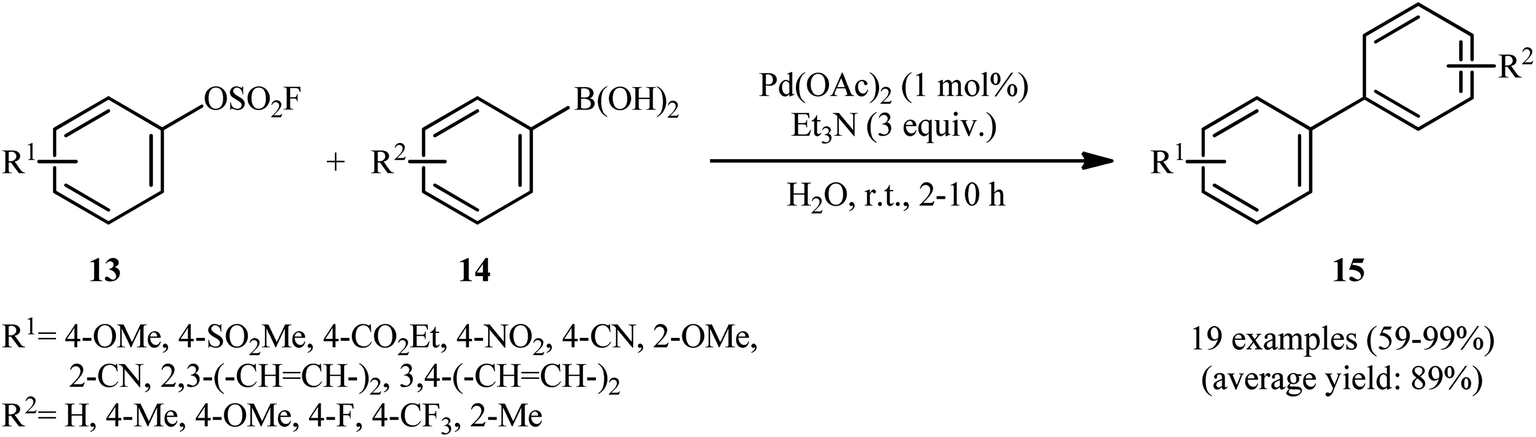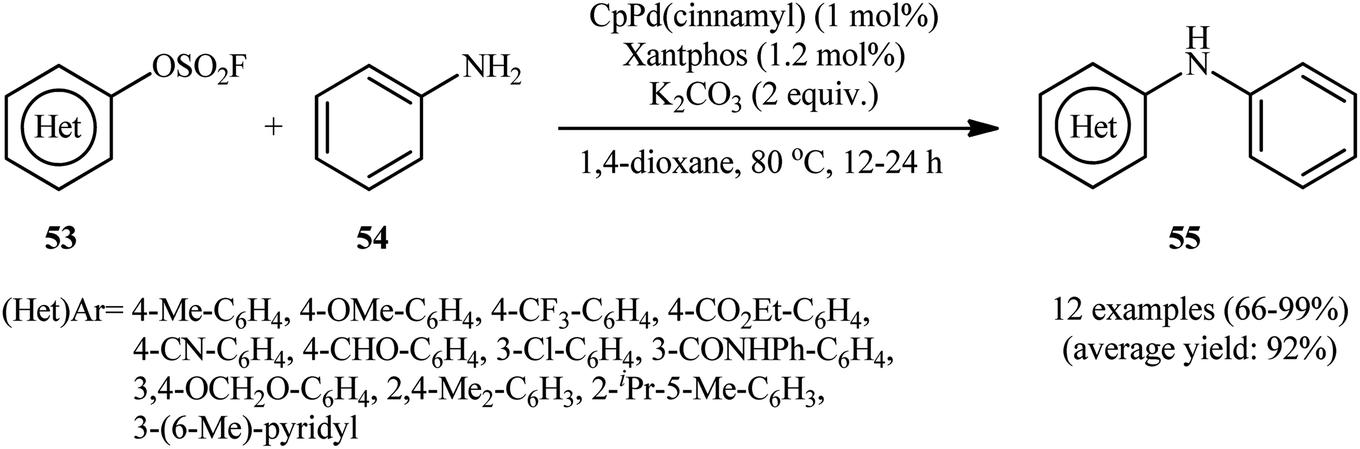DOI:
10.1039/D3RA01791E
(Review Article)
RSC Adv., 2023,
13, 13642-13654
Aryl fluorosulfates: powerful and versatile partners in cross-coupling reactions
Received
18th March 2023
, Accepted 20th April 2023
First published on 3rd May 2023
Abstract
Aryl fluorosulfates are versatile building blocks in organic synthesis and have gained increasing attention in SuFEx (Sulfur Fluoride Exchange) click chemistry. They are easily and conveniently prepared from phenols using sulfuryl fluoride SO2F2 as a low-cost sulfonyl fluoride provider. Recently, they served as less toxic and more atom economical alternatives to triflates in an impressive number of carbon–carbon and carbon-heteroatom cross-coupling reactions. In this review, we summarize the current advances and developments in applying aryl fluorosulfates as electrophilic partners in cross-coupling reactions.
1 Introduction
Organic halides are extensively used as electrophilic partners for transition-metal-catalyzed cross-coupling reactions.1 However, their environmental toxicity (due to the generation of stoichiometric quantities of halide waste) and high costs limited their utility in large-scale syntheses in industrial applications.2 Therefore, considerable attention has been paid to phenol derivatives as easily accessible and naturally abundant alternative electrophiles.3
Due to their superior performance as electrophilic coupling partners, triflates have long been used as alternatives and/or replacements for halogens in cross-coupling reactions.4 However, despite their excellent reactivity, they suffer from several disadvantages, such as instability, environmental toxicity, high cost of preparation, and poor atom economy.2 These problems ultimately limit their application profile on larger scales. Consequently, many efforts have been spotted in seeking alternatives to triflates and several complementary O-based pseudohalides such as tosylates, mesylates, nonaflates, and fluorosulfates has been developed as viable electrophilic partners.5 Although tosylates and mesylates are less expensive and more stable than triflates; however, they are considerably less reactive electrophiles than triflates.6 Nonaflates are not only more cost effective and stable than triflates but also have comparable if not greater levels of reactivity.7 However, they are less atom economic and creating more toxic long-chain fluorocarbon waste. On the other hand aryl fluorosulfates have become increasingly popular as coupling partners in organic synthesis due to several advantages over traditional aryl halides, such as bromides and chlorides. One of the main benefits of using aryl fluorosulfates is their higher reactivity and selectivity towards transition metal-catalyzed coupling reactions.8 This makes them attractive for efficient and selective transformations. Additionally, the reaction conditions for aryl fluorosulfates are often milder compared to aryl halides, which reduces the occurrence of unwanted side reactions and allows for the use of more sensitive functional groups. Furthermore, aryl fluorosulfates are less toxic and more environmentally friendly than aryl halides, making them a more appealing choice for large-scale industrial applications.8,9 Overall, the use of aryl fluorosulfates as coupling partners offers significant advantages over traditional aryl halides and has become a valuable tool in modern organic synthesis. Moreover, they can be easily prepared from the reactions of phenol or alcohol derivatives with various sulfonyl fluoride (SO2F) sources such as sulfuryl fluoride, fluorosulfonic acid, sulfuryl chloride fluoride, and fluorosulfonic anhydride.8
As early as 1991, the first report on the usefulness of aryl fluorosulfates as electrophilic partners in cross-coupling reactions was published by Roth et al.9 However, since then, this page of cross-coupling reactions did not attract the attention of chemists for nearly 25 years. Since 2015, several research groups investigated the scope and limitation of these new classes of electrophilic components in various carbon–carbon and carbon-heteroatom cross-coupling reactions. In 2018, Qin and co-workers published an interesting review paper entitled “Synthesis and Chemical Transformations of Fluorosulfates” that highlights some of the advances in this interesting research topics; albeit with only 7 examples.8 Since a number of remarkable advances and developments in this domain have occurred during the past few decades, seems it is an appropriate time to summarize those discoveries in a comprehensive review. In continuation of our preceding works on cross-coupling reactions10 and modern organic synthesis,11 in this review, we intend to highlight the most important explorations and developments in the cross-coupling reactions using fluorosulfates from 1991 till today. For clarity, the topic is divided into two major parts. The first section covers the available literature on carbon–carbon cross-coupling reactions using fluorosulfates, while the second focuses exclusively on the carbon-heteroatom (nitrogen, oxygen, phosphorus) cross-coupling reactions. We hope that this review will inspire researchers to make further progress in this attractive research arena.
2 Carbon–carbon cross-coupling reactions
In this section, we describe the current literature on C–C cross-coupling reactions utilizing aryl fluorosulfonates as electrophilic partners. Cross-coupling reactions with organometallic nucleophiles are discussed first. This is followed by Suzuki–Miyaura and Sonogashira cross-coupling reactions. Finally, reported examples on carbonylative cross-coupling reactions will be covered at the end of the section.
2.1. Negishi cross-coupling
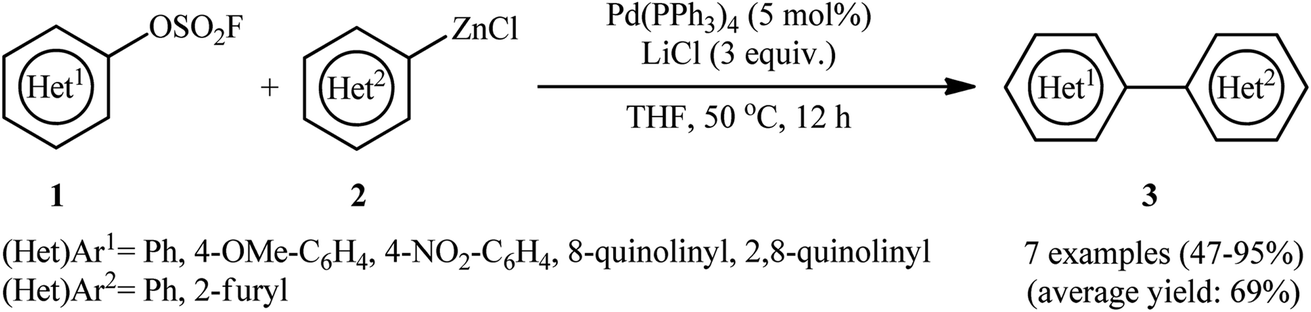
The Negishi cross-coupling can be best described as the reaction between organic (pseudo)halides with organozinc compounds to construct new carbon–carbon bonds with the aid of a transition metal catalyst, mainly palladium complexes.12 This synthetic method has gained increasing popularity among synthetic chemists especially in the field of natural products total synthesis.13 In 1991, Roth and Fuller reported the first examples of the Negishi coupling utilizing fluorosulfonates in the place of the halide component.9 They showed that the reaction of (hetero)aryl fluorosulfonates 1 with a small library of organozinc chlorides 2 in the presence of a catalytic amount of Pd(Ph3)4 in THF afforded corresponding bi(hetero)aryls 3 in moderate to excellent yields (Scheme 1). Notably, the reaction was not limited to using organozincs as the nucleophilic coupling partner. Organostannanes were also competent coupling partners, providing biaryl and styrene derivatives in good yields.
