
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new class of anti-proliferative activity and apoptotic inducer with molecular docking studies for a novel of 1,3-dithiolo[4,5-b]quinoxaline derivatives hybrid with a sulfonamide moiety†
Mostafa A. Ismaila,
Moustafa S. Abusaif b,
Mohamed S. A. El-Gaby*b,
Yousry A. Ammar*b and
Ahmed Ragab*b
b,
Mohamed S. A. El-Gaby*b,
Yousry A. Ammar*b and
Ahmed Ragab*b
aChemistry Department, Faculty of Science, Al-Azhar University, Assiut, 71524, Egypt
bChemistry Department, Faculty of Science (Boys), Al-Azhar University, Nasr City, Cairo, 11884, Egypt. E-mail: m_elgaby@azhar.edu.eg; yossry@azhar.edu.eg; Ahmed_ragab@azhar.edu.eg; Ahmed_ragab7@ymail.com
First published on 24th April 2023
Abstract
A new series of 6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxaline-2-ylidines 10a–f, 12, 14, 16, and 18 were designed, synthesized, and evaluated for their in vitro anticancer activity. The structures of the novel compounds were systematically characterized by 1H NMR, 13C NMR, and elemental analysis. The synthesized derivatives were evaluated for their in vitro antiproliferative activity against three human cancer cell lines (HepG-2, HCT-116, and MCF-7) with more sensitivity to MCF-7. Moreover, three derivatives 10c, 10f, and 12 were the most promising candidates with sub-micromole values. These derivatives were further evaluated against MDA-MB-231, and the results displayed significant IC50 values ranging from 2.26 ± 0.1 to 10.46 ± 0.8 μM and showed low cellular cytotoxicity against WI-38. Surprisingly, the most active derivative 12 revealed sensitivity towards the breast cell lines MCF-7 (IC50 = 3.82 ± 0.2 μM) and MDA-MB-231 (IC50 = 2.26 ± 0.1 μM) compared with doxorubicin (IC50 = 4.17 ± 0.2 and 3.18 ± 0.1 M). Cell cycle analysis showed that compound 12 arrests and inhibits the growth of MCF-7 cells in the S phase with values of 48.16% compared with the untreated control 29.79% and exhibited a significantly higher apoptotic effect in MCF-7 with a value of 42.08% compared to control cell at 1.84%. Furthermore, compound 12 decreased Bcl-2 protein 0.368-fold and activation on pro-apoptotic genes Bax and P53 by 3.97 and 4.97 folds, respectively, in MCF-7 cells. Compound 12 exhibited higher inhibitory activity to EGFRWt, EGFRL858R, and VEGFR-2 with IC50 values (0.19 ± 0.009, 0.026 ± 0.001, and 0.42 ± 0.021 μM) compared with erlotinib (IC50 = 0.037 ± 0.002 and 0.026 ± 0.001 μM) and sorafenib (IC50 = 0.035 ± 0.002 μM). Finally, in silico ADMET prediction presented that 1,3-dithiolo[4,5-b]quinoxaline derivative 12 obeys the Lipinski rule of five and the Veber rule with no PAINs alarms and moderately soluble properties. Additionally, toxicity prediction revealed that compound 12 demonstrated inactivity to hepatotoxic carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. Moreover, molecular docking studies showed good binding affinity with lower binding energy inside the active site of Bcl-2 (PDB: 4AQ3), EGFR (PDB: 1M17), and VEGFR (PDB: 4ASD).
1. Introduction
Cancer is a leading cause of death and a worldwide public health issue. Numerous potent anticancer medications are available, including classic chemotherapy drugs that prevent cell proliferation and DNA replication.1 The greatest cause of mortality worldwide is cancer, which claims 1.61 million lives annually (19.41%) including 0.79 million from liver cancer (9.2%), and 0.79 million from stomach cancer (9.2%).2,3 In addition, breast cancer is the most commonly diagnosed cancer in women, with 287![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 women expected to receive a diagnosis by 2022.4 Despite early detection and treatment advances, patients with distant metastases of breast cancer often get poor results due to their low incomes and the high cost of therapies.5 Most breast cancer-related deaths result from metastasis to distant organs,6 highlighting the need to find pathways or cell populations that promote and accelerate breast cancer metastasis. In breast tumors, a small percentage of cells, known as breast cancer stem cells (BCSCs), are still capable of self-renewing and regenerating the heterogeneous tumor lesions, which is referred to as tumor recurrence.7,8 These BCSCs are rare, often quiescent, highly ATP-binding cassette transporter-expressed, maintain an increased DNA-repair capability, and resist high concentrations of reactive oxygen species (ROS), contributing to therapeutic resistance and poor patient prognoses.4 The clinical need to research and therapeutically target these cell types is supported by the fact that BCSCs cause metastasis and contribute to therapy resistance.9 One of the most important signaling proteins regulating the tumor angiogenesis process is the vascular endothelial growth factor (VEGF).10 VEGF overexpression was found in several cancers, including breast carcinoma.11,12 Tyrosine kinase receptors VEGFR-1, VEGFR-2, and VEGFR-3 are involved in the angiogenic process through their interactions with VEGF ligands. VEGFR-2 mediates all VEGF responses in endothelial cells.13,14 Therefore, developing effective medications for human angiogenesis-dependent cancers should focus on targeting VEGFR-2.15 The FDA has licensed a number of VEGFR-2 suppressors for treating different malignancies.16,17 In general, VEGFR-2 kinase inhibitors are divided into two major categories. Type I kinase inhibitors compete with ATP for ATP-binding sites (ATP competitive inhibitors). In contrast, the second type induces the DFG-out (inactive) conformation of the enzyme to enter an allosteric region (near ATP binding site), which is only visible in the inactive DFG-out conformation.18–20 However, several downsides, such as bleeding complications, have been noted during clinical use; as a result, there is still an urgent need to develop safe VEGFR-2 inhibitors.21
000 women expected to receive a diagnosis by 2022.4 Despite early detection and treatment advances, patients with distant metastases of breast cancer often get poor results due to their low incomes and the high cost of therapies.5 Most breast cancer-related deaths result from metastasis to distant organs,6 highlighting the need to find pathways or cell populations that promote and accelerate breast cancer metastasis. In breast tumors, a small percentage of cells, known as breast cancer stem cells (BCSCs), are still capable of self-renewing and regenerating the heterogeneous tumor lesions, which is referred to as tumor recurrence.7,8 These BCSCs are rare, often quiescent, highly ATP-binding cassette transporter-expressed, maintain an increased DNA-repair capability, and resist high concentrations of reactive oxygen species (ROS), contributing to therapeutic resistance and poor patient prognoses.4 The clinical need to research and therapeutically target these cell types is supported by the fact that BCSCs cause metastasis and contribute to therapy resistance.9 One of the most important signaling proteins regulating the tumor angiogenesis process is the vascular endothelial growth factor (VEGF).10 VEGF overexpression was found in several cancers, including breast carcinoma.11,12 Tyrosine kinase receptors VEGFR-1, VEGFR-2, and VEGFR-3 are involved in the angiogenic process through their interactions with VEGF ligands. VEGFR-2 mediates all VEGF responses in endothelial cells.13,14 Therefore, developing effective medications for human angiogenesis-dependent cancers should focus on targeting VEGFR-2.15 The FDA has licensed a number of VEGFR-2 suppressors for treating different malignancies.16,17 In general, VEGFR-2 kinase inhibitors are divided into two major categories. Type I kinase inhibitors compete with ATP for ATP-binding sites (ATP competitive inhibitors). In contrast, the second type induces the DFG-out (inactive) conformation of the enzyme to enter an allosteric region (near ATP binding site), which is only visible in the inactive DFG-out conformation.18–20 However, several downsides, such as bleeding complications, have been noted during clinical use; as a result, there is still an urgent need to develop safe VEGFR-2 inhibitors.21
Furthermore, the epidermal growth factor receptor (EGFR) is an attractive therapeutic target for cancer treatment due to its association with the regulation of cell survival, proliferation, metastasis, and angiogenesis, as well as its widespread overexpression in a wide range of solid tumors.22 Since the first-generation EGFR-TKI, gefitinib, was introduced in 2002 (Japan), EGFR-TKIs have evolved into three generations, involving more than a dozen medications.23 EGFR-TKIs have evolved into three generations in the past two decades, involving more than a dozen medications since the first-generation EGFR-tyrosine kinase inhibitor (TKI), Gefitinib, was introduced in 2002 (Japan). The first generation of EGFR-TKIs, which includes Gefitinib and erlotinib, inhibits the binding of ATP to the TK domain.24,25 Despite the effectiveness of the first therapy, after 9–14 months of clinical therapy, patients developed acquired drug resistance, and at least 50% of them had the T790M mutation.26 The second generation of inhibitors, which were released in 2013 to combat drug resistance, interacted with Michael receptors and the sulfhydryl groups of Cys in mutant kinase to create covalent binding complexes such as afatinib, which has substantial negative effects in the clinic and is infrequently utilized due to its poor selectivity for mutant versus wild-type EGFR27 (Fig. 1). Moreover, the third generation was approved by the FDA in November 2015 to treat non-small-cell lung cancer (NSCLC) patients who were using EGFR kinase inhibitors and had a metastatic EGFR T790M mutation as osimertinib and olmutinib. Osimertinib received additional FDA approval in April 2018 as a first-line treatment for people with metastatic NSCLC who had EGFR mutations (EGFR 19Del or EGFR L858R).28–30 These substances exhibit specific toxicity toward the tumor endothelial cells needed for the malignancy formation and significant cytotoxicity against a wide range of human cancer cell lines. They thus represent a novel family of vascular disrupting medicines that significantly shut off the blood supply to tumors, resulting in the necrosis and apoptosis of cancer cells.31–33
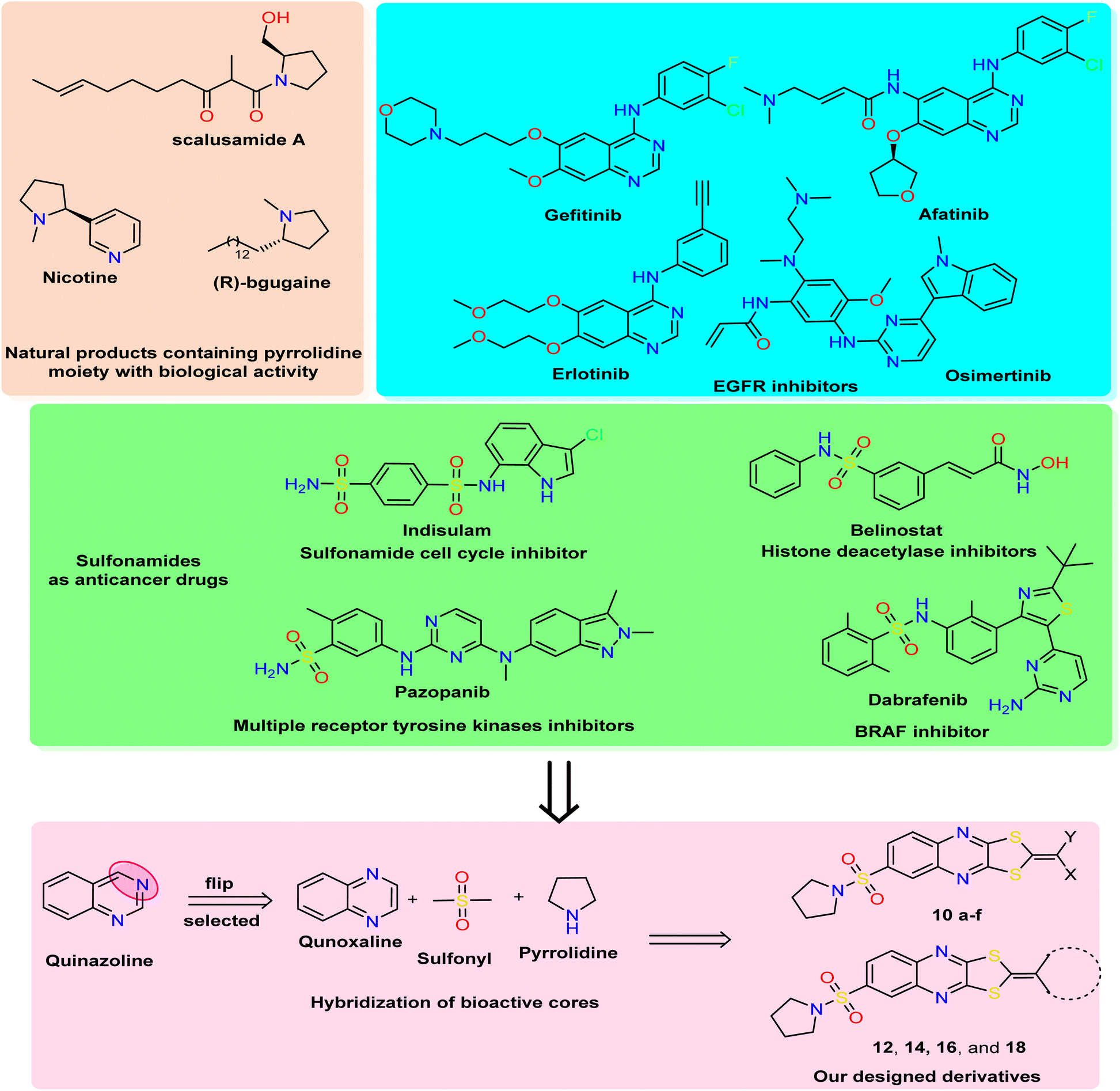 | ||
| Fig. 1 Rational study involved the most bioactive cores as quinazoline (quinoxaline analog), pyrrolidine, and sulfonamide moiety, and our newly designed 1,3-dithiolo[4,5-b]quinoxaline derivatives. | ||
In medicinal chemistry, quinoxalines and quinoxalinones are desirable chemical candidates and are considered analogies for quinazoline by flipping carbon–nitrogen atoms at positions three and four. The quinoxaline derivatives exhibited a variety of biological targets due to their ability to cause biological reactions with biological targets. They, therefore, displayed antiviral,34 herbicidal,35 antimicrobial,36 anticancer,37 and anti-inflammatory38 effects. Additionally, several antibiotics, such as echinomycin, levomycin, and actinoleutin, have a quinoxaline moiety in their structures and are known to impede the development of Gram-positive bacteria.39,40 The quinoxaline scaffold also provides the foundation for many essential aspects in addition to these medical applications. The pharmacological potential of quinoxalines as anticancer drugs has just come to light,41,42 and several theoretical research on quinoxaline and its derivatives have been carried out to discover new antineoplastic molecules. Porter and collaborators' research recently identified the quinoxaline scaffold as a template for creating c-Met kinase inhibitors.43
The polythia-heterocyclic molecules are uncommon and interesting heterocyclic classes that exhibited a wide range of biological activities, especially 1,2-dithiole and 1,3-dithiole.44,45 The 1,2-dithiole derivatives have been isolated from cruciferous vegetables and are protective phytochemicals.46,47 Moreover, 1,3-dithiol-2-ylidenes is a building block for electronic materials.48 The importance of 1,2-dithiol and 1,3-dithiol-2-ylidenes is related to their ability to donate electrons.49 Additionally, it was reported that 2-ylidene-1,3-dithiolane derivatives revealed a variety of biological activities as antimicrobial,50 insecticidal,51 and anticancer activities.52 For drug design and pharmaceutical industries, a pyrrolidine scaffold is a preferred scaffold53 and is ranked within the top five common five-membered nitrogen heterocyclic compounds. Additionally, the pyrrolidine moiety was approved by nearly 37 FDA drugs in the United States.54 Pyrrolidines have various pharmacological activities, including cholinesterase inhibitory,55 anti-HIV,56 antimicrobial,57 anti-inflammatory,58 antioxidant,59 and anticancer properties.60 In natural products with saturated ring systems, the pyrrolidine moiety is typically found in plants or microbially derived alkaloids61 that exhibit a variety of bioactivities, including scalusamides A (antimicrobial activities), nicotine (have anti-inflammatory, antioxidant, and antihyperglycemic), and (R)-bgugaine (anticancer activity)62 The sulfonamide moiety is a key component of numerous clinical drugs with broad-spectrum applications in medicine, pharmaceuticals, and pharmacology, such as antiviral, protease inhibitor, anti-inflammatory, anti-epileptic, anti-diabetic, anti-tumor, and antibacterial activities.63 One aryl sulfonamide medication with anticancer efficacy is indisulam, which produced cell cycle arrest in the G1 phase and cell death64 (Fig. 1). Indisulam's SAR study revealed that its sulfonamide group is crucial for its anticancer effects. More critically, the mitotic arrest phenotype is strongly influenced by the aryl group and substituent.65 Many anticancer drugs containing sulfonamide moiety, such as pazopanib inhibit tumor growth and angiogenesis by targeting multiple receptor tyrosine kinases.66 Additionally, belinostat is a histone deacetylase inhibitor used to treat cancers of the blood and solid tissues.67 The drug dabrafenib is used in the treatment of certain types of cancer, including melanoma, non-small-cell lung cancer, and thyroid cancer (BRAF inhibitor)68 (Fig. 1).
