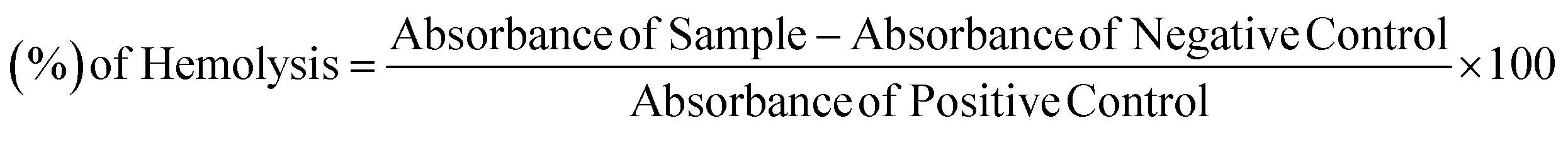DOI:
10.1039/D3RA01348K
(Paper)
RSC Adv., 2023,
13, 13798-13808
Convergent synthesis, kinetics insight and allosteric computational ascriptions of thiazole-(5-aryl)oxadiazole hybrids embraced with propanamides as alkaline phosphatase inhibitors†
Received
28th February 2023
, Accepted 20th April 2023
First published on 15th May 2023
Abstract
Considering the varied pharmacological prominence of thiazole and oxadiazole heterocyclic moieties, a unique series of bi-heterocyclic hybrids, 8a–h, was synthesized in a convergent manner. The structures of newly synthesized compounds were characterized by 1H-NMR, 13C-NMR, and IR spectral studies. The structure–activity relationship of these compounds was predicted by examining their inhibitory effects against alkaline phosphatase, whereby all these molecules exhibited superb inhibitory potentials relative to the standard used. The kinetics mechanism was determined by Lineweaver–Burk plots which revealed that 8g inhibited the studied enzyme non-competitively by forming an enzyme-inhibitor complex. The inhibition constant Ki calculated from Dixon plots for this compound was 0.42 μM. The allosteric computational study was coherent with the experimental records and these ligands exhibited good binding energy values (kcal mol−1). The hemolytic analysis revealed their mild cytotoxicity towards red blood cell membranes and hence, these molecules have potential to be nontoxic medicinal scaffolds for the treatment of alkaline phosphate-associated ailments.
1. Introduction
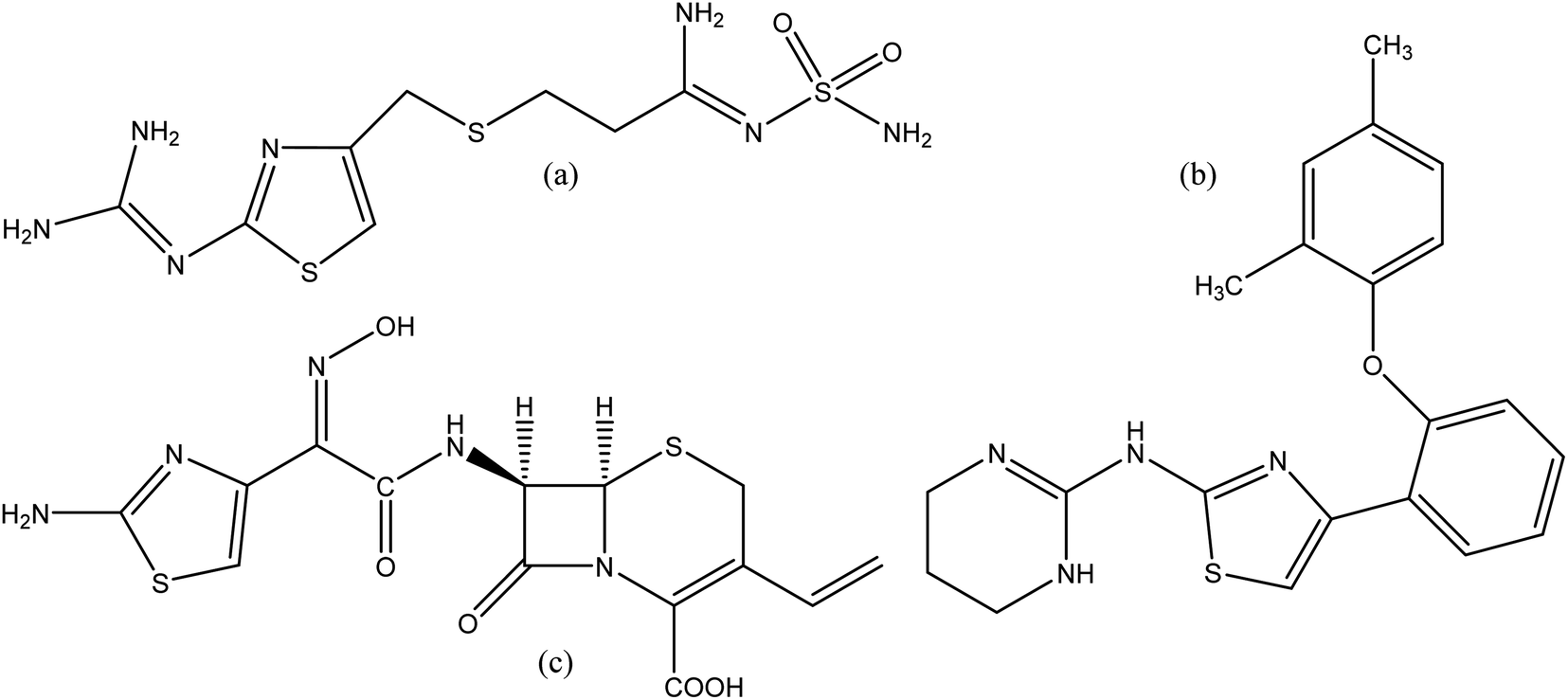
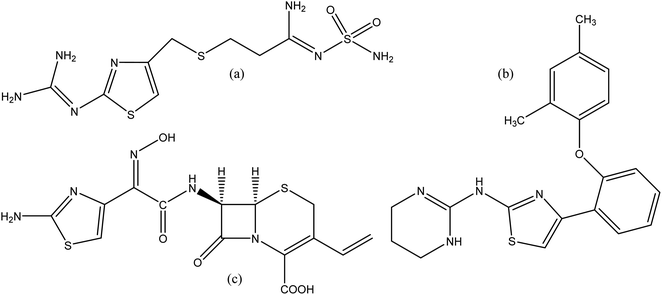
Heterocyclic compounds are expansively distributed in nature as they accomplish a major part in the working of the metabolic system, which is important for living things. Approximately 90% of new drugs contain heterocyclic moieties. Furthermore, due to their different properties, these compounds are practical in many divisions, including medicine, agriculture, polymer and material science.1 2-Aminothiazole (2-AT) is an important and versatile scaffold which is applied extensively in different branches of medicinal chemistry. The thiazole core, a five-membered heterocyclic unit with sulfur and nitrogen at 1,3 positions, is found in natural products such as a component of the vitamin thiamine (B1).2 Derivatives of 2-AT are anti-inflammatory,3 anticancer,4 antimicrobial,5 anti-HIV,6 antifungal,7 anti-bacterial,8 and analgesics.9 Moreover, thiazole derivatives were investigated in many works as corrosion inhibitors for copper and its alloys in chloride solutions.10–12 Thiazole ring also finds applications in other fields, such as polymers,13 liquid crystals,14 photo nucleases,15 fluorescent dyes,16 insecticides,17 and antioxidants.18 A few drugs containing 2-AT core, have been launched in the market. Famotidine (Fig. 1a) is used in the treatment of peptic ulcers and controls gastroesophageal reflux.19 Abafungin (Fig. 1b) is an antimicrobial agent used for the treatment of dermatomycoses.20 Cefdinir (Fig. 1c) is a semi-synthetic third-generation broad-spectrum cephalosporin antibiotic used for the treatment of pneumonia, chronic bronchitis, sinusitis, pharyngitis, and tonsillitis.21 Some of the thiazole derivatives have also been reported as enzyme inhibitors.22
 |
| | Fig. 1 Structures of famotidine (a), abafungin (b) and cefdinir (c). | |
1,3,4-Oxadiazole is a five-membered ring heterocyclic molecule composed of an oxygen atom and two nitrogen atoms. It is derivative by substitution of two methylene groups of furan with two pyridine-type nitrogens. They contain three known isomers: 1,2,3-oxadiazole, 1,2,4-oxadiazole and 1,2,5-oxadiazole. 1,2,4-oxadiazole and 1,3,4-oxadiazole are well-known, and widely studied by researchers due to their imperative chemical and biological properties.23,24 1,3,4-oxadiazole moiety has exposed better binding to the enzyme's active site through its oxygen.25 It can also certainly partake in hydrogen bonding interactions. Furthermore, recent reports exposed that this class of compounds could be potent tyrosinase inhibitors.24 Different biological activities related to these compounds are anti-analgesic, antimalarial, anticonvulsant, antimicrobial, anticancer, anti-HIV, anti-mycobacterial, and antidepressant.26
Alkaline phosphatases are membrane-bound ectonucleotidases which are broadly spread from bacteria to mammals. Alkaline phosphatase is a dimeric enzyme that catalyzes a variety of physiological and non-physiological substrates through dephosphorylation and transphosphorylation reactions.27,28 The enzymatic activity of alkaline phosphatases (APs) is due to the existence of two Zn2+ ions and one Mg2+ ion within their catalytic site.29 In humans, alkaline phosphatases are further classified into two classes; the tissue-specific alkaline phosphatases that cover intestinal alkaline phosphatase, placental alkaline phosphatase and germ cell alkaline phosphatase isoenzymes and tissue-non specific alkaline phosphatase which is encoded by various genes.30 Hydrolysis of phosphoesters, phosphate transferase activity, protein phosphatase activity, phosphate transport, modulation of organic cation transport, and involvement in cell proliferation have been suggested as possible functions of alkaline phosphatases.31 The biological action of the alkaline phosphatase in serum is related to metabolic bone and liver diseases and also is used as a marker of osteoblastic differentiation.32
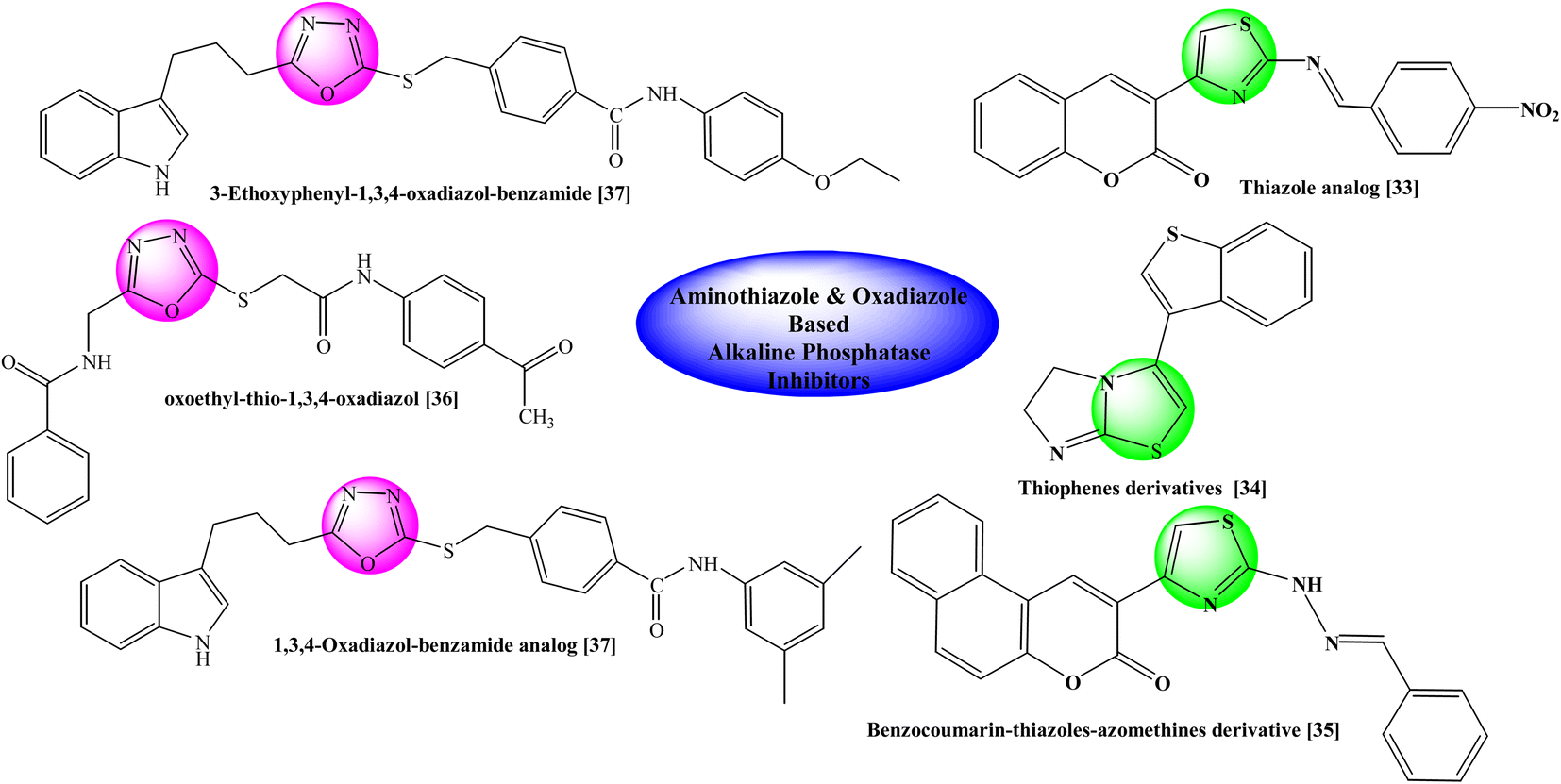
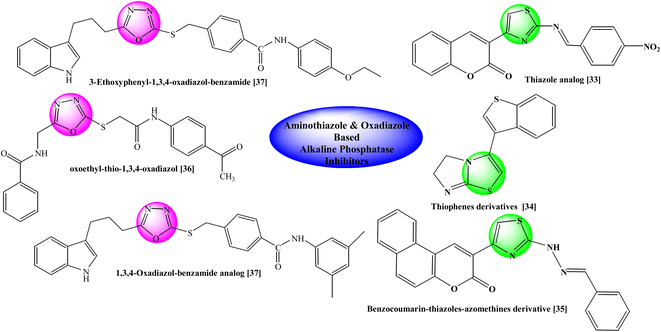
