
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutron diffraction and DFT studies of oxygen defect and transport in higher-order Ruddlesden–Popper phase materials†
Mudasir A. Yatoo *ab,
Ieuan D. Seymoura and
Stephen J. Skinnerab
*ab,
Ieuan D. Seymoura and
Stephen J. Skinnerab
aImperial College London, Department of Materials, Faculty of Engineering, Exhibition Road, London, SW7 2AZ, UK. E-mail: m.yatoo15@imperial.ac.uk
bEPSRC Centre for Doctoral Training in Advanced Characterisation of Materials, Exhibition Road, London, SW7 2AZ, UK
First published on 5th May 2023
Abstract
A series of higher-order Ruddlesden–Popper phase materials – La3PrNi3O10−δ, La2Pr2Ni3O10−δ and LaPr3Ni3O10−δ – were synthesised and investigated by neutron powder diffraction to understand the oxygen defect structure and propose possible pathways for oxygen transport in these materials. Further complimentary DFT calculations of the materials were performed to support the experimental analysis. All of the phases were hypostoichiometric and it was observed that the majority of the oxygen vacancies were confined to the perovskite layers, with a preference for equatorial oxygen sites. A particular preference for vacancies in O(1) and O(5) sites at high temperatures was observed from neutron diffraction measurements which were further complimented by DFT calculations wherein the vacancy formation energy was found to be lowest at the O(1) site. Also, a preference for a curved oxygen transport pathway around the NiO6 octahedra was observed which agrees with the published literature for Ruddlesden–Popper phase materials. Lattice parameters for all three compositions showed a linear increase with increasing temperature, but the increase was greatest in the c parameter while the b parameter showed only a slight increase when compared to the a parameter. The thermal expansion coefficient was calculated for all compositions and was found to be in the range 13.0–13.4 × 10−6 °C−1, which is compatible with the commonly used electrolyte materials for solid oxide fuel cells.
1 Introduction
Mixed ionic–electronic conductivity has been identified as one of the important properties of materials to be used as electrodes in solid oxide fuel cells (SOFCs) and electrolysers (SOECs), collectively termed Solid Oxide Cells (SOCs). As such, ionic conductivity plays an important role in determining the performance of oxygen ion conductors as electrodes in SOCs. Ionic conductivity is the movement of an ion from one site to another through defects in the crystal. In oxygen ion conductors such as the Ruddlesden–Popper (RP) phase materials where the composition is An+1BnO3n+1, A = Ln and B = transition metals (n = 1–3), it is the oxygen defects (vacancies and/or interstitials) which determine the ionic conductivity because ionic conductivity of a material is directly proportional to the concentration of lattice defects. In this context, the investigation of the oxygen defects in these materials assumes importance.It is here that neutron powder diffraction (NPD) plays a crucial role because the neutron scattering length of an oxygen atom is sizeable, 5.805 ± 0.004 fm (ref. 1) and thus easily detectable by neutron powder diffraction. Neutron diffraction is a reliable method to determine the crystallographic positions of the oxide ions, their atomic displacement parameters and their occupancies, while X-rays have a low sensitivity to oxygen, particularly in a matrix of heavy atoms. This could be later used to understand the oxygen conduction mechanisms. All of these parameters are of particular interest in materials with high oxygen mobility such as the Ruddlesden–Popper phases. NPD can also be used to understand the lattice parameter evolution of electrode materials to obtain information such as the thermal expansion coefficient (TEC) of materials, a critical mechanical aspect for the use of these materials in high-temperature devices such as SOCs.
Ruddlesden–Popper phases were first synthesised by Ruddlesden and Popper in 1958.2 The structure consists of nABO3 perovskite layers which are sandwiched between two AO rock-salt layers.3 The lower-order (n = 1) phases such as La2NiO4+δ (LNO) and Pr2NiO4+δ (PNO) have been studied extensively.4–7 The main motivation for intense research focus on the n = 1 Ruddlesden–Popper phase materials stemmed from their superior capability of storing a substantial amount of interstitial oxygen in their structure which bestowed the materials with significant oxide ion conductivity, thereby rendering the materials mixed ionic–electronic conductors (MIEC) at intermediate temperatures.8,9
There has been an interesting and long debate on the nature of ionic conductivity in these materials. Therefore, detailed studies have been carried out on the LNO material and its transport properties are well understood. There is broad agreement regarding the dominant participation of interstitial oxygen atoms in ionic conductivity.10 The neutron diffraction studies by Demourgues et al. predicted that interstitial oxygen atoms are accommodated by the LaO rock salt layer.11 This was further confirmed by Jorgensen12 and co-workers and they determined the location of the oxygen interstitial site to be at 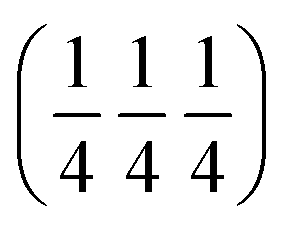 in the Fmmm structure. These studies had further support from Paulus et al. who studied single crystals of LNO and confirmed the location of excess oxygen in the rock salt LaO layer, thereby lending further support to the initial studies.13 In light of these conclusions, Skinner observed the lengthening of apical Ni–O bonds on heating LNO, which is a direct consequence of the loss of excess oxygen from interstitial sites located in the La–O plane.14
in the Fmmm structure. These studies had further support from Paulus et al. who studied single crystals of LNO and confirmed the location of excess oxygen in the rock salt LaO layer, thereby lending further support to the initial studies.13 In light of these conclusions, Skinner observed the lengthening of apical Ni–O bonds on heating LNO, which is a direct consequence of the loss of excess oxygen from interstitial sites located in the La–O plane.14
Higher-order Ruddlesden–Popper phases, however, tend to be understoichiometric and, therefore, oxygen vacancies are dominant defects. In comparison to lower-order Ruddlesden–Popper phases, there are very few neutron diffraction studies of lanthanide-based higher-order Ruddlesden–Popper phases such as La0.3Sr2.7CoFeO7−δ (n = 2), and LaSr3Co1.5Fe1.5O10−δ (n = 3) available in the literature.15–17 This report will, therefore, detail our recent neutron diffraction studies and complimentary DFT calculations of three n = 3 Ruddlesden–Popper compositions – La3PrNi3O10−δ (L3P1N3), La2Pr2Ni3O10−δ (L2P2N3) and LaPr3Ni3O10−δ (L1P3N3) – to provide a comprehensive study of oxygen defect structure and transport properties in these materials. The higher-order phases tend to be oxygen understoichiometric in nature and therefore oxygen vacancies are predominant defects in these materials. The three phases under discussion too were previously reported by our group to be oxygen deficient in nature.6,18,19
2 Methods
2.1 Syntheses and characterisation
All three compositions – L3P1N3, L2P2N3 and L1P3N3 – were synthesised using the sol–gel route.20 Stoichiometric quantities, depending on the material composition, of La(NO3)3·6H2O (Sigma Aldrich, 99.0%), Pr(NO3)3·6H2O (Sigma Aldrich, 99.99%) and Ni(NO3)2·6H2O (Sigma Aldrich, 99.0%) were dissolved in an aqueous solution of 10% (by weight) of citric acid (Sigma Aldrich, 99.99%). The solution was heated at 250 °C, under constant stirring for three hours, until a gel was obtained. The gel, after being decomposed in the air for 12 hours at 600 °C, was ground and the resultant powder was annealed for 24 hours in air at 950 °C (L1P3N3) and 1000 °C (L2P2N3 and L3P1N3) prior to phase determination by X-ray powder diffraction. The XRD patterns of the material were collected using a PANalytical X'Pert Pro MPD (Cu Kα source) and Rietveld refinement of the XRD data, performed with the GSAS/EXPGUI software package21,22 was used to confirm the phase identification. We recently reported the initial XRD studies detailing all three compositions.232.2 Neutron powder diffraction of L2P2N3 at POLARIS
In situ neutron powder diffraction data were collected on the high-flux medium resolution POLARIS diffractometer at ISIS, the UK spallation source at the Rutherford Appleton Laboratory, Oxfordshire, UK.24 A powder sample of L2P2N3 was loaded in a 4 cm high vanadium can and initial measurements were recorded at ambient temperature. Two thermocouples were attached to opposite sides of the sample container to control and monitor the furnace temperature. After calibration, diffraction data were collected at room temperature (25 °C) and 600 °C for ∼8 hours (150 μAmps), and every 50 degrees from 100 °C to 550 °C for ∼2 hours (25 μAmps) after equilibrating the temperature at each measurement. The sample was kept under a vacuum at all times. All the data were collected in the d-spacing range of 0.40–4.50 Å and the data were corrected for absorption before analysing by Rietveld refinement.2.3 Neutron powder diffraction of L1P3N3 and L3P1N3 at POWGEN
In situ neutron powder diffraction measurements were also collected for the other two compositions, L1P3N3 and L3P1N3 at the POWGEN beamline (0.0008 < Δd/d < 0.025), SNS, Oak Ridge National Laboratory, USA.25,26 Powder samples were placed in a quartz sample holder with a 0.5 cm radius and 4 cm length. A fused silica quartz tube furnace, with vanadium foil elements operating under vacuum conditions, was used to heat the sample. A quartz line was used as a vertical hanger for holding the quartz sample holder, which was then lowered into the quartz tube until the sample was in the path of the beam and the furnace hot zone.27 The furnace temperature was calibrated using a ZnO reference sample.28 Powder samples were equilibrated under a continuous flow of simulated air (20% O2) for at least one hour before measurement at each temperature. Diffraction patterns were also collected every 100 °C from room temperature to 800 °C in the d-spacing range of 0.45–5.35 Å. Several patterns with shortened collection times were collected and examined prior to the 2 hour-long measurements to ensure that the sample had reached equilibrium, monitored by means of observing any shifts in lattice parameters.It should be noted that the neutron powder diffraction data recorded on the POLARIS instrument were in a vacuum, whilst the data collected on the POWGEN diffractometer were in a synthetic air atmosphere. Evidently, the vacuum atmosphere, particularly at elevated temperatures, could induce reduction (oxygen loss), and hence direct comparisons of the oxygen contents of the three materials are not possible.