Based on all the above facts and in continuation of our work in the design and synthesis of new heterocyclic bioactive cores by hybridization approach for use in medicinal chemistry.69–73 Our work involved synthesizing a new series of 1,3-dithiolo[4,5-b]quinoxaline derivatives tagged with pyrrolidinosulfonyl moiety in one bioactive scaffold. Additionally, the designed derivatives were evaluated as antiproliferative activity against three human cancer cell lines (HepG2, HCT-116, and MCF-7) using the MTT assay. Moreover, the most active derivatives were screened for other breast cancer cells (MDA-MB-231) using a non-tumorigenic normal cell line (WI-38). Besides, the most active member was selected to determine the effect on apoptosis detection studies, including cell-cycle and apoptosis analysis, mitochondrial apoptosis pathway proteins (BAX, Bcl-2, and p53), and tyrosine kinases enzymes (EGFR and VEGFR). Finally, the in silico ADMET and docking simulation were determined and discussed.
2. Results and discussion
2.1. Chemistry
The designed target compounds 1,3-dithiolo[4,5-b]quinoxalin-2-ylidenes 10a–f, 12, 14, 16, and 18 are depicted in (Schemes 2–4). The key intermediate 2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonyl chloride 3 was obtained by condensing o-phenylenediamine 1 with oxalic acid in the presence of aqueous hydrochloric acid to form 1,4-dihydroquinoxaline-2,3-dione 2, which was subsequently treated with chlorosulfonic acid according to the previously reported procedure74–76 (Scheme 1).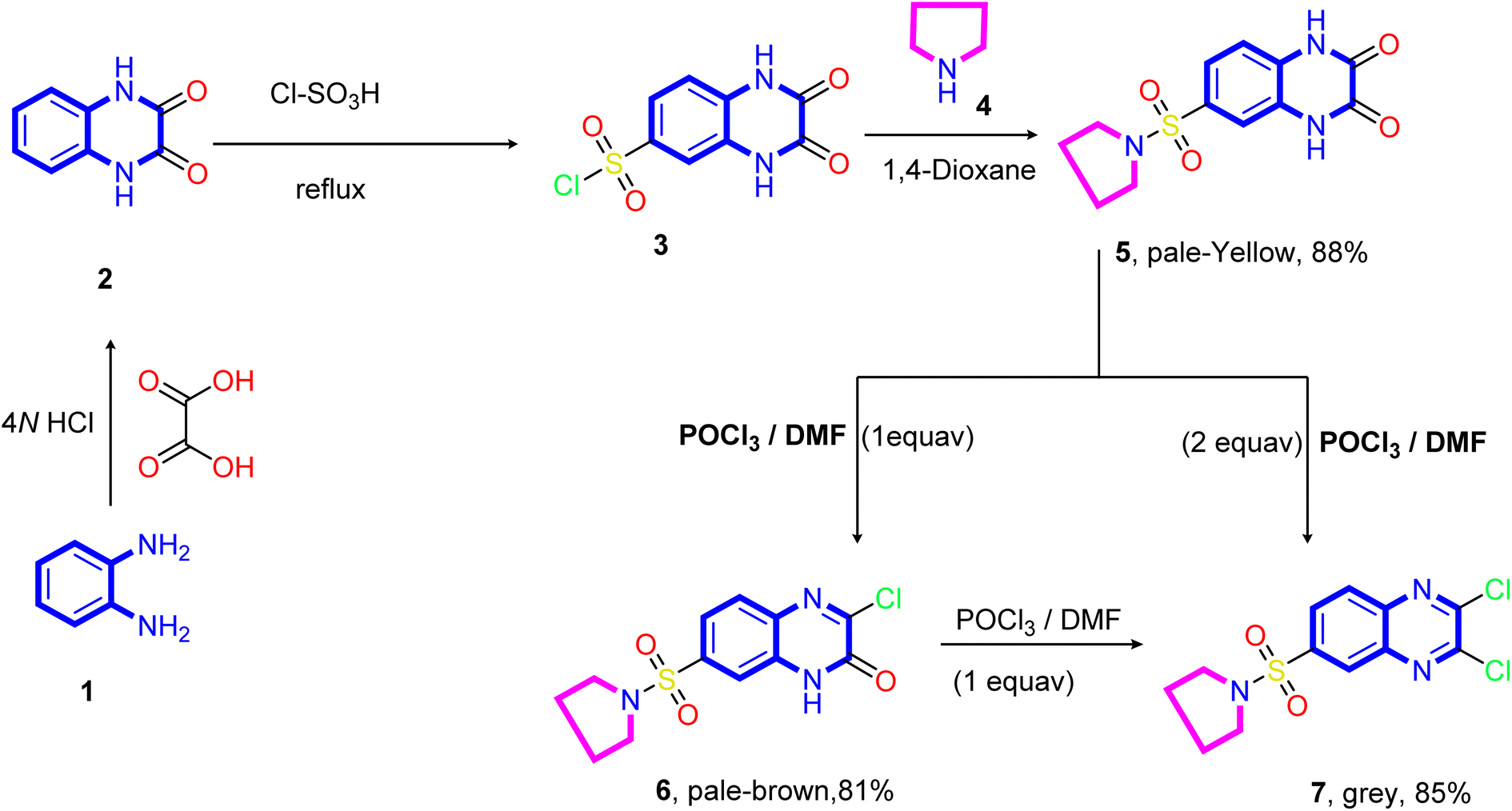 | ||
| Scheme 1 Synthetic pathways of the 2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonyl chloride 3, 3-chloro-6-(pyrrolidin-1-ylsulfonyl)quinoxalin-2(1H)-one 6, and 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline 7. | ||
Treatment of sulfonyl chloride derivative 3 with pyrrolidine 4 in refluxing 1,4-dioxane afforded the corresponding 6-(pyrrolidin-1-ylsulfonyl)-1,4-dihydro quinoxaline-2,3-dione 5 in good yield (88%). The structure of compound 5 was established on the basis of its elemental analysis and spectral data. Its infrared spectrum showed characteristic absorption band at ν 3502 and 3366 cm−1 for the NH stretching, 1692 cm−1 for the two carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1613 cm−1 for the CN stretching, and (ν 1330 and 1148 cm−1) for the SO2 stretching vibrations. Moreover, the quinoxalinedione intermediate 5 was subjected to react with an equimolar amount of phosphorus oxychloride in N,N-dimethylformamide, and 3-chloro-6-(pyrrolidin-1-ylsulfonyl)quinoxalin-2(1H)-one 6 was obtained. The structure of compound 5 was confirmed on the basis of microanalysis and spectral data. The infrared spectrum of compound 5 indicated characteristic absorption bands at ν 3401 cm−1 and 1694 cm−1 due to NH and carbonyl stretching vibrations, respectively. The band observed at 1555 cm−1 is due to CN stretching vibration of the quinoxaline pharmacophore. On the other hand, refluxing of quinoxalinedione 5 with two equivalents of phosphorus oxychloride in N,N-dimethylformamide afforded the 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl) quinoxaline 7.
O stretching), 1613 cm−1 for the CN stretching, and (ν 1330 and 1148 cm−1) for the SO2 stretching vibrations. Moreover, the quinoxalinedione intermediate 5 was subjected to react with an equimolar amount of phosphorus oxychloride in N,N-dimethylformamide, and 3-chloro-6-(pyrrolidin-1-ylsulfonyl)quinoxalin-2(1H)-one 6 was obtained. The structure of compound 5 was confirmed on the basis of microanalysis and spectral data. The infrared spectrum of compound 5 indicated characteristic absorption bands at ν 3401 cm−1 and 1694 cm−1 due to NH and carbonyl stretching vibrations, respectively. The band observed at 1555 cm−1 is due to CN stretching vibration of the quinoxaline pharmacophore. On the other hand, refluxing of quinoxalinedione 5 with two equivalents of phosphorus oxychloride in N,N-dimethylformamide afforded the 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl) quinoxaline 7.
Further, the structure of compound 7 was confirmed on the basis of its elemental analysis, spectral data, and an independent synthesis by reacting an equimolar amount of phosphorus oxychloride in N,N-dimethylformamide under reflux. The infrared spectrum of compound 7 indicated the absence of the carbonyl absorption band and the presence of the characteristic absorption band at ν 1615 cm−1 for the CN group. Its 1H NMR spectrum (DMSO-d6) revealed a quintet at δ 1.63 ppm for the methylene protons at 3- and 4-positions of the pyrrolidine ring and a triplet at δ 3.26 ppm, which are readily assigned to (CH2–N–CH2) protons present in the pyrrolidine ring. The two doublets at δ 8.22 and 8.28 ppm with coupling constant (J = 8.0 Hz) assigned to the hydrogen attached at C5 and C6 of the quinoxaline ring, and a sharp singlet at δ 8.42 assigned to the hydrogen at C8 of the quinoxaline ring. Moreover, the 13C NMR spectrum showed the presence of two signals at δ 25.10 and 48.31 assigned to two methylene groups at C3, C4, and two CH2 at (C2 and C4) of the pyrrolidine ring, respectively. In addition, six signals at δ 125.99, 127.68, 130.03, 138.99, 139.53, and 141.78 ppm (C–SO2) equivalent to the C6H3 group, and two downfield signals at δ 147.09 and 147.77 ppm for the two CN are attached to the chlorine atom.
Our approach to annulate the 1,3-dithiol-2-ylidene group to the quinoxaline ring was based on the good nucleophilicity of the potassium ethene-1,1-dithiolates anion and the fact that the nucleophilic aromatic substitution reactions are known to occur on 2,3-dichloroquinoxaline core.77 Initially, potassium ethene-1,1-bis-(thiolates) 9 are prepared by reacting an acyclic active methylene compound 8 with carbon disulfide in the presence of two equivalents of the base at room temperature. As viewed in Scheme 2, the treatment of 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl) quinoxaline 7 with potassium ethene-1,1-bis-(thiolates) 9 at room temperature in N,N-dimethylformamide afforded the novel 2-ylidene-6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalines 10a–f in good yields. The structure of the reaction products is established on the basis of their elemental analysis and spectral data. The infrared spectra of compounds 10a–f showed the characteristic absorption bands for the -ylidene group (CC). The IR spectrum of compound 10a taken as a representative example of the series, showed three absorption bands at ν 2219, 2193, and 1621 cm−1 due to two cyano and CN, respectively, besides absorption bands at ν 1550 cm−1 assignable for the CC group. The representative 1H NMR spectrum of compound 10b (DMSO-d6) revealed a triplet at δ 1.33 ppm, a quartet at δ 4.37 ppm assigned to the ethyl group, a quintet at δ 1.33 ppm assigned to the methylene protons at 3- and 4-positions of the pyrrolidine ring, and a triplet signal at δ 3.27 ppm, which was readily assigned to CH2–N–CH2 protons present in the pyrrolidine ring. The signals appeared at δ 8.15 ppm (d, 1H, J = 8.0 Hz), at δ 8.29 ppm (d,1H, J = 8.0 Hz), and a sharp singlet at δ 8.29 ppm can be assigned to C7, C8, and C5 of the quinoxaline ring, respectively. The 13C NMR spectrum revealed signals at δ 14.02 for methyl, signals at δ 24.94 and 48.17 ppm for four methylene groups of the pyrrolidine motif, a signal at δ 62.72 ppm for OCH2 of the ethoxy group, two signals at δ 92.39 and 114.45 for the two cyano groups (CN), and signals at δ 122.33, 125.06, 126.40, 136.68, 137.17, 138.16, 149.95, and 151.26 ppm are assigned to the corresponding different types of carbon atoms present in the compound. The most downfield signals appeared at δ 167.57 and 165.86 ppm, which could be assigned to the carbonyl and ethylenic groups.
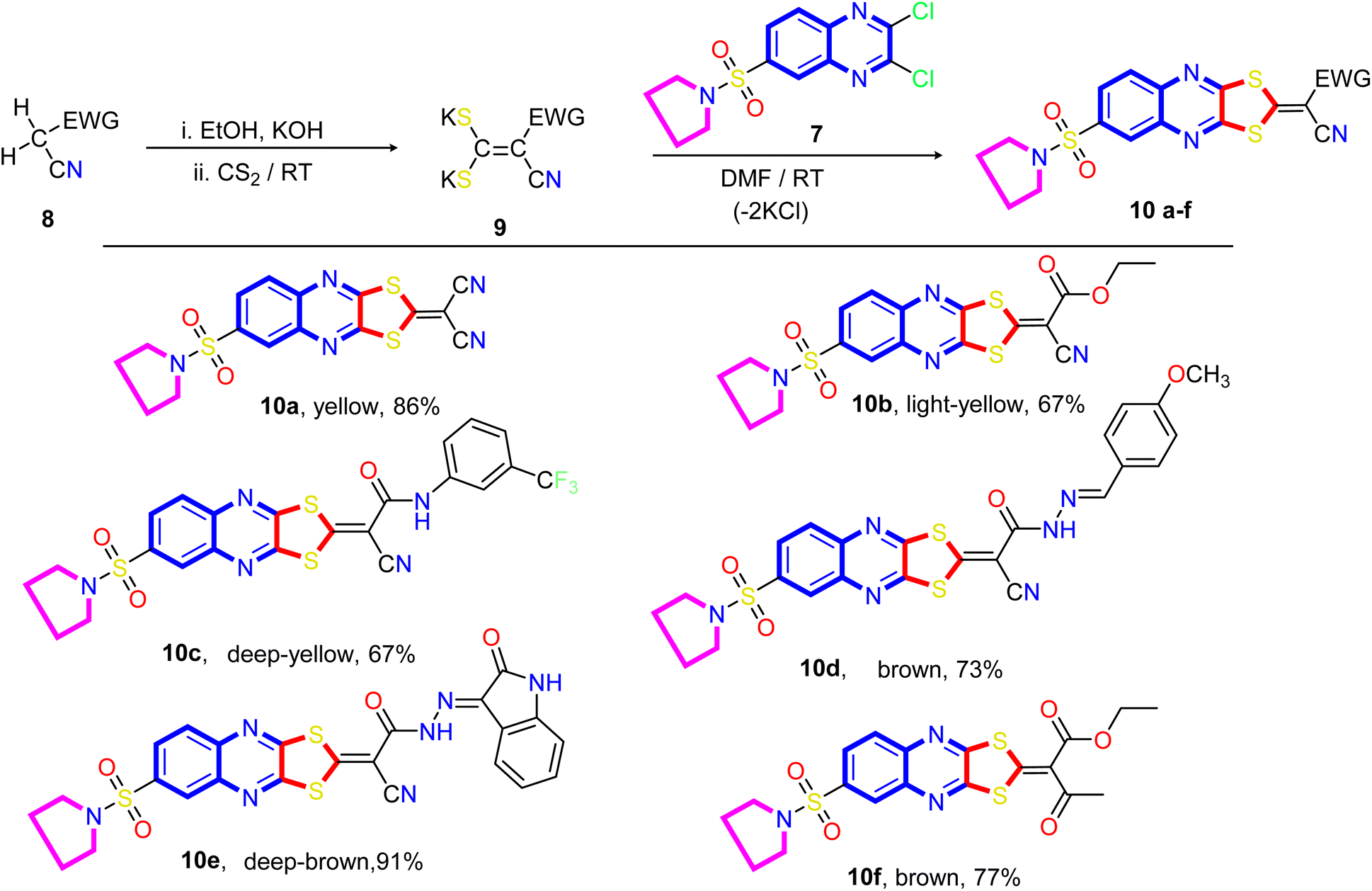 | ||
| Scheme 2 Synthetic pathways of the target 6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxaline-2-ylidines (10a–f). | ||
A proposed mechanism for forming 2-ylidene-[1,3]dithiolo[4,5-b]quinoxalines 10a–f is described in Scheme 3. The formation of [1,3]dithiolo[4,5-b]quinoxalines 10a–f are assumed to proceed via nucleophilic addition of ethene-1,1-dithiolates anion 9 to the activated double bond at position-3 in 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline 7 to give the non-isolable intermediate A and B, followed by intramolecular cyclization by elimination of potassium chloride to afford the final product 10, Scheme 3. The structures of [1,3]dithiolo[4,5-b]quinoxalines 10a–f have been further confirmed from independent work via a one-pot, three-component, reaction of active methylene compounds with carbon disulfide in the presence of a base, followed by in situ coupling of the resulting dithioate salts with 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl) quinoxaline 7 to afford a product identical in all respect (m.p., mixed M.p., TLC, and spectra).
