
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of Sb–pyromellitic acid metal–organic framework material and its sodium storage properties†
Zhiyan Hea,
Wei Zhanga and
Mingqi Li *ab
*ab
aCollege of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637009, China. E-mail: lmingq888@aliyun.com
bChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, Nanchong 637009, China
First published on 2nd June 2023
Abstract
Developing electrode materials with high capacity and low cost is crucial for promoting the application of sodium-ion batteries. In this paper, a new Sb–PMA-300 metal–organic framework (MOF) material is synthesized by chelation of Sb3+ and pyromellitic acid (PMA) followed by a heat treatment at 300 °C. As anodes for sodium-ion batteries, the Sb–PMA-300 composite exhibits a stable capacity of 443 mA h g−1 at a current density of 0.1 A g−1. At a current density of 1 A g−1, the discharge capacity is maintained at 326.4 mA h g−1 after 200 cycles. The electrode process dynamics of this material are mainly controlled by diffusion. The values of the diffusion coefficient of Na+ are between 10−12 and 3.0 × 10−10 cm2 s−1 during discharging, while they are between 10−12 and 5.0 × 10−11 cm2 s−1 during charging. The excellent cycle stability is attributed to the special structure of the MOF material, where the organic ligand prevents the aggregation of Sb alloy particles and buffers the tension resulting from volume variation.
1. Introduction
Due to high specific capacity (660 mA h g−1), suitable working potential (−0.8 V vs. Na+/Na) and low cost, metal Sb is regarded as a promising anode material for sodium-ion batteries.1–4 However, its large volume changes during alloying/dealloying lead to poor cycle stability, which severely hinders its application.5 In recent years, researchers have developed a series of strategies to overcome this problem, such as nanocrystallinity,6–8 alloying,9–11 carbon-based coating,12–14 and mixing with carbon.15–17Metal–organic frameworks (MOFs) have been extensively explored and studied due to their unique microstructure,18–20 which is very attractive as an electrode for secondary batteries.21–23 For instance, Wang formed Sb-MOF by liquid-phase reaction of C6H9O6Sb and organic ligand methylimidazole, and then further heat-treated the precursor to obtain a new type of nitrogen–carbon-doped polyhedral structure Sb/Sb2O3-NC nanoparticles.24 When used as an anode for Na-ion batteries, it exhibited a capacity of 318 mA h g−1 after 100 cycles at a current density of 5 A g−1 and exhibited an excellent rate performance of 183 mA h g−1 at 2 A g−1. Li used Cu-MOF as a precursor to embed Sb into porous carbon nanomaterials through in situ substitution.25 This non-destructive substitution reaction maintains the porous octahedral structure of Cu-MOF and induces the formation of ultrafine Sb nanocrystals in close contact with the carbon matrix. The formed Sb@PC composite has excellent sodium storage performance. It can still maintain a specific capacity of about 635 mA h g−1 after 200 cycles at a current density of 0.1 A g−1, and the reversible capacity can still reach 517 mA h g−1 at a current density of 2 A g−1. Yu synthesized Sb⊂CTHN derived from MOF, fully embedding tiny Sb nanoparticles in MOF-derived carbon and TiO2 nanotubes.26 The as-prepared Sb⊂CTHN has good stability as an anode for sodium-ion batteries. After 2000 cycles at a current density of 2 A g−1, it still has a specific capacity of ∼345 mA h g−1, with a capacity retention of about 93%.
MOF material is very attractive as an electrode for secondary batteries. Its advantages include: (1) it is easily synthesized with a controllable structure; (2) metal ions are confined to the organic framework, which can effectively inhibit the growth of alloy particles during cycling;27 (3) electroactive groups in organic ligands can provide partial capacity. Although antimony-based MOF materials have been extensively studied as battery anode materials, most of the research focuses on composite materials after high-temperature carbonization using MOFs as the precursor. So far, there are very few reports on the direct use of Sb-based MOF materials as electrodes for sodium-ion batteries. In this work, it is found that Sb–PMA (MOF) material synthesized by chelation of Sb3+ and pyromellitic acid (PMA) followed by a heat treatment at 300 °C showed high specific capacity and good cycle stability. The Sb–PMA-300 composite exhibits a stable capacity of 443 mA h g−1 at a current density of 0.1 A g−1. At a current density of 1 A g−1, the discharge capacity is maintained at 326.4 mA h g−1 after 200 cycles. In addition, the reason for the improvement of electrochemical performance and the electrode process dynamics of Sb–PMA-300 is also investigated. This work not only develops a new anode material for sodium-ion batteries, but also provides a simple and low-cost method for improving Sb anode materials.
2. Experiment
2.1. Materials
All chemical reagents were purchased from Aladdin Reagent Co., Ltd. and directly used without further purification. First, 0.441 g of C6H9O6Sb (97%) was dissolved in 10 mL methanol to obtain solution A, and 0.445 g of pyromellitic acid (PMA, 98%) was dissolved in 10 mL methanol to obtain solution B. Then, solution A was slowly dropped into solution B with stirring. After stirring for 24 hours at room temperature, the solid product was collected by centrifugation and washed with methanol twice. The collected solid was dried in vacuum at 60 °C for 6 h and then transferred to a tubular furnace. The dried solid was labeled as Sb–PMA. Subsequently, the Sb–PMA was heated to 300 °C at a ramp of 2 °C min−1 under a flow of 30 mL min−1 of N2 and kept for 5 hours. After natural cooling, the final product was obtained, which was labeled as Sb–PMA-300. For comparison, the Sb–PMA was heated at 300 °C for 2 h and 8 h, respectively, and the obtained products were denoted as Sb–PMA-300-2 and Sb–PMA-300-8. Then, we change the heat treatment temperature of Sb–PMA to 400 °C to get Sb–PMA-400.The synthesis of pure Sb was as follows: 1.45 g of C8H10O15Sb2K2 powder was dissolved in 50 mL of distilled water. Subsequently, NaBH4 aqueous solution (0.75 g, 10 mL) was dropwise added to the above system at 60 °C with stirring for 1 h. Then, the Sb product was rinsed with distilled water five times and dried at 60 °C under vacuum for 10 h. To construct a full cell, a cathode material, i.e. C coated Na3V2(PO4)3/rGO (C coated NVP/rGO), was prepared according to our previous work.28