2.4 DFT calculations
Spin polarised density functional theory (DFT) calculations were performed in this work with the Vienna Ab initio Simulation Package (VASP), using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.29,30 Projector-augmented wave (PAW) pseudopotentials were used for all species.31 Two sets of pseudopotentials were used throughout this work. For the convex hull analysis of LaxPr4−xNi3O10 structures, a softer set of PAW pseudopotentials was used, labelled La, Ni, and O_s in the VASP 5 distribution, using a plane wave cut off of 400 eV. Previous studies have shown that Pr adopts the Pr3+ oxidation state in Pr4Ni3O10.32 The Pr_3 pseudopotential in VASP was used in which the 4f electrons are moved to the core to alleviate the known issues with overbinding associated with f electrons. For the calculation of structural properties, defect formation energies and activation energies, p valence electrons were included for Ni (Ni_pv) and the normal oxygen pseudopotential (O) was used. For this later pseudopotential set, a plane wave cut off of 520 eV was used. An electronic convergence of 1 × 10−6 eV was used for all calculations. A Hubbard U parameter was added to Ni using the rotationally invariant approach of Dudarev et al., to improve the description of electron correlation.33 A U value of 6.2 eV was used for Ni, taken from the Materials Project database.34 This value is calculated based on the approach of Wang et al.35 using the experimental formation energies of the reaction Li2O + 2NiO + 1/2O2 → LiNiO2,31 which captures the Ni2+/Ni3+ redox couple. Gamma centred k-point meshes were used for all calculations to sample the Brillouin zone with a k-point grid density of at least 20 Å. A collinear ferromagnetic spin alignment was used for all calculations in this study.A Bmab unit cell structure was used for all calculations of LaxPr4−xNi3O10 structures. Initial test calculations were performed for La4Ni3O10 in the higher symmetry I4/mmm structure, however, the structure relaxed to the Bmab structure upon full optimisation of the atomic positions and unit cell parameters. All distinct La/Pr orderings within the primitive Bmab unit cell structure (2 formula units) of LaxPr4−xNi3O10 for x = 0, 1, 2, 3 and 4 were enumerated using the CASM package, to predict the formation energy of phases on the convex energy hull.36 The atomic positions and unit cell parameters of all structures were fully optimised without symmetry constraints until the force on any atom fell below 0.05 eV Å−1.
The Pr occupancy (XM(j)) on the two lanthanide sites within the structure, M(1) and M(2), was approximated by taking a Boltzmann average of all configurations at each composition, x:
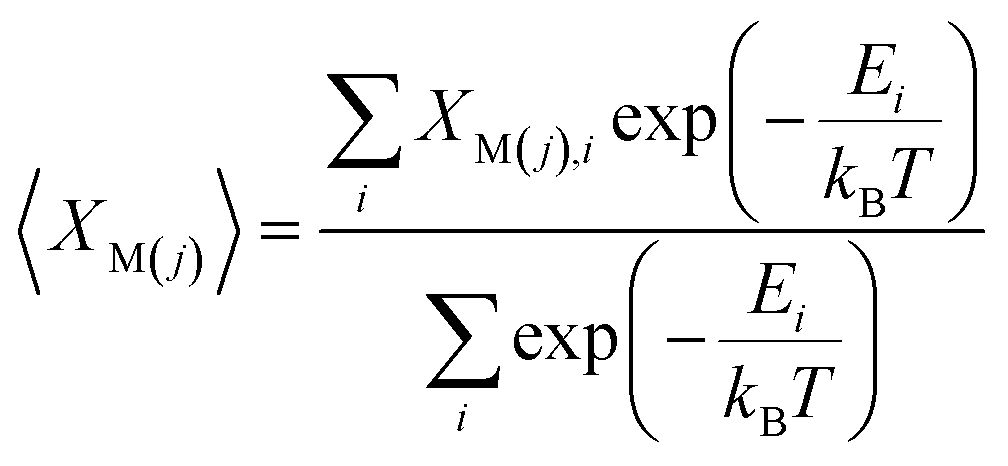
For defect formation energy calculations, a single oxygen vacancy defect was introduced into 5 distinct oxygen positions (O1–O5) of the primitive Bmab unit cell structures of La8Ni6O20±1, Pr8Ni6O20±1 and the lowest energy structure of La4Pr4Ni6O20±1 found from the convex hull analysis. An oxygen interstitial defect was also introduced into each structure within the (La,Pr)O rocksalt layer. The atomic positions and lattice parameters for each structure were allowed to fully relax to a force tolerance of 0.01 eV Å−1. The chemical potential of oxygen (μO2) in the defect calculations was referenced to O2 gas via the calculation of the energy of and isolated triplet O2 molecule (EO2) in a box (12 × 12 × 12 Å). To correct for the known over binding of O2 molecules with the PBE functional, a correction of +1.374 eV was added to the energy of O2, based on the procedure previous described by Wang et al.35 The impact of temperature on the oxygen chemical potential, μO2(T,pO2), was approximated as:37
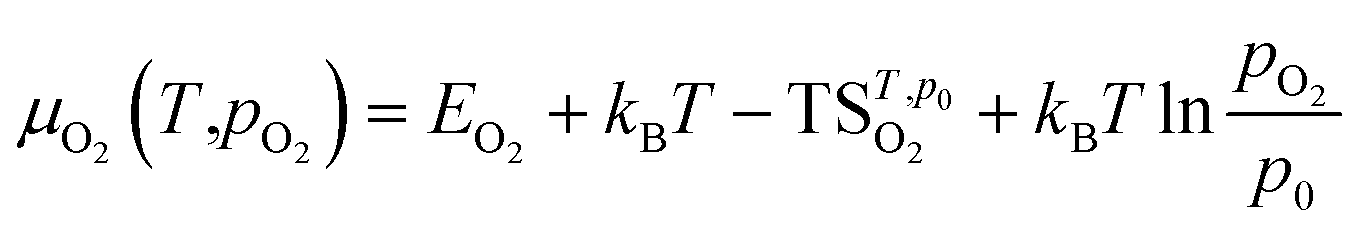
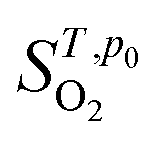 is the entropy of the O2 molecule taken from the JANAF thermochemical tables,38 pO2 is the partial pressure of O2 gas and p0 is the partial pressure of oxygen in the standard state. Structure figures were produced using the VESTA software package.39
is the entropy of the O2 molecule taken from the JANAF thermochemical tables,38 pO2 is the partial pressure of O2 gas and p0 is the partial pressure of oxygen in the standard state. Structure figures were produced using the VESTA software package.39
The activation barriers for vacancy diffusion between adjacent O1 sites within the La4Ni3O10, Pr4Ni3O10 and L2P2N3 structure were calculated using the climbing image nudged elastic band approach (CI-NEB).40 To minimise the interactions between vacancies in adjacent cells, 2 × 2 × 1 supercells of the initial primitive Bmab unit cells were created. A single vacancy was introduced into the supercell (M32Ni24O79) into neighbouring O1 sites, and the atomic positions were optimised under fixed cell conditions to a force tolerance of 0.01 eV Å−1. CI-NEB40 were performed with 5 intermediate images under fixed cell condition using same force tolerance.
3 Results and discussion
3.1 Symmetry and evolution of the lattice constants
The evolution of unit cell constants, and thereby the unit cell volume is an important consideration for these materials as they are considered for potential use in high-temperature SOC devices. Therefore, Rietveld refinements, to be discussed in detail in the next section, were performed using the GSAS/EXPGUI software package15,16 and it was found that L1P3N3 and L3P1N3 compositions studied by POWGEN crystallised in the low-symmetry orthorhombic space group Bmab at all temperatures except the L3P1N3 composition which adopts the high-symmetry tetragonal space group I4/mmm at 800 °C. L2P2N3 studied with the POLARIS diffractometer, however, crystallised with monoclinic symmetry. Table 1 summarises the room-temperature lattice constants obtained from Rietveld refinement of neutron diffraction data for the L3P1N3, L2P2N3 and L1P3N3 compositions and compares them with the two end members as reported by Zhang et al.41 The cell constant data obtained from the NPD measurements are in excellent agreement with our previous XRD studies.23| Composition | a (Å) | b (Å) | c (Å) | Space group | Reference | Instrument |
|---|---|---|---|---|---|---|
| La4Ni3O10−δ | 5.415(1) | 5.465(1) | 27.959(9) | Fmmm | Zhang et al.36 | XRD |
| L3P1N3 | 5.41027(7) | 5.46975(7) | 27.8955(4) | Bmab | This work | POWGEN |
| L2P2N3 | 5.38773(8) | 5.46047(8) | 27.7533(5) | P21/a | This work | POLARIS |
| L1P3N3 | 5.38686(1) | 5.46975(8) | 27.7463(7) | Bmab | This work | POWGEN |
| Pr4Ni3O10−δ | 5.370(1) | 5.462(1) | 27.528(3) | Fmmm | Zhang et al.36 | XRD |
As is evident from the data in Table 1, a decrease in unit cell constants with increasing Pr content was observed. This is consistent with the decrease in the ionic radius of Pr leading to an overall decrease in the cell volume. This agrees with our X-ray diffraction studies of these materials6,23 and the other published literature41,42 for the end member compositions. The significant decrease observed in the c parameter with increasing Pr content is explained by the fact that there is a cumulative effect of reduced lanthanide radii along the crystallographic c direction where the layers are stacked. The overall decrease in cell parameters in going from L4N3 to P4N3 is reflected in the unit cell volume as a function of composition, Fig. 1. There, however, appears to be a very slight increase in b parameter in L3P1N3 and L1P3N3 compositions (shown in bold) when compared with the respective preceding compositions which go against the expected trend. This could be because the L3P1N3 and L1P3N3 measurements (POWGEN) presented above were obtained at slightly higher temperatures, 70 °C while L2P2N3 was measured at 25 °C (POLARIS).