 |
| | Scheme 1 Roth's synthesis of bi(hetero)aryls 3. | |
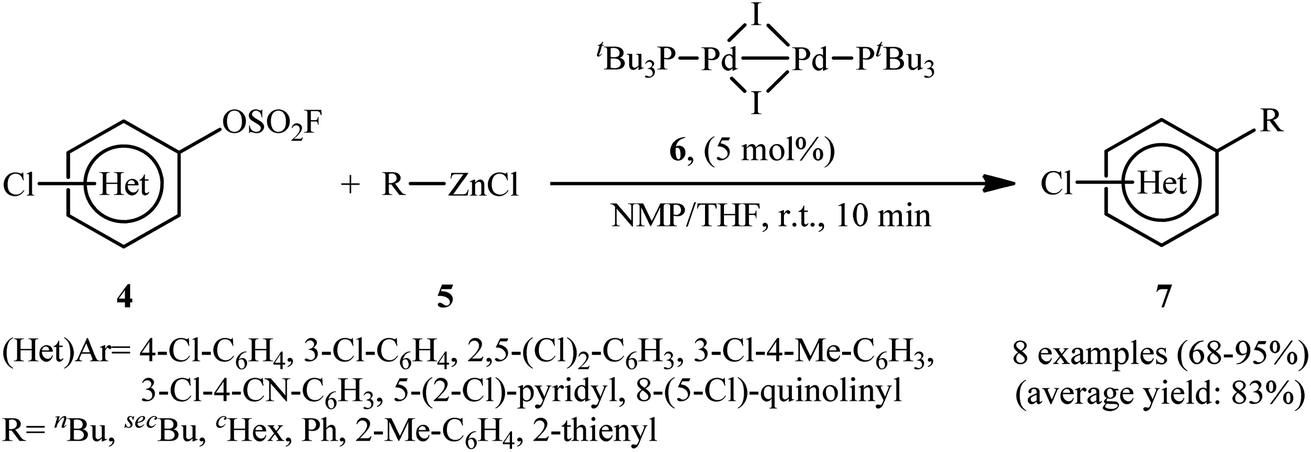
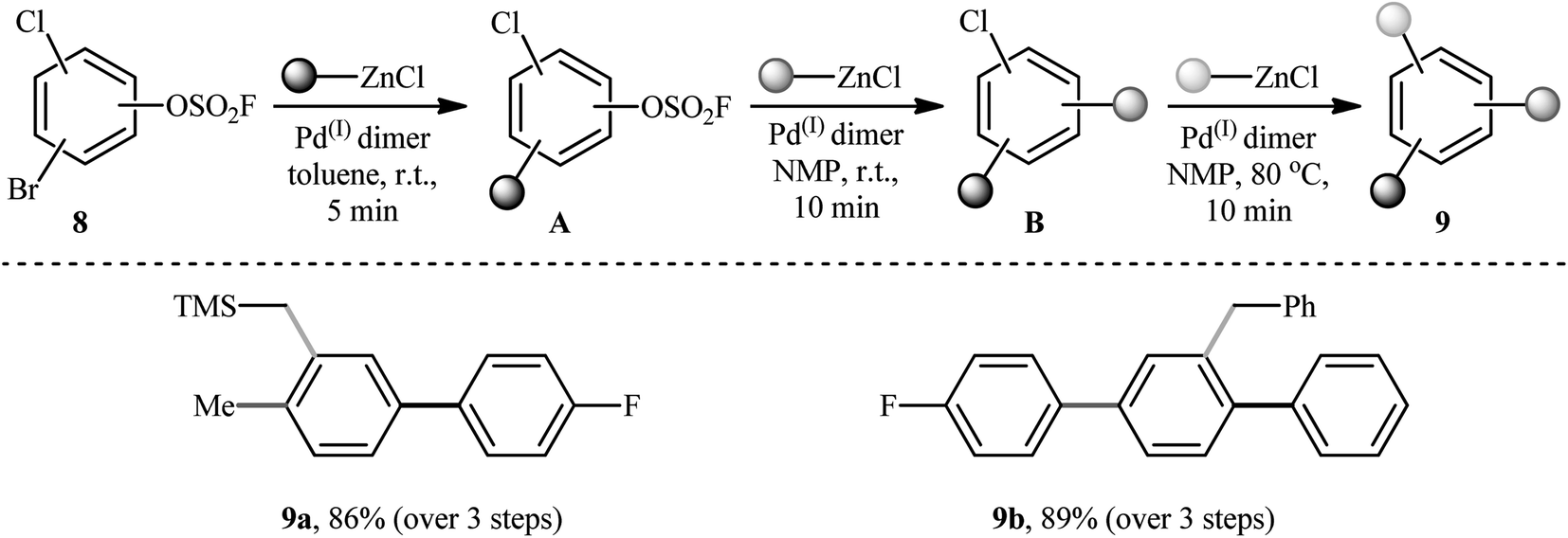
Three decades later, in 2020, Schoenebeck's research group described an effective site-selective coupling of Cl-substituted (hetero)aryl fluorosulfonates 4 with various aliphatic, aromatic, and heteroaromatic organozinc reagents 5 using a dinuclear Pd(I)-iodo-dimer catalyst 6.14 This synthetic transformation exhibited an efficient and attractive method for the high yielding synthesis of Cl-substituted bi(hetero)aryl and (hetero)aryl-alkyl derivatives 7 at room temperature under additive-free conditions within minutes (Scheme 2). Of note, the reaction exhibited extremely high degree of site-selectivity, in which functionalization is exclusively took place on the carbon atom attached to the fluorosulfonate group in arenes (C–OSO2F vs. C–Cl). Interestingly, when Cl was replaced with Br, exclusive coupling at C–Br was seen under the identical conditions (C–Br vs. C–OSO2F). The authors, nicely applied these principles for the diversification of (hetero)arenes 8 with multiple competing coupling sites (–OSO2F, –Cl, –Br) with organozinc chlorides by doubly and triply selective sequential functionalization in the sequence C–Br, then C–OSO2F, then C–Cl (Scheme 3). In this report, the authors also undertook computational studies regarding the reactivity scale for oxidative addition of Ar–OSO2R derivatives (R = OMs, OTs, OFs, OTf, and ONf). Therefore, M06 density functional theory studies on the oxidative addition of PhOSO2R derivatives with Pd(0)P(tBu)3 as a model catalyst, suggested a 5-membered neutral transition states arrangement to be favored for C–OSO2F activation and predicted virtually identical activation barriers as for triflates and nonaflates.
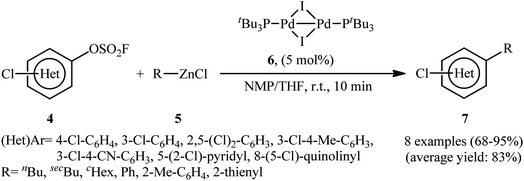 |
| | Scheme 2 Dinuclear Pd-catalyzed coupling of (hetero)aryl fluorosulfonates 4 with organozinc reagents 5. | |
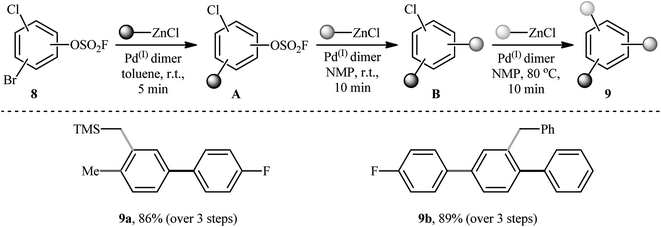 |
| | Scheme 3 Triply selective sequential functionalization of (hetero)arenes 8 developed by Schoenebeck et al. | |
2.2. Stille cross-coupling
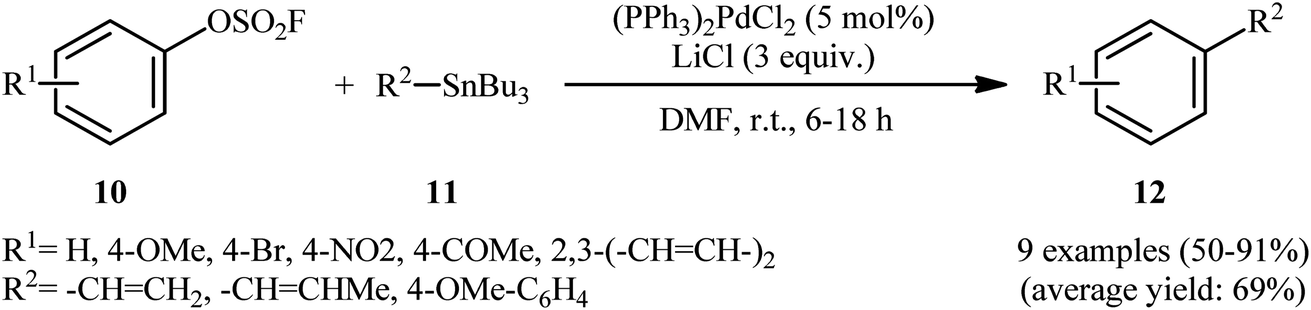
In 1991, in the same paper describing the first example of Negishi cross-coupling utilizing aryl fluorosulfonates as coupling partner in the presence of catalytic amounts of palladium, Roth and Fuller also reported the usefulness of these electrophilic components in the Stille cross-coupling.9 Thus, in the presence of a combination of (PPh3)2PdCl and LiCl in DMF at ambient temperature, the reaction of various aryl fluorosulfonates 10 bearing both electron-withdrawing and electron-donating groups with a range of aryl- and vinyl-stannanes 11 furnished the corresponding coupling products 12 in moderate to excellent isolated yields, ranging from 50% to 91% (Scheme 4). Notably, in the case of internal vinyl-stannanes substrates, the preferential formation of the (Z)-isomers was observed as evidenced by 1H NMR. To the best of our knowledge this is the only example on the Stille coupling employing aryl fluorosulfonates reported till date.
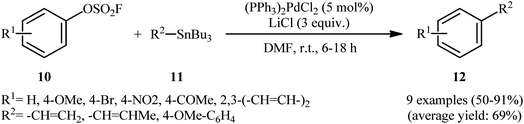 |
| | Scheme 4 Pd-catalyzed coupling of aryl fluorosulfonates 10 with aryl- and vinyl-stannanes 11. | |
2.3. Suzuki–Miyaura cross-coupling
The Suzuki–Miyaura cross-coupling involves the coupling of an organoboron reagent with a (pseudo)halide for the construction of C–C bonds.15 This coupling is one of the widely used reactions in the manufacture of pharmaceuticals and is the most common biaryl bond forming reaction.16
In 2015, Sharpless and Jiang along with their co-workers published one of the earliest reports on the Suzuki–Miyaura coupling reaction using aryl fluorosulfates as electrophilic coupling partners.17 They showed that the treatment of various aryl fluorosulfates 13 with aryl boronic acids 14 in the presence of a combination of Pd(OAc)2 and Et3N in the most environmentally benign solvent, water, resulted in the formation of the corresponding biaryls 15 in good to quantitative yields (Scheme 5). The reaction is noteworthy in that both electron-rich and electron-poor aryl boronic acids were well tolerated. However, due to lower reactivity of electron-rich aryl fluorosulfates in compared to electron-deficient substrates, a higher catalyst and base loading as well as longer reaction time were required to obtain satisfactory results. It should be mentioned that compared with other traditional electrophilic coupling partners, including aryl halides, triflates, tosylates and mesylates, aryl fluorosulfates gave much better yields in Suzuki–Miyaura reaction with boronic acids under the identical conditions. Intriguingly, the catalytic system also showed good reactivity in double Suzuki couplings of diaryl-OSO2F to afford the corresponding products in excellent yields. The authors also disclosed that aryl fluorosulfates are amenable coupling partners in other types of coupling reactions, including Heck, Sonogashira, and homocoupling reactions. However, only single examples have been reported for each of those reactions.