Previously some bi-heterocyclic compounds have been reported as alkaline phosphatase inhibitors shown in Fig. 2.33–37 However over major objective is to discover some unique and safe inhibitors of this enzyme to overcome the problems related to this enzyme. So, the current study was expanded to explore new therapeutic potentials of some new hybrid molecules as alkaline phosphatase inhibitors. In silico studies were also carried out to ascertain different types of interactions with the binding site of the enzyme to explore the therapeutic potentials of some oxadiazole thiazole hybrids as alkaline phosphatase inhibitors. Moreover, their cytotoxicity was also assessed to find their utility as harmless drug candidates in drug discovery and development.
 |
| | Fig. 2 Rationale of the current study. | |
2. Experimental
2.1 Chemistry
Chemicals were obtained from Sigma Aldrich & Alfa Aesar (Germany), and analytical grade solvents were provided by local suppliers. Melting points (uncorrected) were measured on Griffin and George apparatus with the help of capillary tubes. The initial purity of the compound was sensed in thin-layer chromatography (TLC) by means of pre-coated silica gel Al-plates. Ethyl acetate and n-hexane were used as solvent systems (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70). The spots were identified by UV254. Jasco-320-A spectrometer was used to record the IR spectra (ν, cm−1) using KBr pellet method. Bruker Advance III 600 Ascend spectrometer using a BBO probe was used for measuring the Signals in DMSO-d6 of 1H-NMR (δ, ppm) at 600 MHz and 13C-NMR at 150 MHz. For elemental analyses, the Foss Heraeus CHN-O-Rapid instrument was used and the theoretical values were within ±0.4%. Chemical shift (δ) is given in ppm and the coupling constant (J) in Hz. The different abbreviations used in the elucidation of 1H-NMR spectra are as follows: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; br. t, broad triplet; q, quartet; quint., quintet; m, multiplet; dist., distorted.
:70). The spots were identified by UV254. Jasco-320-A spectrometer was used to record the IR spectra (ν, cm−1) using KBr pellet method. Bruker Advance III 600 Ascend spectrometer using a BBO probe was used for measuring the Signals in DMSO-d6 of 1H-NMR (δ, ppm) at 600 MHz and 13C-NMR at 150 MHz. For elemental analyses, the Foss Heraeus CHN-O-Rapid instrument was used and the theoretical values were within ±0.4%. Chemical shift (δ) is given in ppm and the coupling constant (J) in Hz. The different abbreviations used in the elucidation of 1H-NMR spectra are as follows: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; br. t, broad triplet; q, quartet; quint., quintet; m, multiplet; dist., distorted.
2.2 Preparation of 3-bromo-N-(1,3-thiazol-2-yl)propanamide (3a) and 3-bromo-N-(5-methyl-1,3-thiazol-2-yl)propanamide (3b)
The required two electrophiles, 3a and 3b, were prepared by the reacting 1,3-thiazol-2-amine (1a) and 5-methyl-1,3-thiazol-2-amine (1b) separately with 3-bromopropanoyl chloride (2) in the equimolar amount by manual shaking in 5% aqueous solution of Na2CO3. Solid precipitates of respective 3a and 3b were formed in purified form, after stirring 10–20 minutes. The precipitates were filtered, washed with the help of cold distilled water and dried to use further.
2.3 General procedure for synthesis of 5-aryl-1,3,4-oxadiazole-2-thiols (7a–e)
The synthesis of five nucleophiles, 5-aryl-1,3,4-oxadiazole-2-thiols (7a–e) was carried out in multi-steps. In the first step, formation of respective esters was done. The reaction was initiated by using different types of aryl acids (4a–e). The corresponding acids (one by one) were refluxed with ethyl alcohol for 3–4 hours, by adding little quantity of sulfuric acid, which resulted in the formation of esters (5a–e). In the next step, esters converted into acid hydrazides (6a–e) by using MeOH/N2H4·H2O/refluxing for 4–6 hours. In the third step, cyclization was carried out to obtain the corresponding 5-aryl-1,3,4-oxadizole-2-thiols (7a–e) with the help of ethyl alcohol/CS2/KOH and refluxing for 3–6 h.
2.4 Procedure for synthesis of 3-[(5-aryl-1,3,4-oxadiazole-2-yl)sulfanyl]-N-(5-H/methyl-1,3-thiazol-2-yl)propanamides (8a–h)
The respective 5-aryl-1,3,4,oxadiazole-2-thiol (0.2 g; 1.0 mmol) was mixed in about 5 ml DMF. Then a pinch of lithium hydride was added almost in a 50 ml round-bottomed (RB) flask. This mixture was stirred for about 25–30 minutes. Then the equimolar amount of either electrophile (i.e. 3-bromo-N-(1,3-thiazol-yl)propanamide (3a)) and 3-bromo-N-(5-methyl-1,3-thiazol-2-yl)propanamide (3b) was added separately (one in each reaction) and the reaction mixture was stirred for 14–16 hours. The reaction completion was observed with TLC. A single spot was obtained through this process. Ice-chilled distilled water was added dropwise to the reaction mixture until the product was precipitated. The corresponding precipitates were filtered, washed with distilled water and dried to obtain the desired products (8a–h). All the compounds were re-crystallized from methanol to obtain the purified forms.
2.4.1 3-[(5-Phenyl-1,3,4-oxadiazol-2-yl)sulfanyl]-N-(1,3-thiazol-2-yl)propanamide (8a). Light brown amorphous solid; Yield: 82%; m.p.:120–121 °C; Mol. Formula: C14H12N4O2S2; Mol. Mass: 332 g mol−1. IR (KBr, ν, cm−1): 3334 (N–H, stretching), 3048 (C–H, str. of aromatic ring), 1665 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 2890 (–CH2 stretching), 1674 (aromatic CC stretching),1581 (CN stretching), 1198 (C–N–C bond stretching), 612 (C–S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.23 (s, 1H, CO–NH), 7.98 (dd, J = 1.5, 8.4, 2H, H-2′′ & H-6′′), 7.62 (tt, J = 1.5, 7.2, 1H, H-4′′), 7.58 (dist. br. t, J = 7.4, 2H, H-3′′ & H-5′′), 7.46 (d, J = 3.5, 1H, H-4′′′), 7.21 (d, J = 3.5, 1H, H-5′′′), 3.58 (t, J = 6.7, 2H, CH2-3), 3.05 (t, J = 3.5, 2H, CH2-2); 13C-NMR (150 MHz, DMSO-d6, δ, ppm):168.86 (C-1), 165.20 (C-5′), 163.38 (C-2′), 157.64 (C-2′′′), 137.59 (C-4′′′), 131.95 (C-4′′), 129.37 (C-3′′ & C-5′′), 126.37 (C-2′′ & C-6′′), 123.05 (C-1′′), 113.48 (C-5′′′), 34.77 (C-2), 27.51 (C-3). Anal. Calc. for C14H12N4O2S2 (332.40): C, 50.59; H, 3.64; N, 16.86. Found: C, 50.70; H, 3.94; N, 16.66.
