, and in silico ADME predictions, a set of targeted hit molecules with suitable pharmacokinetic properties was produced as representative example compounds (56–63)122 (Fig. 34).
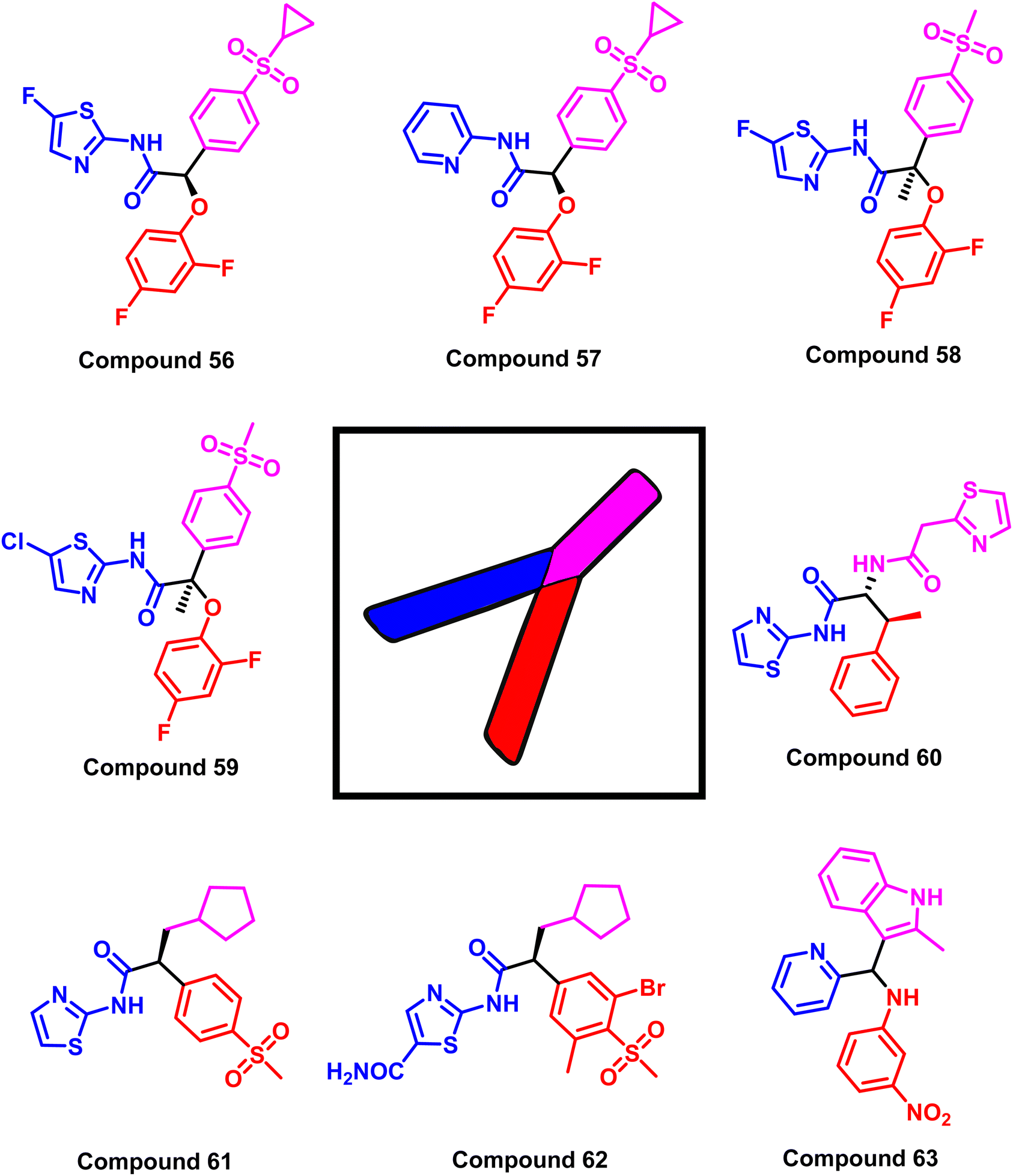 | ||
| Fig. 34 Compounds (56–63) as examples of structures of EGFR inhibitors with Y-shaped configuration. | ||
Ninety-two compounds were chosen in the previously mentioned study for biological evaluation by high-throughput virtual screening of recently released structures of the EGFR kinase domain (wild-type and mutant L858R/T790M EGFRs). Compound (63) exhibited the most promising activity to inhibit the wild-type and double mutant EGFRL858R/T790M, with an allosteric mechanism and EC50 values of 4.7 μM and 6.2 μM, respectively. It can prevent the progression of non-small cell lung cancer at the micromolar level (IC50 = 14.5 μM against wild-type EGFR H1299 cell line, 19 μM against mutant EGFR H1650 cell line, and 24 μM against double mutant H1975 cell line).122
Targeting ATP binding site by the first-, second-, and third-generation EGFR TKIs was not suffering from mutation problems only, which reduced the efficacy of the used EGFR TKIs within the treatment.123 In addition, because EGFR is predominantly expressed on epithelial cells, such as those of the skin and gastrointestinal tract, rashes and diarrhea are the most common adverse effects of EGFR inhibitors.