2.2. Physical characterization
The functional groups and structures of the synthesized materials were examined using Fourier transform infrared spectroscopy (FTIR, Thermo scientific ATR-FTIR) and Raman spectroscopy (RM, Witec alpha300 access). The crystal structure of the materials was examined by X-ray diffractometry (XRD; D8 Discover, Bruker), where Cu Kα was used as the radiation source with a wavelength of 1.5406 Å and the scanning rate was 5° min−1. The morphology and microstructure of the materials were examined by field emission scanning electron microscopy (FESEM, GeminiSEM 500) and transmission electron microscopy (TEM). The composition of the materials was determined by energy dispersive X-ray spectroscopy (EDS, Oxford) and elemental analysis instrument (Vario EL III). The elemental chemical information of the material surface was tested by X-ray photoelectron spectroscopy (XPS, ESCALAB 250 Xi). The specific surface area and porosity of the materials were examined by Brunauer–Emmett–Teller (BET, Micromeritics ASAP 2460) at 77 K, using N2 as the adsorption/desorption gas. Thermogravimetric analysis of samples was conducted using a TGA 209F3 at Ar atmosphere and the heating rate was 10 °C min−12.3. Electrochemical measurement
To prepare the working electrode, the synthetic material was the active substance, Super P was the conductive agent, and sodium alginate was the binder, in which distilled water acted as a solvent and mass ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15. Subsequently, the formed paste was coated uniformly on copper foil and then dried in vacuum at 110 °C for 12 h. The dried sheet was stamped into a disc-shaped electrode. The mass loading of the active substance was about 0.8 mg cm−2. The 2032 button batteries were assembled in a glove box filled with argon to evaluate the electrochemical performance of the prepared material as anodes for sodium-ion batteries. The separator was Whatman glass fiber (GF/A), and the counter/reference electrode was metal sodium. For the full cell, C coated NVP/rGO served as the cathode material. The cathode electrodes were prepared by casting a slurry mixture of C coated NVP/rGO, Super-P carbon black and polyvinylidene fluoride (PVDF) with a mass ratio of 83:10:7 onto a piece of aluminum foil. To preferably reveal the full cell properties, the anodes were first discharged and charged for 5 cycles to form a stable SEI and remove the irreversible capacity. The electrolyte for all cells was 1 mol L−1 NaClO4 in which the solvent is EC:DMC:EMC = 1:1:1 vol% with 5.0% FEC additive. The constant current charge and discharge test was conducted on a Shenzhen Neware battery test system at room temperature. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed on an EC-Lab electrochemical workstation. The sweep speed of CV was 0.2 mV s−1. The frequency range of the electrochemical impedance test was 100 kHz to 100 mHz over an excitation signal of 10 mV.
:15:15. Subsequently, the formed paste was coated uniformly on copper foil and then dried in vacuum at 110 °C for 12 h. The dried sheet was stamped into a disc-shaped electrode. The mass loading of the active substance was about 0.8 mg cm−2. The 2032 button batteries were assembled in a glove box filled with argon to evaluate the electrochemical performance of the prepared material as anodes for sodium-ion batteries. The separator was Whatman glass fiber (GF/A), and the counter/reference electrode was metal sodium. For the full cell, C coated NVP/rGO served as the cathode material. The cathode electrodes were prepared by casting a slurry mixture of C coated NVP/rGO, Super-P carbon black and polyvinylidene fluoride (PVDF) with a mass ratio of 83:10:7 onto a piece of aluminum foil. To preferably reveal the full cell properties, the anodes were first discharged and charged for 5 cycles to form a stable SEI and remove the irreversible capacity. The electrolyte for all cells was 1 mol L−1 NaClO4 in which the solvent is EC:DMC:EMC = 1:1:1 vol% with 5.0% FEC additive. The constant current charge and discharge test was conducted on a Shenzhen Neware battery test system at room temperature. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed on an EC-Lab electrochemical workstation. The sweep speed of CV was 0.2 mV s−1. The frequency range of the electrochemical impedance test was 100 kHz to 100 mHz over an excitation signal of 10 mV.
3. Results and discussion
3.1. Morphology and structural characterization
The morphology and microstructure of Sb–PMA-300 were examined by SEM and TEM. As shown in Fig. 1(a) and (b), Sb–PMA-300 is irregular particles with diameters ranging from 10 to 160 nm. From the TEM image in Fig. 1(c), the adjacent particles are connected to each other to form chains, which is conducive to the transfer of electrons and ions. The HRTEM image in Fig. 1(d) shows that the diffraction fringes are short-range ordered but long-range disordered, while the selected area electron diffraction (SAED) pattern also does not show obvious diffraction rings, indicating mainly an amorphous structure.29 | ||
| Fig. 1 (a and b) SEM images, (c and d) TEM image and HRTEM image with SAED pattern, (e) element distribution maps of Sb–PMA-300. | ||
The element maps from EDS analysis show that it contains Sb, C and O elements, which are evenly distributed throughout the material (Fig. 1(e)). Combined with the results of elemental analysis and EDS analysis, it can be calculated that Sb–PMA-300 is composed of 68.3 wt% Sb, 14.7 wt% C, 16.0 wt% O and 1 wt% H.