O stretching), 2890 (–CH2 stretching), 1674 (aromatic CC stretching),1581 (CN stretching), 1198 (C–N–C bond stretching), 612 (C–S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.23 (s, 1H, CO–NH), 7.98 (dd, J = 1.5, 8.4, 2H, H-2′′ & H-6′′), 7.62 (tt, J = 1.5, 7.2, 1H, H-4′′), 7.58 (dist. br. t, J = 7.4, 2H, H-3′′ & H-5′′), 7.46 (d, J = 3.5, 1H, H-4′′′), 7.21 (d, J = 3.5, 1H, H-5′′′), 3.58 (t, J = 6.7, 2H, CH2-3), 3.05 (t, J = 3.5, 2H, CH2-2); 13C-NMR (150 MHz, DMSO-d6, δ, ppm):168.86 (C-1), 165.20 (C-5′), 163.38 (C-2′), 157.64 (C-2′′′), 137.59 (C-4′′′), 131.95 (C-4′′), 129.37 (C-3′′ & C-5′′), 126.37 (C-2′′ & C-6′′), 123.05 (C-1′′), 113.48 (C-5′′′), 34.77 (C-2), 27.51 (C-3). Anal. Calc. for C14H12N4O2S2 (332.40): C, 50.59; H, 3.64; N, 16.86. Found: C, 50.70; H, 3.94; N, 16.66.
2.4.2 3-{[5-(4-Hydroxyphenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(1,3-thiazol-2-yl)propanamide (8b). Light grey amorphous solid; Yield: 82%; m.p.:168–169 °C; Mol. Formula: C14H12N4O3S2; Mol. Mass: 348 g mol−1. IR (KBr, ν, cm−1): 3351 (N–H, stretching), 3098 (C–H, str. of aromatic ring), 1678 (CO str.), 2919 (–CH2 stretching), 16896(aromatic CC stretching), 1589 (CN stretching), 1203 (C–N–C bond stretching), 632 (C–S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.22 (s, 1H, CO–NH), 10.32 (s, 1H, –OH), 7.81 (br. d, J = 8.7 Hz, 2H, H-2′′ & H-6′′), 7.46 (d, J = 3.5, 1H, H-4′′′), 7.21 (d, J = 3.5, 1H, H-5′′′), 6.94 (br. d, J = 8.7, 2H, H-3′′ & H-5′′), 3.54 (t, J = 6.7, 2H, CH2-3), 3.03 (t, J = 6.7, 2H, CH2-2); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.79 (C-1), 165.35 (C-5′), 162.05 (C-2′), 160.66 (C-4′′), 157.57 (C-2′′′), 137.50 (C-4′′′), 128.29 (C-2′′ & C-6′′), 116.40 (C-3′′ & C-5′′), 113.73 (C-1′′), 113.39 (C-5′′′), 34.73 (C-2), 27.40 (C-3); Anal. Calc. for C14H12N4O3S2 (348.40): C, 48.26; H, 3.47; N, 16.08. Found: C, 48.70; H, 3.94; N, 16.66.
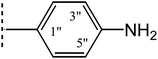
2.4.3. 3-{[5-(4-Aminophenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(1,3-thiazol-2-yl)propanamide (8c). Light grey amorphous solid; Yield: 88%; m.p.:148–149 °C; Mol. Formula: C14H13N5O2S2; Mol. Mass: 347 g mol−1. IR (KBr, ν, cm−1): 3346 (N–H, stretching), 3105 (C–H, str. of aromatic ring), 1688 (CO str.), 2932 (–CH2 stretching), 1693 (aromatic CC stretching), 1558 (CN stretching), 1171 (C–N–C bond stretching), 619 (C-S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.22 (s, 1H, CO–NH′), 7.62 (br. d, J = 8.7, 2H, H-2′′ & H-6′′), 7.46 (d, J = 3.5, 1H, H-4′′′), 7.21 (d, J = 3.5, 1H, H-5′′′), 6.66 (br. d, J = 8.7, 2H, H-3′′ & H-5′′), 3.51 (t, J = 6.7, 2H, CH2-3), 3.01 (t, J = 6.7, 2H, CH2-2); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.87 (C-1), 166.03(C-5′), 161.01 (C-2′), 157.65 (C-2′′′), 152.31 (C-4′′), 137.58(C-4′′′), 127.90 (C-2′′ & C-6′′), 113.49 (C-3′′ & C-5′′),113.46 (C-5′′′), 109.35 (C-1′′), 34.82 (C-2), 27.45 (C-3); Anal. Calc. for C14H13N5O2S2 (347.42): C, 48.40; H, 3.77; N, 20.16. Found: C, 48.62; H, 3.74; N, 20.34.
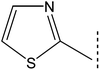
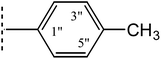
2.4.4. 3-{[5-(4-Methylphenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(1,3-thiazol-2-yl)propanamide (8d). Buffer-colored amorphous solid; Yield: 83%; m.p.:135–136 °C; Mol. Formula: C15H14N4O2S2; Mol. Mass: 346 g mol−1. IR (KBr, ν, cm−1): 3358 (N–H, stretching), 3090 (C–H, str. of aromatic ring), 1682 (CO str.), 2920 (–CH2 stretching), 1658 (aromatic CC stretching), 1578 (CN stretching), 1212 (C–N–C bond stretching), 621 (C–S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.22 (s, 1H, CO–NH), 7.87 (br. d, J = 8.1, 2H, H-2′′ & H-6′′), 7.46 (d, J = 3.5, 1H, H-4′′′), 7.39 (br. d, J = 8.0, 2H, H-3′′ &H-5′′), 7.21 (d, J = 3.5, 1H, H-5′′′), 3.57 (t, J = 6.7, 2H, CH2-3), 3.05 (t, J = 6.7, 2H,CH2-2), 2.39 (s, 3H, CH3-7′′); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.86(C-1), 165.29(C-5′),162.99 (C-2′), 157.64 (C-2′′′), 142.10 (C-4′′), 137.58 (C-4′′′), 129.90 (C-2′′ & C-6′′), 126.33 (C-3′′ & C-5′′), 120.31 (C-1′′), 113.46 (C-5′′′), 34.77 (C-2), 27.49(C-3), 21.10 (C-7′′); Anal. Calc. for C15H14N4O2S2 (346.43): C, 52.00; H, 4.07; N, 16.17. Found: C, 52.12; H, 4.28; N, 16.24.
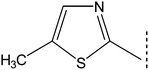
2.4.5. N-(5-Methyl-1,3-thiazol-2-yl)-3-[(5-phenyl-1,3,4-oxadiazol-2-yl)sulfanyl]propanamide (8e). Off-white colored amorphous solid; Yield: 88%; m.p.:117–118 °C; Mol. Formula:C15H14N4O2S2; Mol. Mass.: 346 g mol−1. IR (KBr, ν, cm−1): 3355 (N–H stretching), 3049 (C–H str. of aromatic ring), 2945 (–CH2– stretching), 1592 (aromatic CC stretching), 1584 (CN str.), 1188 (C–N–C bond str.), 637 (C–S str.); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.03 (s, 1H, –CO–NH), 7.97 (dist. dd, J = 1.5, 7.0, 2H, H-2′′ & H-6′′), 7.63 (br. t, J = 7.1, 1H, H-4′′), 7.58 (br. t, J = 7., H-3′′ & H-5′′), 7.11 (dist. d, J = 1.1, 1H, H-4′′′, due to allylic coupling), 3.56 (t, J = 6.7, 2H, CH2-3), 3.02 (t, J = 6.6, 2H, CH2-2), 2.32 (dist. d, J = 1.0 Hz, 3H, CH3-6′′′, due to allylic coupling); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.54 (C-1), 165.19 (C-5′), 163.38 (C-2′), 155.81 (C-2′′′), 134.68 (C-4′′′), 131.93 (C-4′′), 129.36 (C-3′′ & C-5′′), 126.36 (C-2′′ & C-6′′), 126.17 (C-5′′′), 123.06 (C-1′′), 34.76 (C-2), 27.58 (C-3), 11.03 (C-6′′′). Anal. Calc. for C15H14N4O2S2 (346.43): C, 52.00; H, 4.07; N, 16.17. Found: C, 52.22; H, 4.52; N, 16.20.
2.4.6. 3-{[5-(4-Hydroxyphenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(5-methyl-1,3-thiazol-2-yl)propanamide (8f). Light brown colored amorphous solid; Yield: 86%; m.p.:132–133 °C; Mol. Formula: C15H14N4O3S2; Mol. Mass.: 362 g mol−1. IR (KBr, ν, cm−1): 3352 (N–H str.), 3041 (C–H stretching of aromatic ring), 2949 (–CH2– str.), 1593 (CC stretching of aromatic ring), 1584 (CN stretching), 1191 (C–N–C bond str.), 634 (C–S str.); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 7.67 (br. d, J = 8.7, 2H, H-2′′ & H-6′′), 7.10 (dist. d, J = 1.3 Hz, 1H, H-4′′′, due to allylic coupling), 6.91 (br. d, J = 8.7, 2H, H-3′′ & H-5′′), 4.36 (t, J = 6.9, 2H, CH2-3), 2.98 (t, J = 6.9, 2H, CH2-2), 2.33 (dist. d, J = 1.1 Hz, 3H, CH3-6′′′, due to allylic coupling); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 174.93 (C-5′), 167.76 (C-1), 161.37 (C-2′), 158.96 (C-4′′), 155.90 (C-2′′′), 134.64 (C-4′′′), 128.17 (C-2′′ & C-6′′), 126.16 (C-5′′′), 116.28 (C-3′′ & C-5′′), 112.47 (C-1′′), 44.63 (C-3), 32.63 (C-2), 11.03 (C-6′′′). Anal. Calc. for C15H14N4O3S2 (362.43): C, 49.71; H, 3.89; N, 15.46. Found: C, 49.78; H, 3.99; N, 15.51.