For patients with advanced NSCLC, strategies to enhance the assessment and management of EGFR-TKI-related adverse events, such as rash and diarrhea, should result in superior clinical outcomes, greater adherence, and improved quality of life.124 One of the most important clinical findings was an acneiform eruption and complicated skin xerosis.125 Additionally, in vivo, the binding of osimertinib (8) to non-target EGFR receptors has resulted in severe adverse effects, including diarrhea, itching, nausea, decreased appetite, hyperglycemia, prolongation of the corrected QT interval, and pneumonia.126 On the other hand, the fourth-generation EGFR TKIs that target the allosteric binding site can assist in overcoming “undruggable” resistance (C797S) with expected negligible side effects.86
4. Cost-effectiveness of using EGFR-TKI in treatment for EGFR-mutated advanced NSCLC
In non-small cell lung cancer patients harboring oncogenic EGFR mutations, tyrosine-kinase inhibitors (TKIs) have emerged as the treatment of choice.123 However, these medications are counterbalanced by the financial burden they impose, which frequently impedes treatment access in developing nations.126 Therefore, a prospective survey was conducted in 2014 to evaluate the availability of EGFR-TKI in 192 countries. Erlotinib (2), gefitinib (1), afatinib (5), and icotinib (4) were routinely accessible in 86%, 73%, 38%, and 5% of the 74 countries that responded. However, in these countries, the monthly cost of erlotinib (2), gefitinib (1), and afatinib (5) exceeded $1000 for 39%, 35%, and 50% of patients covered by public or private mandatory health insurance systems.127A study compared the cost-effectiveness of three distinct groups of patients who received first- and second-generation TKIs as first-line therapy for patients with NSCLC that harbor EGFR mutations. There was a statistically significant difference between the average costs of TKIs, with afatinib (5) being the most costly treatment. This difference was observed in the daily and total treatment costs. In addition, a cost-effectiveness analysis determined that afatinib (5) was more cost-effective than its alternative of the first-generation TKIs (erlotinib (2) and gefitinib (1)).128
Another study on the cost-effectiveness of using EGFR TKIs in treating mutated NSCLC was conducted recently.129,130 This study's objective was to estimate the cost-effectiveness of osimertinib (8) for the first-line treatment of patients with advanced EGFR-mutated NSCLC within the framework of the Chinese healthcare system. A Markov model was developed to simulate the outcomes and direct medical costs of osimertinib (8) or standard EGFR-TKI in the initial treatment of patients with EGFR-mutated advanced NSCLC who had not been previously treated. The study revealed that based on its current market price, first-line osimertinib (8) therapy may not be cost-effective in China for patients with EGFR-mutated advanced NSCLC compared to standard EGFR-TKI. However, when the price of osimertinib (8) is decreased by 5%, the cost-effectiveness is significantly improved.129,130 Unfortunately, there is no data or expected data until now for the cost-effective impact of using the fourth-generation EGFR TKIs in treating mutated NSCLC patients.
5. Combination therapies versus multi-target agents overcome resistance C797S
The development of small molecule targeted therapies has undoubtedly led to a significant advancement in lung cancer treatment.131 Due to the development of a new drug resistance mechanism for EGFRC797S, the pace of clinical treatment must, regrettably, slow down. Effective C797S medications are still in the research and development stage and have not yet had the chance to go on the market. Therefore, in clinical practice, conservative treatment is thought to be preferred.The main focus of lung cancer resistance is the allelic link between C797S and T790M. Lung cancer cells are shown to be resistant to both the first- and third-generations of EGFR-TKIs when the C797S and T790M mutations are in cis (located at the same allele). However, lung cancer cells are shown to be effective for the combination therapy of the first and third-generation EGFR-TIK when the C797S and T790M mutations are in trans (located in different alleles). Therefore, in the in vitro and clinical practice, the analysis of the allelic relationship between C797S and T790M has crucial therapeutic value83,132 (Fig. 35).
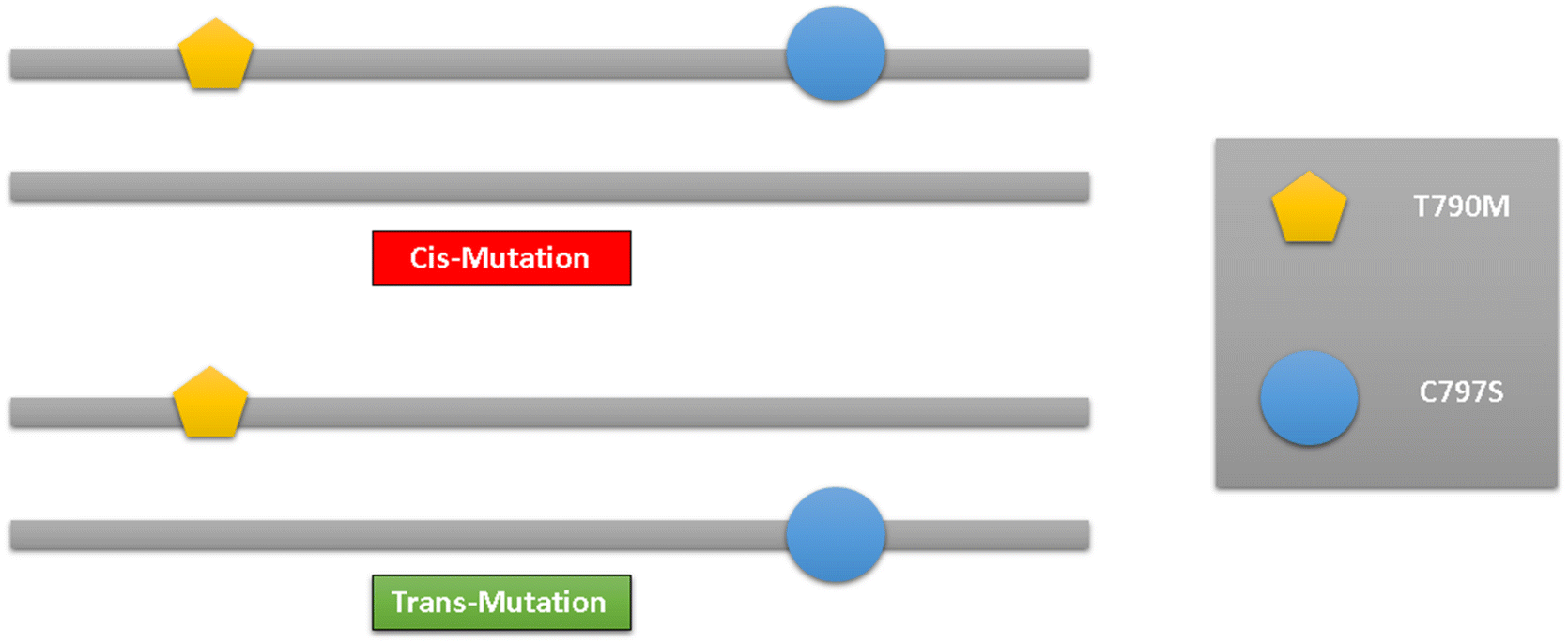 | ||
| Fig. 35 cis and trans of the T790M/C797S mutations. | ||
Clinical studies have evaluated a variety of EGFR inhibitors; however, the mono-therapy outcomes were underwhelming, given the development of epigenetic resistance. Two strategies to combat resistance include combination therapy and multi-target molecules were included. But compared to combination therapy, a single multi-target molecule gives more benefits.133
Combination therapy is a beneficial treatment method involving multiple medications to manage a specific condition.134 Combining EGFR TKIs with other therapies effectively combats treatment resistance and boosts antitumor efficacy.135 In this regard, inhibiting the EGFR signaling pathway in combination with traditional cytotoxic medications or radiotherapy may increase the cytotoxicity, induction of apoptotic cell death, and anti-proliferative of cancer cells.136 Combining medications that target several signaling pathways involved in cancer growth is another critical tactic in this area for increasing cytotoxic therapy. Addressing many paths at once could produce more effective anticancer outcomes.137
