
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective C-alkylation of aldimines, derived from chiral α-carbon heteroatom-substituted aldehydes, with triethylborane. Application to the synthesis of benzylisoquinolines†
David Fuentes-Ríos,
Carmen Muñoz,
Amelia Díaz*,
Francisco Sarabia and
J. Manuel López-Romero*
and
J. Manuel López-Romero*
Department of Organic Chemistry, Faculty of Sciences, University of Malaga, Campus de Teatinos s/n, 29071-Málaga, Spain. E-mail: jmromero@uma.es
First published on 17th March 2023
Abstract
The one-pot reaction of a chiral aldehyde, p-methoxyaniline or p-fluoroaniline, and triethylborane produces the corresponding alkylated chiral amine with high yields and diastereoisomeric ratios. Stereocontrol is induced by the presence of a heteroatom in the α-position to the aldehyde. In the case of alkylation of imines derived from chiral aliphatic amines, good yields and moderate to high diastereoselectivity are obtained: yields are significantly better when the preformed imine is used in the reaction with triethyl borane, and diastereoselectivity of the reactions largely depends on the structure of the chiral aliphatic amine. The methodology is successfully applied to the synthesis of romneine, a natural benzylisoquinoline.
1 Introduction
The alkylamine is a fundamental group in organic chemistry, representing a common subunit of a large variety of natural products and bioactive compounds. Particularly, chiral α-branched amines play a crucial role as biologically active materials and hence, their chemistry attracts great interest in the areas of bioorganic and medicinal chemistry, as well as in the synthesis of natural products.1 There are numerous methods for chiral amine synthesis, such as asymmetric reductive amination of carbonyl compounds, catalytic asymmetric reduction of ketimines, enamines or enamides, catalyzed direct reductive amination of alkyl aryl ketones, or the asymmetric addition reaction of organometallic reagents to imines. Most of the strategies use activated imines bearing electron-withdrawing groups on the nitrogen, for example, oxime ethers,2 hydrazones,3 and tosylimines.4 The 1,2-radical addition of alkyl groups to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond has also emerged as a valuable option for the preparation of a variety of both non-chiral and chiral amines.5,2c Moreover, reports have shown that the control of the stereochemistry is also feasible in these reactions, even when acyclic systems are involved.6 For example, catalytic anionic procedures have now improved the stereocontrol during reaction,7 and also carbanion methods are available for the preparation of chiral amines.8 On the other hand, the Petasis reaction provides an efficient, stereoselective and powerful access to α-(hetero)aryl-, vinyl-, alkynyl-, and allyl-functionalized amine derivatives.9
N bond has also emerged as a valuable option for the preparation of a variety of both non-chiral and chiral amines.5,2c Moreover, reports have shown that the control of the stereochemistry is also feasible in these reactions, even when acyclic systems are involved.6 For example, catalytic anionic procedures have now improved the stereocontrol during reaction,7 and also carbanion methods are available for the preparation of chiral amines.8 On the other hand, the Petasis reaction provides an efficient, stereoselective and powerful access to α-(hetero)aryl-, vinyl-, alkynyl-, and allyl-functionalized amine derivatives.9
Boron derivatives are commonly used as Lewis Acid catalysts in alkylation reactions. For example, Et3B or Et3B/RI have been investigated over activated chiral substrates such as oxime ethers,10 nitrones,11 hydrazones,12 or glyoxylate imines.13 However, low yields and/or the stereoinduction level attained in these reactions, are not always satisfactory. Furthermore, in case of radical mechanisms, intermolecular additions to CN bonds with acyclic stereocontrol are restricted to secondary and tertiary radicals, and stabilized alkyl radicals.
This paper focuses on the 1,2-addition of alkyl groups to the CN bond as a suitable methodology for the preparation of chiral alkylamines. One of the research interests of our group is the development of new synthetic strategies to isoquinoline alkaloids, and we have previously described a one-pot alkylative amination reaction involving three components, which are an aromatic aldehyde, an aniline derivative, and a trialkylborane as alkylating agent.14 In this reaction, the trialkylborane acts both as a Lewis acid catalyst in the formation of the imine and as a chain donor to the CN bond. The foremost advantage of this procedure is the possibility of adding a wide variety of alkyl groups (either primary or secondary) to the CN bond by using the properly selected trialkylborane. The broad applicability of this procedure was ascertained by extending the reaction to enolizable aldehydes and aliphatic amines. We have now addressed our efforts to study the one-pot alkylative amination using enantiomeric substrates, particularly those with heteroatom at the α-carbonyl position. We propose here the preparation of chiral alkylamines by a one-pot process employing a chiral aldehyde and p-methoxyaniline or p-fluoroaniline as imine precursors, and Et3B as imine alkylating catalyst. nBu3B was also tested. To the best of our knowledge, the one-pot procedure for the alkylation of non-having electron-withdrawing groups of chiral imines, employing a trialkylborane as alkylating agent, has not been described so far. Finally, we extend the methodology to the preparation of benzylisoquinoline alkaloids by using tris(3,4-dimethoxy benzyl)borane.
2 Results and discussion
We have focused our study on the reaction of a selection of cyclic aldehydes having a heteroatom in α position, as the non-chiral (±)-1 and the chiral 2–4, with p-methoxyaniline (5), to obtain the corresponding amines (6–9, Scheme 1). | ||
| Scheme 1 Synthesis of the amines 6–9 by three-component reaction. Data of the reactions in Tables 1 and 2. | ||
The reaction is made with the purpose of assessing the effect that the heteroatom located at the vicinal position to the carbonyl exerts on the efficiency and stereoselectivity of the reaction. Reaction was carried out by using Et3B as catalyst and dichloromethane as solvent (see Experimental).
The addition to (±)-tetrahydrofuran-2-carboxaldehyde (1) was firstly examined, paying particular attention to the effect of temperature on the reaction stereoselectivity. As seen in Table 1, a high level of stereoinduction with formation of a 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 mixture of the two diastereomeric pairs (R,R/S,S and R,S/S,R) of ethylated amine (±)-6 (Table 1, entry 1) was obtained at 20 °C. The stereoselectivity was slightly favoured at 0 °C, although a longer period was needed to reach a complete conversion at this temperature (Table 1, entry 2). At lower temperatures the diastereomeric ratio did not increase further and conversion remained far from completion, even after 3–5 h of reaction (Table 1, entries 3 and 4).