2.4.7. 3-{[5-(4-Aminophenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(5-methyl-1,3-thiazol-2-yl)propanamide (8g). Dark brown colored amorphous solid; Yield: 86%; m.p.:142–143 °C; Mol. Formula: C15H15N5O2S2; Mol. Mass.: 361 g mol−1. IR (KBr, ν, cm−1): 3333 (N–H str.), 3082 (C–H stretching of aromatic ring), 2960 (–CH2– stretching), 1599 (CC stretching of aromatic ring), 1548 (CN stretching), 1194 (C-N-C bond stretching), 645 (C–S str.); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.02 (s, 1H, –CO–NH), 7.61 (br. d, J = 8.6, 2H, H-2′′ & H-6′′), 7.11 (dist. d, J = 1.2, 1H, H-4′′′, due to allylic coupling), 6.66 (br. d, J = 8.7, 2H, H-3′′ & H-5′′), 5.94 (br. s, 2H, NH2-4′′), 3.50 (t, J = 6.7, 2H, CH2-3), 2.98 (t, J = 6.7, 2H, CH2-2), 2.33 (dist. d, J = 1.1, 3H, CH3-6′′′, due to allylic coupling); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.56 (C-1), 166.02 (C-5′), 161.01 (C-2′), 155.83 (C-2′′′), 152.31 (C-4′′), 134.67 (C-4′′′), 127.89 (C-2′′ & C-6′′), 126.16 (C-5′′′), 113.48 (C-3′′ & C-5′′), 109.36 (C-1′′), 34.80 (C-2), 27.51 (C-3), 11.04 (C-6′′′). Anal. Calc. for C15H15N5O2S2 (361.44): C, 49.84; H, 4.18; N, 19.38. Found: C, 49.97; H, 4.22; N, 19.50.
2.4.8. N-(5-Methyl-1,3-thiazol-2-yl)-3-{[5-(3-nitrophenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}propanamide (8h). Orange colored amorphous solid; Yield: 83%; m.p.: 158–159 °C; Mol. Formula: C15H13N5O4S2; Mol. Mass.: 391 g mol−1. IR (KBr, ν, cm−1): 3357 (N–H str.), 3032 (C–H stretching of aromatic ring), 2928 (–CH2– stretching), 1589 (CC str. of aromatic ring), 1558 (CN stretching), 1177 (C–N–C bond stretching), 621 (C–S stretching); 1H-NMR (600 MHz, DMSO-d6, δ, ppm, J in Hz): 12.02 (s, 1H, –CO–NH), 8.63 (dist. t, J = 1.9, 1H, H-2′′), 8.45 (dist. dd, J = 1.4, 8.3, 1H, H-4′′), 8.39 (dist. dd, J = 1.1, 8.2, 1H, H-6′′), 7.89 (br. t, J = 8.0, 1H, H-5′′), 7.09 (dist. d, J = 1.3, 1H, H-4′′′, due to allylic coupling), 3.60 (t, J = 6.7 Hz, 2H, CH2-3), 3.02 (t, J = 6.6, 2H, CH2-2), 2.31 (dist. d, J = 1.1, 3H, CH3-6′′′, due to allylic coupling); 13C-NMR (150 MHz, DMSO-d6, δ, ppm): 168.52 (C-1), 164.41 (C-5′), 163.70 (C-2′), 155.79 (C-2′′′), 148.20 (C-3′′), 134.66 (C-4′′′), 132.42 (C-6′′), 131.27 (C-5′′), 126.25 (C-4′′), 126.16 (C-5′′′), 124.56 (C-1′′), 120.84 (C-2′′), 34.79 (C-2), 27.71 (C-3), 11.00 (C-6′′′). Anal. Calc. for C15H13N5O4S2 (391.43): C, 46.03; H, 3.35; N, 17.89. Found: C, 46.37; H, 3.52; N, 17.78.
2.5 Alkaline phosphatase assay
The activity of calf intestinal alkaline phosphatase (CIALP) was measured by the spectrophotometric assay as previously described by.38,39 The reaction mixture comprised of 50 mM Tris-HCl buffer (5 mM MgCl2, 0.1 mM ZnCl2 pH 9.5), the compound (0.1 mM with final DMSO 1% (v/v)) and the mixture was pre-incubated for 10 min by adding 5 μl of CIALP (0.025 U ml−1). Then, 10 μl of a substrate (0.5 mM p-NPP (p-nitrophenylphosphate disodium salt) was added to initiate the reaction and the assay mixture was incubated again for 30 min at 37 °C. The change in absorbance of released p-nitrophenolate was monitored at 405 nm, using a 96-well microplate reader (SpectraMax ABS, USA). All the experiments were repeated three times in a triplicate manner. KH2PO4 was used as the reference inhibitor of CIALP.
The Alkaline Phosphatase activities (Table 1) were calculated according to the following formula:
| Alkaline phosphatase activity (%) = (ODcontrol − ODsample × 100)/ODcontrol |
where OD
control and OD
sample represents the optical densities in the absence and presence of the sample, respectively.
Table 1 Alkaline phosphatase inhibitory activity of bi-heterocyclic propanamide (8a–h)a
| Compounds |
1,3-thiazole part |
Phenyl/substituted aryl part |
Alkaline phosphatase inhibitory activity IC50 ± SEM (μM) |
Hemolysis (%) (Mean ± SEM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. IC50 values were calculated by nonlinear regression using GraphPad Prism 5.0. |
| 8a |
 |
 |
3.977 ± 0.356 |
7.79 ± 0.02 |
| 8b |
 |
 |
5.987 ± 0.417 |
5.68 ± 0.04 |
| 8c |
 |
 |
8.731 ± 0.664 |
7.58 ± 0.01 |
| 8d |
 |
 |
1.878 ± 0.071 |
8.32 ± 0.03 |
| 8e |
 |
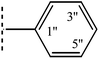 |
6.316 ± 0.551 |
6.84 ± 0.03 |
| 8f |
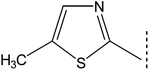 |
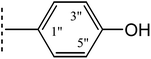 |
2.157 ± 0.118 |
7.47 ± 0.05 |
| 8g |
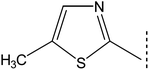 |
 |
0.211 ± 0.019 |
8.82 ± 0.02 |
| 8h |
 |
 |
3.745 ± 0.981 |
8.63 ± 0.02 |
| KH2PO4 (Standard) |
5.242 ± 0.473 |
— |
| Triton X (Standard) |
— |
89.00 ± 0.01 |
2.6 Kinetic analysis
On the basis of IC50 results, it was rational to select the most potent inhibitor 8g for determining the mechanism of enzyme inhibition, by following our reported method.38 The inhibitor concentrations were used at 0.00, 0.106, 0.211 and 0.422 μM. Substrate p-NPP concentrations were 10, 5, 2.5, 1.25, 0.625 and 0.3125 mM. Pre-incubation time and other conditions were the same as described in the alkaline phosphatase inhibition assay section. Maximal initial velocities were determined from the initial linear portion of absorbances up to 10 minutes after the addition of enzyme at minute's interval. The inhibition type on the enzyme was assayed by Lineweaver–Burk plot of inverse of velocities (1/V) versus the inverse of substrate concentration 1/[S] mM−1. The EI dissociation constant Ki was determined by a secondary plot of 1/V versus inhibitor concentration.
2.7 Computational methodology
2.7.1. Protein preparation. The X-ray Crystal Structure of human placental alkaline phosphatase (PDB ID: 1ZED) was retrieved from a protein data bank (https://www.rcsb.org/) with a resolution of 1.57 Å. The protein preparation was carried out through MGL tools. The water molecules were removed, and polar hydrogens were added and Kollmann charges were added. The attached p-nitrophenyl-phosphate ligand was removed.
2.7.2. Binding energy and intermolecular analysis. The popular technique used for the analysis of drug and protein interaction is molecular docking. Molecular docking was performed through CB-Dock (http://cao.labshare.cn/cb-dock/) to analyze the binding regions of protein with synthesized compounds. The best pose of the ligands was selected and docked with the allosteric site of human placental alkaline phosphatase. The binding centre for an allosteric site is taken as x = 27, y = 27 z = 27. The visualization tool Discovery Studio Visualizer (DSV) was used for the analysis of docked complex.
2.8 Hemolytic activity
Bovine blood samples were collected in EDTA diluted with salt (0.9% NaCl), and centrifuged at 1000×g for 10 minutes. Extracted erythrocytes in phosphate saline buffer of pH 7.4 and a suspension were made. Add 20 μl of solution (10 mg ml−1) of a synthetic compound in 180 μl suspension of RBCs and incubate for 30 minutes at room temperature. PBS was used as negative control and 100-X Triton was taken as a positive control.40,41 Hemolysis percentage was calculated by using the formula:
3. Results and discussion
3.1 Chemistry
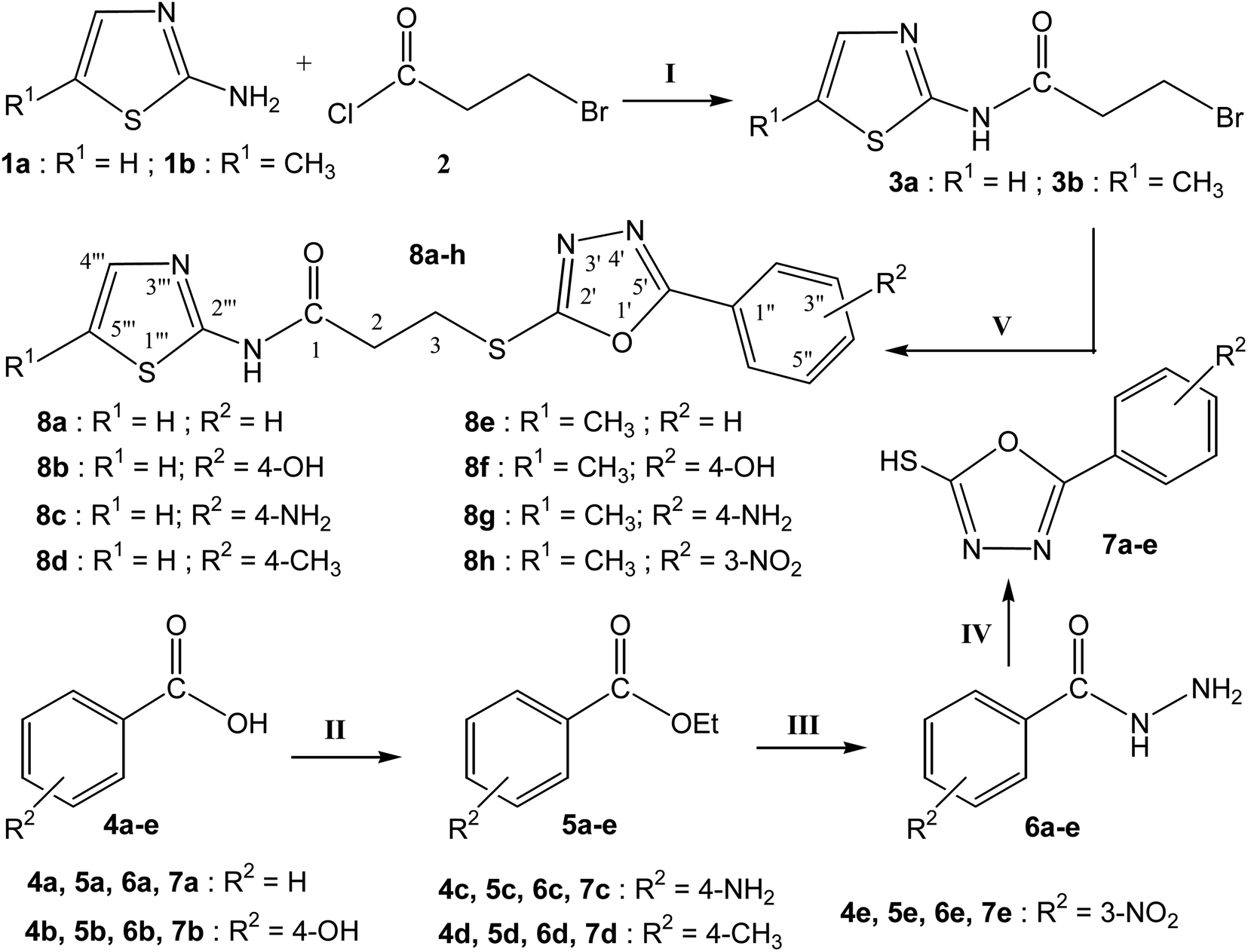
The purpose of the study presented here was to produce new bi-heterocyclic molecules containing a thiazole and an oxadiazole in the sole skeleton, linked with a propanamide moiety. The synthesis of the designed 3-[(5-aryl-1,3,4-oxadiazol-2-yl)sulfanyl]-N-(1,3-thiazol-2-yl) propanamides (8a–d)and 3-[(5-aryl-1,3,4-oxadiazol-2-yl)sulfanyl]-N-(5-methyl-1,3-thiazol-2-yl) propanamides (8e–h) have been outlined in the Scheme 1. The convergent process was initiated by reacting 1,3-thiazol-2-amine (1a) and 5-methyl-1,3-thiazol-2-amine (1b), separately, with 3-bromopropanoyl chloride (2) to obtain two electrophiles, namely, 3-bromo-N-(1,3-thiazol-2-yl)propanamide (3a) and 3-bromo-N-(5-methyl-1,3-thiazol-2-yl)propanamide (3b). In a side-wise set of reactions, the sequential conversion of aryl acids (4a–e) to ethyl esters (5a–e), and hydrazides (6a–e) were carried out followed by cyclization to respective 5-aryl-1,3,4-oxadizole-2-thiols (7a–e). These five nucleophiles were coupled with newly synthesized two electrophiles (3a and 3b) in a polar aprotic medium to acquire the required eight bi-heterocyclic hybrids, 8a–h.
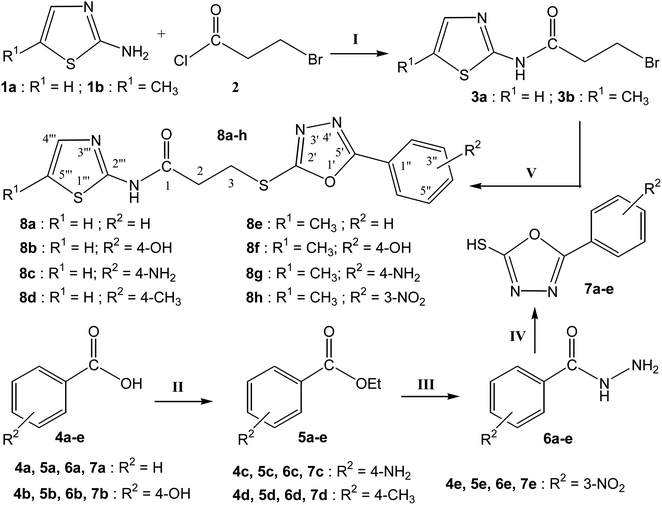 |
| | Scheme 1 Outline for the synthesis of 3-[(5-aryl-1,3,4-oxadiazole-2-yl)sulfanyal]-N-(5-H/methyl-1,3-thiazol-2-yl)propanamides (8a–h). Reagents & Conditions: (I) aq. 5% Na2CO3 soln/stirring. (II) EtOH/H2SO4/refluxing for 3–4 h (III) MeOH/N2H4·H2O/refluxing for 4–6 h (IV) EtOH/CS2/KOH/refluxing for 3–6 h (V) DMF/LiH/stirring for 1–2 h. R1 & R2 = Different substituents as depicted in the final derivatives (8a–h). | |
For the lucidity of the reader, a comprehensive structural analysis of a compound is discussed hereby. The molecular formula, C15H15N5O2S2, of compound 8g was recognized by its CHN analysis data and by counting the number of resonances in its 1H-NMR spectrum and 13C-NMR spectrum. The IR spectrum of this molecule was helpful to ascertain the different functionalities, by virtue of the absorption bands seen at 3333 cm−1 for stretching of N–H, 3082 cm−1 for aromatic C-H stretching, 2960 cm−1 for the stretching of the CH2 molecule and 1599 cm−1 for aromatic CC stretching. A broad singlet in the most downfield region of its 1H-NMR spectrum δ 12.02 ppm was assigned to a hetero-atom proton (–N–H) of the amide group. The 4-aminophenyl group in this molecule was clearly identified by two ortho-coupled doublets, at δ 7.61 (br. d, J = 8.6, 2H, H-2′′ & H-6′′) and 6.66 (br. d, J = 8.7, 2H, H-3′′ & H-5′′) along with a broad signal at δ 5.94 which was assignable to an amino group (–NH2) present at para-position of this aromatic ring. The 5-methyl-1,3-thiazol-2-yl group attached to the nitrogen atom was characterized by two allylically coupled distorted doublets at δ 7.11 (J = 1.2, 1H, H-4′′′), and 2.33 (J = 1.1, 3H, CH3-6′′′). The propanamide unit in the molecule was coherent by two adjacent methylene groups, identified by two triplets at δ 3.50 (J = 6.7, 2H, CH2-3) and 2.98 (J = 6.7, 2H, CH2-2), whereby the chemical shift of the former methylene was rational for its connectivity to a sulfur atom. The 1H-NMR spectrum of this molecule has been shown in Fig. S1-A† while Fig. S1-B† displayed the expanded aromatic region. The expanded aliphatic region of this spectrum has been shown in Fig. S1-C.†
The 13C-NMR spectrum (Fig. S2†) of this molecule exhibited overall thirteen carbon resonances, owing to two sets of symmetrical duplet carbons in 4-aminophenyl group, which was characterized with overall four signals, two quaternary at δ 152.31 (C-4′′)&109.36 (C-1′′), along with two sets of duplet methines at 127.89 (C-2′′ & C-6′′), and 113.48 (C-3′′ & C-5′′). The 1,3,4-oxadiazole heterocycle was clearly signified by two quaternary signals at δ 166.02 (C-5′) and 161.01 (C-2′). Similarly, the other heterocycle, 5-methyl-1,3-thiazol-2-yl, attached to a nitrogen atom, was recognized by two quaternary signals at δ 155.83 (C-2′′′) and 126.16 (C-5′′′), one methine signal at 134.67 (C-4′′′), and one methyl carbon at 11.04 (CH3-6′′′). The two heterocycles were embraced with a propanamide unit, which was manifested by an amidic carbonyl carbon at δ 168.56 (C-1), along with two linearly connected methylenes at δ 34.80 (C-1) and 27.51 (C-3). The chemical shift of the latter methylene was and articulated for its connectivity to the sulfur atom in the molecule.42 Thus, based upon all aforementioned collective evidences, the structure of 8g was confirmed and it was named as is 3-{[5-(4-aminophenyl)-1,3,4-oxadiazol-2-yl]sulfanyl}-N-(5-methyl-1,3-thiazol-2-yl)propanamide. Likewise, the structures of all other synthesized compounds were affirmed by a similar structural analysis (Fig. S3–S20†).
3.2 Alkaline phosphatase inhibitory activity and structure–activity relationship (SAR)
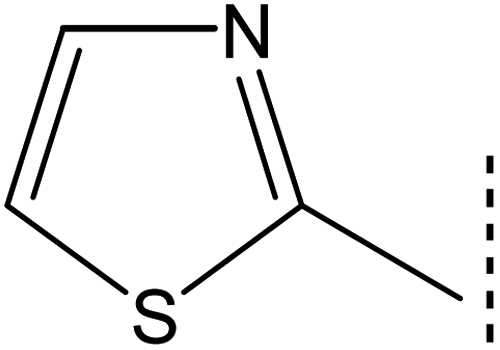
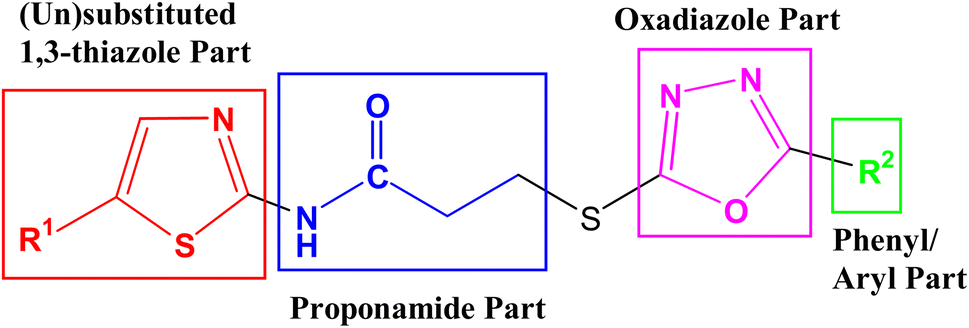
The intended 1,3-thiazole-oxadiazole bi-heterocyclic hybrids (8a–h), connected with a propanamide unit, were subjected to alkaline phosphatase inhibition to find out their potential. A substantial inhibition was demonstrated by all these molecules and the results obtained thereafter have been presented in Table 1. It was noteworthy that all molecules exhibited much lower IC50 (μM) values relative to the standard (KH2PO4), having an IC50 value of 5.242 ± 0.473 μM. It is rational that the presented activity is mainly the characteristic of the whole molecule; nevertheless, a fractional SAR might be predicted by examining the impact of varying substituent on the inhibitory potential. In our molecules, it was the phenyl/aryl part only, which was undergoing substitutional variations and all other parts were remaining intact in all molecules. Therefore, the SAR contemplation was focused primarily on different groups lying in the phenyl/aryl part. The general structural units of our designed compounds are demarcated in Fig. 3.
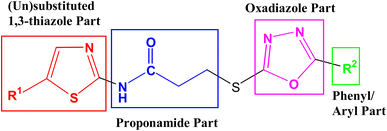 |
| | Fig. 3 General structural parts of compounds, 8a–h. | |
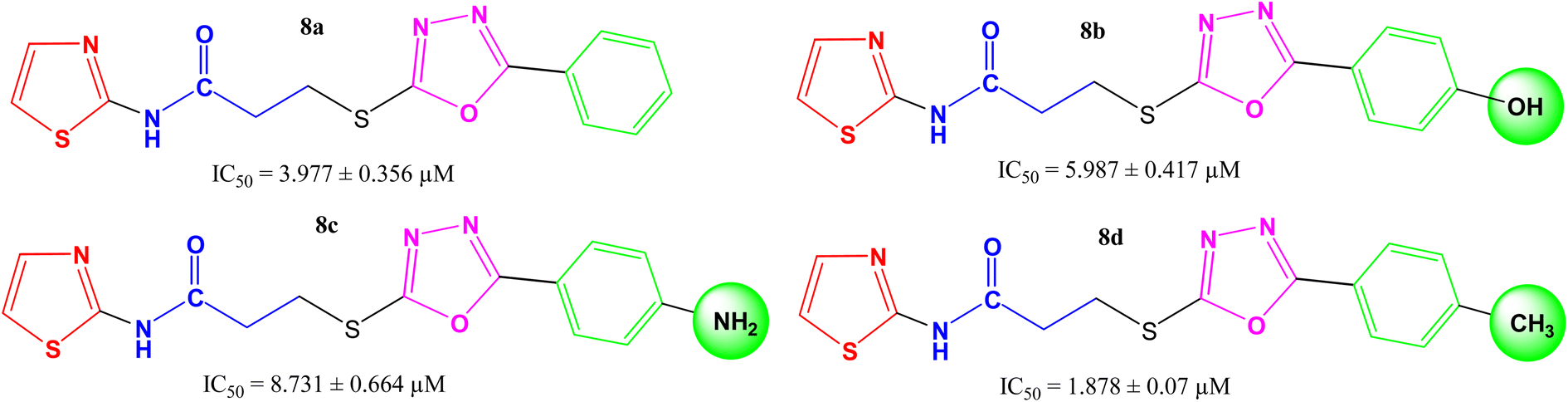
Amongst the four N-(1,3-thiazol-2-yl) bearing propanamides (8a–d), the compound 8d in which the methyl groups was present on para position in aryl part (4-methylphenyl) exhibited better inhibitory activity (IC50 = 1.878 ± 0.07 μM) as compared to other analogues. However, 8a having (un-substituted) phenyl group, had good activity (IC50 = 3.977 ± 0.356 μM), as compared to other analogues 8b (IC50 = 5.987 ± 0.417 μM) and 8c (8.731 ± 0.664 μM) in which 4-hydroxy and 4-amino groups, respectively, were present at the para positions of aryl part. It means, a non-polar small-sized group at the para-position of aryl entity, as –CH3, in 8d, was prone to enhance the inhibitory activity and in fact, this compound was identified as the second most potent molecule among the series (Fig. 4).
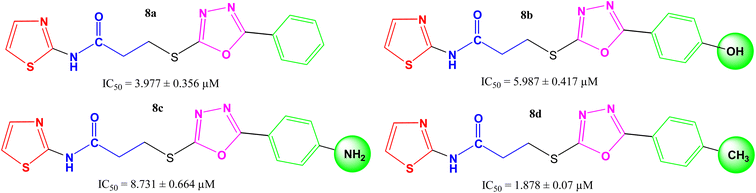 |
| | Fig. 4 Structure–activity relationships of compounds, 8a–8d. | |
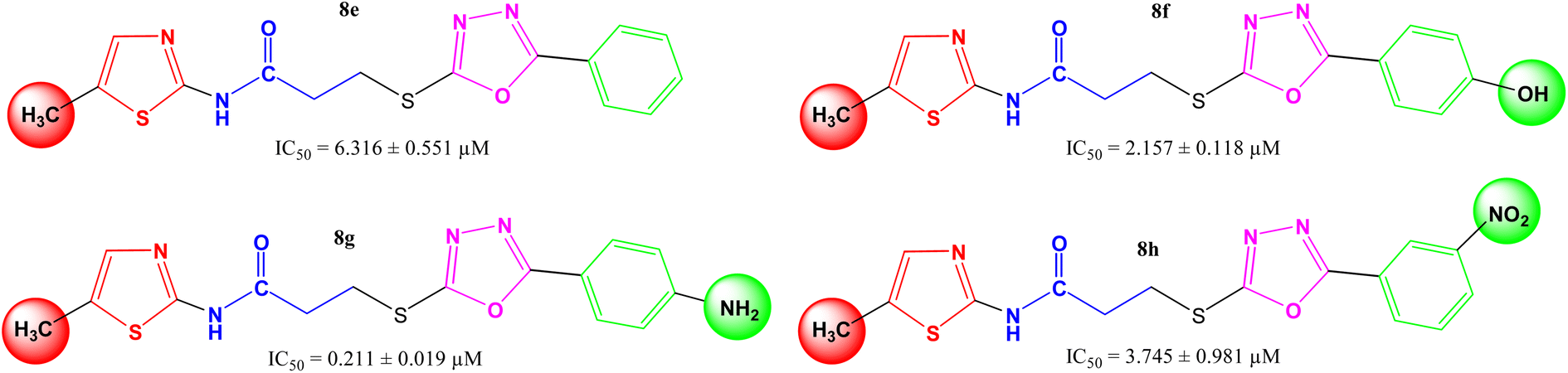
In other four derivatives (8e–8h), the non-polar methyl group was embedded in the thiazole part viz. N-(5-methyl-1,3-thiazol-2-yl) entity (Fig. 5), and hereby, it was inferred that now the compound 8g bearing a polar 4-amino group in the aryl part behaved the most potent molecule in the synthetic series, having IC50 = 0.211 ± 0.019 μM. It means, in such skeletons, a non-polar methyl group in the thiazole part and a polar group with considerable −I and +R effects in the aryl part might be rationalized as suitable components for the enhanced inhibition of the studied enzyme by the molecule as a whole.
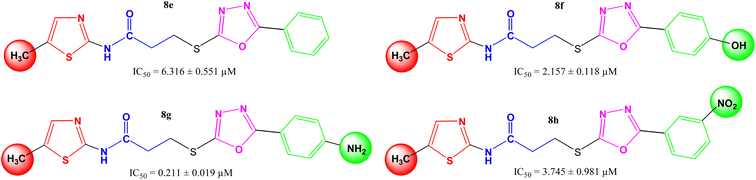 |
| | Fig. 5 Structure–activity relationships of compounds, 8e–8h. | |
3.3 Kinetic mechanism
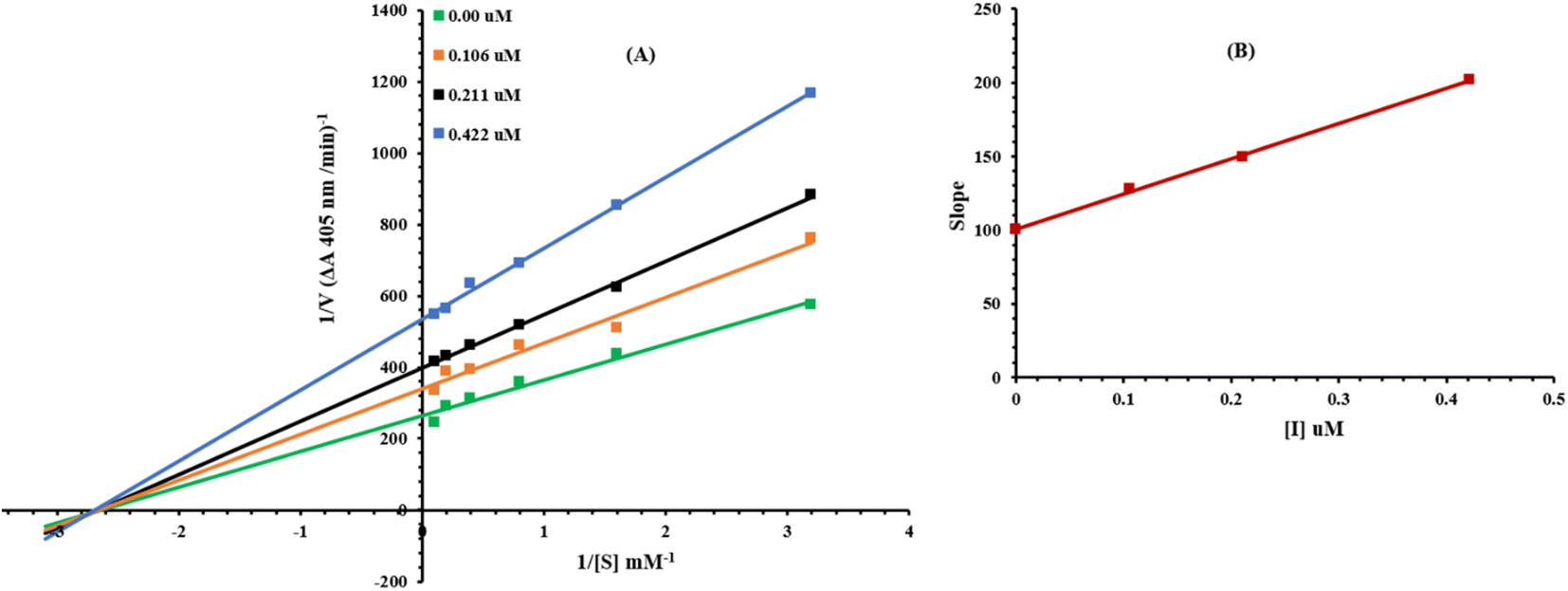
Presently, the most potent compound 8g was studied for their mode of inhibition against alkaline phosphatase. The potential of the compound to inhibit the free enzyme and enzyme–substrate complex was determined in terms of EI and ESI constants respectively. The kinetic studies of the enzyme by the Lineweaver–Burk plot of 1/V versus substrate para-nitrophenyl phosphate disodium salt 1/[S] in the presence of different inhibitor concentrations gave a series of straight lines as shown in Fig. 6A. The results of compound 8g showed that it intersected within the second quadrant. The analysis showed that Vmax decreased to new increasing doses of inhibitors on the other hand Km remained the same. This behavior indicated that 8g inhibited the alkaline phosphatase non-competitively to form an enzyme inhibitor complex. The secondary plot of slope against the concentrations of inhibitor showed enzyme-inhibitor dissociation constant (Ki) Fig. 6B. The kinetic results are presented in Table 2.