Fig. 2(a) shows the XRD patterns of Sb–PMA-300. To examine the effects of annealing at 300 °C, the XRD pattern of Sb–PMA is also given. Sb–PMA presents only a broad diffraction peak at 2θ = 28°, indicating that it is a completely amorphous structure. In comparison, Sb–PMA-300 presents an obvious peak at 2θ = 28° and two small peaks at 2θ = 45.9 and 54.5°, respectively, which can be indexed to crystalline Sb2O3 (PDF# 43-1071).30 However, the intensity of these peaks is still very weak, indicating that the content of Sb2O3 is very low. FTIR and Raman spectroscopy were also used to characterize Sb–PMA-300. From Fig. 2(b), at 3000 cm−1, Sb–PMA showed a very weak O–H vibration peak, while not any characteristic of O–H peak is observed in the FTIR of Sb–PMA-300. In addition, both have absorption peaks of Sb–O–C at 599 cm−1.29 These results indicate that PMA successfully chelates with Sb3+ and the free carboxyl group is also removed after heat treatment. The thermogravimetric curve of Sb–PMA is shown in Fig. S1.† After heat treatment at 300 °C, the weight loss is approximately 4.7%, which originates from the decomposition or polymerization of uncoordinated carboxyl groups. In the Raman spectrum of Fig. 2(c), for Sb–PMA and Sb–PMA-300 samples, Sb–O vibration peak can be observed between 350 and 518 cm−1.31–34 The difference is that the peak of Sb–PMA-300 is slightly sharper. The peak at 1173 cm−1 is assigned to a C–C stretch distortion mode, including the coupling of carboxylic acid groups.7,35
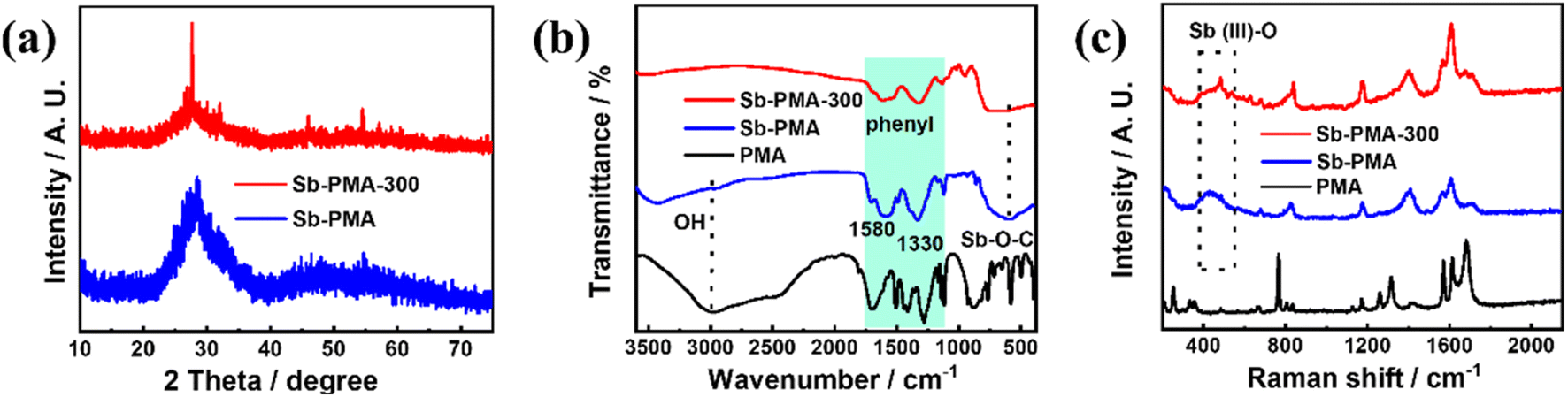 | ||
| Fig. 2 (a) XRD patterns, (b) FTIR spectra and (c) Raman spectra of Sb–PMA-300. | ||
To understand the chemical information of the elements on the material surface, the Sb–PMA-300 composite was characterized using XPS. As seen in Fig. 3(a), Sb, C and O elements are detected. Fig. 3(b) and (c) shows C 1s, O 1s and Sb 3d HRXPS. Since the XPS spectra of O 1s and Sb 3d have a large part of overlap, they are put together in Fig. 3(c). C 1s HRXPS can be separated into four peaks at 288.6, 285.4, 284.7 and 284.4 eV, which belong to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, C–C and CC bonds, respectively (Fig. 3(b)).31,36 Sb 3d and O 1s HRXPS can be deconvoluted into four peaks (Fig. 3(c)). The two high intensity peaks at 540.1 and 530.6 eV are assigned to Sb 3d3/2 and Sb 3d5/2 in the Sb–O bond,37,38 respectively. The peaks at 532.3 and 530.8 eV originate from O 1s and correspond to C–O and CO bonds, respectively. These results indicate that Sb3+ and organic ligand PMA successfully coordinated to form MOFs. Fig. 3(d) shows the N2 adsorption and desorption isotherms of Sb–PMA-300. There is a clear hysteresis loop in the medium-pressure section, indicating the presence of mesopores. The adsorption volume of N2 is very small in the low-pressure section, indicating that there are very few micropores. After the relative pressure exceeds 0.8, the adsorption volume of N2 increases rapidly with increasing pressure, indicating that the material contains a number of macropores.39 These conclusions are further supported by the pore size distribution curves (Fig. 3(e)). Calculation shows that the specific surface area and pore volume of Sb–PMA-300 are 35.92 m2 g−1 and 0.126 cm3 g−1, respectively. The large specific surface area and rich pores help to increase the contact between electrolyte and material, thus improving the kinetics of the electrode process.
O, C–O, C–C and CC bonds, respectively (Fig. 3(b)).31,36 Sb 3d and O 1s HRXPS can be deconvoluted into four peaks (Fig. 3(c)). The two high intensity peaks at 540.1 and 530.6 eV are assigned to Sb 3d3/2 and Sb 3d5/2 in the Sb–O bond,37,38 respectively. The peaks at 532.3 and 530.8 eV originate from O 1s and correspond to C–O and CO bonds, respectively. These results indicate that Sb3+ and organic ligand PMA successfully coordinated to form MOFs. Fig. 3(d) shows the N2 adsorption and desorption isotherms of Sb–PMA-300. There is a clear hysteresis loop in the medium-pressure section, indicating the presence of mesopores. The adsorption volume of N2 is very small in the low-pressure section, indicating that there are very few micropores. After the relative pressure exceeds 0.8, the adsorption volume of N2 increases rapidly with increasing pressure, indicating that the material contains a number of macropores.39 These conclusions are further supported by the pore size distribution curves (Fig. 3(e)). Calculation shows that the specific surface area and pore volume of Sb–PMA-300 are 35.92 m2 g−1 and 0.126 cm3 g−1, respectively. The large specific surface area and rich pores help to increase the contact between electrolyte and material, thus improving the kinetics of the electrode process.