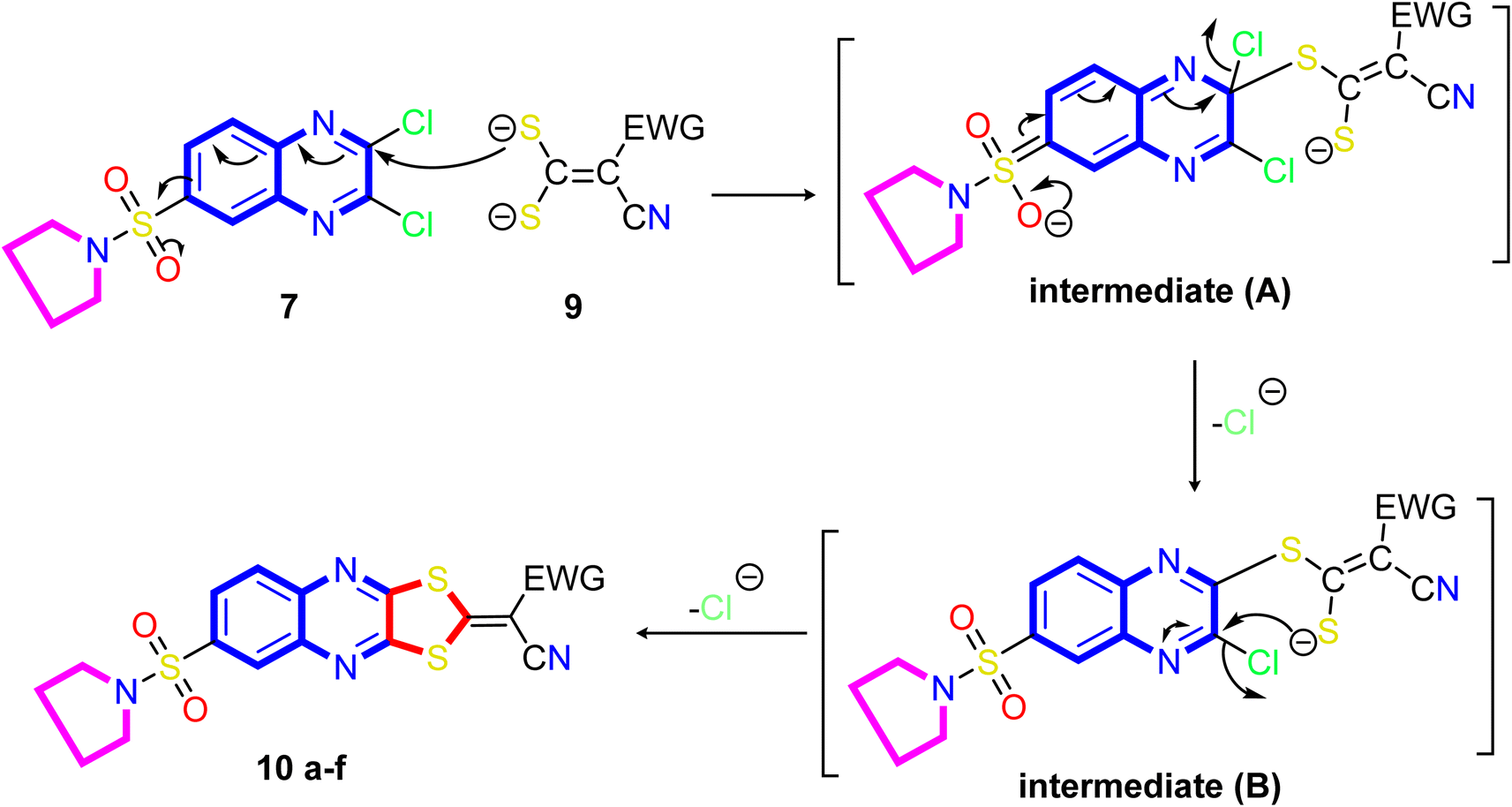 | ||
| Scheme 3 Illustration on the resection mechanism of bi-nucleophile 1,3-dithiolo with unsymmetrical from bi-electrophile and 2,3-dichloro derivative 7. | ||
Our study was extended to include synthesizing novel [1,3]dithiolo[4,5-b]quinoxaline linked to cyclohexyl, pyrazolyl, and pyrimidinyl moieties. Thus, the reaction of the dithioate salts derived from cyclic active methylene compounds 11, 13, 15, and 17 with compound 7 in N,N-dimethylformamide at room temperature gave the corresponding [1,3]dithiolo[4,5-b]quinoxalines 12, 14, 16 and 18, respectively (Scheme 4).
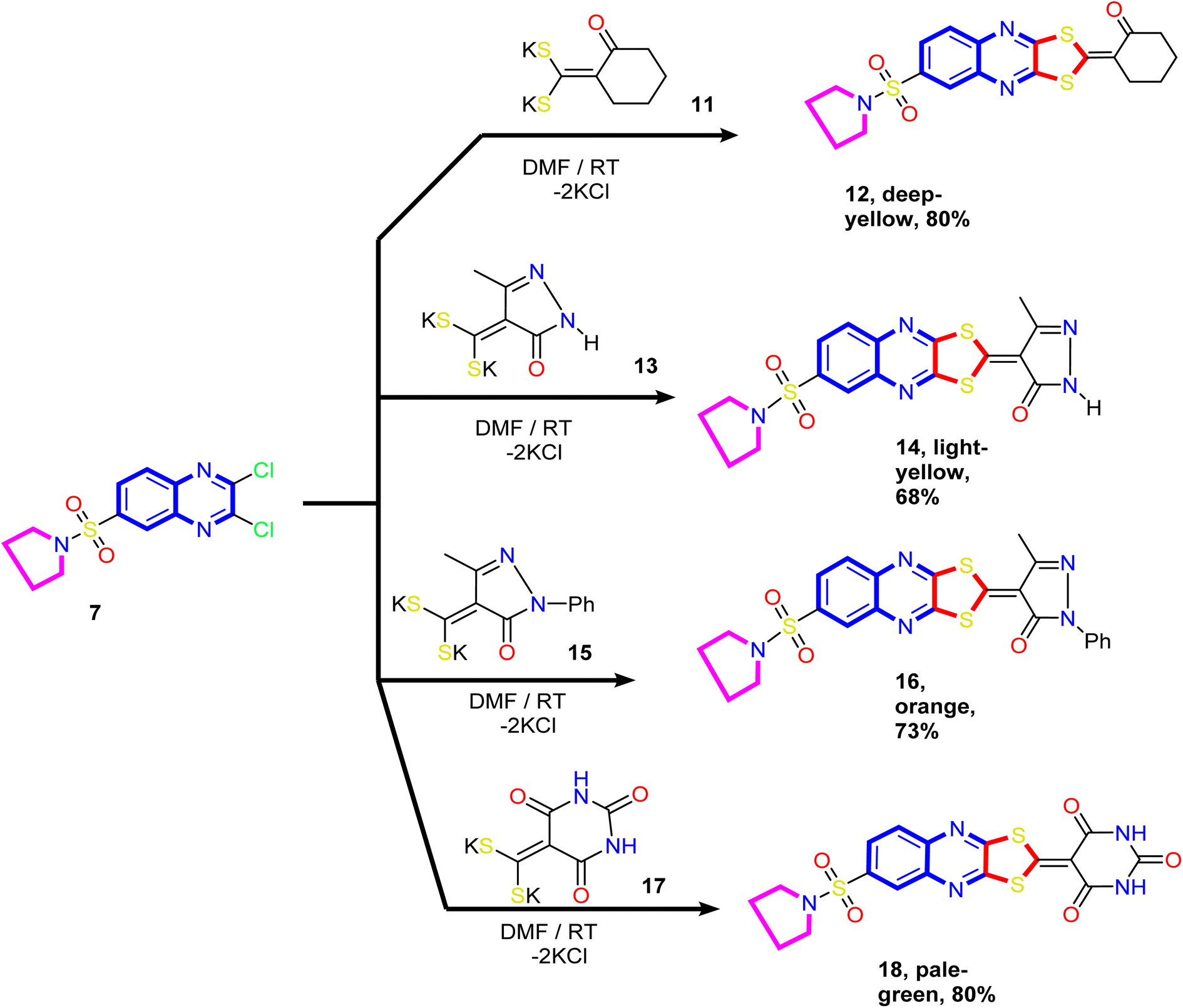 | ||
| Scheme 4 Synthesis of novel [1,3]dithiolo[4,5-b]quinoxalines 12, 14, 16, and 18 linked to cyclohexyl, pyrazolyl, and pyrimidinyl moieties. | ||
The analytical and spectral data of compounds 12, 14, 16, and 18 were in agreement with their proposed structure. Thus, the 1H NMR spectrum of compound 14 showed a quintet at δ 1.69 ppm assigned to methylene protons (C3 and C4) of the pyrrolidine ring, a triplet at δ 3.27 ppm assigned to methylene protons (C2 and C5) of the pyrrolidine ring, and a singlet at δ 2.39 ppm assigned to methyl protons at position-4 of the pyrazole ring. The two doublets at δ 8.19 ppm and 8.27 ppm and a sharp singlet at δ 8.30 ppm were readily assigned to the hydrogen attached at C5, C6, and C8 of the quinoxaline ring, respectively. The downfield singlet signal at δ 11.58 ppm corresponds to the pyrazole ring's NH. In addition, the 13C NMR spectrum revealed signals at δ 14.54 (CH3), 25.74 (2CH2; pyrrolidine), 50.61 (2CH2; pyrrolidine), and signals at δ 103.72, 128.34, 130.73, 132.22, 136.37, 137.46, 140.62, 145.02, 149.27, and 152.61 ppm corresponding to a different type of carbon atoms present in the compound. The most downfield signal appeared at δ 177.76 and 169.17 ppm attributed to the CO and CC groups, respectively.
2.2. Biological activity
| Cpd no | In vitro cytotoxicity activity represented by IC50 (μM)b ± SD/(SI)c | ||||
|---|---|---|---|---|---|
| Cancer Cells | Normal cell | ||||
| HepG2 | HCT-116 | MCF-7 | MDA-MB-231 | WI-38 | |
| a (—) = not tested.b Averaging three independent results is used.c Selectivity index = (IC50 of WI-38)/(IC50 of cancer cell line). | |||||
| 7 | 48.74 ± 2.8 | 38.13 ± 2.2 | 31.04 ± 2.1 | — | — |
| 10a | 89.72 ± 4.5 | 93.76 ± 4.7 | 79.42 ± 4.1 | — | — |
| 10b | 80.30 ± 4.1 | 87.41 ± 4.2 | 74.49 ± 3.9 | — | — |
| 10c | 17.01 ± 1.3 (3.82) | 9.26 ± 0.8 (7.01) | 6.13 ± 0.4 (10.61) | 7.66 ± 0.5 (8.48) | 65.06 ± 3.5 |
| 10d | 32.12 ± 2.2 | 18.38 ± 1.4 | 11.29 ± 0.9 | — | — |
| 10e | 41.90 ± 2.5 | 22.02 ± 1.7 | 24.30 ± 1.8 | — | — |
| 10f | 26.77 ± 1.9 (1.76) | 12.51 ± 1.0 (3.69) | 8.78 ± 0.6 (5.27) | 10.46 ± 0.8 (4.41) | 46.18 ± 2.4 |
| 12 | 8.25 ± 0.6 (9.89) | 7.95 ± 0.5 (10.26) | 3.82 ± 0.2 (21.37) | 2.26 ± 0.1 (36.12) | 81.64 ± 4.1 |
| 14 | 68.97 ± 3.9 | 71.18 ± 3.8 | 70.11 ± 3.7 | — | — |
| 16 | >100 | >100 | 85.65 ± 4.3 | — | — |
| 18 | 83.38 ± 4.3 | >100 | 62.84 ± 3.4 | — | — |
| DOX | 4.50 ± 0.2 | 5.23 ± 0.3 | 4.17 ± 0.2 | 3.18 ± 0.1 | — |
Firstly, modification of 1,3-dithiolo[4,5-b]quinoxaline derivative with acyclic acetonitrile moiety displayed moderate activity, where the presence of cyan acetonitrile moiety as compound 10a causes a decrease in the activity over all cell lines (IC50 ranging between 79.42 ± 4.1–93.76 ± 4.7 μM) and replacing one cyano group by ethyl ester group as compound 10b does not enhance the activity (IC50 ranging from 74.49 ± 3.9 to 87.41 ± 4.2 μM). At the same time, introducing 2-cyanoacetohydrazone with an electron-withdrawing group (CF3) at the phenyl group to 1,3-dithiolo[4,5-b]quinoxaline pharmacophore, as represented in compound 10c, revealed antiproliferative activity with IC50 values ranging from 6.13 ± 0.4 to 17.01 ± 1.3 μM relative to doxorubicin (4.17 ± 0.2–5.23 ± 0.3 μM) against the tested cell lines and this activity might be related to the presence of N-(3-trifloromethylphenyl) group. Moreover, combining the 1,3-dithiolo[4,5-b]quinoxaline derivative with 2-cyanoacetohydrazones with different fragments in hydrazone as compounds 10d and 10e exhibited good to moderate activity with IC50 ranging from 11.29 ± 0.9 to 41.90 ± 2.5 μM. In addition, the 2-cyanoacetohydrazone derivative 10d that involved 4-methoxybenzylidene core revealed antiproliferative activity with IC50 values ranging from 11.29 ± 0.9 to 32.12 ± 2.2 μM better than 2-oxoindolin-3-ylidene derivative 10e (IC50 range from 22.02 ± 1.7 to 41.90 ± 2.5 μM). Meanwhile, compound 10f demonstrated good antiproliferative activity against (HepG2 = 26.77 ± 1.9 μM and HCT-116 = 12.51 ± 1.0 μM). Besides, compound 10f showed promising activity against MCF-7 with an IC50 value of 8.78 ± 0.6 μM relative to doxorubicin (IC50 = 4.17 ± 0.2 μM).
For the second series that appears in Scheme 4, incorporating the 2-oxo-cyclohexan-2-ylidene to 1,3-dithiolo[4,5-b]quinoxaline derivative as compound 12 was revealed to be the most potent candidate with IC50 values 8.25 ± 0.6, 7.95 ± 0.5, and 3.82 ± 0.2 μM compared with doxorubicin 4.50 ± 0.2, 5.23 ± 0.3, and 4.17 ± 0.2 μM against HepG2, HCT-116, and MCF-7, respectively. Moreover, replacing the cyclohexan-2-ylidene in compound 12 with heterocyclic cores as 1H-pyrazol-4-ylidene (compounds 14 and 16) and pyrimidin-5-ylidene derivative (compound 18) dramatically reduced the overall activity of the cell lines (IC50 = >68 μM), indicating that the antireflective activity preferred lipophilic group (CH2) than hydrophilic atoms and groups (N and NH) in pyrazole and pyrimidine.
Moreover, the results found that the most active derivatives 10c, 10f, and 12 exhibited better cytotoxicity activity with IC50 values lower than the remaining compounds, especially against the MCF-7 (IC50 ≤ 8.78 μM). Moreover, our work was extended to determine the most active three derivatives 10c, 10f, and 12, against other breast cancer cells (MDA-MB-231) to confirm sensitivity against breast cancer. As described in Table 1, the results on these derivatives showed significant IC50 values ranging from 2.26 ± 0.1 to 10.46 ± 0.8 μM.
Furthermore, compound 12 was the most potent derivative with an IC50 value of 2.26 ± 0.1 μM followed by compound 10c (IC50 = 7.66 ± 0.5 μM) and compound 10f (IC50 = 10.46 ± 0.8 μM) relative to doxorubicin (IC50 = 3.18 ± 0.1 μM). Additionally, to determine the safety of these derivatives, the cytotoxic activity against non-tumorigenic normal cell line (WI-38) was investigated. The result displayed that these three derivatives 10c, 10f, and 12 showed low cellular cytotoxicity with IC50 values of 65.06 ± 3.5, 46.18 ± 2.4, and 81.64 ± 4.1 μM, respectively. Surprisingly, the selectivity index revealed that the highest value was observed by compound 12 against MDA-MB-231 with a value of 36.12, followed by MCF-7 (SI = 21.37) and confirmed selectivity and sensitivity to breast cancer.
Finally, for the screening study, it can be concluded that among the tested compounds, three derivatives 10c, 10f, and 12 showed promising activity against the tested cell line with safe cytotoxicity against WI-38. Moreover, compound 12 revealed the most active derivative and displayed sensitivity towards the breast cell lines MCF-7 and MDA-MB-231 with IC50 3.82 ± 0.2 μM (1.09-fold increase) and 2.26 ± 0.1 μM (1.41-fold increase), as compared with doxorubicin (IC50 = 4.17 ± 0.2 and 3.18 ± 0.1 M), respectively.
2.2.2.1 Cell cycle analysis and apoptosis induction by annexin-V assay. Antiproliferative agents often work by arresting the cell cycle at specific points, causing apoptosis.79 The effect of the most potent 1,3-dithiolo[4,5-b]quinoxaline derivative 12 on the MCF-7 cell line at its IC50 value (3.82 μM) was studied, and a significant change was observed in the phases of the cell cycle, as described in Fig. 2. The target compound causes cell cycle arrest and inhibits the growth of MCF-7 cells in the S phase with values of 48.16% compared with untreated control at 29.79%. Meanwhile, a simultaneous decrease in the percentage of cells in the G0-G1 and G2/M phases with values of 48.16% and 7.32% compared with untreated breast cancer cells at 57.66 and 12.55%, respectively.