Combination therapy involving anti-EGFR monoclonal antibodies and various EGFR TKIs is an excellent example of how the extracellular and intracellular domains of the EGFR receptor can be dually inhibited.138 It is necessary to employ and develop novel therapeutic regimens, and numerous trials of this combination therapy have been planned and reported. According to preliminary clinical studies, the potential of osimertinib (8) will enhance when paired with ramucirumab.139 Some preclinical models showed that the combination of second-generation EGFR TKIs and the monoclonal antibody cetuximab is a promising regime that improved efficacy in mutant EGFRT790M positive tumors; however, the high incidence of adverse events (rash and diarrhea) represented a significant barrier to the clinical application of this combination.140
In recent years, multi-target agents, also known as “hybrid” compounds, have received much attention due to their ability to combine two or more different pharmacophore moieties into a single molecule directly through a covalent bond or the use of a linker. Additionally, to increase the solubility of the final compounds, some structural alterations, such as the insertion of solubilizing functionalities, should be taken into account.141 Most new hybrid anticancer compounds were developed and tested as dual-selective inhibitors of the EGFR and other signaling pathways.142,143
Multi-target anticancer drugs have a more comprehensive range of activity than conventional agents because they contain two different pharmacological features and target multiple signaling pathways.144,145 The multi-target anticancer agents have shown many advantages over medication combinations, including a reduced risk of potential drug interactions and side effects, accelerated drug transport, and simplified drug metabolism. In addition, the difficulties with dosage optimization and drug combination sequences have been resolved using multi-target agents. These advantages allow the multi-target compounds to be promising candidates for developing the upcoming anticancer medications.146 Most of the multi-kinase inhibitors that have received clinical approval and are currently being used in clinics, including sorafenib (64), sunitinib (65), lapatinib (3), afatinib (5), and dacomitinib (7), were not initially intended to target multiple ligands147–149 (Fig. 4, 6 and 36).
 | ||
| Fig. 36 Examples of multi-kinase inhibitors. | ||
6. Conclusion and future prospects
EGFR TKIs increased the survival rate of several cancer patients with EGFR mutations. Numerous EGFR TKIs have been evaluated in clinical studies; however, the findings of long-term use are disappointing in terms of the fast genetic changes in the tyrosine kinase domain of EGFR. In patients receiving EGFR TKI therapy, epigenetic changes in the tyrosine kinase domain of EGFR (T790M and C797S mutations) are considered the most common source of resistance. Many efforts have been made to rationally design particular small compounds that act as EGFR TK inhibitors and specifically target the mutated tyrosine kinase domain of EGFR.In this regard, many compounds were developed and evaluated for their potential to be EGFR TK inhibitors, with encouraging outcomes in both in vivo and in vitro experiments. Research is still going on to develop more effective agents in this area. To overcome resistance brought on by T790M and C797S mutations, it is crucial to identify and develop novel candidates as the next-generation small molecule EGFR TKIs with various scaffolds and binding interactions. A practical solution to these resistance issues is proposed by further research in small inhibitors via the EGFR allosteric site.
In conclusion, this review explored the structural classification of small molecule EGFR TKIs that have recently revealed considerable cytotoxic and kinase inhibitory activity against C797S mutation. The interaction between these molecules and the active site's amino acid residues is also investigated. The rational design of the prior literature and the optimization of inhibitor structure also play essential roles in this review. The development of more potent anti-tumor EGFR as small molecule inhibitors aimed at treating the rising drug-resistant mutations is what the authors seek to achieve by introducing these methodological approaches. In the interim, some novel chemical entities were described in this review to help scientists and researchers focusing on studying pharmaceutical chemistry to design and build effective small-molecule EGFR TKIs combating resistance obstacles in NSCLC patients.
Conflicts of interest
The authors affirm that they have no known financial or interpersonal conflicts that would have appeared to impact the research presented in this paper.References
- S. Warnakulasuriya, O. Kujan, J. M. Aguirre-Urizar, J. V. Bagan, M. Á. González-Moles, A. R. Kerr, G. Lodi, F. W. Mello, L. Monteiro and G. R. Ogden, Oral Dis., 2021, 27, 1862–1880 CrossRef PubMed.
- R. K. Sain, R. Chouhan, L. P. Bagri and A. Bajpa, J. Cancer Res. Updates, 2012, 1, 129–152 Search PubMed.