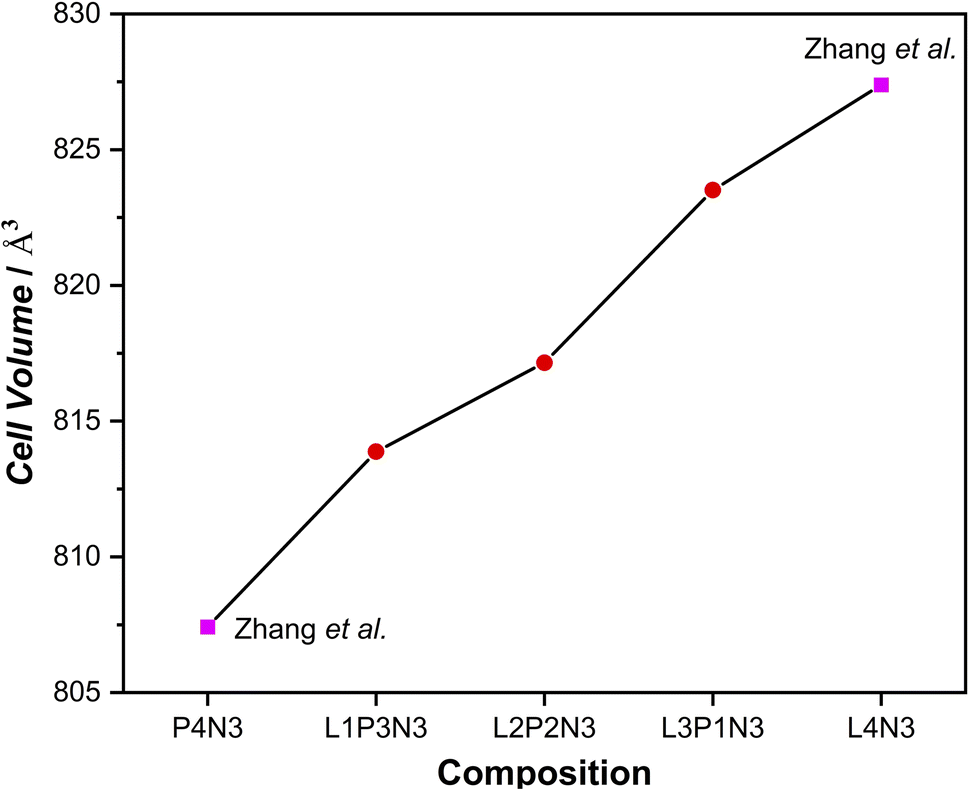 | ||
| Fig. 1 Cell volume increase with the increase in La content of the composition, reflecting the trend observed in the unit cell constant evolution. (The cell volumes plotted run up to 2 significant digits and the estimated std. dev. lie within the data markers.) | ||
In recent high-resolution X-ray and neutron diffraction studies,43–46 higher-order Ruddlesden–Popper phases have been shown to adopt monoclinic symmetry. With this in mind, we investigated the POLARIS high-resolution detector, bank 5 (Δd/d = 3 × 10−3) L2P2N3 composition data set and found that the material adopts the monoclinic P21/a space group symmetry at room temperature, Fig. S1.† The lattice parameters obtained were a = 5.38773(8) Å, b = 5.46047(8) Å, c = 27.7533(5) Å with the cell angles obtained being: α = γ = 90.0 and β = 90.235(2). It is evident that the monoclinic distortion of the cell is slight and is only observed when the highest resolution data are obtained. It is therefore likely that the orthorhombic assignment of the lower temperature data reported here and in the literature is a feature of the instrument resolution and that the true symmetry is monoclinic. For consistency, however, all further data are related to the orthorhombic cell identified with the lower resolution instruments.
3.2 Thermal expansion coefficients
Fig. 2a–c illustrate the increase of the a, b and c lattice parameters as a function of increasing temperature and Fig. 2d shows the calculated thermal expansion coefficients (TEC) for all three compositions. As expected, the unit cell parameters show a linear increase throughout the temperature range. On close examination of the data, it is evident that the c parameter showed a significant increase when compared to both the a and b parameters, thereby reflecting the cumulative effect of stacking of layers across the crystallographic c direction. The c parameter was more responsive to the changes in temperature than the a parameter because the expansion along the c axis is less restricted by the expansion of the rock-salt layers.16,47 Also evident from Fig. 2 is the relatively higher increase in a lattice parameter compared with the b lattice parameter, which points towards an increase in orthorhombic strain with increasing temperature as argued by us in our earlier reports.23,48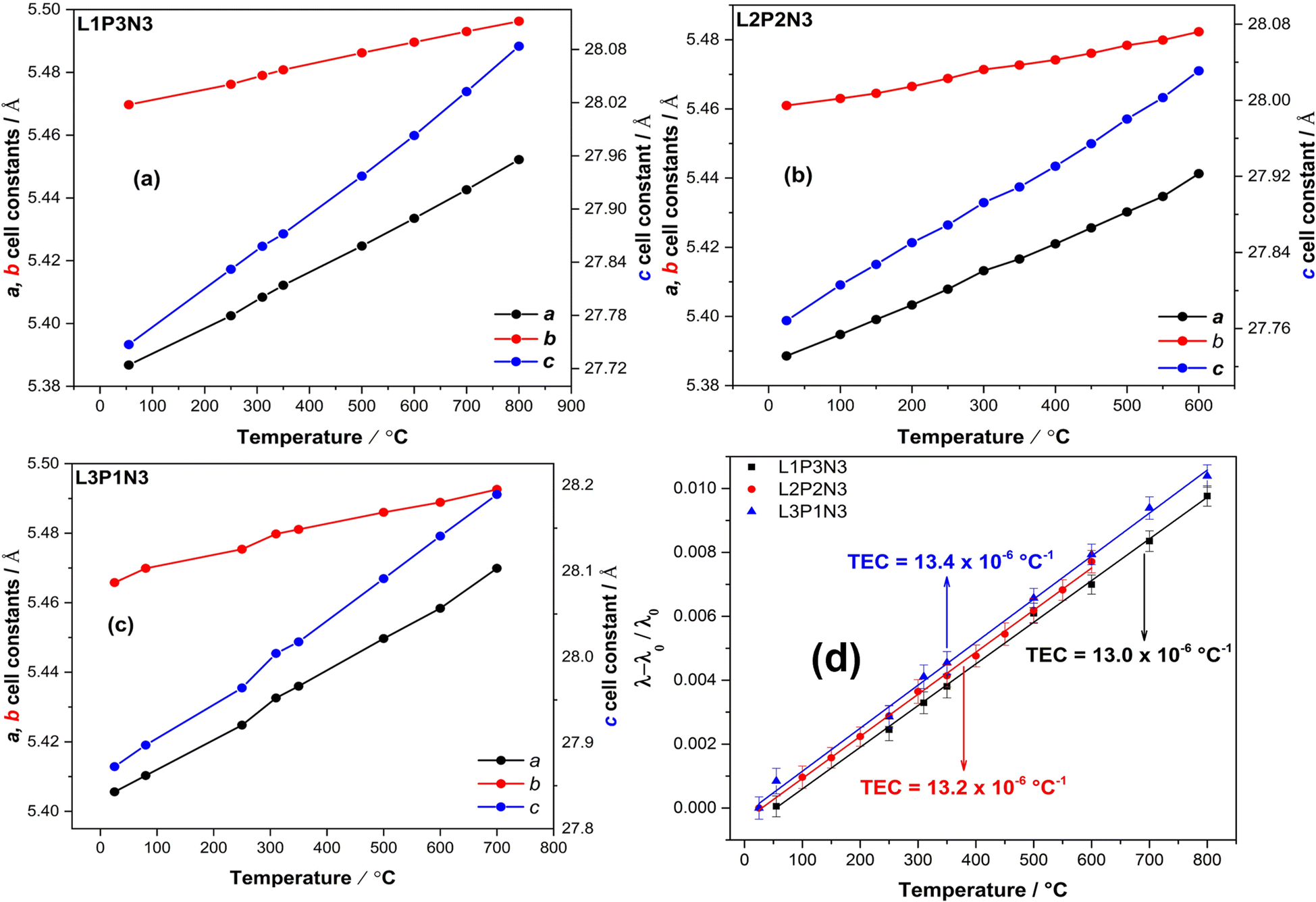 | ||
| Fig. 2 Increase of the unit cell parameters with the increase in temperature for (a) L1P3N3 composition (b) L2P2N3 composition and (c) L3P1N3 composition. Unit cell constant c shows a significant change, reflective of the cumulative effect of stacking of layers across crystallographic c direction. (d) (λ–λ0/λ0) as a function of temperature for all the three compositions showing the compatible TEC's. (Note: standard deviations for the unit cell parameters lie within the data markers.) | ||
The slope of (λ–λ0/λ0) vs. T (Fig. 2d), where λ and λ0 are the cubic roots of the volume at a particular temperature and room temperature respectively were used to calculate thermal expansion coefficients. The calculated values of the TEC were found to be 13.0 × 10−6 °C−1 for L1P3N3; 13.2 × 10−6 °C−1 for L2P2N3 and 13.4 × 10−6 °C−1 for L3P1N3, of the same order of magnitude as those of the other components of SOFCs, and therefore compatible with commonly used electrolytes such as Y2O3-stabilised ZrO2 (YSZ), Ce0.8Gd0.2O2−δ (CGO) and La0.8Sr0.2Ga0.8Mg0.2O3−δ (LSGM).49,50 These results, however, vary slightly from the value, 10 × 10−6 °C−1, calculated from XRD measurements and reported by Vaibhav et al. for P4N3 composition.51 It could possibly of different La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pr ratio and the fact that they measured the TEC from room temperature to 1000 °C while the results reported here were measured over the temperature range of RT to 800 °C for L1P3N3 and L3P1N3 compositions and RT to 600 °C for L2P2N3 composition.