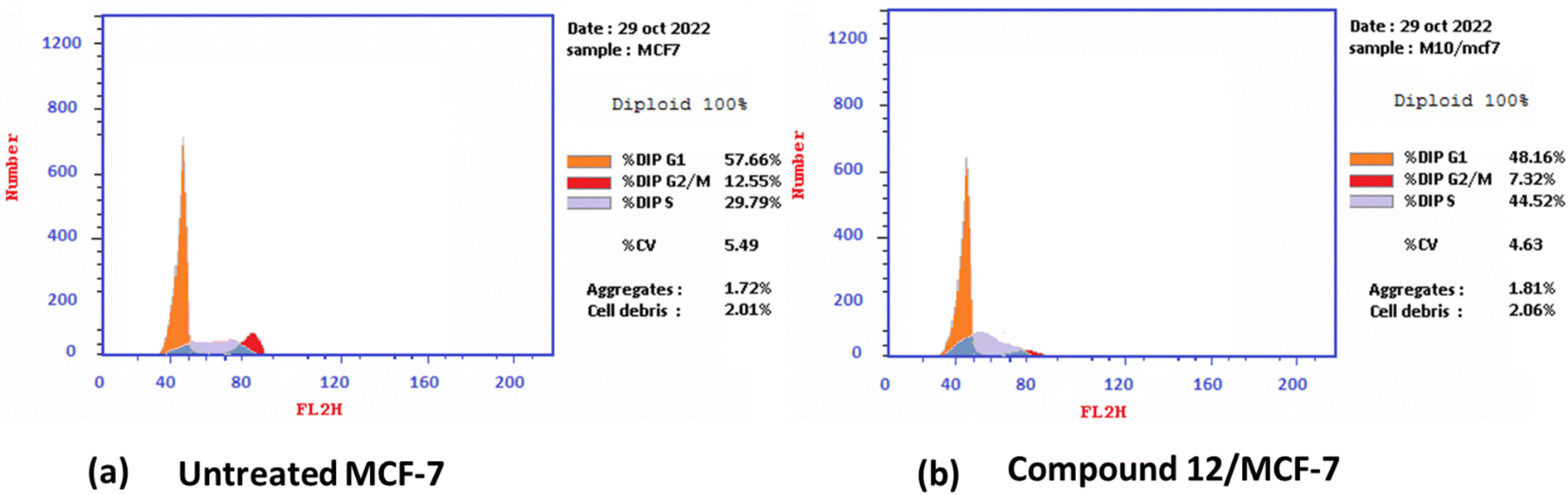 | ||
| Fig. 2 Cell cycle distribution% assessment using FACS analysis (a) untreated cell; (b) compound 12 treated with MCF-7 at (IC50 = 3.82 μM). | ||
Furthermore, the ability of the most active derivative to induce apoptosis was confirmed by applying double staining of annexin-V/propidium iodide (PI) to stain DNA and, therefore, stated the dead cells. The most active derivative 1,3-dithiolo[4,5-b]quinoxaline derivative 12 exhibited a significantly higher apoptotic effect in MCF-7 with a value of 42.08% compared to that of control cell at 1.84%. Additionally, compound 12 displayed a remarkable increase in DNA content in early apoptosis with a value of 24.02 (54.59-folds) and late apoptosis with a value of 11.9 (79.33-folds) compared to the control, as described in Table 2 and Fig. 3.
| Cpd no. | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 12/MCF-7 | 42.08 | 24.02 | 11.9 | 6.16 |
| Cont.MCF-7 | 1.84 | 0.44 | 0.15 | 1.25 |
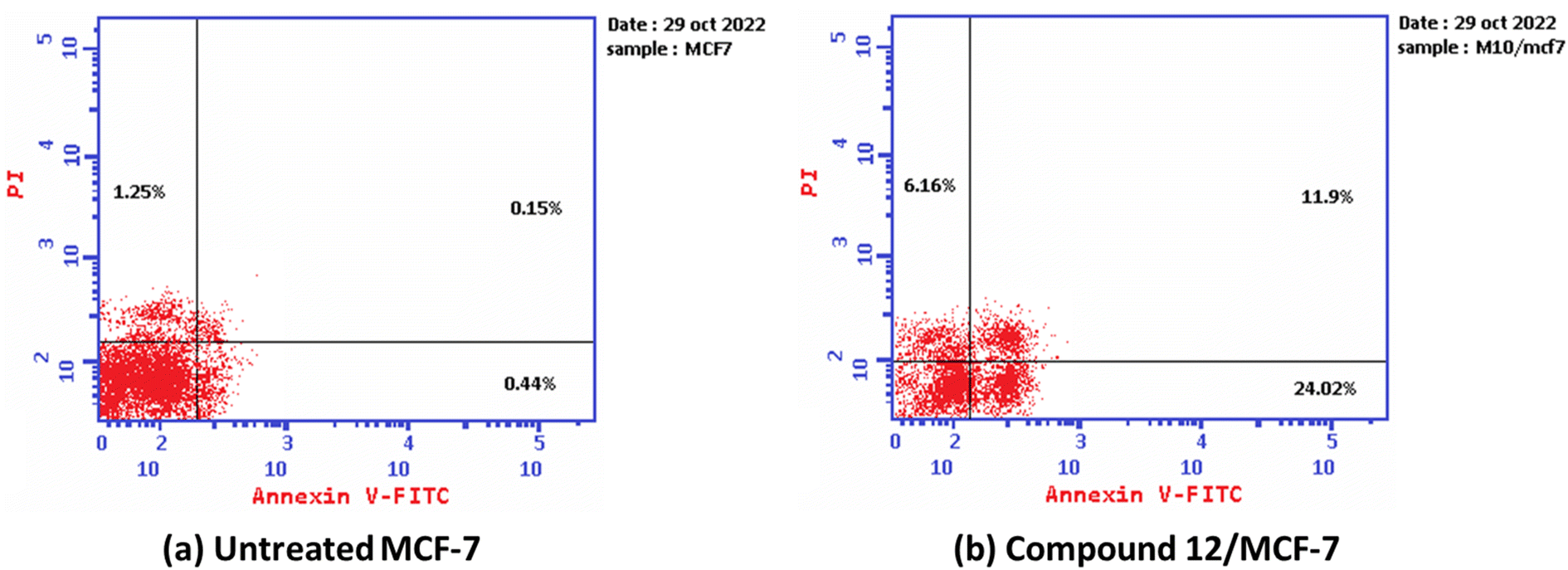 | ||
| Fig. 3 The graph shows the apoptosis-inducing effects of the most active derivative 12 in MCF-7 when treated at its IC50 for 24 h (a) untreated cell; (b) compound 10/MFC-7. | ||
2.2.2.2 Effect of the most active derivative compound 12 on mitochondrial apoptosis pathway proteins (BAX, Bcl-2, and p53). 2-(cyclohexan-2-ylidene)-[1,3]dithiolo[4,5-b]quinoxaline derivative 12, which displayed the best cytotoxic activity on MCF-7 and MDA-MB-231, was further evaluated to some apoptosis marker, such as Bax and P53 (pro-apoptotic) and Bcl-2 (anti-apoptotic) using the RT-PCR technique. Additionally, it is known that Bcl-2 family members, particularly the pro-apoptotic Bax and anti-apoptotic Bcl-2 genes, play a crucial role in controlling apoptosis.80,81
As described in Table 3, the 1,3-dithiolo[4,5-b]quinoxaline derivative 12 exhibited down-regulation to Bcl-2 protein with 0368 folds compared to untreated MCF-7 cells. At the same time, it revealed activation to pro-apoptotic genes Bax and P53 with 3.97 and 4.97 folds, respectively. Finally, MCF-7 cells' growth rate was significantly reduced by these changes in expression profiles when treated with compound 12.
| Tested cpd | RT-PCR results fold change | ||
|---|---|---|---|
| pro-apoptotic | anti-apoptotic | ||
| Bax | p53 | Bcl-2 | |
| 12/MCF7 | 3.792 | 4.976 | 0.368 |
| Cont.MCF7 | 1 | 1 | 1 |
2.2.2.3 Study the activity of compound 12 on receptor tyrosine kinase proteins (EGFR and VEGFR). To prove the mechanism of the antiproliferative activity, the in vitro inhibitory activity against tyrosine kinase proteins: epidermal growth factor EGFR (wide and mutant), and vascular epidermal growth factor receptor VEGFR2 were evaluated for the most active derivative 12 using ELISA analysis. The results of inhibitory activity for the target compound 12 and positive controls against receptor tyrosine kinase proteins expressed by IC50 (μM) and the inhibitory percentage at 10 μM are summarized in Table 4. All the results of the target compound 12 against tested proteins displayed values with sub-micromolar (<0.5 μM).
| Cpd no | Enzyme inhibitory activity IC50a (μM) | |||||
|---|---|---|---|---|---|---|
| EGFRWt | Inhibitory%b | EGFR(L858R) | Inhibitory%b | VEGFR2 | Inhibitory%b | |
| a The data is an average of three independent tests.b Inhibitory% at 10 μM. | ||||||
| 12 | 0.19 ± 0.009 | 85.41 | 0.121 ± 0.007 | 87 | 0.42 ± 0.021 | 78.92 |
| Doxorubicin | 0.349 ± 0.016 | 77.59 | — | — | — | — |
| Erlotinib | 0.037 ± 0.002 | 92.48 | 0.026 ± 0.001 | 93 | — | — |
| Sorafenib | — | — | — | — | 0.035 ± 0.002 | 91.68 |
Firstly, 1,3-dithiolo[4,5-b]quinoxaline derivative 12 exhibited the highest potential inhibitory activity against EGFRWt with IC50 value 0.19 ± 0.009 μM and inhibitory% = 85.41, compared to doxorubicin (IC50 = 0.349 ± 0.016 μM and inhibitory% = 77.59) but still slightly higher than Erlotinib (IC50 = 0.037 ± 0.002 μM and inhibitory% = 92.48). Moreover, compound 12 showed a decrease in the suppression effect with an inhibitory percentage value of 87 and the IC50 value of 0.121 ± 0.007 μM against mutant EGFR(L858R) related to erlotinib (IC50 = 0.026 ± 0.001 μM and inhibitory% = 93). At the same time, compound 12 effectively inhibited VEGFR2 activity with the IC50 value of 0.42 ± 0.021 μM and inhibitory% = 78.92, compared with sorafenib (IC50 = 0.035 ± 0.002 μM and inhibitory% = 91.68) Fig. 4.
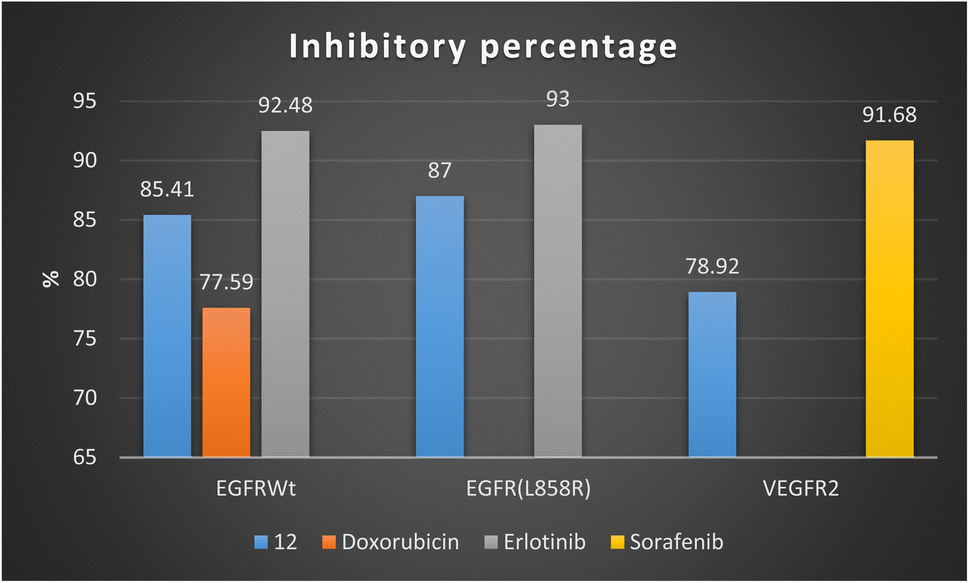 | ||
| Fig. 4 Inhibitory percentage of 1,3-dithiolo[4,5-b]quinoxaline derivative 12 and positive controls (doxorubicin, erlotinib, and sorafenib) against EGFR and VEGFR-2 enzymes. | ||
Finally, the sub-micromolar level of inhibition revealed by the assessment results might indicate that the 1,3-dithiolo[4,5-b]quinoxaline derivative 12 may benefit EGFR and VEGFR-2 inhibitory activities with higher inhibitory potency to EGFR-wide and mutant type than VEGFR-2.
2.3. In silico ADME and toxicity predictions
| Test items | Most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12 and positive controls | ||||
|---|---|---|---|---|---|
| 12 | Dox.a | Erl.a | Sor.a | ||
| a DOX. = doxorubicin, Erl. = erlotinib, and Sor. = sorafenib. | |||||
| SwissADME | Molecular properties | ||||
| MlogP |
1.75 | −2.10 | 1.89 | 2.91 | |
| TPSA (Å2) | 139.21 | 206.07 | 74.73 | 92.35 | |
| M. Wt | 433.57 | 543.52 | 393.44 | 464.82 | |
| nHBA (NO) | 6 | 12 | 6 | 7 | |
| nHBD (OHNH) | 0 | 6 | 1 | 3 | |
| NRB | 2 | 5 | 10 | 9 | |
|
|||||
| Pharmacokinetics | |||||
| GI absorption | Low | Low | High | Low | |
| BBB permeant | No | No | Yes | No | |
| P-gp substrate | Yes | Yes | No | No | |
| Skin permeation (logKp) cm s−1 |
−6.69 | −8.71 | −6.35 | −6.25 | |
|
|||||
| Drug likeness and medicinal chemistry | |||||
| logS (ESOL) |
−4.66 | −3.91 | −4.11 | −5.11 | |
| Solubility | Mod. Soluble | Soluble | Mod. soluble | Mod. soluble | |
| PAINS | 0 | 1 (quinone) | 0 | 0 | |
| Synthetic accessibility | 3.75 | 5.81 | 3.19 | 2.87 | |
| Bioavailability score | 0.55 | 0.17 | 0.55 | 0.55 | |
| Lipinski rule (violation) | Yes (0) | No (3) | Yes (0) | Yes (0) | |
| Veber rule (violation) | Yes (0) | No (1) | Yes (0) | Yes (0) | |
The result represented that 1,3-dithiolo[4,5-b]quinoxaline derivative 12, erlotinib, and sorafenib obey both the Lipinski rule of five and Veber rule without any violations, except the doxorubicin that unfollowed the Lipinski rule due to three violations including M. wt over 500 Dalton, hydrogen bond acceptors = 12 (two higher than standard), and hydrogen bond donor = 6 (one more than standard). Additionally, doxorubicin does not follow the Veber rule because the topological polar surface area (TPSA) is higher than 140. Moreover, all the tested compounds exhibited soluble to moderately soluble behaviors with logS (ESOL) values ranging from −5.31 to −3.91. Additionally, the most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12, erlotinib, sorafenib demonstrated no pan-assay interference compounds (PAINs) alarms in their structure, while doxorubicin showed only one PAINs alarm due to the quinone ring.
Furthermore, the tested derivative and positive controls revealed acceptable bioavailability scores with values of 0.17 for doxorubicin and 0.55 for the rest of the derivatives. Additionally, compound 12 showed easy synthetic accessibility = 3.75, which is very close to that of erlotinib (3.19) and sorafenib (2.87), as well as less than that of doxorubicin (5.81). Moreover, for pharmacokinetic prediction, all tested derivatives displayed a low gastrointestinal (GI) tract and did not pass the BBB, except erlotinib, which showed high GI and passed the BBB. Also, compound 12 and doxorubicin were revealed to be a substrate for P-gp, which does not cause problems with drug excretion. In contrast, erlotinib and sorafenib are not substrates for P-gp. Based on these data, compound 12 appears to be a promising drug candidate for further research and development.
Fig. 5 shows the calculation of the radar charts obtained from the SissADME web tool to predict the accessibility of the tested derivatives to be oral bioavailability. These charts involve six parameters as insaturation (INSATU), polarity (POLAR), insolubility (INSOLU), flexibility (FLEX), lipophilicity (LIPO), and size, and the tested compound is represented by a red line integrated into the pink area. Molecules that fall within the pink region of the radar are considered drug-like. Compound 12 exhibited six of the six rules. At the same time, doxorubicin demonstrated a violation in polarity that related to a number of hydrogen bond donors and acceptors, as well as TPSA > 140, which showed a slight violation to flexibility and that related to the number of rotatable bonds. Moreover, sorafenib demonstrated INSAT violation, which refers to the ratio of hybridized sp3 atoms to the total number of C atoms.