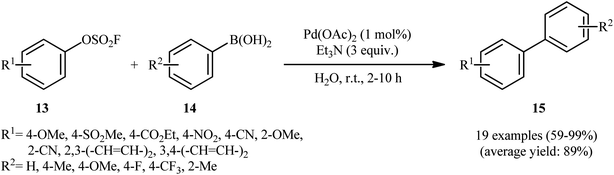 |
| | Scheme 5 Suzuki–Miyaura coupling of aryl fluorosulfates 13 with aryl boronic acids 14 in water. | |
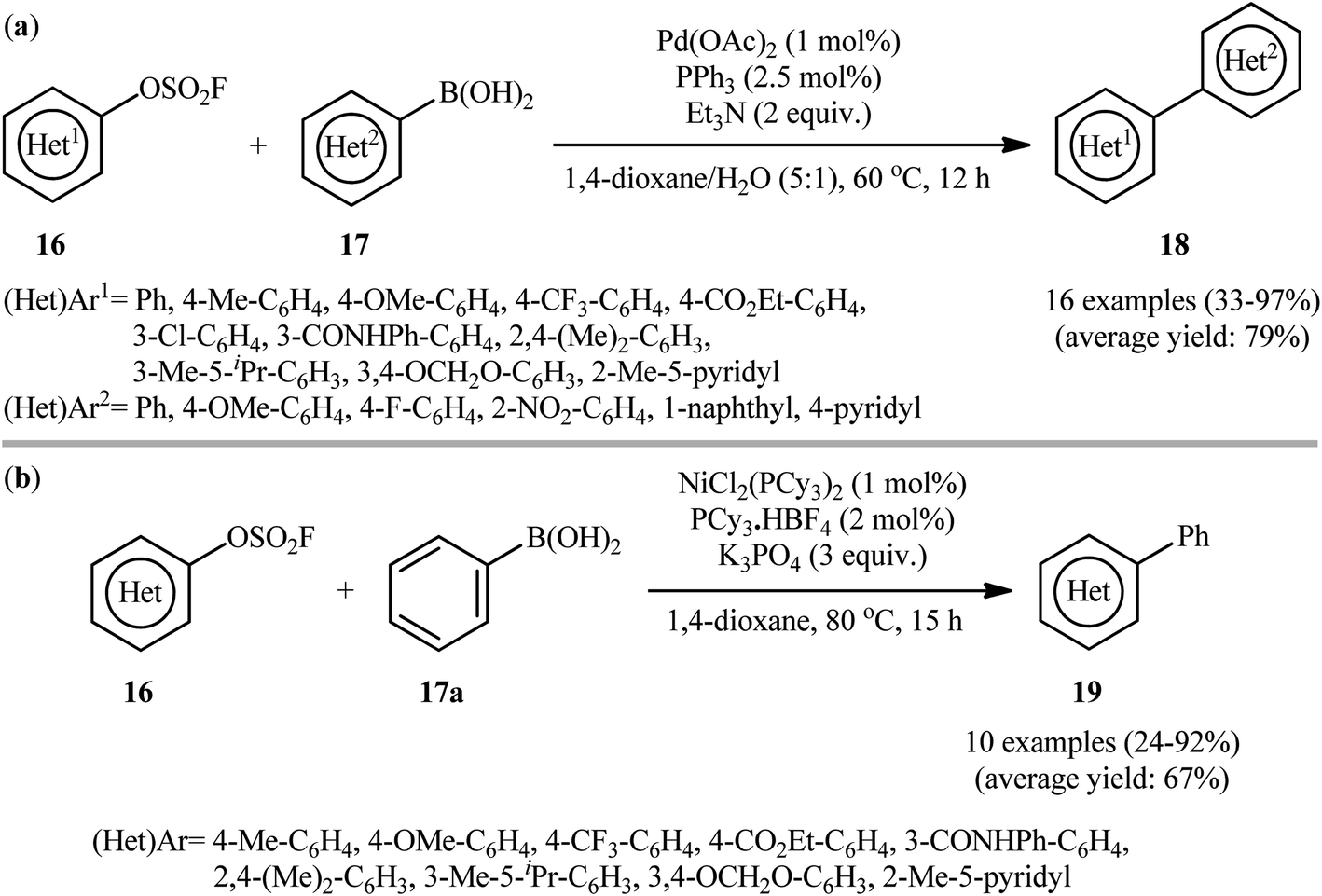
Concurrently, Hanley and co-workers reported a closely related coupling between aryl fluorosulfonates 16 and (hetero)aryl boronic acids 17 employing the combination of Pd(OAc)2, PPh3, and Et3N as catalytic system.18 The reaction was conducted in the binary solvent 1,4-dioxane/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), tolerated various important functional groups (e.g., OMe, F, Cl, NO2, CONHPh), and provided the desired biaryls 18 in moderate to excellent yields (Scheme 6a). However, amino group was incompatible in this system. Interestingly, one-pot version of this transformation using in situ generated aryl fluorosulfonates from the corresponding phenols was also examined under the optimized conditions and the desired products were obtained in satisfactory yields. It is noteworthy that a series of competition experiments between phenyl boronic acid, tolyl fluorosulfonate and traditional electrophilic coupling partners (1:1:1 mixture) under the standard conditions revealed that the relative reactivity of examined electrophiles follows the trend I > Br > OTf ≈ OFs ≫ Cl, OTs, OMs. In this study, the authors also disclosed the usefulness of nickel catalysts as cheaper alternatives to palladium-based catalysts for this transformation. Thus, with the NiCl2(PCy3)2/PCy3·HBF4/K3PO4 catalytic system, the same set of aryl fluorosulfonates 16 efficiently reacted with phenyl boronic acid 17a to give the corresponding biaryl products 19 in good yields (Scheme 6b). Of note, in contrast to reactions performed with palladium catalysts, higher yields of products were obtained for electron-rich aryl fluorosulfonates than for electron-poor ones in reactions catalyzed by nickel. However, the nickel system was less tolerant to hierarchy sterically hindered fluorosulfonates than the palladium system. More importantly, in comparison to the complete lack of activity of the Pd-catalyst system with the primary amine containing fluorosulfonate, the Ni-catalyst system showed good compatibility with amino-group.
:1), tolerated various important functional groups (e.g., OMe, F, Cl, NO2, CONHPh), and provided the desired biaryls 18 in moderate to excellent yields (Scheme 6a). However, amino group was incompatible in this system. Interestingly, one-pot version of this transformation using in situ generated aryl fluorosulfonates from the corresponding phenols was also examined under the optimized conditions and the desired products were obtained in satisfactory yields. It is noteworthy that a series of competition experiments between phenyl boronic acid, tolyl fluorosulfonate and traditional electrophilic coupling partners (1:1:1 mixture) under the standard conditions revealed that the relative reactivity of examined electrophiles follows the trend I > Br > OTf ≈ OFs ≫ Cl, OTs, OMs. In this study, the authors also disclosed the usefulness of nickel catalysts as cheaper alternatives to palladium-based catalysts for this transformation. Thus, with the NiCl2(PCy3)2/PCy3·HBF4/K3PO4 catalytic system, the same set of aryl fluorosulfonates 16 efficiently reacted with phenyl boronic acid 17a to give the corresponding biaryl products 19 in good yields (Scheme 6b). Of note, in contrast to reactions performed with palladium catalysts, higher yields of products were obtained for electron-rich aryl fluorosulfonates than for electron-poor ones in reactions catalyzed by nickel. However, the nickel system was less tolerant to hierarchy sterically hindered fluorosulfonates than the palladium system. More importantly, in comparison to the complete lack of activity of the Pd-catalyst system with the primary amine containing fluorosulfonate, the Ni-catalyst system showed good compatibility with amino-group.
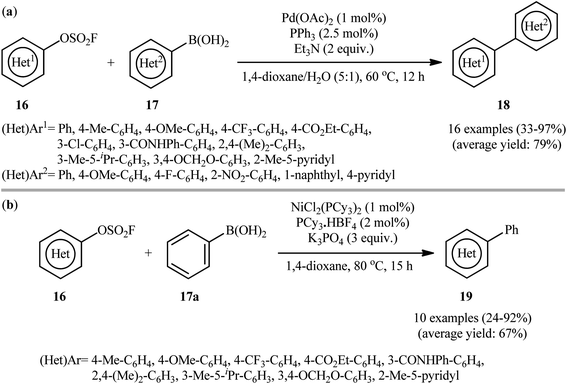 |
| | Scheme 6 (a) Hanley's synthesis of biaryls 18; (b) Ni-catalyzed Suzuki reaction using aryl fluorosulfates 16. | |
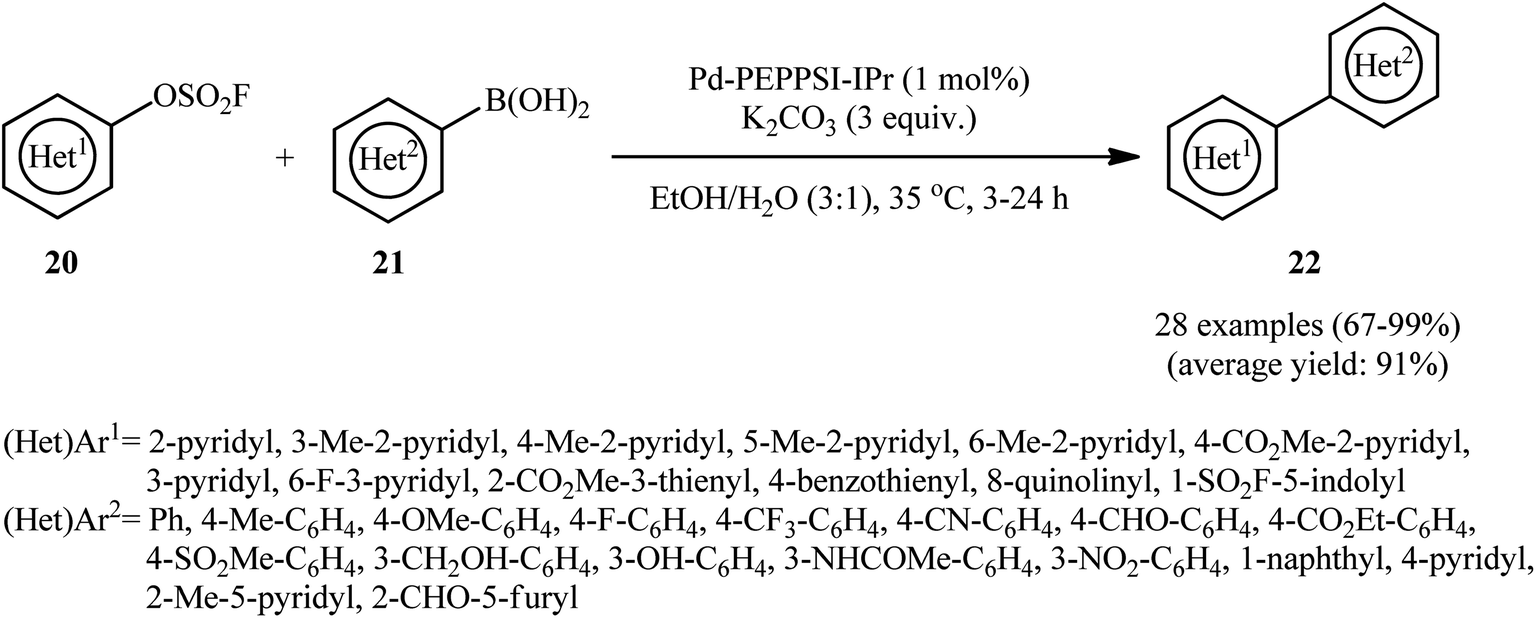
Drawing inspiration from these elegant works, Zhang, Sharpless and colleagues discovered that treatment of various nitrogen- and sulfur-containing heteroaromatic fluorosulfonates 20 with (hetero)aryl boronic acids 21 in the presence of Pd-PEPPSI-IPr/K2CO3 combination as the catalytic system in EtOH/H2O (3:1) afforded the corresponding bi(hetero)aryls 22 in good to quantitative yields, ranging from 67% to 99% (Scheme 7); in addition, a tolerance for vinyl boronic acid was also demonstrated.19 A series of important competition studies demonstrated the relative reactivity of examined leaving groups on substituted pyridines to be Br ≥ OTf > OSO2F > Cl, which was in agreement with the finding of Hanley et al.18 In order to further value the applicability of their methodology, the authors successfully synthesized Etoricoxib, an anti-inflammatory drug, from 5-bromo-6-chloropyridin-3-yl fluorosulfate through chemoselective sequential Suzuki cross-coupling reactions in an overall yield of 40.3%.