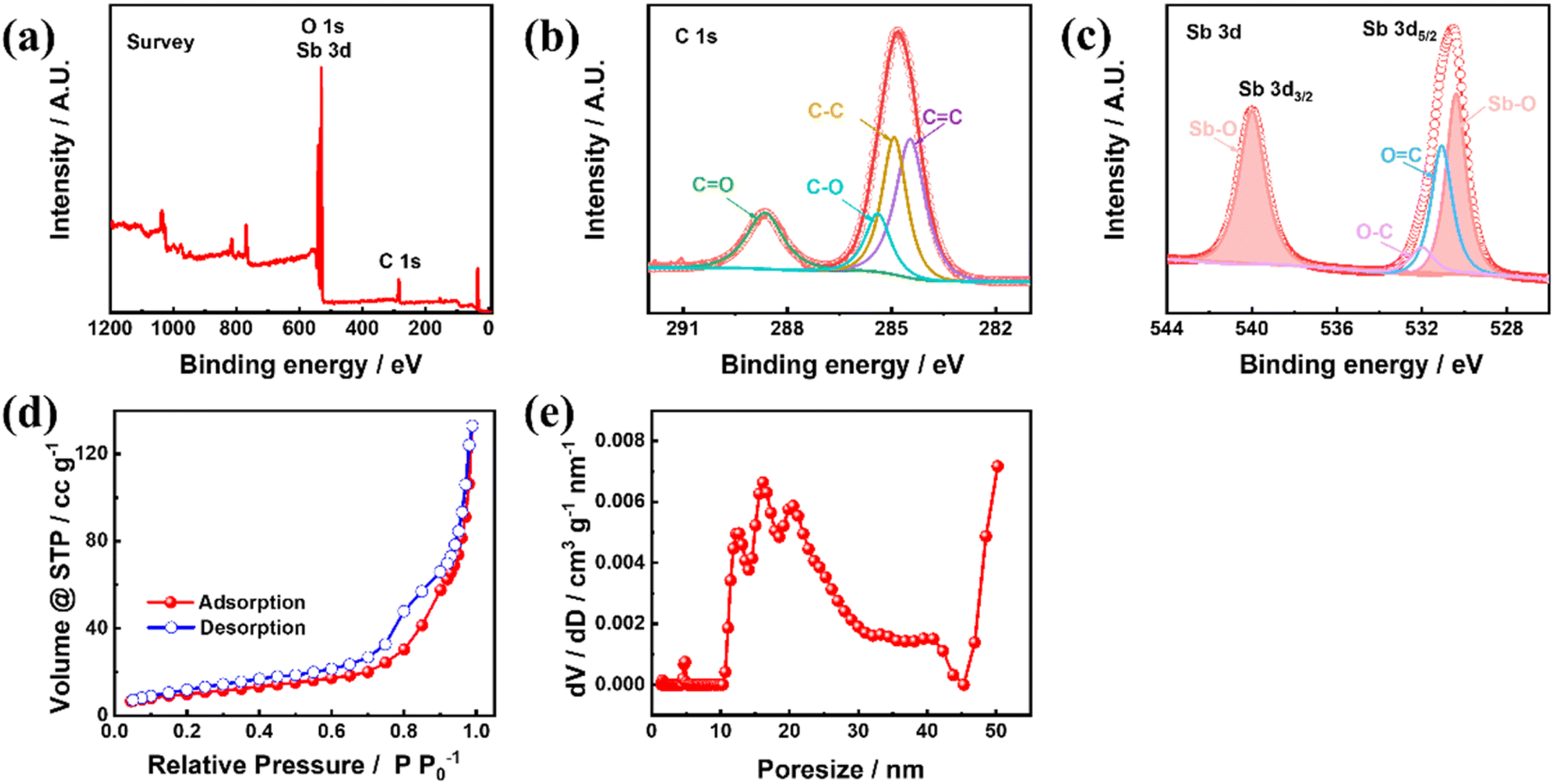 | ||
| Fig. 3 (a) XPS survey, (b) C 1s HRXPS, (c) O 1s and Sb 3d HRXPS, (d) N2 adsorption and desorption isotherms and (e) pore size distribution curve of Sb–PMA-300. | ||
3.2. Electrochemical properties
CR2032 was assembled to evaluate the electrochemical performance of Sb–PMA-300 as anodes for sodium-ion batteries. Fig. 4(a) shows the initial five CV curves at 0.2 mV s−1. In the first CV curve, this pair of redox peaks at 1.03 and 1.54 V correspond to a reversible reaction of the active groups in PMA ligands (Fig. 4(b)), while the two reduction peaks at 0.35 and 0.05 V are attributed to the reduction of Sb3+ → Sb0 and the alloying process of Sb0 → Na3Sb.12,40 From the second cycle, the alloying process of Sb contains five reduction peaks, which are located at 0.92, 0.74, 0.42, 0.33 and 0.05 V, indicating that the alloying process of Sb0 → Na3Sb is transformed into multi-step reactions.41 In all CV curves, the oxidation peak positions of the anode branches remain relatively stable, but the peak current increases to a certain extent with the activation of the electrode. Fig. 4(c) shows the first three charge and discharge profiles of Sb–PMA-300 at 0.1 A g−1. The charge and discharge profiles present multiple plateaus with different slopes, which accord with the multiple peaks in the CV curves. The first discharge and charge capacities are 702.8 and 370.6 mA h g−1 respectively, with an initial coulomb efficiency of 52.7%. The low initial efficiency mainly arises from the irreversible reduction of Sb3+ to Sb0, and the irreversible consumption of SEI film formation.42 As shown in Fig. 4(d), the coulombic efficiency increases to 98.6% after two cycles and remains relatively stable in the subsequent cycles. In the first seven cycles, the specific capacity gradually increases, indicating that the electrode experiences an activation process. The discharge capacity in the eighth cycle reaches 443 mA h g−1, and the attenuation was very slow in subsequent cycles. After 120 cycles, the discharge capacity is maintained at 417 mA h g−1, with a capacity retention of 94.1%. Fig. 4(e) shows the rate capability of Sb–PMA-300 at current densities of 0.1, 0.5, 1, and 2 A g−1. With the increase of the current density, the discharge capacity decreases. The stable discharge capacity at 2 A g−1 is about 216 mA h g−1, which is 47.2% of that at 0.1 A g−1, indicating that Sb–PMA-300 has a good rate capability. When the current density returns to 0.1 A g−1, the stable discharge capacity can be completely restored, indicating that the structure of the electrode is not destroyed after cycling at big current densities. Fig. 4(f) shows the cycle stability of Sb–PMA-300 at big current densities. After 200 cycles, the discharge capacities are kept at 327 and 184.52 mA h g−1 at 1 and 2 A g−1, respectively, confirming that the material can work at a large current density.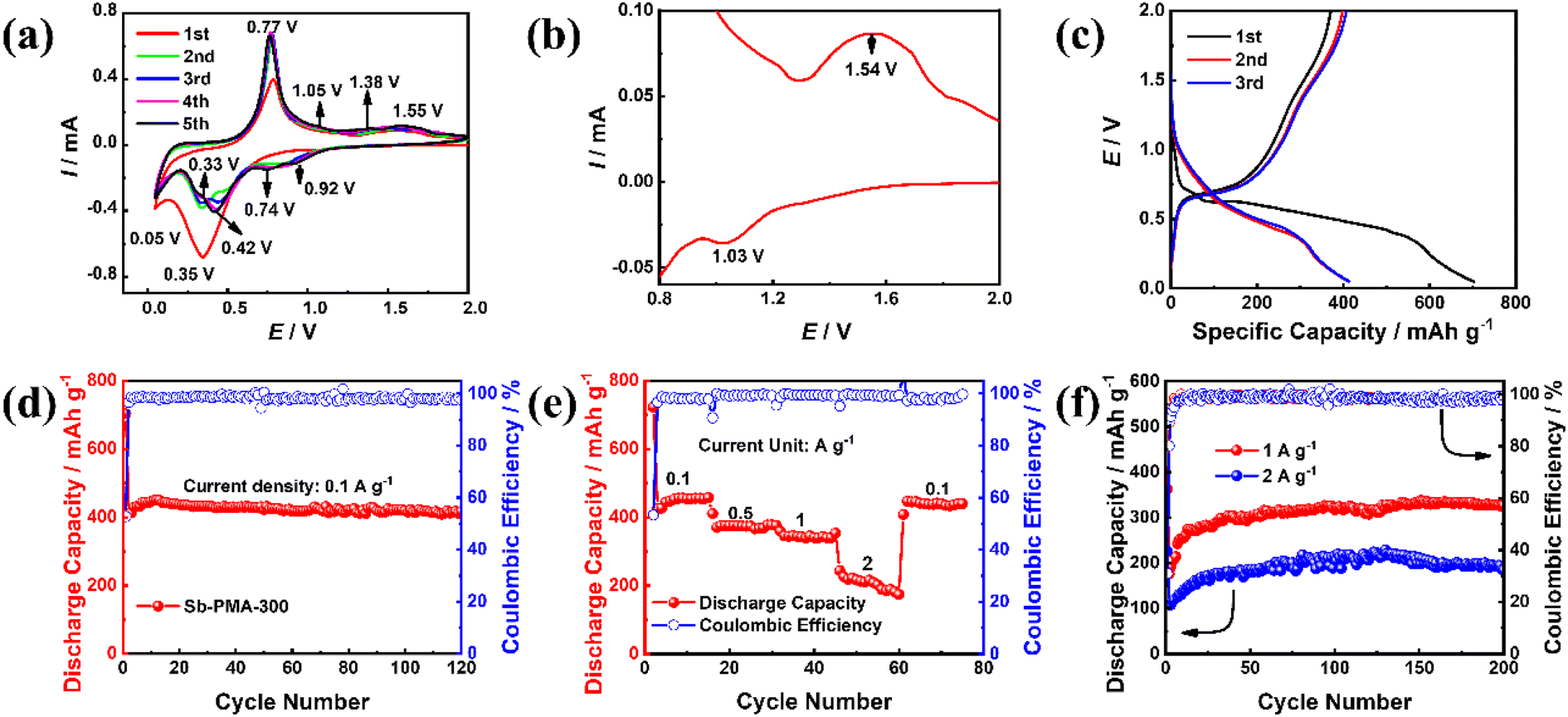 | ||
| Fig. 4 Electrochemical properties of Sb–PMA-300: (a) the first five CV curves at 0.2 mV s−1, (b) partial amplification of the first CV curve in (a), (c) the first three charge and discharge profiles at 0.1 A g−1, (d) cycle performance at 0.1 A g−1, (e) rate capability, and (f) cycle performance at 1 A g−1 and 2 A g−1. | ||