:Pr ratio and the fact that they measured the TEC from room temperature to 1000 °C while the results reported here were measured over the temperature range of RT to 800 °C for L1P3N3 and L3P1N3 compositions and RT to 600 °C for L2P2N3 composition.
3.3 La/Pr ordering: neutron diffraction and DFT calculations
Complimentary DFT calculations were performed to support the experimental analysis. DFT calculations on unit cell structures of LaxPr4−xNi3O10 (x = 0–4) were used to investigate the energetic preference for La/Pr ordering.The energies of all symmetrically distinct La/Pr orderings on the M(1) (perovskite) and M(2) (rocksalt) sites were calculated with DFT+U using the Bmab unit cell of LaxPr4−xNi3O10. The resulting convex energy hull is shown in Fig. 3a. Compositions of LaPr3Ni3O10 (x = 1), La2Pr2Ni3O10 (x = 2) and La3PrNi3O10 (x = 3) are observed as stable phases on the hull. The lowest energy ordering for each structure are shown in Fig. 3b. In all of the low energy structures on the hull, Pr preferentially occupies the M(2) site in the rocksalt layer.
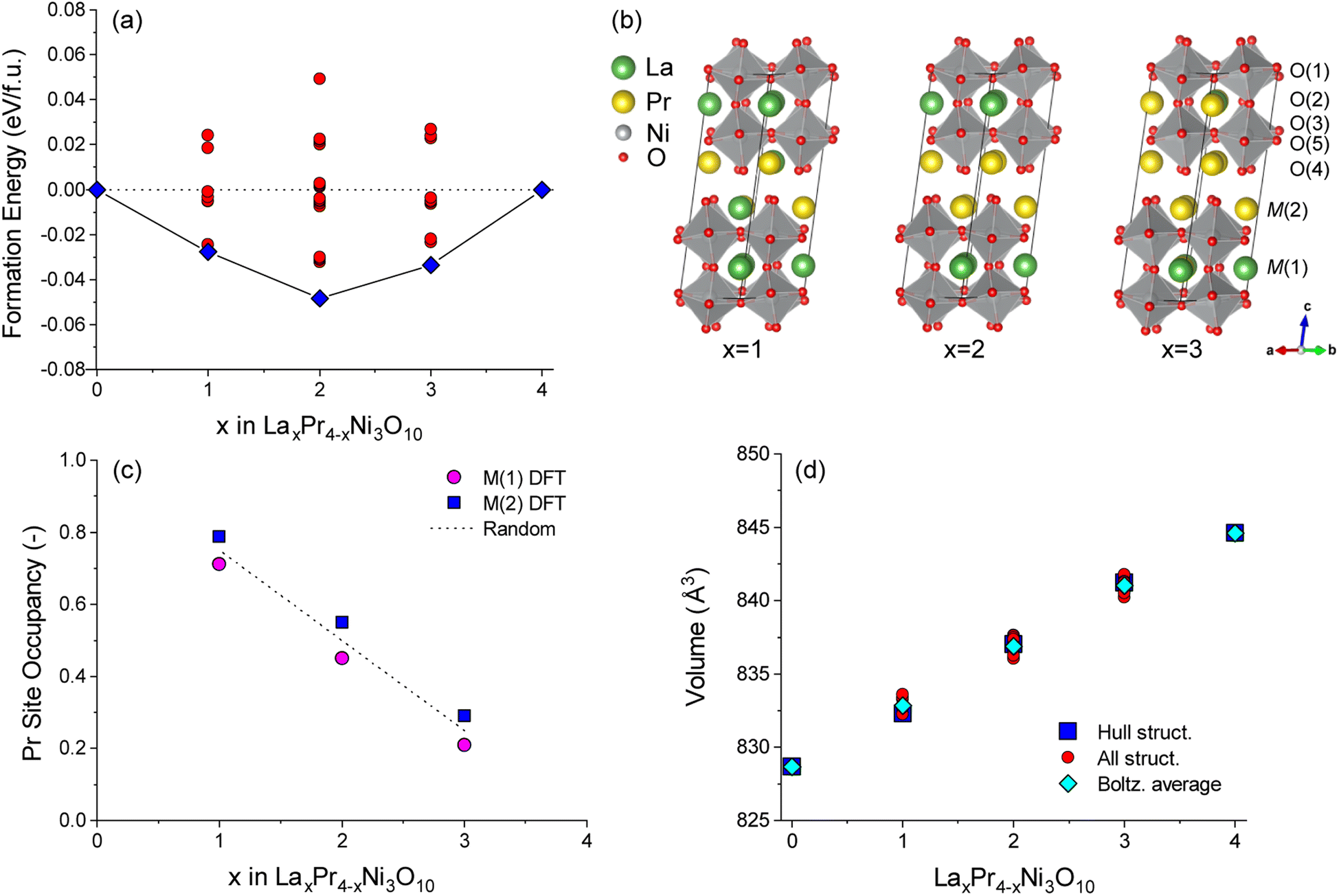 | ||
| Fig. 3 (a) Convex energy hull for LaxPr4−xNi3O10 structures calculated with DFT. Both the lowest energy structures on the hull (blue squares) and configurations above the hull (red circles) are shown. (b) Lowest energy hull structures for x = 1, 2 and 3. (c) Boltzmann averaged Pr occupancy on M(1) (perovskite) and M(2) (rocksalt) sites as a function of composition, x. Occupancies are calculated at 800 °C from all convex hull structures at each composition. The random solution limit is shown as a dashed line. (d) Comparison of unit cell volume with composition, x, for all structures in the convex hull (red circles). The unit cell volumes of the lowest energy structures as shown as blue squares and the Boltzmann averaged unit cell volume for each composition at 800 °C is shown as cyan diamonds. | ||
To gauge the preference for La/Pr ordering at the experimental measurement temperature of 800 °C, a Boltzmann average of the Pr site occupancy was calculated from the cell energies of all structures at each composition, as shown in Fig. 3c. For all x compositions, at 800 °C there is only a weak preference for Pr ordering on the M(2) site, as the thermal energy (kBT = 0.107 eV) is larger than the energy difference between most of the configurations. The occupancy of the M(1) and M(2) sites is therefore close to the random solution limit of 0.5:0.5.
The Rietveld refinements of the neutron diffraction measurements revealed that the fractional occupancy for the A-site cations, La and Pr, were slightly different, than the nominal compositions; La3.26Pr0.74Ni3O10−δ, La1.9Pr2.1Ni3O10−δ and La1.04Pr2.96Ni3O10−δ. Furthermore, the examination of the data for 50:50 composition (L2P2N3) in Tables 2 and 3 shows that Pr preferentially orders into the 9-coordinated M(2) site in the rocksalt layer as compared to the 12-coordinated M(1) in the perovskite layer. This further agrees with our DFT calculations discussed above (Fig. 3c). However, in contrast to the DFT calculations showing only a weak preference for Pr ordering on the M(2) site for all the compositions, the neutron diffraction measurements of L1P3N3 and L3P1N3 compositions at elevated temperatures showed that Pr has a weak preference for M(1) site.
| Atom | Atomic displacement parameters (Å2) × 100 | Wyckoff position | Fractional occupancy | ||||
|---|---|---|---|---|---|---|---|
| U11/Uiso | U22 | U33 | U12 | U23 | |||
| La(1) | 0.6(9) | 8f | 0.57(3) | ||||
| Pr(1) | 0.6(9) | 8f | 0.43(3) | ||||
| La(2) | 0.4(1) | 8f | 0.38(4) | ||||
| Pr(2) | 0.4(1) | 8f | 0.62(4) | ||||
| Ni(1) | 0.4(1) | 4a | 1.0 | ||||
| Ni(2) | 0.4(1) | 8f | 1.0 | ||||
| O(1) | 4.0(5) | 3.8(4) | 0.8(4) | 4.7(3) | 8e | 0.98(3) | |
| O(2) | 0.4(2) | 0.3(2) | 2.5(3) | 0.5(4) | 8f | 1.0 | |
| O(3) | 0.1(3) | 0.8(3) | 1.3(3) | 0.4(2) | 8e | 1.0 | |
| O(4) | 1.6(3) | 3.5(5) | 0.4(2) | 1.8(6) | 8f | 1.0 | |
| O(5) | 1.5(3) | 1.0(3) | 0.2(2) | 0.2(2) | 8e | 1.0 | |
| Atom | Atomic displacement parameters (Å2) × 100 | Wyckoff position | Fractional occupancy | ||||
|---|---|---|---|---|---|---|---|
| U11/Uiso | U22 | U33 | U12 | U23 | |||
| La(1) | 2.2(1) | 8f | 0.57(3) | ||||
| Pr(1) | 2.2(1) | 8f | 0.43(3) | ||||
| La(2) | 0.9(1) | 8f | 0.38(4) | ||||
| Pr(2) | 0.9(1) | 8f | 0.62(4) | ||||
| Ni(1) | 1.4(9) | 4a | 1.0 | ||||
| Ni(2) | 1.2(6) | 8f | 1.0 | ||||
| O(1) | 5.1(6) | 4.1(6) | 7.5(8) | 5.4(4) | 8e | 0.92(2) | |
| O(2) | 2.9(4) | 2.3(4) | 3.6(4) | 0.5(4) | 8f | 0.95(2) | |
| O(3) | 0.5(3) | 1.7(4) | 1.3(3) | 0.3(2) | 8e | 1.0 | |
| O(4) | 4.0(4) | 4.8(7) | 2.0(3) | 1.8(6) | 8f | 0.99(1) | |
| O(5) | 3.0(6) | 2.9(5) | 0.6(4) | 0.2(3) | 8e | 0.91(3) | |
The variation in the unit cell volume was calculated for each structure as shown in Fig. 3d. An approximately linear increase in the unit cell volume is observed from compositions of x = 0 to x = 4 in LaxPr4−xNi3O10, which is consistent with the experiment (Fig. 1). A slightly larger unit cell volume is predicted computationally compared to the experiment, as is commonly observed with DFT(+U) calculations using the PBE functional. For all configurations studied, it can be seen from Fig. 3d that the change in the chemical composition (x) has a bigger influence on the unit cell volume than the La/Pr ordering at fixed composition.