:13 mixture of the two diastereomeric pairs (R,R/S,S and R,S/S,R) of ethylated amine (±)-6 (Table 1, entry 1) was obtained at 20 °C. The stereoselectivity was slightly favoured at 0 °C, although a longer period was needed to reach a complete conversion at this temperature (Table 1, entry 2). At lower temperatures the diastereomeric ratio did not increase further and conversion remained far from completion, even after 3–5 h of reaction (Table 1, entries 3 and 4).
| Entry | T (°C) | t (h) | Yieldb (%) | drb (%) |
|---|---|---|---|---|
| a Reactions were carried out using equimolar amounts of aldehyde and amine, and 3 eq. of Et3B.b Yields and diastereomeric ratios were determined by 1H NMR spectroscopy with an estimated detection limit of ∼5%. | ||||
| 1 | 20 | 0.33 | 100 | 87:13 |
| 2 | 0 | 1 | 100 | 89:11 |
| 3 | −10 | 3 | 80 | 90:10 |
| 4 | −20 | 5 | 50 | 90:10 |
In all cases, the major diastereomeric pair was isolated from the reaction mixture by column chromatography, whereas attempts to isolate the minor pair systematically failed. The major diastereomer configuration was determined by NOE experiments (Fig. 1). The relative configuration between C-2′ and C-1 is ascertained by H,H-NOESY data. The R,R (or S,S) diastereomer in the most probable conformation (Fig. 1) exhibits intense NOE effects between H-1 and H-5′a, CH3 and H-3′b, H-2′, and H-5′b, H-5′a and H-2′′. In addition, strong cross peaks are observed between H-2 and the ethyl group signals. Furthermore, a Molecular Dynamics Simulated Annealing (MDSA) study of the two diastereomers (R,R and R,S) structure corroborated the observed NOE, confirming the preferred configuration and conformation in the major isomer.
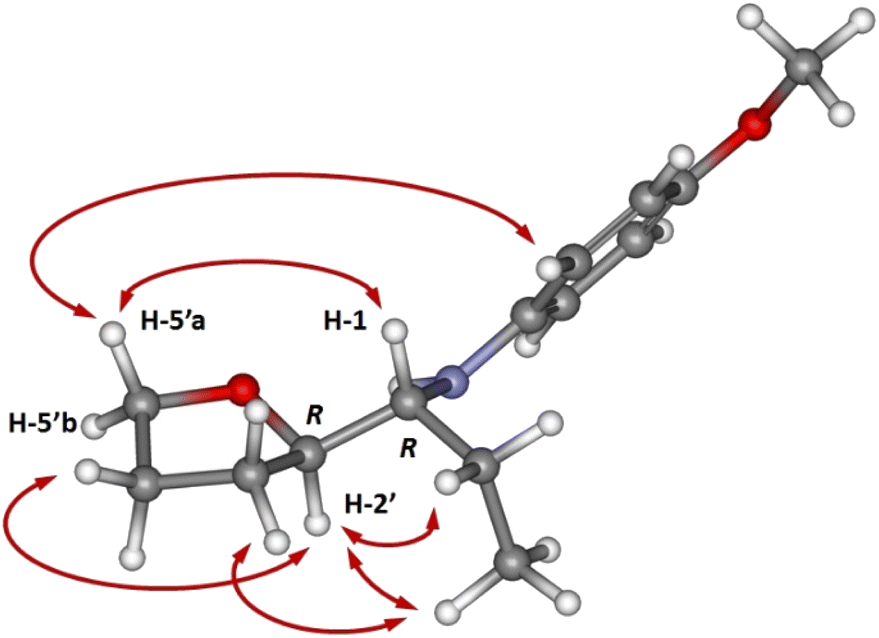 | ||
| Fig. 1 3D Model structure of the most stable conformer of 6 major with indication of the observed NOE effects. | ||
Stereoselectivity of the addition of a Grignard reagent to (1R)-tetrahydrofuran-2-carboxaldehyde has been previously studied by Amoroux.15 A preferential nucleophilic attack of EtMgX on the less hindered side of the chelate formed by coordination of the carbonyl oxygen, the heterocyclic oxygen, and the magnesium atom (re-attack) determined the major diastereomer obtained. However, in our case, asymmetric induction cannot be explained based on the highly ordered nature of transition structures (“closed” of “chelate” transition models), since triethylborane, as trifluoroborane, are monodentate Lewis acids. More likely, the reaction follows an “open”-transition state pathway, where the predominating diastereomer is formed by the approach of the entering group through the least hindered side of the imine double bond. This favoured conformation is reached when the rotational conformation of the C–C bond is such that the double bond is flanked by the two least bulky groups attached to the adjacent asymmetric center (Scheme 2). Consequently, as, for example, in aldol condensations, in our case the chelating ability of the Lewis acid is not relevant to the stereochemical outcome of the reaction.16
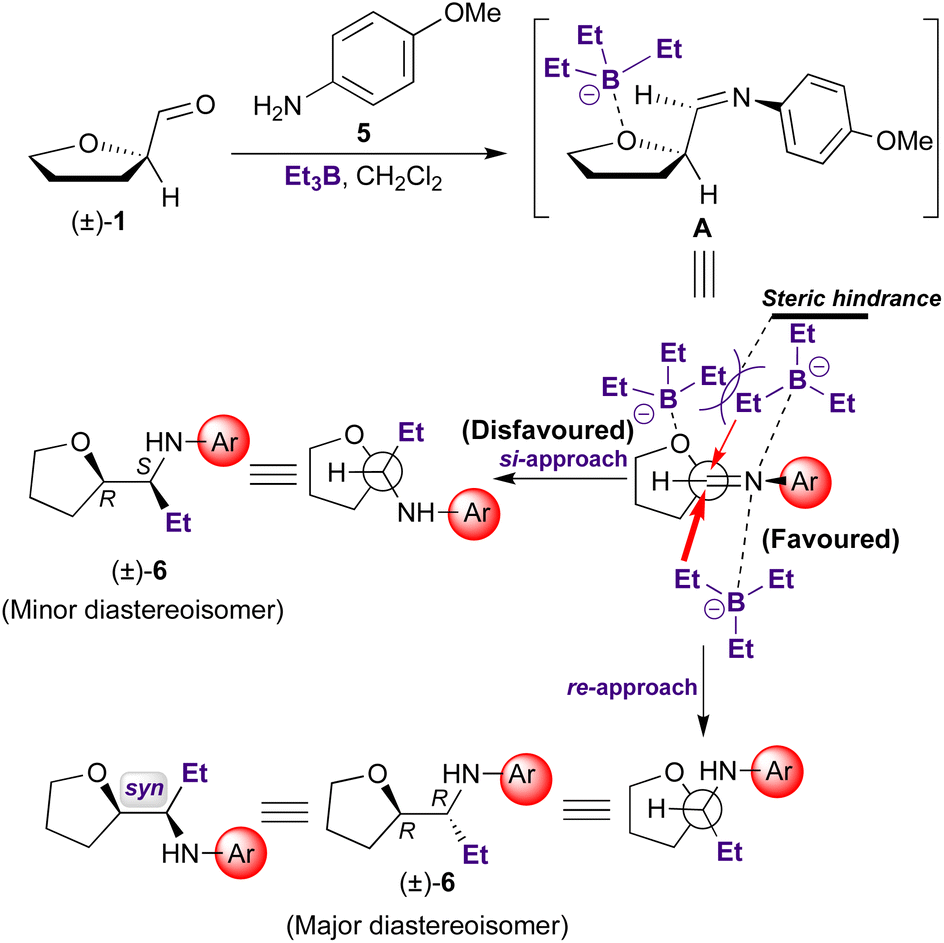 | ||
| Scheme 2 Mechanistic proposal for the rationale of the stereocontrol during the 3C-reaction. | ||
The aldehyde (±)-1 presents an oxygen atom at the vicinal position to the imine group. This fact would explain the observed stereoinduction, based on the less-hindered open-transition state (Scheme 2). According to Scheme 2, the attack would take place preferentially on the less-hindered side of the carbon-nitrogen double bond, with generation of the (R,R) and (S,S) stereoisomeric pair, reaching the 87:13 diastereomeric ratio (Table 1, entry 1). Moreover, interaction of Et3B with furane oxygen may contribute to increase the steric hindrance on the si-face of the imine double bond.