To investigate the effect of annealing temperature, Fig. S2† shows the cycling performance of Sb–PMA and Sb–PMA-400 electrodes. Compared with the Sb–PMA electrode, although the Sb–PMA-300 electrode presents a close specific capacity, its cycle stability is obviously better. After 50 cycles, the capacity of Sb–PMA begins to decline. After 120 cycles, the discharge capacity decays to 177 mA h g−1, with a 42.4% capacity retention. In addition, the charge–discharge efficiency of Sb–PMA has been slightly lower than that of Sb–PMA-300, indicating that heat treatment can significantly improve the electrochemical stability of Sb–PMA. Heat treatment improves the binding force of coordination bonds and removes uncoordinated carboxyl groups. However, when the annealing temperature increases to 400 °C, due to the onset of severe carbonization, although the obtained Sb–PMA-400 displays a similar specific capacity to pure Sb, but its cycling stability is very poor. Therefore, 300 °C is selected as the annealing temperature. The effect of annealing time is also investigated. Fig. S3† shows the cycling performance and rate capability of Sb–PMA annealed at 300 °C for 2, 5 and 8 h, respectively. From the perspective of cycle stability and rate performance, the electrochemical performance of Sb–PMA annealed at 300 °C for 5 h is the best. From Fig. S4,† long term heat treatment leads to the formation of more Sb2O3, which seriously affects the coordination structure.
To gain more insights into the redox mechanism of the Sb–PMA-300, ex situ FT-IR spectra are conducted upon redox processes. The peak assigned to the CO groups at 1636 cm−1 disappears and the C–O group at 1250 cm−1 increases after discharging to 0.05 V (Fig. S5†), and a reverse trend could be observed after recharging, indicating the transformation between CO and C–O groups.43 This result indicates that the carbonyl functional group in the organic ligand participates in the reaction during charge and discharge, providing part of the capacity.
To investigate the reasons for the excellent electrochemical performance, EIS and ex situ SEM were used to characterize the cycled Sb–PMA-300 electrodes. Fig. 5(a) shows the EIS variation during cycling. All EIS spectra are composed of two partially overlapping semicircles and a slash. The semicircle in the high-frequency region corresponds to the resistance of the SEI film, while the semicircle in the middle-frequency region corresponds to the charge transfer resistance. The slash in the low-frequency region is related to the ion diffusion of Na+ in the electrode. Equivalent circuit (Fig. S6†) simulation analysis shows that after the first cycle, the impedance values of SEI film and charge transfer are 2.2 and 84.1 Ω, respectively, while after 200 cycles, their values are 1.9 and 93.1 Ω, respectively, indicating SEI film has high stability and the conductive network of the electrode is well maintained during cycling. The SEM images of the electrodes after different cycles are shown in Fig. 5(b)–(d). Compared with that before cycling, although the electrochemical fusion of nanomaterials at the beginning of the cycle leads to the increase of particles, the phenomena of material pulverization and falling off from the current collector have not been observed. In addition, the morphology of the electrode has little change in the subsequent cycles. The high specific capacity and structural stability of the electrode should be attributed to the special structure of the MOF material. Sb confined in the organic skeleton contributes main capacity through alloying reaction, while the organic skeleton can not only prevent the growth of Sb particles and buffer the stress caused by volume change, but also provide partial capacity through the redox of active groups in PMA ligands.