3.4 Phase transition
The neutron diffraction pattern obtained at 250 °C in flowing air for the L3P1N3 composition fitted best to an orthorhombic structure adopting the Bmab space group. The lattice parameters were determined to be a = 5.42478(2) Å, b = 5.47534(2) Å and c = 27.9632(1) Å, and are in close agreement with the previously reported results for the end member materials.6,18,19,41,52 A phase transition was observed on further heating and the diffraction pattern collected at 800 °C was refined with the tetragonal space group I4/mmm. The lattice parameters obtained after refinement again agree well with the literature for the end member composition and were a = b = 3.87876(1) Å, and c = 28.2287(2) Å.16 An impurity of ∼2 wt% NiO was also detected in the neutron diffraction data, which was not observed by X-ray diffraction measurements. The refinements of the data obtained at both temperatures, 250 and 800 °C, along with χ2 and R factors are illustrated in Fig. 4. The transition to the tetragonal I4/mmm high symmetry structure at 800 °C in flowing air is clearly visible after scrutiny of the 2.3 to 2.6 Å d-spacing range (insets, Fig. 4a and b). A close inspection of the insets confirms the assignment of the two structural models (Bmab and I4/mmm) at low and high temperatures respectively. The low-symmetry Bmab structure can be seen to show seven distinct reflections in the d-spacing range of 2.3–2.6 Å while the high-symmetry I4/mmm structure only shows three reflections in the same d-spacing range, thereby confirming the completion of the phase transition. The values of x, y, z and Uiso parameters determined by Rietveld refinement are provided in Tables S1 (250 °C) and S2 (800 °C) of the ESI.†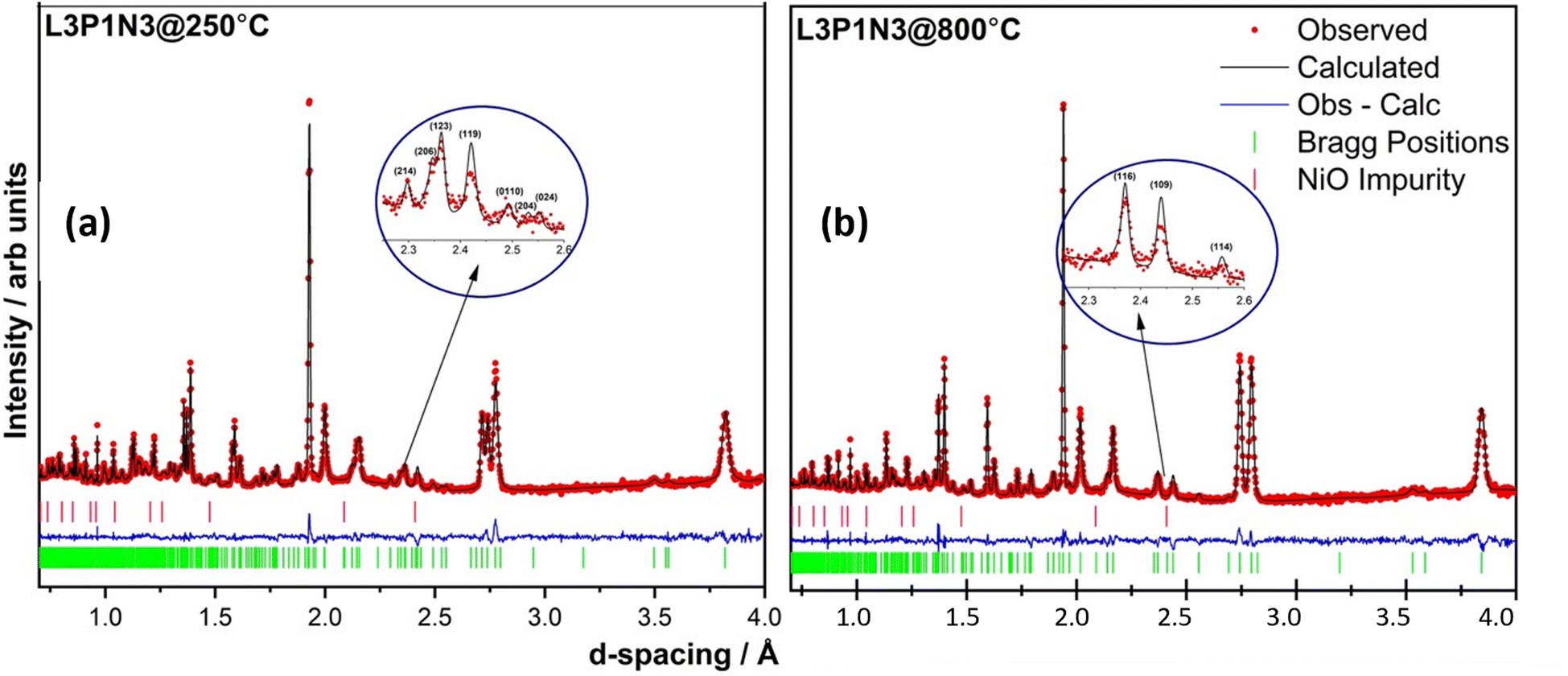 | ||
Fig. 4 Rietveld refinement of the two data sets obtained at (a) 250 °C (χ2 = 4.297; Rp = 5.2% and Rwp = 4.3%) and (b) 800 °C (χ2 = 5.03; Rp = 5.8% and Rwp = 4.9%). A ∼2% impurity of NiO was detected and was refined in cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The transition to a tetragonal high symmetry structure is easily discernible after examining the 2.3–2.6 Å d-spacing range as shown in the insets. m space group. The transition to a tetragonal high symmetry structure is easily discernible after examining the 2.3–2.6 Å d-spacing range as shown in the insets. | ||
It is instructive to examine the refined structures in detail; the structural changes on the transition from the orthorhombic to tetragonal structure primarily reflect changes in the NiO6 octahedra as demonstrated in Fig. 5. The tilt in the NiO6 octahedra for the sample measured at 250 °C is clearly visible when compared to the tetragonal structure obtained from the data set obtained at 800 °C. Similarly, the other two compositions, L2P2N3 (Fig. S2†) and L3P1N3 (Fig. S3†) were best refined in orthorhombic Bmab symmetry at both low and high temperature, and no transition to higher symmetry structures on heating was observed in either of the two compositions. However, as mentioned above L2P2N3 adopts monoclinic symmetry at RT, so it appears that there is a transition to higher symmetry orthorhombic structure at 250 °C, at least for the L2P2N3 composition.