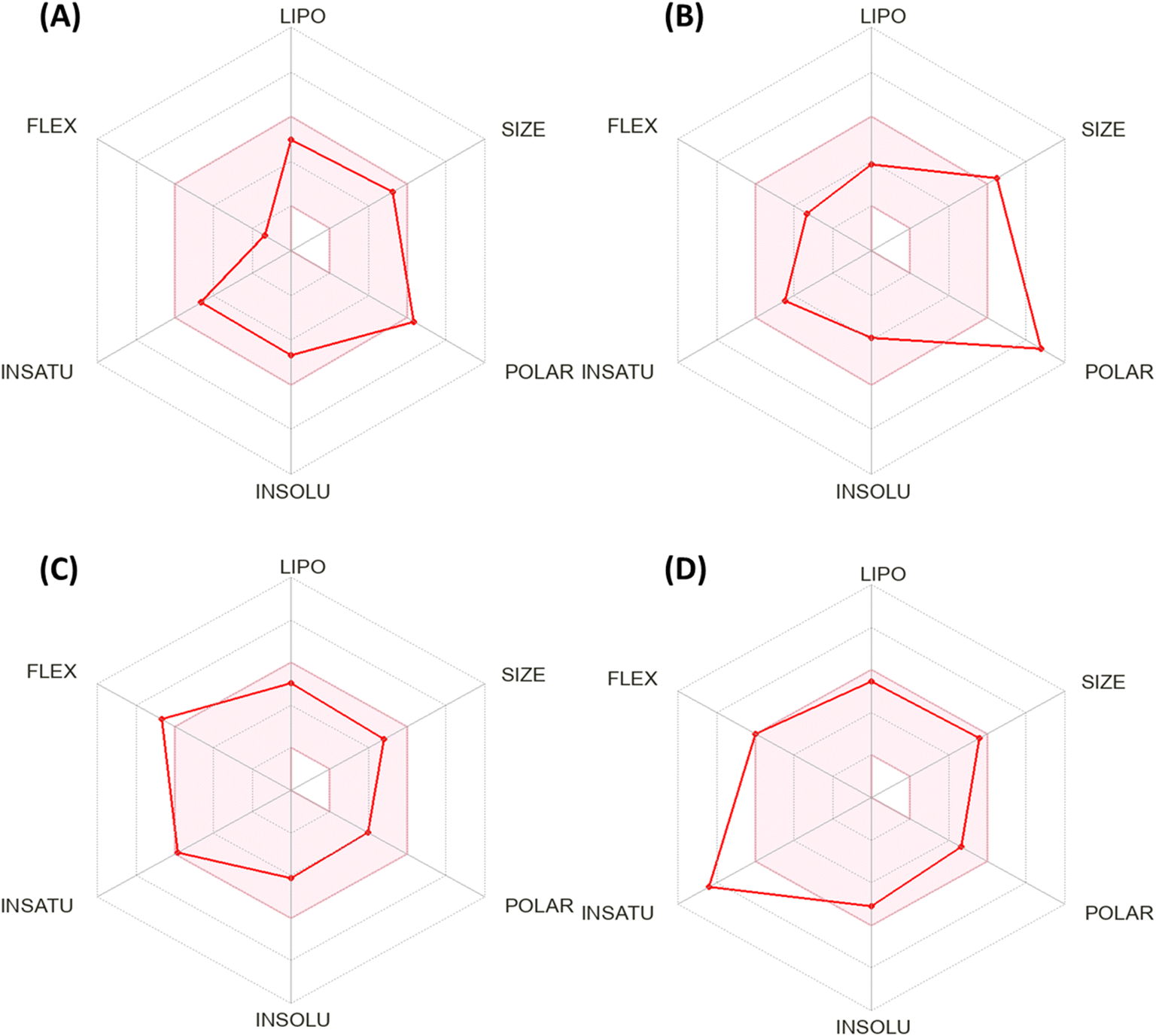 | ||
| Fig. 5 Bioavailability radar chart generated by Swiss-ADME for (A) the most active compound 12, (B) doxorubicin, (C) erlotinib, and (D) sorafenib. | ||
On the other hand, the positive controls showed activity to immunotoxin, mutagenic, and cytotoxicity, except sorafenib, which displayed inactive to mutagenicity. Additionally, all the tested derivatives were inactive to heat shock factor response element (HSE) with probability values ranging between 0.91 and 0.96 and inactive to mitochondrial membrane potential (MMP) with a probability value ranging between 0.56–0.78, except sorafenib, which displayed activity with a probability of 0.79. In addition, compound 12 and erlotinib depicted inactive tumor suppressor phosphoprotein (p53) with probabilities of 0.88 and 0.89, respectively, and showed activity for doxorubicin and sorafenib (Table 6).
| Oral toxicity prediction | Most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12 and positive controls | ||||
|---|---|---|---|---|---|
| 12 | Dox.a | Erl.a | Sor.a | ||
| Oral toxicity prediction | |||||
| a DOX. = doxorubicin, Erl. = erlotinib, and Sor. = sorafenib. | |||||
| ProTox-II prediction | LD50 mg kg−1 | 420 | 205 | 125 | 800 |
| Toxicity class | IV | III | III | IV | |
| Hepatotoxicity | Inactive | Inactive | Active | Active | |
| 0.59 | 0.86 | 0.78 | 0.82 | ||
| Carcinogenicity | Inactive | Inactive | Inactive | Inactive | |
| 0.67 | 0.90 | 0.51 | 0.50 | ||
| Immunotoxicity | Inactive | Active | Active | Active | |
| 0.99 | 0.99 | 0.91 | 0.92 | ||
| Mutagenicity | Inactive | Active | Active | Inactive | |
| 0.68 | 0.98 | 0.55 | 0.79 | ||
| Cytotoxicity | Inactive | Active | Active | Active | |
| 0.65 | 0.94 | 0.75 | 0.77 | ||
| Heat shock factor response element (HSE) | Inactive | Inactive | Inactive | Inactive | |
| 0.91 | 0.98 | 0.96 | 0.96 | ||
| Mitochondrial Membrane Potential (MMP) | Inactive | Inactive | Inactive | Active | |
| 0.78 | 0.56 | 0.68 | 0.79 | ||
| Phosphoprotein | Inactive | Active | Inactive | Active | |
| (Tumor suppressor) p53 | 0.88 | 0.52 | 0.89 | 0.57 | |
| pkCSM prediction | AMES toxicity | No | Yes | No | No |
| Skin sensitisation | No | No | No | No | |
| hERG I inhibitor | No | No | No | No | |
| hERG II inhibitor | No | No | Yes | Yes | |
| Max. tolerated dose (human) | −0.042 | 0.654 | 0.654 | 0.677 | |
| (logmg kg−1 per day) |
|||||
| Oral rat chronic toxicity (LOAEL) | 0.973 | 3.296 | 1.404 | 1.054 | |
| (logmg kg−1_bw per day) |
|||||
| Oral rat acute toxicity (LD50) (mol kg−1) | 3.344 | 3.978 | 2.757 | 2.595 | |
For the pkCSM prediction, all tested derivatives featured non-AMES except doxorubicin, non-skin sensitivity (was not able to elicit an allergic response), and non-inhibitors for hERG I and II (except erlotinib and sorafenib that displayed inhibitor to hERG II). Moreover, the most promising compound 12 expressed the lowest Max. tolerated dose (human) (−0.042logmg kg−1 per day) and oral rat chronic toxicity (LOAEL = 0.973 log mg kg−1_bw per day). Besides, compound 12 showed oral rat acute toxicity (LD50 = 3.344 mol kg−1) higher than erlotinib (LD50 = 2.757 mol kg−1) and sorafenib (LD50 = 2.595 mol kg−1) and lower than doxorubicin (LD50 = 3.978 mol kg−1).
Finally, based on the previous toxicity profile, it can be concluded that the most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12 exhibited a non-toxic profile on organ toxicity and toxicity endpoints with good LD50 value.
2.4. Molecular docking simulations
To determine the suitable anticancer mechanism activity and to explain the experimental result obtained previously, the molecular docking simulation for the most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12 was performed inside the active sites of Bcl-2 (PDB: 4AQ3), EGFR (PDB: 1M17), and VEGFR-2 (PDB: 4ASD). All these proteins were downloaded from the protein data bank (https://www.rcsb.org/access 14/1/2023).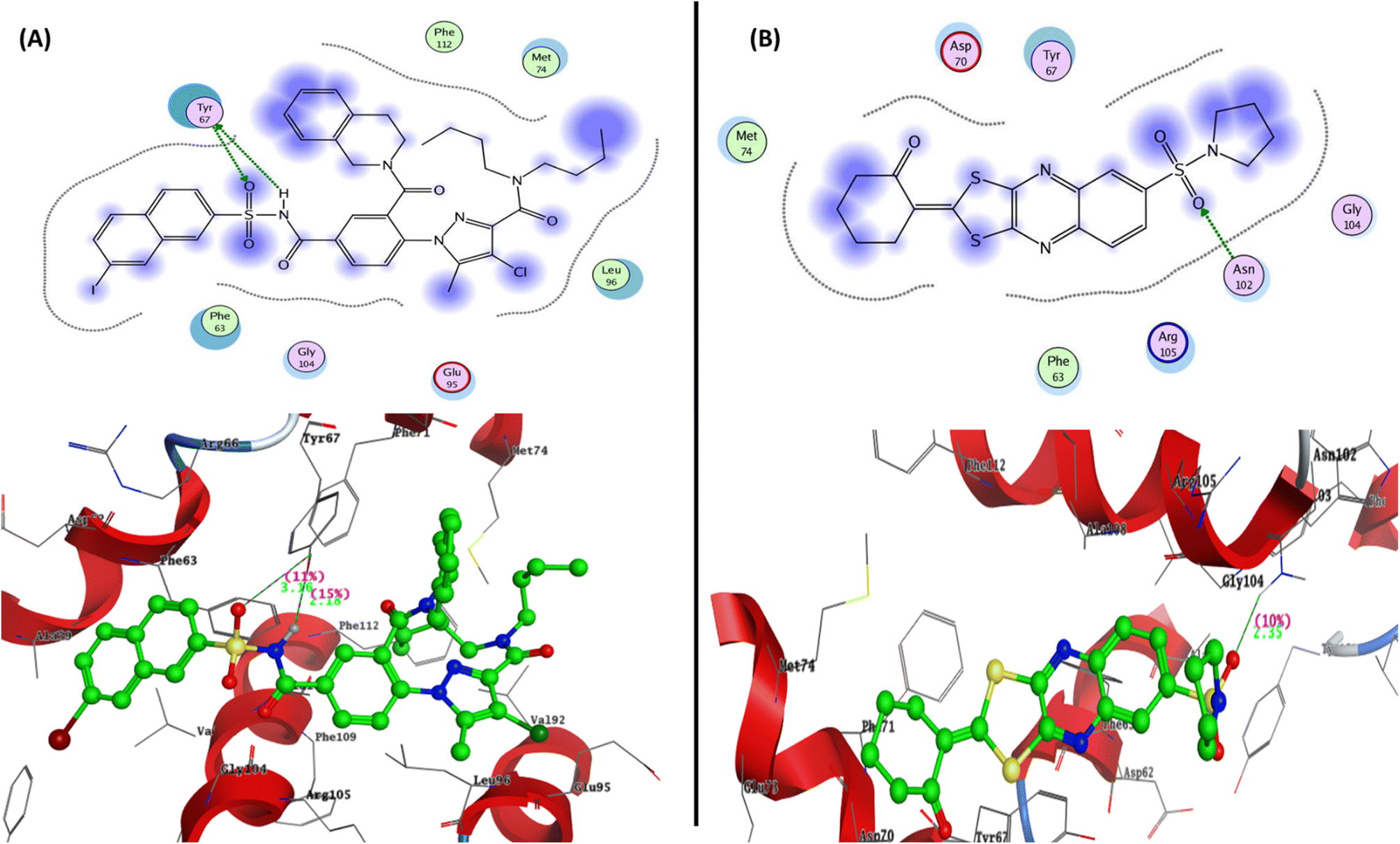 | ||
| Fig. 6 Represented the 2D and 3D binding modes of (A) co-crystallized ligand and (B) compound 12 inside Bcl-2 binding pocket (PDB: 4AQ3). | ||
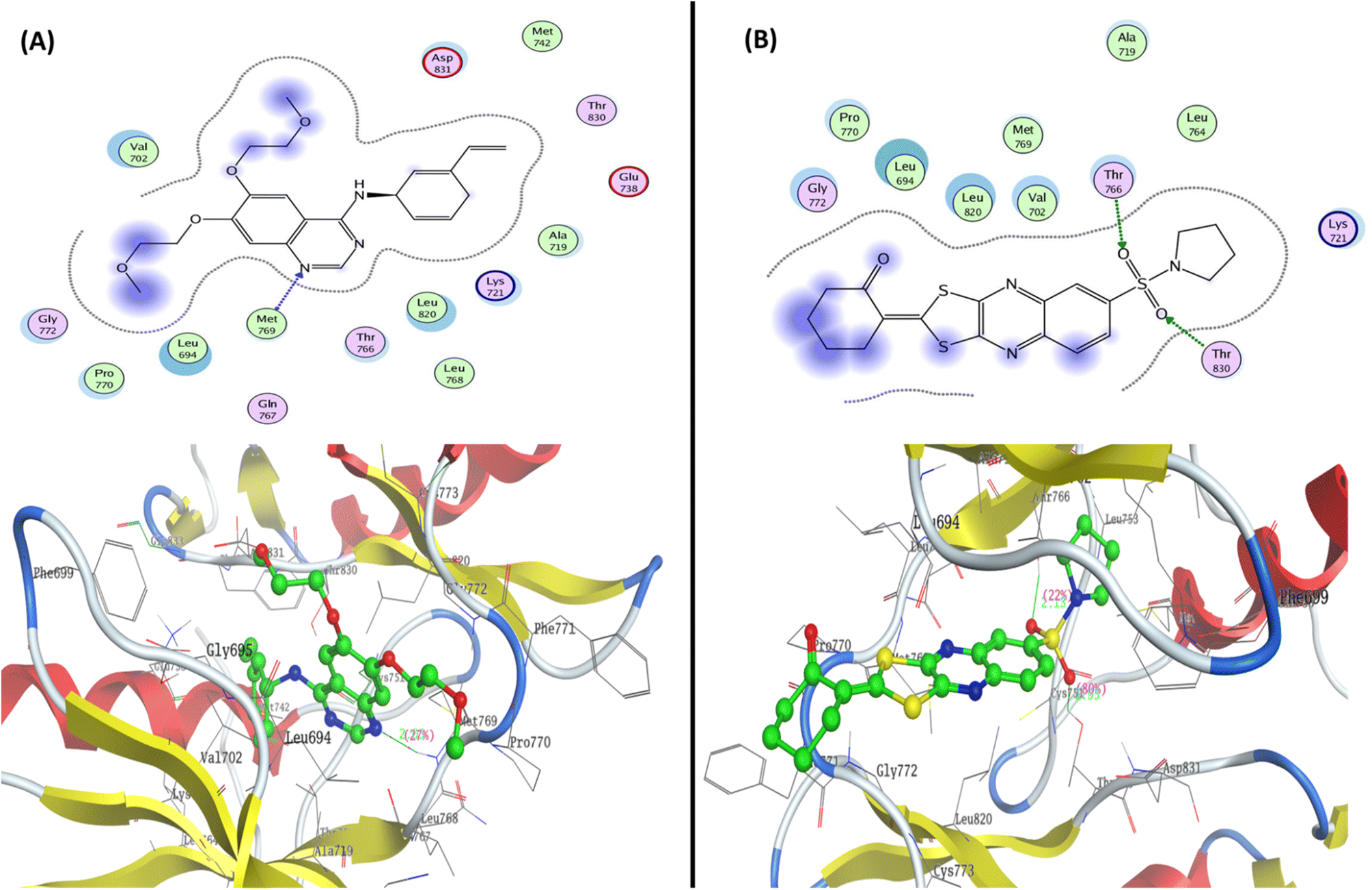 | ||
| Fig. 7 Represented the 2D and 3D binding modes of (A) co-crystallized ligand and (B) compound 12 inside the EGFR binding pocket (PDB: 1M17). | ||
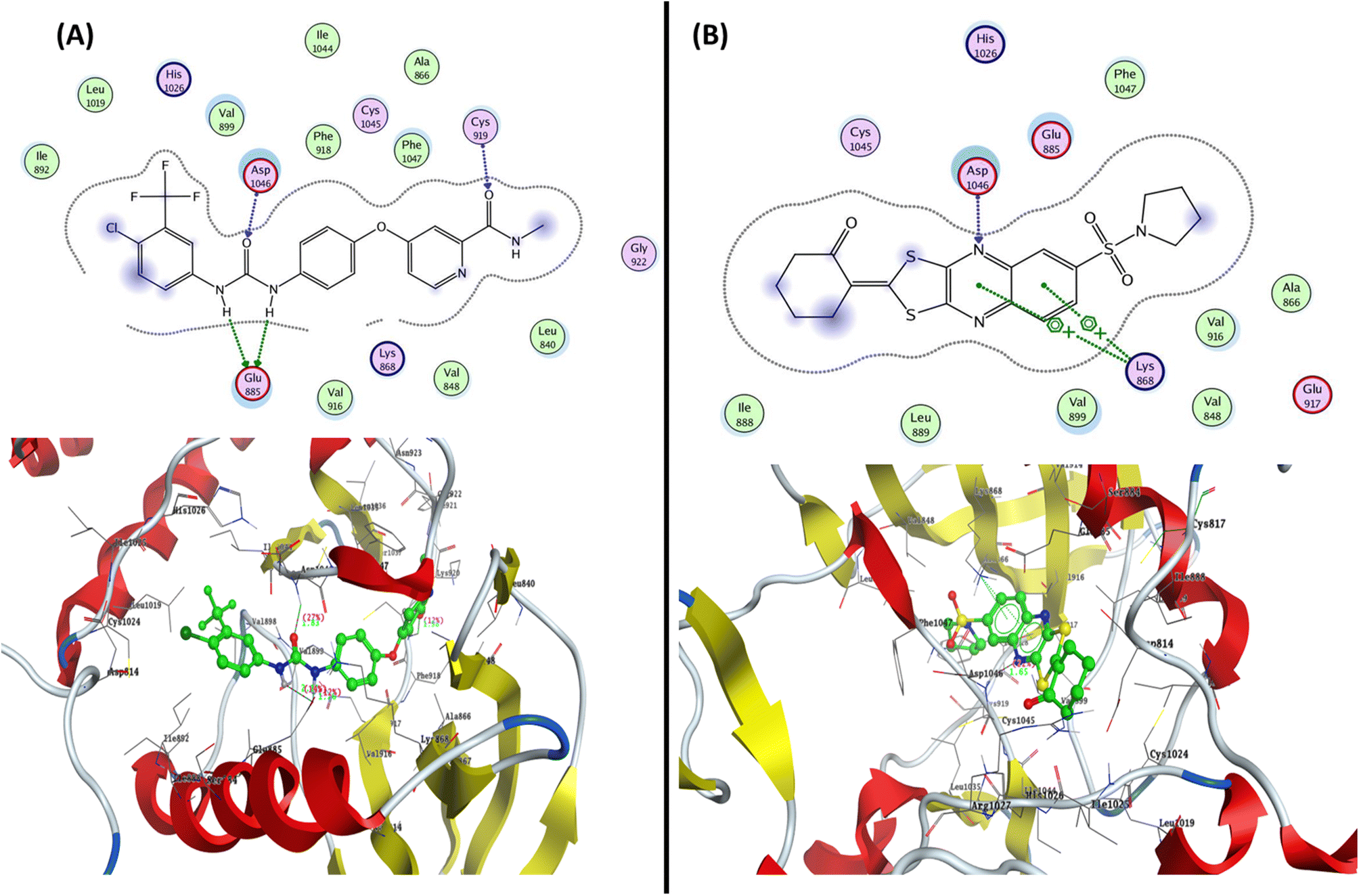 | ||
| Fig. 8 Represented the 2D and 3D binding modes of (A) co-crystallized ligand and (B) compound 12 inside the VEGFR-2 binding pocket (PDB: 4ASD). | ||
3. Conclusion
A series of 1,3-dithiolo[4,5-b]quinoxaline derivatives were designed and synthesized based on the hybridization approach between the 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline 7 and potassium salt of ethene-1,1-dithiolate derivatives. The designed derivatives contain 1,3-dithiolo[4,5-b]quinoxaline derivatives with different fragments and pharmacophores in position two of 2-ylidene-1,3-dithiolanes as the cyclic core and acyclic groups. All the newly designed derivatives were confirmed on the basis of elemental analysis and spectroscopic data. In vitro anticancer activity was determined by evaluating the half-maximal inhibitory concentration (IC50) of the synthesized derivative was measured and expressed in μM against HepG-2, HCT-116, and MCF-7 for all the synthesized derivatives. Generally, the tested derivatives exhibited activity in the breast cancer cell line (MCF-7) rather than the colon cell line (HCT-116) and liver cell line (HepG2), with three derivatives exhibiting low micromole against MCF-7 (IC50 ≤ 8.78 μM). Additionally, the most active derivatives 10c, 10f, and 12 were tested against other breast cancer cells (MDA-MB-231) and showed significant IC50 values ranging from 2.26 ± 0.1 to 10.46 ± 0.8 μM. Moreover, these derivatives showed high cellular cytotoxicity against WI-38 cell lines with IC50 values ranging from 46.18 ± 2.4 to 81.64 ± 4.1 μM. Surprisingly, the most active compound 12 exhibited down-regulation to Bcl-2 protein with 0368 folds and caused an activation to pro-apoptotic genes Bax and P53 with 3.97 and 4.97 folds, respectively, compared to untreated MCF-7 cells. Additionally, compound 12 revealed EGFRWt and EGFR(L858R) with inhibition percentages of 85.41% and 87% at 10 μM, as well as inhibited VEGFR-2 activity with the IC50 value of 0.42 ± 0.021 μM and inhibitory% = 78.92, compared with sorafenib (IC50 = 0.035 ± 0.002 μM and inhibitory% = 91.68). Further, compound 12 inhibited the growth of MCF-7 cells at its IC50 value in the S phase with values of 48.16% compared with the untreated control at 29.79%. Finally, some in silico ADMET prediction was performed for the most active compound 12, and the results exhibited a safety profile with drug-likeness properties. Additionally, the docking simulation inside the active site of Bcl-2, EGFR, and VEGFR-2 was performed and discussed.4. Experimental
4.1. Chemistry