- M. Q. Mahmood, S. D. Shukla, C. Ward and E. H. Walters, Biomolecules, 2021, 11, 1394 CrossRef CAS PubMed.
- A. Lepucki, K. Orlińska, A. Mielczarek-Palacz, J. Kabut, P. Olczyk and K. Komosińska-Vassev, J. Clin. Med., 2022, 11, 1250 CrossRef CAS PubMed.
- M. Hoteit, Z. Oneissi, R. Reda, F. Wakim, A. Zaidan, M. Farran, E. Abi-Khalil and M. El-Sibai, Oncol. Lett., 2021, 22, 1–18 Search PubMed.
- M. Glumac, V. Čikeš Čulić, I. Marinović-Terzić and M. Radan, Cancers, 2023, 15, 981 CrossRef CAS PubMed.
- R. Sharma, Int. J. Clin. Oncol., 2022, 27, 665–675 CrossRef PubMed.
- A. Thai, B. Solomon, L. Sequist, J. Gainor and R. Heist, Lancet, 2021, 398, 535–554 CrossRef PubMed.
- X. Wang, Z. Liu, X. Sui, Q. Wu, J. Wang and C. Xu, Phytomedicine, 2019, 59, 152787 CrossRef CAS PubMed.
- A.-M. Dingemans, M. Früh, A. Ardizzoni, B. Besse, C. Faivre-Finn, L. Hendriks, S. Lantuejoul, S. Peters, N. Reguart and C. Rudin, Ann. Oncol., 2021, 32, 839–853 CrossRef PubMed.
- J. Remon, M. Aldea, B. Besse, D. Planchard, M. Reck, G. Giaccone and J.-C. Soria, Ann. Oncol., 2021, 32, 698–709 CrossRef CAS PubMed.
- D. N. Carney, Mass Medical Soc., 2002, 346, 126–128 Search PubMed.
- A. Pallis, L. Serfass, R. Dziadziuszko, J. Van Meerbeeck, D. Fennell, D. Lacombe, J. Welch and C. Gridelli, Eur. J. Cancer, 2009, 45, 2473–2487 CrossRef CAS PubMed.
- R. M. Aly, R. A. Serya, A. M. El-Motwally, A. Esmat, S. Abbas and D. A. Abou El Ella, Bioorg. Chem., 2017, 75, 368–392 CrossRef CAS PubMed.
- V. Schirrmacher, Int. J. Oncol., 2019, 54, 407–419 CrossRef CAS PubMed.
- S. Rayego-Mateos, R. Rodrigues-Diez, J. L. Morgado-Pascual, F. Valentijn, J. M. Valdivielso, R. Goldschmeding and M. Ruiz-Ortega, Mediators Inflammation, 2018, 2018, 1–22 CrossRef PubMed.
- U. Jarouliya and R. K. Keservani, in Medicinal Chemistry with Pharmaceutical Product Development, Apple Academic Press, 2019, pp. 1–32 Search PubMed.
- K. Umezawa and I. Kii, Molecules, 2021, 26, 651 CrossRef CAS PubMed.
- M. Okay and I. C. Haznedaroglu, Protein Kinase-mediated Decisions Between Life and Death, 2021, pp. 383–393 Search PubMed.
- T. Hölting, A. E. Siperstein, O. H. Clark and Q.-Y. Duh, Eur. J. Endocrinol., 1995, 132, 229–235 Search PubMed.
- E. Ifeanyi Obeagu, Q. Babar, C. CN Vincent, C. Lawrence Udenze, R. Eze, C. J Okafor, B. I Ifionu, A. Amaeze Amaeze and F. Ngozi Amaeze, J. Pharm. Res. Int., 2021, 82–99 CrossRef.
- X. Shi, M. Sun, H. Liu, Y. Yao, R. Kong, F. Chen and Y. Song, Mol. Carcinog., 2015, 54, E1–E12 CrossRef CAS PubMed.
- K. Nakai, M. C. Hung and H. Yamaguchi, Am. J. Cancer Res., 2016, 6, 1609 CAS.
- F. Chen, N. Chen, Y. Yu and J. Cui, Front. Oncol., 2020, 10, 904 CrossRef PubMed.
- A. Markovic and C. H. Chung, Expert Rev. Anticancer Ther., 2012, 12, 1149–1159 CrossRef CAS PubMed.
- A. Dokala and S. Thakur, Oncogene, 2017, 36, 2337–2344 CrossRef CAS PubMed.
- N. Seebacher, A. Stacy, G. Porter and A. Merlot, J. Exp. Clin. Cancer Res., 2019, 38, 1–39 Search PubMed.
- L. M. M. Jaramillo, Agricultural sciences, Université Claude Bernard - Lyon I, 2014, pp. 2–293 Search PubMed.
- V. Ahire, D. Das, K. P. Mishra and D. G. R. Kulkarni, European J. Biotechnol. Biosci., 2014, 2, 09–14 Search PubMed.
- T. Amelia, A. N. Setiawan, R. E. Kartasasmita, T. Ohwada and D. H. Tjahjono, Int. J. Mol. Sci., 2022, 23, 15828 CrossRef CAS PubMed.
- S. Yoshikawa, M. Kukimoto-Niino, L. Parker, N. Handa, T. Terada, T. Fujimoto, Y. Terazawa, M. Wakiyama, M. Sato and S. Sano, Oncogene, 2013, 32, 27–38 CrossRef CAS PubMed.
- A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri and A. Foroumadi, Bioorg. Chem., 2020, 99, 103811 CrossRef CAS PubMed.
- A. Unnisa, A. K. Chettupalli, T. Hussain and M. A. Kamal, Anti-Cancer Agents Med. Chem., 2022, 22, 3370–3381 CrossRef CAS PubMed.
