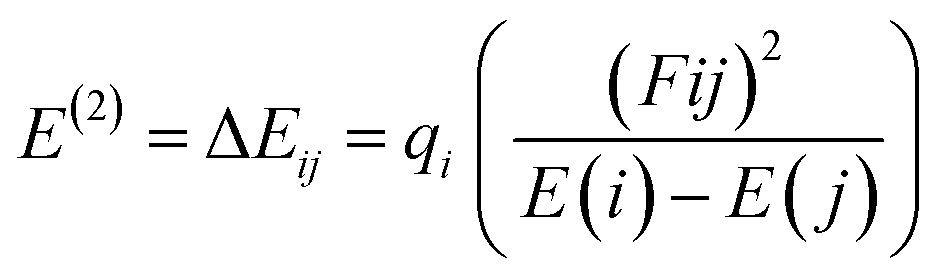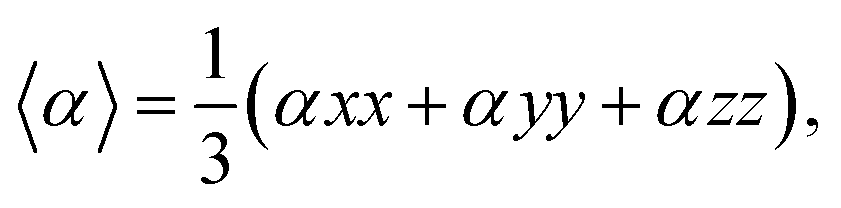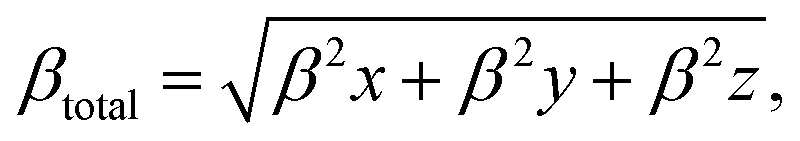DOI:
10.1039/D3RA01279D
(Paper)
RSC Adv., 2023,
13, 13624-13641
Modeling of magnesium-decorated graphene quantum dot nanostructure for trapping AsH3, PH3 and NH3 gases†
Received
24th February 2023
, Accepted 18th April 2023
First published on 3rd May 2023
Abstract
A magnesium-decorated graphene quantum dot (C24H12-Mg) surface has been examined theoretically using density functional theory (DFT) computations at the ωB97XD/6-311++G(2p,2d) level of theory to determine its sensing capability toward XH3 gases, where X = As, N and P, in four different phases: gas, benzene solvent, ethanol solvent and water. This research was carried out in different phases in order to predict the best possible phase for the adsorption of the toxic gases. Analysis of the electronic properties shows that in the different phases the energy gap follows the order NH3@C24H12-Mg < PH3@C24H12-Mg < AsH3@C24H12-Mg. The results obtained from the adsorption studies show that all the calculated adsorption energies are negative, indicating that the nature of the adsorption is chemisorption. The adsorption energies can be arranged in an increasing trend of NH3@C24H12-Mg < PH3@C24H12-Mg < AsH3@C24H12-Mg. The best adsorption performance was noted in the gas phase compared to the other studied counterparts. The interaction between the adsorbed gases and the surfaces shows a non-covalent interaction nature, as confirmed by the quantum theory of atoms-in-molecules (QTAIM) and non-covalent interactions (NCI) analysis. The overall results suggest that we can infer that the surface of the magnesium-decorated graphene quantum dot C24H12-Mg is more efficient for sensing the gas AsH3 than PH3 and NH3.
1 Introduction
Recent advances in nanomaterial science have improved the concept of gas sensors1 and furthermore created an increase in the potential for designing efficient, effective and selective gas sensors for identifying and sensing volatile and hazardous gases.2–4 Several nanostructured materials have been considered by researchers due to their outstanding features, such as high selectivity, good stability, low toxicity, high surface activity, quick recovery, high sensitivity, fast response, low operating temperature, and pronounced quantum confinement.5–7 Graphene quantum dots, which are graphene nanoparticle materials having a size of less than 100 nm, have drawn the attention of researchers owing to their excellent sensing properties.8,9 Graphene quantum dots possesses good structural, electronic, spin optical and photoelectric features.10 In addition to their uses in sensors, they are also applicable in solar cells, photodetectors, drug delivery, bioimaging and photoluminescent materials.11–13 The promising properties of graphene quantum dots in sensing have been reviewed and reported. As reported by Meixiu Li et al.,14 graphene quantum dots exhibit high photostability against blinking and photo-leaching, as well as low toxicity, and thus have greater biocompatibility. Graphene quantum dot materials for sensing, bioimaging and energy storage applications have been reviewed by Kumar et al.15
Recently, the density functional theory (DFT) method has been invoked for theoretical calculations to examine and predict the properties of materials under investigation.16,17 This method is an important method in the scientific community today, and it can help to determine whether a nanomaterial could be considered for sensing applications.18 DFT methodology can be also employed to gain insight into a gas sensor material to understand the molecular electronic and structural properties, mechanistic behavior, conductivity, and sensitivity of the material to detect and identify hazardous gases such as AsH3, NH3, PH3.19–21 Arsine (AsH3), ammonia (NH3) and phosphine (PH3) are toxic colorless hydride gases with a pungent smell.22,23 They are highly irritating gases that are also flammable.23 Exposure to these gases can cause skin irritation, headache, vomiting, nausea, and pains.24–26 Inhalation of large concentrations of these gases can cause liver, kidney or nerve damage and can lead to death.27–29 Hence, it is of utmost importance that these gases should be sensed or detected.30 Numerous efforts have been made through the density functional theory (DFT) method towards the sensing and adsorption of XH3 (X = As, N, P) gases using a graphene or graphene-like structure. NH3, PH3, and AsH3 adsorption and sensing on alkaline earth metal (AEM = Be, Mg, Ca and Sr)-doped graphene was conducted by Singsen et al.31 Their results revealed that the gases were weakly absorbed on intrinsic graphene. On the doped structures, significant increases in the charge transfer, adsorption energies and orbital hybridization of the systems were observed. The Mg-doped graphene showed sensitivity towards PH3 and AsH3 in the sensitivity response and recovery time analysis, while the Sr-doped graphene revealed sensitivity towards NH3. Luo et al.32 also studied the sensing and adsorption of AsH3, NH3, and PH3 on rare-earth-metal-doped graphene through the DFT method. Their study showed that the graphene exhibited physisorption towards the gases. No significant changes were observed in the geometry, density of states or charge population of the structure. However, upon modification by a rare-earth metal, enhanced chemisorption of the gases was observed Luo et al.33 conducted a DFT investigation of NH3, PH3 and AsH3 adsorption on Sc-, Tl-, V-, and Cr-doped single-wall carbon nanotubes (SWCNT). Their adsorption ability followed the order NH3 > PH3 > AsH3. Their study showed that the transition metal (TM)-doped SWCNTs were more favorable for gas sensing and adsorption than the pristine SWCNTs. Based on various studies, it can be deduced that structural modification such as doping, encapsulation or decoration of a material can enhance its properties to make it a better gas sensing material.
However, to the best of our knowledge there have been no studies involving the theoretical investigation of the sensing of hydride gases using graphene quantum dots in different phases and solvation states. Additionally, the detection and removal of toxic gases such as AsH3, PH3, and NH3 is of critical importance due to their harmful effects on the environment and human health. Magnesium-decorated graphene quantum dot nanostructures have shown promising potential for trapping these gases, but a comprehensive understanding of their behavior and efficacy is lacking. Therefore, the research problem is to model the interaction of AsH3, PH3, and NH3 with magnesium-decorated graphene quantum dot nanostructures and investigate the factors influencing their gas sensing performance. In this paper, the density functional theory (DFT) method with the WB97XD functional and 6-311++G(2p,2d) basis set has been employed to study and analyze the selectivity, sensitivity, conductivity and efficacy of a graphene quantum dot and its decorated magnesium atom in sensing the toxic hydride gases XH3, where X = As, N and P. This study was carried out in four different phases, namely, gas, benzene, ethanol and water, to provide insights into the effect of solvation and to determine the best possible phase for the adsorption of the gases. The following analyses were carried out in this study: structural properties, frontier molecular orbital (FMO), density of states (DOS), natural bond orbital (NBO), quantum theory of atoms-in-molecules (QTAIM), non-covalent interactions (NCI), adsorption study, sensor mechanics, electrical conductivity, recovery time and work function, dipole moment and charge transfer, nonlinear optics (NLO) analysis and thermodynamics properties. The FMO parameters and the DOS were invoked to study the molecular electronic properties of the magnesium-decorated graphene quantum dots. QTAIM and NCI were utilized to verify the non-covalent interactions existing within the adsorbate and adsorbent. Comparative adsorption energy and sensor mechanisms were employed in this study to determine the sensing ability of the studied surfaces and the adsorbate.
2 Computational details
The density functional theory (DFT) method was employed to handle all theoretical calculations performed on the studied surface of the graphene quantum dot with its decorated magnesium atom. The calculations were achieved with the Gaussview 6.0.16 (ref. 34) and Gaussian 16 suite of programs35 using the hybrid long-range separated empirical-correlated dispersion ωB97XD functional and the 6-311++G(2p,2d) basis set. The calculations were first carried out in vacuum, which is the gas phase, before further calculations were made for solvation with benzene, ethanol and water. Local minima on the potential energy surface were achieved by considering the frequency calculation at the same level of theory. Thus, the absence of imaginary frequencies confirmed the absolute correspondence of the optimized geometry to a local minimum on the potential energy surface.36,37 The structural properties were achieved via the software Chemcraft 16.38 The frontier molecular orbital (FMO), which analyses the conductivity and stability of the magnesium-decorated graphene quantum dots, was carried out. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy values were obtained through Gaussian 16 and further calculations of the quantum descriptors were calculated with the aid of Koopmans' approximation.39 The pictorial representations of the HOMO and LUMO iso-surfaces were obtained via Chemcraft. The software Origin40 was used to plot the graphical representation of the density of states (DOS). Using the same functional and basis set, the natural bond orbital (NBO) calculations were carried out using the NBO 3.0 (ref. 41) embedded in the Gaussian 16 software. The quantum theory of atoms in molecules and non-covalent interaction analysis was achieved via the multifunctional wave function analyzer program multiwfn 3.0.42 Pictorial representations of the QTAIM and NCI were obtained using Visual Molecular Dynamics (VMD).43 The absorption energies of the gases adsorbed on the C24H12-Mg quantum dot surface were calculated using eqn (1):| | |
Eads = EC24H12-Mg/gas − (Egas + EC24H12-Mg)
| (1) |
where Eads = absorption energy of the gases on the magnesium-decorated graphene quantum dot surface. EC24H12-Mg/gas = Total energy of the structure after absorption of the XH3 (X = As, N, P) on the magnesium-decorated graphene quantum dots surface. Egas = energy of each of the isolated gases. EC24H12-Mg = energy of the magnesium-decorated graphene quantum dots surface.