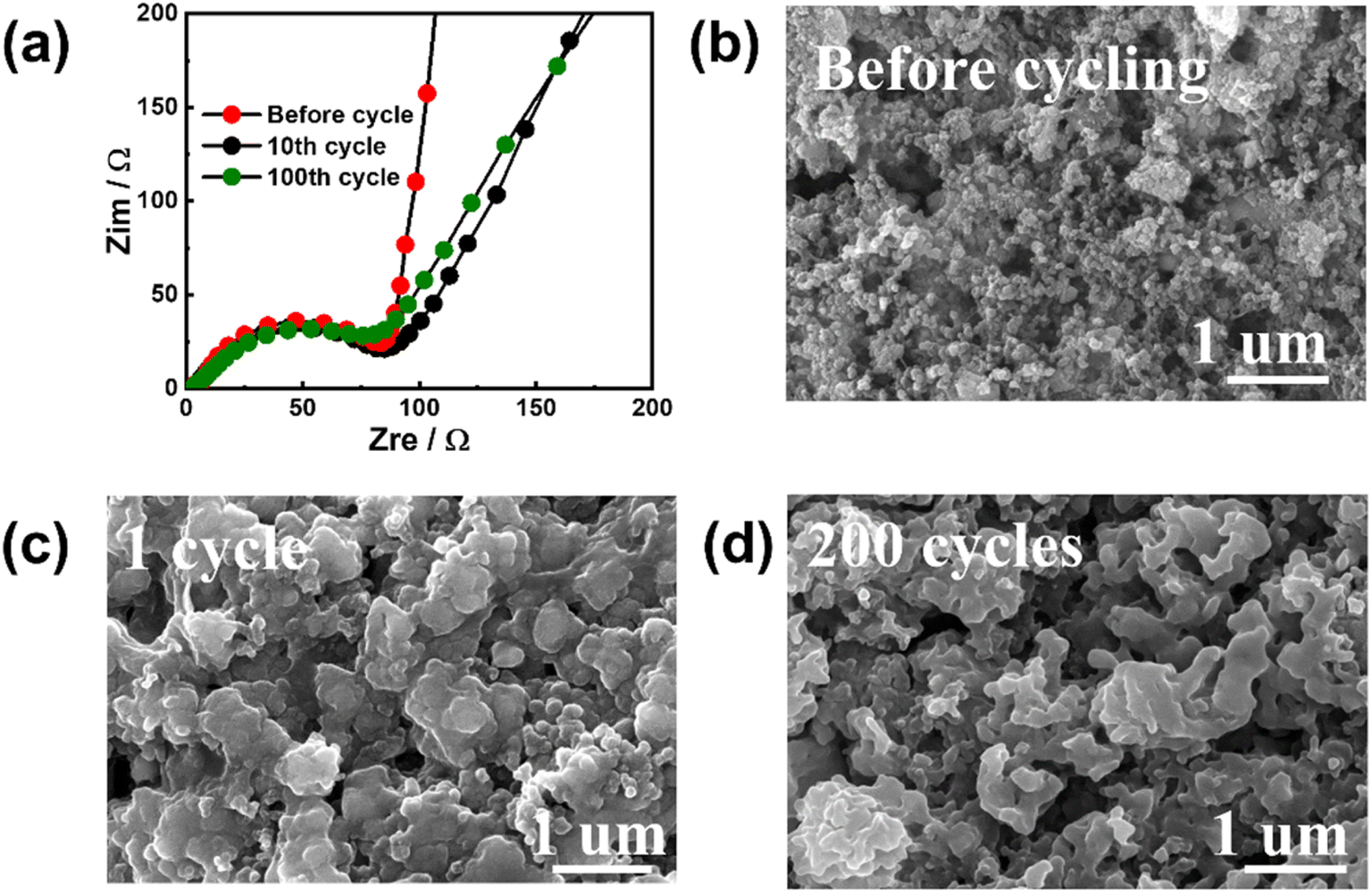 | ||
| Fig. 5 (a) EIS variations of Sb–PMA-300 electrode during cycling; Morphology of Sb–PMA-300 electrodes (b) before cycling, after (c) 1 cycle and (d) 200 cycles. | ||
To study the electrode process dynamics, Fig. 6(a) shows the CV curve of the stabilized Sb-MOF-300 electrode at 0.2–1.0 mV s−1. The peak current increases with the increase of the scanning rate. The obtained peak current (Ip, mA) and scan rate (v, mV s−1) follow the following power-law relationship.44,45
| Ip = avb | (1) |
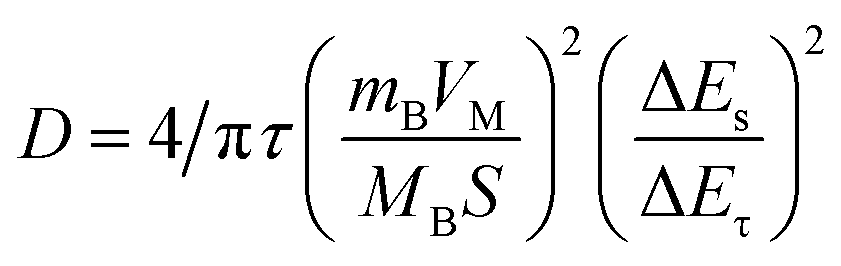 | (2) |
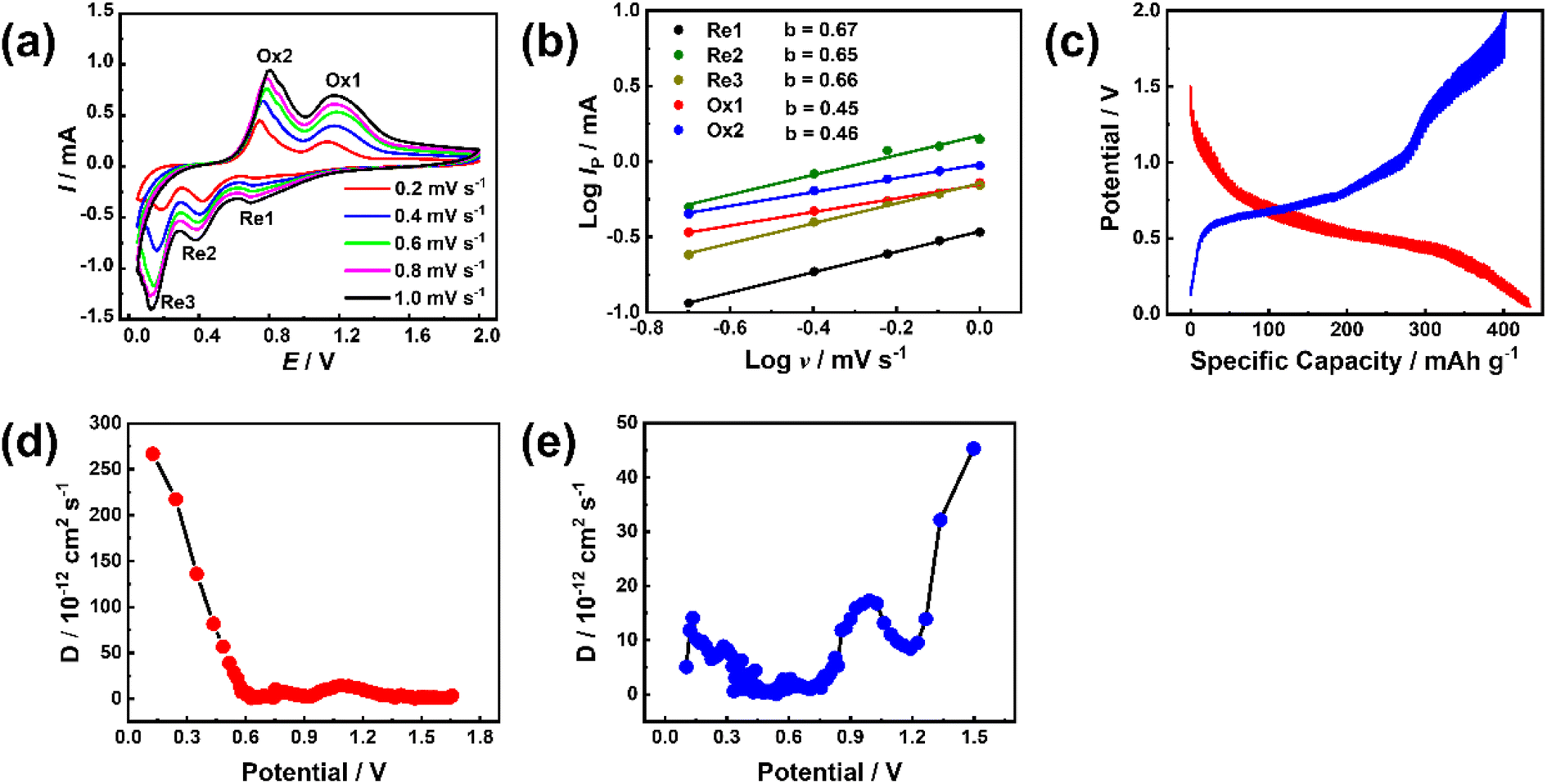 | ||
| Fig. 6 (a) CV curves and (b) linear relationship between log(Ip) versus log(v) of Sb-MOF-300 at 0.2–1.0 mV s−1; (c) GITT curves, variation of D value of Sb-MOF-300 during (d) discharging and (e) charging. | ||
To examine the availability of the Sb–PMA-300 as an anode fuel cells, Sb–PMA-300 ‖C coated NVP/rGO cells were assembled, in which the capacity ratio of anode and cathode is 2:1. The charge–discharge curves of Sb–PMA-300 ‖C coated NVP/rGO are shown in Fig. S7(a).† A long voltage plateau can be observed from the charge–discharge curves, which originate from the potential plateau characteristics of the anode. Based on the weight of the active material in the cathode, the full cell delivers a reversible capacity for the first week of 120.81 mA h g−1 at 1C (based on the cathode). After 40 cycles, the discharge capacity remains at 100.70 mA h g−1 (Fig. S7(b)†). As shown in Fig. S7(c),† even at a high current density of 8C (1C = 100 mA h g−1), a discharge capacity of 61.74 mA h g−1 can be achieved. These results indicate that the synthesized Sb–PMA is a promising anode material for sodium ion batteries.
4. Conclusions
A new Sb–PMA MOF material as anodes for sodium-ion batteries is synthesized by a simple and low-cost method. It is found that the Sb-MOF composite annealed at 300 °C exhibits significantly improve cycle stability. The stable capacity of the synthesized Sb-MOF-300 composite is about 443 mA h g−1 at 0.1 A g−1, and the discharge capacity remains at 326.4 mA h g−1 after 200 cycles at 1 A g−1. The excellent electrochemical performance is ascribed to the following