4.1.2.1 Synthesis of 6-(pyrrolidin-1-ylsulfonyl)-1,4-dihydroquinoxaline-2,3-dione (5). A solution of sulfonyl chloride derivative, 3 (1 mmol) in 1,4-dioxane (25 mL), and pyrrolidine 4 (1.5 mmol) as a secondary amine was added dropwise for 15 min at room temperature. The solution mixture was stirred 15 min at room temperature for a further 5 h (monitored by TLC). After the new product was precipitated, the precipitate was collected by filtration and crystallized from EtOH to obtain the required product.
Pale-white powder (EtOH); 88% Yield; M.p. = 275–277 °C; IR (KBr): νmax = 3502, 3366 (2NH), 3046 (CHar), 2959, 2922 (CHalip), 1692 (br. 2CO), 1613 (CN), 1330, 1148 (SO2) cm−1; 1H NMR (δ, ppm) = 1.67 (4H, qu, 2CH2·pyrolidine), 3.25 (4H, t, 2N–CH2·pyrolidine), 8.24 (d, 1H, J = 8.0 Hz, H7·quinox), 8.28 (d, 1H, J = 8.0 Hz, H8·quinox), 8.41 (s, 1H, H5·quinox), 11.97 (2H, s, 2NH: D2O exchangeable); 13C NMR (δ, ppm) = 25.10 (2CH2·pyrolidine), 48.31 (2N–CH2·pyrolidine), 124.94, 126.16, 127.68, 130.03, 138.99, 139.53 (Ar.Cs), 167.44 (2CO); Anal. Calcd for C12H13N3O4S (295.31): C, 48.81; H, 4.44; N, 14.23; Found: C, 48.65; H, 4.12; N, 14.54.
4.1.2.2 Synthesis of 2,3-disubstituted-6-(pyrrolidin-1-ylsulfonyl)quinoxaline (6 and 7). DMF (2 mL) was added drop by drop to a solution of 6-(pyrrolidin-1-ylsulfonyl)-1,4-dihydroquinoxaline-2,3-dione 2 (4 mmol) and trichloro-phosphate (10 mmol), the solution was stirred at 80 °C for 2 h to precipitate the monochloro derivatives 6. After adding excess trichloro-phosphate (10 mmol) and stirring at 80 °C for 4 h, the previously formed precipitate disappeared and the solution becomes viscous (monitored by TLC). After the reaction was completed, the solution was added portion-wise to ice water and neutralized with an ammonia solution, 30%. The formed precipitate was collected by filtration and crystallized from CH3CN to obtain the dichloro derivatives 7.
4.1.2.3 3-Chloro-6-(pyrrolidin-1-ylsulfonyl)quinoxalin-2(1H)-one (6). Pale-brown powder (CH3CN); 81% Yield; M.p. = 240–242 °C; IR (KBr): νmax = 3401 (NH), 3063 (CHar), 2956, 2897 (CHalip), 1694 (CO), 1555(C
N), 1344, 1150 (SO2) cm−1; 1H NMR (δ, ppm) = 1.91 (4H, qu, 2CH2·pyrolidine), 3.75 (4H, t, 2N–CH2·pyrolidine), 7.94 (d, 1H, J = 9.7 Hz, H7·quinox), 8.08 (d, 1H, J = 9.2 Hz, H8·quinox), 8.11 (s, 1H, H5·quinox); 13C NMR (δ, ppm) = 25.29 (2CH2·pyrolidine), 50.19 (2N–CH2·pyrolidine), 125.00, 127.93, 130.33, 131.79, 135.98, 140.06 (Ar.Cs), 144.65 (NC–Cl), 164.59 (CO); Anal. Calcd. for C12H12ClN3O3S (313.76): C, 45.94; H, 3.86; N, 13.39; Found: C, 45.78; H, 3.92; N, 13.42.
4.1.2.4 2,3-Dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline (7). Grey powder (CH3CN); 85% Yield; M.p. = 190–192 °C; IR (KBr): νmax = 3051(CHar), 2970, 2875 (CHalip), 1615 (C
N), 1336, 1151 (SO2) cm−1; 1H NMR (δ,ppm) = 1.63 (4H, qu, 2CH2·pyrrolidine), 3.26 (4H, t, 2N–CH2·pyrolidine), 8.22 (d, 1H, J = 8.0 Hz, H7·quinox), 8.28 (d, 1H, J = 8.0 Hz, H8·quinox), 8.42 (s, 1H, H5·quinox); 13C NMR (δ, ppm) = 25.10 (2CH2·pyrrolidine), 48.31 (2N–CH2·pyrolidine), 125.99, 127.68, 130.03, 138.99, 139.53 (Ar.Cs), 141.78 (C–SO2), 147.09 (NC–Cl), 147.77 (NC–Cl); Anal. Calcd for C12H11ClN3O2S (332.20): C, 43.39; H, 3.34; N, 12.65; Found: C, 43.46; H, 3.78; N, 12.21.
4.1.2.5 A general method for the preparation of potassium ethene-1,1-dithiolates anion (9). A mixture of active methylene compound 8 (0.01 mmol), carbon disulfide (0.01 mmol), and potassium hydroxide (0.02 mmol) in absolute ethanol (15 mL) was stirred at room temperature for 30 min. The resulting solid product was collected by filtration, and washed with ether to obtain the desired disulphide anion (9).
4.1.2.6 Synthesis of substituted 2-ylidene-6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalines (10a–f). General method: In a 100 mL bottom conical flask, a solution of 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline (2 mmol) (7) in a little amount of dimethylformamide as a solvent, and substituted potassium ethene-1,1-bis(thiolates) 9 (2 mmol) were added. The reaction mixture was stirred at room temperature for 2–6 h (monitored by TLC). After the reaction was completed, the precipitate was filtered off and recrystallized from the proper solvent to produce the desired pure solid 10a–i. The spectroscopic data of all prepared compounds are listed below.
4.1.2.7 2-(6-(Pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)malononitrile (10a). Yellow powder (CH3CN); 86% Yield; M.p. = 230–232 °C; IR (KBr): νmax = 3072 (CHar), 2971, 2878 (CHalip), 2219, 2193 (2CN), 1621 (C
N), 1550 (CC), 1334, 1150 (SO2) cm−1; 1H NMR (δ, ppm) 1.65 (4H, qu, 2CH2·pyrolidine), 3.25 (4H, t, 2N–CH2·pyrrolidine), 8.24 (1H, dd, J = 6.4, 2.0 Hz, H7·quinox), 8.31 (1H, d, J = 8.8 Hz, H8·quinox), 8.43 (1H, s, H5·quinox); 13C NMR (δ, ppm) = 25.53 (2CH2·pyrolidine), 48.44 (2N–CH2·pyrolidine), 88.87 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –CN), 111.92 (2CN), 128.14, 128.90, 130.45, 138.80, 139.19, 141.27, 149.74, 150.42 (Carom), 173.46 (S–CC); MS: (Mwt: 401): m/z, 401 [M+, (16.41%)], 317.44 (100%); Anal. Calcd for C16H11N5O2S3 (401.48): C, 47.87; H, 2.76; N, 17.44; Found: C, 47.61; H, 2.55; N, 17.33.
–CN), 111.92 (2CN), 128.14, 128.90, 130.45, 138.80, 139.19, 141.27, 149.74, 150.42 (Carom), 173.46 (S–CC); MS: (Mwt: 401): m/z, 401 [M+, (16.41%)], 317.44 (100%); Anal. Calcd for C16H11N5O2S3 (401.48): C, 47.87; H, 2.76; N, 17.44; Found: C, 47.61; H, 2.55; N, 17.33.
4.1.2.8 Ethyl 2-cyano-2-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)acetate (10b). Light-yellow powder (CH3CN); 67% Yield; M.p. = 233–235 °C; IR (KBr): νmax = 3075 (CHar), 2977, 2875 (CHalip), 2211 (CN), 1691 (CO), 1605 (C
N), 1553 (CC), 1345, 1152 (SO2) cm−1; 1H NMR (δ, ppm) = 1.33 (3H, t, CH3·ester), 1.67 (4H, qu, 2CH2·pyrrolidine), 3.27 (4H, t, 2N–CH2·pyrolidine), 4.37 (2H, q, CH2·ester), 8.15 (1H, dd, J = 8.8, 2.4 Hz, H7·quinox), 8.29 (1H, d, J = 8.8 Hz, H8·quinox), 8.29 (1H, s, H5·quinox); 13C NMR (δ, ppm) = 14.02 (CH3·ester), 24.94 (2CH2·pyrolidine), 48.17 (2N–CH2·pyrolidine), 62.72 (CH2·ester), 92.39 (–CN), 114.45 (CN), 122.33, 125.06, 126.40, 136.68, 137.17, 138.16, 149.95, 151.26 (Carom), 165.86 (CO. ester), 167.57 (S–CC); Anal. Calcd for C18H16N4O4S3 (448.53): C, 48.20; H, 3.60; N, 12.49; Found: C, 48.10; H, 3.55; N, 12.30.
4.1.2.9 2-Cyano-2-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)-N-(3-(trifluoromethyl)phenyl)acetamide (10c). Deep-yellow powder (CH3CN); 67% Yield; M.p. = 161–163 °C; IR (KBr): νmax = 3342 (br. OH), 3046 (CHar), 2950, 2922 (CHalip), 2224 (CN), 1592 (C
O), 1511 (CC), 1329, 1150 (SO2) cm−1; 1H NMR (δ, ppm) = 1.67 (4H, qu, 2CH2·pyrolidine), 3.18 (4H, t, 2N–CH2·pyrrolidine), 7.17 (1H, d, J = 8.0, Harom), 7.69–7.71 (1H, m, Harom), 7.81 (1H, d, J = 9.6 Hz, Harom), 7.88 (dd, 1H, J = 8.7, 2.1 Hz, H7·quinox), 7.92 (1H, s, Harom), 8.07 (1H, d, J = 6.8 Hz, H8·quinox), 8.10 (1H, s, H5·quinox), 9.87 (1H, s, NH: exchangeable by D2O); 13C NMR (δ, ppm) = 24.80 (2CH2·pyrolidine), 48.33 (2N–CH2·pyrolidine), 88.27 (–CN), 115.01 (CN), 123.37, 123.50, 124.65 (C–F3), 125.57, 127.37, 129.66, 132.89, 133.40, 134.04, 136.58, 137.92, 139.30, 151.23, 151.42 (Carom), 163.44 (C–NH), 170.67 (S–CC); MS: m/z, 563 [M+, (23.45%)], 198.35 (100%); Anal. Calcd for C23H16F3N5O3S3 (563.59): C, 49.02; H, 2.86; N, 12.43; Found: C, 49.01; H, 2.76; N, 12.35.