3 Results and discussions
3.1 Structural properties
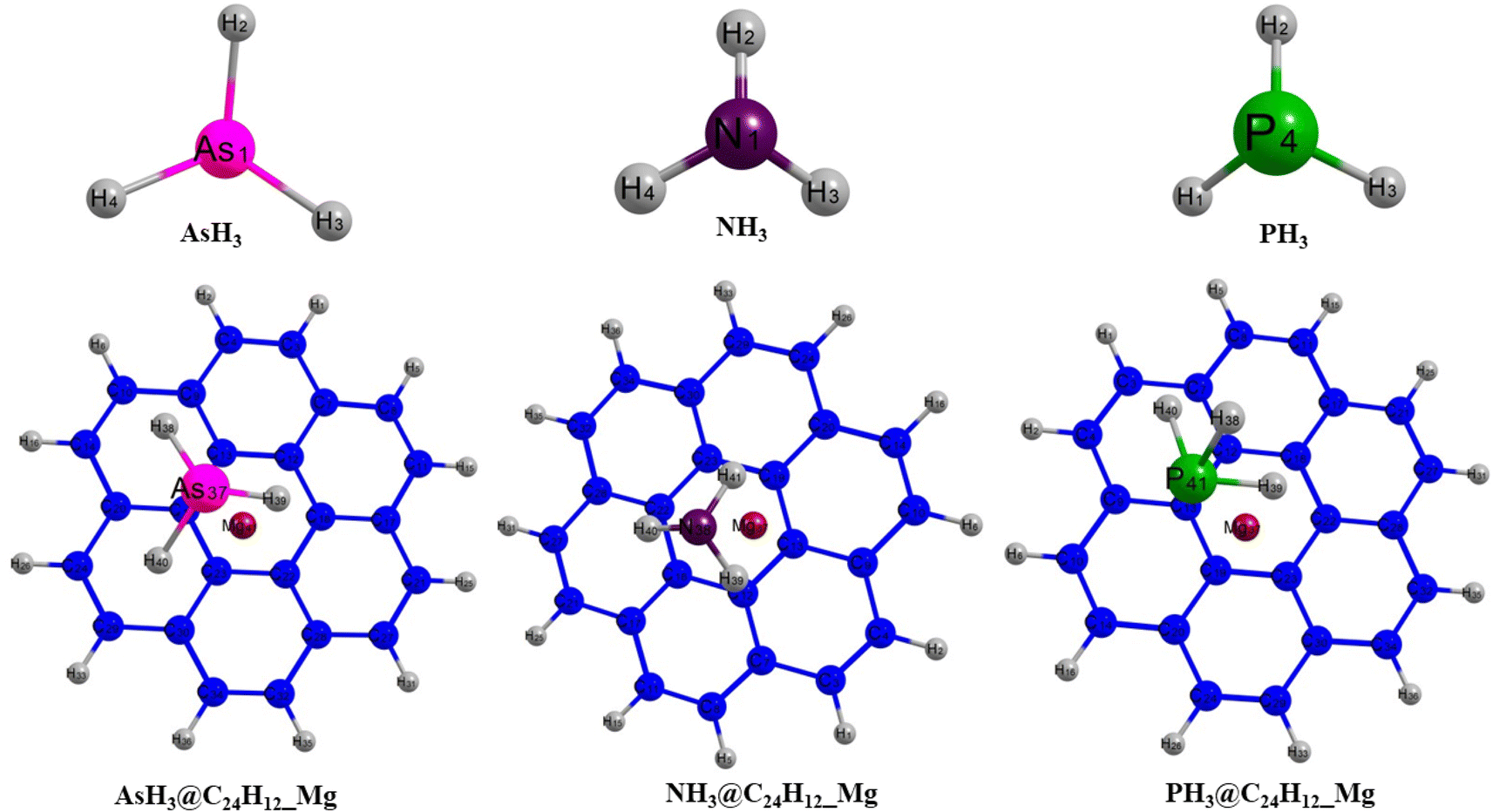
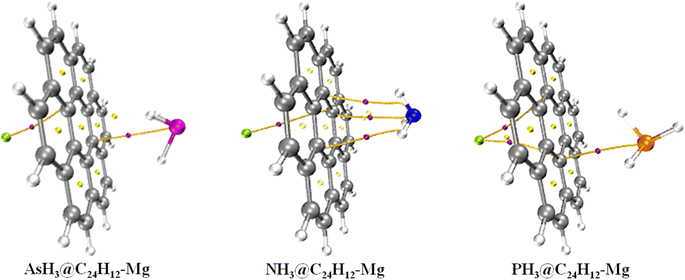
Insight into the structural configuration of studied structures can be obtained by proper structural and geometrical optimization.44 In this work, all the structures of the adsorbed gases on the magnesium-decorated graphene quantum dot in the four different phases (gas, benzene, ethanol and water) were optimized using the DFT/ωB97XD/6-311++G(2p,2d) method. The optimized structures of magnesium-decorated graphene quantum dot as absorbent and the studied gases AsH3, NH3, and PH3 are presented in Fig. 1. The graphene quantum dot comprises seven hexagons, which makes it a hexagonal structure.45 AsH3, NH3 and PH3 have a tetrahedral geometry and trigonal pyramidal shapes.46 The bond distance between the gases and the quantum dot is an important parameter that reveals details about the strength of the material and can also determine the kind of bond formation.47 The bond lengths of the quantum dot before and after the adsorption of the gases have been evaluated and computed in Table 1. Before adsorption of the gases on the adsorbent, it is observed that the bond length of magnesium to carbon (Mg–C) is in the range of 3.7–6.1 Å. For the gas phase, the shortest bond length between the magnesium atom and carbon atom is 3.745 Å, occurring for Mg37–C18. For the benzene phase, the bond length between the quantum dot and the magnesium atom is observed to be 3.963 Å for Mg37–C22, while it is found to be 3.981 Å and 3.986 Å for the ethanol and water phases, respectively, for Mg37–C18. After the interaction of the bare surface of the quantum dot and the investigated gas, it is noted that there is an effect on the geometrical bond length of the surface, especially between the magnesium and carbon atom. It is observed that there is a slight difference in the magnesium–carbon bond length in all phases. Upon the adsorption of AsH3 on the surface, the bond length of magnesium to carbon 18, Mg37–C18, exhibited a shift in value from 3.745 Å to 3.757 Å, 3.963 Å to 3.954 Å, 3.981 Å to 3.977 Å, and from 3.986 Å to 3.977 Å in the gas, benzene, ethanol and water phase, respectively. For NH3, it is observed that the shortest bond length of magnesium to carbon atom was recorded to be 3.825 Å, 3.966 Å, 3.972 Å, 4.016 Å corresponding to gas, benzene, ethanol, and water phase, respectively. Similarly, for PH3, there is also a change in the values of the length of the bond existing between the magnesium and carbon atom, which is found to be 3.789 Å for the gas phase, 3.963 Å for benzene, 4.000 Å for ethanol and 3.999 Å for water. The differences occurring upon adsorption of the investigated gases on the surface of the magnesium-decorated graphene quantum dot are the result of charge transfer occurring between the adsorbate (investigated gases) and the adsorbent (the quantum dot). There is the formation of a bond in the structure between the gases and the surface after the adsorption process. In the gas phase, AsH3@C24H12-Mg is noted to have a bond length of 3.734 Å for As37–C19. The shortest bond length formed between the nitrogen and carbon atom in the case of NH3@C24H12-Mg for the gas phase is recorded to be 3.565 Å. For PH3@C24H12-Mg in the gas phase, the bond label of P41–C13 corresponds to a bond length of 3.470 Å. Examining the bond length between the different gases and the surface in the gas phase, it can be deduced that PH3@C24H12-Mg has the shortest bond length. Observations from the benzene, ethanol and water phases show AsH3@C24H12-Mg to have the longest bond length and PH3@C24H12-Mg to have the shortest bond length. Therefore, for all the phases, it can be said that the structure in terms of bond length follows an increasing trend of PH3@C24H12-Mg < NH3@C24H12-Mg < AsH3@C24H12-Mg. The bond lengths between As, N and P and their hydrogen atoms, as well as the shortest carbon-to-carbon bonds, are also recorded in Table 1.
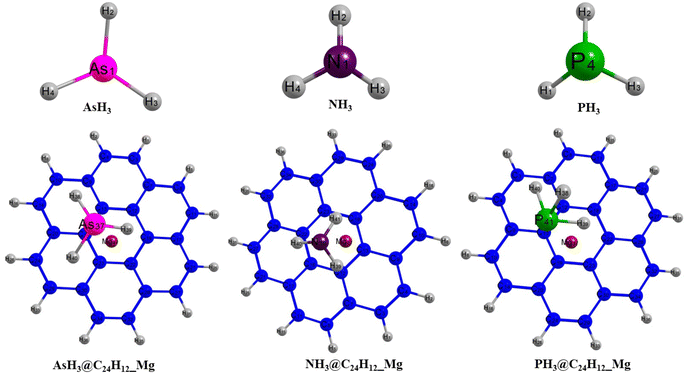 |
| | Fig. 1 Pictorial representation of AsH3, NH3 and PH3, and the gas-adsorbed surfaces of AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg. | |
Table 1 Selected bond lengths of the investigated system with DFT/ωB97XD/6-311++G(2p,2d) method
| Phases |
System |
| C24H12-Mg |
AsH3@C24H12-Mg |
NH3@C24H12-Mg |
PH3@C24H12-Mg |
| Bond label |
Bond length (Å) |
Bond label |
Bond length (Å) |
Bond label |
Bond length (Å) |
Bond label |
Bond length (Å) |
| Gas |
Mg37–C18 |
3.745 |
As37–H39 |
1.517 |
N38–H39 |
1.012 |
P41–H39 |
1.414 |
| C18–C17 |
1.406 |
As37–C19 |
3.734 |
N38–C13 |
3.565 |
P41–C13 |
3.470 |
| C18–C12 |
1.423 |
C19–C20 |
1.405 |
C13–C9 |
1.405 |
C13–C9 |
1.405 |
| C18–C22 |
1.423 |
Mg41–C18 |
3.757 |
Mg37–C18 |
3.825 |
Mg37–C22 |
3.789 |
| Benzene |
Mg37–C22 |
3.963 |
As37–H39 |
1.516 |
N38–H39 |
1.013 |
P41–H39 |
1.415 |
| C22–C28 |
1.406 |
As37–C20 |
4.066 |
N38–C13 |
3.594 |
P41–C13 |
3.471 |
| C22–C18 |
1.422 |
C20–C24 |
1.420 |
C13–C9 |
1.406 |
C13–C9 |
1.406 |
| C22–C28 |
1.422 |
Mg41–C18 |
3.954 |
Mg37–C19 |
3.966 |
Mg37–C23 |
3.963 |
| Ethanol |
Mg37–C18 |
3.981 |
As37–H39 |
1.516 |
N38–H39 |
1.014 |
P41–H39 |
1.413 |
| C18–C17 |
1.406 |
As37–C19 |
3.737 |
N38–C18 |
3.651 |
P41–C13 |
3.460 |
| C18–C12 |
1.422 |
C19–C20 |
1.405 |
C18–C17 |
1.406 |
C13–C9 |
1.406 |
| C18–C22 |
1.422 |
Mg41–C18 |
3.977 |
Mg37–C13 |
3.972 |
Mg37–C23 |
4.000 |
| Water |
Mg37–C18 |
3.986 |
As37–H39 |
1.516 |
N38–H40 |
1.014 |
P41–H39 |
1.413 |
| C18–C17 |
1.406 |
As37–C23 |
3.945 |
N38–C13 |
3.593 |
P41–C13 |
3.460 |
| C18–C12 |
1.423 |
C23–C30 |
1.406 |
C13–C9 |
1.406 |
C13–C9 |
1.406 |
| C18–C22 |
1.423 |
Mg41–C18 |
3.977 |
Mg37–C22 |
4.016 |
Mg37–C23 |
3.999 |
3.2 Frontier molecular orbital (FMO)
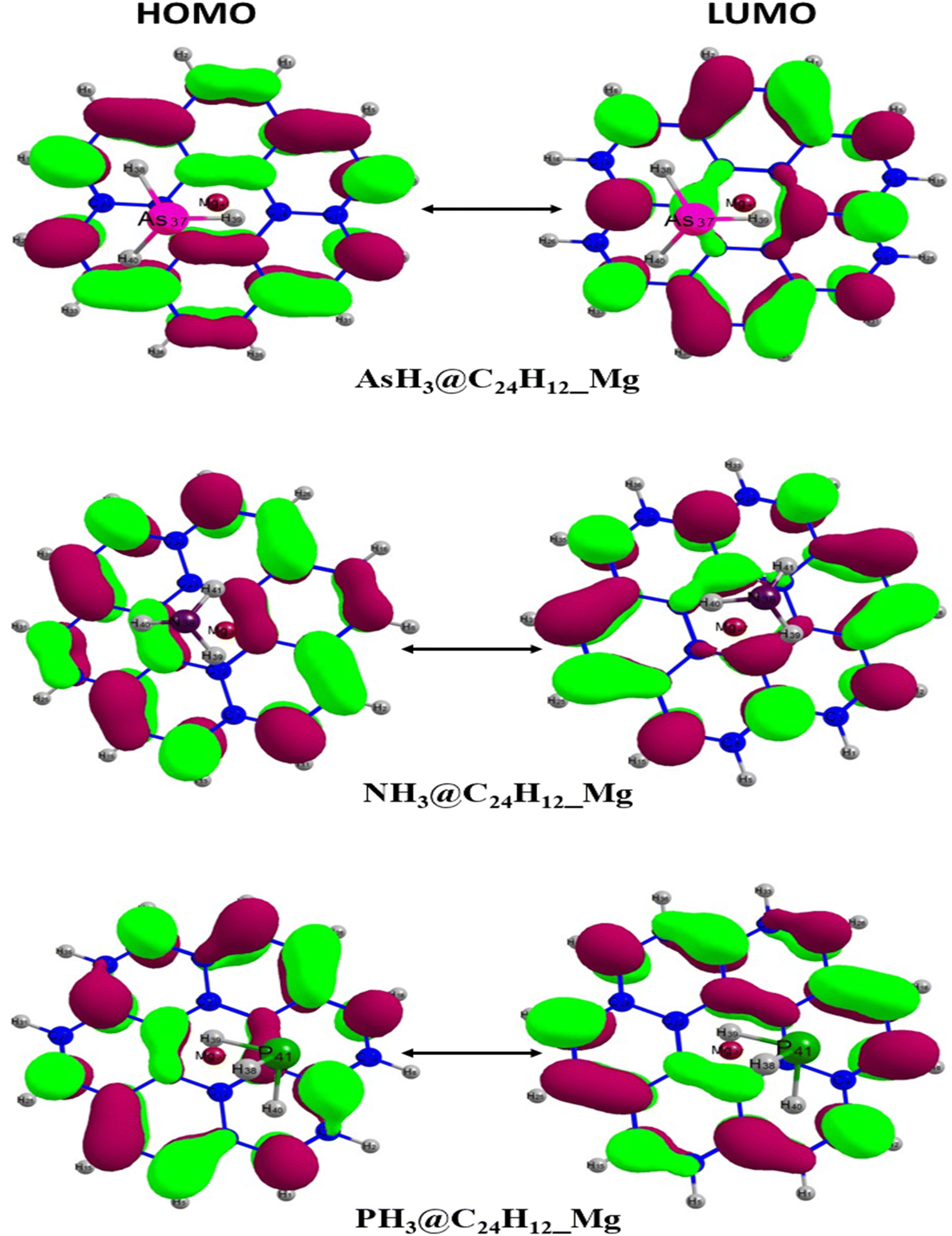
For the purpose of analyzing the electron transfer, kinetic stability, conductivity and sensing ability of the magnesium-decorated graphene quantum dot, the frontier molecular orbital (FMO) has been computed.48,49 The FMO serves as an approach for illustrating and understanding the electronic properties of a system.50 The FMO is constituted by two important orbitals: the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).51,52 The highest occupied molecular (HOMO) and lowest unoccupied molecular orbital (LUMO) help to depict the chemical activity of molecules and are also closely related to the donation and acceptance of an electron within a molecule.53,54 By default, the HOMO has higher tendency to donate an electron, while the LUMO has a higher tendency to accept an electron.55 The HOMO and LUMO give rise to a very important parameter, which is called the energy gap.56 The energy gap reveals details regarding the electron transfer, stability and electrical conductivity displayed by a structure.57 The energy gap is obtained by calculating the difference between the HOMO and the LUMO, and can be expressed mathematically as eqn (2):where Eg = energy gap, ELUMO = energy of the LUMO and EHOMO = energy of the HOMO.