- J. Ferlay, P. Autier, M. Boniol, M. Heanue, M. Colombet and P. Boyle, Ann. Oncol., 2007, 18, 581–592 CrossRef CAS PubMed.
- S. A. Kenfield, E. K. Wei, M. J. Stampfer, B. A. Rosner and G. A. Colditz, Tob. Control, 2008, 17, 198–204 CrossRef CAS PubMed.
- K. B. Duggirala, Y. Lee and K. Lee, Biomol. Ther., 2022, 30, 19 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, N. Engl. J. Med., 2008, 358, 1160–1174 CrossRef CAS PubMed.
- V. Gristina, U. Malapelle, A. Galvano, P. Pisapia, F. Pepe, C. Rolfo, S. Tortorici, V. Bazan, G. Troncone and A. Russo, Cancer Treat. Rev., 2020, 85, 101994 CrossRef CAS PubMed.
- E. Grassilli and M. G. Cerrito, Cancer Drug Resist., 2022, 5, 36 CAS.
- M. L. Uribe, I. Marrocco and Y. Yarden, Cancers, 2021, 13, 2748 CrossRef CAS PubMed.
- W.-Z. Zhong, Q. Zhou and Y.-L. Wu, Oncotarget, 2017, 8, 71358 CrossRef PubMed.
- L. Landi and F. Cappuzzo, Transl. Lung Cancer Res., 2013, 2, 40 CAS.
- Z.-F. Lim and P. C. Ma, J. Hematol. Oncol., 2019, 12, 1–18 CrossRef PubMed.
- S.-G. Wu and J.-Y. Shih, Mol. Cancer, 2018, 17, 1–14 Search PubMed.
- L. Saldaña-Rivera, M. Bello and D. Méndez-Luna, J. Biomol. Struct. Dyn., 2019, 4671–4684 CrossRef PubMed.
- X. Du, B. Yang, Q. An, Y. G. Assaraf, X. Cao and J. Xia, Innovation, 2021, 2, 100103 CAS.
- Y.-C. Zhang, Q. Zhou and Y.-L. Wu, Cancer Lett., 2019, 459, 240–247 CrossRef CAS PubMed.
- X. Zhai, R. A. Ward, P. Doig and A. Argyrou, Biochemistry, 2020, 59, 1428–1441 CrossRef CAS PubMed.
- C.-S. Tan, N. B. Kumarakulasinghe, Y.-Q. Huang, Y. L. E. Ang, J. R.-E. Choo, B.-C. Goh and R. A. Soo, Mol. Cancer, 2018, 17, 1–14 Search PubMed.
- A. Mathialagan, T. Visnumukkala, K. Kademane, I. Agus, B. Mathialagan and T. Palaniyappan, J. Adv. Pharm. Educ. Res., 2018, 8, 115–120 CAS.
- R. A. Kardile, A. P. Sarkate, D. K. Lokwani, S. V. Tiwari, R. Azad and S. R. Thopate, Eur. J. Med. Chem., 2023, 245, 114889 CrossRef CAS PubMed.
- K. Ohashi, L. V. Sequist, M. E. Arcila, C. M. Lovly, X. Chen, C. M. Rudin, T. Moran, D. R. Camidge, C. L. Vnencak-Jones and L. Berry, Clin. Cancer Res., 2013, 19, 2584–2591 CrossRef CAS PubMed.
- M. Patel, A. Eckburg, S. Gantiwala, Z. Hart, J. Dein, K. Lam and N. Puri, Cancers, 2021, 13, 1115 CrossRef CAS PubMed.
- I. Ahmad, M. Shaikh, S. Surana, A. Ghosh and H. Patel, J. Biomol. Struct. Dyn., 2022, 40, 3046–3059 CrossRef CAS PubMed.
- Z. Du and C. M. Lovly, Mol. Cancer, 2018, 17, 1–13 CrossRef PubMed.
- N. Karachaliou, C. Mayo-de las Casas, C. Queralt, I. de Aguirre, B. Melloni, F. Cardenal, R. Garcia-Gomez, B. Massuti, J. M. Sánchez and R. Porta, JAMA Oncol., 2015, 1, 149–157 CrossRef PubMed.
- J. Rawluk and C. F. Waller, Small molecules in oncology, 2018, pp. 235–246 Search PubMed.
- Q. He, J. Liu, X. Cai, M. He, C. Li, H. Liang, B. Cheng, X. Xia, M. Guo and P. Liang, Transl. Lung Cancer Res., 2021, 10, 4120 CrossRef PubMed.
- S. Johnston and A. Leary, Drugs Today, 2006, 42, 441–453 CrossRef CAS PubMed.
- C.-H. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS PubMed.
- S. S. Fois, P. Paliogiannis, A. Zinellu, A. G. Fois, A. Cossu and G. Palmieri, Int. J. Mol. Sci., 2021, 22, 612 CrossRef CAS PubMed.
- T.-H. Tsai, S.-G. Wu, Y.-L. Chang, C.-T. Wu, M.-F. Tsai, P.-F. Wei, C.-H. Yang, C.-J. Yu, P.-C. Yang and J.-Y. Shih, J. Thorac. Oncol., 2012, 7, 993–1000 CrossRef CAS PubMed.
- V. D. Cataldo, D. L. Gibbons, R. Pérez-Soler and A. Quintás-Cardama, N. Engl. J. Med., 2011, 364, 947–955 CrossRef CAS PubMed.
- K. Miyazaki, T. Tamura, T. Kaburagi, K. Saito, M. Inagaki, T. Yamashita, H. Ichimura, T. Nawa, T. Endo and K. Hayashihara, Anticancer Res., 2018, 38, 5409–5415 CrossRef CAS PubMed.
- L. Huang and L. Fu, Acta Pharm. Sin. B, 2015, 5, 390–401 CrossRef PubMed.
- C. Ma, S. Wei and Y. Song, J. Thorac. Dis., 2011, 3, 10 CAS.