4.1.2.10 2-Cyano-N′-(4-methoxybenzylidene)-2-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)acetohydrazide (10d). Yellow powder (CH3CN); 73% Yield; M.p. = 145–147 °C; IR (KBr): νmax = 3442, 3333 (br. OH), 3046 (CHar), 2950, 2881 (CHalip), 2198 (CN), 1603 (C
O), 1552 (CC), 1332, 1148 (SO2) cm−1; 1H NMR (δ, ppm) 1.66 (4H, qu, 2CH2·pyrolidine), 3.18 (4H, t, 2N–CH2·pyrrolidine), 3.69 (3H, s, CH3·methoxy), 7.60 (2H, d, J = 9.2H, Harom), 7.69–7.73 (2H, m, Harom), 7.90–7.93 (1H, m, H7·quinox), 8.07 (1H, d, J = 7.2 Hz, H8·quinox), 8.16 (1H, s, H5·quinox), 11.38 (1H, s, NH: exchangeable); 13C NMR (δ, ppm) = 24.60 (2CH2·pyrolidine), 47.84 (2N–CH2·pyrolidine), 54.63 (OCH3) 86.10 (–CN), 115.56 (CN), 122.90, 123.59, 124.51, 125.56, 127.14, 129.84, 130.25, 130.63, 134.21, 136.04, 139.24, 144.07, 151.24, 151.72 (Carom), 161.31 (C–NH), 165.53 (C–OMe), 170.90 (S–CC); Anal. Calcd for C25H20N5O4S3 (550.65): C, 54.53; H, 3.66; N, 12.72; Found: C, 54.43; H, 3.54; N, 12.47.
4.1.2.11 2-Cyano-N′-(2-oxoindolin-3-ylidene)-2-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)acetohydrazide (10e). Brown powder (CH3CN); 91% Yield; M.p. = 310–312 °C; IR (KBr): νmax = 3418 (2NH), 3046 (CHar), 2955 (CHalip), 2202 (CN), 1718 (CO), 1675 (CO), 1618 (C
N), 1545 (CC), 1331, 1149 (SO2) cm−1; 1H NMR (δ, ppm) 1.66 (4H, qu, 2CH2·pyrolidine), 3.16 (4H, t, 2N–CH2·pyrrolidine), 7.35 (1H, t, J = 8.4 Hz, Harom), 7.47 (1H, t, J = 6.8 Hz, Harom), 7.55–7.70 (2H, m, Harom), 7.72 (1H, d, J = 8.8 Hz, H7·quinox), 7.95 (1H, d, J = 10.8 Hz, H8·quinox), 8.50 (1H, s, H5·quinox), 11.24 (1H, s, NH: exchangeable with D2O), 12.42 (1H, s, NH: exchangeable with D2O); 13C NMR (δ, ppm) = 25.27 (2CH2·pyrolidine), 50.21 (2N–CH2·pyrolidine), 95.17 (–CN), 114.24 (CN), 120.04, 121.24, 124.96, 125.40, 127.15, 130.30, 131.77, 133.73, 135.89, 137.06, 141.58, 142.62, 144.58, 148.48, 148.79 (Carom), 160.61 (CO), 164.04 (CO), 168.66 (S–CC); Anal. Calcd for C24H17N7O4S3 (563.63): C, 51.14; H, 3.04; N, 17.40; Found: C, 51.01; H, 3.00; N, 17.12.
4.1.2.12 Ethyl 3-oxo-2-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)butanoate (10f). Brown powder (CH3CN); 77% Yield; M.p. = 217–219 °C; IR (KBr): νmax = 3046 (CHar), 2984, 2957 (CHalip), 1714 (br.CO), 1626 (C
N), 1576 (CC), 1328, 1149 (SO2) cm−1; 1H NMR (δ, ppm) = 1.43 (3H, t, CH3·ester), 1.65 (4H, qu, 2CH2·pyrolidine), 2.49 (3H, s, CH3·acetyl), 3.20 (4H, t, 2N–CH2·pyrolidine), 4.51 (2H, q, CH2·ester), 7.85 (1H, dd, J = 4.7, 1.6 Hz, H7·quinox), 7.87 (1H, d, J = 8.4 Hz, H8·quinox), 8.02 (1H, s, H5·quinox); 13C NMR (δ, ppm) = 14.07 (CH3·ester), 24.28 (2CH2·pyrolidine), 28.77 (CH3·acetyl), 48.11 (2N–CH2·pyrolidine), 63.15 (CH2·ester), 109.60, 124.52, 125.61, 127.35, 132.89, 136.11, 139.37, 150.64, 150.98 (Carom), 165.18 (CO.ester), 170.00 (S–CC), 187.32 (CO.acetyl); MS: m/z, 465 [M+, (16.99%)], 246.20 (100%); Anal. Calcd for C19H19N3O5S3 (465.56): C, 49.02; H, 4.11; N, 9.03; Found: C, 49.01; H, 4.10; N, 9.01.
4.1.2.13 Synthesis of novel [1,3]dithiolo[4,5-b]quinoxaline linked to cyclohexyl, pyrazolyl, and pyrimidinyl moieties (12, 14, 16, and 18). In a 100 mL bottom conical flask, a solution of the starting material 2,3-dichloro-6-(pyrrolidin-1-ylsulfonyl)quinoxaline (7) (2 mmol) in a little amount of DMF as a solvent, and substituted potassium ethene-1,1-bis(thiolates) (2 mmol) specific namely, potassium (2-oxocyclohexylidene)methanebis(thiolate) (11), potassium (3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)methanebis(thiolate) (13), potassium (3-methyl-5-oxo-1-phenyl-1,5-dihydro-4H-pyrazol-4-ylidene)methanebis(thiolate) (15), and potassium (2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methanebis(thiolate) (17) were added. The reaction mixture was stirred at room temperature for 3–7 h (monitored by TLC). After the reaction was completed, the precipitate was filtered off and recrystallized from the proper solvent to produce the desired pure solid. The spectroscopic data of all prepared compounds are listed below.
4.1.2.14 2-(6-(Pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)cyclohexan-1-one (12). Deep-yellow (CH3CN); 80% yield; M.p. 176–178 °C; IR (KBr): νmax = 3073 (CHar), 2972, 2923, 2880 (CHalip), 1654 (CO), 1602 (C
N), 1545 (CC), 1330, 1151 (SO2) cm−1; 1H NMR (δ, ppm) = 1.43 (2H, t, CH2·hex), 1.62–1.65 (6H, m, 2CH2·pyrrolidine + CH2·hex), 2.61 (2H, s, CH2·hex), 3.16–3.20 (6H, m, 2N–CH2·pyrrolidine + CH2·hex), 7.87 (1H, d, J = 8.0 Hz, H7·quinox), 7.92 (1H, d, J = 10.4, H8·quinox), 8.02 (1H, s, H5·quinox); 13C NMR (δ, ppm) = 18.63 (CH2·hex), 22.85 (CH2·hex), 24.92 (2CH2·pyrolidine), 27.83 (CH2·hex), 37.66 (CH2·hex), 47.93 (2N–CH2·pyrolidine), 117.40 (–CO), 123.78, 124.44, 125.57, 127.32, 130.12, 134.18, 136.09, 139.24, 151.20, 151.53 (Carom), 163.37 (S–CC), 178.00 (CO); MS: m/z, 433 [M+, (30.54%)], 73.30 (100%); Anal. Calcd for C19H19N3O3S3 (433.56): C, 52.64; H, 4.42; N, 9.69; Found: C, 52.44; H, 4.27; N, 9.23.
4.1.2.15 5-Methyl-4-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)-2,4-dihydro-3H-pyrazol-3-one (14). Light-yellow (CH3CN); 68% Yield; M.p. = 225–227 °C; IR (KBr): νmax = 3117 (NH), 3064 (CHar), 2956, 2925 (CHalip), 1670 (CO), 1525 (C
C), 1340, 1150 (SO2) cm−1. 1H NMR (δ, ppm) = 1.69 (4H, qu, 2CH2·pyrolidine), 2.39 (3H, s, CH3·pyrazole), 3.27 (4H, t, 2N–CH2·pyrrolidine), 8.19 (1H, dd, J = 11.6, 3.6, H7·quinox), 8.27 (1H, d, J = 8.8 Hz, H8·quinox), 8.30 (1H, s, H5·quinox), 11.58 (1H, s, NH: exchangeable with D2O); 13C NMR (δ, ppm) = 14.54 (CH3·pyrazole), 25.74 (2CH2·pyrolidine), 50.61 (2N–CH2·pyrolidine), 103.72 (–CO), 128.34, 130.73, 132.22, 136.37, 137.46, 140.62, 145.02, 149.27, 152.61 (Carom), 169.17 (S–CC), 177.76 (CO.pyrazole); Anal. Calcd for C17H15N5O3S3 (433.52): C, 47.10; H, 3.49; N, 16.16; Found: C, 47.01; H, 3.38; N, 16.05.
4.1.2.16 5-Methyl-2-phenyl-4-(6-(pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)-2,4-dihydro-3H-pyrazol-3-one (16). Orange powder (CH3CN); 73% Yield; M.p. = 240–242 °C; IR (KBr): νmax = 3062, 3045 (CHar), 2980, 2948 (CHalip), 1705 (CO), 1641 (C
N), 1593 (CC), 1320, 1150 (SO2) cm−1; 1H NMR (δ, ppm) 1.68 (4H, qu, 2CH2·pyrolidine), 2.44 (3H, s, CH3, pyrazole), 3.25 (4H, t, 2N–CH2·pyrrolidine), 7.20–7.27 (1H, m, Harom), 7.47 (2H, t, J = 7.2 Hz, Harom), 7.92 (1H, d, J = 8.0, H7·quinox), 8.12 (2H, d, J = 6.8, Harom), 8.18 (1H, d, J = 8.4, H8·quinox), 8.30 (1H, s, H5·quinox); 13C NMR (δ, ppm) = 15.19 (CH3·pyrazole), 26.48 (2CH2·pyrolidine), 50.63 (2N–CH2·pyrrolidine), 103.05 (–CO), 124.24, 124.44, 125.65, 126.60, 126.82, 128.05, 128.76, 131.10, 132.30, 136.58, 139.88, 140.57, 147.58, 149.36, 149.81 (Carom), 166.17 (S–CC), 175.51 (CO.pyrazole); MS: m/z, 509 [M+, (22.35%)], 229.15 (100%); Anal. Calcd for C23H19N5O3S3 (509.62): C, 54.21; H, 3.76; N, 13.74; Found: C, 54.08; H, 3.55; N, 13.69.
4.1.2.17 5-(6-(Pyrrolidin-1-ylsulfonyl)-[1,3]dithiolo[4,5-b]quinoxalin-2-ylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (18). Pale-green powder (CH3CN); 80% Yield; M.p. = 238–240 °C; IR (KBr): νmax = 3502, 3115 (2NH), 3046 (CHar), 2985, 2917 (CHalip), 1640 (br. 3CO), 1615 (C
N), 1488 (CC), 1370, 1150 (SO2) cm−1; 1H NMR (δ, ppm) = 1.65 (4H, s, 2CH2·pyrolidine), 3.25 (4H, s, 2N–CH2·pyrrolidine), 8.25 (1H, d, J = 9.6, H7·quinox), 8.30 (1H, d, J = 8.8, H8·quinox), 8.42 (1H, s, H5·quinox), 11.12 (1H, s, 2NH: exchangeable with D2O); 13C NMR (δ, ppm) = 25.37 (2CH2·pyrolidine), 48.30 (2N–CH2·pyrolidine), 105.37 (–CO), 127.79, 128.91, 130.30, 136.03, 139.44, 140.00, 142.15, 147.37, 147.81, 152.13 (Carom), 157.97 (CO-pyrimidine), 160.41 (2CO-pyrimidine), 168.13 (S–CC); Anal. Calcd for C17H13N5O5S3 (463.50): C, 44.05; H, 2.83; N, 15.11; Found: C, 44.01; H, 2.66; N, 15.04.
4.2. Biological activities
The in vitro cytotoxicity screening of the newly designed 6-(pyrrolidin-1-ylsulfonyl)quinoxaline derivatives 7, 10, 12, 14, 16, and 18 was performed against three human cancer cell lines (Hep G2, HCT-116, and MCF-7) using MTT assay through incubation period 24 h, as described previously.37,78 Moreover, the most active derivatives 10c, 10f, and 12 were further evaluated against MDA-MB-231 and non-tumorigenic normal cell line (WI-38) using MTT assay. The cell lines were obtained from ATCC via the Holding company for biological products and vaccines (VACSERA), Cairo, Egypt (the principle of MTT assay and standard protocol are shown in the ESI,† including all steps). Moreover, the in vitro flow cytometry cell cycle analysis, and apoptosis annexin V-FITC Apoptosis Detection BioVision Kit were performed at the VACSERA Tissue Culture Unit, Cairo, Egypt, as described previously.33 The effect of most active derivative 12 on the gene expression of Bcl-2, Bax, and P53 was determined using qRT-PCR using the Bio-Rad Laboratories iScript TM One-Step RT-PCR Kit with SYBR® Green according to manufacturer's instructions and as described previously.88 Additionally, the in vitro EGFR wt, EGFR L858R, and VEGFR for the most active derivative 12 were performed using the BPS-Bioscience EGFR Kinase Assay Kit Catalog #40321, EGFR(L858R) Kinase Assay Kit Catalog #40324, and VEGFR-2(KDR) Kinase Assay Kit Catalog #40325, respectively, according to the manufacturer's instructions.4.3. Molecular docking simulations
The molecular docking studies for the most active 1,3-dithiolo[4,5-b]quinoxaline derivative 12 inside the active sites of the Bcl-2 (PDB: 4AQ3), EGFR (PDB: 1M17), and VEGFR-2 (PDB: 4ASD) were performed using Molecular Operating Environmental (MOE)89–92 version 2009.10. All these proteins were downloaded from the protein data bank (https://www.rcsb.org/access 14/1/2023). The structure of compound 12 was constructed in 2D using ChemBioDraw. 2014 and exported to MOE. Additionally, the structure of compound 12 was protonated and then the structure was minimized using forcefield MMFF94x, as described previously.93–96 For Bcl-2 (PDB: 4AQ3), the validation process was carried out by selecting only one chain and deleting all other chains. Moreover, the redocking process was performed, and the co-crystallized ligand (sulfonamide molecule) exhibited binding energy S = −23.39 kcal mol−1 with RMSD = 1.49 Å, where the alpha triangle placement and London dG as rescoring functions were selected. For EGFR (PDB: 1M17), the validation process reported that the erlotinib (co-crystallized ligand) showed binding energy S = −17.84 kcal mol−1 with RMSD = 1.73 Å, where the triangle matcher placement and London dG as rescoring functions were selected. Moreover, For VEGFR-2 (PDB: 4ASD), the redocking process of co-crystallized ligand (sorafenib) showed binding energy S = −16.169 kcal mol−1 with RMSD = 1.484 Å through four hydrogen bonds, where the triangle matcher placement and London dG as rescoring functions were selected.Abbreviations
| BCSCs | Breast cancer stem cells |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
| FAD | Food and Drug Administration |
| EGFR | Epidermal growth factor receptor |
| TKI | Tyrosine kinase inhibitor |
| NSCLC | Non-small-cell lung cancer |
| IC50 | Half-maximal inhibitory concentration |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| Bcl-2 | B-cell lymphoma 2 |
| BAX gene | Bcl-2 associated X-protein |
| p53 | Tumor protein P53 or cellular tumor antigen p53 |
| RT-PCR | Real-time reverse transcription–polymerase chain reaction |
| ADME | Absorption, distribution, metabolism, and excretion |
| TPSA | Topological polar surface area |
| PAINS | Pan-assay interference compounds |
| BBB | Blood–brain barrier |
| GI | Gastrointestinal |
| P-gp | Permeability glycoprotein |
| HSE | Heat shock factor response element |
| MMP | Mitochondrial membrane potential |
| PDB | Protein Data Bank |
Data availability
All data that support the finding of this study are available in the ESI† data of this article.Conflicts of interest
The authors declare no conflicts of interest.References
- Y. Sun, Y. Liu, X. Ma and H. Hu, Int. J. Mol. Sci., 2021, 22, 6923 CrossRef CAS PubMed.
- M. M. Rahman, M. R. Islam, S. Akash, M. Harun-Or-Rashid, T. K. Ray, M. S. Rahaman, M. Islam, F. Anika, M. K. Hosain, F. I. Aovi, H. A. Hemeg, A. Rauf and P. Wilairatana, Biomed. Pharmacother., 2022, 153, 113305 CrossRef CAS PubMed.
- M. M. Rahman, M. R. Islam, S. Akash, S. Shohag, L. Ahmed, F. A. Supti, A. Rauf, A. S. M. Aljohani, W. Al Abdulmonem, A. A. Khalil, R. Sharma and M. Thiruvengadam, Chem.-Biol. Interact., 2022, 368, 110198 CrossRef CAS PubMed.
- G. L. Wong, S. G. Manore, D. L. Doheny and H.-W. Lo, Semin. Cancer Biol., 2022, 86, 84–106 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs, A. Jemal and C. A. Cancer, J. Clin., 2022, 72, 7–33 Search PubMed.
- Q. Wu, J. Li, S. Zhu, J. Wu, C. Chen, Q. Liu, W. Wei, Y. Zhang and S. Sun, Oncotarget, 2017, 8, 27990–27996 CrossRef PubMed.
- M. T. Chen, H. F. Sun, Y. Zhao, W. Y. Fu, L. P. Yang, S. P. Gao, L. D. Li, H. L. Jiang and W. Jin, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3983–3988 CrossRef CAS PubMed.