In this study, the energies of the HOMO and LUMO were established using the DFT functional WB97XD with the 6-311++G (2d,2p) basis set, and their values are reported in Table 2. Here, the FMO analysis was computed on the bare surface of the magnesium-decorated graphene quantum dot and also on the gas-adsorbed magnesium-decorated graphene quantum dot for the four different phases, which are gas, benzene, ethanol and water. Before the adsorption of the studied gases on the surface, the bare surface of the magnesium-decorated graphene quantum dot was observed to have an energy gap of 6.340 eV with a HOMO value of −6.531 eV and LUMO of −0.190 eV for the gas phase. The energy gap for the benzene phase is 5.850 eV, and similarly, for the ethanol and the water phase, the energy gap of the bare surface of the quantum dot is 5.292 eV and 5.297 eV, respectively. Looking closely at Table 2, it can be observed that the surface of the magnesium-decorated graphene quantum dot has high energy gap values, indicating better stability of the surface. As presented in Table 2, it is observed that upon adsorption of the three gases in the different phases, there is a slight decrement in the values of the energy gap, except in the ethanol phase. According to some literature review, a decrease in energy gap indicates a good effect in conductivity of the surface, which is essential for the selective and effective sensing of the studied gases.58 In the gas phase, it is noted that the highest energy gap value was obtained upon adsorption of phosphorus hydride (PH3) on the magnesium-decorated graphene quantum dot, while the lowest energy gap was obtained for ammonia (NH3). In the gas phase, the HOMO–LUMO gap (energy gap) increases in the order 6.302 eV < 6.313 eV < 6.317 eV for NH3@C24H12-Mg < AsH3@C24H12-Mg < PH3@C24H12-Mg, respectively. For the benzene phase, upon adsorption of the studied gases, the lowest energy gap was found for NH3@C24H12-Mg with a value of 5.818 eV. In the benzene phase, both AsH3@C24H12-Mg and PH3@C24H12-Mg have the same energy gap of 5.834 eV. In the ethanol phase, the highest energy gap was observed in AsH3@C24H12-Mg, with NH3@C24H12-Mg and PH3@C24H12-Mg having the same energy gap value of 5.297 eV. In the water phase, the studied gases adsorbed on C24H12-Mg show a decreasing trend of 5.273 eV > 5.262 eV > 5.254 eV for AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg, respectively. It is only in the water phase that the sensing of NH3 on C24H12-Mg does not have the lowest energy gap value. The lowest energy gap values for the studied gases are observed in the water phase, while the highest are attributed to the gas phase. The different phases in which the adsorption process of AsH3, NH3 and PH3 on C24H12-Mg were analyzed show an increasing trend in terms of the energy gap of water < ethanol < benzene < gas with values within the range of 5.2–6.4 eV. Hence, it can be deduced that since AsH3@C24H12-Mg has the highest energy gap in all investigated phases, it has the most stable structure, since a higher energy gap corresponds to better stability of a structure. Depictions of the HOMO and LUMO plots are presented in Fig. 2. The HOMO and LUMO plots are similar in the different phases; hence, only the representation of the gas phase is presented in this paper. Furthermore, the HOMO and LUMO energy values can be used to obtain the quantum descriptors. These quantum descriptors include the ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical potential (μ), chemical hardness (η), chemical softness (σ) and electrophilicity (ω). According to Koopmans' hypothesis, these quantum descriptors can be obtained from eqn (3)–(9).
| |
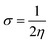 | (8) |
| |
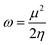 | (9) |
Table 2 Quantum chemical parameters for the studied gases adsorbed on the C24H12-Mg quantum dot calculated using the DFT/ωB97XD/6-311++G(2p,2d) method
| Phase |
System |
HOMO (eV) |
LUMO (eV) |
Energy gap |
IP |
EA |
χ |
μ |
η |
σ |
ω |
| Gas |
C24H12-Mg |
−6.531 |
−0.190 |
6.340 |
6.531 |
0.190 |
3.361 |
−3.361 |
3.170 |
0.158 |
1.781 |
| AsH3@C24H12-Mg |
−6.553 |
−0.240 |
6.313 |
6.553 |
0.240 |
3.397 |
−3.397 |
3.157 |
0.158 |
1.827 |
| NH3@C24H12-Mg |
−6.617 |
−0.315 |
6.302 |
6.617 |
0.315 |
3.466 |
−3.466 |
3.151 |
0.159 |
1.906 |
| PH3@C24H12-Mg |
−6.551 |
−0.235 |
6.317 |
6.551 |
0.235 |
3.393 |
−3.393 |
3.158 |
0.158 |
1.822 |
| Benzene |
C24H12-Mg |
−6.025 |
−0.174 |
5.850 |
6.025 |
0.174 |
3.099 |
−3.099 |
2.925 |
0.171 |
1.642 |
| AsH3@C24H12-Mg |
−6.033 |
−0.198 |
5.834 |
6.033 |
0.198 |
3.116 |
−3.116 |
2.917 |
0.171 |
1.664 |
| NH3@C24H12-Mg |
−6.064 |
−0.247 |
5.818 |
6.064 |
0.247 |
3.155 |
−3.155 |
2.909 |
0.172 |
1.711 |
| PH3@C24H12-Mg |
−6.034 |
−0.200 |
5.834 |
6.034 |
0.200 |
3.117 |
−3.117 |
2.917 |
0.171 |
1.666 |
| Ethanol |
C24H12-Mg |
−5.524 |
−0.232 |
5.292 |
5.524 |
0.232 |
2.878 |
−2.878 |
2.646 |
0.189 |
1.565 |
| AsH3@C24H12-Mg |
−5.544 |
−0.230 |
5.314 |
5.544 |
0.230 |
2.887 |
−2.887 |
2.657 |
0.188 |
1.568 |
| NH3@C24H12-Mg |
−5.546 |
−0.250 |
5.297 |
5.546 |
0.250 |
2.898 |
−2.898 |
2.648 |
0.189 |
1.586 |
| PH3@C24H12-Mg |
−5.546 |
−0.250 |
5.297 |
5.546 |
0.250 |
2.898 |
−2.898 |
2.648 |
0.189 |
1.586 |
| Water |
C24H12-Mg |
−5.546 |
−0.250 |
5.297 |
5.546 |
0.250 |
2.898 |
−2.898 |
2.648 |
0.189 |
1.586 |
| AsH3@C24H12-Mg |
−5.510 |
−0.237 |
5.273 |
5.510 |
0.237 |
2.873 |
−2.873 |
2.637 |
0.190 |
1.566 |
| NH3@C24H12-Mg |
−5.513 |
−0.252 |
5.262 |
5.513 |
0.252 |
2.883 |
−2.883 |
2.631 |
0.190 |
1.579 |
| PH3@C24H12-Mg |
−5.512 |
−0.259 |
5.254 |
5.512 |
0.259 |
2.885 |
−2.885 |
2.627 |
0.190 |
1.585 |
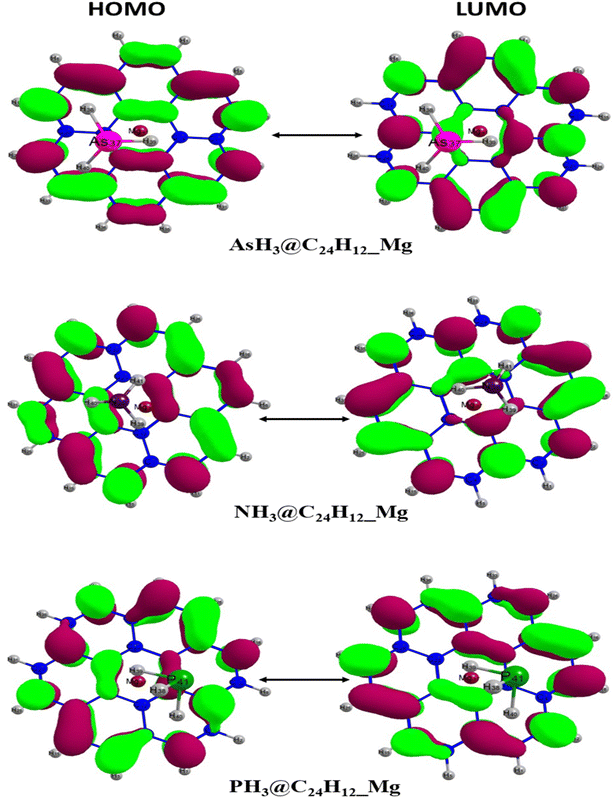 |
| | Fig. 2 Optimized HOMO and LUMO isosurface plots of AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg. | |
These quantum descriptors were studied to further authenticate the results explained by the energy gap. The ionization potential (IP) is the negative of the EHOMO while the electron affinity (EA) is the negative of the ELUMO.59 The ionization potential (IP) and the chemical hardness (η) help to further determine the stability of the surfaces.60 High energy values of the ionization potential (IP) and the chemical hardness (η) correspond to better stability of the surfaces being studied.61,62 In all scenarios of the adsorption process of AsH3, NH3 and PH3 on C24H12-Mg, high ionization potential (IP) and chemical hardness (η) values are observed, which further indicate the good stability properties of the surfaces.
3.3 Density of states
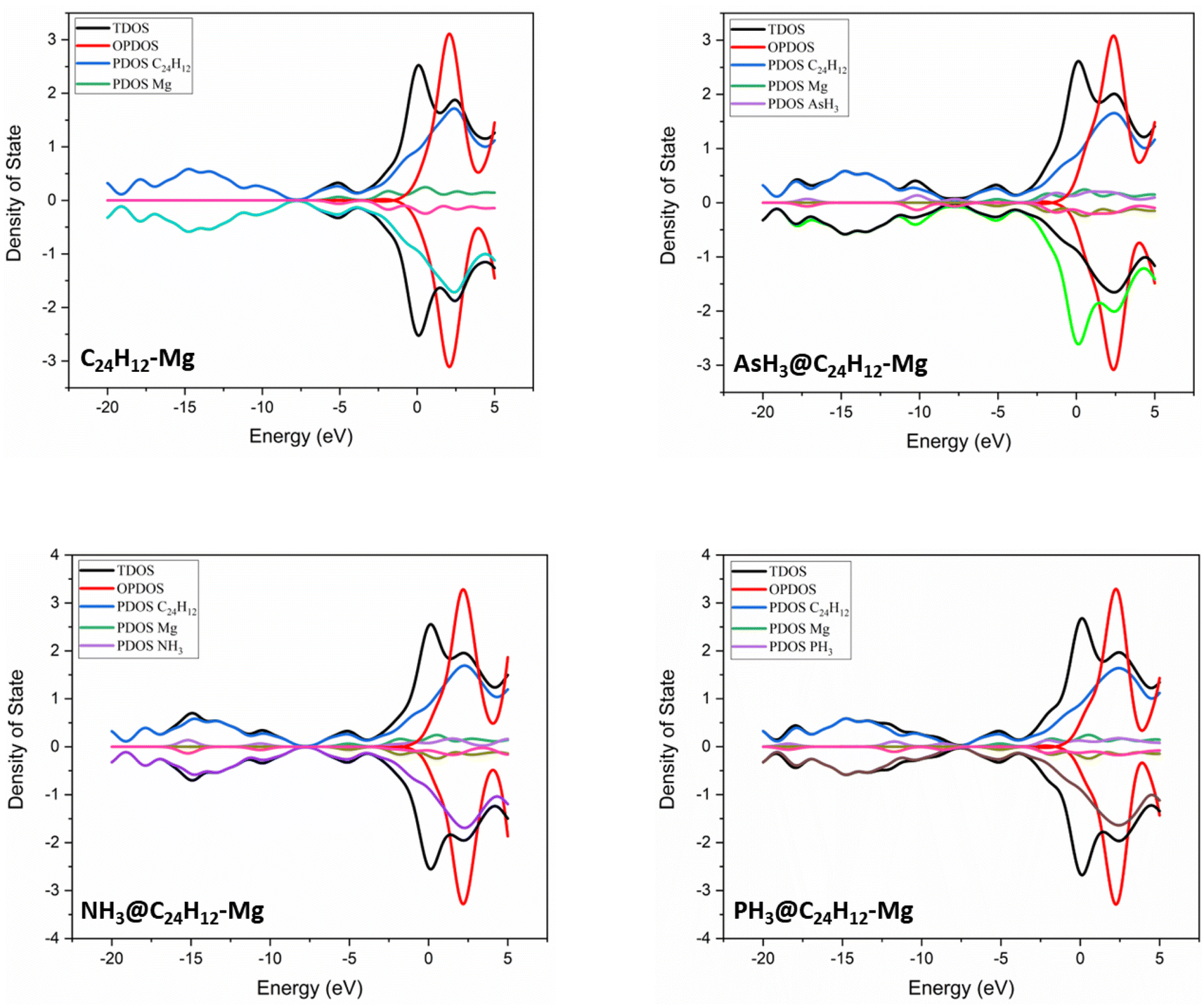
To gain further insight into the electronic properties and to visualize the change in the energy gap resulting from the XH3 (X = As, N, P) gas adsorption on the magnesium-decorated graphene surface, the corresponding density of states (DOS) plots are presented in Fig. 3.63 As shown in Fig. 3, the adsorption of XH3 (X = As, N, P) in the gas phase results in new energy states appearing close to the Fermi energy level for the XH3@C24H12-Mg (X = As, N, P) complexes. This led to the decrease in Eg from 6.331 eV (C24H12-Mg) to 6.313, 6.302, and 6.317 eV for the AsH3@C24H12-Mg, NH3@C24H12-Mg, and PH3@C24H12-Mg complexes, respectively (see Table 2). The reduction in Eg indicates that the C24H12-Mg surface can detect the three gases.64 However, comparing the total density of states (TDOS) plots in Fig. 3, the alteration of the TDOS plots is more pronounced in the NH3@C24H12-Mg plot when compared with that of the magnesium-decorated graphene quantum dot surface, indicating that the C24H12-Mg surface has sensitivity for NH3 gas.