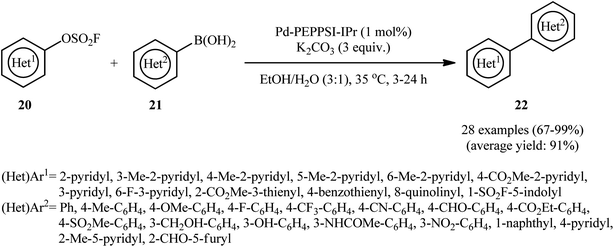 |
| | Scheme 7 Suzuki reaction of heteroaromatic fluorosulfonates 20 with (hetero)aryl boronic acids 21 catalyzed by Pd-PEPPSI-IPr. | |
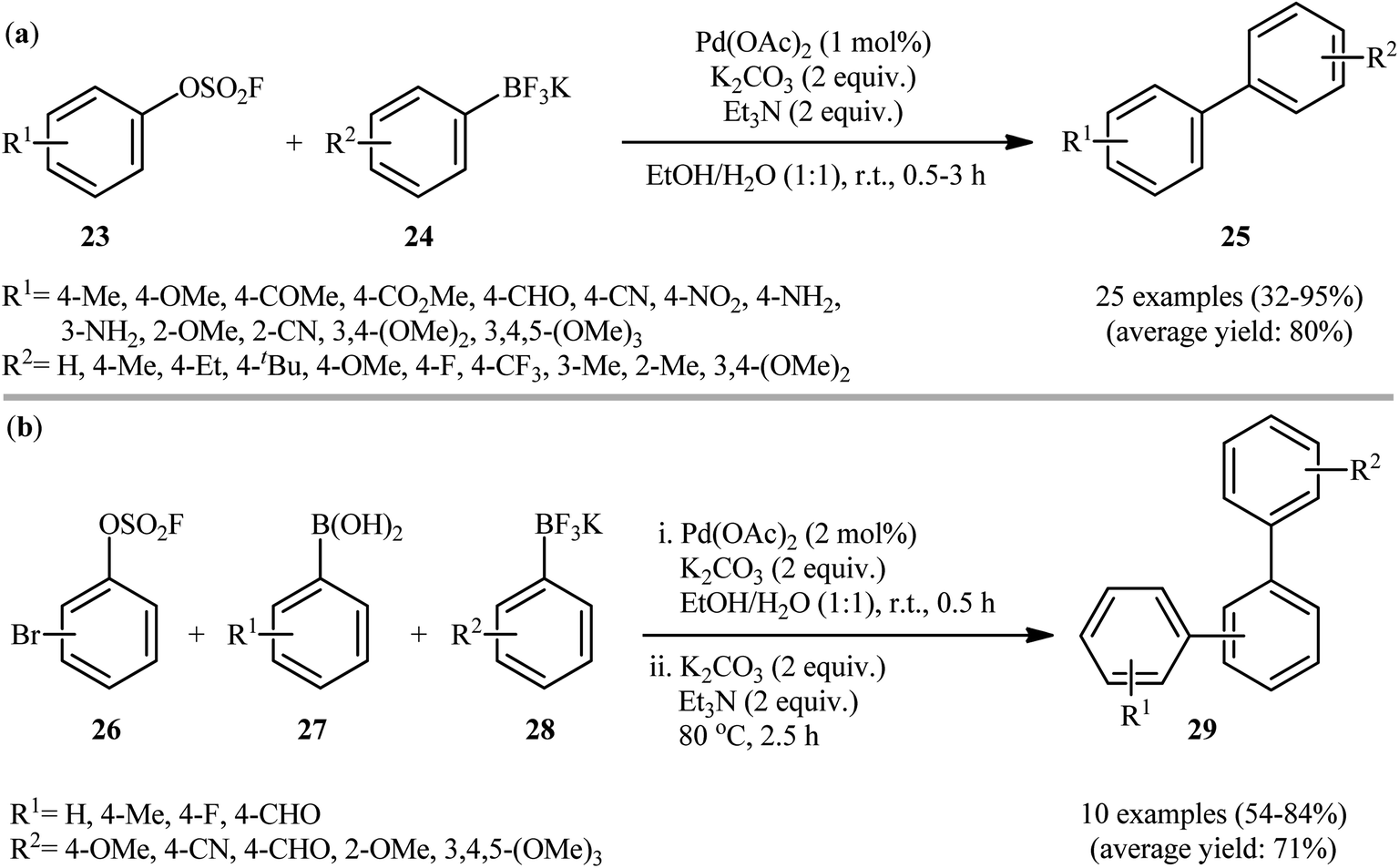
Subsequently, Li-Yuan's research group extended the substrates scope of organoboron compounds in the above procedure to potassium aryl trifluoroborates employing Pd(OAc)2 as the catalyst and a combination of Et3N and K2CO3 as the mix base.20 The binary solvent EtOH/H2O (1:1) was found to be the best medium for the reaction and, among several solvents tested, MeCN was found to be less effective. Apparently, the outcome of reaction was also dependent on the selected atmosphere. Also the same product yields were obtained under air and O2 atmosphere. The reaction rate was quite slower under N2 atmosphere. Therefore, it can be concluded that oxygen has a promoting effect on this transformation. Under optimized conditions, a wide range of aryl fluorosulfonates 23 bearing both electron withdrawing and donating groups including primary amine coupled with a variety of aryl trifluoroborates 24 to give the desired biaryls 25 in moderate to excellent yields (Scheme 8a). Additionally, this synthetic strategy was extended to one-pot double Suzuki–Miyaura reactions of bromophenyl fluorosulfates 26, aryl boronic acids 27 and potassium aryltrifluoroborates 28, allowing the synthesis of various biologically important unsymmetrical terphenyl derivatives 29 (Scheme 8b).
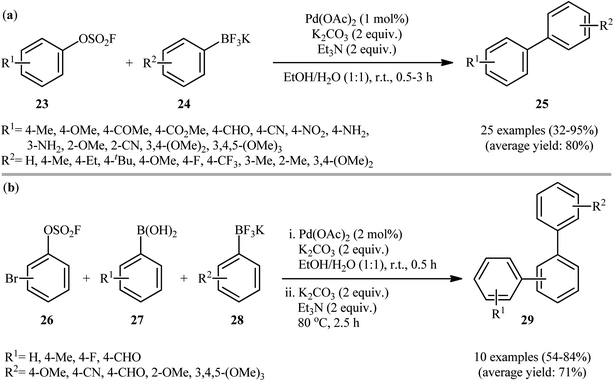 |
| | Scheme 8 (a) Suzuki reaction of aryl fluorosulfates 23 and potassium aryltrifluoroborates 24; (b) Li-Yuan's synthesis of terphenyls 29. | |
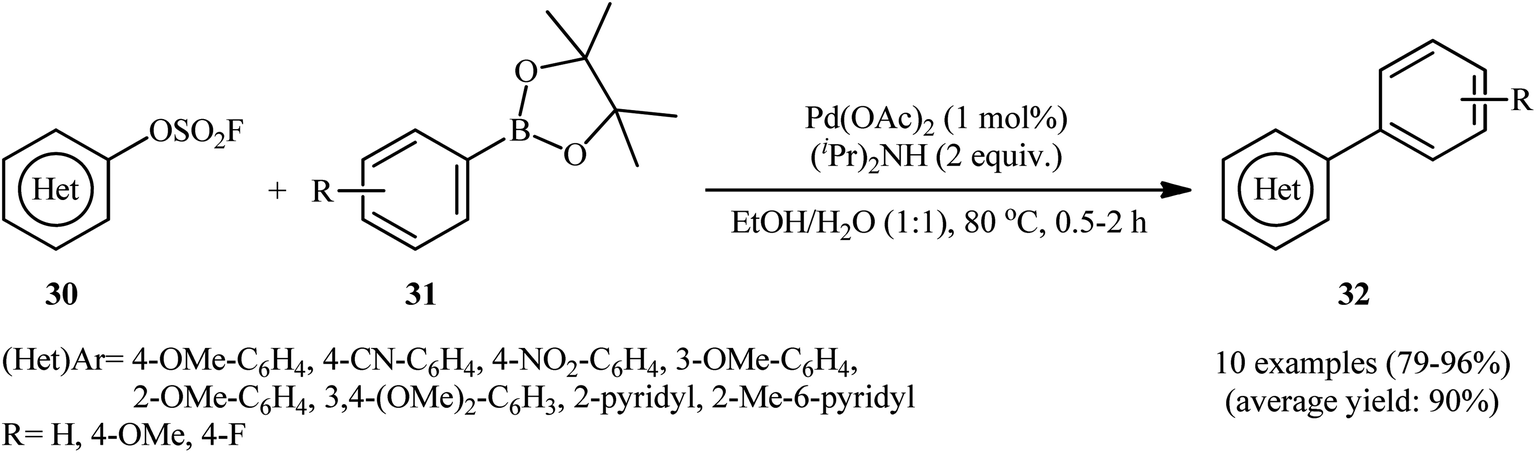
Shortly afterwards, the same research group reported the usefulness of arylboronic acid esters as coupling partners in the titled reaction.21 Thus, by using a closely similar system [Pd(OAc)2, (iPr)2NH, EtOH/H2O], the reaction of a series of (hetero)aryl fluorosulfonates 30 with arylboronic acid pinacol (BPin) esters 31 furnished the expected bi(hetero)aryls 32 within 30–120 min (Scheme 9). Notably, compare to the corresponding aryl trifluoroborates, pinacol arylboronates afforded higher yield of the target products under the identical conditions. The coupling of aryl N-methyliminodiacetic acid (MIDA) boronates was also feasible in this system, albeit in diminished efficiency.
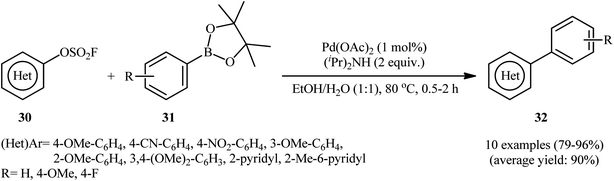 |
| | Scheme 9 Suzuki reaction of (hetero)aryl fluorosulfates 30 and arylboronic acid pinacol esters 31. | |
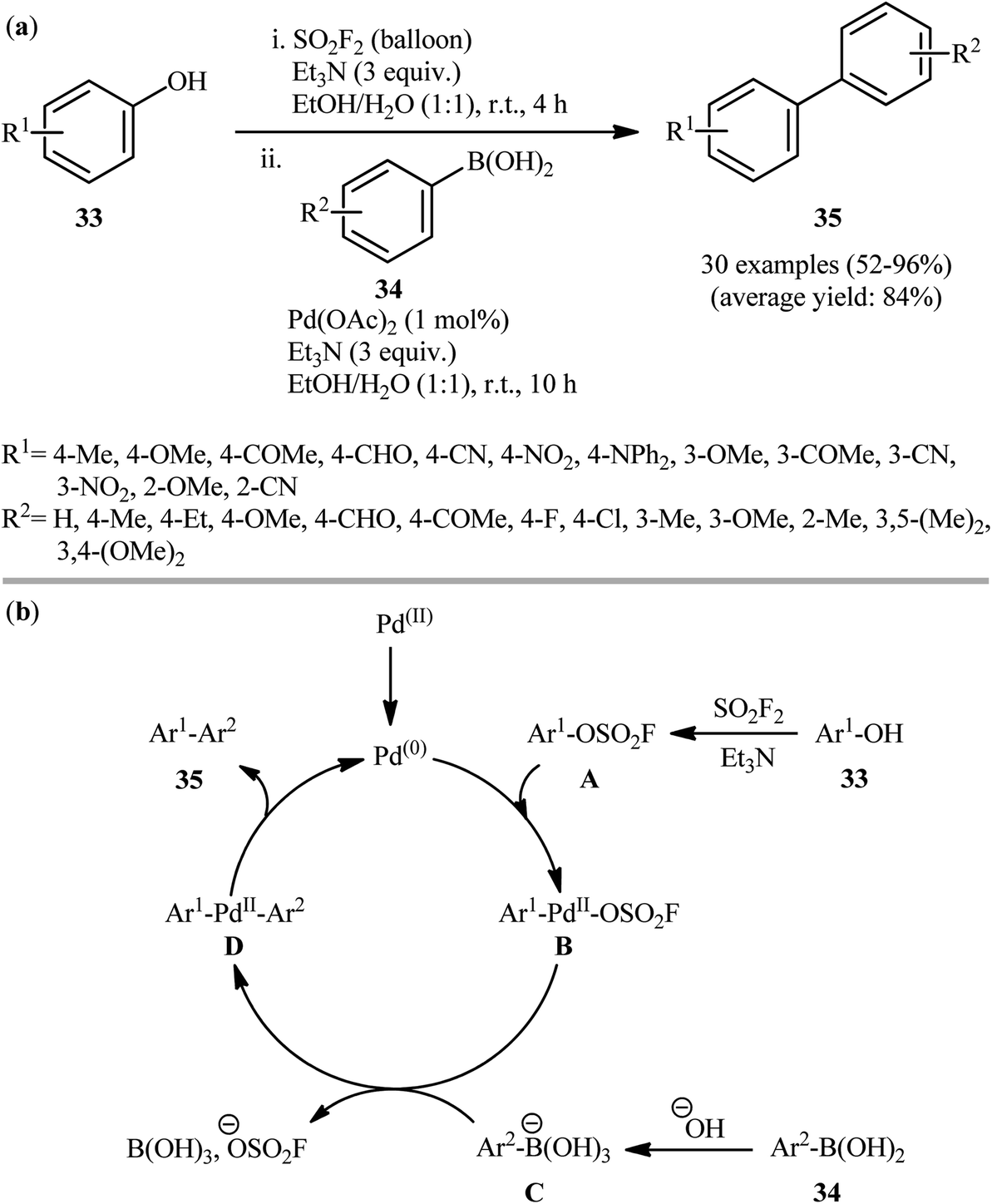
Recently, in another important development, the same research group demonstrated that phenols 33 can be converted to biaryls 35 in a one-pot process via Suzuki coupling of in situ formed fluorosulfonates with aryl boronic acids 34.22 The authors identified a combination of Pd(OAc)2 and Et3N as the optimal system for this transformation. As shown in Scheme 10a, the reactions proceed well with both electron-rich and electron-poor partners; however, pyridin-ol derivatives and highly hindered aryl boronic acids (e.g., 2,6-dimethylbenzeneboronic acid) are not suitable substrates for this cross-coupling. This tandem reaction could also be easily scaled up to the gram-scale as exemplified by the formation of 4′-methyl-[1,1′-biphenyl]-2-carbonitrile on a 1.26 g scale (81.5%). Noteworthy, the authors demonstrated the applicability of their methodology in the preparation of terphenyls from biaryl fluorosulfates. The authors proposed mechanistic pathway for this sequential reaction is depicted in Scheme 10b. Initially, an aryl fluorosulfate intermediate A was formed via a base-promoted reaction of phenols 33 with SO2F2 gas, which subsequently underwent an oxidative addition with Pd0 to furnish Ar–Pd–OSO2F B. The transmetallation of trihydroxyboronate C (generated from boronic acid 34) with complex B then gave Ar1–Pd(II)–Ar2 complex D. Finally, the reductive elimination of this complex D resulted in the formation of the target biaryl 35 while regenerating the Pd0. It should be mentioned