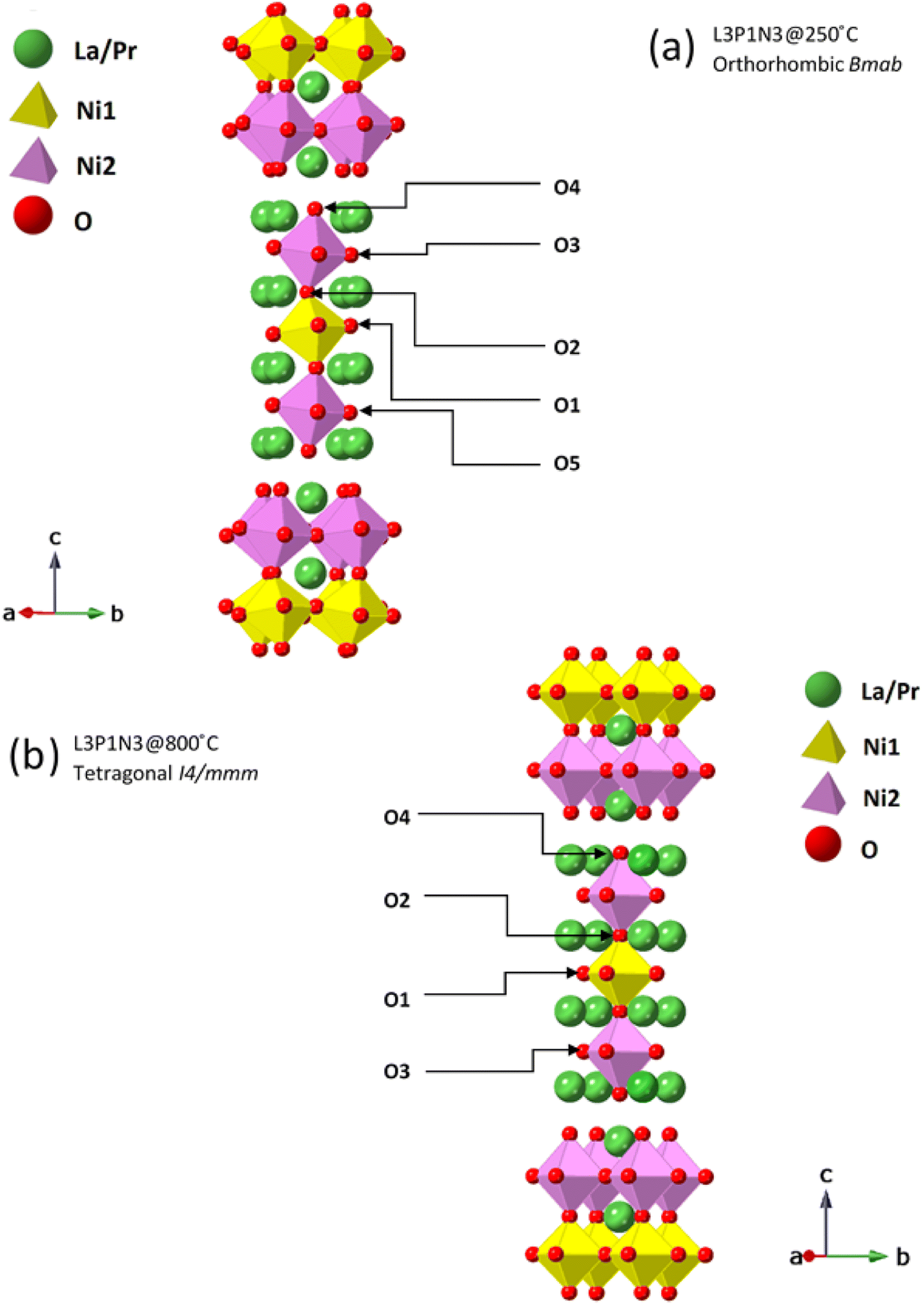 | ||
| Fig. 5 Ruddlesden–Popper unit cells for L3P1N3 structure. These structures were derived from Rietveld refinement of the neutron diffraction data collected at (a) 250 °C and (b) 800 °C. The tilt in the NiO6 octahedra in the 250 °C data set is clearly visible. Oxygen positions are further marked to guide the reader to visualise the oxygen transport pathways discussed. | ||
The Ni(1)–O(2) and Ni(2)–O(4) bond lengths are instructive; the bond lengths determined from the data obtained at 250 °C for the L3P1N3 composition were 1.938(9) Å and 2.121(9) Å respectively. The main reason for this large difference in the bond distances is the fact that O(4) is the apical O atom shared between both the perovskite layer and the rock salt layer structural units in these RP phases and is therefore exposed to competing crystal-chemical driving forces. In fact, the elongation of the Ni–O bond involving the apical O atom which projects into the rock-salt type layer is found throughout the Ruddlesden–Popper series. This further points to the preference of Ni3+ occupying the Ni(1) site and Ni2+ occupying the Ni(2) site and this kind of charge ordering have indeed been reported earlier for Ruddlesden–Popper phase materials.53 Similar behaviour was observed in this work for the L2P2N3 and L1P3N3 compositions.
3.5 Oxygen defects and transport: neutron diffraction and DFT calculations
Atomic displacement parameters and fractional occupancies obtained after refinement of neutron diffraction data of the representative L2P2N3 composition, along with χ2 and R factors are illustrated in Tables 2 and 3 below.The occupancies of all the oxygen sites were refined and it was observed that vacancies at room temperature were limited with a total oxygen stoichiometry of 9.96. These vacancies were confined to the O(1) site for the sample measured at 25 °C agreeing well with the DFT calculations (Fig. 6a). In cases where occupancies refined to 1.0, these were fixed in subsequent refinement cycles. As was expected the total oxygen stoichiometry decreased with increasing temperature, due to the reduction of the B-site cation, pointing towards the expected decrease in the B-site cation average oxidation state. The oxygen stoichiometry of 9.54 at 600 °C agrees well with previously published reports in related higher-order Ruddlesden–Popper materials by our group and others in the community.16,18,19 At 600 °C, the oxygen vacancies were observed in the O(1), O(2) and O(5) sites in the order O(1) ≈ O(5) < O(2) ≪ O(3) ≈ O(4), meaning that the vacancies predominantly prefer perovskite units of the structure.48 The atomic displacement parameters for the oxygen atoms were best fit with an anisotropic model and the thermal parameters show a considerable increase with the increase in temperature, which is expected (Table 3). The large ab plane anisotropy of the apical O(1) sites, particularly at high temperatures, points towards faster oxygen diffusion in these materials. This apical oxygen position is indeed known to exhibit an anisotropic Debye–Waller factor and has been widely reported for lower-order Ruddlesden–Popper K2NiF4 materials.12,14,54
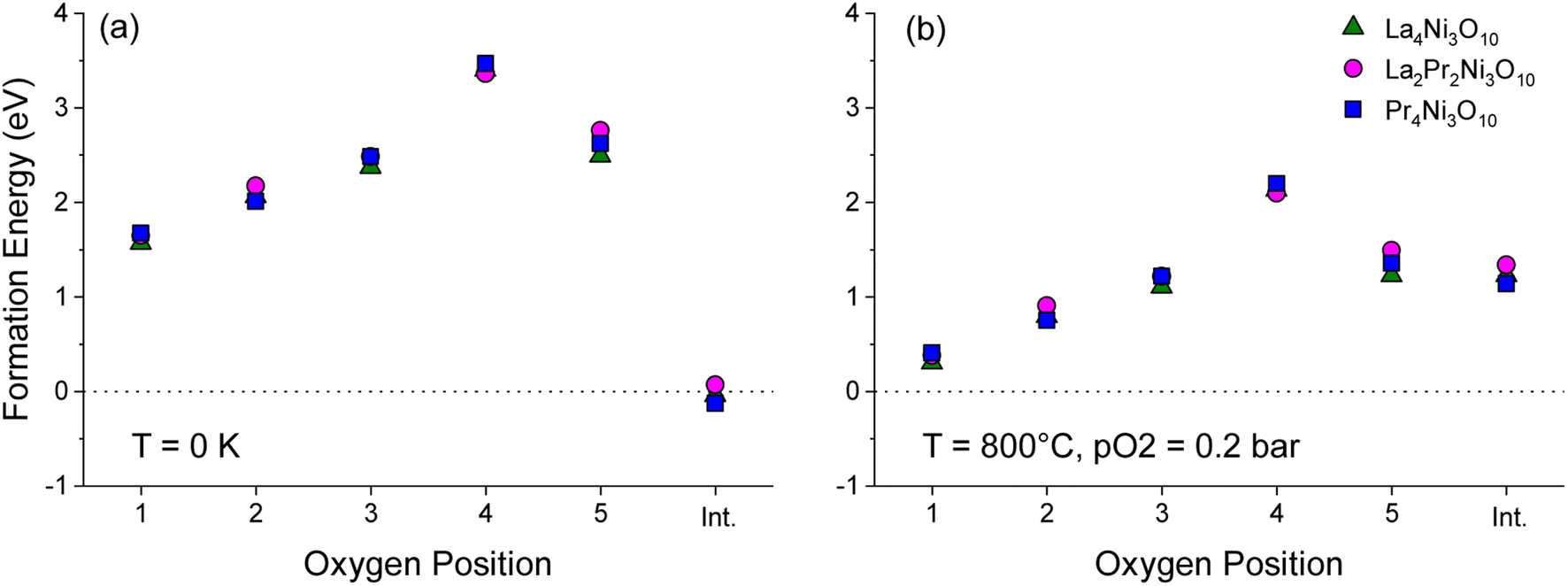 | ||
| Fig. 6 Plot of formation energy for oxygen vacancies at the O1–O5 sites in the Bmab primitive cell structures of La4Ni3O10, La2Pr2Ni3O10 and Pr4Ni3O10, with 2 formula units per cell (M8Ni6O20±1) at (a) T = 0 K and pO2 = 0 bar and (b) T = 800 °C and pO2 = 0.2 bar. The energy to form an oxygen interstitial (int.) in the rocksalt layer is also shown. | ||
To aid the neutron diffraction results, DFT calculations were used to investigate the formation energy of oxygen vacancies and oxygen interstitials. The energy to form an oxygen vacancy at each of the 5 distinct O sites (O1–O5) in the lowest energy La2Pr2Ni3O10 and end member La4Ni3O10 and Pr4Ni3O10 structures was calculated as shown in Fig. 6.
At 0 K, the oxygen interstitial is the lowest energy defect for all structures with a small negative formation energy for La4Ni3O10 (−0.127 eV) and Pr4Ni3O10 (−0.041 eV) and a small positive formation energy for La2Pr2Ni3O10 (0.067 eV). The formation energy for oxygen vacancies is above 1.5 eV for all sites indicating that there is a strong preference for interstitials in the (M)4Ni3O10 structure at low temperatures.
The sensitivity of the defect formation energies to the values of the U correction on Ni was tested by calculating the oxygen vacancy and interstitial formation energies without a U correction as shown in Fig. S4.† Consistent with Fig. 6a, the interstitial sites is the most favourable defect at 0 K. Without a U correction, the formation energy of all oxygen vacancies increased, and the O(2) site has a smaller formation energy than the O(1) site. However, the overall trend is consistent with the O(4) site in the rocksalt layer having the largest formation energy.
The variation in the formation energy of oxygen interstitials and O(1) oxygen vacancies as a function of temperature is shown in Fig. S5.† For the La4Ni3O10, La2Pr2Ni3O10 and Pr4Ni3O10 structures, the temperature below which oxygen interstitials have lower formation energy than O(1) vacancies are predicted to be 723, 708 and 795 K, respectively at pO2 = 0.2 bar. This indicates that the defect chemistry of the LaxPr4−xNi3O10 structures can be subtly tuned via the x composition and that the thermal history of the sample and cooling conditions, such as the oxygen partial pressure and cooling rate may have a large impact.