facts are: (1) Sb limited in the organic skeleton contributes high capacity through alloying reaction, while the organic skeleton blocks off the aggregation of Sb particles and buffers the stress caused by huge volume change during cycling; (2) heat treatment strengthens the interaction between PMA ligands and Sb cations. This work demonstrates that Sb MOF material with suitable organic ligand can achieve high sodium storage capacity and good cycle stability without carbonization treatment.Conflicts of interest
The authors declared no competing financial interest.Acknowledgements
This research was financially supported by Sichuan Science and Technology Program (No. 2021YJ0051), Open Program of Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province (No. CSPC202106) and the Fundamental Research Funds of China West Normal University (No. CXTD2020-1).References
- Q. Li, W. Zhang, J. Peng, W. Zhang, Z. Liang, J. Wu, J. Feng, H. Li and S. Huang, ACS Nano, 2021, 15, 15104–15113 CrossRef CAS PubMed.
- B. Chen, M. Liang, Q. Wu, S. Zhu, N. Zhao and C. He, Trans. Tianjin Univ., 2021, 28, 6–32 CrossRef.
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC.
- X. Liu, J. Zhu, L. Yue, X. Wang, W. Wang, T. Zheng and Y. Li, Small, 2022, 18, 2204552 CrossRef CAS PubMed.
- W. T. Jing, Y. Zhang, Y. Gu, Y. F. Zhu, C. C. Yang and Q. Jiang, Matter, 2019, 1, 720–733 CrossRef CAS.
- W. Luo, P. Zhang, X. Wang, Q. Li, Y. Dong, J. Hua, L. Zhou and L. Mai, J. Power Sources, 2016, 304, 340–345 CrossRef CAS.
- Z. Yi, Q. Han, P. Zan, Y. Wu, Y. Cheng and L. Wang, J. Power Sources, 2016, 331, 16–21 CrossRef CAS.
- X. Zheng, K. Chen, Y. Hsieh and H. Tuan, ACS Sustainable Chem. Eng., 2020, 8, 18535–18544 CrossRef CAS.
- W. Zhang, W. Yan, H. Jiang, C. Wang, Y. Zhou, F. Ke, H. Cong and H. Deng, ACS Sustainable Chem. Eng., 2020, 8, 335–342 CrossRef CAS.
- M. W. Orzech, F. Mazzali, J. D. McGettrick, C. Pleydell-Pearce, T. M. Watson, W. Voice, D. Jarvis and S. Margadonna, J. Mater. Chem. A, 2017, 5, 23198–23208 RSC.
- X. Liu, B. Xiao, A. Daali, X. Zhou, Z. Yu, X. Li, Y. Liu, L. Yin, Z. Yang, C. Zhao, L. Zhu, Y. Ren, L. Cheng, S. Ahmed, Z. Chen, X. Li, G. Xu and K. Amine, ACS Energy Lett., 2021, 6, 547–556 CrossRef CAS.