that this catalytic platform was also elegantly applied by Yang, Lerner, and co-workers in on-DNA Suzuki–Miyaura cross-coupling reaction of a series of DNA-conjugated aryl fluorosulfonates with various boronic acids.23 Noteworthy, the presence of Et3N is crucial for the success of this reaction. Replacing Et3N with some other bases (e.g., DIPEA, Na2CO3, K2CO3, NaOH) led to much lower yields or even no product at all. Following these works, Zhao et al. investigated the coupling of genetically encoded fluorosulfate-L-tyrosine with various boronic acid substrates for protein modification.24 The reaction was run at pH 8.0 in phosphate buffer using Pd(OAc)2 and aminopyrimidine-4,6-diol (L1) as effective and water-soluble catalyst and ligand, respectively. The authors showed that this protein modification strategy can be used for protein fluorogenic labeling that enables in vitro and in vivo imaging of proteins with minimal background noises. It is worthwhile to note that beside Pd(OAc)2 and Pd-PEPPSI-IPr, other palladium catalysts were also successfully applied in the coupling of fluorosulfonates with boronic acids, such as Pd2(dba)3 (ref. 25) and [Pd(NHC)(μ-Cl)Cl]2 precatalysts.26
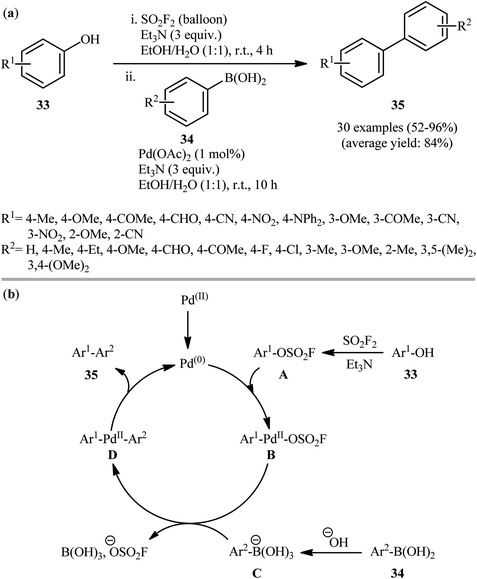 |
| | Scheme 10 (a) Synthesis of biaryls 35 through a one-pot sequential fluorosulfonation-Suzuki coupling approach developed by Li-Yuan's group; (b) mechanistic explanation for the formation of biaryls 35. | |
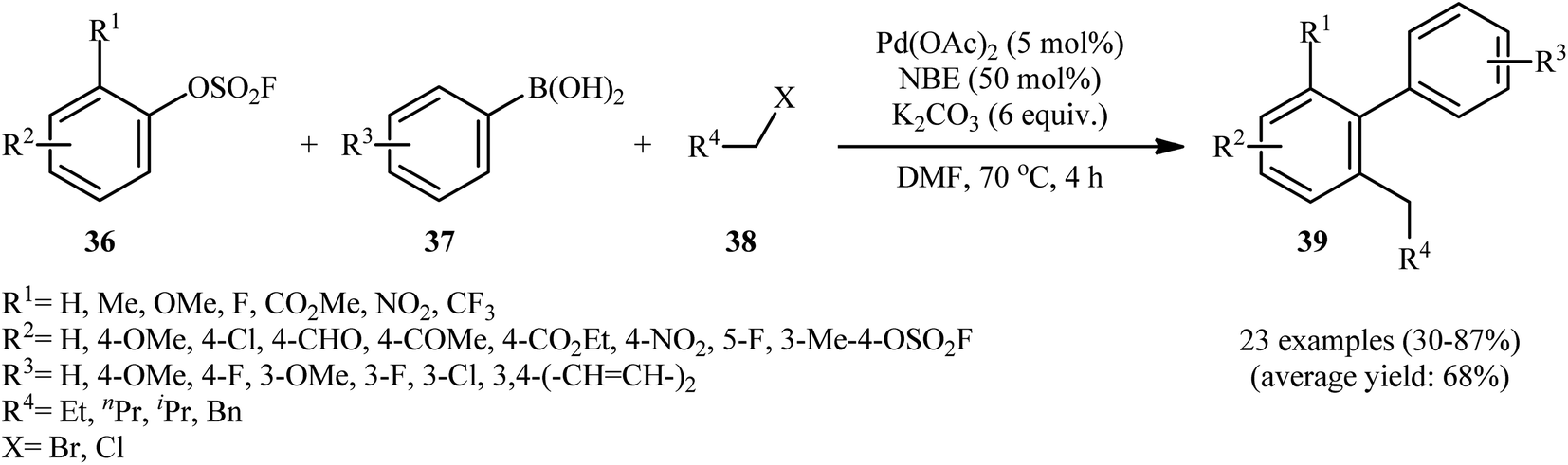
In a significant contribution in this field, Bieliūnas and De Borggraeve found that treatment of 2-substituted aryl fluorosulfonates 36 with aryl boronic acids 37 and primary alkyl halides 38 in the presence of Pd(OAc)2/norbornene (NBE)/K2CO3 combination as catalytic system in DMF resulted in corresponding multisubstituted arenes 39 in modest to high yields (Scheme 11).27 The results indicated that the presence of at least one relatively strong electron-withdrawing substituents in the phenyl ring periphery of aryl fluorosulfonates was crucial for this Catellani-type reaction, while the use of compounds bearing weakly electron-withdrawing substituents led to poor yields. Incompatibility of the reaction with substrates possessing aldehyde or nitrile moieties at the 2-position, was another limitation which was reported by the authors for their methodology. Notably, under the standard conditions, a symmetrical bis(fluorosulfate) reacted once, while the second fluorosulfate moiety served as an activating group and remained intact. Interestingly, this cascade chemistry was successfully extended to S-heterocyclic fluorosulfonates; however, N-heterocyclic systems let to low yields or none at all.
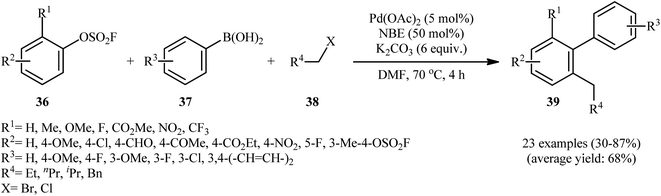 |
| | Scheme 11 Synthesis of multisubstituted arenes 39 via three-component reaction between aryl fluorosulfonates 36, boronic acids 37, and alkyl halides 38. | |
2.4. Sonogashira cross-coupling
About half a century ago, Sonogashira et al. developed coupling reaction of terminal alkynes with aryl halides in the presence of a Pd(II)/Cu(I) system.28 Today this reaction is one of the most versatile and powerful processes to generate aryl alkynes29 and is an essential tool within the pharmaceutical industries.30
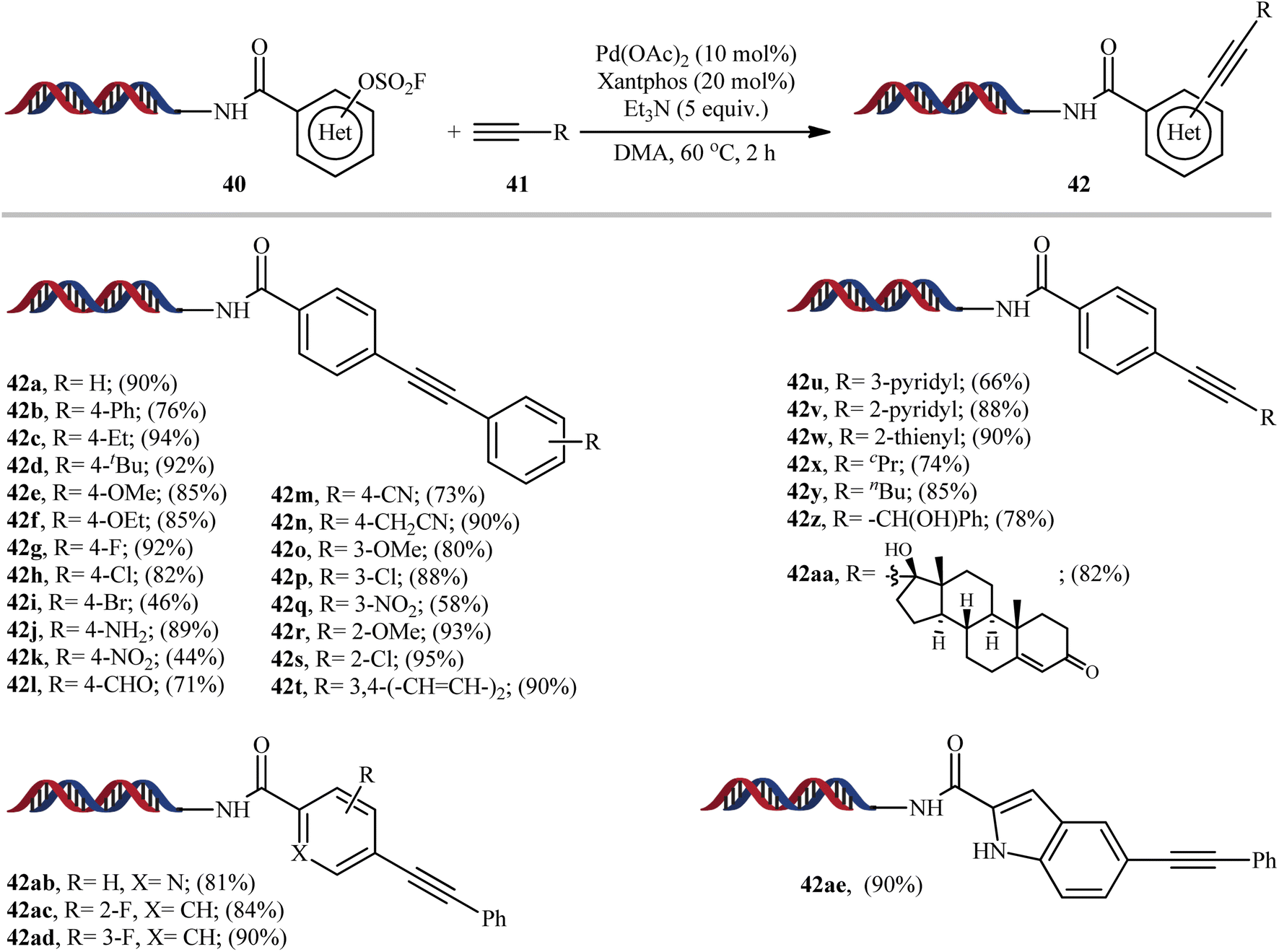
In 2019, Yang-Lerner's research group reported an interesting Pd-catalyzed Sonogashira cross-coupling of DNA-encoded (hetero)aryl fluorosulfates 40 with various terminal alkynes 41 (Scheme 12).23 This represents the first Sonogashira reaction using aryl fluorosulfates. Various aliphatic, aromatic and heteroaromatic alkynes were employed successfully in this system and good yields of the expected C(sp2)–C(sp) coupling products are obtained (44–95%). The results indicated that both electron-neutral and electron-rich terminal aryl alkynes afforded better yields compared to electron-deficient ones. Unfortunately, the reaction failed in the case of a CF3-substituted aryl alkynes. Interestingly, the outcome of reaction almost was not dependent on the electronic nature of the aryl fluorosulfates.