At the measurement temperature of 800 °C, oxygen vacancies are the lowest energy defects. The oxygen vacancy formation energy in Fig. 6b increases layer by layer from the centre of the perovskite layer O(1) to the apical oxygen within the rocksalt layer (O4) in the sequence: O(1) < O(2) < O(3) ≈ O(5) ≪ O(4). Importantly, from Fig. 6a and b, it can be seen that the oxygen site (O1–O5) has a significantly larger influence on the vacancy formation energy than the x composition in the LaxPr4−xNi3O10 structure.
At 800 °C, the low oxygen vacancy formation energy of the O(1) sites (0.304–0.407 eV) for all structures indicates that these sites should have a significant concentration of oxygen vacancies, with a smaller fraction of vacancies on the O(2), O(3) and O(5) sites. This is consistent with the experimental results of the L2P2N3 composition discussed above. The neutron diffraction refinement further shows full occupancy for O(4) sites, again agreeing well with DFT calculations which show the highest oxygen vacancy formation energy for the O(4) site among the five oxygen sites. A difference in L2P2N3 composition is that while DFT calculations show a majority of vacancy concentration at the O(1) site, the neutron data refinement shows that O(1) and O(5) sites almost have the same oxygen vacancy concentration.
Taking into consideration the site occupancies, distances between these vacant O sites and the atomic displacement parameters, it is possible to envision the possible transport pathways in these materials. The distances between the oxygen atoms at 600 °C for L2P2N3 were found to be: 2.72 Å between O(5) sites; 2.72 Å between O(1) and O(2) sites and 2.77 Å between O(1) sites. This suggests predominant participation of fractionally vacant sites via pathways such as O(1)–O(2)–O(1), O(1)–O(1)–O(1) and O(5)–O(5)–O(2) with a possible preference to the transport pathways involving O(1) and O(5) sites because of the relatively shorter distances and high vacancy concentration at these sites. This preference for a curved oxygen transport pathway such as O(1)–O(1)–O(1) around the NiO6 octahedra is expected and in agreement with the published literature for materials such as La0.3Sr2.7CoFeO7−δ, LaSr3Co1.5Fe1.5O10−δ, LaSrCo0.5Fe0.5O4−δ and La1−xSrxBO3 (B = Cr, Mn, Fe and Co).16,48,55,56
Similarly, for the L3P1N3 and L1P3N3 compositions (Tables S3 and S4† respectively), neutron data shows oxygen vacancies are spread over O(1), O(2) and O(3) sites and oxygen sites O(4) and O(5) were fully occupied. This agrees with our DFT calculations which show the highest oxygen vacancy formation energy for O(4) and O(5) sites. However, in both compositions instead of the O(1) site being the most vacant site as per DFT calculations, neutron data shows O(3), another equatorial site, as the most vacant site.
Again, considering the distances between neighbouring O sites which at 800 °C for L3P1N3 were found to be: 2.74 Å between O(3) sites, 2.74 Å between O(1) sites and 2.72 Å between O(2) and O(1) sites. This suggests three major transport pathways; O(3)–O(3)–O(3), O(1)–O(1)–O(1) and O(1)–O(2)–O(1) with O(3)–O(3)–O(3) being the dominant pathway because of the considerable vacancies present at the O(3) oxygen site. Similarly, the distance between oxygen sites for L1P3N3 at 800 °C was found to be 2.73 Å between O(1) sites; 2.78 Å between O(1) and O(2) sites and 2.72 Å between O(2) and O(3) sites suggesting the predominant participation of O(1), O(2) and O(3) sites visa transport pathways such O(3)–O(2)–O(3) and O(1)–O(1)–O(1).
The activation energy for O(1)–O(1) vacancy diffusion was calculated using the CI-NEB approach for supercells of La4Ni3O10, Pr4Ni3O10 and the lowest energy convex hull structure of La2Pr2Ni3O10. The diffusion pathway of a vacancy between two O1 sites in the La4Ni3O10 structure is shown in Fig. 7a. The activation energy as a function of x in LaxPr4−xNi3O10 is shown in Fig. 7b. The activation energy is lowest for the La4Ni3O10 (x = 0) end member (0.60 eV) and increases almost linearly as a function of x from La2Pr2Ni3O10 (0.63 eV) to Pr4Ni3O10 (0.65 eV). The very low activation barrier for O(1) oxygen vacancies within the centre of the perovskite blocks of the M4Ni3O10 structure is consistent with the low activation energies for O vacancy diffusion predicted computationally for the LaNiO3 structure (0.69–0.78 eV) in previous studies.57,58
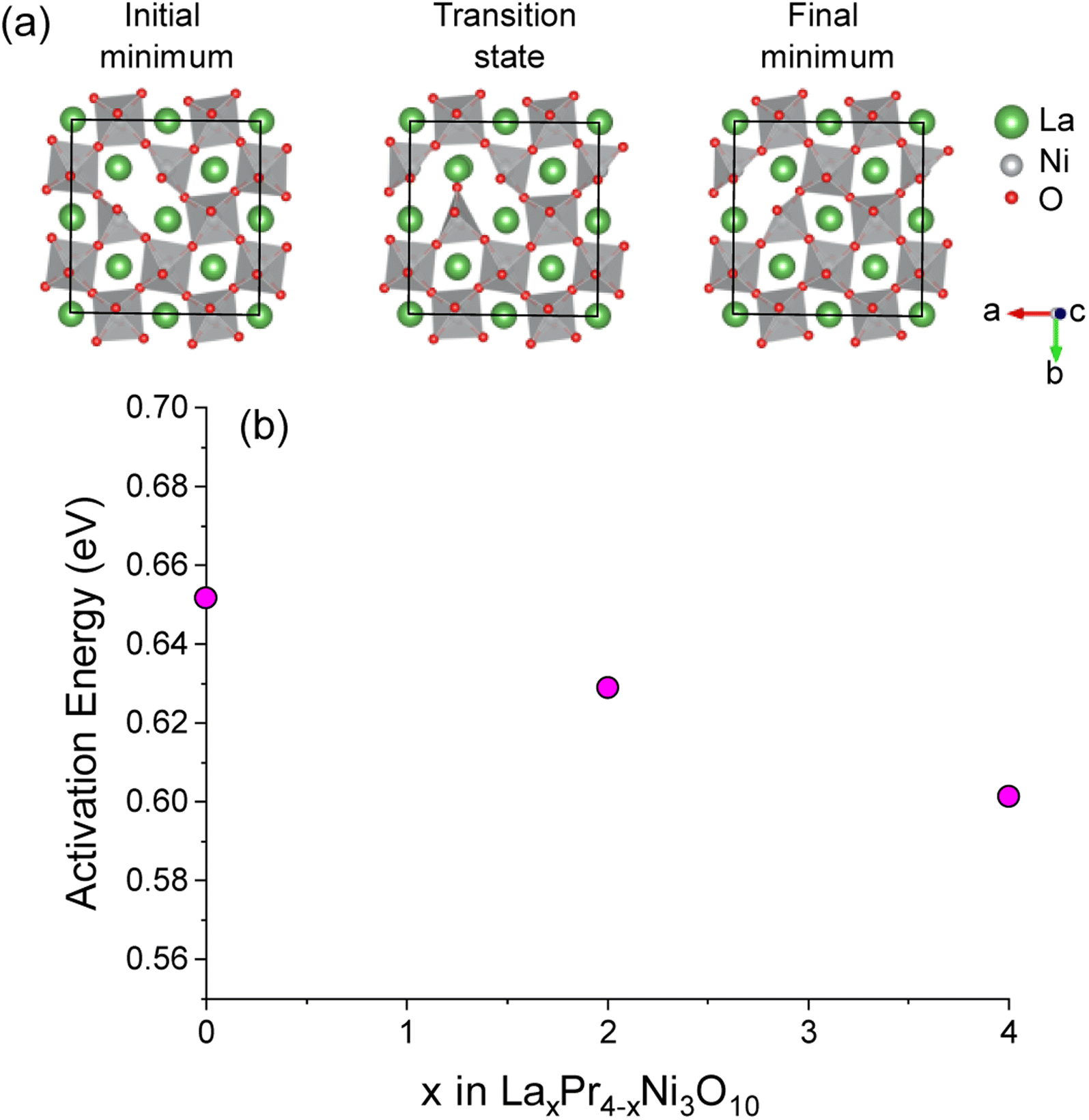 | ||
| Fig. 7 (a) Diagram of the initial and final minima and transition state along the O(1)–O(1) pathway in the perovskite layer of the La32Ni24O79 supercell structure. Only the central perovskite layer of the structure is shown for clarity. (b) Activation energies for O(1)–O(1) diffusion as a function of x composition in LaxPr4−xNi3O10 calculated with NEB. | ||
4 Conclusions
Neutron diffraction studies and DFT calculations of three novel higher-order Ruddlesden–Popper phases were discussed. Across these newly synthesised compositions, it was observed that the lattice parameters show a systematic decrease with an increase in the Pr content of the composition, which agrees with the conception of effective ionic radii of Pr3+ being less than the corresponding La3+. L1P3N3 and L2P2N3 compositions were found to crystallise in the orthorhombic Bmab/Fmmm space group, with only L3P1N3 showing transition to higher symmetry tetragonal I4/mmm space group at 800 °C. L2P2N3 composition, however, was found to crystallise in monoclinic symmetry. It was also observed that orthorhombic strain in these materials increases with the increase in the Pr content of the composition. This is expected and is because of the decrease in ionic radii of Pr.Lattice parameters were shown to increase linearly with the increase in temperature, with c parameter showing a significant increase than a and b cell parameters. This is attributed to the cumulative effect of stacking of layers along the c crystallographic direction and c axis being less restricted by the expansion of the rock-salt layers. A further observation of the b parameter showing only a slight increase as compared to a parameter is interpreted in terms of an increase in orthorhombic strain with the increase in temperature. The calculated TEC based on neutron diffraction measurements was found to be around 13.0 × 10−6 °C−1, which is of the same order as that of commonly used electrolyte materials in SOFC devices.