We next investigated this one-pot reaction with several enantiomerically pure cyclic aldehydes, namely (R)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde (2), L-N-benzyl prolinal (3) and 3-O-methyl-1,2-O-isopropylidene-α-D-xylodialdo-1,4-furanose (4), showed in Scheme 1. As can be seen in Table 2, the alkylated amines 7–9 (Scheme 1) coming from aldehydes 2–4 were obtained with good yields and diastereomeric ratios.
| Entry | Et3B (mmol) | t (h) | Amine | Yieldb (%) | drc (%) |
|---|---|---|---|---|---|
| a Reactions were carried out at room temperature (T = 20 °C) using equimolar amounts of aldehyde and amine and 3 eq. of Et3B.b Yield after purification by silica gel chromatography.c Diastereomeric ratio determined by GC-MS and 1H NMR of the product.d Six eq. of Et3B were used in this reaction. | |||||
| 1 | 3 | 0.75 | 7 | 70 | 84:16 |
| 2 | 3 | 4 | 8 | 75 | 83:17 |
| 3 | 6 | 14 | 9d | 87 | 92:8 |
The mixture of p-methoxyaniline, (R)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde (2) and triethylborane produced amine 7 in 70% yield and with a diastereomeric ratio of 84:16 after 45 min of reaction at 20 °C. The configuration of the major diastereomer could be assigned according to the results described for addition reaction to imines,17 hydrazones,18 and nitrones19 derived from aldehyde 2. The attack of the ethyl radical would take place mainly on the less hindered side (re-face), favouring therefore the formation of the 7 stereoisomer with R,S configuration (Scheme 2). The configuration of C-1 in the two 7 diastereomers was evidenced by NOE experiments. In agreement with the assigned configurations, intense H-1/H5′b and H-1/H-4′ NOE correlations (Fig. 2) were observed for the major and minor diastereomers, respectively.
 | ||
| Fig. 2 NOE effects displayed by the two diastereoisomers of 7 and the major diastereoisomer of 9. | ||
The one-pot reaction between p-methoxyaniline, triethylborane and N-benzyl L-prolinal (3) led to amine 8 in 75% yield and with a diastereomeric ratio of 83:17. The assignment of the configuration of the major diastereomer was also established by assuming that the attack takes place on the less hindered side (Scheme 2), with formation of the S,S diastereomer as the main product. Such preferential attack has been previously described in addition reactions to derivatives of L-prolinal.20
With the purpose of evaluating the effect of a more complex chiral structure on the reaction efficiency, we studied the one-pot alkylative amination of 3-O-methyl-1,2-O-isopropylidene-α-D-xylodialdo-1,4-furanose (4). This alkylation reaction required longer time for completion and three additional equivalents of Et3B to produce a 92:8 diastereomeric mixture of the alkylated amine 9 in 87% yield, also at 20 °C. As can be seen in the mechanistic proposal for the stereocontrol showed in Scheme 2, a second interaction between a second molecule of Et3B and the oxygen atom probably happens and may contribute to increase the steric hindrance of the less favoured face, and consequently improving the diastereomeric excess. At the same time, this fact can explain the need of longer reaction times and additional equivalents in the reaction of 4: the presence of more oxygen atoms in the cyclic moiety of 4 if compared to 1–3 will passivate a part of the Et3B added. On the other hand, due to the C-4′ and C-5′ configurations of the tetrahydrofuran ring, the attack must preferentially occur at the less hindered re-face of the imine group, with formation of the R,R diastereomer as the main product. Such preferential attack has been already described for additions to imines derived from α-D-xylodialdo-1,4-furanose.21 As expected for R,R-configuration, intense NOE correlations between H-1 and the methoxy group, as well as between H-5′ and the ethyl group, were observed for 9 (Fig. 2).
Regarding the alkylation reaction over chiral aldehydes, it is worth to mention that the α chiral stereogenic center of the employed chiral aldehydes 2–4 do not epimerize during the reaction.
Regarding the use of other alkylating boranes, tri(n-butyl)borane was used in the one-pot reaction of (±)-1 with 2. However, no alkylation products were found after 2 h reaction at 20 °C.
On the other hand, to generalize the method to other anilines, we study of the one-pot alkylation reaction using p-fluoroaniline (10), which has an electron-withdrawing group in para-position. Aniline 10 was made to react with aldehyde 2 in presence of Et3B in dichloromethane at 20 °C (Scheme 3), affording the amine 11 in 58% yield. The configuration of the major diastereomer could be assigned according to the results described for addition reaction to imines.
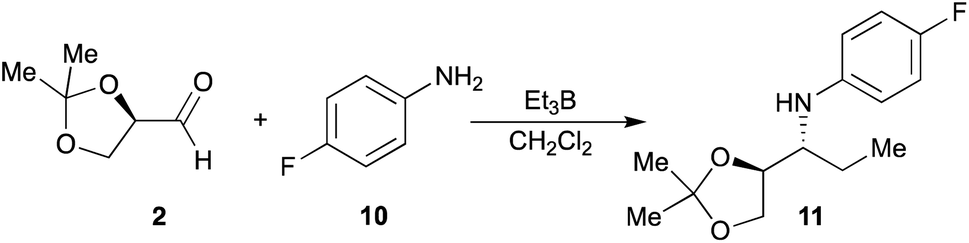 | ||
| Scheme 3 One-pot alkylation of aniline 10. | ||
To appraise the applicability of the procedure, we extended the study of the one-pot alkylation reaction to enantiomerically pure amines. Chiral amines (1S)-1-phenylethanamine (13) and L-valine methyl ester (14) were made to react with p-anisaldehyde (12) and Et3B (Scheme 4), in dichloromethane at 20 °C.