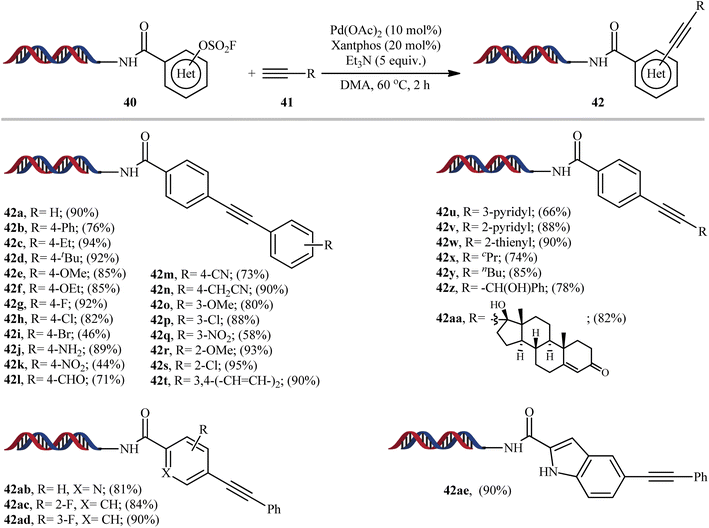 |
| | Scheme 12 Selected examples of Pd-catalyzed Sonogashira cross-coupling of DNA-encoded (hetero)aryl fluorosulfates 40 with alkynes 41 reported by Yang-Lerner's research group. | |
2.5. Carbonylative cross-coupling reactions
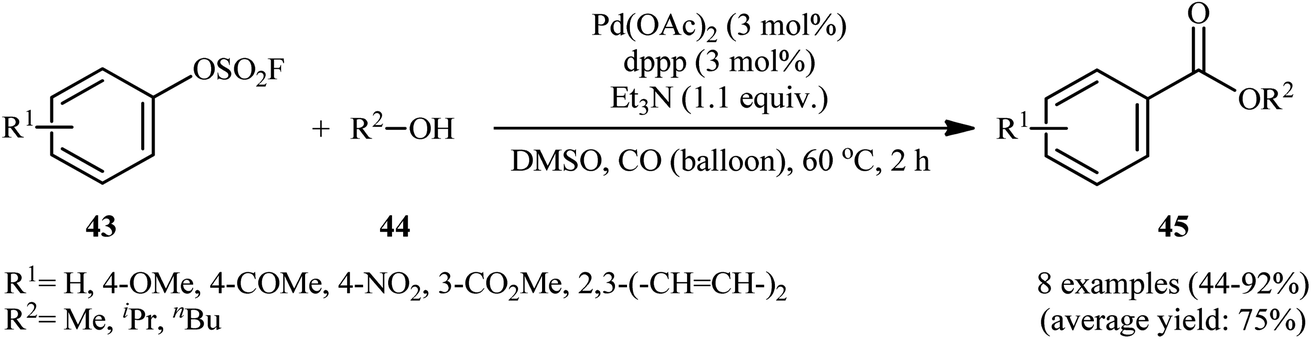
In 1992, aryl fhorosulfonates 43 as activated phenol derivatives were employed in palladium-catalyzed carbonylative reactions with simple alcohols 44 by Roth and Thomas.31 This represents the first Pd-catalyzed alkoxycarbonylation of aryl fhorosulfonates. The reactions were carried out under carbon monoxide (CO) atmosphere, tolerated the presence of various functional groups, and provided the desired esters 45 in moderate to high isolated yields (Scheme 13). The results demonstrated that the nature of ligand had a major impact on the success of this alkoxycarbonylation. When 1,3-bis(diphenylphosphino)propane (dppp) ligand was replaced with 1,1′-bis(diphenylphosphino)ferrocene (dppf), the desired products were obtained in much lower yields.
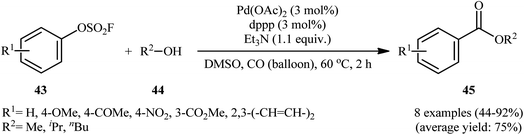 |
| | Scheme 13 Pd-catalyzed alkoxycarbonylation of aryl fhorosulfonates 43 developed by Roth. | |
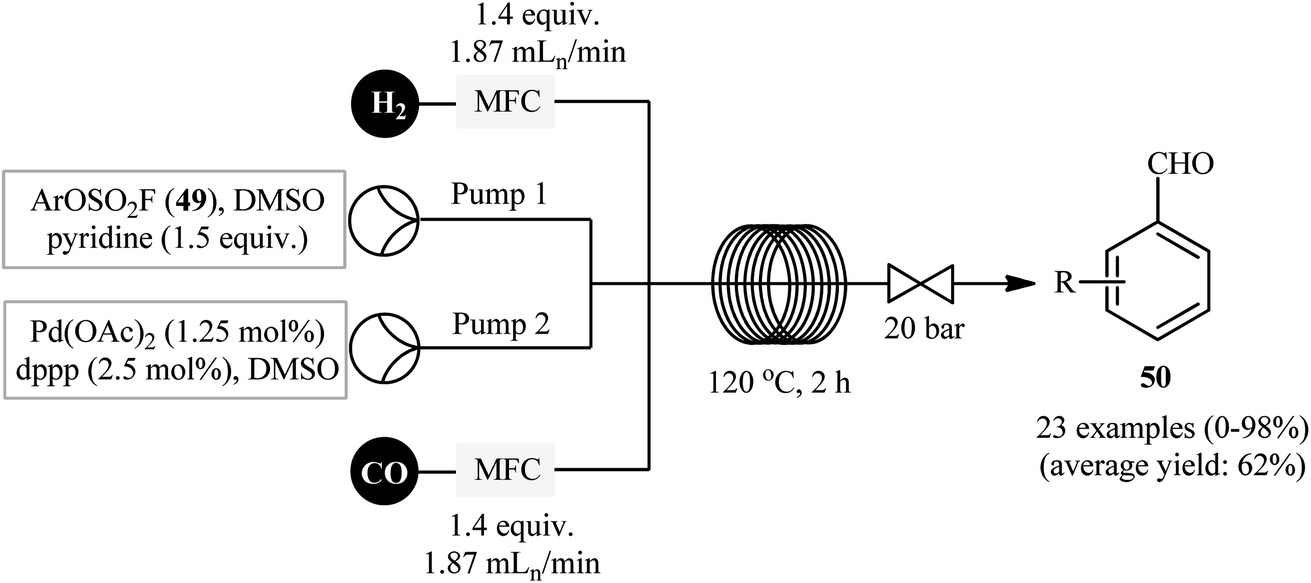
Drawing inspiration from this preliminary study, Qin's research team developed a general and efficient methodology for the synthesis of arylcarboxylic amide derivatives through in situ conversion of the phenols into their aryl fluorosulfonates and the subsequent carbonylative cross-coupling reaction with amines in a single pot.32 In this study, twenty-nine amides 48 were synthesized via C–O bond activation of phenols 46 using sulfuryl fluoride (SO2F2) follow by treatment of in situ generated aryl fhorosulfonates with various amines 47 under CO atmosphere, in the presence of Pd(OAc)2/dppp combination as the catalytic system in DMSO at 60 °C (Scheme 14). These authors demonstrated significant scope of the amines, but limited scope of the phenols substrate. The scope of amines that underwent coupling was broad enough to include acyclic aliphatic, benzylic, aromatic, and heteroaromatic derivatives, and all of which were found to be highly suitable amino sources. However, the same reaction provided sulfonamides if cyclic amines were used. Guided by the same principle, recently Hone, Kappe, and co-workers synthesized a library of aryl aldehydes 50 by Pd-catalyzed formylation of the corresponding aryl fluorosulfonates 49 under continuous-flow conditions (Scheme 15).33
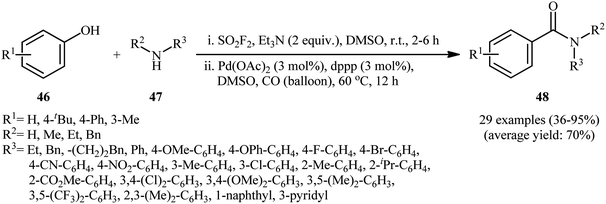 |
| | Scheme 14 Qin's synthesis of amides 48. | |
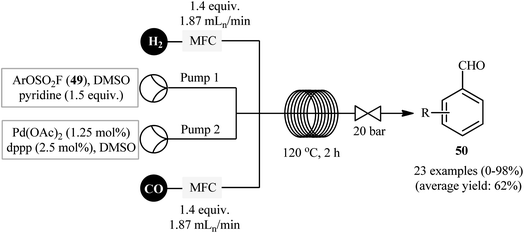 |
| | Scheme 15 Continuous flow synthesis of aryl aldehydes 50 from aryl fluorosulfonates 49. | |
2.6. Carboxylation reactions
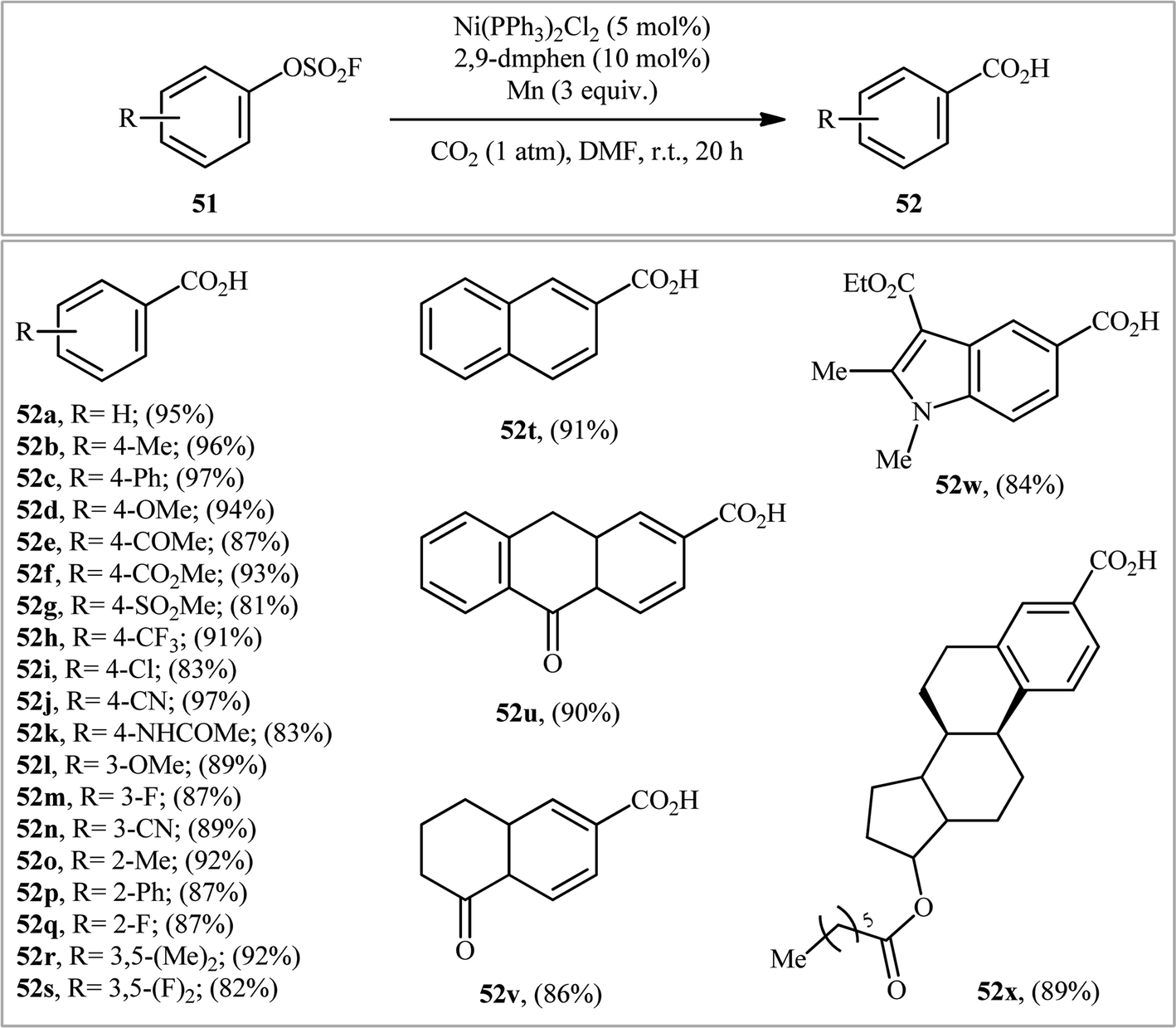
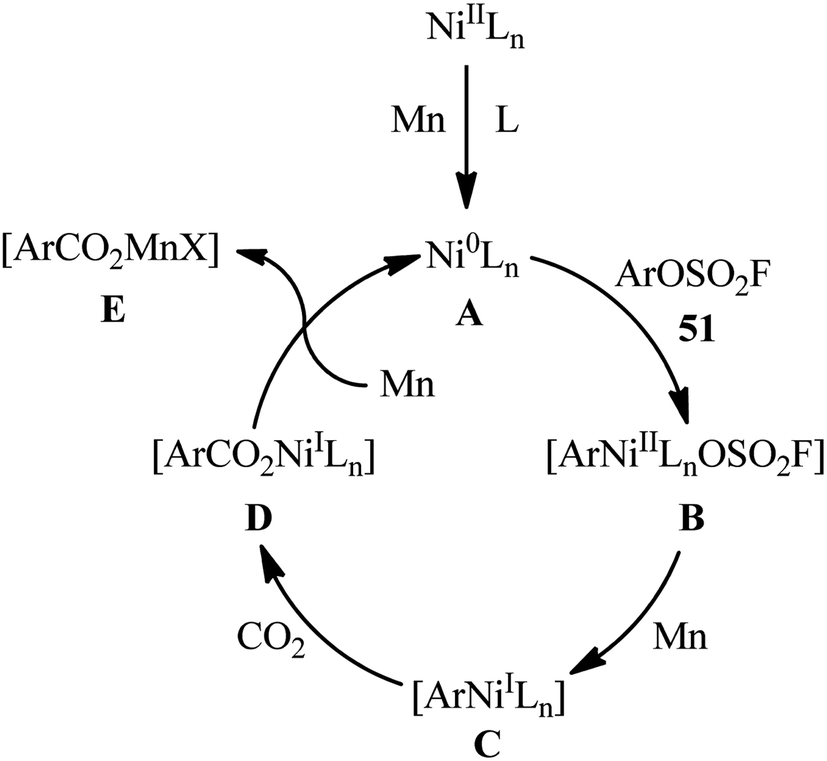
The application of aryl fluorosulfates as aryl sources in carboxylation reactions has been scarcely investigated (Scheme 16). In fact, to the best of our knowledge, only one practical example of such a reaction was reported in literature till date (Scheme 17). In this study, Mei and colleagues disclosed that the treatment of various (hetero)aromatic fluorosulfates 51 containing electron-donating groups (e.g., Me, OMe) and electron-withdrawing groups (e.g., F, Cl, CN, COMe, CO2Me) with atmospheric CO2 in the presence of Ni(PPh3)2Cl2/2,9-dimethyl-1,10-phenanthroline (2,9-dmphen)/Mn combination as a catalytic system in DMF slowly afforded the corresponding carboxylic acids 52 in good to excellent yields (Scheme 18).34 It is noteworthy that both Ni catalyst and Mn are essential for this carboxylation. No product was observed in the absence of any of them. In the lack of a ligand, the reaction furnished the carboxylated products, albeit with considerable reduced yields. Besides 2,9-dmphen, other ligands such as PPh3 and bpy were also found to promote this carboxylation reaction; albeit, in lower yields. Interestingly, under the optimized conditions, twelve aromatic carboxylic acids were also synthesized in good yields (60–79%) through the one-pot