- E. Batlle and H. Clevers, Nat. Med., 2017, 23, 1124–1134 CrossRef CAS PubMed.
- A. F. Karamysheva, Biochem, 2008, 73, 751–762 CAS.
- X. Yuan, Q. Yang, T. Liu, K. Li, Y. Liu, C. Zhu, Z. Zhang, L. Li, C. Zhang, M. Xie, J. Lin, J. Zhang and Y. Jin, Eur. J. Med. Chem., 2019, 179, 147–165 CrossRef CAS PubMed.
- L. Hlatky, C. Tsionou, P. Hahnfeldt and C. N. Coleman, Cancer Res., 1994, 54, 6083–6086 CAS.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Bioorg. Chem., 2020, 104, 104350 CrossRef CAS PubMed.
- Y. Huang, X. Chen, M. M. Dikov, S. V Novitskiy, C. A. Mosse, L. Yang and D. P. Carbone, Blood, 2007, 110, 624–631 CrossRef CAS PubMed.
- C. Fontanella, E. Ongaro, S. Bolzonello, M. Guardascione, G. Fasola and G. Aprile, Ann. Transl. Med., 2014, 2(12), 123–133 Search PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, Trends Pharmacol. Sci., 2015, 36, 422–439 CrossRef CAS PubMed.
- J. Y. Hsu and H. A. Wakelee, BioDrugs, 2009, 23, 289–304 CrossRef CAS PubMed.
- F. Zuccotto, E. Ardini, E. Casale and M. Angiolini, J. Med. Chem., 2010, 53, 2681–2694 CrossRef CAS PubMed.
- J. Blanc, R. Geney and C. Menet, Anti-Cancer Agents Med. Chem., 2013, 13, 731–747 CrossRef CAS PubMed.
- Y. Liu and N. S. Gray, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS PubMed.
- A. M. Srour, D. H. Dawood, E. S. Nossier, R. A. El-Shiekh, A. E. Mahmoud, A. G. Hussien, M. M. Omran and M. M. Ali, J. Mol. Struct., 2023, 1271, 134130 CrossRef CAS.
- A. R. Y. A. Ammar, A. M. Sh El-Sharief, A. Belal, S. Y. Abbas, Y. A. Mohamed and A. B. M. Mehany, Eur. J. Med. Chem., 2018, 156, 918–932 CrossRef PubMed.
- L. Wang, X. Ding, K. Wang, R. Sun, M. Li, F. Wang and Y. Xu, J. Mol. Struct., 2023, 1274, 134499 CrossRef CAS.
- J. G. Paez, P. A. Jänne, J. C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F. J. Kaye, N. Lindeman, T. J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M. J. Eck, W. R. Sellers, B. E. Johnson and M. Meyerson, Science, 2004, 304, 1497–1500 CrossRef CAS PubMed.
- R. Montanari, D. Capelli, K. Yamamoto, H. Awaishima, K. Nishikata, A. Barendregt, A. J. R. Heck, F. Loiodice, F. Altieri, A. Paiardini, A. Grottesi, L. Pirone, E. Pedone, F. Peiretti, J. M. Brunel, T. Itoh and G. Pochetti, J. Med. Chem., 2020, 63, 4811–4823 CrossRef CAS PubMed.
- W. Zhou, D. Ercan, L. Chen, C.-H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob, R. Padera, J. R. Engen, K.-K. Wong, M. J. Eck, N. S. Gray and P. A. Jänne, Nature, 2009, 462, 1070–1074 CrossRef CAS PubMed.
- H. A. Yu, M. E. Arcila, N. Rekhtman, C. S. Sima, M. F. Zakowski, W. Pao, M. G. Kris, V. A. Miller, M. Ladanyi and G. J. Riely, Clin. Cancer Res., 2013, 19, 2240–2247 CrossRef CAS PubMed.
- S. S. Ramalingam, J. Vansteenkiste, D. Planchard, B. C. Cho, J. E. Gray, Y. Ohe, C. Zhou, T. Reungwetwattana, Y. Cheng, B. Chewaskulyong, R. Shah, M. Cobo, K. H. Lee, P. Cheema, M. Tiseo, T. John, M.-C. Lin, F. Imamura, T. Kurata, A. Todd, R. Hodge, M. Saggese, Y. Rukazenkov and J.-C. Soria, N. Engl. J. Med., 2020, 382, 41–50 CrossRef CAS PubMed.
- C.-C. Lan, P.-C. Hsieh, C.-Y. Huang, M.-C. Yang, W.-L. Su, C.-W. Wu and Y.-K. Wu, World J. Clin. Cases, 2022, 10, 6360–6369 CrossRef PubMed.
- R. T. Dungo and G. M. Keating, Drugs, 2013, 73, 1503–1515 CrossRef CAS PubMed.
- N. M. Jackson and B. P. Ceresa, Exp. Cell Res., 2017, 356, 93–103 CrossRef CAS.
- P. Shi, S. Zhang, L. Zhu, G. Qian, H. Ren, S. S. Ramalingam, M. Chen and S.-Y. Sun, Transl. Oncol., 2019, 12, 705–713 CrossRef PubMed.
- M. M. S. Wassel, Y. A. Ammar, G. A. M. Elhag Ali, A. Belal, A. B. M. Mehany and A. Ragab, Bioorg. Chem., 2021, 110, 104794 CrossRef CAS PubMed.
- A. Furlan, F. Colombo, A. Kover, N. Issaly, C. Tintori, L. Angeli, V. Leroux, S. Letard, M. Amat, Y. Asses, B. Maigret, P. Dubreuil, M. Botta, R. Dono, J. Bosch, O. Piccolo, D. Passarella and F. Maina, Eur. J. Med. Chem., 2012, 47, 239–254 CrossRef CAS PubMed.
- F. Colombo, C. Tintori, A. Furlan, S. Borrelli, M. S. Christodoulou, R. Dono, F. Maina, M. Botta, M. Amat, J. Bosch and D. Passarella, Bioorg. Med. Chem. Lett., 2012, 22, 4693–4696 CrossRef CAS.
- A. Ragab, D. M. Elsisi, O. A. Abu Ali, M. S. Abusaif, A. A. Askar, A. A. Farag and Y. A. Ammar, Arabian J. Chem., 2022, 15, 103497 CrossRef CAS.
- E. A. Fayed, Y. A. Ammar, M. A. Saleh, A. H. Bayoumi, A. Belal, A. B. M. Mehany and A. Ragab, J. Mol. Struct., 2021, 1236, 130317 CrossRef CAS.
- A. Burguete, E. Pontiki, D. Hadjipavlou-Litina, R. Villar, E. Vicente, B. Solano, S. Ancizu, S. Pérez-Silanes, I. Aldana and A. Monge, Bioorg. Med. Chem. Lett., 2007, 17, 6439–6443 CrossRef CAS PubMed.
- R. Ingle, R. Marathe, D. Magar, H. M. Patel and S. J. Surana, Eur. J. Med. Chem., 2013, 65, 168–186 CrossRef CAS.
- D. M. Elsisi, A. Ragab, A. A. Elhenawy, A. A. Farag, A. M. Ali and Y. A. Ammar, J. Mol. Struct., 2022, 1247, 131314 CrossRef CAS.
- X. Jiang, K. Wu, R. Bai, P. Zhang and Y. Zhang, Eur. J. Med. Chem., 2022, 229, 114085 CrossRef CAS PubMed.
- E. A. Fayed, Y. A. Ammar, A. Ragab, N. A. Gohar, A. B. M. Mehany and A. M. Farrag, Bioorg. Chem., 2020, 100, 103951 CrossRef CAS PubMed.
- A. Levitzki, Acc. Chem. Res., 2003, 36, 462–469 CrossRef CAS PubMed.
- Z. H. Chohan, A. U. Shaikh and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2006, 21, 733–740 CrossRef CAS PubMed.
- S. T. Bhaskaran and P. Mathew, J. Mol. Struct., 2022, 1251, 132071 CrossRef CAS.
- T. Rescigno, M. F. Tecce and A. Capasso, Open Biochem. J., 2018, 12, 46–64 CrossRef CAS PubMed.
- V. Kartsev, K. S. Shikhaliev, A. Geronikaki, S. M. Medvedeva, I. V Ledenyova, M. Y. Krysin, A. Petrou, A. Ciric, J. Glamoclija and M. Sokovic, Eur. J. Med. Chem., 2019, 175, 201–214 CrossRef CAS.
- J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS PubMed.
- A. Bazgir and A. M. Astaraki, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1916–1921 CrossRef CAS.
- A. A. Fadda, A. Fekri and N. M. Bayoumy, RSC Adv., 2015, 5, 80844–80852 RSC.
- A. A. Fadda, M. A. El Salam, E. H. Tawfik, E. M. Anwar and H. A. Etman, RSC Adv., 2017, 7, 39773–39785 RSC.
- S. A. Shaker and M. I. Marzouk, Molecules, 2016, 21, 155 CrossRef PubMed.
- X. C. Liu, D. Lai, Q. Z. Liu, L. Zhou, Q. Liu and Z. L. Liu, Molecules, 2016, 21, 1665 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. S. Kumar, A. I. Almansour, N. Arumugam, D. M. Q. Althomili, M. Altaf, A. Basiri, D. Kotresha, T. Sai Manohar and S. Venketesh, Bioorg. Chem., 2018, 77, 263–268 CrossRef CAS PubMed.
- B. Li, E. D. Jones, E. Zhou, L. Chen, D. C. Baylis, S. Yu, M. Wang, X. He, J. A. V. Coates, D. I. Rhodes, G. Pei, J. J. Deadman, X. Xie and D. Ma, Bioorg. Med. Chem. Lett., 2010, 20, 5334–5336 CrossRef CAS PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. Elhag Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- M. S. Jan, S. Ahmad, F. Hussain, A. Ahmad, F. Mahmood, U. Rashid, O.-R. Abid, F. Ullah, M. Ayaz and A. Sadiq, Eur. J. Med. Chem., 2020, 186, 111863 CrossRef CAS PubMed.
- P. Devi, A. Bishnoi and V. Singh, ChemistrySelect, 2019, 4, 629–632 CrossRef CAS.
- Y. Arun, K. Saranraj, C. Balachandran and P. T. Perumal, Eur. J. Med. Chem., 2014, 74, 50–64 CrossRef CAS PubMed.
- G. Li Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef CAS PubMed.
- C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405–5452 RSC.
- Z. Guo, Y. Xu, Y. Peng, H. ur Rashid, W. Quan, P. Xie, L. Wu, J. Jiang, L. Wang and X. Liu, Bioorg. Med. Chem. Lett., 2019, 29, 1133–1137 CrossRef CAS PubMed.
- T. Owa, H. Yoshino, T. Okauchi, K. Yoshimatsu, Y. Ozawa, N. H. Sugi, T. Nagasu, N. Koyanagi and K. Kitoh, J. Med. Chem., 1999, 42, 3789–3799 CrossRef CAS PubMed.
- C. T. Supuran, Expert Opin. Invest. Drugs, 2003, 12, 283–287 CrossRef CAS PubMed.
- B. Sloan and N. S. Scheinfeld, Curr. Opin. Invest. Drugs, 2008, 9, 1324–1335 CAS.
- J. A. Plumb, P. W. Finn, R. J. Williams, M. J. Bandara, M. R. Romero, C. J. Watkins, N. B. La Thangue and R. Brown, Mol. Cancer Ther., 2003, 2, 721–728 CAS.
- A. B. Suttle, K. F. Grossmann, D. Ouellet, L. E. Richards-Peterson, G. Aktan, M. S. Gordon, P. M. LoRusso, J. R. Infante, S. Sharma, K. Kendra, M. Patel, S. Pant, H.-T. Arkenau, M. R. Middleton, S. C. Blackman, J. Botbyl and S. W. Carson, J. Clin. Pharmacol., 2015, 55, 392–400 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Drug Dev. Res., 2022, 84, 3–24 CrossRef PubMed.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- Y. A. Ammar, G. A. M. Elhagali, M. S. Abusaif, M. R. Selim, M. A. Zahran, T. Naser, A. B. M. Mehany and E. A. Fayed, Med. Chem. Res., 2021, 30, 1649–1668 CrossRef CAS.
- M. S. Abusaif, M. Fathy, M. A. Abu-Saied, A. A. Elhenawy, A. B. Kashyout, M. R. Selim and Y. A. Ammar, J. Mol. Struct., 2021, 1225, 129297 CrossRef CAS.
- Y. A. Ammar, A. A. Farag, A. M. Ali, S. A. Hessein, A. A. Askar, E. A. Fayed, D. M. Elsisi and A. Ragab, Bioorg. Chem., 2020, 99, 103841 CrossRef CAS PubMed.
- Y. A. Ammar, A. A. Farag, A. M. Ali, A. Ragab, A. A. Askar, D. M. Elsisi and A. Belal, Bioorg. Chem., 2020, 104, 104164 CrossRef CAS PubMed.
- J. Lin, P. Wang, Z. Zhang, G. Xue, D. Zha, J. Wang, X. Xu and Z. Li, Synth. Commun., 2020, 50, 823–830 CrossRef CAS.
- J. M. Neri, L. N. Cavalcanti, R. M. Araújo and F. G. Menezes, Arabian J. Chem., 2020, 13, 721–739 CrossRef CAS.
- S. A. Ibrahim, A. Ragab and H. A. El-Ghamry, Appl. Organomet. Chem., 2022, 36, 1–17 Search PubMed.
- M. M. S. Wassel, A. Ragab, G. A. M. Elhag Ali, A. B. M. Mehany and Y. A. Ammar, J. Mol. Struct., 2021, 1223, 128966 CrossRef CAS.
- R. Z. Batran, S. M. El-Daly, W. A. El-Kashak and E. Y. Ahmed, Chem. Biol. Drug Des., 2022, 99, 470–482 CrossRef CAS PubMed.
- M. Eldeeb, E. F. Sanad, A. Ragab, Y. A. Ammar, K. Mahmoud, M. M. Ali and N. M. Hamdy, Biomedicines, 2022, 10, 722 CrossRef CAS PubMed.
- A. Ragab, Y. A. Ammar, A. Ezzat, A. M. Mahmoud, M. Basseem, I. Mohamed, A. S. El-tabl and R. S. Farag, Comput. Biol. Med., 2022, 145, 105473 CrossRef CAS PubMed.
- A. Y. Alzahrani, Y. A. Ammar, M. Abu-Elghait, M. A. Salem, M. A. Assiri, T. E. Ali and A. Ragab, Bioorg. Chem., 2022, 119, 105571 CrossRef CAS PubMed.
- R. Ayman, A. M. Radwan, A. M. Elmetwally, Y. A. Ammar and A. Ragab, Arch. Pharm., 2023, 356, e2200395 CrossRef PubMed.
- K. E. Saadon, N. M. H. Taha, N. A. Mahmoud, G. A. M. Elhagali and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 3899–3917 CrossRef CAS.
- A. Ragab, S. A. Fouad, Y. A. Ammar, D. S. Aboul-magd and M. S. Abusaif, Antibiotics, 2023, 12, 128 CrossRef CAS PubMed.
- E. A. Fayed, M. Mohsen, S. M. A. El-Gilil, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2022, 1262, 133028 CrossRef CAS.
- A. E. Azab, M. S. Alesawy, W. M. Eldehna, A. Elwan and I. H. Eissa, Arch. Pharm., 2022, 355, 2200133 CrossRef CAS PubMed.
- E. S. A. E. H. Khattab, A. Ragab, M. A. Abol-Ftouh and A. A. Elhenawy, J. Biomol. Struct. Dyn., 2021, 40, 13291–13309 CrossRef PubMed.
- S. A. El-Kalyoubi, A. Ragab, O. A. Abu Ali, Y. A. Ammar, M. G. Seadawy, A. Ahmed and E. A. Fayed, Pharmaceuticals, 2022, 15, 376 CrossRef CAS PubMed.
- H. F. Rizk, M. A. El-Borai, A. Ragab, S. A. Ibrahim and M. E. Sadek, Polycyclic Aromat. Compd., 2023, 43, 500–522 CrossRef CAS.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, RSC Adv., 2023, 13, 9281–9303 RSC.
- A. Ezzat, M. B. I. Mohamed, A. M. Mahmoud, R. S. Farag, A. S. El-Tabl and A. Ragab, J. Mol. Struct., 2022, 1251, 132004 CrossRef CAS.
- A. Y. Alzahrani, Y. A. Ammar, M. A. Salem, M. Abu-Elghait and A. Ragab, Arch. Pharm., 2022, 355, e2100266 CrossRef PubMed.
- H. Ali Mohamed, Y. A. Ammar, G. A. m. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, ACS Omega, 2022, 7, 4970–4990 CrossRef CAS PubMed.
- R. R. Raslan, Y. A. Ammar, S. A. Fouad, S. A. Hessein, N. A. M. Shmiess and A. Ragab, RSC Adv., 2023, 13, 10440–10458 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01635h |
| This journal is © The Royal Society of Chemistry 2023 |