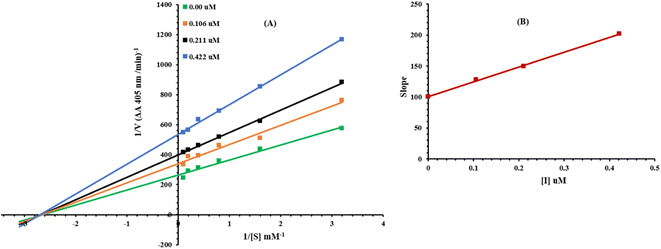 |
| | Fig. 6 Lineweaver–Burk plots for inhibition of alkaline phosphatase in the presence of compound 8g (A). Concentrations of 8g were 0.00, 0.106, 0.211 and 0.422 μM. (B) The insets represent the plot of the slope versus inhibitor 8g concentrations to determine inhibition constant. The lines were drawn using linear least squares fit. | |
Table 2 Kinetic parameters of the alkaline phosphatase for p-nitrophenylphosphate disodium salt activity in the presence of different concentrations of 8ga
| Concentration (μM) |
Vmax (ΔA min−1) |
Km (mM) |
Inhibition type |
Ki (μM) |
| Vmax = the reaction velocity; Km = Michaelis–Menten constant; Ki = EI dissociation constant. |
| 0.00 |
0.004082697 |
0.36 |
Non-competitive |
0.42 |
| 0.106 |
0.003003636 |
0.36 |
| 0.211 |
0.002405612 |
0.36 |
| 0.422 |
0.001825994 |
0.36 |
3.4 Molecular docking results
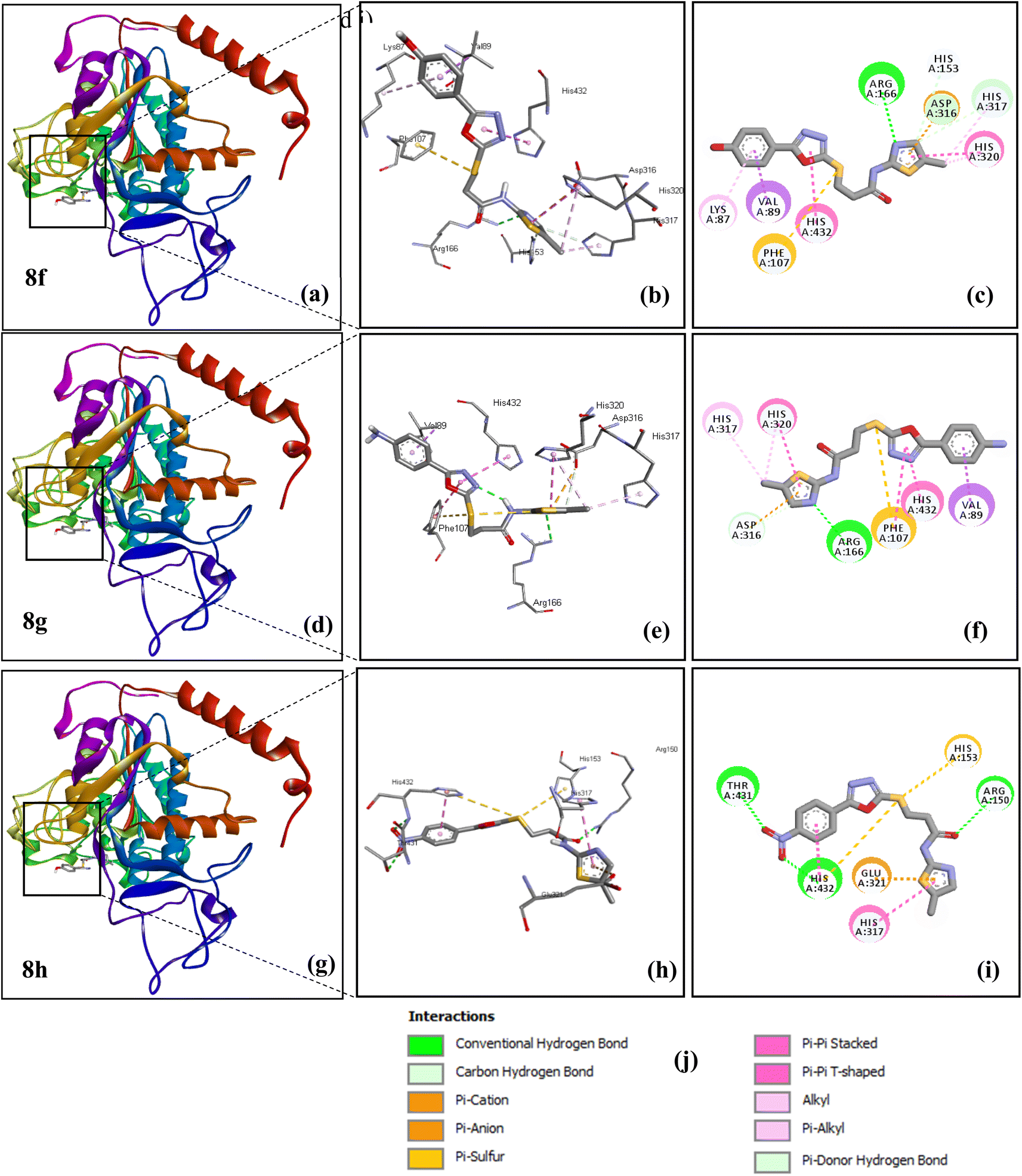
3.4.1. Compounds complexed with allosteric site. The binding energies of all the complexes are given in Table 3 and their illustration corresponding to the inhibitory activity of derivatives (8f, 8g and 8h) for an allosteric site is exhibited in Fig. 7. Results showed that all the ligands, 8a–h, exhibited good docking energy values and showed their interaction within the active region of target protein (Table 3). Among all of the docked ligands 8f, 8g and 8h showed lowest binding energy. The binding energy of compound 8f is −7.1 Kcal mol−1 with allosteric site. The best docked ligand pose showed conventional hydrogen bond and carbon hydrogen bond with ARG166, HIS153 and HIS317 amino acid residues. Different types of hydrophobic interactions (π-sulphur, π-cation, π-anion, π-alkyl and π–π stacked) with ASP316, HIS320, HIS317, VAL89, LYS87, PHE107 and HIS432 amino acid residues were shown in allosteric site. The docked pose of compound 8f in the allosteric site in terms of 3D and 2D interactions is shown in Fig. 7a–c.
Table 3 Binding energies of derivatives (8a–8h) for Allosteric site of human placental alkaline phosphatase
| Compounds |
Protein |
Binding energy (Kcal mol−1) |
| 8a |
Human placental alkaline phosphatase |
−6.8 |
| 8b |
−6.9 |
| 8c |
−6.9 |
| 8d |
−7.1 |
| 8e |
−7.1 |
| 8f |
−7.1 |
| 8g |
−7.2 |
| 8h |
−7.4 |
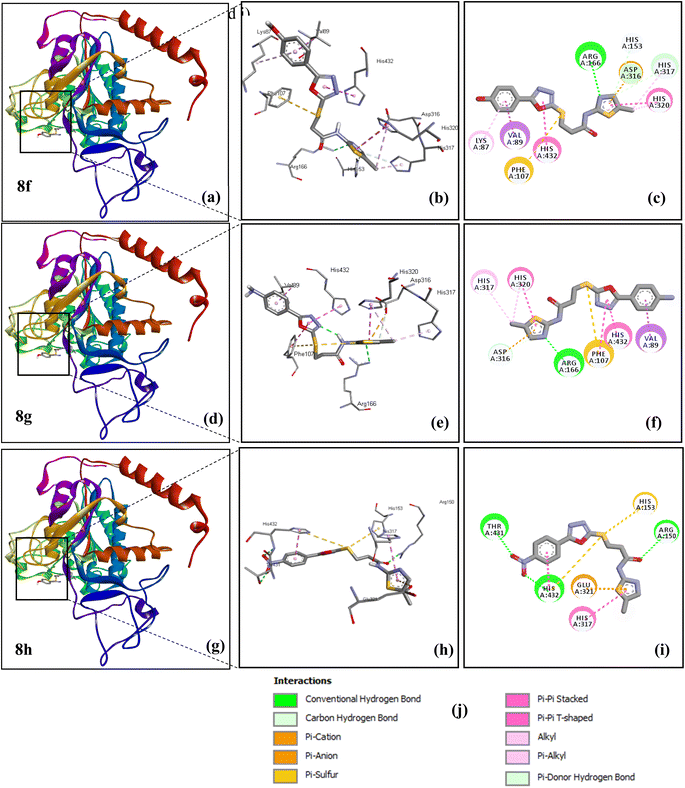 |
| | Fig. 7 The diagram shows the 3D structure and. 2D binding interactions of compounds 8f (a, b and c), 8g (d, e and f), 8h (g, h and i) with allosteric site of human alkaline phosphatase. The type of interactions shown by these compounds is given (j). | |
The binding energy of compound 8g is −7.2 Kcal mol−1 with allosteric site. The different type of hydrophobic interactions (π-sulphur, π-sigma, π-anion, π–π stacked, π–π T-shaped, π-alkyl) with ASP316, HIS320, HIS317, VAL89, PHE107 and HIS432 amino acid residues. However amino acid residues ARG166 and ASP316 showed conventional hydrogen bond and carbon hydrogen bond, respectively. The docked pose of compound 8g in terms of 3D and 2D interactions is shown in Fig. 7d–f.