version of this carboxylation using in situ generated aryl fluorosulfonates from the corresponding phenols. Furthermore, this carboxylation strategy was also successfully applied to a range of biologically and synthetically important pyridyl substrates. In this case, CH2N2 was used to methylate the carboxylic acid products to avoid the difficult separation of the pyridine carboxylic acids from water. In the proposed mechanistic pathway (Scheme 19), the authors suggested that this intramolecular C–C bond forming reaction proceeds via generation of nickel(0) complex A through reduction of the nickel(II) catalyst by Mn. Next, oxidative addition of this complex with an aryl fluorosulfate 51 generates Ni(II) species B. Subsequently, single-electron reduction of intermediate B by Mn produces intermediate C, which after reaction with CO2 affords complex D. The subsequent single-electron reduction of intermediate D delivers the desired carboxylated product E and regenerates Ni(0) catalyst.
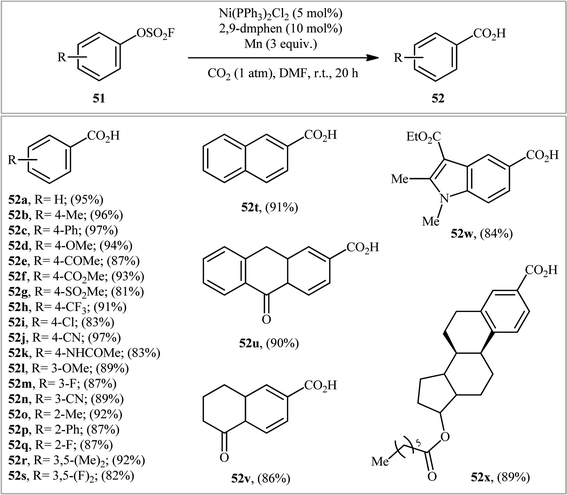 |
| | Scheme 16 Ni-catalyzed carboxylation of (hetero)aryl fluorosulfates 51 using CO2. | |
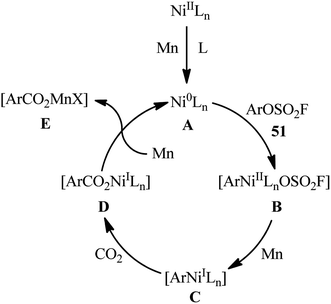 |
| | Scheme 17 Proposed mechanistic pathways for the reaction in Scheme 17. | |
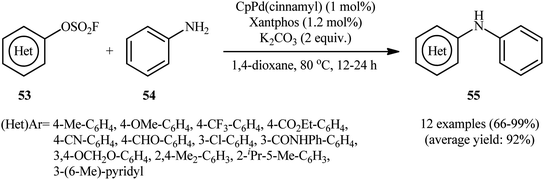 |
| | Scheme 18 Buchwald–Hartwig C–N coupling reaction between (hetero)aryl fluorosulfonates 53 with aniline 54. | |
 |
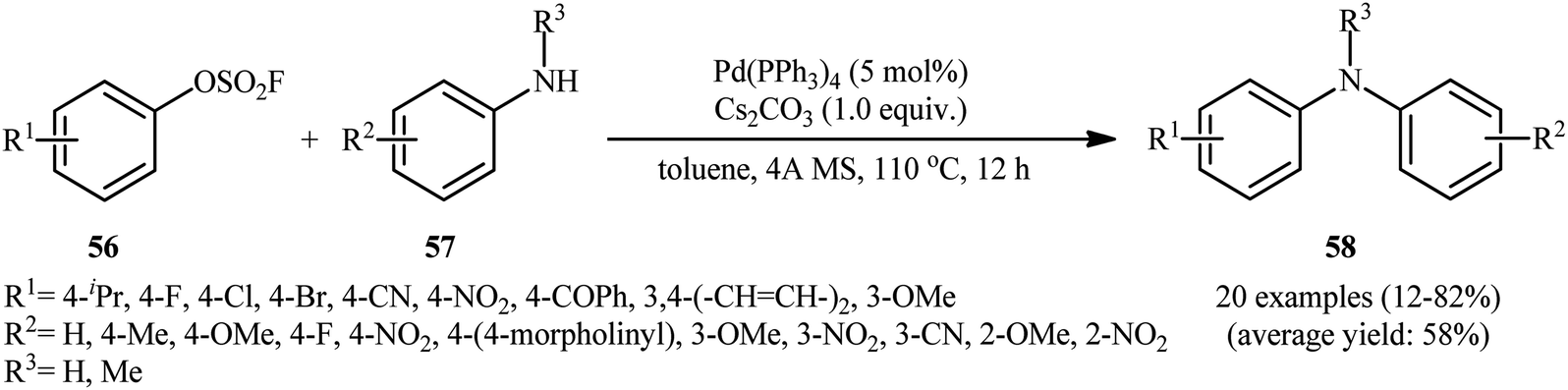
| | Scheme 19 Pd(PPh3)4-catalyzed amination of aryl fluorosulfonates 56 with aryl amines 57. | |
3 Carbon-heteroatom cross-coupling reactions
3.1. C–N cross-coupling
The Pd-catalyzed cross-coupling between aryl halides and amines is known as the Buchwald–Hartwig reaction which represents a powerful tool for the synthesis of arylamines.35
One of the earliest reports on the utilization of aryl fluorosulfonates as electrophilic partners in the Buchwald–Hartwig C–N coupling reaction was published by Hanley and co-workers in 2016,36 who showed that the treatment of (hetero)aryl fluorosulfonate derivatives 53 with aniline 54 in the presence of catalytic amounts of CpPd(cinnamyl) and Xantphos in 1,4-dioxane, resulted in the formation of the corresponding diarylamines 55 in good to quantitative yields. As shown in Scheme 18, the reaction displayed good reactivity and tolerance to aryl fluorosulfonates with functional groups both electron-rich and electron-deficient, including methoxy, chloro, trifluoromethyl, cyano, ester, and aldehyde functionalities. Apart from aniline, benzyl amine was also compatible with this scenario. However, like aniline, the substrate scope of benzyl amine was not exported in this study. With the aim of development of cheaper and less toxic catalytic system, in this study, the authors developed a Ni-based catalytic system. They showed that merge of 5.0 mol% of Ni(COD)2 with dppf could effectively catalyze this C–N bond forming reaction; albeit in lower efficiency than their Pd-based catalytic system.
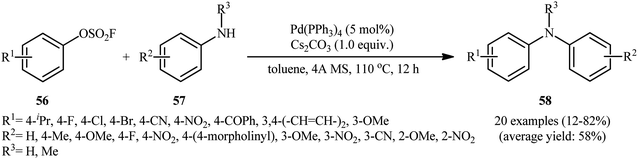
Subsequently, in an attempt to further demonstrate the strength of this attractive page of diarylamine synthesis, Lim, Byun, and Kim documented an elegant Pd(OAc)2-catalyzed amination of aryl fluorosulfonates 56 using a range of functionalized primary and secondary aniline derivatives 57, which allowed high yielding access to the corresponding diarylamine products 58 under ligand-free conditions (Scheme 19).37 Through exploration and optimization of this C–N coupling reaction, the author identified that the reaction rate is strongly dependent to the nature of base and solvent. Among various bases tested (e.g., Cs2CO3, K2CO3, Na2CO3, K3PO4), Cs2CO3 dispensed the excellent result, whereas MeCN was found to be the most effective solvent among the solvents tested (e.g., toluene, 1,4-dioxane, DMF, DMA, THF). Notably, the reactivity of aryl fluorosulfonates were compared with other common aryl electrophiles under the standard conditions. Overall the relative reaction rates of tested electrophilic partners followed the order: Ar–OSO2F > Ar–OTf > Ar–Cl ≥ Ar–Br ≥ Ar–I ≫ Ar–F.