- T. Kosaka, J. Tanizaki, R. M. Paranal, H. Endoh, C. Lydon, M. Capelletti, C. E. Repellin, J. Choi, A. Ogino and A. Calles, Cancer Res., 2017, 77, 2712–2721 CrossRef CAS PubMed.
- D.-D. Li, Y.-J. Qin, J. Sun, J.-R. Li, F. Fang, Q.-R. Du, Y. Qian, H.-B. Gong and H.-L. Zhu, PLoS One, 2013, 8, e69427 CrossRef CAS PubMed.
- S. Pimple, Synthesis and biological evaluation of EGFR/HER-2 inhibitors: analogs of 5-substituted-4-anilinoquinazoline and 6,7-disubstituted-4-anilinoquinoline-3-carbonitrile: screening for development of novel PET tracers, Corpus ID: 91200718, University of Geneva, 2014 Search PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- M. Zou, J. Li, B. Jin, M. Wang, H. Chen, Z. Zhang, C. Zhang, Z. Zhao and L. Zheng, Bioorg. Chem., 2021, 114, 105200 CrossRef CAS PubMed.
- M. Joshi, S. M. Rizvi and C. P. Belani, Cancer Manage. Res., 2015, 75–82 CrossRef PubMed.
- J. He, P. Ma, D. Zhao, X. Shi, R. Guo, W. Gao and Y. Shu, Cancer, 2023, 1513–1522 CrossRef CAS PubMed.
- D. R. Camidge, W. Pao and L. V. Sequist, Nat. Rev. Clin. Oncol., 2014, 11, 473–481 CrossRef CAS PubMed.
- M. Nagasaka, V. W. Zhu, S. M. Lim, M. Greco, F. Wu and S.-H. I. Ou, J. Thorac. Oncol., 2021, 16, 740–763 CrossRef CAS PubMed.
- W. Han and Y. Du, Chem. Biodiversity, 2017, 14, e1600372 CrossRef PubMed.
- H. Cheng, S. K. Nair and B. W. Murray, Bioorg. Med. Chem. Lett., 2016, 26, 1861–1868 CrossRef CAS PubMed.
- R. A. Ward, M. J. Anderton, S. Ashton, P. A. Bethel, M. Box, S. Butterworth, N. Colclough, C. G. Chorley, C. Chuaqui and D. A. Cross, J. Med. Chem., 2013, 56, 7025–7048 CrossRef CAS PubMed.
- J. W. Carlisle and S. S. Ramalingam, Future Oncol., 2019, 15, 805–816 CrossRef CAS PubMed.
- W. Cheng, J. Zhou, X. Tian and X. Zhang, Curr. Med. Chem., 2016, 23, 3343–3359 CrossRef CAS PubMed.
- R. Bao, P. Gao, F. Zhang, Z. Tong, H. Yu and Y. Xu, Cancer Res., 2016, 76, 3063 CrossRef.
- D. Planchard, Y. Loriot, F. Andre, A. Gobert, N. Auger, L. Lacroix and J. Soria, Ann. Oncol., 2015, 26, 2073–2078 CrossRef CAS PubMed.
- M. J. Niederst, H. Hu, H. E. Mulvey, E. L. Lockerman, A. R. Garcia, Z. Piotrowska, L. V. Sequist and J. A. Engelman, Clin. Cancer Res., 2015, 21, 3924–3933 CrossRef CAS PubMed.
- D. Ercan, H. G. Choi, C.-H. Yun, M. Capelletti, T. Xie, M. J. Eck, N. S. Gray and P. A. Jänne, Clin. Cancer Res., 2015, 21, 3913–3923 CrossRef CAS PubMed.
- C. Ricordel, L. Friboulet, F. Facchinetti and J.-C. Soria, Ann. Oncol., 2018, 29, i28–i37 CrossRef CAS PubMed.
- I. Ahmad, R. Pawara, A. Pathan and H. Patel, in Natural Products as Enzyme Inhibitors: An Industrial Perspective, Springer, 2022, pp. 1–23 Search PubMed.
- Y. Jia, C.-H. Yun, E. Park, D. Ercan, M. Manuia, J. Juarez, C. Xu, K. Rhee, T. Chen and H. Zhang, Nature, 2016, 534, 129–132 CrossRef CAS PubMed.
- K. Kashima, H. Kawauchi, H. Tanimura, Y. Tachibana, T. Chiba, T. Torizawa and H. Sakamoto, Mol. Cancer Ther., 2020, 19, 2288–2297 CrossRef CAS PubMed.
- S. K. Tripathi and B. K. Biswal, Drug Discovery Today, 2021, 26, 1466–1472 CrossRef CAS PubMed.
- S. Wang, Y. Song and D. Liu, Cancer Lett., 2017, 385, 51–54 CrossRef CAS PubMed.
- M. Shaikh, Y. Shinde, R. Pawara, M. Noolvi, S. Surana, I. Ahmad and H. Patel, J. Med. Chem., 2021, 65, 1008–1046 CrossRef PubMed.
- X. Lu, L. Yu, Z. Zhang, X. Ren, J. B. Smaill and K. Ding, Med. Res. Rev., 2018, 38, 1550–1581 CrossRef CAS PubMed.
- Y.-Y. Hei, Y. Shen, J. Wang, H. Zhang, H.-Y. Zhao, M. Xin, Y.-X. Cao, Y. Li and S.-Q. Zhang, Bioorg. Med. Chem., 2018, 26, 2173–2185 CrossRef CAS PubMed.
- H. Lei, S. Fan, H. Zhang, Y.-J. Liu, Y.-Y. Hei, J.-J. Zhang, A.-Q. Zheng, M. Xin and S.-Q. Zhang, Eur. J. Med. Chem., 2020, 186, 111888 CrossRef CAS PubMed.