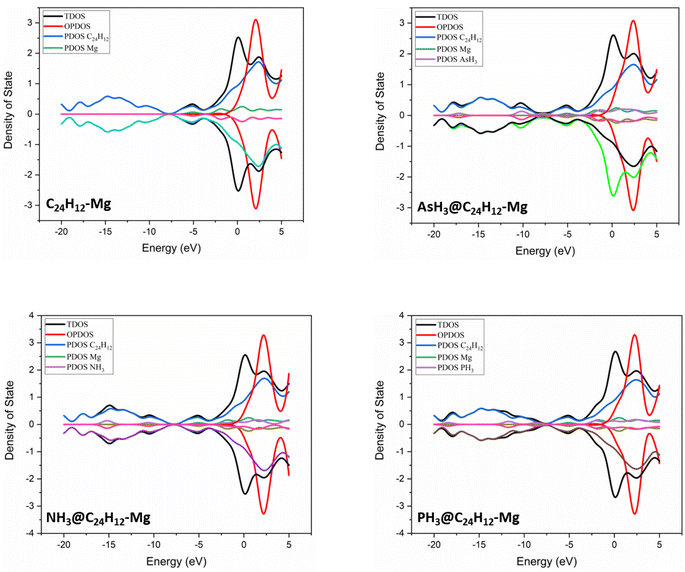 |
| | Fig. 3 Density of states plots for XH3 (X = As, N, P) on the magnesium-decorated graphene quantum dot surface. | |
3.4 Natural bond orbital (NBO) analysis
Natural bond orbital (NBO) analysis of the gas-adsorbed surface was undertaken in this study to chemically understand the distribution of the charges between the donor and acceptor orbitals.65 In addition to investigating the charge distribution between orbitals, natural bond orbital analysis is a tool that explains the bond type and nature of bonding interactions, as well as intermolecular and intramolecular interaction.66,67 The DFT method used earlier for evaluating the optimized structure was also employed in the NBO analysis. The nature of the interaction between the donor and the acceptor orbitals denoted by the stabilization energy is often referred to as the second-order perturbation energy, and it can be represented as eqn (10):| |
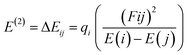 | (10) |
where qi represents the donor occupancy, F(i,j) is the off-diagonal NBO Fock Matrix elements, and Ei and Ej are the diagonal elements. Through literature reviews, it can be understood that when the values of the stabilization energy are large, it indicates the existence of a strong interaction between the donor and acceptor orbital.68 It also indicates a higher degree of conjugation of the whole system under study.69 In this paper, the estimated stabilization energy for the gas-adsorbed surfaces for the different phases have been computed and are tabulated in Table 3 alongside their respective donors (i), acceptors (j) and their diagonal elements with the off-diagonal NBO Fock Matrix elements. Close examination of Table 3 shows that the major transitions attributed to the system under study are BD (1) → BD* (1) which is the σ → σ* bond, BD (1) → BD* (2) [σ → π*], BD (2) → BD* (1) [π → σ*], BD (2) → BD* (2) [π → π*] and BD* (2) → BD* (1) [π* → σ*]. Other transitions observed in NBO analysis but not tabulated include the Rydberg (RY) and the center core pair (CR). The computed stabilization energies presented in Table 3 are in the range of 79–100 kcal mol−1. From the displayed stabilization energies, in the gas phase, it can be observed that the gas-adsorbed surfaces follow the trend AsH3@C24H12-Mg > NH3@C24H12-Mg > PH3@C24H12-Mg in terms of stabilization energy. Their values are 98.83 kcal mol−1, 96.60 kcal mol−1 and 93.36 kcal mol−1, respectively. In the benzene phase, the π → σ* transition is prominent. AsH3@C24H12-Mg is observed to have higher values of second-order perturbation energy compared to the other gas-adsorbed surfaces. The other two phases, ethanol and water, also support the results obtained in the other phases. The values of the stabilization energy are similar to each other because of the similarly in the structure of the surfaces being studied. From the NBO analysis, it can also be noted that the σ → σ* transition contributed more to the stabilization of the gas-adsorbed surfaces.
Table 3 Donors (i), acceptors (j), second-order perturbation energy and their diagonal elements with the off-diagonal NBO Fock matrix elements for the investigated surfaces calculated using the DFT/ωB97XD/6-311++G(2p,2d) method
| Phase |
System |
Donor (i) |
Acceptor (j) |
E(2) kcal mol−1 |
E(j) − E(i) a.u. |
F(i,j) a.u. |
| Gas |
AsH3@C24H12-Mg |
σ (C3–C4) |
π* (C10–C14) |
99.78 |
3.52 |
0.515 |
| σ (C17–C21) |
σ* (C12–C18) |
98.85 |
1.72 |
0.366 |
| NH3@C24H12-Mg |
σ (C3–C7) |
σ* (C9–C13) |
97.72 |
0.68 |
0.230 |
| σ (C11–C17) |
σ* (C24–H26) |
97.94 |
0.91 |
0.267 |
| PH3@C24H12-Mg |
π* (C32–C34) |
σ* (C22–C28) |
84.98 |
1.41 |
0.895 |
| π* (C9–C13) |
σ* (C24–C29) |
95.48 |
0.51 |
0.208 |
| Benzene |
AsH3@C24H12-Mg |
σ (C17–C21) |
σ* (C30–C34) |
85.46 |
0.03 |
0.046 |
| π (C19– C20) |
σ* (C4–C9) |
98.83 |
10.73 |
1.740 |
| NH3@C24H12-Mg |
σ (C19– C20) |
σ* (C7–C8) |
83.05 |
0.39 |
0.160 |
| π (C19– C20) |
σ* (C17–C21) |
96.60 |
2.56 |
0.497 |
| PH3@C24H12-Mg |
π (C8–C11) |
π* (C19–C20) |
82.92 |
2.50 |
0.382 |
| π (C8–C11) |
σ* (C29–C33) |
93.36 |
4.11 |
0.586 |
| Ethanol |
AsH3@C24H12-Mg |
σ (C3–C4) |
σ* (C30–C34) |
94.15 |
0.16 |
0.110 |
| σ (C21–C27) |
σ* (C30–C34) |
95.72 |
0.12 |
0.097 |
| NH3@C24H12-Mg |
σ (C19–C23) |
π* (C19–C20) |
92.62 |
1.69 |
0.392 |
| σ (C21–C27) |
π* (C19–C20) |
92.81 |
14.99 |
1.623 |
| PH3@C24H12-Mg |
π (C8–C11) |
π* (C19–C20) |
83.40 |
0.30 |
0.149 |
| σ (C7–C12) |
σ* (C19–C20) |
92.85 |
0.95 |
0.265 |
| Water |
AsH3@C24H12-Mg |
σ (H2–C4) |
σ* (C20–C24) |
93.11 |
0.19 |
0.120 |
| σ (C7–C12) |
σ* (C20–C24) |
94.61 |
0.36 |
0.166 |
| NH3@C24H12-Mg |
π* (C10–C14) |
σ* (C8–C11) |
79.96 |
0.36 |
0.448 |
| σ (C27–C28) |
σ* (H2–C4) |
93.98 |
0.40 |
0.174 |
| PH3@C24H12-Mg |
σ (H6–C10) |
σ* (C19–C23) |
89.36 |
0.75 |
0.232 |
| σ (C7–C12) |
σ* (C12–C18) |
93.75 |
0.93 |
0.265 |
3.5 Topological analysis – quantum theory of atoms-in-molecules (QTAIM)
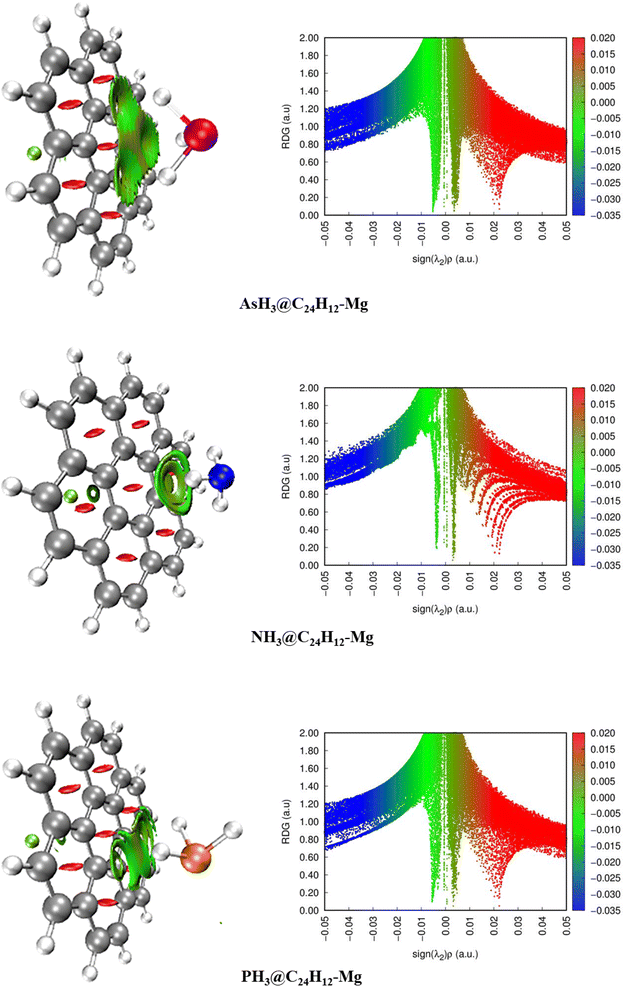
Using the optimized XH3@C24H12-Mg (X = As, N, P) structures, we carried out Bader's QTAIM analysis to investigate the bond type and inter- or intra-molecular nature of the interactions that exist between the interacting atoms of the XH3 gases and the magnesium-decorated graphene quantum dot surface.70 Based on QTAIM theory, the chemical bond existing between two interacting atoms is characterized by a topological (3, −1) point through-space known as the bond critical points (BCPs).71 Topological parameters at the BCPs, such as the density of electrons ρ(r), Laplacian electron density ∇2ρ(r), Lagrangian kinetic energy G(r), potential electron energy density V(r), total electron energy density H(r), Eigenvalues (λ1, λ2, λ3), and the ellipticity ε are vital parameters in determining the nature of interactions.72,73 The values of these parameters as calculated at the WB97XD/6-311++ (2d,2p) level of theory are presented in Table 4, and the corresponding QTAIM graph is shown in Fig. 4. The small values of electron density with ρ(r) < 0.1 a.u. signify non-covalent interaction between the XH3 (X = As, N, P) gases and magnesium-decorated graphene quantum dot surface.74 Also, positive values for the Laplacian electron density (∇2ρ(r) > 0) and the total electron energy density (H(r) > 0) imply non-covalent interactions.75 As shown in Table 4, the ρ(r) values range between 0.00058 to 0.00564 a.u., whereas the ∇2ρ(r) and H(r) values range between 0.00025 to 0.01953 a.u. The ρ(r) values are less than 0.1 a.u. and both the ∇2ρ(r) and H(r) values are positive for all interactions, which indicates the dominance of non-covalent forces between the interacting systems.76 Furthermore, calculating the values of the ratio −G(r)/V(r) gives further insight into the nature of the interactions.77 As shown in Table 4, the −G(r)/V(r) values of >1 for all studied interactions confirm the non-covalent nature of the interactions. From the discussion above, it is evident that the formation of the XH3@C24H12-Mg (X = As, N, P) complexes occurs essentially through non-covalent interactions.
Table 4 Study of weak interactions on the basis of bond critical point (BCP) and topological analysis parameters
| System |
Bond |
ρ(r) |
∇2(r) |
G(r) |
H(r) |
V(r) |
−G(r)/V(r) |
λ1 |
λ2 |
λ3 |
| AsH3@C24H12-Mg |
C12–Mg41 |
0.00061 |
0.00323 |
0.00055 |
0.00025 |
−0.00029 |
1.89655 |
−0.00023 |
−0.00003 |
0.00350 |
| |
C23–As37 |
0.00528 |
0.01586 |
0.00295 |
0.00100 |
−0.00195 |
1.51282 |
−0.00090 |
−0.00002 |
0.01678 |
| NH3@C24H12-Mg |
C13–Mg37 |
0.00062 |
0.00326 |
0.00055 |
0.00026 |
−0.00029 |
1.89655 |
−0.00031 |
−0.00012 |
0.00370 |
| |
C19–H41 |
0.00406 |
0.01382 |
0.00263 |
0.00081 |
−0.00018 |
1.46111 |
−0.00214 |
−0.00092 |
0.01689 |
| |
C13–N38 |
0.00388 |
0.01362 |
0.00257 |
0.00083 |
−0.00173 |
1.48554 |
−0.00202 |
−0.00021 |
0.01585 |
| |
C22–H40 |
0.00372 |
0.01253 |
0.00239 |
0.00073 |
−0.00166 |
1.43975 |
−0.00203 |
−0.00080 |
0.01537 |
| PH3@C24H12-Mg |
C22–Mg37 |
0.00059 |
0.00318 |
0.00053 |
0.00025 |
−0.00028 |
1.89285 |
−0.00018 |
−0.00007 |
0.00343 |
| |
C13–Mg37 |
0.00058 |
0.00317 |
0.00053 |
0.00025 |
−0.00028 |
1.89285 |
−0.00015 |
−0.00005 |
0.00338 |
| |
C13–P41 |
0.00564 |
0.01953 |
0.00371 |
0.00117 |
−0.00254 |
1.46063 |
−0.00143 |
−0.00072 |
0.02169 |
 |
| | Fig. 4 Pictorial representation of the quantum theory of atoms-in-molecules (QTAIM) analysis. | |
3.6 Noncovalent interactions (NCI)
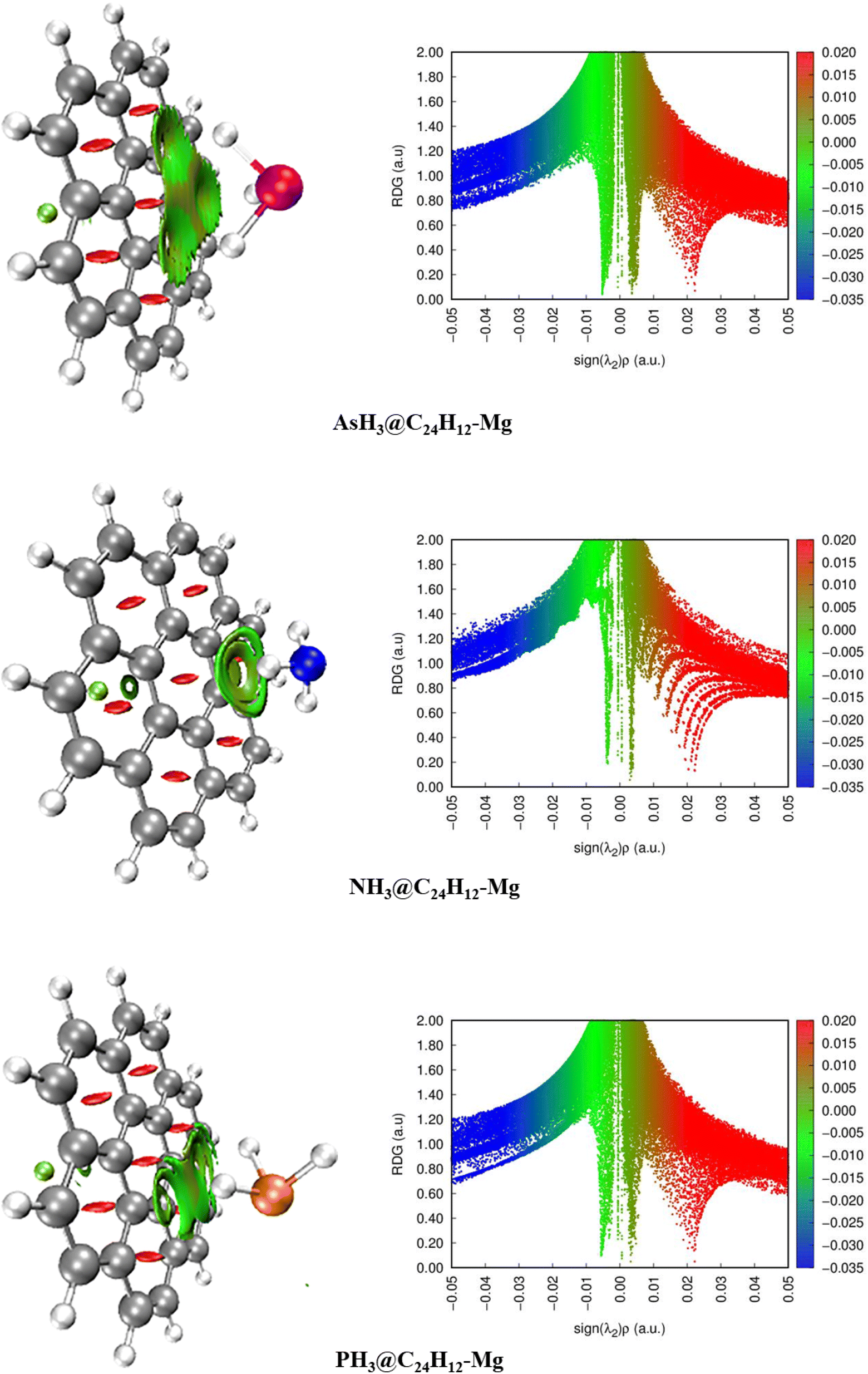
The QTAIM analysis shows that non-covalent forces play the major role in the interactions between the XH3 (X = As, N, P) gases and the magnesium-decorated graphene quantum dot surface. Therefore, it is important to further study the weak interactions using non-covalent interaction (NCI) analysis.78 The NCI analysis is based on studying the plots of the reduced density gradient (RDG) against the product of the second Eigenvalue of the Hessian matrix (λ2) with the electron density ρ(sign(λ2)ρ).79 Visualization of the weak interactions is carried out through 2D-RDG scatter maps and 3D iso-surface plots, which are presented in Fig. 5. The 2D-RDG scatter maps are interpreted using the peaks observed in the low-RDG and low-electron-density regions. The regions in which sign(λ2)ρ < 0 indicates strong bonding, and sign(λ2)ρ > 0 indicates strong repulsive forces, whereas the point where sign(λ2)ρ ≈ 0 indicates van der Waals (vdW) forces.80 Similarly, NCI plots are interpreted using the blue-green-red color scale. Thus, the blue, green and red regions correspond to strong attraction, van der Waals (vdW), and steric repulsion forces, respectively.81 From the 3D iso-surface plots in Fig. 5, green patches between the XH3 (X = As, N, P) gases and the surface show that van der Waals (vdW) forces are the driving force for the adsorption of these gases onto the magnesium-decorated graphene quantum dot surface. Meanwhile, the red patches within the graphene surface result from repulsive steric effect from the rings forming the quantum dot structure. This was confirmed by the green and red spikes observable in the 2D-RDG scatter maps for all the studied XH3@C24H12-Mg (X = As, N, P) complexes.
 |
| | Fig. 5 Pictorial representation of the non-covalent interaction analysis. | |
3.7 Adsorption study
In order to ensure a stable adsorption configuration, the structures were optimized separately before adsorption and optimized again upon adsorption. The cluster, initial structural equilibration has been carried out by employing the def2svp basis set. All calculations were carried out using the DFT/ωB97XD/6-311++G (2d,2p) method. To gain more insight into the effect of solvation on the adsorptive behavior of C24H12-Mg, calculations were carried out in four distinct phases: benzene, ethanol, water and gas. The adsorption behavior of the Mg-decorated graphene quantum dot (C24H12-Mg) surface in the adsorption of AsH3, NH3 and PH3 was carried out using the calculation of adsorption energies of the respective complexes formed. Eqn (1) was employed in obtaining the different adsorption energies, and the results obtained are summarized in Table 5. Previous theoretical studies on the adsorption of gases agree that greater adsorption energy is favorable in comparative studies among different systems. That is, the greater the adsorption energy, the more likely that an adsorbent material best adsorbs the adsorbate under study.82 From the data presented in Table 5, it is apparent that all the calculated adsorption energies are of a negative magnitude. Thus, this adsorption phenomenon is best described as chemisorption.83 The adsorption energies can be arranged in an increasing trend of NH3@C24H12-Mg < PH3@C24H12-Mg < AsH3@C24H12-Mg. This pattern was observed to apply for all phases under consideration. Among the considered phases, the best adsorption performance was noted in the gas phase due to its highest values of adsorption energy as compared to its studied counterparts in benzene, ethanol and water. The adsorption of AsH3 on the magnesium-decorated graphene quantum dot (C24H12-Mg) corresponding to AsH3@C24H12-Mg complex reflects the highest Ead value of −1.87819, indicating that AsH3 is better adsorbed on the C24H12-Mg surface than NH3 or PH3. In addition, NH3 and PH3 were adsorbed similarly on the Mg-decorated graphene quantum dot (C24H12-Mg), which is accounted for by their similar adsorption energies. The adsorption energy values obtained here were in agreement with previously reported literature.84,85 However, PH3@C24H12-Mg exhibits a relatively higher energy, indicating that the adsorption of PH3 is more favored than that of NH3. Hence, the studied Mg-decorated graphene quantum dot (C24H12-Mg) surface exhibits stronger adsorption potency for the AsH3 gas molecule than its studied counterparts. The idea of deformation energy is used to provide a thorough explanation of a substrate's capacity to accommodate an adsorbate. According to a literature review, when a molecule adheres to a surface, it may result in energy-intensive surface deformation. The adsorption energy, which is the energy released or absorbed when a molecule is adsorbed onto a surface, can be impacted by this deformation energy. The total stability of the adsorbed state can be impacted by the magnitude of the deformation energy. For instance, the adsorption energy will be reduced and the adsorption may be less stable if the deformation energy is high. The adsorption may be more stable if the deformation energy is low, and vice versa.85 Although energy can impact the effectiveness of materials used in a variety of applications, including catalysis, gas separation, and energy storage, it is essential for forecasting and optimizing adsorption processes. From the results presented in Table 5, it can be seen that the studied systems have higher deformation energy in the gas phase compared to in the other solvents, and these higher values can be explained based on solvent effects.
Table 5 Adsorption energies of the three studied systems in four distinct solvents: benzene, ethanol, water and gas, as calculated using the DFT/ωB97XD/6-311++G (2d, 2p) method
| System |
Benzene |
Ethanol |
Water |
Gas |
| AsH3@C24H12-Mg |
−1.87601 |
−1.87192 |
−1.8737 |
−1.87819 |
| NH3@C24H12-Mg |
−0.00641 |
−0.0056 |
−0.0056 |
−0.0067 |
| PH3@C24H12-Mg |
−0.00755 |
−0.00746 |
−0.0075 |
−0.0076 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Deformation energy |
| AsH3@C24H12-Mg |
−2.312 |
−2.317 |
−2.315 |
−2.321 |
| NH3@C24H12-Mg |
−2.123 |
−2.154 |
−2.165 |
−2.323 |
| PH3@C24H12-Mg |
−2.212 |
−2.214 |
−2.132 |
−2.232 |
3.8 Sensor mechanisms
The properties capable of describing the sensing performance of a material are generally regarded as sensor mechanisms. These mechanisms provide necessary insights from the interactions existing between the adsorbate and adsorbent materials through measuring the electrical conductivity, recovery time, charge transfer, dipole moment and work function. These mechanisms have been theoretically explored in this section to strengthen the theoretical investigation of the adsorption of the three different gases on the Mg-decorated graphene quantum dot (C24H12-Mg).
3.8.1. Electrical conductivity. The ability of a molecule to conduct electricity can be attributed and traced to the flow of electrons between the HOMO and LUMO energies.86 It is paramount to note that the energy gap is an essential parameter with the potential of predicting the electrical conductivity of a material. Therefore, it is safe to say that the reusability and stability of a material depend on the HOMO–LUMO energy gap. Previous theoretical studies have shown that electrical conductivity can be related to the energy gap in such a way that an increase in the energy gap induces a decrease in the conductivity of a material.87 The inverse relationship in the conductivity can be demonstrated in eqn (11) as:where the electrical conductivity, Boltzmann constant, temperature and constant are denoted by σ, K, T and A, respectively. From the results obtained in Section 3.2 for the quantum descriptors, it is evident that NH3@C24H12-Mg with the smallest energy gap possesses the lowest electrical conductivity. Furthermore, the relatively higher energy gap in PH3@C24H12-Mg indicates the conducting nature of the material corresponding to the C24H12-Mg surface upon the adsorption of PH3. Among the three gases, the highest conductivity was observed for the adsorption of AsH3, and this is due to its high energy gap.
3.8.2. Recovery time and work function. One of the key parameters in investigating materials suitable for gas sensing is their reusability and stability.88 The strength of an adsorption can be predicted by the interaction strength obtained from the adsorption energy, which is the chief parameter among other mechanisms.89 The recovery time can be predicted from the adsorption energy using eqn (12).| |
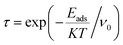 | (12) |
T, ν0, K and Ead are the temperature, attempted frequency, Boltzmann constant and adsorption energy, respectively. In the calculation, the Boltzmann constant can be approximated as ∼2.0 × 10−3 kcal mol−1, and the attempted frequency was obtained as 1.380649 × 10−23 m2 kg s−2 K−1.To obtain the recovery time experimentally, the electric field and thermal effects are generally considered and utilized. Furthermore, the recovery time can be theoretically calculated from the transition state theory. In eqn (12), it can be noted that higher adsorption energy will increase the recovery time of a material, and thus, this concept is employed as follows. The adsorption follows an increasing trend of NH3@C24H12-Mg < PH3@C24H12-Mg < AsH3@C24H12-Mg. From this trend, the order of increasing recovery time can likewise be illustrated. For example, the adsorption of AsH3 on the surface reflects the greatest Ead value of −1.87819. This implies that the AsH3@C24H12-Mg system has the relatively longest recovery time.
The work function (φ), which is the minimum energy required to move the electron from the internal part of a solid to the surface, is an essential parameter in monitoring the electronic and optical characteristics of a material.90 It is evident in Table 6 that the complexes resulting upon adsorption exhibited relatively more similar work function values of 3.3417, 3.4218 and 3.3284 eV for AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg, respectively. It has been observed that the material indicated excellent adsorption potency in use for the adsorption of the gases.
Table 6 Dipole moment (μD) in four distinct phases, and charge transfer (Q) and work function (φ) in gas calculated using the DFT/ωB97XD/6-311++G(2d,2p) methoda
| System |
Dipole moment |
Charge transfer |
φ |
| Benzene |
Ethanol |
Gas |
Water |
QC |
Qads |
Qt |
| QC and Qads are the respective charges on carbon and adsorbates. |
| C24H12-Mg |
0.4283 |
0.9537 |
1.0203 |
0.5935 |
— |
— |
— |
3.3605 |
| AsH3@C24H12-Mg |
0.2106 |
1.0191 |
0.4231 |
1.1459 |
−0.0785 |
−1.1180 |
1.03945 |
3.3417 |
| NH3@C24H12-Mg |
1.6430 |
2.4986 |
0.9091 |
2.6565 |
−0.1874 |
0.0162 |
−0.236 |
3.4218 |
| PH3@C24H12-Mg |
0.6902 |
1.0753 |
0.9549 |
1.1755 |
0.0760 |
0.3343 |
−0.2584 |
3.3284 |
3.8.3. Dipole moment and transfer. As one of the essential mechanisms of sensing, the dipole moment (μD) is used to gain insight into the charge separation of materials.91 Necessary information on the dipole moment is important in tracking the interaction strength of studied systems. It is often seen that the quantity of charge transferred has a direct way of affecting the dipole moment.92 The variability in the studied surface before and after the adsorption of AsH3, NH3 and PH3 molecules is summarized in Table 6. Also, the effect of solvation on the adsorption of gases has been investigated from the perspective of the dipole moment. The dipole moment was calculated in benzene, ethanol, water and gas in order to investigate the adsorption behavior in the aforementioned solvents. Upon adsorption, higher dipole moments are observed in ethanol and water, with the dipole moments of 1.019102, 2.498643 and 1.075318 D in ethanol and 1.145908, 2.656500 and 1.175505 D in water for AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg, respectively. In addition, relatively higher dipole moments are obtained in gas as compared to benzene. The greater dipole moments observed in these solvents are an indication of greater charge separation, which further implies strong interaction. The interactions in the four distinct solvents increase in the order benzene < gases < ethanol < water. A thorough literature search indicates that higher dipole moment is an indication of strong interactions.93 Herein, NH3@C24H12-Mg exhibits the highest dipole moment, followed by PH3@C24H12-Mg. This indicates that NH3@C24H12-Mg exhibits the strongest interaction in benzene and ethanol. However, a slight change in the behavior of adsorption was observed in gas, where the strongest interaction was noted for PH3@C24H12-Mg as a result of its highest μD of 0.954942 D.Milliken charge analysis has been employed in tracing the charge transfer of the respective atoms present in the decorated C24H12 surface. The electronic charge transfer occurring between the two interacting compounds, such as the adsorbate and adsorbent, was investigated via the natural charge on the C24H12-Mg surface before and upon interaction, and the results are summarized in Table 6. Changes in charge transfer (Qt) of 1.03945, −0.236 and −0.2584 are attributed to AsH3@C24H12-Mg, NH3@C24H12-Mg and PH3@C24H12-Mg, respectively. From this, the strongest charge transfer was encountered in the adsorption of AsH3 on the C24H12-Mg surface. This is evident in the AsH3@C24H12-Mg system with the highest Qt value. Furthermore, relatively strong charge transfer is noted in the PH3@C24H12-Mg system, indicating strong charge transfer upon the adsorption of PH3 on the Mg-decorated graphene quantum dot (C24H12-Mg).