The compound 8h is −7.4 Kcal mol−1 with an allosteric site. This complex showed conventional hydrogen bonds with THR431, ARG150 and HIS432. The different types of hydrophobic interactions (π-sulphur,π-anion, π–π stacked, π–π T-shaped) with HIS432, GLU321, HIS153 and HIS317 amino acid residues. The docked pose of compound 8h in terms of 3Dand 2D interactions is shown in Fig. 7g–i. The docking information of 8a–8e is given in supplementary part (Fig. S21–S23†).
3.5 Hemolytic activity
All the synthesized derivatives, 8a–h, were also imperiled to hemolysis to find out their cytotoxicity profile. The results of percentage hemolysis are shown in Table 1. Our results showed that all compounds of this series exhibited mild toxicity towards red blood cell membrane and hence might be rationalized as safe agents.
4. Conclusions
In conclusion, a new series of thiazole–oxadiazole hybrids, embraced with propanamide entity, was synthesized with a convergent approach, as a promising inhibitor of alkaline phosphatase. The in silico computational ascriptions were also in full agreement with in vitro inhibition results and showed good interaction within the active region of the target protein, having a good binding energy value. All these compounds exhibited mild hemolysis of red blood cells; therefore, these bi-heterocyclic molecules might be utilized as safe and suitable therapeutic agents for the treatment of alkaline phosphatase associated ailments.
Author contributions
Abdul Rehman Sadiq Butt: investigation, writing – original draft. Muhammad Athar Abbasi: supervision, writing – review & editing. Aziz-ur-Rehman: investigation, methodology. Sabahat Zahra Siddiqui: methodology, data curation. Shabbir Muhammad: software. Hussain Raza: investigation, formal analysis. Syed Adnan Ali Shah: investigation, validation. Muhammad Shahid: methodology, data curation. Abdullah G. Alsehemi: visualization, formal analysis. Song Ja Kim: supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors from Pakistan are thankful to HEC Pakistan for facilitation in spectral analysis. The authors from Kongju National University acknowledge the National Research Foundation of Korea (NRF) funded by the Korean Government (MEST) (2020R1I1A306969914). The authors from King Khalid University extend their appreciation to the Deanship of Scientific Research at King Khalid University for support through the Research Project R.G.P.2/272/44.
References
- B. D. Vanjare, P. G. Mahajan, N. C. Dige, H. Raza, M. Hassan, Y. Han, S. J. Kim, S.-Y. Seo and K. H. Lee, Mol. Diversity, 2021, 25, 2089–2106 CrossRef CAS PubMed
 .
. - D. Das, P. Sikdar and M. Bairagi, Eur. J. Med. Chem., 2016, 109, 89–98 CrossRef CAS PubMed .
- P. Y. Lin, R. S. Hou, H. M. Wang, I. J. Kang and L. J. Chen, J. Chin. Chem. Soc., 2009, 56, 455–458 CrossRef CAS .
- S. M. Gomha, S. M. Riyadh, I. M. Abbas and M. A. Bauomi, Heterocycles, 2013, 87, 341–356 CrossRef CAS .
- R. S. Koti, G. D. Kolavi, V. S. Hegde and I. M. Khaji, Indian J. Chem., 2006, 45, 1900–1904 Search PubMed .
- S. J. Kashyap, V. K. Garg, P. K. Sharma, N. Kumar, R. Dudhe and J. K. Gupta, Med. Chem. Resear., 2012, 21, 2123–2132 CrossRef CAS .
- K. M. Khan, N. Ambreen, A. Karim, S. Saied, A. Amyn, A. Ahmed and S. Perveen, J. Pharm. Res., 2012, 5, 651–656 Search PubMed .
- K. Tsuji and H. Ishikawa, Bioorg. Med. Chem. Lett., 1994, 4, 1601–1606 CrossRef CAS .
- J. S. Cartner, S. Kramer, J. J. Talley, T. Penning, P. Collins, M. J. Graneto, K. Seibert, C. M. Koboldt, J. Masferrer and B. Zweifel, Bioorg. Med. Chem. Lett., 1999, 1171–1174 CrossRef PubMed .
- L. Tommesani, G. Brunoro, A. Frignani, C. Monticelli and M. Dal Colle, Corros. Sci., 1997, 39, 1221–1237 CrossRef CAS .
- K. Es-Salah, M. Keddam, K. Rahmouni, A. Srhiri and H. Takenouti, Electrochim. Acta, 2004, 49, 2771–2778 CrossRef CAS .
- A. Nagiub and F. Mansfeld, J. Corros. Sci., 2001, 43, 2147–2171 CrossRef CAS .
- L. Y. Wang, C. X. Zhang, Z. Q. Liu, D. Z. Lio, Z. H. Jang and S. P. Yan, Inorg. Chem. Comm., 2003, 6, 1255–1258 CrossRef CAS .
- A. H. Al-Dujali, A. T. Atto and A. M. Al-Kurde, Eur. Polym. J., 2001, 37, 927–932 CrossRef .
- Y. Li, Y. Xu, X. Qian and B. Qu, Tetrahedron Lett., 2004, 45, 1247–1251 CrossRef CAS .
- V. C. Rucker, S. Foister, C. Melander and P. B. Dervan, J. Am. Chem. Soc., 2003, 125, 1195–1202 CrossRef CAS PubMed .
- Q. Wang, H. Li, Y. Li and R. Huang, J. Agric. Food Chem., 2004, 52, 1918–1922 CrossRef CAS PubMed .
- K. Yanagimoto, K. G. Lee, H. Ochi and T. Shibamoto, J. Agric. Food Chem., 2002, 50, 5480–5484 CrossRef CAS PubMed .
- L. Laine, A. J. Kivitz, A. E. Bello, A. Y. Grahn, M. H. Schiff and A. S. Taha, Am. J. Gastroenterol., 2012, 107, 379–386 CrossRef CAS PubMed .
- C. Borelli, M. Schaller, M. Niewerth, K. Nocker, B. Baasner, D. Berg, R. Tiemann, K. Tietjen, B. Fugmann, S. Lang-Fugmann and H. C. Korting, Chemotherapy, 2008, 54, 245–259 CrossRef CAS PubMed .
- D. R. P. Guay, Clin. Ther., 2002, 24, 473–489 CrossRef CAS PubMed .
- A. R. S. Butt, M. A. Abbasi, A.-U.-Rehman, S. Z. Siddiqui, M. Hassan, H. Raza, S. A. A. Shah and S.-Y. Seo, Bioorg. Chem., 2019, 86, 197–209 CrossRef CAS PubMed .
- M. Arshad, Int. J. Pharma Sci. Res., 2014, 5, 1124–1137 Search PubMed .
- M. F. F. M. Aluwi, K. Rullah, T. H. Huan, C. K. Meng, T. S. Jie, L. S. Wei, A. H. Mansor, B. M. Yamine and L. K. Wai, RSC Adv., 2016, 6, 72177–72184 RSC .
- S. Rasool, A.-U.-Rehman, M. A. Abbasi, S. Z. Siddiqui and S. A. A. Shah, Braz. J. Pharm. Sci., 2016, 52, 471–482 CrossRef CAS .
- K. Nafeesa, A.-U.-Rehman, M. A. Abbasi, S. Z. Siddiqui, S. Rasool and S. A. A. Shah, Bull. Fac. Pharm. (Cairo Univ.), 2017, 55, 333–343 Search PubMed .
- J. L. Millan, Signal, 2006, 2, 335–341 CAS .
- J. P. Lalles, Nutr. Rev., 2014, 72, 82–94 CrossRef PubMed .
- E. A. Sergienko and J. L. Millán, Nat. Protoc., 2010, 5, 1431–1439 CrossRef CAS PubMed .
- K. A. Lomashvili, P. Garg, S. Narisawa, J. L. Millan and W. C. O'Neill, Kidney Int., 2008, 73, 1024–1030 CrossRef CAS PubMed .
- M. Muda, N. N. Rao and A. Torriani, J. Bacteriol., 1992, 174, 8057–8064 CrossRef CAS PubMed .
- M. Li, W. Ding, B. Baruah, D. C. Crans and R. Wang, J. Inorg. Biochem., 2008, 102, 1846–1853 CrossRef CAS PubMed .
- A. Saeed, S. A. Ejaz, M. Shehzad, S. Hassan, M. al-Rashida, J. Lecka and J. Iqbal, RSC Adv., 2016, 6(25), 21026–21036 RSC .
- M. Al-Rashida and J. Iqbal, Mini-Rev. Med. Chem., 2015, 15(1), 41–51 CrossRef CAS PubMed .
- P. A. Channar, H. Irum, A. Mahmood, G. Shabir, S. Zaib, A. Saeed and J. Iqbal, Bioorg. Chem., 2019, 91, 103137 CrossRef PubMed .
- Z. Iqbal, A. Iqbal, Z. Ashraf, M. Latif, M. Hassan and H. Nadeem, Drug Dev. Res., 2019, 80(5), 646–654 CAS .
- M. A. Abbasi, M. Nazir, A.-U.-Rehman, S. Z. Siddiqui, M. Hassan, H. Raza, S. A. A. Shah, M. Shahid and S.-Y. Seo, Arch. Pharm. Chem. Life Sci., 2019, e1800278 CrossRef PubMed .
- M. Rafiq, M. Saleem, M. Hanif, M. R. Maqsood, N. H. Rama, K. H. Lee and S.-Y. Seo, Bull. Korean Chem. Soc., 2012, 33(12), 3943–3949 CrossRef CAS .
- A. Saeed, G. Saddique, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan, H. Raza, T. A. Fattah and S.-Y. Seo, Bioorg. Med. Chem., 2018, 26(12), 3707–3715 CrossRef PubMed .
- P. Sharma and J. D. Sharma, J. Ethnopharmacol., 2001, 74(3), 239–243 CrossRef CAS PubMed .
- W. A. Powell, C. M. Catranis and C. A. Maynard, Lett. Appl. Microbiol., 2000, 31(2), 163–168 CrossRef CAS PubMed .
- M. A. Abbasi, M. S. Ramzan, A.-U.-Rehman, S. Z. Siddiqui, M. Hassan, S. A. A. Shah, M. Ashraf, M. Shahid and S.-Y. Seo, Iran. J. Pharm. Res., 2020, 19(1), 487–506 CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Aziz-ur-Rehmana,
Sabahat Zahra Siddiquia,
Shabbir Muhammad
*a,
Aziz-ur-Rehmana,
Sabahat Zahra Siddiquia,
Shabbir Muhammad