3.2. C–P cross-coupling
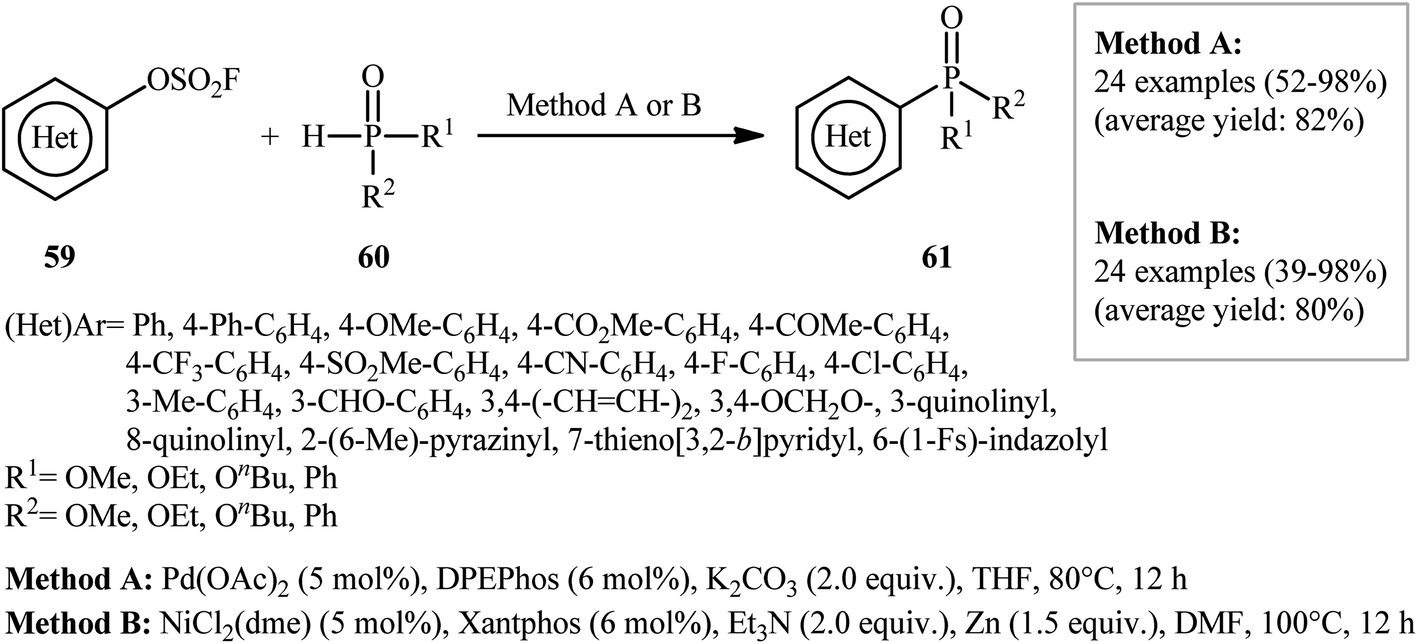
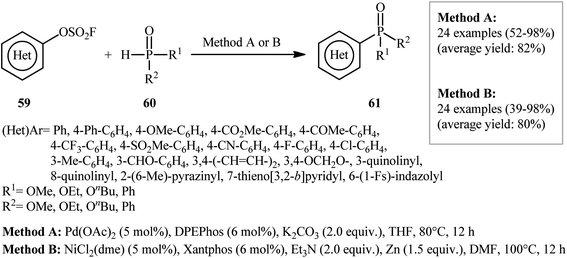
Very recently, Ding's research group studied the possibility of synthesis of aryl phosphonates through the transition metal-catalyzed C–P cross-coupling of aryl fluorosulfonates and hydrogen phosphoryl compounds.38 By employing p-biphenyl fluorosulfonate and dibutyl phosphite as the model substrates, the reaction variables such as catalysts, ligands, bases, and solvents were carefully screened. The results indicated that the merge of 5 mol% of Pd(OAc)2 with 6 mol% of DPEPhos and 2.0 equiv. of K2CO3 was the most appropriate catalytic system for this conversion and among the various aprotic solvents (e.g., toluene, 1,4-dioxane, THF, DMF, DMSO); THF was found to be the most suitable solvent. Under the optimized conditions, 24 (hetero)aryl phosphonate derivatives 61 were obtained in moderate to excellent yields by reaction of various (hetero)aryl fluorosulfonates 59 with hydrogen phosphoryl compounds 60 (Scheme 20). A wide range of important functional groups including OMe, CF3, F, Cl, CN, CHO, COMe, CO2Me and SO2Me are tolerated by the reaction conditions employed. Thus this procedure offers a versatile synthetic handle for further manipulation of products. Interestingly, all the three kinds of P(O)–H compounds (H-phosphonates, H-phosphinates, and secondary phosphine oxides) were applicable to this reaction. In this study, the authors also developed an alternative Ni-based catalytic system for this transformation. Thus, in the presence of NiCl2(dme)/Xantphos/Et3N/Zn combination as a catalytic system in DMF, the same set of (hetero)aryl phosphonate derivatives were obtained in comparable yields. They also reported one-pot version of the same reaction where the requisite aryl fluorosulfonates were prepared in situ from the corresponding phenols and SO2F2 (Scheme 21).
 |
| | Scheme 20 Ni- and Pd-catalyzed cross-coupling of (hetero)aryl fluorosulfonates 59 and hydrogen phosphoryl compounds 60. | |
 |
| | Scheme 21 Direct conversion of phenols to the corresponding aryl phosphonates though a sequential fluorosulfonation/C–P coupling approach. | |
4 Conclusion
Aryl fluorosulfates as more stable, more atom economical, and less hazardous alternatives of aryl triflates have drawn great attention over the past few years from organic chemists as powerful and versatile electrophilic partners in cross-coupling reactions. As illustrated, these easy accessible O-based pseudohalides have been successfully employed as electrophilic arylation agents in various carbon–carbon and carbon-heteroatom (N, O, P) cross-coupling reactions. Interestingly, some comparative studies disclosed superior activity of aryl fluorosulfates than the corresponding triflates in various coupling reactions. Challenges that remain to be faced in the future include: (i) identification of catalytic systems based on cheaper and less toxic metals; (ii) exploration of metal-free procedures; (iii) development of the coupling of aliphatic fluorosulfates; (iv) extension of the heteroatom coupling partners beyond simple amines, alcohols, and P(O)–H compounds; and (v) further investigation of the scope and limitation of existed couplings (e.g., Sonogashira coupling).
Ethical approval
This article does not contain any studies with human participants or animals performed by the authors.
Ethics approval and consent to participate
We comply with the ethical standards. We provide our consent to take part.
Consent for publication
All of the authors are giving consent to publish.
Conflicts of interest
There is no conflict of interest by any author.
Acknowledgements
The authors would like to thank Scientific Research Deanship at King Khalid University, Abha, Saudi Arabia through the Large Research Group Project under grant number (RGP.02/219/43).
References
-
(a) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed;
(b) J. I. Ayogu and E. A. Onoabedje, Catal. Sci. Technol., 2019, 9, 5233–5255 RSC;
(c) X. Pang, X. Peng and X. Z. Shu, Synthesis, 2020, 52, 3751–3763 CrossRef CAS.
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS.
- C. Zarate, M. van Gemmeren, R. J. Somerville and R. Martin, Adv. Organomet. Chem., 2016, 66, 143–222 CrossRef.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963–4972 RSC.
- J. Albaneze-Walker, R. Raju, J. A. Vance, A. J. Goodman, M. R. Reeder, J. Liao, M. T. Maust, P. A. Irish, P. Espino and D. R. Andrews, Org. Lett., 2009, 11, 1463–1466 CrossRef CAS PubMed.
- P. Batsomboon, B. A. Gold, I. V. Alabugin and G. B. Dudley, Synthesis, 2012, 44, 1818–1824 CrossRef CAS.
- L. Revathi, L. Ravindar, J. Leng, K. P. Rakesh and H. L. Qin, Asian J. Org. Chem., 2018, 7, 662–682 CrossRef CAS.
- G. P. Roth and C. E. Fuller, J. Org. Chem., 1991, 56, 3493–3496 CrossRef CAS.
- Selected reviews:
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(c) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(d) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(e) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef PubMed;
(f) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(g) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(h) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef;
(i) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(j) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(k) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS PubMed;
(l) I. Söğütlü, E. A. Mahmood, S. A. Shendy, S. Ebrahimiasl and E. Vessally, RSC Adv., 2021, 11, 2112–2125 RSC.
- Selected reviews:
(a) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(b) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2021, 11, 470–483 RSC;
(c) B. Azizi, M. R. P. Heravi, Z. Hossaini, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 13138–13151 RSC;
(d) L. Yan-mei, F. Jin-feng, H. Long-qiang, L. Wei-na and E. Vessally, RSC Adv., 2021, 11, 24474–24486 RSC;
(e) Y. Zhang and E. Vessally, RSC Adv., 2021, 11, 33447–33460 RSC;
(f) Y. Cao, S. Soleimani-Amiri, R. Ahmadi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 32513–32525 RSC;
(g) Z. Jiang, E. A. Mahmood, N. Z. Harofteh, A. G. Ebadi, M. Toughani and E. Vessally, Top. Curr. Chem., 2022, 380, 27 CrossRef CAS PubMed;
(h) M. M. Kadhim, M. E. Abdulkareem, M. R. Poor Heravi, S. Soleimani-Amiri, A. G. Ebadi and E. Vessally, J. Sulfur Chem., 2022, 1–15 Search PubMed.
-
(a) E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821–1823 CrossRef CAS;
(b) V. B. Phapale and D. J. Cárdenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC;
(c) B. Wei and P. Knochel, Synthesis, 2022, 54, 246–254 CrossRef CAS.
-
(a) M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed;
(b) W. D. Brittain and S. L. Cobb, Org. Biomol. Chem., 2018, 16, 10–20 RSC.
- M. Mendel, I. Kalvet, D. Hupperich, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed. Engl., 2020, 59, 2115–2119 CrossRef CAS PubMed.
- S. Kotha, K. Lahiri and K. Dhurke, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- B. S. Takale, F. Y. Kong and R. R. Thakore, Organics, 2022, 3, 1–21 CrossRef CAS.
- Q. Liang, P. Xing, Z. Huang, J. Dong, K. B. Sharpless, X. Li and B. Jiang, Org. Lett., 2015, 17, 1942–1945 CrossRef CAS PubMed.
- P. S. Hanley, M. S. Ober, A. L. Krasovskiy, G. T. Whiteker and W. J. Kruper, ACS Catal., 2015, 5, 5041–5046 CrossRef CAS.
- E. Zhang, J. Tang, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Chem. Eur. J., 2016, 22, 5692–5697 CrossRef CAS PubMed.
- X. Li, Y. Ma, Q. Hu, B. Jiang, Q. Wu and Z. Yuan, Catal. Commun., 2018, 117, 57–62 CrossRef CAS.
- X. Li, H. Zhang, F. Feng, Q. Hu and Z. Yuan, ChemistrySelect, 2018, 3, 12287–12290 CrossRef CAS.
- X. Li, T. Zhang, R. Hu, H. Zhang, C. Ren and Z. Yuan, Org. Biomol. Chem., 2020, 18, 4748–4753 RSC.
- H. Xu, F. Ma, N. Wang, W. Hou, H. Xiong, F. Lu, J. Li, S. Wang, P. Ma, G. Yang and R. A. Lerner, Adv. Sci., 2019, 6, 1901551 CrossRef CAS PubMed.
- Q. Zhao, G. Guo, W. Zhu, L. Zhu, Y. Da, Y. Han, H. Xu, S. Wu, Y. Cheng, Y. Zhou and X. Cai, Chem. Eur. J., 2020, 26, 15938–15943 CrossRef CAS PubMed.
- J. Leng and H. L. Qin, Org. Biomol. Chem., 2019, 17, 5001–5008 RSC.
- S. Yang, H. Li, X. Yu, J. An and M. Szostak, J. Org. Chem., 2022, 87, 15250–15260 CrossRef CAS PubMed.
- V. Bieliūnas and W. M. De Borggraeve, J. Org. Chem., 2019, 84, 15706–15717 CrossRef PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
-
(a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed;
(b) M. Karak, L. C. Barbosa and G. C. Hargaden, RSC Adv., 2014, 4, 53442–53466 RSC.
- J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Org. Chem. Front., 2021, 8, 384–414 RSC.
- G. P. Roth and J. A. Thomas, Tetrahedron Lett., 1992, 33, 1959–1962 CrossRef CAS.
- W. Y. Fang, Y. M. Huang, J. Leng and H. L. Qin, Asian J. Org. Chem., 2018, 7, 751–756 CrossRef CAS.
- M. Köckinger, P. Hanselmann, G. Hu, C. A. Hone and C. O. Kappe, RSC Adv., 2020, 10, 22449–22453 RSC.
- C. Ma, C. Q. Zhao, X. T. Xu, Z. M. Li, X. Y. Wang, K. Zhang and T. S. Mei, Org. Lett., 2019, 21, 2464–2467 CrossRef CAS PubMed.
-
(a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed;
(b) M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS;
(c) R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed. Engl., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- P. S. Hanley, T. P. Clark, A. L. Krasovskiy, M. S. Ober, J. P. O'Brien and T. S. Staton, ACS Catal., 2016, 6, 3515–3519 CrossRef CAS.
- T. Lim, S. Byun and B. M. Kim, Asian J. Org. Chem., 2017, 6, 1222–1225 CrossRef CAS.
- G. Zhang, J. Wang, C. Guan, Y. Zhao and C. Ding, Eur. J. Org. Chem., 2021, 810–813 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c,
Halim Zainid,
Nabeel Ahmad
*c,
Halim Zainid,
Nabeel Ahmad