3.9 Nonlinear optics (NLO) analysis
Nonlinear optics (NLO) studies the deflection of high-intensity lasers via interaction with a material.94 A material is polarized when charged particles are dislodged within the material and a dipole moment is set up; on the other hand, static polarizabilities are utilized to investigate intramolecular and intermolecular interactions in a molecule.95 It accurately predicts excited states in density functionals and represents the initial response of the electron density to electric fields. The primary goal of this objective is to conduct theoretical research on the complexes' optical properties. Strongly hyper-polarizable molecules, which have a wide range of technological applications, can be used to create NLO materials. Nonlinear optical materials have grown in prominence recently because of their prospective applications in fields such as laser technology, optical communication, optical data storage, and optical signal processing.96 There are numerous optoelectronic and photonic applications for them as well. Organic chromophores may be a good substitute for inorganic materials because of their potent and rapid nonlinearities. This occurs as a result of their propensity to contribute to electron delocalization. Asymmetric polarization caused by electron donor and acceptor groups on each side of the molecule at the proper places in molecular systems is used to create effective organic materials for non-linear optical effects.97 The Gaussian 16 program was used to perform the calculations. The polarizabilities were calculated during the geometry optimization, and the NLO descriptors were generated using the coordinate values taken from the log file. Calculations of the frequency-dependent electronic hyper-polarizability and static polarizability, denoted by the symbols βxyz and αxyz, were made using the coupled-perturbed Hartree–Fock method. Calculation of the polarizabilities of molecules can be done using the basis set. The Multiwfn tool was used to calculate the NLO descriptors from the output of the Gaussian log file. The dipole moments (μ), polarizability anisotropies (α), isotropically averaged polarizabilities (Δαtotal), and isotopic first-order hyperpolarizabilities (βtotal), which were calculated using eqn (13)–(19) below, were among the parameters obtained.| |
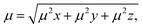 | (13) |
| |
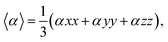 | (14) |
| |
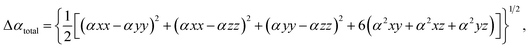 | (15) |
| |
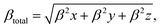 | (16) |
where,| | |
βx = βxxx + βxyy + βxzz
| (17) |
| | |
βy = βyyy + βxxy + βyzz
| (18) |
| | |
βz = βzzz + βxzz + βyyz
| (19) |
The results predicted the dipole moment (μ), polarizability anisotropy 〈α〉, average polarizability, isotopically averaged polarizability (Δαtotal) and first-order hyperpolarizabilities (βtotal) of the complexes. As presented in Table 7, the non-linear optic properties of C24H12-Mg, NH3@C24H12-Mg, PH3@C24H12-Mg and AsH3@C24H12-Mg showed an increase in the dipole in the order C24H12-Mg: 1.0202μ > PH3@C24H12-Mg: 0.9547μ > NH3@C24H12-Mg: 0.9092μ > AsH3@C24H12-Mg: 0.4231μ. The polarizability anisotropy increased in the order NH3@C24H12-Mg: 69.528 > C24H12-Mg: 47.858 > PH3@C24H12-Mg: 46.919 > AsH3@C24H12-Mg: 44.105 a.u., and the averaged polarizability increased in the order C24H12-Mg: −139.912 > NH3@C24H12-Mg: −150.275 > PH3@C24H12-Mg: −155.865 > AsH3@C24H12-Mg: −161.856 a.u., while the first-order hyperpolarizability increased in the order AsH3@C24H12-Mg: 237.450 > PH3@C24H12-Mg: 75.399 > C24H12-Mg: 67.989 > NH3@C24H12-Mg: 20.147 a.u. The high first-order hyperpolarizability of AsH3@C24H12-Mg makes it a suitable material for optoelectronic applications.
Table 7 NLO descriptors of magnesium-decorated graphene quantum dot structure and interactions with the adsorbed gases calculated at DFT/ωB97XD/6-311++G(2p,2d) method
| Structure |
Dipole moment (μ) |
Polarizability anisotropies (αtotal) a.u. |
Averaged polarizabilities 〈α〉 a.u. |
First-order hyperpolarizabilities (βtotal) a.u. |
| C24H12-Mg |
1.0202 |
47.85854359 |
−139.91287 |
67.98918423 |
| NH3@C24H12-Mg |
0.9092 |
69.52853744 |
−150.27543 |
20.14768183 |
| PH3@C24H12-Mg |
0.9549 |
46.91934464 |
−155.86527 |
75.39993366 |
| AsH3@C24H12-Mg |
0.4231 |
44.1052877 |
−161.8569 |
237.4505579 |
3.10 Thermodynamics properties
To fully understand the dynamics of heat and energy transfer present in the magnesium-decorated graphene quantum dot, as well as the spontaneity of the reaction involving graphene in sensing toxic gases like AsH3, NH3, and PH3 in the surroundings, the thermodynamics properties were studied in the gas phase and in the solvents benzene, ethanol and water to enable us to ascertain the effects of the above-mentioned solvents on the thermodynamics of the sensor material.98 The enthalpy (H) which is the sum total of a system's internal energy (Q) in addition to its constant pressure and total change in volume99 ΔH = Q + pΔV was calculated for all the optimized studied complexes in both gas phase and solution using the equations below:| | |
ΔH* (298 K) = ∑ΔfH*product (298 K) − ∑ΔfH* reactants (298 K)
| (20) |
| | |
ΔfH* (298 K) = ∑(ε0 + Hcorr) product − ∑(ε0 + Hcorr) reactants
| (21) |
where ε0 is the electronic energy and Hcorr is sum of the electronic energy and thermal correction to H. To estimate the enthalpy of a system, the difference between the products and corresponding reactants should be obtained in order to ascertain whether the reaction is exothermic (ΔH > 0) or endothermic (ΔH < 0).100 All the thermodynamic parameters are shown in Table 8. Also, detailed analysis of the thermodynamic calculations in different solvents has been presented in the ESI (Tables (1–3)†). As can be observed from the tabulated results, in the gas phase, which is the point of comparison, the reactions between the Mg-decorated graphene quantum dot were endothermic when interacting with NH3 gas and exothermic when interacting with PH3 and AsH3. In the solvation-optimized structures in benzene, ethanol and water, all the interactions were noted to be exothermic; this is justified by the all-negative values of enthalpy of the systems that took part in the reaction. Similarly, the Gibbs free energy (ΔG) of a system undergoing a chemical reaction is described as the sum of the enthalpy and the product obtained from the summation of the system's temperature (K) and the entropy change (ΔS).101 Thermodynamically, the Gibbs free energy of a system must be negative for a spontaneous reaction to occur, that is ΔG < 0, while positive values of ΔG indicate non-spontaneity, that is, ΔG > 0. The Gibbs free energy was calculated in this study using the equation below:102| | |
ΔfG* (298 K) = ∑(ε0 + Gcorr) product − ∑(ε0 + Gcorr) reactants
| (22) |
where ε0 is the electronic energy and Gcorr is the sum of electronic energy and the thermal correction to G. The results obtained in these comparative thermodynamic studies show that in the gaseous phase, all interactions of the Mg-decorated graphene quantum dot (C24H12-Mg) with the studied gases, namely, AsH3, NH3 and PH3, were spontaneous in the order NH3@C24H12-Mg > AsH3@C24H12-Mg > PH3@C24H12-Mg. In the benzene phase, AsH3@C24H12-Mg and PH3@C24H12-Mg were spontaneous, while NH3@C24H12-Mg was non-spontaneous. In ethanol, AsH3@C24H12-Mg and NH3@C24H12-Mg were spontaneous in reaction, while PH3@C24H12-Mg showed non-spontaneity. Finally, in water, the reaction between AsH3@C24H12-Mg and PH3@C24H12-Mg was spontaneous, whereas NH3@C24H12-Mg was non-spontaneous. The results in all the studied phases suggest that the free energy of reaction in the studied systems for the formation of the product is most favorable for AsH3@C24H12-Mg compared to other systems, and so the reaction of the Mg-decorated graphene quantum dot to sense AsH3 gas will occur in a much easier and suitable state. These results are in agreement with the adsorption energy analysis as well as the HOMO–LUMO energy gap analysis as obtained in these studies.
Table 8 Thermodynamic parameters of the optimized structures, including the enthalpy and Gibbs free energy in the gas phase, calculated using the DFT/ωB97XD/6-311++G(2p,2d) method
| Parameter |
C24H12-Mg |
AsH3 |
AsH3@C24H12-Mg |
NH3 |
NH3@C24H12-Mg |
PH3 |
PH3@C24H12-Mg |
| ε0 |
−1121.867 |
−2237.627 |
−3359.566 |
−56.564 |
−1178.438 |
−343.150 |
−1465.025 |
| εZPE |
0.283 |
0.021 |
0.306 |
0.035 |
0.318 |
0.024 |
0.308 |
| Etot |
0.299 |
0.024 |
0.327 |
0.037 |
0.337 |
0.027 |
0.329 |
| Hcorr |
0.300 |
0.025 |
0.328 |
0.038 |
0.338 |
0.028 |
0.330 |
| Gcorr |
0.235 |
0.000 |
0.254 |
0.016 |
0.268 |
0.003 |
0.255 |
| ε0 + εZPE |
−1121.585 |
−2237.606 |
−3359.259 |
−56.530 |
−1178.120 |
−343.126 |
−1465.717 |
| ε0 + Etot |
−1121.568 |
−2237.603 |
−3359.239 |
−56.527 |
−1178.101 |
−343.123 |
−1465.696 |
| ε0 + Hcorr |
−1121.567 |
−2237.602 |
−3359.238 |
−56.526 |
−1178.100 |
−343.122 |
−1465.695 |
| ε0 + Gcorr |
−1121.632 |
−2237.627 |
−3359.312 |
−56.547 |
−1178.170 |
−343.147 |
−1465.770 |
| ΔfH* (298 K) |
|
|
−0.038 |
|
0.007 |
|
−1.081 |
| ΔfG* (298 K) |
|
|
−0.053 |
|
−0.009 |
|
−0.991 |
4 Conclusions
Theoretical calculations were performed using the DFT/WB97XD/6-311++G(2p,2d) approach to investigate the selectivity, sensitivity, conductivity, and adsorption process of the graphene quantum dot and its decorated magnesium atom in sensing the hazardous hydride gases XH3, where X = As, N, and P. This work was carried out in four different phases to comparatively study the effect of solvation and to deduce the best possible phase for the adsorption of the gases. From the structural properties, PH3@C24eH12-Mg was observed to have the shortest bond length and AsH3@C24H12-Mg to have the longest bond length in all phases. The bond lengths increased in the order PH3@C24H12-Mg < NH3@C24H12-Mg < AsH3@C24H12-Mg. Comparing the four phases, the shortest bond length was found in the gas phase, while the longest was observed in the water phase. Upon adsorption of the gases on the magnesium-decorated graphene quantum dot, there was a slight reduction in the energy gaps, which enhances the conductivity and suggests the stability of the investigated surfaces. AsH3@C24H12-Mg had the highest energy gap in all phases. The gas phase was observed to be the phase with the highest energy gap, followed by the benzene phase, the ethanol phase and finally the water phase. From the NBO analysis, the σ → σ* transition contributed more to the stabilization of the gas-adsorbed surfaces. The interaction between the gas-adsorbed surfaces is a non-covalent interaction, as was confirmed through the QTAIM and NCI analyses. The nature of the adsorption is chemisorption, as indicated by the negative values of the adsorption energy. AsH3@C24H12-Mg exhibited the highest adsorption energy, thus indicating that AsH3@C24H12-Mg is best sensed on the magnesium-decorated graphene quantum dot. Based on our findings, the quantum dot under investigation has good sensing properties towards gases, and it has a higher sensing potency for the AsH3 gas molecule than its examined counterparts. In comparison to the other phases, the gas phase is preferable.
Data availability
All data are contained within the manuscript and ESI.†
Author contributions
Ernest C. Agwamba and Hitler Louis: project conceptualization, design, and supervision. Praise O. Olagoke and Terkumbur E. Gber: writing, results extraction, analysis, and manuscript first draft. Gideon A. Okon: manuscript revision, review, and proofreading. Chidera F. Fedelis: manuscript proofreading. Adedapo S. Adeyinka: resources, review, and editing.
Conflicts of interest
All authors declare zero financial or inter-personal conflicts of interest that could have influenced the research work or results reported in this research paper.
Acknowledgements
This research was not funded by any Governmental or Non-governmental agency. The authors would like to acknowledge the centre for high performance computing (CHPC), at the University of Johannesburg, South Africa for providing computational resources for this research project.
References
- S. Steinhauer, Gas sensors based on copper oxide nanomaterials: A review, Chemosensors, 2021, 9(3), 51 CrossRef CAS.
- D. Lun and K. Xu, Recent Progress in Gas Sensor Based on Nanomaterials, Micromachines, 2022, 13(6), 919 CrossRef PubMed.
- F. Yin, W. Yue, Y. Li, S. Gao, C. Zhang, H. Kan and Y. Guo, Carbon-based nanomaterials for the detection of volatile organic compounds: A review, Carbon, 2021, 180, 274–297 CrossRef CAS.
- N. Jamil, F. Jameel, S. Z. Bajwa, A. Rehman, R. F. H. Khan, A. Mahmood and W. S. Khan, Potential carbon nanotube–metal oxide hybrid nanostructures for gas-sensing applications, in Metal Oxide-Carbon Hybrid Materials, Elsevier, 2022, pp. 459–474 Search PubMed.
- R. Ahmad, S. M. Majhi, X. Zhang, T. M. Swager and K. N. Salama, Recent progress and perspectives of gas sensors based on vertically oriented ZnO nanomaterials, Adv. Colloid Interface Sci., 2019, 270, 1–27 CrossRef CAS PubMed.
- N. M. Noah, Design and synthesis of nanostructured materials for sensor applications, J. Nanomater., 2020, 1–20 Search PubMed.
- C. Hou, G. Tai, Y. Liu and X. Liu, Borophene gas sensor, Nano Res., 2022, 15(3), 2537–2544 CrossRef CAS.
- Y. Ghahramani, F. Fallahinezhad and M. Afsa, Graphene Quantum Dots and their applications: A mini-review, Adv. Appl. NanoBio-Technol., 2021, 53–59 Search PubMed.
- R. K. Singh, R. Kumar, D. P. Singh, R. Savu and S. A. Moshkalev, Progress in microwave-assisted synthesis of quantum dots (graphene/carbon/semiconducting) for bioapplications: a review, Mater. Today Chem., 2019, 12, 282–314 CrossRef CAS.
- M. T. Hasan, R. Gonzalez-Rodriguez, C. Ryan, K. Pota, K. Green, J. L. Coffer and A. V. Naumov, Nitrogen-doped graphene quantum dots: Optical properties modification and photovoltaic applications, Nano Res., 2019, 12(5), 1041–1047 CrossRef CAS.
- T. K. Henna and K. Pramod, Graphene quantum dots redefine nanobiomedicine, Mater. Sci. Eng., C, 2020, 110, 110651 CrossRef CAS PubMed.
- A. Ghaffarkhah, E. Hosseini, M. Kamkar, A. A. Sehat, S. Dordanihaghighi, A. Allahbakhsh and M. Arjmand, Synthesis, applications, and prospects of graphene quantum dots: a comprehensive review, Small, 2022, 18(2), 2102683 CrossRef CAS PubMed.
- P. Kumar, C. Dhand, N. Dwivedi, S. Singh, R. Khan, S. Verma and A. K. Srivastava, Graphene quantum dots: A contemporary perspective on scope, opportunities, and sustainability, Renewable Sustainable Energy Rev., 2022, 157, 111993 CrossRef CAS.
- M. Li, T. Chen, J. J. Gooding and J. Liu, Review of carbon and graphene quantum dots for sensing, ACS Sens., 2019, 4(7), 1732–1748 CrossRef CAS PubMed.
- Y. R. Kumar, K. Deshmukh, K. K. Sadasivuni and S. K. Pasha, Graphene quantum dot based materials for sensing, bio-imaging and energy storage applications: a review, RSC Adv., 2020, 10(40), 23861–23898 RSC.
- Q. He, B. Yu, Z. Li and Y. Zhao, Density functional theory for battery materials, Energy Environ. Mater., 2019, 2(4), 264–279 CrossRef CAS.
- P. Verma and D. G. Truhlar, Status and challenges of density functional theory, Trends Chem., 2020, 2(4), 302–318 CrossRef CAS.
- Q. Wang, Y. Zhou, X. Zhao, K. Chen, G. Bingni, T. Yang and J. Chen, Tailoring carbon nanomaterials via a molecular scissor, Nano Today, 2021, 36, 101033 CrossRef CAS.
- T. E. Gber, H. Louis, A. E. Owen, B. E. Etinwa, I. Benjamin, F. C. Asogwa and E. A. Eno, Heteroatoms (Si, B, N, and P) doped 2D monolayer MoS 2 for NH 3 gas detection, RSC Adv., 2022, 12(40), 25992–26010 RSC.
- Z. Li, L. Jia, J. Chen, X. Cui, W. Zeng and Q. Zhou, Ag-modified hexagonal GaN monolayer as an innovative gas detector toward SF6 decomposed species: Insights from the first-principles computations, Appl. Surf. Sci., 2022, 589, 153000 CrossRef CAS.
- A. Kumar, N. Sharma, A. P. Gutal, D. Kumar, P. Kumar, M. Paranjothy and M. Kumar, Growth and NO2 gas sensing mechanisms of vertically aligned 2D SnS2 flakes by CVD: Experimental and DFT studies, Sens. Actuators, B, 2022, 353, 131078 CrossRef CAS.
- A. Nematollahzadeh, H. Basharnavaz, A. Habibi-Yangjeh, and S. H. Kamali, Enhancing AsH3 Gas Adsorption, 2021 Search PubMed.
- A. Nematollahzadeh, H. Basharnavaz, A. Habibi-Yangjeh and S. H. Kamali, Enhancing AsH3 Gas Adsorption Potentials of Graphitic Carbon Nitride by Codoping Cr/P, Mo/P, and W/P Atoms: A DFT Investigation, J. Nanostructures, 2021, 11(4), 638–646 CAS.
- A. Vouzavalis, Fabrication And Characterization Of Electrochemical Ammonia Sensors, Doctoral dissertation, University of Thessaly, 2018 Search PubMed.
- S. V. T. Coalition, Toward a just and sustainable solar energy industry, San Jose, 400, 2009 Search PubMed.
- A. T. M. Mustafa Kamal, Occurrence and Characterization of Atmospheric Trace Gases in Dhaka City, Bangladesh, Doctoral dissertation, University of Dhaka, 2018 Search PubMed.
- D. Penalva Milagro, Chemical sensing of dangerous gases based on a 4G drone platform, Bachelor's thesis, Universitat Politècnica de Catalunya, 2022 Search PubMed.
- V. A. Ranea, P. L. D. Quiña and N. M. Yalet, General adsorption model for H2S, H2Se, H2Te, NH3, PH3, AsH3 and SbH3 on the V2O5 (0 0 1) surface including the van der Waals interaction, Chem. Phys. Lett., 2019, 720, 58–63 CrossRef CAS.
- C. Winder, Toxicity of gases, vapours and particulates, in Occupational toxicology, CRC Press, 2004, pp. 399–424 Search PubMed.
- V. Vincent, The Place of Gas Analysis in Forensic Toxicology, J. Forensic Leg. Investig. Sci., 2015, 1(005) DOI:10.24966/flis-733x/100005.
- S. Singsen, N. Thatsami, P. Tangpakonsab, H. Bae, H. Lee, T. Hussain and T. Kaewmaraya, Transition-Metals Graphdiyne Monolayer as Efficient Sensor toward Phosphide (PH3) and Arsine (AsH3), Phys. Chem. Chem. Phys., 2022, 26622–26630 RSC.
- H. Luo, L. Zhang, S. Xu, M. Shi, W. Wu and K. Zhang, NH3, PH3 and AsH3 adsorption on alkaline earth metal (Be-Sr) doped graphenes: Insights from DFT calculations, Appl. Surf. Sci., 2021, 537, 147542 CrossRef CAS.
- H. Luo, K. Xu, Z. Gong, N. Li, K. Zhang and W. Wu, NH3, PH3, AsH3 adsorption and sensing on rare earth metal doped graphene: DFT insights, Appl. Surf. Sci., 2021, 566, 150390 CrossRef CAS.
- P. Buasaeng, W. Rakrai, B. Wanno and C. Tabtimsai, DFT investigation of NH3, PH3, and AsH3 adsorptions on Sc-, Ti-, V-, and Cr-doped single-walled carbon nanotubes, Appl. Surf. Sci., 2017, 400, 506–514 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, 6.0.16, Semichem Inc., Shawnee Mission, KS, USA, 2001 Search PubMed; T. HyperChem, HyperChem, 8.07, HyperChem Professional Program, Gainesville, Hypercube, 2016 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Conte, P. L. Houston, C. Qu, J. Li and J. M. Bowman, Full-dimensional, ab initio potential energy surface for glycine with characterization of stationary points and zero-point energy calculations by means of diffusion Monte Carlo and semiclassical dynamics, J. Chem. Phys., 2020, 153(24), 244301 CrossRef CAS PubMed.
- M. K. Hema, C. S. Karthik, K. J. Pampa, P. Mallu and N. K. Lokanath, Solvent induced mononuclear and dinuclear mixed ligand Cu (II) complex: structural diversity, supramolecular packing polymorphism and molecular docking studies, New J. Chem., 2020, 44(41), 18048–18068 RSC.
- G. A. Zhurko and D. A. Zhurko, Chemcraft, Version 1.7 (Build 132), http://www.chemcraftprog.com/ Search PubMed.
- Origin(Pro), Version 2018, OriginLab Corporation, Northampton, MA, USA Search PubMed.
- F. Weinhold, The Path to Natural Bond Orbitals, Isr. J. Chem., 2022, 62(1–2), e202100026 CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- S. Shittu, G. Li, X. Zhao and X. Ma, Review of thermoelectric geometry and structure optimization for performance enhancement, Appl. Energy, 2020, 268, 115075 CrossRef.
- R. Rabeya, S. Mahalingam, A. Manap, M. Satgunam, M. Akhtaruzzaman and C. H. Chia, Structural defects in graphene quantum dots: A review, Int. J. Quantum Chem., 2022, 122(12), e26900 CrossRef CAS.
- G. Chen, L. Gan, H. Xiong and H. Zhang, Density functional theory study of B, N, and Si doped penta-graphene as the potential gas sensors for NH3 detection, Membranes, 2022, 12(1), 77 CrossRef CAS PubMed.
- A. N. Sosa, J. E. Santana, Á. Miranda, L. A. Pérez, A. Trejo, F. Salazar and M. Cruz-Irisson, NH3 capture and detection by metal-decorated germanene: a DFT study, J. Mater. Sci., 2022, 57(18), 8516–8529 CrossRef CAS.
- B. J. Cid, J. E. Santana, L. G. Arellano, Á. Miranda, S. E. Pérez-Figueroa, M. I. Iturrios and M. Cruz-Irisson, Metal-decorated siligene as work function type sensor for NH3 detection: A DFT approach, Appl. Surf. Sci., 2023, 610, 155541 CrossRef CAS.
- E. Salih and A. I. Ayesh, First principle investigation of
H2Se, H2Te and PH3 sensing based on graphene oxide, Phys. Lett. A, 2020, 384(29), 126775 CrossRef CAS.
- E. A. Eno, F. A. Patrick-Inezi, H. Louis, T. E. Gber, T. O. Unimuke, E. C. Agwamba and S. A. Adalikwu, Theoretical investigation and antineoplastic potential of Zn (II) and Pd (II) Complexes of 6-Methylpyridine-2-carbaldehyde-N (4)-ethylthiosemicarbazone, Chem. Phys. Impact, 2022, 5, 100094 CrossRef.
- Q. S. Obu, H. Louis, J. O. Odey, I. J. Eko, S. Abdullahi, T. N. Ntui and O. E. Offiong, Synthesis, spectra (FT-IR, NMR) investigations, DFT study, in silico ADMET and Molecular docking analysis of 2-amino-4-(4-aminophenyl) thiophene-3-carbonitrile as a potential anti-tubercular agent, J. Mol. Struct., 2021, 1244, 130880 CrossRef CAS.
- H. Louis, T. E. Gber, F. C. Asogwa, E. A. Eno, T. O. Unimuke, V. M. Bassey and B. I. Ita, Understanding the lithiation mechanisms of pyrenetetrone-based carbonyl compound as cathode material for lithium-ion battery: Insight from first principle density functional theory, Mater. Chem. Phys., 2022, 278, 125518 CrossRef CAS.
- D. M. Mamand and H. M. Qadr, Density Functional Theory and Computational Simulation of the Molecular Structure on Corrosion of Carbon Steel in Acidic Media of Some Amino Acids, Russ. J. Phys. Chem. A, 2022, 96(10), 2155–2165 CrossRef CAS.
- H. O. Edet, H. Louis, I. Benjamin, M. Gideon, T. O. Unimuke, S. A. Adalikwu and A. S. Adeyinka, Hydrogen storage capacity of C12X12 (X= N, P, and Si), Chem. Phys. Impact, 2022, 5, 100107 CrossRef.
- H. Louis, I. O. Amodu, T. O. Unimuke, T. E. Gber, B. B. Isang and A. S. Adeyinka, Modeling of Ca12O12, Mg12O12, and Al12N12 nanostructured materials as sensors for phosgene (Cl2CO), Mater. Today Commun., 2022, 32, 103946 CrossRef CAS.
- H. Louis, D. Etiese, T. O. Unimuke, A. E. Owen, A. O. Rajee, T. E. Gber and E. N. Nfor, Computational design and molecular modeling of the interaction of nicotinic acid hydrazide nickel-based complexes with H 2 S gas, RSC Adv., 2022, 12(47), 30365–30380 RSC.
- M. D. Mohammadi, H. Y. Abdullah, H. Louis and G. E. Mathias, 2D Boron Nitride Material as a sensor for H2SiCl2, Comput. Theor. Chem., 2022, 113742 CrossRef.
- H. Louis, G. E. Mathias, O. J. Ikenyirimba, T. O. Unimuke, D. Etiese and A. S. Adeyinka, Metal-doped Al12N12X (X= Na, Mg, K) nanoclusters as nanosensors for carboplatin: insight from first-principles computation, J. Phys. Chem. B, 2022, 126(27), 5066–5080 CrossRef CAS PubMed.