- Z. Su, T. Yang, J. Wang, M. Lai, L. Tong, G. Wumaier, Z. Chen, S. Li, H. Li and H. Xie, Bioorg. Med. Chem. Lett., 2020, 30, 127327 CrossRef CAS PubMed.
- H.-Y. Zhao, X.-X. Xi, M. Xin and S.-Q. Zhang, Bioorg. Chem., 2022, 106057 CrossRef CAS PubMed.
- A. A. Romu, Z. Lei, B. Zhou, Z.-S. Chen and V. Korlipara, Bioorg. Med. Chem. Lett., 2017, 27, 4832–4837 CrossRef CAS PubMed.
- J. Lategahn, M. Keul, P. Kloevekorn, H. L. Tumbrink, J. Niggenaber, M. P. Mueller, L. Hodson, M. Flasshoff, J. Hardick and T. Grabe, Chem. Sci., 2019, 10, 10789–10801 RSC.
- L. Tan, Z. Zhang, D. Gao, J. Luo, Z.-C. Tu, Z. Li, L. Peng, X. Ren and K. Ding, J. Med. Chem., 2016, 59, 6807–6825 CrossRef CAS PubMed.
- Q. Xun, Z. Zhang, J. Luo, L. Tong, M. Huang, Z. Wang, J. Zou, Y. Liu, Y. Xu and H. Xie, J. Med. Chem., 2018, 61, 2353–2371 CrossRef CAS PubMed.
- S. Chang, L. Zhang, S. Xu, J. Luo, X. Lu, Z. Zhang, T. Xu, Y. Liu, Z. Tu and Y. Xu, J. Med. Chem., 2012, 55, 2711–2723 CrossRef CAS PubMed.
- J. Shen, T. Zhang, S.-J. Zhu, M. Sun, L. Tong, M. Lai, R. Zhang, W. Xu, R. Wu and J. Ding, J. Med. Chem., 2019, 62, 7302–7308 CrossRef CAS PubMed.
- H. Zhang, J. Wang, Y. Shen, H.-Y. Wang, W.-M. Duan, H.-Y. Zhao, Y.-Y. Hei, M. Xin, Y.-X. Cao and S.-Q. Zhang, Eur. J. Med. Chem., 2018, 148, 221–237 CrossRef CAS PubMed.
- S. Lee, J. Kim, K. B. Duggirala, A. Go, I. Shin, B. C. Cho, G. Choi, C. H. Chae and K. Lee, Bull. Korean Chem. Soc., 2018, 39, 895–898 CrossRef CAS.
- Q. Li, T. Zhang, S. Li, L. Tong, J. Li, Z. Su, F. Feng, D. Sun, Y. Tong and X. Wang, ACS Med. Chem. Lett., 2019, 10, 869–873 CrossRef CAS PubMed.
- H. Park, H. Y. Jung, S. Mah and S. Hong, Angew. Chem., Int. Ed., 2017, 56, 7634–7638 CrossRef CAS PubMed.
- K. S. Karnik, A. P. Sarkate, S. V. Tiwari, R. Azad and P. S. Wakte, Bioorg. Chem., 2021, 115, 105226 CrossRef CAS PubMed.
- C. To, J. Jang, T. Chen, E. Park, M. Mushajiang, D. J. De Clercq, M. Xu, S. Wang, M. D. Cameron and D. E. Heppner, Cancer Discovery, 2019, 9, 926–943 CrossRef CAS PubMed.
- R. Selig, M. Goettert, V. Schattel, D. Schollmeyer, W. Albrecht and S. Laufer, J. Med. Chem., 2012, 55, 8429–8439 CrossRef CAS PubMed.
- M. Günther, M. Juchum, G. Kelter, H. Fiebig and S. Laufer, Angew. Chem., Int. Ed., 2016, 55, 10890–10894 CrossRef PubMed.
- M. Gunther, J. Lategahn, M. Juchum, E. Doring, M. Keul, J. Engel, H. L. Tumbrink, D. Rauh and S. Laufer, J. Med. Chem., 2017, 60, 5613–5637 CrossRef PubMed.
- H. Engelhardt, D. Böse, M. Petronczki, D. Scharn, G. Bader, A. Baum, A. Bergner, E. Chong, S. Döbel and G. Egger, J. Med. Chem., 2019, 62, 10272–10293 CrossRef CAS PubMed.
- T. Rao, Z. Tan, J. Peng, Y. Guo, Y. Chen, H. Zhou and D. Ouyang, Pharmacol. Res., 2019, 146, 104283 CrossRef CAS PubMed.
- M. Facompré, C. Tardy, C. Bal-Mahieu, P. Colson, C. Perez, I. Manzanares, C. Cuevas and C. Bailly, Cancer Res., 2003, 63, 7392–7399 Search PubMed.
- N. Nishiya, Y. Oku, C. Ishikawa, T. Fukuda, S. Dan, T. Mashima, M. Ushijima, Y. Furukawa, Y. Sasaki and K. Otsu, Cancer Sci., 2021, 112, 1963–1974 CrossRef CAS.
- S.-L. Paiva and C. M. Crews, Curr. Opin. Chem. Biol., 2019, 50, 111–119 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87.e75 CrossRef CAS.
- A. D. Buhimschi, H. A. Armstrong, M. Toure, S. Jaime-Figueroa, T. L. Chen, A. M. Lehman, J. A. Woyach, A. J. Johnson, J. C. Byrd and C. M. Crews, Biochemistry, 2018, 57, 3564–3575 CrossRef CAS PubMed.
- H. Zhang, R. Xie, H. Ai-Furas, Y. Li, Q. Wu, J. Li, F. Xu and T. Xu, ACS Med. Chem. Lett., 2022, 13, 278–283 CrossRef CAS.
- Z. Song, Y. Ge, C. Wang, S. Huang, X. Shu, K. Liu, Y. Zhou and X. Ma, J. Med. Chem., 2016, 59, 6580–6594 CrossRef CAS PubMed.