 | ||
| Scheme 4 General alkylation scheme for aldimines derived from chiral amines. | ||
We found that the presence of BF3·OEt2 (2 eq.) was required in these one-pot alkylations due to the low reactivity of the starting amines (13 and 14). As can be seen in Table 3, both 15 and 16 could be prepared in modest yields, 43 and 47%, respectively, and 75 and 80%, respectively, when the reaction is carried out on the preformed imine. In the synthesis of 15 a moderate stereocontrol (60:40) was observed (Table 3, entry 1), even when the reaction was carried out with the corresponding preformed imine 17 (Table 3, entry 2). The low stereocontrol found in the reaction of 12 with 13 to obtain 15 can be explained by the small difference in the steric hindrances between si-face and re-face in the open-transition structure to produce 15 (Scheme 4). If compared to that of 16, [A]‡ (Scheme 4, reaction of 12 with 14 and to the preformed imine 18), the last present a bulky isopropyl group that almost avoids the approach of Et3B though the re-face, thus favouring the si-approach to deliver the S,S diastereomer with high degree of stereocontrol (90:10, Table 3, entries 3 and 4). As it can be seen, diastereoselectivity of the reactions is largely depending on the structure of the amine.
| Entry | t (min) | Starting compounds | Yieldc (%) | drd |
|---|---|---|---|---|
| a One-pot reactions were carried out using equimolar amounts of aldehyde and imine, 2 eq. of BF3·OEt2, 3 eq. of Et3B and 1 eq. of H2O2.b Stepwise reactions were carried out using imine, 2 eq. of BF3·OEt2 3 eq. of Et3B and 1 eq. of H2O2 (entries 2 and 4).c Yields after purification by silica gel chromatography.d Diastereomeric ratio determined by GC-MS and 1H NMR of the product. | ||||
| 1 | 60 | 12+13 | 15, 43a | 60:40 |
| 2 | 15 | 17 | 15, 75b | 60:40 |
| 3 | 60 | 12+14 | 16, 47a | 90:10 |
| 4 | 20 | 18 | 16, 80b | 90:10 |
It is worth to mention that there is not a second heteroatom involved in the open-transition structure as occurred in cyclic aldehydes 1–4, and consequently, no interaction of a second molecule of Et3B can be expected, decreasing the steric hindrance of the non-preferred face of the imine double bond.
Based on these results, the configuration of the major diastereomer of 15 was tentatively assigned assuming that the attack must preferentially occur at the less hindered re-face of the imine group, with formation of the R,S diastereomer as the main product (Scheme 4). Similarly, the configuration for the major diastereomer of amine 16 was assigned assuming that the attack would take place on the less hindered imine si-face, as proposed in the literature (Scheme 4).22 Results are in agreement with the previously described by Torii,22a where the preferential attack on the less hindered side of the formed chelate could provide the S,S diastereomer as the main product. For amine 16 the assignment of the two diastereomers was confirmed by 1H NMR spectroscopy analysis. Clear differences in the chemical shifts of the proton signals arising from COOMe and NCHC = O were observed for the two diastereomers, which are in good agreement with the data previously reported for other amines derived from L-valine methyl ester.21 Whereas the COOMe signal appears for the S,S configuration at lower field than for the R,S configuration, 3.68 and 3.67 ppm, respectively. The opposite is observed when the NCHC = O signals are compared, 2.72 ppm for S,S configuration and 2.97 ppm for the R,S configuration.
Moreover, when we carried out the alkylation addition with the preformed imines 17 and 18, and Et3B in the presence of 2 eq. of BF3·OEt2, the yield of alkylation was increased significantly, reaching 75% for 15 and 80% for 16 (Table 3). Reactions were carried out at 20 °C in dichloromethane as solvent. As expected, stereoselectivity of the reaction is the same to that obtained in the one-pot reaction (Scheme 4, Table 3, entries 2 and 4).
As mentioned, for many years our group has been interested in the synthesis of isoquinoline alkaloids.23 Nevertheless, the negative results of alkylation using nBu3B, probably due to steric hindrances, and to illustrate the synthetic applicability of the developed procedure, we decided to study the reactivity of other borane derivatives with synthetic interest.
We focus on the synthesis of the benzylisoquinoline skeleton. Benzylisoquinolines alkaloids are a diverse class of plant secondary metabolites that exhibit a myriad of pharmacological activities, including antimicrobial, antitussive, antispasmodic, and anticancer properties.24 We planned the synthesis of romneine type substitution by reaction of the imine 6,7-methylenedioxy-3,4-dihydroisoquinoline (19) with an appropriate borane, tris(3,4-dimethoxy benzyl)borane (20) (Scheme 5). Borane 20 was prepared according to ref. 25. Surprisingly, the reaction of imine 19 with borane 20 in the presence of 2 eq. of BF3·OEt2 in dichloromethane at 20 °C, followed by methylation of nitrogen atom,26 afforded the benzylisoquinoline (±)-romneine (21), in good yield (28%, two steps). As expected, no chirality was induced in the C-1 of the benzylisoquinoline, since not heteroatom is present in the α-position of the starting imine. However, this reaction opens a new synthetic strategy to these natural alkaloids and the possibility of chiral synthesis of natural benzylisoquinolines when substituted in ring B (Scheme 5).
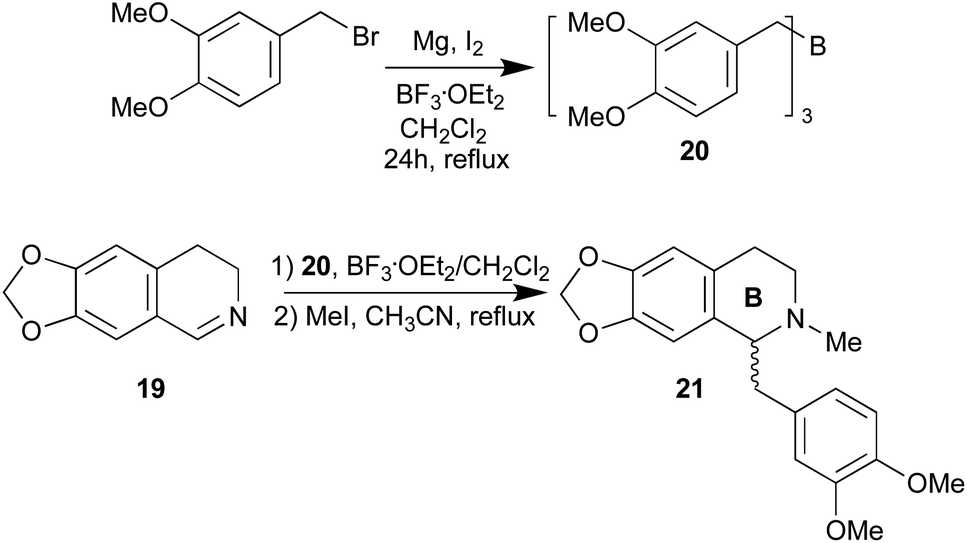 | ||
| Scheme 5 Application to the synthesis of (±)-romneine (21). | ||
3 Conclusions
We have developed an effective procedure for the alkylation of non-activated chiral imines based on the use of triethylborane as the alkylating agent. This procedure has allowed to synthesize optically active amines in good yields and with good diastereoselectivity. The one-pot procedure is applicable to a variety of chiral aldehydes. In the case that starting amines are chiral, the yield is significantly increased when the alkylation addition is performed on the already preformed imine. The presence of a heteroatom located at the vicinal position to the carbonyl plays an important role in the efficiency and stereoselectivity of the reaction, driving the reaction higher diastereoselectivity ratios, but longer reaction times are needed. The methodology has been performed over p-methoxyaniline and p-fluoroaniline, with the trialkyl boranes nBu3B and Et3B, and finally, successfully applied to the synthesis of the natural benzylisoquinoline romneine.4 Experimental
4.1. General
MS (EI) data were recorded with an HP-MS 5988A spectrometer operating at 70 eV and HRMS data with a VG Autospec spectrometer. GC were registered with a DB5 column using an initial temperature of 80 °C, a single-ramp temperature of 20 °C min−1 and a final temperature of 250 °C. IR spectra were recorded with ATR accessory (MIRacle ATR, PIKE Technologies, USA) coupled to an FTIR spectrometer (FT/IR-4100, JASCO). 1H and 13C NMR spectra were recorded with a 400 MHz ARX 400 Bruker spectrometer by using the residual solvent peak in CDCl3 (δH 7.24 ppm for 1H and δC = 77.0 ppm for 13C). TLC analyses were performed on Merck silica gel 60 F 254 plates, and column chromatography was performed on silica gel 60 (0.040–0.063 mm). Reagents were purchased from commercial sources and were used as received unless mentioned otherwise. All experiments were performed under an inert atmosphere (Ar) in oven-dried glassware, sealed with a rubber septum and using anhydrous solvents. Triethylborane, tri(n-butyl)borane, 3,4-dimethoxy benzyl bromide, 6,7-methylenedixoy-3,4-dihydroisoquinoline (19), 3-O-methyl-1,2-O-isopropylidene-α-D-xylopentodialdo-1,4-furanose (4) and (R)-(+)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde (2) were purchased from Aldrich. (±)-Tetrahydrofuran-2-carboxaldehyde (1) was obtained from (±)-tetrahydrofuran-2-methanol, according to the previously described procedure.27 N-Benzyl L-prolinal (3) was obtained using the Swern oxidation procedure from N-benzyl L-prolinol.28 Imine 17 was synthesized from the (S)-1-phenylethanamine and the p-anisaldehyde in Et2O in the presence of molecular sieves according to Taguchi's protocol.29 The analytical and spectroscopic data observed were in agreement with the reported in literature for de same imine.30 L-Valine methyl ester was condensed with p-anisaldehyde in the presence of Et3N and MgSO4 to obtain the L-valinate 18.314.2. General procedure for the one-pot reaction of chiral aldehyde, p-methoxyaniline and triethylborane. Synthesis of amines 6–9, 11
1 M Solution of triethylborane in hexane (3 mL, 3 mmol) was added to a solution of aldehyde (1 mmol) and p-methoxyaniline (5, 123 mg, 1 mmol) or p-fluoroaniline (10, 111 mg, 1 mmol) in dichloromethane and the reaction mixture was stirred at temperatures and periods showed in Tables 1 and 2 and Schemes 1 and 3. When the reaction was completed the crude reaction was concentrated to dryness under vacuum. The 1H NMR spectra of the residue exhibited signals corresponding to the almost exclusive presence of the expected amine diastereomers. The residue was purified by column chromatography (SiO2, 10% ethyl acetate in hexane) to afford the alkylated amines 6–9 or 11 in 50–87% yields (Tables 1 and 2, Schemes 1 and 3).(1R)-7 Major: yield: 157 mg, 59%, colourless oil. Rf 0.35 (silica gel, 30% ethyl acetate in hexane). +6 (c 0.9, CH2Cl2). IR (neat) νmax (cm−1) 3374, 3010, 2983, 2961, 1617, 1509, 1229. 1H NMR (400 MHz, CDCl3, 25 °C) δ 6.74 (d, J = 8.5 Hz, 2H, 3′′-H, 5′′-H), 6.58 (d, J = 8.5 Hz, 2H, 2′′-H, 6′′-H), 4.1–4.0 (m, 2H, 4′-H, 5′a-H), 3.82 (dd, J = 7.4, 4.6 Hz, 1H, 5′b-H), 3.72 (s, 3H, OMe), 3.25 (dt, J = 7.0, 4.0 Hz, 1H, 1-H), 3.25 (brs, 1H, NH), 1.76 (m, 1H, CH2–CH3), 1.47–1.38 (m, 1H, CH2–CH3), 1.41, 1.32 (2 × s, 3H each, CMe2), 0.94 (t, J = 7.5 Hz, 3H, CH2–CH3) ppm. 13C NMR (100 MHz, CDCl3, 25 °C) δ 152.0 (C-4′′), 142.1 (C-1′′), 114.7 (C-2′′, C-6′′, C-3′′, C-5′′), 109.2 (C-2′), 78.0 (C-4′), 67.2 (C-5′), 58.1 (C-1), 55.7 (OMe), 26.7 (CMe2), 25.2 (CMe2), 24.5 (CH2–CH3), 10.0 (CH2–CH3) ppm. GC Rt 4.36 min. EI-MS m/z (%) 265 (16) [M+], 190 (16), 164 (100). HR-MS calcd. for C15H23NO3 265.1678, found 265.1673.
(1S)-8 Major: yield: 203 mg, 58%, colourless oil. Rf 0.15 (silica gel, 30% ethyl acetate in hexane). −25 (c 0.3, CH2Cl2). IR (neat) νmax (cm−1) 3343, 3026, 2957, 1617, 1508, 1453, 1408, 1373, 1230. 1H NMR (200 MHz, CDCl3, 25 °C) δ 7.33–7.26 (m, 5H, CHAr), 6.72 (d, J = 8.5 Hz, 2H, 3′′-H, 5′′-H), 6.49 (d, J = 8.5 Hz, 2H, 2′′-H, 6′′-H), 4.03 (d, J = 12.8 Hz, 1H, α-H), 3.78 (s, 3H, OMe), 3.40 (d, J = 12.8 Hz, 1H, α′-H), 3.23–3.10 (m, 1H, 1-H), 2.95–2.87 (m, 2H, 5′a-H, 2′-H), 2.21 (q, J = 8.0 Hz, 1H, 5′b-H), 1.92–1.74 (m, 1H, CH2–CH3), 1.71–1.61 (m, 4H, 3′-H, 4′-H), 1.44–1.34 (m, 1H, CH2–CH3), 0.95 (t, J = 7.3 Hz, 3H, CH2–CH3) ppm. 13C NMR (50 MHz, CDCl3, 25 °C) δ 151.3 (C-4′′), 143.0 (C-1′′), 140.3 (Cquat,Ar), 128.5, 128.2, 126.8 (5 × CHAr), 114.9 (C-3′′, C-5′′), 113.9 (C-2′′, C-6′′), 65.2 (C-2′), 60.7, (C-α), 58.6 (C-1), 55.8 (OMe), 55.4 (C-5′), 27.7, 24.0 (C-3′, C-4′), 24.4 (CH2–CH3), 11.5 (CH2–CH3) ppm. GC Rt 6.47 min. EI-MS m/z (%) 324 (4) [M+], 160 (100), 91 (32). HR-MS calcd. for C21H28N2O 324.2202, found 324.2216.
(1S)-9 Minor: yield: 23 mg, 7%, colourless oil. Rf 0.38 (silica gel, 30% ethyl acetate in hexane). Spectroscopic data for an enriched mixture of this isomer: 1H NMR (200 MHz, CDCl3, 25 °C): δ = 6.72 (d, J = 8.5 Hz, 2H, 2′′-H, 6′′-H), 6.62 (d, J = 8.5 Hz, 2H, 3′′-H, 5′′-H), 5.89 (d, J = 4.0 Hz, 1H, 2′-H), 4.57 (d, J = 4.0 Hz, 1H, 3′-H), 4.08 (dd, J = 8.5, 2.4 Hz, 1H, 5′-H), 3.70 (s, 3H, OMeAr), 3.7–3.5 (m, 2H, 1-H, 4′-H), 3.37 (s, 3H, OMe), 1.81–1.64 (m, 1H, CH2–CH3), 1.53–1.34 (m, 1H, CH2–CH3), 1.47, 1.30 (2 × s, 3H each, CMe2), 0.94 (t, J = 7.3 Hz, 3H, CH2–CH3) ppm. GC Rt 5.26 min.
(1R)-9 Major: yield: 270 mg, 80%, colourless oil. Rf 0.42 (silica gel, 30% ethyl acetate in hexane). +20 (c 0.8, CH2Cl2). IR (neat): νmax (cm−1) 3379, 3010, 2961, 2933, 1617, 1509, 1458, 1410, 1374, 1230. 1H NMR (200 MHz, CDCl3, 25 °C) δ 6.72 (d, J = 8.5 Hz, 2H, 3′′-H, 5′′-H), 6.60 (d, J = 8.5 Hz, 2H, 2′′-H, 6′′-H), 5.87 (d, J = 4.0 Hz, 1H, 2′-H), 4.51 (d, J = 4.0 Hz, 1H, 3′-H), 3.93 (dd, J = 9.0, 3.0 Hz, 1H, 5′-H), 3.70 (s, 3H, OMeAr), 3.72–3.69 (m, 1H, 1-H), 3.60 (d, J = 3.0 Hz, 1H, 4′-H), 3.18 (s, 3H, OMe), 1.98–1.75 (m, 1H, CH2–CH3), 1.55–1.37 (m, 1H, CH2–CH3), 1.48, 1.30 (2 × s, 3H each, CMe2), 0.93 (t, J = 7.3 Hz, 3H, CH2–CH3) ppm. 13C NMR (50 MHz, CDCl3, 25 °C) δ 151.7 (C-4′′), 142.3 (C-1′′), 114.8 (C-2′′, C-6′′), 114.6 (C-3′′, C-5′′), 111.3 (CMe2), 104.8 (C-2′), 83.5 (C-4′), 82.3 (C-5′), 81.1 (C-3′), 57.4 (OMe), 55.7 (OMeAr), 53.4 (C-1), 26.7, 26.1 (CMe2), 25.9 (CH2–CH3), 9.7 (CH2–CH3) ppm. GC tR 5.30 min. EI-MS m/z (%) 337 (12) [M+], 164 (100). HR-MS calcd. for C18H27NO5 337.1889, found 337.1896.
4.3. General procedure for the reaction of chiral imines 17 and 18, boron trifluoride-diethyl ether and triethylborane. Synthesis of amines 15 and 16
Boron trifluoride diethyl etherate (250 μL, 2 mmol) was added to a solution of the corresponding imine 17 or 18 (1 mmol) in dichloromethane. The reaction mixture was stirred at 20 °C for 2 h and then a 1 M solution of triethylborane in hexane (3 mL, 3 mmol) was added. Subsequently, an aqueous solution of hydrogen peroxide (0.1 mL, 33%) was added and the mixture stirred. When the reaction was completed (15–20 min) the crude reaction was concentrated to dryness under vacuum. The 1H NMR of the residue exhibited signals corresponding to the almost exclusive presence of the expected amine diastereomers. The residue was purified by column chromatography (SiO2, 10% ethyl acetate in hexane) to afford the alkylated amines 15 or 16 in 75 and 80% yield respectively.(1R)-15 Major: yield: 121 mg, 45%, colourless oil. Rf 0.44 (silica gel, 20% ethyl acetate in hexane). −19 (c 0.3, CH2Cl2). IR (neat) νmax (cm−1) 3400, 3019, 2971, 2925, 1611, 1513, 1407, 1367, 1224. 1H NMR (200 MHz, CDCl3, 25 °C) δ 7.33–7.17 (m, 5H, CHPh), 7.12 (d, J = 8.5 Hz, 2H, 2′-H, 6′-H), 6.83 (d, J = 8.5 Hz, 2H, 3′-H, 5′-H), 3.78 (s, 3H, OMe), 3.67 (q, J = 6.4 Hz, 1H, NCHPh), 3.48 (dd, J = 8.5, 4.8 Hz, 1H, 1-H), 1.83–1.48 (m, 2H, CH2–CH3), 1.30 (d, J = 6.4 Hz, 3H, CH3CHPh), 0.73 (t, J = 7.5 Hz, 3H, CH2–CH3) ppm. 13C NMR (50 MHz, CDCl3, 25 °C) δ 158.5 (C-4′), 145.9 (Cquat,Ar), 136.0 (C-1′), 128.3, 128.2, 126.7, 126.6 (CHPh, C-2′, C-6′), 113.6 (C-3′, C-5′), 61.1 (C-1), 55.2 (MeO), 54.6 (NCHPh), 30.0 (CH2–CH3), 22.3 (CH3CHPh), 10.7 (CH2–CH3) ppm. GC Rt 4.55 min. EI-MS m/z (%) = 241 (18), 240 (100), 136 (22). HR-MS calcd. for C18H24NO 270.1857, found 270.1852.
O), 158.7 (C-4′), 135.6 (C-1′), 128.5 (C-2′, C-6′), 113.5 (C-3′, C-5′), 65.2, 63.6 (NCHC = O, C-1), 55.2, 51.2 (2 × OMe), 31.7, 29.6 (CH2–CH3, CHMe2), 19.0, 18.8 (CHMe2), 10.5 (CH2–CH3) ppm. GC Rt 4.22 min.(1S)-16 Major: yield: 223 mg, 78%, colourless oil. Rf 0.75 (silica gel, 20% ethyl acetate in hexane). −117 (c 0.7, CH2Cl2). IR (neat) νmax (cm−1) 3338, 3020, 2961, 2930, 1733, 1612, 1586, 1512, 1457, 1407, 1387, 1200. 1H NMR (200 MHz, CDCl3, 25 °C) δ 7.16 (d, J = 8.5 Hz, 2H, 2′-H, 6′-H), 6.80 (d, J = 8.5 Hz, 2H, 3′-H, 5′-H), 3.77 (s, 3H, OMe), 3.68 (s, 3H, COOMe), 3.25 (t, J = 7.0 Hz, 1H, 1-H), 2.72 (d, J = 7.0 Hz, 1H, NCHC = O), 1.89–1.69 (m, 1H, CHMe2), 1.68–1.48. (m, 2H, CH2–CH3), 0.86 (d, J = 6.7 Hz, 3H, CHMe2), 0.80 (d, J = 6.7 Hz, 3H, CHMe2), 0.76 (t, J = 7.3 Hz, 3H, CH2–CH3) ppm. 13C NMR (50 MHz, CDCl3, 25 °C) δ 176.4 (CO), 158.7 (C-4′), 135.6 (C-1′), 128.7 (C-2′, C-6′), 113.4 (C-3′, C-5′), 64.3 (NCHC = O), 62.8 (C-1), 55.1 (OMe), 51.2 (COOMe), 31.9 (CH2–CH3), 31.7 (CHMe2), 19.4, 18.5 (CHMe2), 10.6 (CH2–CH3) ppm. GC Rt 4.17 min. EI-MS m/z (%) = 278 (1), 251 (15), 250 (100), 190 (13), 149 (25). HR-MS calcd. for C16H25NO3 279.1834, found 279.1830.
4.4. Synthesis of (±)-romneine (21)
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Junta de Andalucía (FQM209, FQM397 and UMA20-FEDERJA84).Notes and references
- (a) Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, WILEY-VCH Verlag GmbH Co. KGaA, Weinheim, 2010 Search PubMed; (b) B.-L. Chen, B. Wang and G.-Q. Lin, J. Org. Chem., 2010, 75, 941–944 CrossRef CAS PubMed; (c) C. Roe, H. Hobbs and R. A. Stockman, J. Org. Chem., 2011, 76, 9452–9459 CrossRef CAS PubMed; (d) Stereoselective Formation of Amines, ed. W. Li and X. Zhang, Springer, Berlin, 2014 Search PubMed; (e) X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024–2027 CrossRef CAS PubMed.
- (a) H. Miyabe, M. Ueda and T. Naito, J. Org. Chem., 2000, 65, 5043–5047 CrossRef CAS PubMed; (b) M. P. Bertrand, L. Feray, R. Nouguier and P. Perfetti, Synlett, 1999, 1148–1151 CrossRef CAS; (c) G. K. Friestad, Top. Curr. Chem., 2014, 343, 1–32 CAS.
- (a) G. K. Friestad, A. Ji, C. S. Korapala and J. Qin, Org. Biomol. Chem., 2011, 9, 4039–4043 RSC; (b) G. K. Friestad, Top. Curr. Chem., 2012, 320, 61–92 CrossRef CAS PubMed.
- (a) D. A. Gutierrez, J. Fettinger, K. N. Houk, K. Ando and J. T. Shaw, Org. Lett., 2022, 24, 1164–1168 CrossRef CAS PubMed; (b) K. Yamada, T. Konishi, M. Nakano, S. Fujii, R. Cadou, Y. Yamamoto and K. Tomioka, J. Org. Chem., 2012, 77, 1547–1553 CrossRef CAS PubMed; (c) L. C. Moore, A. Lo, J. S. Fell, M. R. Duong, J. A. Moreno, B. E. Rich, M. Bravo, J. C. Fettinger, L. W. Souza, M. M. Olmstead, K. N. Houk and J. T. Shaw, Eur. J. Chem., 2019, 25, 12214–12220 CrossRef CAS PubMed.
- (a) G. K. Friestad, Eur. J. Org. Chem., 2005, 3157–3172 CrossRef CAS; (b) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541–2569 CrossRef CAS; (c) N. Pastori, C. Gambarotti and C. Punta, Mini-Rev. Org. Chem., 2009, 6, 184–195 CrossRef CAS; (d) T. Akindele, K. Yamada and K. Tomioka, Acc. Chem. Res., 2009, 42, 345–355 CrossRef CAS PubMed; (e) H. Miyabe, E. Yoshioka and S. Kohtani, Curr. Org. Chem., 2010, 14, 1254–1264 CrossRef CAS.
- (a) D. P. Curran, N. A. Porter and B. Giese, in Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, VCH, Weinheim, 1996 Search PubMed; (b) G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175–9206 CrossRef CAS PubMed.
- (a) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394–1401 CrossRef CAS PubMed; (b) R. L. Patman, J. F. Bower, I. S. Kim and M. J. Krische, Aldrichimica Acta, 2008, 41, 95–104 CAS.
- (a) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS; (b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- P. Wu, M. Givskov and T. E. Nielsen, Chem. Rev., 2019, 119, 11245–11290 CrossRef CAS PubMed.
- (a) H. Miyabe, C. Ushiro, M. Ueda, K. Yamakawa and T. Naito, J. Org. Chem., 2000, 65, 176–185 CrossRef CAS PubMed; (b) H. Miyabe, C. Konishi and T. Naito, Org. Lett., 2000, 2, 1443–1445 CrossRef CAS PubMed; (c) S. B. McNabb, M. Ueda and T. Naito, Org. Lett., 2004, 6, 1911–1914 CrossRef CAS PubMed; (d) M. Ueda, H. Miyabe, H. Sugino, O. Miyata and T. Naito, Angew. Chem., Int. Ed., 2005, 44, 6190–6193 CrossRef CAS PubMed.
- (a) M. Ueda, H. Miyabe, M. Teramachi, O. Miyata and T. Naito, Chem. Commun., 2003, 426–427 RSC; (b) M. Ueda, H. Miyabe, M. Teramachi, O. Miyata and T. Naito, J. Org. Chem., 2005, 70, 6653–6660 CrossRef CAS PubMed.
- (a) G. K. Friestad and J. Qin, J. Am. Chem. Soc., 2001, 123, 9922–9923 CrossRef CAS PubMed; (b) G. K. Friestad, Y. Shen and E. L. Ruggles, Angew. Chem., Int. Ed., 2003, 42, 5061–5063 CrossRef CAS PubMed.
- (a) M. P. Bertrand, S. Coantic, L. Feray, R. Nouguier and P. Perfetti, Tetrahedron, 2000, 56, 3951–3961 CrossRef CAS; (b) N. Singh, R. D. Anand and S. Trehan, Tetrahedron Lett., 2004, 45, 2911–2913 CrossRef CAS; (c) N. Halland and K. A. Jørgensen, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 1290–1295 RSC.
- M. Valpuesta, C. Muñoz, A. Díaz, G. Torres and R. Suau, Eur. J. Org. Chem., 2010, 1934–1942 CrossRef CAS.
- R. Amoroux, S. Ejjiyar and M. Chastrette, Tetrahedron Lett., 1986, 27, 1035–1038 CrossRef.
- R. Mahrwald, Chem. Soc. Rev., 1999, 99, 1095–1120 CrossRef CAS PubMed.
- (a) R. Badorrey, C. Cativiela, M. D. Diaz de Villegas and J. A. Galvez, Tetrahedron, 2002, 58, 341–354 CrossRef CAS; (b) R. Badorrey, C. Cativiela, M. D. Diaz de Villegas and J. A. Galvez, Tetrahedron Lett., 2003, 44, 9189–9192 CrossRef CAS; (c) D. Díez, A. B. Antón, P. García, M. G. Nuñez, N. M. Garrido, R. F. Moro, I. S. Marcos, P. Basabe and J. G. Urones, Tetrahedron: Asymmetry, 2006, 17, 2260–2264 CrossRef; (d) R. Díez, R. Badorrey, M. D. Díaz de Villegas and J. A. Gálvez, Eur. J. Org. Chem., 2007, 2114–2120 CrossRef.
- (a) D. A. Claremon, P. K. Lumma and B. T. Phillips, J. Am. Chem. Soc., 1986, 108, 8265–8266 CrossRef CAS; (b) J. M. Lassaletta, R. Fernandez, E. Martin-Zamora and C. Pareja, Tetrahedron Lett., 1996, 37, 5787–5790 CrossRef CAS; (c) P. Remuzon, C. Dussy, J.-P. Jacquet, P. Roty and D. Bouzard, Tetrahedron: Asymmetry, 1996, 7, 1181–1188 CrossRef CAS.
- (a) A. Dondoni, S. Franco, F. L. Merchan, P. Merino and T. Tejero, Tetrahedron Lett., 1993, 34, 5475–5478 CrossRef CAS; (b) P. Merino, S. Anoro, E. Castillo, F. Merchan and T. Tejero, Tetrahedron: Asymmetry, 1996, 7, 1887–1890 CrossRef CAS; (c) P. Merino, S. Franco, F. L. Merchan and T. Tejero, Tetrahedron: Asymmetry, 1997, 8, 3489–3496 CrossRef CAS; (d) P. Merino, V. Mannucci and T. Tejero, Tetrahedron, 2005, 61, 3335–3347 CrossRef CAS; (e) P. Merino, I. Delso, V. Mannucci and T. Tejero, Tetrahedron Lett., 2006, 47, 3311–3314 CrossRef CAS.
- (a) J. M. Andrés, R. Pedrosa, A. Pérez and A. Pérez-Encabo, Tetrahedron, 2001, 57, 8521–8530 CrossRef; (b) J. M. Andrés, R. Pedrosa, A. Pérez-Encabo and M. Ramírez, Tetrahedron, 2006, 62, 7783–7792 CrossRef.
- (a) G. Casiraghi, L. Colombo, G. Rassu and P. Spanu, J. Org. Chem., 1990, 55, 2565–2567 CrossRef CAS; (b) A. Bordier, P. Compain, O. R. Martin, K. Ikeda and N. Asano, Tetrahedron: Asymmetry, 2003, 14, 47–51 CrossRef CAS.
- (a) H. Tanaka, K. Inoue, U. Pokorski, M. Taniguchi and S. Torii, Tetrahedron Lett., 1990, 31, 3023–3026 CrossRef CAS; (b) A. Bocoum, D. Savoia and A. Umani-Ronchi, J. Chem. Soc., Chem. Commun., 1993, 1542–1544 RSC; (c) T. Basile, A. Bocoum, D. Savoia and A. Umani-Ronchi, J. Org. Chem., 1994, 59, 7766–7773 CrossRef CAS; (d) A. Bocoum, C. Boga, D. Savoia and A. Umani-Ronchi, Tetrahedron Lett., 1991, 32, 1367–1370 CrossRef CAS; (e) G. Alvaro and D. Savoia, Tetrahedron: Asymmetry, 1996, 7, 2083–2092 CrossRef CAS; (f) C. Fiorelli and D. Savoia, J. Org. Chem., 2007, 72, 6022–6028 CrossRef CAS PubMed.
- For example: (a) R. Suau, R. Rico, J. M. López-Romero, F. Nájera, A. Ruiz and F. J. O. López, Arkivok, 2005, 5, 62–72 Search PubMed; (b) R. Suau, R. Rico, F. Nájera, F. J. Ortiz, J. M. López-Romero, M. Moreno-Mañas and A. Roglans, Tetrahedron, 2004, 60, 5725–5735 CrossRef CAS; (c) J. Hierrezuelo, E. Guillén, J. M. López-Romero, R. Rico and R. Martínez-Mallorquin, Tetrahedron, 2011, 67, 2555–2561 CrossRef.
- S. E. O'Connors, Benzylisoquinoline Alkaloids in Comprehensive Natural Products III, ed. H. W. Liu and T. P. Begley, Elsevier, 2020 Search PubMed.
- H. C. Brown and U. S. Racherla, J. Org. Chem., 1986, 51, 427–432 CrossRef CAS.
- B. Gabet, P.-C. Kuo, S. Fuentes, Y. Patel, A. Adow, M. Alsakka, P. Avila, T. Beam, J.-H. Yen and D. A. Brown, Bioorg. Med. Chem., 2018, 26, 5711–5717 CrossRef CAS PubMed.
- P. Bianchi, G. Roda, S. Riva, B. Danieli, A. Zabelinskaja-Mackova and H. Griengl, Tetrahedron, 2001, 57, 2213–2220 CrossRef CAS.
- K. Thai, L. Wang, T. Dudding, F. Bilodeau and M. Gravel, Org. Lett., 2010, 12, 5708–5711 CrossRef CAS PubMed.
- L. Troisi, S. De Lorenzis, M. Fabio, F. Rosato and C. Granito, Tetrahedron: Asymmetry, 2008, 19, 2246–2251 CrossRef CAS.
- (a) G. Alvaro, C. Boga, D. Savoia and A. Umani-Ronchi, J. Chem. Soc., Perkin Trans. 1, 1996, 1, 875–882 RSC; (b) R. I. Kureshy, K. J. Prathap, S. Agrawal, N. H. Khan, S. H. R. Abdi and R. V. Jasra, Eur. J. Org. Chem., 2008, 3118–3128 CrossRef CAS.
- A. Leggio, A. Le Pera, P. A. Liguori, A. Napoli, C. Romeo, C. Siciliano and G. Sindona, Synth. Commun., 2003, 33, 4331–4338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01397a |
| This journal is © The Royal Society of Chemistry 2023 |