Furthermore, neutron diffraction and DFT calculations both suggested that vacancies prefer the perovskite block – O(1) and O(3) sites – and it, in fact, has been reported earlier that majority of the oxygen vacancies in these materials are confined to the perovskite layers, with a particular preference to equatorial oxygen sites. A further strong preference for the curved oxygen transport pathways such as O(1)–O(1)–O(1) around the NiO6 octahedra was recorded, in consonance with the earlier reports in these materials.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
M. A. Y. and S. J. S. would like to thank the Imperial College President's PhD Scholarships, EU Horizon 2020 (HERMES; Grant No. 952184) and EPSRC Advanced Characterisation of Materials Centre for Doctoral Training for funding this research. I. D. S. and S. J. S. acknowledge funding for their research from EPSRC Platform Grant EP/R002010/1. I. D. S. acknowledges the Imperial College Research Computing Service (10.14469/hpc/2232), and associated support services used during this work. A portion of this research used resources at the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. Experiments at the ISIS Neutron and Muon Source were supported by a beamtime allocation RB1710304 from the Science and Technology Facilities Council. Data is available here: https://doi.org/10.5286/ISIS.E.RB1710304. Thanks, are also due to Dr Helen Playford (ISIS) and Dr Cheng Li (SNS) for their assistance in recording the high temperature in situ data.References
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- S. N. Ruddlesden and P. Popper, Acta Crystallogr., 1958, 11, 54–55 CrossRef CAS.
- B. V. Beznosikov and K. S. Aleksandrov, Crystallogr. Rep., 2000, 45, 792–798 CrossRef.
- M. A. Yatoo and S. J. Skinner, Mater. Today: Proc., 2022, 56, 3747–3754 CAS.
- M. A. Yatoo, S. S. Kawale and S. J. Skinner, in Intermediate Temperature Solid Oxide Fuel Cells, ed. G. Kaur, Elsevier, 2020, pp. 315–346, DOI:10.1016/B978-0-12-817445-6.00010-7.
- M. Yatoo, PhD thesis, Imperial College London, 2019, DOI:10.25560/89551.
- G. Kaur, Intermediate Temperature Solid Oxide Fuel Cells: Electrolytes, Electrodes and Interconnects, Elsevier Science, 2019, DOI:10.1016/C2018-0-00575-7.
- V. V. Kharton, A. P. Viskup, E. N. Naumovich and F. M. B. Marques, J. Mater. Chem., 1999, 9, 2623–2629 RSC.
- J. A. Kilner and S. J. Skinner, Solid State Ionics, 2000, 135, 709–712 CrossRef.
- T. Nakamura, R. Oike, Y. Ling, Y. Tamenori and K. Amezawa, Phys. Chem. Chem. Phys., 2016, 18, 1564–1569 RSC.
- A. Demourgues, A. Wattiaux, J. C. Grenier, M. Pouchard, J. L. Soubeyroux, J. M. Dance and P. Hagenmuller, J. Solid State Chem., 1993, 105, 458–468 CrossRef CAS.
- J. D. Jorgensen, B. Dabrowski, S. Pei, D. R. Richards and D. G. Hinks, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 2187–2199 CrossRef CAS PubMed.
- W. Paulus, A. Cousson, G. Dhalenne, J. Berthon, A. Revcolevschi, S. Hosoya, W. Treutmann, G. Heger and R. Le Toquin, Solid State Sci., 2002, 4, 565–573 CrossRef CAS.
- S. J. Skinner, Solid State Sci., 2003, 5, 419–426 CrossRef CAS.
- M. A. Tamimi and S. McIntosh, J. Mater. Chem. A, 2014, 2, 6015–6026 RSC.
- A. C. Tomkiewicz, M. Tamimi, A. Huq and S. McIntosh, J. Mater. Chem. A, 2015, 3, 21864–21874 RSC.
- A. C. Tomkiewicz, M. A. Tamimi, A. Huq and S. McIntosh, Faraday Discuss., 2015, 182, 113–127 RSC.
- M. A. Yatoo, A. Aguadero and S. J. Skinner, APL Mater., 2019, 7, 8 Search PubMed.
- M. A. Yatoo, Z. Du, H. Zhao, A. Aguadero and S. J. Skinner, Solid State Ionics, 2018, 320, 148–151 CrossRef CAS.
- P. Pechini Maggio, US Pat., 3330697, 1967 Search PubMed.
- B. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. V. Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748, 2004 Search PubMed.
- M. A. Yatoo, Z. Du, Z. Yang, H. Zhao and S. J. Skinner, Crystals, 2020, 10, 428, DOI:10.3390/cryst10060428.
- R. I. Smith, S. Hull, M. G. Tucker, H. Y. Playford, D. J. McPhail, S. P. Waller and S. T. Norberg, Rev. Sci. Instrum., 2019, 90, 115101 CrossRef CAS PubMed.
- A. Huq, M. Kirkham, P. F. Peterson, J. P. Hodges, P. S. Whitfield, K. Page, T. Hűgle, E. B. Iverson, A. Parizzi and G. Rennich, J. Appl. Crystallogr., 2019, 52, 1189–1201 CrossRef CAS PubMed.
- M. Kirkham, L. Heroux, M. Ruiz-Rodriguez and A. Huq, Rev. Sci. Instrum., 2018, 89, 092904 CrossRef PubMed.
- R. A. Cox-Galhotra, A. Huq, J. P. Hodges, J. H. Kim, C. F. Yu, X. Q. Wang, A. J. Jacobson and S. McIntosh, J. Mater. Chem. A, 2013, 1, 3091–3100 RSC.
- Y. Touloukian, S. Saxena and P. Hestermans, Thermophysical Properties of Matter, Thermal Expansion (Nonmetallic Solids), Purdue Research Foundation, 1977, vol. 13 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. Zhang, H. Zheng, Y.-S. Chen, Y. Ren, M. Yonemura, A. Huq and J. F. Mitchell, Phys. Rev. Mater., 2020, 4, 83402 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- CASM, v1.1.1, 2021, https://github.com/prisms-center/CASMcode Search PubMed.
- S. P. Ong, A. Jain, G. Hautier, B. Kang and G. Ceder, Electrochem. Commun., 2010, 12, 427–430 CrossRef CAS.
- D. Richard Stull, JANAF_Thermochemical_Tables, Clearinghouse, 1965 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga, H. Jónsson and H. Jó, J. Chem. Phys., 2000, 113, 2082 Search PubMed.
- Z. Zhang and M. Greenblatt, J. Solid State Chem., 1995, 117, 236–246 CrossRef CAS.
- O. A. Lavrova, V. A. Somenkov, A. K. Tkalich, S. S. Shul'shtein, I. D. Damm and G. A. Ivanov-Smolenski, Superconductivity, 1991, 4, 1743 Search PubMed.
- J. Song, D. Ning, B. Boukamp, J.-M. Bassat and H. J. M. Bouwmeester, J. Mater. Chem. A, 2020, 8, 22206–22221 RSC.
- C. Y. Tsai, A. Aguadero and S. J. Skinner, J. Solid State Chem., 2020, 289, 121533 CrossRef CAS.
- A. Olafsen, H. Fjellvåg and B. C. Hauback, J. Solid State Chem., 2000, 151, 46–55 CrossRef CAS.
- M. U. Nagell, W. A. Sławiński, P. Vajeeston, H. Fjellvåg and A. O. Sjåstad, Solid State Ionics, 2017, 305, 7–15 CrossRef CAS.
- C. Chatzichristodoulou, B. C. Hauback and P. V. Hendriksen, J. Solid State Chem., 2013, 201, 164–171 CrossRef CAS.
- M. A. Yatoo and S. J. Skinner, ECS Transactions, The Electrochemical Society, New Jersey, 2023, pp. 1–7 Search PubMed.
- A. Flura, S. Dru, C. Nicollet, V. Vibhu, S. Fourcade, E. Lebraud, A. Rougier, J.-M. Bassat and J.-C. Grenier, J. Solid State Chem., 2015, 228, 189–198 CrossRef CAS.
- T. Ishihara, H. Matsuda and Y. Takita, J. Am. Chem. Soc., 1994, 116, 3801–3803 CrossRef CAS.
- V. Vibhu, A. Rougier, C. Nicollet, A. Flura, S. Fourcade, N. Penin, J.-C. Grenier and J.-M. Bassat, J. Power Sources, 2016, 317, 184–193 CrossRef CAS.
- J. M. Bassat, C. Allancon, P. Odier and J. P. Loup, Eur. J. Solid State Inorg. Chem., 1998, 35, 173–188 CrossRef CAS.
- C. D. Ling, D. N. Argyriou, G. Wu and J. J. Neumeier, J. Solid State Chem., 2000, 152, 517–525 CrossRef CAS.
- J. Song, D. Ning, B. Boukamp, J.-M. Bassat and H. J. M. Bouwmeester, J. Mater. Chem. A, 2020, 8, 22206–22221 RSC.
- M. Saiful Islam, J. Mater. Chem., 2000, 10, 1027–1038 RSC.
- M. Cherry, M. S. Islam and C. R. A. Catlow, J. Solid State Chem., 1995, 118, 125–132 CrossRef CAS.
- H. Kwon, J. Park, B.-K. Kim and J. W. Han, J. Korean Ceram. Soc., 2015, 52, 331 CrossRef CAS.
- T. Mayeshiba and D. Morgan, Phys. Chem. Chem. Phys., 2015, 17, 2715 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01772a |
| This journal is © The Royal Society of Chemistry 2023 |