- Y. Yuan, S. Jan, Z. Wang and X. Jin, J. Mater. Chem. A, 2018, 6, 5555–5559 RSC.
- T. Wu, C. Zhang, H. Hou, P. Ge, G. Zou, W. Xu, S. Li, Z. Huang, T. Guo, M. Jing and X. Ji, Adv. Funct. Mater., 2018, 28, 1705744 CrossRef.
- J. Tian, H. Yang, C. Fu, M. Sun, L. Wang and T. Liu, Compos. Commun., 2020, 17, 177–181 CrossRef.
- N. Cheng, J. Zhao, L. Fan, Z. Liu, S. Chen, H. Ding, X. Yu, Z. Liu and B. Lu, Chem. Commun., 2019, 55, 12511–12514 RSC.
- J. Han, D. Liu, S. Liu, W. Shao, E. Yang, T. Zhang, X. Jin, X. Jian and F. Hu, J. Alloys Compd., 2022, 921, 166054 CrossRef CAS.
- B. Chen, H. Qin, K. Li, B. Zhang, E. Liu, N. Zhao, C. Shi and C. He, Nano Energy, 2019, 66, 104133 CrossRef CAS.
- N. Javed, T. Noor, N. Iqbal and S. R. Naqvi, RSC Adv., 2023, 13, 1137–1161 RSC.
- R. Mehek, N. Iqbal, T. Noor, M. Z. B. Amjad, G. Ali, K. Vignarooban and M. A. Khan, RSC Adv., 2021, 11, 29247–29266 RSC.
- R. Mori, RSC Adv., 2017, 7, 6389–6395 RSC.
- J. Liu, D. Xie, X. Xu, L. Jiang, R. Si, W. Shi and P. Cheng, Nat. Commun., 2021, 12, 3131 CrossRef CAS PubMed.
- L. Yu, J. Liu, X. Xu, L. Zhang, R. Hu, J. Liu, L. Yang and M. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 2516–2525 CrossRef CAS PubMed.
- J. Wang, X. Yue, Z. Xie, A. Abudula and G. Guan, Energy Storage Mater., 2021, 41, 404–426 CrossRef.
- Z. Wang, Z. Fanming, Z. Dongyu, W. Xinyu, Y. Weiling, C. Li, Y. Cheng and W. Limin, Electrochim. Acta, 2021, 395, 139210 CrossRef CAS.
- Q. Li, W. Zhang, J. Peng, W. Zhang, Z. Liang, J. Wu, J. Feng, H. Li and S. Huang, ACS Nano, 2021, 15, 15104–15113 CrossRef CAS PubMed.
- L. Yu, L. Zhang, J. Fu, J. Yun and K. H. Kim, Chem. Eng. J., 2021, 417, 129106 CrossRef CAS.
- Z. Chen, A. Soltani, Y. Chen, Q. Zhang, A. Davoodi, S. Hosseinpour, W. Peukert and W. Liu, Adv. Energy Mater., 2022, 12, 2200924 CrossRef CAS.
- S. Cai, F. Yan, Y. Zhao, M. Li, Y. Chen, X. He and C. Wang, Chem. Eng. J., 2022, 430, 132938 CrossRef CAS.
- J. Fei, Y. Cui, J. Li, Z. Xu, J. Yang, R. Wang, Y. Cheng and J. Hang, Chem. Commun., 2017, 53, 13165–13167 RSC.
- O. A. Jaramillo-Quintero, R. V. Barrera-Peralta, A. Baron-Jaimes, R. A. Miranda-Gamboa and M. E. Rincon, RSC Adv., 2021, 11, 31566–31571 RSC.
- B. Chen, L. Yang, X. Bai, Q. Wu, M. Liang, Y. Wang, N. Zhao, C. Shi, B. Zhou and C. He, Small, 2021, 17, e2006824 CrossRef PubMed.
- X. Yan, C. Fan, X. Yang, Y. Wang, B. Hou, W. Pang and X. Wu, Mater. Today. Energy, 2019, 13, 302–307 CrossRef.
- S. Bahfenne and R. L. Frost, Spectrosc. Lett., 2010, 43, 486–490 CrossRef CAS.
- K. Ramakrishnan, C. Nithya, B. Kundoly Purushothaman, N. Kumar and S. Gopukumar, ACS Sustainable Chem. Eng., 2017, 5, 5090–5098 CrossRef CAS.
- A. Nguyen, H. T. T. Le, R. Verma, D. Vu and C. Park, Chem. Eng. J., 2022, 429, 132359 CrossRef CAS.
- A. Xu, C. Huang, G. Li, K. Zou, H. Sun, L. Fu, J. Ju, S. Yang, S. Wu, Z. Xu and Y. Yan, J. Mater. Chem. A, 2021, 9, 12169–12178 RSC.
- J. Li, N. Zhuang, J. Xie, X. Li, W. Zhuo, H. Wang, J. B. Na, X. Li, Y. Yamauchi and W. Mai, Adv. Energy Mater., 2019, 10, 1903455 CrossRef.
- X. Xu, L. Si, X. Zhou, F. Tu, X. Zhu and J. Bao, J. Power Sources, 2017, 349, 37–44 CrossRef CAS.
- L. Liang, Y. Xu, C. Wang, L. Wen, Y. Fang, Y. Mi, M. Zhou, H. Zhao and Y. Lei, Energy Environ. Sci., 2015, 8, 2954–2962 RSC.
- J. Du, Y. Zhang, H. Lv and A. Chen, Adv. Powder Technol., 2021, 32, 1294–1299 CrossRef CAS.
- M. Li, T. Qiu, A. C. Foucher, J. Fu, Z. Wang, D. Zhang, A. M. Rappe, E. A. Stach and E. Detsi, ACS Appl. Energy Mater., 2020, 3, 11231–11241 CrossRef CAS.
- A. Amardeep, R. C. Shende, P. Gandharapu, M. S. Wani and A. Mukhopadhyay, ACS Appl. Mater. Interfaces, 2022, 14, 45296–45307 CrossRef CAS.
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Angew. Chem., 2021, 133, 18917–18924 CrossRef.
- W. Meng, M. Guo, X. Liu, J. Chen, Z. Bai and Z. Wang, J. Alloys Compd., 2019, 795, 141–150 CrossRef CAS.
- Y. Li, Z. Song, T. Sun, Y. Shen, X. Lv, D. Xu and H. Wang, Int. J. Hydrogen Energy, 2021, 46, 26308–26317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02132g |
| This journal is © The Royal Society of Chemistry 2023 |