- M. D. Mohammadi, F. Abbas, H. Louis, G. E. Mathias and T. O. Unimuke, Trapping of CO, CO2, H2S, NH3, NO, NO2, and SO2 by polyoxometalate compound, Comput. Theor. Chem., 2022, 1215, 113826 CrossRef CAS.
- H. Louis, T. C. Egemonye, T. O. Unimuke, B. E. Inah, H. O. Edet, E. A. Eno and A. S. Adeyinka, Detection of Carbon, Sulfur, and Nitrogen Dioxide Pollutants with a 2D Ca12O12 Nanostructured Material, ACS Omega, 2022, 7(39), 34929–34943 CrossRef CAS PubMed.
- H. Louis, D. E. Charlie, I. O. Amodu, I. Benjamin, T. E. Gber, E. C. Agwamba and A. S. Adeyinka, Probing the reactions of thiourea (CH4N2S) with metals (X= Au, Hf, Hg, Ir, Os, W, Pt, and Re) anchored on fullerene surfaces (C59X), ACS Omega, 2022, 7(39), 35118–35135 CrossRef CAS PubMed.
- R. Hussain, M. Saeed, M. Y. Mehboob, S. U. Khan, M. U. Khan, M. Adnan and K. Ayub, Density functional theory study of palladium cluster adsorption on a graphene support, RSC Adv., 2020, 10(35), 20595–20607 RSC.
- J. A. Agwupuye, H. Louis, T. O. Unimuke, P. David, E. I. Ubana and Y. L. Moshood, Electronic structure investigation of the stability, reactivity, NBO analysis, thermodynamics, and the nature of the interactions in methyl-substituted imidazolium-based ionic liquids, J. Mol. Liq., 2021, 337, 116458 CrossRef CAS.
- V. Nagarajan and R. Chandiramouli, Detection of trace level of hazardous phosgene gas on antimonene nanotube based on first-principles method, J. Mol. Graphics Modell., 2019, 88, 32–40 CrossRef CAS PubMed.
- H. Louis, O. J. Ikenyirimba, T. O. Unimuke, G. E. Mathias, T. E. Gber and A. S. Adeyinka, Electrocatalytic activity of metal encapsulated, doped, and engineered fullerene-based nanostructured materials towards hydrogen evolution reaction, Sci. Rep., 2022, 12(1), 1–21 CrossRef PubMed.
- E. Nemati-Kande, R. Karimian, V. Goodarzi and E. Ghazizadeh, Feasibility of pristine, Al-doped and Ga-doped Boron Nitride nanotubes for detecting SF4 gas: A DFT, NBO and QTAIM investigation, Appl. Surf. Sci., 2020, 510, 145490 CrossRef CAS.
- W. Emori, G. J. Ogunwale, H. Louis, E. C. Agwamba, K. Wei, T. O. Unimuke, C. Cheng, E. U. Ejiofor, F. C. Asogwa and A. S. Adeyinka, Spectroscopic (UV-vis, FT-IR, FT-Raman, and NMR) analysis, structural benchmarking, molecular properties, and the in-silico cerebral anti-ischemic activity of 2 amino-6-ethoxybenzothiazole, J. Mol. Struct., 2022, 133318 CrossRef CAS.
- M. Doust Mohammadi and H. Y. Abdullah, The adsorption of chlorofluoromethane on pristine, and Al-and Ga-doped boron nitride nanosheets: a DFT, NBO, and QTAIM study, J. Mol. Model., 2020, 26(10), 1–15 CrossRef PubMed.
- W. Abisha, D. A. Dhas, S. Balachandran and I. H. Joe, Molecular Structure, Spectroscopic Elucidation (FT-IR, FT-Raman, UV-visible and NMR) with NBO, ELF, LOL, RDG, Fukui, Drug Likeness and Molecular Docking Analysis on Dimethomorph, Polycyclic Aromat. Compd., 2022, 1–44 Search PubMed.
- E. A. Eno, J. I. Mbonu, H. Louis, F. S. Patrick-Inezi, T. E. Gber, T. O. Unimuke and O. E. Offiong, Antimicrobial activities of 1-phenyl-3-methyl-4-trichloroacetyl-pyrazolone: Experimental, DFT studies, and molecular docking investigation, J. Indian Chem. Soc., 2022, 99(7), 100524 CrossRef CAS.
- C. H. Suresh, G. S. Remya and P. K. Anjalikrishna, Molecular electrostatic potential analysis: A powerful tool to interpret and predict chemical reactivity, Wiley Interdisciplinary Reviews: Computational Molecular Science, 2022, p. e1601 Search PubMed.
- O. V. Rudnitskaya, T. A. Tereshina, E. V. Dobrokhotova, E. K. Kultyshkina, A. S. Novikov, A. G. Tskhovrebov and V. N. Khrustalev, Monoprotonated Dimethyl Sulfoxide,[HOSMe2]+: Synthesis, Crystal Structure, Spectroscopic and Theoretical Studies of [HOSMe2] 2 [OsCl6]·2H2O, ChemistrySelect, 2021, 6(21), 5211–5217 CrossRef CAS.
- A. G. Tskhovrebov, A. S. Novikov, A. S. Kritchenkov, V. N. Khrustalev and M. Haukka, Attractive halogen halogen interactions in crystal structure of trans-dibromogold (III) complex, Z. Kristallogr. Cryst. Mater., 2020, 235(10), 477–480 CrossRef CAS.
- D. K. Miller, I. Y. Chernyshov, Y. V. Torubaev and S. V. Rosokha, From weak to strong interactions: structural and electron topology analysis of the continuum from the supramolecular chalcogen bonding to covalent bonds, Phys. Chem. Chem. Phys., 2022, 24(14), 8251–8259 RSC.
- N. Kumar, S. Saha and G. N. Sastry, Towards developing a criterion to characterize non-covalent bonds: a quantum mechanical study, Phys. Chem. Chem. Phys., 2021, 23(14), 8478–8488 RSC.
- M. Doust Mohammadi and H. Y. Abdullah, Non-covalent interactions of Cysteine onto C60, C59Si, and C59Ge: A DFT study, J. Mol. Model., 2021, 27(11), 1–14 CrossRef PubMed.
- G. J. Ogunwale, H. Louis, T. E. Gber and A. S. Adeyinka, Modeling of pristine, Ir-and Au-decorated C60 fullerenes as sensors for detection of Hydroxyurea and Nitrosourea drugs, J. Environ. Chem. Eng., 2022, 108802 CrossRef CAS.
- T. N. Lohith, M. K. Hema, C. S. Karthik, S. Sandeep, L. Mallesha, N. S. Alsaiari and S. R. Kumaraswamy, Persistent prevalence of non-covalent interaction in pyrimidine containing sulfonamide derivative: A quantum computational analysis, J. Mol. Struct., 2022, 133378 CrossRef CAS.
- A. G. Tskhovrebov, A. S. Novikov, B. S. Tupertsev, A. A. Nazarov, A. A. Antonets, A. A. Astafiev and V. N. Khrustalev, Azoimidazole gold (III) complexes: Synthesis, structural characterization and self-assembly in the solid state, Inorg. Chim. Acta, 2021, 522, 120373 CrossRef CAS.
- H. Li, J. Guo, Y. Zhang, L. Zhao, J. Gao and C. Xu, Research on separation of aromatics from FCC diesel using organic solvent: A combination of experiments and quantum chemical calculations, Fuel, 2022, 308, 121982 CrossRef CAS.
- P. Peng, N. Ding, C. Zhao, Y. Li, J. Liu, S. Li and S. Pang, Improving the Stability of All-Carbon-Nitrated Azoles through Cocrystallization, Cryst. Growth Des., 2022, 22(4), 2158–2167 CrossRef CAS.
- A. H. Birniwa, A. S. Abubakar, H. N. M. E. Mahmud, S. R. M. Kutty, A. H. Jagaba, S. S. A. Abdullahi and Z. U. Zango, Application of agricultural wastes for cationic dyes removal from wastewater, in Textile Wastewater Treatment, Springer, Singapore, 2022, pp. 239–274 Search PubMed.
- H. Louis, O. J. Ikenyirimba, T. O. Unimuke, G. E. Mathias, T. E. Gber and A. S. Adeyinka, Electrocatalytic activity of metal encapsulated, doped, and engineered fullerene-based nanostructured materials towards hydrogen evolution reaction, Sci. Rep., 2022, 12(1), 1–21 CrossRef PubMed.
- H. Luo, K. Xu, Z. Gong, N. Li, K. Zhang and W. Wu, NH3, PH3, AsH3 adsorption and sensing on rare earth metal doped graphene: DFT insights, Appl. Surf. Sci., 2021, 566, 150390 CrossRef CAS.
- S. Singsen, N. Thasami, P. Tangpakonsab, H. Bae, H. Lee, T. Hussain and T. Kaewmaraya, Transition-metal decorated graphdiyne monolayer as an efficient sensor toward phosphide (PH 3) and arsine (AsH 3), Phys. Chem. Chem. Phys., 2022, 24(43), 26622–26630 RSC.
- C. G. Apebende, H. Louis, A. E. Owen, I. Benjamin, I. O. Amodu, T. E. Gber and F. C. Asogwa, Adsorption properties of metal functionalized fullerene (C59Au, C59Hf, C59Ag, and C59Ir) nanoclusters for application as a biosensor for hydroxyurea (HXU): insight from theoretical computation, Z. Phys. Chem., 2022, 236(11–12), 1515–1546 CrossRef CAS.
- R. Dong, Synthesis of Asymmetric Heterocycle-Fused Indacenes for Organic Electronic Application, Doctoral dissertation, University of Oregon, 2022, https://scholarsbank.uoregon.edu/xmlui/handle/1794/27512 Search PubMed.
- R. Woods-Robinson, Y. Han, H. Zhang, T. Ablekim, I. Khan, K. A. Persson and A. Zakutayev, Wide band gap chalcogenide semiconductors, Chem. Rev., 2020, 120(9), 4007–4055 CrossRef CAS PubMed.
- M. V. Nikolic, V. Milovanovic, Z. Z. Vasiljevic and Z. Stamenkovic, Semiconductor gas sensors: Materials, technology, design, and application, Sensors, 2020, 20(22), 6694 CrossRef CAS PubMed.
- M. Rakhshi, M. Mohsennia and H. Rasa, DFT Study of PH 3 Physisorption and Chemisorption on Boron Nitride Nanotubes, Russ. J. Phys. Chem. A, 2018, 92, 540–546 CrossRef.
- S. Sardarzadeh, J. Karamdel and P. Nayebi, Adsorption of SO2, H2S, NH3, PH3, and AsH3 Gas Molecules on Pristine Armchair Phosphorene Nanoribbon: A First-Principles Study, Phys. Status Solidi B, 2020, 257(9), 2000120 CrossRef CAS.
- M. Chauhan, R. Kashyap, M. Singh, B. C. Choudhary and R. K. Sharma, A DFT Modulated Analysis of Manganese doped Graphene Nanoribbons as a potential material for sensing of highly toxic gases CO, PH3 and SbH3, Phys. Scr., 2023, 045803 Search PubMed.
- J. Luo, D. Yu, K. D. Hristovski, K. Fu, Y. Shen, P. Westerhoff and J. C. Crittenden, Critical review of advances in engineering nanomaterial adsorbents for metal removal and recovery from water: Mechanism identification and engineering design, Environ. Sci. Technol., 2021, 55(8), 4287–4304 CrossRef CAS PubMed.
- P. Jiménez-Calvo, V. Caps and V. Keller, Plasmonic Au-based junctions onto TiO2, gC3N4, and TiO2-gC3N4 systems for photocatalytic hydrogen production: Fundamentals
and challenges, Renewable Sustainable Energy Rev., 2021, 149, 111095 CrossRef.
- D. Cortes-Arriagada, and D. E. Ortega, Phosphorene–Fullerene nanostructures: A first-principles study, 2021 Search PubMed.
- M. Veit, D. M. Wilkins, Y. Yang, R. A. DiStasio Jr and M. Ceriotti, Predicting molecular dipole moments by combining atomic partial charges and atomic dipoles, J. Chem. Phys., 2020, 153(2), 024113 CrossRef CAS PubMed.
- E. V. Kulakov, C. J. Sprain, P. V. Doubrovine, A. V. Smirnov, G. A. Paterson, L. Hawkins and A. J. Biggin, Analysis of an updated paleointensity database (QPI-PINT) for 65–200 Ma: Implications for the long-term history of dipole moment through the Mesozoic, J. Geophys. Res.: Solid Earth, 2019, 124(10), 9999–10022 CrossRef.
- S. Ravi, R. Sreedharan, K. R. Raghi, T. M. Kumar and K. Naseema, Linear–nonlinear optical and quantum chemical studies on Quinolinium 3, 5-dinitrobenzoate: a novel third order non-linear optical material for optoelectronic applications, Spectrochim. Acta, Part A, 2021, 249, 119304 CrossRef CAS PubMed.
- L. Mydlova, S. Taboukhat, K. Waszkowska, N. Ibrahim, A. Migalska-Zalas, B. Sahraoui and M. Makowska-Janusik, Selected molecules based on (-1-cyanovinyl) benzonitrile as new materials for NLO applications—Experimental and computational studies, J. Mol. Liq., 2020, 314, 113622 CrossRef CAS.
- S. Ahmed, X. Jiang, C. Wang, U. E. Kalsoom, B. Wang, J. Khan and H. Zhang, An Insightful Picture of Nonlinear Photonics in 2D Materials and their Applications: Recent Advances and Future Prospects, Adv. Opt. Mater., 2021, 9(11), 2001671 CrossRef CAS.
- D. H. Williams, E. Stephens, D. P. O’Brien and M. Zhou, Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes, Angew. Chem. Int. Ed., 2004, 43(48), 6596–6616 CrossRef CAS PubMed.
- G. Han, X. Zhao and A. Farajtabar, Equilibrium Solubility and Solvent Effect Study of 3-Nitrosalicylic Acid in Different Monosolvents Covering Temperatures from 278.15 to 323.15 K, J. Chem. Eng. Data, 2021, 66(7), 2882–2894 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *acd,
Hitler Louis
*acd,
Hitler Louis