- H. Patel, R. Pawara and S. Surana, Comput. Biol. Chem., 2018, 74, 167–189 CrossRef CAS PubMed.
- F. Caporuscio, A. Tinivella, V. Restelli, M. S. Semrau, L. Pinzi, P. Storici, M. Broggini and G. Rastelli, Future Med. Chem., 2018, 10, 1545–1553 CrossRef CAS PubMed.
- A. C. Gelatti, A. Drilon and F. C. Santini, Lung Cancer, 2019, 137, 113–122 CrossRef PubMed.
- V. Hirsh, Curr. Oncol., 2011, 18, 126–138 CrossRef CAS PubMed.
- B. Madke, P. Gole, P. Kumar and U. Khopkar, Indian J. Dermatol., 2014, 59, 271 CrossRef PubMed.
- O. Arrieta, Z. L. Zatarain-Barrón, F. Aldaco, F. Barrón, R. Báez-Saldaña, S. Campos-Gómez, R. Trejo and J. De la Garza, J. Thorac. Oncol., 2019, 14, 1695–1700 CrossRef PubMed.
- D. Wu, J. Xie, H. Dai and W. Fang, BMC Health Serv. Res., 2022, 22, 431 CrossRef PubMed.
- O. Arrieta, R. Catalán, S. Guzmán-Vazquez, F. Barrón, L. Lara-Mejía, H. Soto-Molina, M. Ramos-Ramírez, D. Flores-Estrada and J. de la Garza, BMC Cancer, 2020, 20, 1–8 CrossRef PubMed.
- T. Khoo and L. Gao, Expert Rev. Pharmacoeconomics Outcomes Res., 2021, 21, 415–423 CrossRef PubMed.
- Y. Shu, Y. Ding, X. He, Y. Liu, P. Wu and Q. Zhang, Front. Pharmacol., 2022, 3842 Search PubMed.
- S. Yao, X. Zhi, R. Wang, K. Qian, M. Hu and Y. Zhang, Thorac. Cancer, 2016, 7, 543–548 CrossRef CAS PubMed.
- N. Hidaka, E. Iwama, N. Kubo, T. Harada, K. Miyawaki, K. Tanaka, I. Okamoto, E. Baba, K. Akashi and H. Sasaki, Lung Cancer, 2017, 108, 75–82 CrossRef PubMed.
- Z. Wang, J.-J. Yang, J. Huang, J.-Y. Ye, X.-C. Zhang, H.-Y. Tu, H. Han-Zhang and Y.-L. Wu, J. Thorac. Oncol., 2017, 12, 1723–1727 CrossRef PubMed.
- X. Zhu, L. Chen, L. Liu and X. Niu, Front. Oncol., 2019, 9, 1044 CrossRef PubMed.
- N. Girard, Future Oncol., 2019, 15, 2983–2997 CrossRef CAS PubMed.
- Y. Tomizawa, Y. Fujita, A. Tamura, M. Shirai, S. Shibata, T. Kawabata, T. Shibayama, S. Fukai, M. Kawahra and R. Saito, Lung Cancer, 2010, 68, 269–272 CrossRef PubMed.
- S. Leong, R. A. Moss, D. W. Bowles, J. A. Ware, J. Zhou, J. M. Spoerke, M. R. Lackner, G. Shankar, J. L. Schutzman and R. van der Noll, Oncologist, 2017, 22, 1491–1499 CrossRef CAS PubMed.
- G. Mountzios, Ann. Transl. Med., 2018, 6 CAS.
- H. Akamatsu, Y. Koh, Y. Ozawa, D. Fujimoto, A. Hata, N. Katakami, K. Tomii, T. Shimokawa and N. Yamamoto, Clin. Lung Cancer, 2018, 19, e871–e874 CrossRef CAS PubMed.
- L. Regales, Y. Gong, R. Shen, E. de Stanchina, I. Vivanco, A. Goel, J. A. Koutcher, M. Spassova, O. Ouerfelli and I. K. Mellinghoff, J. Clin. Investig., 2009, 119, 3000–3010 CAS.
- E. Kucuksayan and T. Ozben, Curr. Top. Med. Chem., 2017, 17, 907–918 CrossRef CAS PubMed.
- X. Cai, H.-X. Zhai, J. Wang, J. Forrester, H. Qu, L. Yin, C.-J. Lai, R. Bao and C. Qian, J. Med. Chem., 2010, 53, 2000–2009 CrossRef CAS PubMed.
- Y. Li, C. Tan, C. Gao, C. Zhang, X. Luan, X. Chen, H. Liu, Y. Chen and Y. Jiang, Bioorg. Med. Chem., 2011, 19, 4529–4535 CrossRef CAS PubMed.
- S. Mahboobi, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2010, 53, 8546–8555 CrossRef CAS PubMed.
- M. M. Alam, A. H. Hassan, Y. H. Kwon, H. J. Lee, N. Y. Kim, K. H. Min, S.-Y. Lee, D.-H. Kim and Y. S. Lee, Arch. Pharmacal Res., 2018, 41, 35–45 CrossRef CAS PubMed.
- N. M. Raghavendra, D. Pingili, S. Kadasi, A. Mettu and S. Prasad, Eur. J. Med. Chem., 2018, 143, 1277–1300 CrossRef CAS PubMed.
- H. K. Byeon, M. Ku and J. Yang, Exp. Mol. Med., 2019, 51, 1–14 CrossRef CAS PubMed.
- C. L. Bello, L. Sherman, J. Zhou, L. Verkh, J. Smeraglia, J. Mount and K. J. Klamerus, Anti-Cancer Drugs, 2006, 17, 353–358 CrossRef CAS PubMed.
- S. Wilhelm, C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz, R. Simantov and S. Kelley, Nat. Rev. Drug Discovery, 2006, 5, 835–844 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |