
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering the glioblastoma microenvironment with bioactive nanoparticles for effective immunotherapy
Ryan Blanchard and
Isaac Adjei*
and
Isaac Adjei*
Department of Biomedical Engineering, Texas A&M University, TX, USA. E-mail: adjeii@tamu.edu
First published on 27th October 2023
Abstract
While immunotherapies have revolutionized treatment for other cancers, glioblastoma multiforme (GBM) patients have not shown similar positive responses. The limited response to immunotherapies is partly due to the unique challenges associated with the GBM tumor microenvironment (TME), which promotes resistance to immunotherapies, causing many promising therapies to fail. There is, therefore, an urgent need to develop strategies that make the TME immune permissive to promote treatment efficacy. Bioactive nano-delivery systems, in which the nanoparticle, due to its chemical composition, provides the pharmacological function, have recently emerged as an encouraging option for enhancing the efficacy of immunotherapeutics. These systems are designed to overcome immunosuppressive mechanisms in the TME to improve the efficacy of a therapy. This review will discuss different aspects of the TME and how they impede therapy success. Then, we will summarize recent developments in TME-modifying nanotherapeutics and the in vitro models utilized to facilitate these advances.
1. Introduction
Glioblastoma multiforme (GBM) is the most common and severe type of primary brain cancer, with a country-dependent incidence rate of 0.6–5 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 individuals.1,2 GBM develops spontaneously in glial cells, with the highest incidence in persons aged 45–65.3 Even with aggressive treatments comprising surgical resection, radiotherapy, and chemotherapy (temozolomide), the median survival time after diagnosis is only 14.6 months.4 While immunotherapy has improved survival for patients with different cancers, particularly liquid cancers, GBM patients have not benefited from these therapeutic advances, and outcomes have remained stagnant for almost twenty years. Unfortunately, novel immunotherapies for GBM that are successful preclinically have not progressed beyond stage 3 clinical trials.5
000 individuals.1,2 GBM develops spontaneously in glial cells, with the highest incidence in persons aged 45–65.3 Even with aggressive treatments comprising surgical resection, radiotherapy, and chemotherapy (temozolomide), the median survival time after diagnosis is only 14.6 months.4 While immunotherapy has improved survival for patients with different cancers, particularly liquid cancers, GBM patients have not benefited from these therapeutic advances, and outcomes have remained stagnant for almost twenty years. Unfortunately, novel immunotherapies for GBM that are successful preclinically have not progressed beyond stage 3 clinical trials.5
Several factors account for the failure of most GBM therapeutics. GBM tumors are genetically heterogeneous, making the design of targeted therapies challenging.6,7 Moreover, GBM tumors mold their microenvironment to support their growth by altering the phenotype of stromal cells, modulating blood vessel growth, and modifying the composition of the extracellular matrix (ECM). The altered GBM tumor microenvironment (TME) promotes resistance to immunotherapies by modulating hypoxia, oxidation state, and metabolism.8,9 In addition to TME-induced immune evasion, GBM cell niches resistant to immunotherapies arise during treatment that promote recurrence.10 These cell niches, often composed of self-renewing GBM stem-like cells (GSC), are maintained by the TME, which stimulates survival mechanisms.10 Lastly, the blood–brain barrier (BBB), which shields the brain from hazardous substances, creates a physical barrier that prevents the transport of therapeutics into the brain, hampering their success. The BBB and strategies for bypassing it are discussed in these excellent reviews and not covered in detail here.11–13
Nanoparticles (NPs), which can be fine-tuned as precision drug carriers or be bioactive, are promising tools to overcome TME-induced evasion or resistance to immunotherapy. NPs present unique benefits for GBM treatment due to their ability to cross the BBB. By modifying the surface of the NP, therapies can maximize BBB transcytosis and GBM targeting.12–14 While the tunability of nanoparticles has increased their potential for delivering therapeutics into GBMs to improve efficacy, a new class of nanoparticles that act as therapeutics themselves due to their chemical properties offers novel avenues for treatment. These NPs, responsive to biochemical cues in the TME, allow for conventional immunotherapies to be more effective. While numerous reviews discuss immunotherapy for GBM and NP-based GBM therapy, none currently examine this emerging class of NPs and their potential to alter the GBM TME. Therefore, this review will discuss the GBM TME, its role in disease progression and resistance to immunotherapy, and nanotherapeutic platforms designed to overcome these mechanisms and improve therapeutic efficacy.
2. The glioblastoma microenvironment
The GBM tumors are a complex mix of non-malignant cells, matrix, and molecules in addition to the cancer cells. The cancer cells co-opt these other microenvironment components to promote survival, immune evasion, and growth (Fig. 1). Changes in the microenvironment, such as oxygen level, acidity, and oxidation state, shape the phenotype and metabolism of malignant and non-malignant cells. However, unlike genomic changes to GBM cells, which are heterogeneous and vary within and between tumors, alterations to the TME are generally consistent across tumors. This consistency of changes in the TME across tumors makes it an exciting target for therapeutic development.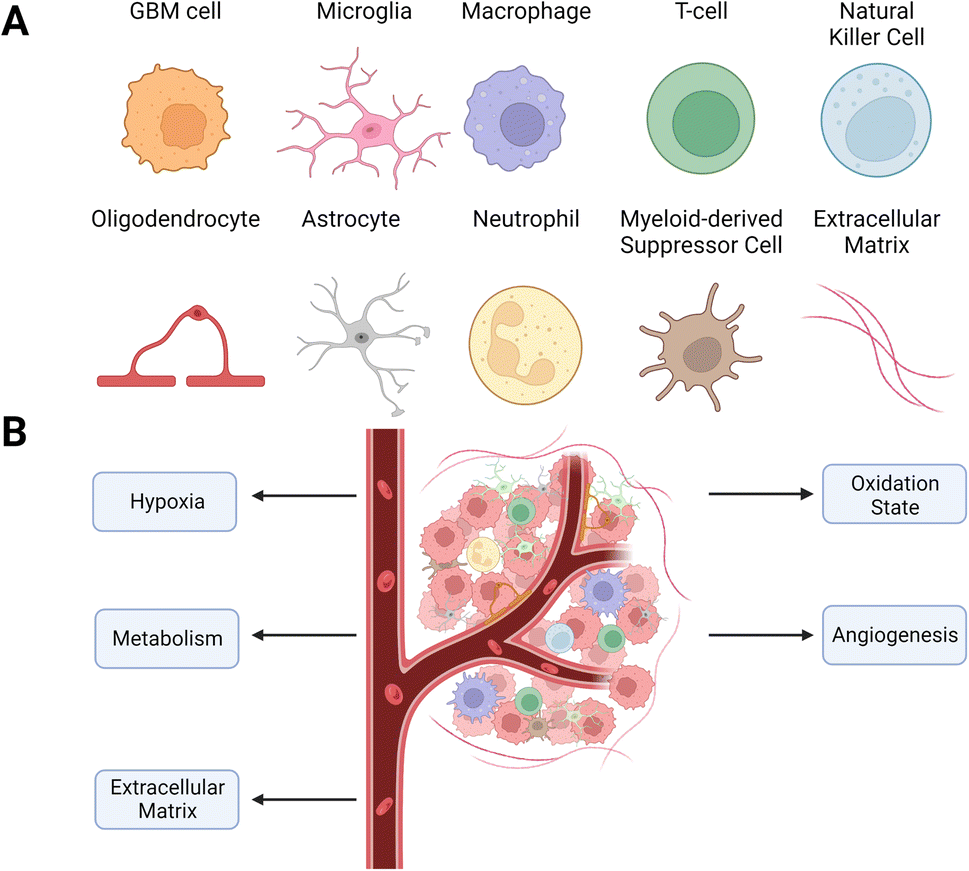 | ||
| Fig. 1 Cellular composition of the GBM TME. (A) Types of cells found within the TME. The TME is highly heterogeneous, containing various stromal and immune cells that enable cancer growth. (B) Schematic of the TME with targetable alterations currently being investigated to improve GBM therapy. | ||
2.1 Cellular composition
2.1.2.1 Glioblastoma-associated stromal cells (GASC). The presence of fibroblast-like cells in GBM tumors is a highly disputed question due to the lack of fibroblasts in healthy brain tissue. However, recent studies have identified GASC populations with myofibroblast properties that drive ECM remodeling and angiogenesis.19–21 These cells are associated with tumor progression and stemness, driving negative patient outcomes.22 One GASC population, which is mesenchymal stem cell-like, constructs a pro-invasive matrix through force-mediated ECM remodeling.21 Another GASC phenotype in the surgical margins of GBM tumors promotes tumor and endothelium development by stimulating angiogenesis.19 GASCs also promote the expansion of the GSC populations through the secretion of hepatocyte growth factor (HGF) and can induce M2 polarization through extra domain A fibronectin.23 These identified cells facilitate cell migration, drug resistance, and angiogenesis through the secretion of paracrine factors such as TGF-β and vascular endothelial growth factor (VEGF).24–27
2.1.2.2 Glial cells. Astrocytes maintain the homeostasis of neuron metabolism and mediate neuronal communication with microglia, monocytes, and T cells.28,29 While the pathophysiology of astrocytes within GBM is not well-defined, recent work shows that reactive astrocytes secrete the anti-inflammatory cytokines TGF-β, IL-10 and granulocyte colony-stimulating factor (G-CSF) that also have immune suppressive properties.30–32
Oligodendrocytes are rare within the TME but increase in number at the tumor border.33,34 There, oligodendrocyte progenitor cells induce stemness and chemoresistance.34
2.1.2.3 Neurons. Neurons stimulate the proliferation of GBM cells through the secretion of neuroligin-3, which activates the PI3K-mTOR pathway.35 The PI3K-mTOR pathway is continuously stimulated in cancer cells, which promotes tumorigenesis and therapy resistance.36 In addition, neuronal signaling can activate calcium signaling in populations of GBM cells, driving microtube formation and invasion.37
Neutrophils accumulate in the TME and promote GBM cell proliferation and epithelial-to-mesenchymal transition behavior by expressing S100A4.42,43 While DCs are absent in healthy brain tissue, they migrate to the CNS after tumorigenesis to support tumor rejection.44 However, GBM cells inhibit DC maturation and thus suppress their antigen-presenting function while promoting their role in anergy by stimulating the release of the redox-sensitive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2).45 MDSCs are a heterogeneous population of myeloid cells that promote the immunosuppression of natural killer (NK) cells and cytotoxic T cells through a variety of pathways, including nitric oxide, prostaglandin E2 (PGE2), and arginase 1 (ARG1).46
2.2 Vascular changes and hypoxia in the TME
The high rates of cellular growth and increased VEGF expression result in a vascular network that is tortuous and disorganized, with larger diameters than normal blood vessels.53,54 Increased permeability and disorganization lead to unequal blood flow in the tumor, which causes the formation of hypoxic and necrotic zones.55Hypoxia is a hallmark of GBM and leads to increased invasiveness and aggressiveness.55,56 Cells in hypoxic zones form hypercellular pseudopalisades, which promote migration and aggressiveness.57 Hypoxia increases the expression of the hypoxia-inducible factor (HIF-1α), which controls angiogenic, transcriptional, and metabolic pathways.58,59 HIF-1α induces the expression of matrix metalloproteinases (MMP) that contribute to invasion by remodeling the ECM.60 Hypoxia increases the expression of multidrug resistance-associated protein 1 (MRP-1), resulting in therapy resistance by pumping drugs out of the cells.61 Hypoxia also increases autophagy or self-digestion, a survival mechanism GBM cells use to avoid apoptosis by activating the PI3K/MAPK pathway.62 Autophagy leads to resistance to TMZ and radiation therapy.63
2.3 Metabolic changes in the TME
Lactate transported out of tumor cells lowers the extracellular pH from approximately 7.4 on the leading edges to approximately 6.0 in the central necrotic zones of the tumor.71,72 Cells re-establish intracellular pH levels by importing additional bicarbonate ions, facilitated by the hypoxia-induced downstream protein carbonic anhydrase IX (CA-IX).73 Acidification of the tumor prompts significantly higher levels of immunosuppression and GSC migration and proliferation.74–76
Molecular building blocks such as amino acids and nucleosides also have increased concentrations within the TME, contributing to greater aggression, invasion, and immunosuppression. Adenosine, a potent immunosuppressant produced in large quantities in hypoxic conditions by GBM cells and exported into the extracellular space, increases cancer aggressiveness and dampens the immune response through A2A receptors (A2AR) on immune cells.77–79 Arginine promotes tumorigenesis and angiogenesis80 and is provided by external sources since the urea cycle, the primary source of arginine, is inhibited in GBM cells through the silencing of argininosuccinate synthetase 1.81
T cells rely primarily on fatty acid oxidation and oxidative phosphorylation in their quiescent state.87 When activated, the energy needs increase, which is met by enhanced requirements of glucose, glutamine, and L-arginine.88 The TME is poor in glucose and amino acids, which hinders proliferation and effector functions such as TCR signaling and inflammatory cytokine secretion.67,89 On the other hand, Treg cells rely primarily on fatty acid oxidation and can proliferate more easily within the TME. Naïve T cells in the TME will preferentially differentiate into Treg cells instead of cytotoxic T cells due to AMP-activated protein kinase (AMPK1), which senses and is expressed in environments of nutrient deficiency.90
TGF-β inhibits the IL-2-induced metabolism of NK cells, decreasing oxidative phosphorylation.91 Excess adenosine in the TME stimulates NK cells through A2AR, decreasing their cytotoxicity.92 In addition, lactic acid blocks IFN-γ expression in cytotoxic T cells and NK cells by inhibiting the nuclear factor of activated T cells (NFAT).93 Like T cells, depletion of glucose and amino acids impairs NK cell function and proliferation.94,95
Cancer cells increase the production of antioxidants such as glutathione (GSH) and catalase to prevent ROS-induced apoptosis.98 Catalase is an enzyme that catalyzes the reaction of hydrogen peroxide into oxygen and water, while GSH is oxidized glutathione disulfide (GSSG), which reduces hydrogen peroxide to oxygen and water. In the TME, these antioxidants induce resistance to the application of therapies. GSH elevates the expression of multidrug resistance protein, and accumulation of GSSG has adverse effects on metabolic regulation and cellular integrity.102,103 While overexpression of catalase reduces levels of H2O2, it also leads to increased proliferation, resistance to temozolomide and radiotherapy, and the formation of neurospheres, a feature of GSCs.104
2.4 Changes to the ECM in the TME
The brain ECM contains a uniquely flexible matrix that structures the parenchyma and guides signaling pathways that regulate cellular functions such as differentiation and migration.105 The main components of the ECM are glycosaminoglycans (GAG) and glycoproteins such as laminins, fibronectin, and tenascins.106 The brain contains less fibrous proteins, such as collagen, than other tissues.107 In GBM, the ECM increases in stiffness and density, leading to a rise in interstitial pressure.108 These changes in the ECM reduce nutrient and oxygen penetration and access to the cancer cells, increasing hypoxia and metabolic stress. The effectiveness of drugs to penetrate the tumor is also reduced, minimizing their efficacy (Fig. 2).109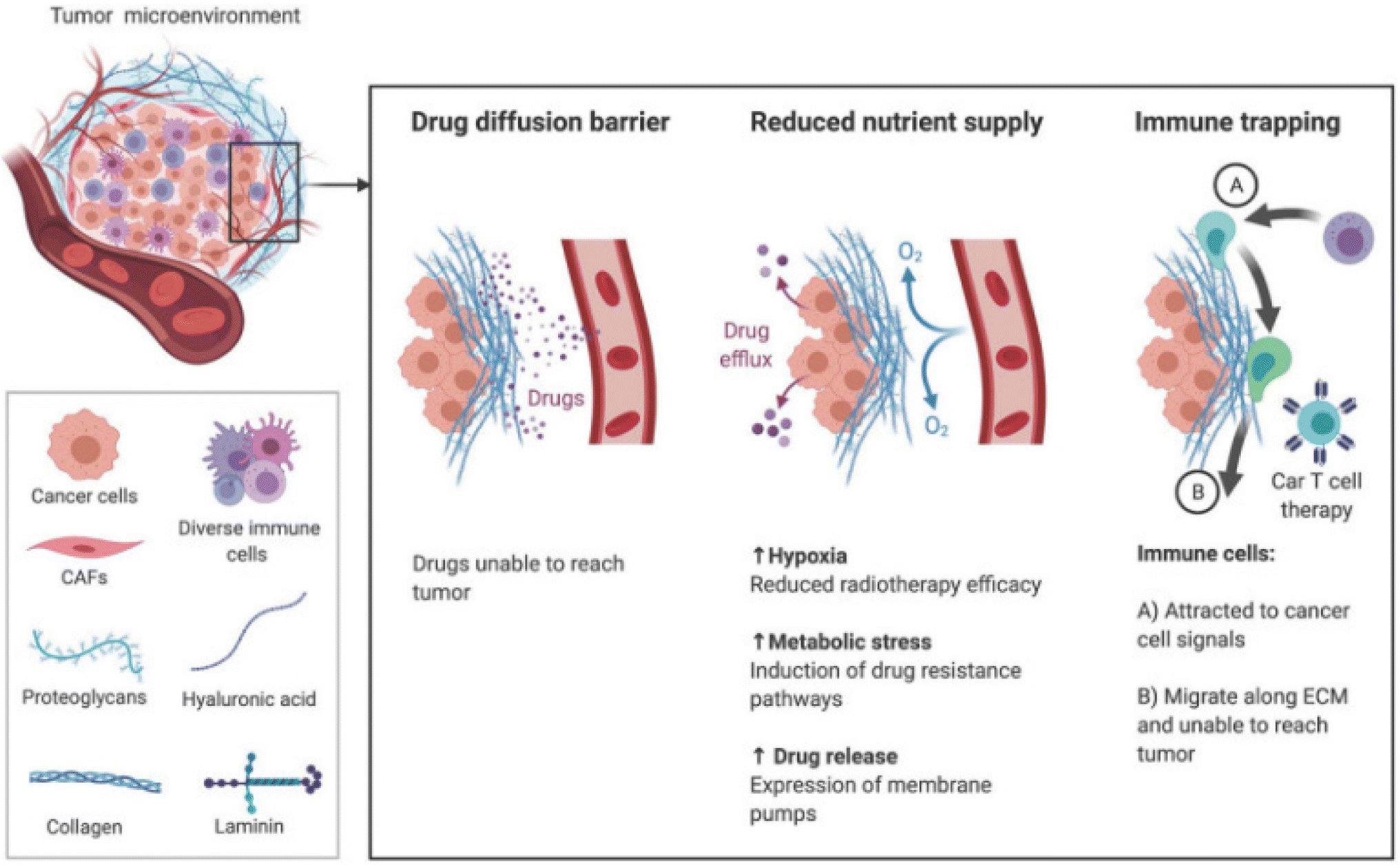 | ||
| Fig. 2 The TME ECM impairs therapy through increased stiffness and density. Increased pressure prevents the transport of drugs and nutrients to tumor and movement of immune cells toward chemokine signals [reproduced from ref. 188 with permission from AJCR, ©2021]. | ||
The GAG hyaluronic acid (HA) is vital in tumor progression. The molecular weight of HA determines if it inhibits or enhances tumor progression. Low molecular weight HA (<250 kDa) triggers the G1 phase to promote proliferation, while high molecular weight HA (>1000 kDa) locks cells in the G1 phase.110,111 The expression of the adhesion receptor CD44, which binds to HA, is associated with increased GBM aggressiveness, proliferation, chemoresistance, and stemness.112 Chondroitin sulfate proteoglycan-4 (CSPG-4) is another GAG upregulated in GBM.113 Tumors expressing CSPG-4 have worse outcomes, as the GAG encourages migration by promoting the expression of β-integrins and growth factors.114 Increased collagen in the TME, which increases its stiffness, leads to immune trapping, where migration of immune cells such as T cells is physically inhibited.115
A variety of glycoproteins are also upregulated in GBM. Tenascin-C (TN-C) is expressed almost exclusively in GBM ECM versus healthy brain ECM, promotes cell migration and angiogenesis, and mediates changes in ECM constituents.116,117 Fibronectin increases the adhesion, proliferation, and resistance of GSCs.118 Fibulin-3, a glycoprotein only found in cancerous brain tissue, promotes tumor progression through the Notch and NF-κB signaling pathways, enhancing the viability of tumor-initiating cells.119,120
One of the most critical components related to the ECM is matrix metalloproteinases (MMP), enzymes that degrade and remodel the ECM. MMP-2 and MMP-9 are upregulated in GBM, with MMP-9 expression changing from undetectable in the healthy brain to significantly expressed in GBM.121,122 Both molecules promote cancer malignancy. MMP-2 promotes the transition to an invasive, undifferentiated phenotype, alters the metabolism, and inhibits apoptosis.123,124 Similarly, MMP-9 is essential in instigating GBM invasion.125
3. In vitro models to study TME
In vitro models are used in conjunction with in vivo models to understand the TME and its effects on therapeutic efficacy better.126 However, standard two-dimensional cell culture models fail to reproduce the unique conditions in the TME, limiting their applications in studying the TME and evaluating new cancer therapeutics.127 In 2D models, cell–ECM and cell–cell interactions are superseded by cell-tissue culture plastic interactions.128 Therefore, three-dimensional tumor models that recapitulate dysfunctional aspects of the TME, such as hypoxia gradients and cell-ECM interactions, are necessary (Fig. 3).129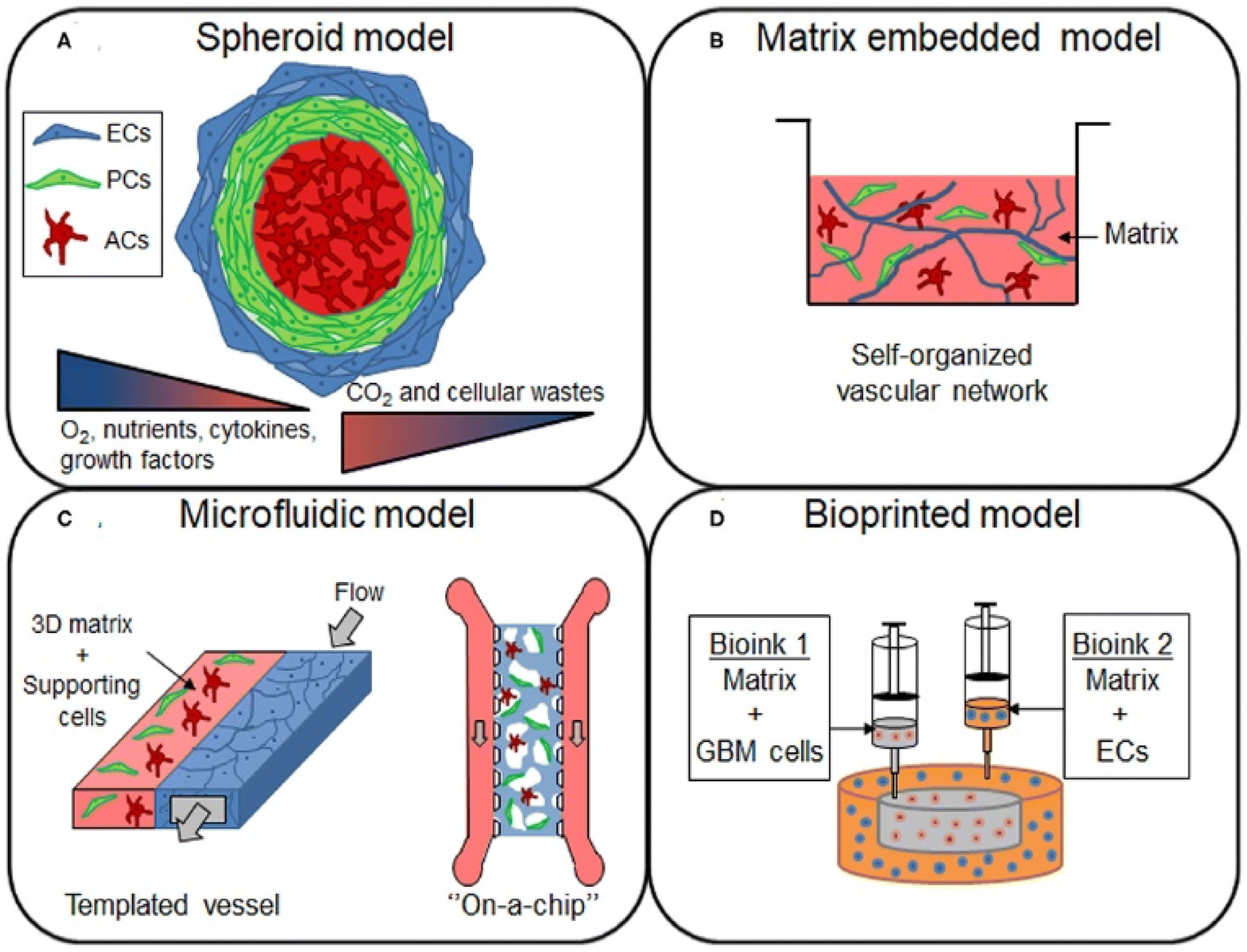 | ||
| Fig. 3 In vitro models for studying the GBM TME. (A) Spheroid models are constructed by using cell–cell interactions to form cell aggregates of one or more cell types in a culture. These models are ideal for the study of hypoxia and nutrient gradients, as spheroids replicate the necrotic cores found in human GBM tumors. (B) Cells or spheroids can be embedded in a matrix containing natural or synthetic materials that replicate the ECM. (C) Microfluidic models, which can be customized for the application, are gaining popularity. These models are ideal for dynamic models, where the effects of flow on the TME can be observed by flowing media across fabricated vessels coated with matrix or cells. (D) Bioinks are used to 3D print models containing various types of cells. This can be utilized to easily construct a custom scaffold that better reflects the structure and function of a GBM tumor. Models can be constructed from commercially available cell lines or patient-derived cells using various model approaches depending on the application [reproduced from ref. 189 with permission from Frontiers, ©2021]. | ||
Spheroids, which are 3D aggregations of cells often used as tumor models, are formed using various techniques, including bioprinting and aggregation on a low attachment surface such as agarose in static or dynamic conditions.130 Once generated, spheroids can be cultured in a scaffold or a scaffold-free environment. Scaffolds, or matrices, allow researchers to mimic the ECM of the TME, replicating some of the organizational structure in vitro.131 For GBM research, the most common materials are natural biomaterials such as Matrigel, HA, collagen, and decellularized ECM or synthetic polymers such as poly(ethylene glycol) (PEG) and poly(urethane).132,133 A critical variable in constructing a model is cell choice. The most common choice is a glioblastoma cell line, such as human U87 or mouse GL261. However, these cell lines lack the heterogeneity in GBM tumors; therefore, many researchers utilize patient-derived organoids.134 By collecting patient-derived GSCs for organoid generation, models can reflect the structure of the tumor rather than that of the normal brain.135 The last primary consideration for organoid models is co-culturing cancer cells with non-malignant tumor cells. For example, organoids constructed of GSCs can be augmented with human cerebral organoids to study invasion into the brain.136,137 These cerebral co-culture models are derived from mouse embryonic stem cells or genetically engineered human embryonic stem cells.138 Additionally, through rapid 3D bioprinting combining GSCs with astrocytes, neural precursor cells, and macrophages in an HA scaffold, researchers were able to recapture cellular interactions and immune functions.139
Although organoids are currently popular for studying the TME, microfluidic lab-on-a-chip models have risen as alternatives. These devices, fabricated mostly from poly (dimethylsiloxane) (PDMS) using soft lithography, offer flexibility in design choices and model complexity that are not available in organoid models. After fabrication, vessels within the chip can be coated with a protein that promotes cell attachment, such as fibronectin, and a matrix to model the TME. Lab-on-a-chip models are ideal for many situations, particularly studies involving fluid flow, cell migration, and drug discovery.140 Like organoids, cell choice and scaffold material are critical decisions for the design of lab-on-a-chip models. Most researchers utilize a human GBM cell line such as U87MG, with other studies also constructing co-cultures with HUVECs, astrocytes, or hCMEC/d3, a brain endothelial cell line.141
4. Techniques to engineer the TME with nanotechnology
The changes in the GBM TME described above have been under investigation with the goal of developing more effective therapeutics. Recently, interest has increased in applying these advances to developing nanotherapeutic platforms targeting specific TME parts.4.1 Hypoxia
Hypoxia is a central focus of TME-targeting therapies. The most direct strategy developed to reduce hypoxia levels utilizes metal oxide NPs. These NPs react with various molecules in the TME to produce oxygen. For example, calcium peroxide (CaO2) NPs form oxygen in the presence of water (eqn (1A)). However, because of the ubiquity of water, this reaction is non-specific, and the NPs react immediately after administration. Coating CaO2 NPs with a pH-sensitive polymer for oxygen production in the acidic environment in pancreatic cancer addresses this drawback.142 These NPs generate oxygen in the acidic tumor but produce minimal oxygen at the normal physiological pH (pH = ∼7.4) in healthy tissues (Fig. 4A). While CaO2 NPs have been demonstrated in some tumors, their effectiveness has not been tested in GBM.| 2CaO2 + 2H2O → Ca(OH)2 + O2 | (1A) |
| MnO2 + 2H2O2 → Mn2+ + 2H2O + 2O2 | (1B) |
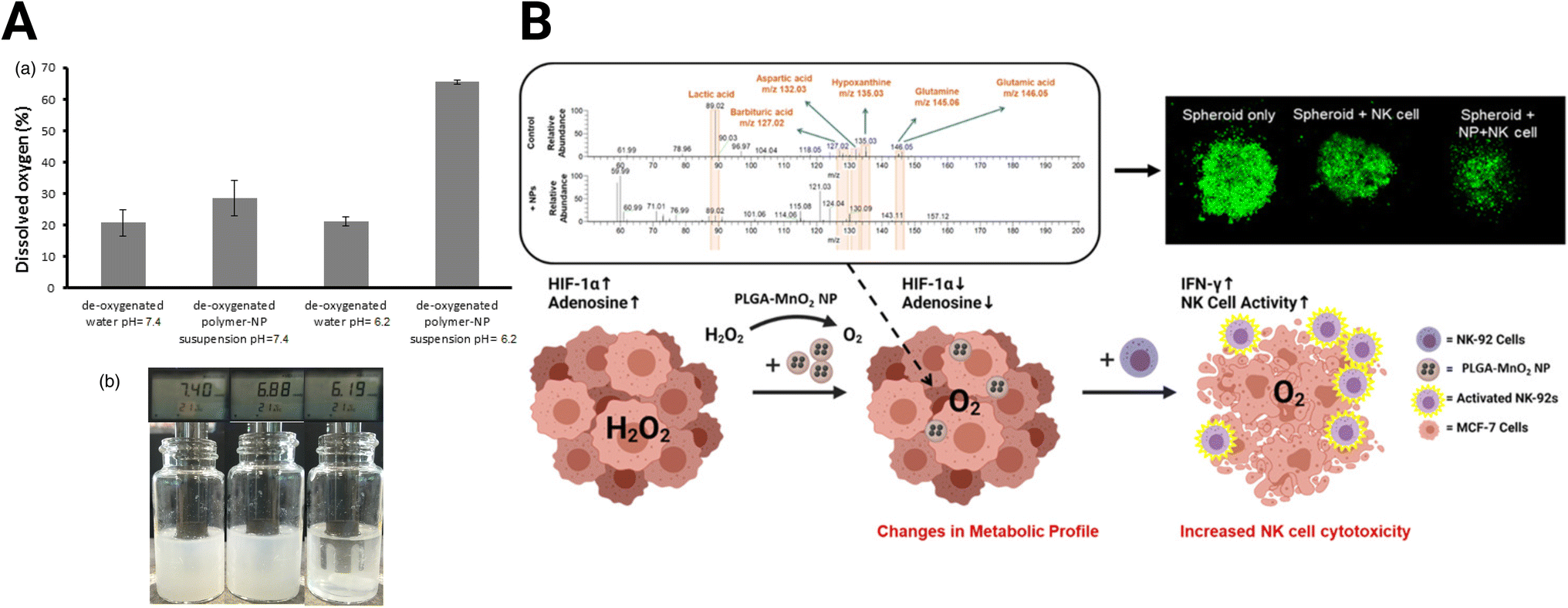 | ||
| Fig. 4 Oxide-based NPs for hypoxia reduction. (A) Dissolved oxygen generated by CaO2 NPs with changes in pH [reproduced from ref. 142 with permission from Elsevier, ©2017]. (B) MnO2 NPs skew TME from immunosuppression and improve adoptive NK cell therapy [reproduced from ref. 144 with permission from American Chemical Society, ©2021]. | ||
Eqn (1A) and (1B): chemical reactions of oxygen-generating NPs. (A) CaO2 NP reaction with H2O (B) MnO2 reaction with H2O2.
Due to the reaction's non-specificity, CaO2 NPs are utilized less than manganese oxide (MnO2) NPs. MnO2 NPs, in contrast to CaO2, react with hydrogen peroxide in the hypoxic tumor core to form oxygen (eqn (1B)). However, MnO2 NPs require stabilization, or they will aggregate.143 PEGylation can improve stability, but the exposed MnO2 NPs react too quickly to adequately shift the expression of hypoxic cancer cells in the immunosuppressive TME. This problem was addressed by encapsulating the MnO2 NPs in poly(lactic-co-glycolic acid) (MnO2-PLGA NP) to extend the oxygen generation and shield cells from non-specific toxicity.144 The authors demonstrated that delivery of the MnO2-PLGA NPs reduced hypoxia without an increase in cytotoxicity, skewing the expression and metabolism of breast cancer 3D spheroid models away from their immunosuppressive phenotype (Fig. 4B). Specifically, MnO2-PLGA NPs reduced expression of HIF-1α and presence of immunosuppressive metabolites, such as lactic acid and adenosine. These changes improved the cytotoxicity of natural killer (NK) cell immunotherapy towards the spheroids. In addition, MnO2 NP-based platforms have been designed specifically for GBM treatment. Liang et al. developed a MnO2 NP system to enhance sonodynamic therapy (SDT), using protoporphyrin as a sonosensitizer that utilizes generated oxygen to enhance efficacy.145 This therapy conjugated a high-affinity transferrin to the NP to boost BBB penetration through receptor-mediated endocytosis and decreased tumor burden in C6 GBM xenografts.
Like MnO2, cerium oxide (CeO2) leverages reacting with H2O2 for hypoxia treatment. While CeO2 NPs have not been utilized for GBM treatment, therapies using this technique as a sensitizer for radiotherapy or photodynamic therapy have shown promise in other cancer types.146,147
Another solution to reduce hypoxia is to encapsulate hypoxia-reducing drugs in NP shells. Wu et al. designed hypoxia-reducing silica NPs that encapsulate catalase, which degrades hydrogen peroxide to produce oxygen.148 These NPs were covered with aptamer-modified macrophage exosomes to improve BBB penetration and reduce uptake by the reticuloendothelial system. In addition, the silica NPs were degradable in the presence of GSH, an antioxidant elevated in the TME, boosting delivery efficiency.149
4.2 Redox
There are two primary, intertwined mechanisms that NPs can use to affect the oxidation state of the tumors. The first is the direct generation of ROS by NPs, generally through Fenton or Fenton-like reactions with metal oxide NPs. In the Fenton reaction, ferrous ions react with hydrogen peroxide to produce hydroxide radicals (eqn (2)).150 Taking advantage of excess H2O2 in the TME, iron oxide NPs create ROS, which are toxic to cancer cells and drive the TME towards an inflammatory phenotype.151 The weakly acidic TME catalyzes the Fenton reaction, effectively localizing its effect to the tumor site.152 Similarly, Fenton-like reactions utilize other metal ions, such as Mn2+, Cu+, and Ti3+, to generate hydroxyl radicals. The second method of affecting the oxidation state impacts the availability of antioxidants such as GSH and catalase.| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (2) |
Eqn (2): Fenton and Fenton-like reactions.
A photo-responsive NP system to produce a Fenton-like reaction was developed using ultrasmall Cu2−xSe NPs (Fig. 5A).153 This system, when activated with near-infrared (NIR) irradiation, generates ROS and O2, inducing a change in polarization of TAMs from the M2 to M1 state within the TME. Combined with the indoleamine 2,3-dioxygenase (IDO) inhibitor indoximod, these NPs improved PD-L1 blockade therapy in GBM through JQ1, which is a small molecule inhibitor of bromodomains that inhibits PD-L1 expression.153 Complexing Fe2+ with the natural anti-inflammatory agent, gallic acid could extend the Fenton reactions (Fig. 5B).154 These NPs strongly absorb NIR light upon exposure, which produces heat and promotes GBM cell ferroptosis, a non-apoptotic cell death dependent on intracellular iron concentration.155,156 Combining the complexed Fe2+ and gallic acid with cisplatin and a glutathione peroxidase-4 small interfering RNA (siRNA) improves the antitumor effect of the system further. Localized heating of Fe3O4 NPs stimulates the production of ROS-producing lipid peroxides, allowing for more effective cell death in prostate cancer by ferroptosis.157
 | ||
| Fig. 5 Schematics of nanomedicines utilizing oxidation state. (A) Cu2−xSe NPs for ROS production to improve checkpoint blockade therapy [reproduced from ref. 153 with permission from KeAi, ©2022]. (B) Extended Fenton reactions with gallic acid/Fe2+ NPs for redox disruption and effective ferroptosis [reproduced from ref. 154 with permission from Elsevier, ©2021]. (C) Ce6-modified MnO2 nanosheets for GSH depletion and reduced radioresistance [reproduced from ref. 158 with permission from Wiley-VGH, ©2016]. | ||
In addition to reducing hypoxia, MnO2 can reduce the concentration of antioxidants, making the TME more susceptible to the generation of ROS. Fan et al. designed a MnO2-photosensitizer system for effective photodynamic therapy (Fig. 5C).158 MnO2 nanosheets protect and carry the photosensitizer chlorin e6 (Ce6) into the cell, where the MnO2 is reduced by GSH, depleting GSH concentrations and releasing Ce6, which generates ROS upon application of light. Yong et al. developed Gd-containing nanospheres with polyoxometalate-conjugated chitosan (GdW10@CS) for reducing hypoxia and depleting GSH to reduce radioresistance.159 While acting as a carrier for HIF-1α siRNA, the nanospheres trigger GSH oxidation to generate ROS through W6+.
4.3 Angiogenesis
While NP systems that target hypoxia affect new blood vessel formation, other strategies are employed to prevent angiogenesis in the TME. Most of the current research in this field focuses on synthesizing targeted and multi-functional NPs using chemotherapeutic agents such as doxorubicin (DOX) and paclitaxel and VEGF-targeting agents such as bevacizumab.160 For example, Lu et al. designed a multi-functional NP to maximize DOX efficacy by employing the anti-angiogenic drug combretastatin A4 (CA4).161 This tubulin-binding agent causes blood vessel regression in the TME and acts in concert with DOX and all-trans retinoic acid within an MRI-traceable iron oxide nanocube for a controlled release system (CARD-B6).162 The CARD-B6 platform drastically decreased blood flow within the tumor compared to free drugs and the nanoparticle platform without functionalization. Another strategy utilizes RNAi targeting VEGF using a dendrigraft poly-L-lysine gene vector NP modified by transferrin receptor for increased BBB penetration.163CXCR4, the receptor for CXCL12, or SDF-1α, plays an essential role in facilitating the proliferation of GSCs and promoting angiogenesis.164,165 Séhédic et al. designed a novel lipid nanocapsule for internal radioimmunotherapy through dual rhenium-188 (188Re) delivery and a function-blocking antibody for CXCR4.166 188Re, a beta radionuclide, acts as an internal vectorized radiotherapy. Therefore, 188Re is accompanied by 12G5, an antibody that blocks the function of CXCR4, to improve efficacy. The NP platform decreased tumor size and improved survival in an orthotopic U87MG mouse compared to mice treated with NP without non-specific IgG2a or saline injection. After study completion, CD31 staining of the tumor showed a significant decrease in blood vessel presence.
4.4 Metabolic alterations
Therapies targeting metabolism seek to improve treatment by exploiting changes in the energy balance and cell metabolite production. The increased dependence of the TME on glucose due to the Warburg effect can be targeted to hinder the energy cycle of GBM cells. Glucose oxidase (GOx) converts glucose into gluconic acid and H2O2 in the presence of O2, cutting off the primary energy source of the tumor and leading to increased tumor stress and cell death.167,168 The effectiveness of GOx is improved when incorporated into a multimodal NP system. For example, Ke et al. designed a red blood cell membrane-cloaked zeolitic imidazolate framework-8 (ZIF-8), a type of metal–organic framework (MOF), for the co-delivery of GOx and DOX (Fig. 6A).169 As the GOx is released from the MOF and increases tumor acidity, the MOF degrades, releasing DOX into the starved TME. In vivo studies with U87 tumors show the improved tumor inhibition capability of the NPs compared to any singular component. Qiao et al. utilized GOx-modified bovine serum albumin (BSA) NPs with the cationic polymer PEG2k-PEI1.8k as a coating to prolong circulation time (Fig. 6B).170 These NPs encapsulated CuO for delivery, combining Fenton-like reactions and starvation therapy for more effective antitumor activity.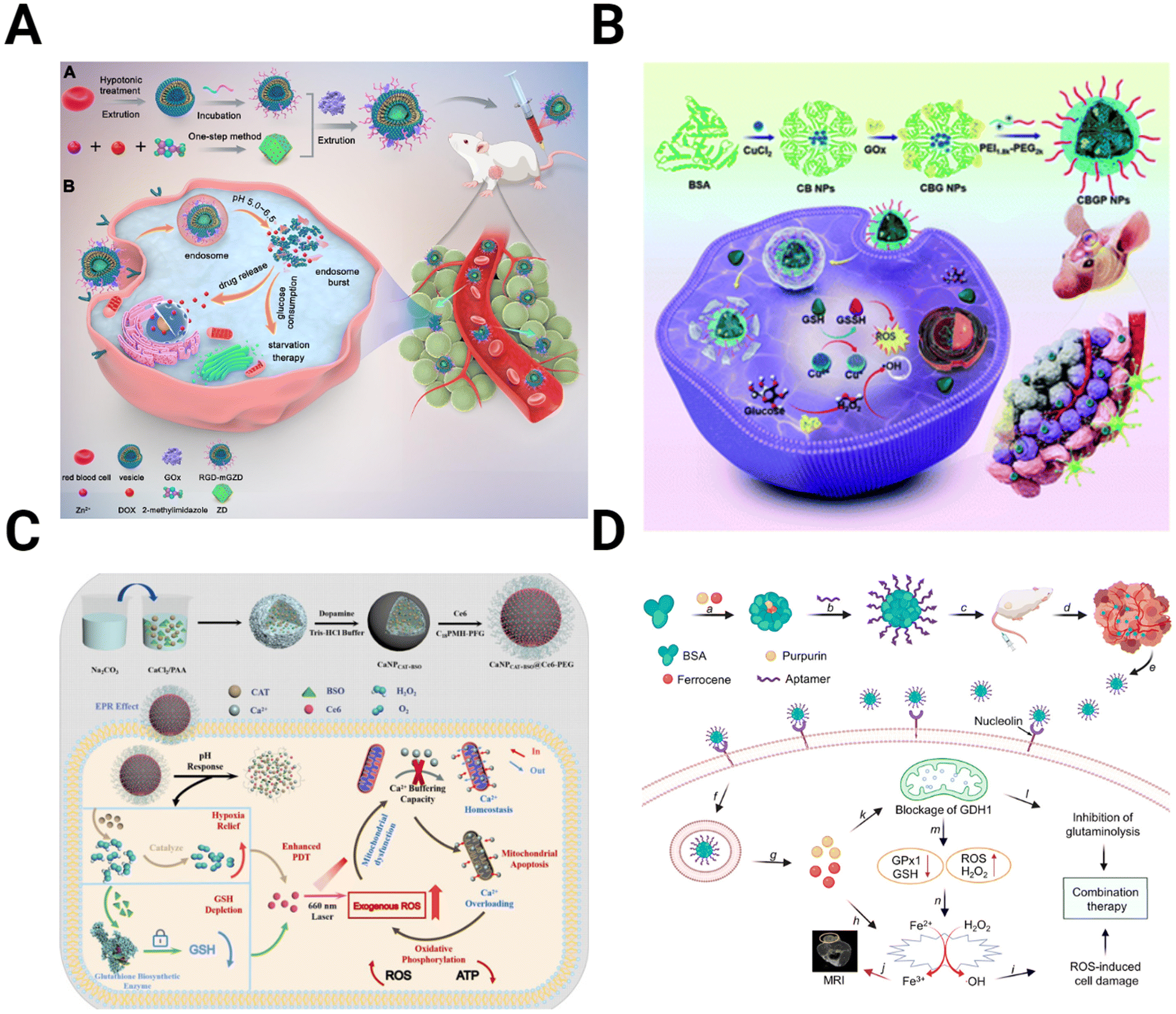 | ||
| Fig. 6 Schematics depicting the preparation and mechanism for NPs targeting metabolic alterations in the TME. (A) ZIF-8 MOFs modified with the red blood cell membrane to deliver GOx and DOX for starvation chemotherapy [reproduced from ref. 169 with permission from Elsevier, ©2022]. (B) BSA NPs containing CuO coated with GOx and PEG2k-PEI1.8k for targeted Fenton-like reaction and starvation therapy [reproduced from ref. 170 with permission from Royal Society of Chemistry, ©2022]. (C) CaCO3 NPs loaded with catalase, Ce6, and buthionine sulfoximine, designed to overwhelm calcium buffering capacity to inhibit ATP production [reproduced from ref. 175 with permission from Elsevier, ©2022]. (D) Targeted BSA NPs encapsulate purpurin to inhibit glutaminolysis and ferrocene for ROS-mediated cell damage [reproduced from ref. 177 with permission from American Chemical Society, ©2021]. | ||
Similarly, a fibrin gel containing IDH1 was used for GBM starvation therapy.171 After tumor resection, the gel containing Mn-doped calcium phosphate NPs encapsulating GOx is sprayed into a surgical defect. These NPs utilized a combination of Fenton-like generation of H2O2 and glucose starvation therapy. GBM starvation therapy is also achieved by inhibiting the overexpressed glucose transporter Glut1. CD44-targeting gold nanorods conjugated with a Glut1 inhibitor to interfere with the energy balance in GBM cells.172 By inhibiting Glut1, ATP levels are reduced, decreasing the expression of heat shock proteins (HSP), which have increased expression in cancer and play an essential role in maintaining thermoresistance.173
An alternative strategy for altering cancer cell metabolism is directly targeting ATP synthesis. In addition to powering function and proliferation of GBM cells, intracellular ATP concentrations drive drug resistance.174 Zhu et al. overloaded the GBM mitochondria with Ca2+ to induce cell apoptosis (Fig. 6C).175 The authors fabricated CaCO3 NPs loaded with catalase, Ce6, and buthionine sulfoximine (BSO) to combine oxygen and GSH depletion with calcium overloading. While Ca2+-related toxicity would be limited on its own, ROS production from GSH and O2 depletion reduce the buffering capacity of the mitochondria, allowing them to be more easily overwhelmed and resulting in greater therapy efficacy.
Another target within the metabolism besides glucose is glutaminolysis. Like glycolysis, glutaminolysis is upregulated in the TME, driving the TCA cycle and the production of antioxidants such as GSH.176 Xu et al. take advantage of this by encapsulating purpurin, the inhibitor of glutamate dehydrogenase 1 (GDH1), an essential enzyme in glutaminolysis, in a BSA NP (Fig. 6D).177 When combined with the H2O2-producing ferrocene, purpurin disrupts cell metabolism and inhibits cancer growth.
4.5 Extracellular matrix
The last area to be discussed is the application of NP systems to modify the ECM. These therapies target components of the ECM with the primary goal of inhibiting those that increase tumor aggressiveness.The first group of ECM-targeting therapies seeks to reduce the density of the ECM within the TME. One method of accomplishing this task is to utilize a proteolytic enzyme to degrade the matrix. Zinger et al. designed a liposome system encapsulating collagenase to enhance the transport of a drug into the dense tumor core of pancreatic ductal carcinoma.178 When administered intravenously, these liposomes decreased the fibrotic tissue mass to approximately 5%, meaning the dense collagen network within the TME had been deconstructed. Collagen depletion should be treated cautiously, as it can induce the release of pro-inflammatory cytokines and growth factors that promote tumorigenesis.179 Losartan, an angiotensin inhibitor, reduces the synthesis of HA and collagen, reducing stress on the tumor and improving perfusion.180 Zhao et al. combined losartan with anti-PD1 therapy to enhance the efficacy of chemo-immunotherapy for triple-negative breast cancer.181 Using liposomes to encapsulate DOX, losartan, and α-PD1, the authors could normalize the TME, reducing stromal density, hypoxia, and immunosuppression. The components within the NP were able to reduce tumor size more than compared to only one or two drugs. Grabowska et al. chose TN-C as their target for inhibition, utilizing double-stranded RNA (dsRNA).182 The dsRNA was encapsulated in a magnetite NP coated with the cationic polymer polyethyleneimine (PEI) for simultaneous drug delivery and MRI imaging. By inhibiting TN-C, the authors reduced the migration and proliferation of GBM cells.
The final strategy is to inhibit MMPs to reduce migration and invasiveness. The most popular drug inhibiting MMP-2 is chlorotoxin (CTX), a peptide derived from scorpion venom.183 Agarwal et al. incorporated CTX onto a morusin-loaded PLGA NP for GBM treatment.184 Combining MMP-2 inhibition through CTX with morusin, which inhibits MMP-2 and MMP-9 in hepatoma SK-Hep1 cells, produced a potent cytotoxic effect in vitro.185 In addition, morusin stimulates apoptosis by inhibiting the NF-κB and STAT3 pathways and antioxidant scavenging.185–187
5. Outlook
There is interest in engineering the TME with nanotechnology, leading to a fast-moving landscape of innovations. One area of particular interest is combination therapies, where one or more TME-altering agents are used in concert with a traditional therapy within an NP system. However, there are still areas where TME therapies have not yet been explored for GBM. For example, while acidity is a core component of the TME, no therapies specifically target it. Hypoxia, oxidation state, metabolism, or immunosuppression are interdependent within the TME. While hypoxia reduction inadvertently reduces acidity, assessing their relative impact would advance our understanding of the TME. In addition, while target molecules like CPSG-4 and fibulin-3 are demonstrated to promote cancer aggressiveness, no nanocarrier therapies have been developed to target them. The field is relatively new, with no therapies successfully tested in clinical trials, but it is rapidly expanding. As NP-based therapies enter clinical trials in more significant volumes, it will be vital to understand more thoroughly the effects these treatments have through the lens of the TME, the healthy brain parenchyma, and systemic function. For this reason, there is still much to learn before GBM treatment can be improved and the survival time extended.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fig. 1 was created using https://www.Biorender.com.References
- A. F. Tamimi and M. Juweid, in Glioblastoma, Codon Publications, 2017, pp. 143–153 Search PubMed.
- N. Grech, T. Dalli, S. Mizzi, L. Meilak, N. Calleja and A. Zrinzo, Cureus, 2020, 12(5), e8195 Search PubMed.
- Q. T. Ostrom, H. Gittleman, J. Fulop, M. Liu, R. Blanda, C. Kromer, Y. Wolinsky, C. Kruchko and J. S. Barnholtz-Sloan, Neurooncology, 2015, 17, iv1–iv62 Search PubMed.
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- M. Lim, Y. Xia, C. Bettegowda and M. Weller, Nat. Rev. Clin. Oncol., 2018, 15(7), 422–442 CrossRef CAS PubMed.
- D. A. Nathanson, B. Gini, J. Mottahedeh, K. Visnyei, T. Koga, G. Gomez, A. Eskin, K. Hwang, J. Wang, K. Masui, A. Paucar, H. Yang, M. Ohashi, S. Zhu, J. Wykosky, R. Reed, S. F. Nelson, T. F. Cloughesy, C. D. James, P. N. Rao, H. I. Kornblum, J. R. Heath, W. K. Cavenee, F. B. Furnari and P. S. Mischel, Science, 2014, 343, 72–76 CrossRef CAS PubMed.
- A. P. Patel, I. Tirosh, J. J. Trombetta, A. K. Shalek, S. M. Gillespie, H. Wakimoto, D. P. Cahill, B. v. Nahed, W. T. Curry, R. L. Martuza, D. N. Louis, O. Rozenblatt-Rosen, M. L. Suvà, A. Regev and B. E. Bernstein, Science, 2014, 344, 1396–1401 CrossRef CAS PubMed.
- Z. Chen and D. Hambardzumyan, Front. Immunol., 2018, 9, 1004 CrossRef.
- E. Fletcher-Sananikone, S. Kanji, N. Tomimatsu, L. F. M. E. di Cristofaro, R. K. Kollipara, D. Saha, J. R. Floyd, P. Sung, R. Hromas, T. C. Burns, R. Kittler, A. A. Habib, B. Mukherjee and S. Burma, Cancer Res., 2021, 81, 5935–5947 CrossRef CAS PubMed.
- J. D. Lathia, S. C. Mack, E. E. Mulkearns-Hubert, C. L. L. Valentim and J. N. Rich, Genes Dev., 2015, 29, 1203 CrossRef CAS PubMed.
- L. Jena, E. McErlean and H. McCarthy, Drug Delivery Transl. Res., 2020, 10, 304 CrossRef PubMed.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- Z. Ebrahimi, S. Talaei, S. Aghamiri, N. H. Goradel, A. Jafarpour and B. Negahdari, IET Nanobiotechnol., 2020, 14, 441 CrossRef PubMed.
- P. Henrich-Noack, D. Nikitovic, M. Neagu, A. O. Docea, A. B. Engin, S. Gelperina, M. Shtilman, P. Mitsias, G. Tzanakakis, I. Gozes and A. Tsatsakis, Nanomedicine, 2019, 17, 359–379 CrossRef CAS PubMed.
- E. Hulleman and K. Helin, Adv. Cancer Res., 2005, 94, 1–27 CrossRef CAS PubMed.
- A. G. M. Mostofa, S. R. Punganuru, H. R. Madala, M. Al-Obaide and K. S. Srivenugopal, Biomolecules, 2017, 7(2), 34 CrossRef.
- S. S. Widodo, M. Dinevska, L. M. Furst, S. S. Stylli and T. Mantamadiotis, Br. J. Cancer, 2021, 125, 1466–1476 CrossRef CAS PubMed.
- M. Butler, S. Prasad and S. K. Srivastava, Adv. Exp. Med. Biol., 2020, 1296, 1–9 CrossRef PubMed.
- A. Clavreul, C. Guette, R. Faguer, C. Tétaud, A. Boissard, L. Lemaire, A. Rousseau, T. Avril, C. Henry, O. Coqueret and P. Menei, J. Pathol., 2014, 233, 74–88 CrossRef CAS PubMed.
- A. Clavreul, A. Etcheverry, A. Chassevent, V. Quillien, T. Avril, M. L. Jourdan, S. Michalak, P. François, J. L. Carré, J. Mosser and P. Menei, J. Neurooncol., 2012, 106, 493–504 CrossRef PubMed.
- E. J. Lim, Y. Suh, S. Kim, S. G. Kang and S. J. Lee, BMB Rep., 2018, 51, 182 CrossRef CAS PubMed.
- Z. Chen, S. Zhuo, G. He, J. Tang, W. Hao, W. Q. Gao, K. Yang and H. Xu, Front. Cell Dev. Biol., 2021, 9, 721897 CrossRef PubMed.
- S. Jain, J. W. Rick, R. S. Joshi, A. Beniwal, J. Spatz, S. Gill, A. C. C. Chang, N. Choudhary, A. T. Nguyen, S. Sudhir, E. J. Chalif, J. S. Chen, A. Chandra, A. F. Haddad, H. Wadhwa, S. S. Shah, S. Choi, J. L. Hayes, L. Wang, G. Yagnik, J. F. Costello, A. Diaz, D. H. Heiland and M. K. Aghi, J. Clin. Invest., 2023, 133(5), e147087 CrossRef PubMed.
- O. E. Franco, A. K. Shaw, D. W. Strand and S. W. Hayward, Semin. Cell Dev. Biol., 2010, 21, 33–39 CrossRef CAS PubMed.
- H. Yoon, C. M. Tang, S. Banerjee, A. L. Delgado, M. Yebra, J. Davis and J. K. Sicklick, Oncogenesis, 2021, 10, 13, DOI:10.1038/S41389-021-00302-5.
- A. Calon, E. Espinet, S. Palomo-Ponce, D. V. F. Tauriello, M. Iglesias, M. V. Céspedes, M. Sevillano, C. Nadal, P. Jung, X. H. F. Zhang, D. Byrom, A. Riera, D. Rossell, R. Mangues, J. Massagué, E. Sancho and E. Batlle, Cancer Cell, 2012, 22, 571 CrossRef CAS PubMed.
- M. Najafi, B. Farhood and K. Mortezaee, J. Cell. Biochem., 2019, 120, 2782–2790 CrossRef CAS PubMed.
- A. Verkhratsky and M. Nedergaard, Physiol. Rev., 2018, 98, 239 CrossRef CAS PubMed.
- B. M. Andersen, C. Faust Akl, M. A. Wheeler, E. A. Chiocca, D. A. Reardon and F. J. Quintana, Nat. Rev. Cancer, 2021, 21, 786 CrossRef CAS PubMed.
- A. Mega, M. Hartmark Nilsen, L. W. Leiss, N. P. Tobin, H. Miletic, L. Sleire, C. Strell, S. Nelander, C. Krona, D. Hägerstrand, P. Enger, M. Nistér and A. Östman, Glia, 2020, 68, 316–327 CrossRef PubMed.
- M. Valiente, A. C. Obenauf, X. Jin, Q. Chen, X. H. F. Zhang, D. J. Lee, J. E. Chaft, M. G. Kris, J. T. Huse, E. Brogi and J. Massagué, Cell, 2014, 156, 1002 CrossRef CAS.
- D. Henrik Heiland, V. M. Ravi, S. P. Behringer, J. H. Frenking, J. Wurm, K. Joseph, N. W. C. Garrelfs, J. Strähle, S. Heynckes, J. Grauvogel, P. Franco, I. Mader, M. Schneider, A. L. Potthoff, D. Delev, U. G. Hofmann, C. Fung, J. Beck, R. Sankowski, M. Prinz and O. Schnell, Nat. Commun., 2019, 10(1), 2541 CrossRef PubMed.
- T. Hide, I. Shibahara and T. Kumabe, Brain Tumor Pathol., 2019, 36, 63–73 CrossRef PubMed.
- T. Hide and Y. Komohara, Adv. Exp. Med. Biol., 2020, 1234, 107–122 CrossRef CAS PubMed.
- H. S. Venkatesh, T. B. Johung, V. Caretti, A. Noll, Y. Tang, S. Nagaraja, E. M. Gibson, C. W. Mount, J. Polepalli, S. S. Mitra, P. J. Woo, R. C. Malenka, H. Vogel, M. Bredel, P. Mallick and M. Monje, Cell, 2015, 161, 803 CrossRef CAS PubMed.
- X. Li, C. Wu, N. Chen, H. Gu, A. Yen, L. Cao, E. Wang and L. Wang, Oncotarget, 2016, 7, 33440 CrossRef PubMed.
- V. Venkataramani, Y. Yang, M. C. Schubert, E. Reyhan, S. K. Tetzlaff, N. Wißmann, M. Botz, S. J. Soyka, C. A. Beretta, R. L. Pramatarov, L. Fankhauser, L. Garofano, A. Freudenberg, J. Wagner, D. I. Tanev, M. Ratliff, R. Xie, T. Kessler, D. C. Hoffmann, L. Hai, Y. Dörflinger, S. Hoppe, Y. A. Yabo, A. Golebiewska, S. P. Niclou, F. Sahm, A. Lasorella, M. Slowik, L. Döring, A. Iavarone, W. Wick, T. Kuner and F. Winkler, Cell, 2022, 185, 2899–2917 CrossRef CAS PubMed.
- A. De Leo, A. Ugolini and F. Veglia, Cells, 2021, 10, 1–20 Search PubMed.
- D. Hambardzumyan, D. H. Gutmann and H. Kettenmann, Nat. Neurosci., 2015, 19(1), 20–27 CrossRef PubMed.
- B. Badie, J. Schartner, S. Prabakaran, J. Paul and J. Vorpahl, J. Neuroimmunol., 2001, 120, 19–24 CrossRef CAS PubMed.
- C. Gratas, Y. Tohma, E. G. van Meir, M. Klein, M. Tenan, N. Ishii, O. Tachibana, P. Kleihues and H. Ohgaki, Brain Pathol., 1997, 7, 863–869 CrossRef CAS PubMed.
- M. Massara, P. Persico, O. Bonavita, V. M. Poeta, M. Locati, M. Simonelli and R. Bonecchi, Front. Immunol., 2017, 8, 1349 CrossRef PubMed.
- J. Liang, Y. Piao, L. Holmes, G. N. Fuller, V. Henry, N. Tiao and J. F. de Groot, Clin. Cancer Res., 2014, 20, 187 CrossRef CAS PubMed.
- P. M. D'Agostino, A. Gottfried-Blackmore, N. Anandasabapathy and K. Bulloch, Acta Neuropathol., 2012, 124, 599 CrossRef PubMed.
- J. Wang, P. Liu, S. Xin, Z. Wang and J. Li, Exp. Cell Res., 2017, 360, 66–73 CrossRef CAS PubMed.
- F. Veglia, V. A. Tyurin, M. Blasi, A. de Leo, A. v. Kossenkov, L. Donthireddy, T. K. J. To, Z. Schug, S. Basu, F. Wang, E. Ricciotti, C. DiRusso, M. E. Murphy, R. H. Vonderheide, P. M. Lieberman, C. Mulligan, B. Nam, N. Hockstein, G. Masters, M. Guarino, C. Lin, Y. Nefedova, P. Black, V. E. Kagan and D. I. Gabrilovich, Nature, 2019, 569, 73 CrossRef CAS PubMed.
- M. M. Grabowski, E. W. Sankey, K. J. Ryan, P. Chongsathidkiet, S. J. Lorrey, D. S. Wilkinson and P. E. Fecci, J. Neurooncol., 2021, 151, 3 CrossRef PubMed.
- A. N. Akbar, S. M. Henson and A. Lanna, Trends Immunol., 2016, 37, 866–876 CrossRef CAS PubMed.
- F. M. Karlhofer, R. K. Ribaudo and W. M. Yokoyama, Nature, 1992, 358, 66–70 CrossRef CAS PubMed.
- C. A. Crane, S. J. Han, J. J. Barry, B. J. Ahn, L. L. Lanier and A. T. Parsa, Neurooncology, 2010, 12, 7 CAS.
- J. Kmiecik, A. Poli, N. H. C. Brons, A. Waha, G. E. Eide, P. Ø. Enger, J. Zimmer and M. Chekenya, J. Neuroimmunol., 2013, 264, 71–83 CrossRef CAS PubMed.
- I. Golán, L. R. de La Fuente and J. A. Costoya, Cancers, 2018, 10(12), 522 CrossRef PubMed.
- R. K. Jain, R. T. Tong and L. L. Munn, Cancer Res., 2007, 67, 2729–2735 CrossRef CAS PubMed.
- R. K. Jain, E. di Tomaso, D. G. Duda, J. S. Loeffler, A. G. Sorensen and T. T. Batchelor, Nat. Rev. Neurosci., 2007, 8(8), 610–622 CrossRef CAS PubMed.
- A. Dimberg, Biochem. Soc. Trans., 2014, 42, 1647–1652 CrossRef CAS PubMed.
- P. Jayaprakash, P. D. A. Vignali, G. M. Delgoffe and M. A. Curran, Annu. Rev. Med., 2022, 73, 251–265 CrossRef PubMed.
- D. J. Brat, A. A. Castellano-Sanchez, S. B. Hunter, M. Pecot, C. Cohen, E. H. Hammond, S. N. Devi, B. Kaur and E. G. van Meir, Cancer Res., 2004, 64, 920–927 CrossRef CAS PubMed.
- W. Gang, J. J. Wang, X. L. Fu, G. Rui and S. S. Tony To, Oncol. Rep., 2017, 37, 657–670 CrossRef PubMed.
- A. Goenka, D. Tiek, X. Song, T. Huang, B. Hu and S. Y. Cheng, Cells, 2021, 10, 1–21 CrossRef PubMed.
- S. Mahase, R. N. Rattenni, P. Wesseling, W. Leenders, C. Baldotto, R. Jain and D. Zagzag, Am. J. Pathol., 2017, 187, 940 CrossRef CAS PubMed.
- J. Chen, Z. Ding, Y. Peng, F. Pan, J. Li, L. Zou, Y. Zhang and H. Liang, PLoS One, 2014, 9(6), e98882 CrossRef PubMed.
- S. Kothari, J. Cizeau, E. McMillan-Ward, S. J. Israels, M. Bailes, K. Ens, L. A. Kirshenbaum and S. B. Gibson, Oncogene, 2003, 22, 4734–4744 CrossRef CAS PubMed.
- F. Lefranc and R. Kiss, Neurosurg Focus, 2006, 20(4), E7 Search PubMed.
- O. Warburg, F. Wind and E. Negelein, J. Gen. Physiol., 1927, 8, 519 CrossRef CAS PubMed.
- C. E. Griguer, C. R. Oliva and G. Y. Gillespie, J. Neurooncol., 2005, 74, 123–133 CrossRef CAS PubMed.
- M. I. Koukourakis, A. Giatromanolaki, A. L. Harris and E. Sivridis, Cancer Res., 2006, 66, 632–637 CrossRef CAS PubMed.
- S. K. Biswas, Immunity, 2015, 43, 435–449 CrossRef CAS PubMed.
- R. J. DeBerardinis, N. Sayed, D. Ditsworth and C. B. Thompson, Curr. Opin. Genet. Dev., 2008, 18, 54 CrossRef CAS PubMed.
- D. C. Liemburg-Apers, P. H. G. M. Willems, W. J. H. Koopman and S. Grefte, Arch. Toxicol., 2015, 89, 1209 CrossRef CAS PubMed.
- G. L. Semenza, P. H. Roth, H. M. Fang and G. L. Wang, J. Biol. Chem., 1994, 269, 23757–23763 CrossRef CAS PubMed.
- A. Honasoge and H. Sontheimer, Front. Physiol., 2013, 4, 316 Search PubMed.
- J. U. Rao, D. Coman, J. J. Walsh, M. M. Ali, Y. Huang and F. Hyder, Sci. Rep., 2017, 7(1), 1–6 CrossRef.
- O. Sedlakova, E. Svastova, M. Takacova, J. Kopacek, J. Pastorek and S. Pastorekova, Front. Physiol., 2014, 4, 400 Search PubMed.
- T. N. Seyfried, G. Arismendi-Morillo, P. Mukherjee and C. Chinopoulos, iScience, 2020, 23, 101761 CrossRef CAS PubMed.
- K. D. Cortés Franco, I. C. Brakmann, M. Feoktistova, D. Panayotova-Dimitrova, S. Gründer and Y. Tian, Pflugers Arch, 2023, 475, 405–416 CrossRef PubMed.
- T. N. Seyfried, G. Arismendi-Morillo, G. Zuccoli, D. C. Lee, T. Duraj, A. M. Elsakka, J. C. Maroon, P. Mukherjee, L. Ta, L. Shelton, D. D’Agostino, M. Kiebish and C. Chinopoulos, Front. Oncol., 2022, 12, 968351 CrossRef CAS PubMed.
- V. Bova, A. Filippone, G. Casili, M. Lanza, M. Campolo, A. P. Capra, A. Repici, L. Crupi, G. Motta, C. Colarossi, G. Chisari, S. Cuzzocrea, E. Esposito and I. Paterniti, Cancers, 2022, 14(16), 4032 CrossRef CAS PubMed.
- D. Pietrobono, C. Giacomelli, L. Marchetti, C. Martini and M. L. Trincavelli, Int. J. Mol. Sci., 2020, 21, 1–22 Search PubMed.
- S. Rafii, A. Ghouzlani, O. Naji, S. Ait Ssi, S. Kandoussi, A. Lakhdar and A. Badou, Int. J. Mol. Sci., 2023, 24, 6688 CrossRef CAS PubMed.
- S. C. S. Nagamani and A. Erez, Mol. Cell. Oncol., 2016, 3(2), e1127314 CrossRef PubMed.
- N. Sun and X. Zhao, Front. Pharmacol, 2022, 13, 2806 Search PubMed.
- B. Burke, A. Giannoudis, K. P. Corke, D. Gill, M. Wells, L. Ziegler-Heitbrock and C. E. Lewis, Am. J. Pathol., 2003, 163, 1233–1243 CrossRef CAS PubMed.
- G. M. Tannahill, A. M. Curtis, J. Adamik, E. M. Palsson-Mcdermott, A. F. McGettrick, G. Goel, C. Frezza, N. J. Bernard, B. Kelly, N. H. Foley, L. Zheng, A. Gardet, Z. Tong, S. S. Jany, S. C. Corr, M. Haneklaus, B. E. Caffrey, K. Pierce, S. Walmsley, F. C. Beasley, E. Cummins, V. Nizet, M. Whyte, C. T. Taylor, H. Lin, S. L. Masters, E. Gottlieb, V. P. Kelly, C. Clish, P. E. Auron, R. J. Xavier and L. A. J. O'Neill, Nature, 2013, 496(7444), 238–242 CrossRef CAS PubMed.
- Z. Zhu, H. Zhang, B. Chen, X. Liu, S. Zhang, Z. Zong and M. Gao, Front. Immunol., 2020, 11, 588552 CrossRef CAS PubMed.
- D. I. Gabrilovich, S. Ostrand-Rosenberg and V. Bronte, Nat. Rev. Immunol., 2012, 12(4), 253–268 CrossRef CAS PubMed.
- M. K. Srivastava, P. Sinha, V. K. Clements, P. Rodriguez and S. Ostrand-Rosenberg, Cancer Res., 2010, 70, 68–77 CrossRef CAS PubMed.
- P. J. Siska and J. C. Rathmell, Trends Immunol., 2015, 36, 257–264 CrossRef CAS PubMed.
- M. Bental and C. Deutsch, Magn. Reson. Med., 1993, 29, 317–326 CrossRef CAS PubMed.
- C. M. Cham, G. Driessens, J. P. O'Keefe and T. F. Gajewski, Eur. J. Immunol., 2008, 38, 2438–2450 CrossRef CAS PubMed.
- R. D. Michalek, V. A. Gerriets, S. R. Jacobs, A. N. Macintyre, N. J. MacIver, E. F. Mason, S. A. Sullivan, A. G. Nichols and J. C. Rathmell, J. Immunol., 2011, 186, 3299–3303 CrossRef CAS PubMed.
- V. Zaiatz-Bittencourt, D. K. Finlay and C. M. Gardiner, J. Immunol., 2018, 200, 3934–3941 CrossRef CAS PubMed.
- A. M. Chambers, K. B. Lupo and S. Matosevic, Front. Immunol., 2018, 9, 2517 CrossRef PubMed.
- A. Brand, K. Singer, G. E. Koehl, M. Kolitzus, G. Schoenhammer, A. Thiel, C. Matos, C. Bruss, S. Klobuch, K. Peter, M. Kastenberger, C. Bogdan, U. Schleicher, A. Mackensen, E. Ullrich, S. Fichtner-Feigl, R. Kesselring, M. Mack, U. Ritter, M. Schmid, C. Blank, K. Dettmer, P. J. Oefner, P. Hoffmann, S. Walenta, E. K. Geissler, J. Pouyssegur, A. Villunger, A. Steven, B. Seliger, S. Schreml, S. Haferkamp, E. Kohl, S. Karrer, M. Berneburg, W. Herr, W. Mueller-Klieser, K. Renner and M. Kreutz, Cell Metab., 2016, 24, 657–671 CrossRef CAS PubMed.
- B. Lamas, J. Vergnaud-Gauduchon, N. Goncalves-Mendes, O. Perche, A. Rossary, M. P. Vasson and M. C. Farges, Cell. Immunol., 2012, 280, 182–190 CrossRef CAS PubMed.
- I. Terrén, A. Orrantia, J. Vitallé, O. Zenarruzabeitia and F. Borrego, Front. Immunol., 2019, 10, 2278 CrossRef PubMed.
- G. Y. Liou and P. Storz, Free Radic Res, 2010, 44, 479–496 CrossRef CAS PubMed.
- H. S. Marinho, C. Real, L. Cyrne, H. Soares and F. Antunes, Redox Biol., 2014, 2, 535 CrossRef CAS PubMed.
- A. Salazar-Ramiro, D. Ramírez-Ortega, V. P. de La Cruz, N. Y. Hérnandez-Pedro, D. F. González-Esquivel, J. Sotelo and B. Pineda, Front. Immunol., 2016, 7, 156 Search PubMed.
- A. S. Baas and B. C. Berk, Circ. Res., 1995, 77, 29–36 CrossRef CAS PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21(7), 363–383 CrossRef CAS PubMed.
- P. Hernansanz-Agustín, E. Ramos, E. Navarro, E. Parada, N. Sánchez-López, L. Peláez-Aguado, J. D. Cabrera-García, D. Tello, I. Buendia, A. Marina, J. Egea, M. G. López, A. Bogdanova and A. Martínez-Ruiz, Redox Biol., 2017, 12, 1040–1051 CrossRef PubMed.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- D. P. Jones, Methods Enzymol., 2002, 348, 93–112 CAS.
- S. Flor, C. R. Oliva, M. Y. Ali, K. L. Coleman, J. D. Greenlee, K. A. Jones, V. Monga and C. E. Griguer, Antioxidants, 2021, 10(12), 1988 CrossRef CAS PubMed.
- E. Mohiuddin and H. Wakimoto, Am. J. Cancer Res., 2021, 11, 3742 CAS.
- L. W. Lau, R. Cua, M. B. Keough, S. Haylock-Jacobs and V. W. Yong, Nat. Rev. Neurosci., 2013, 14(10), 722–729 CrossRef CAS PubMed.
- Y. Yamaguchi, Cell. Mol. Life Sci., 2000, 57, 276–289 CrossRef CAS PubMed.
- C. Wang, S. Sinha, X. Jiang, L. Murphy, S. Fitch, C. Wilson, G. Grant and F. Yang, Tissue Eng., Part A, 2021, 27, 390–401 CrossRef CAS PubMed.
- E. Henke, R. Nandigama and S. Ergün, Front. Mol. Biosci., 2019, 6, 160 CrossRef CAS PubMed.
- A. G. Tavianatou, I. Caon, M. Franchi, Z. Piperigkou, D. Galesso and N. K. Karamanos, FEBS J., 2019, 286, 2883–2908 CrossRef CAS PubMed.
- C. A. Cuff, D. Kothapalli, I. Azonobi, S. Chun, Y. Zhang, R. Belkin, C. Yeh, A. Secreto, R. K. Assoian, D. J. Rader and E. Puré, J. Clin. Invest., 2001, 108, 1031–1040 CrossRef CAS PubMed.
- K. L. Mooney, W. Choy, S. Sidhu, P. Pelargos, T. T. Bui, B. Voth, N. Barnette and I. Yang, J. Clin. Neurosci., 2016, 34, 1–5 CrossRef CAS PubMed.
- D. Schiffer, M. Mellai, R. Boldorini, I. Bisogno, S. Grifoni, C. Corona, L. Bertero, P. Cassoni, C. Casalone and L. Annovazzi, Int. J. Mol. Sci., 2018, 19(9), 2724 CrossRef PubMed.
- W. B. Stallcup and F. J. Huang, Cell Adhes. Migr., 2008, 2, 192–201 CrossRef PubMed.
- N. Hartmann, N. A. Giese, T. Giese, I. Poschke, R. Offringa, J. Werner and E. Ryschich, Clin. Cancer Res., 2014, 20, 3422–3433 CrossRef CAS PubMed.
- N. Brösicke and A. Faissner, Cell Adhes. Migr., 2015, 9, 131 CrossRef PubMed.
- L. W. Lau, R. Cua, M. B. Keough, S. Haylock-Jacobs and V. W. Yong, Nat. Rev. Neurosci., 2013, 14, 722–729 CrossRef CAS PubMed.
- Q. Yu, Y. Xue, J. Liu, Z. Xi, Z. Li and Y. Liu, Front. Mol. Neurosci., 2018, 11, 130 CrossRef PubMed.
- B. Hu, M. S. Nandhu, H. Sim, P. A. Agudelo-Garcia, J. C. Saldivar, C. E. Dolan, M. E. Mora, G. J. Nuovo, S. E. Cole and M. S. Viapiano, Cancer Res., 2012, 72, 3873 CrossRef CAS PubMed.
- M. S. Nandhu, P. Behera, V. Bhaskaran, S. L. Longo, L. M. Barrera-Arenas, S. Sengupta, D. J. Rodriguez-Gil, E. A. Chiocca and M. S. Viapiano, Clin. Cancer Res., 2018, 24, 821–833 CrossRef CAS PubMed.
- M. Nakada, Y. Okada and J. Yamashita, Front. Biosci., 2003, 8(5), 261–269 CrossRef PubMed , https://www.article.imrpress.com.
- H. Könnecke and I. Bechmann, Clin. Dev. Immunol., 2013, 2013, 914104 CrossRef PubMed.
- C. S. Gondi, D. H. Dinh, J. D. Klopffenstein, M. Gujrati and J. S. Rao, Int. J. Oncol., 2009, 35, 257–263 CAS.
- A. S. Ribeiro, A. Albergaria, B. Sousa, A. L. Correia, M. Bracke, R. Seruca, F. C. Schmitt and J. Paredes, Oncogene, 2009, 29(3), 392–402 CrossRef PubMed.
- P. O. Estève, É. Chicoine, O. Robledo, F. Aoudjit, A. Descoteaux, E. F. Potworowski and Y. St-Pierre, J. Biol. Chem., 2002, 277, 35150–35155 CrossRef PubMed.
- S. E. Gould, M. R. Junttila and F. J. de Sauvage, Nat. Med., 2015, 21(5), 431–439 CrossRef CAS PubMed.
- O. Habanjar, M. Diab-Assaf, F. Caldefie-Chezet and L. Delort, Int. J. Mol. Sci., 2021, 22(22), 12200 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8(10), 839–845 CrossRef CAS PubMed.
- L. B. Weiswald, D. Bellet and V. Dangles-Marie, Neoplasia, 2015, 17, 1–15 CrossRef PubMed.
- G. Rijal and W. Li, Biomaterials, 2016, 81, 135–156 CrossRef CAS PubMed.
- H. Dolznig, C. Rupp, C. Puri, C. Haslinger, N. Schweifer, E. Wieser, D. Kerjaschki and P. Garin-Chesa, Am. J. Pathol., 2011, 179, 487–501 CrossRef PubMed.
- C. Zhang, M. Jin, J. Zhao, J. Chen and W. Jin, Am. J. Cancer Res., 2020, 10, 2242 CAS.
- M. Tang, J. N. Rich and S. Chen, Adv. Mater., 2021, 33, e2004776 CrossRef PubMed.
- F. Jacob, R. D. Salinas, D. Y. Zhang, P. T. T. Nguyen, J. G. Schnoll, S. Z. H. Wong, R. Thokala, S. Sheikh, D. Saxena, S. Prokop, D. ao Liu, X. Qian, D. Petrov, T. Lucas, H. I. Chen, J. F. Dorsey, K. M. Christian, Z. A. Binder, M. Nasrallah, S. Brem, D. M. O'Rourke, G. li Ming and H. Song, Cell, 2020, 180, 188 CrossRef CAS PubMed.
- C. G. Hubert, M. Rivera, L. C. Spangler, Q. Wu, S. C. Mack, B. C. Prager, M. Couce, R. E. McLendon, A. E. Sloan and J. N. Rich, Cancer Res., 2016, 76, 2465 CrossRef CAS PubMed.
- T. G. Krieger, S. M. Tirier, J. Park, K. Jechow, T. Eisemann, H. Peterziel, P. Angel, R. Eils and C. Conrad, Neurooncology, 2020, 22, 1138 Search PubMed.
- B. da Silva, R. K. Mathew, E. S. Polson, J. Williams and H. Wurdak, SLAS Discovery, 2018, 23, 862–868 CrossRef CAS PubMed.
- A. Linkous, D. Balamatsias, M. Snuderl, L. Edwards, K. Miyaguchi, T. Milner, B. Reich, L. Cohen-Gould, A. Storaska, Y. Nakayama, E. Schenkein, R. Singhania, S. Cirigliano, T. Magdeldin, Y. Lin, G. Nanjangud, K. Chadalavada, D. Pisapia, C. Liston and H. A. Fine, Cell Rep., 2019, 26, 3203 CrossRef CAS PubMed.
- M. Tang, Q. Xie, R. C. Gimple, Z. Zhong, T. Tam, J. Tian, R. L. Kidwell, Q. Wu, B. C. Prager, Z. Qiu, A. Yu, Z. Zhu, P. Mesci, H. Jing, J. Schimelman, P. Wang, D. Lee, M. H. Lorenzini, D. Dixit, L. Zhao, S. Bhargava, T. E. Miller, X. Wan, J. Tang, B. Sun, B. F. Cravatt, A. R. Muotri, S. Chen and J. N. Rich, Cell Res., 2020, 30, 833 CrossRef CAS PubMed.
- Y. Fan, D. T. Nguyen, Y. Akay, F. Xu and M. Akay, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- A. H. Alves, M. P. Nucci, J. B. Mamani, N. M. E. Valle, E. F. Ribeiro, G. N. A. Rego, F. A. Oliveira, G. Minas, S. Krauss, A. H. Alves, M. P. Nucci, J. B. Mamani, N. M. E. Valle, E. F. Ribeiro, G. N. A. Rego, F. A. Oliveira, M. H. Theinel, R. S. Santos and L. F. Gamarra, Cancers, 2022, 14, 869 CrossRef CAS PubMed.
- Y. Sheng, H. Nesbitt, B. Callan, M. A. Taylor, M. Love, A. P. McHale and J. F. Callan, J. Controlled Release, 2017, 264, 333–340 CrossRef CAS PubMed.
- M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633 CrossRef CAS PubMed.
- D. A. Murphy, H. Cheng, T. Yang, X. Yan and I. M. Adjei, Mol. Pharm., 2021, 18, 2935–2946 CrossRef CAS PubMed.
- K. Liang, Z. Li, Y. Luo, Q. Zhang, F. Yin, L. Xu, H. Chen, H. Wang, K. Liang, Q. Zhang, H. Chen, Z. Li, F. Yin, L. Xu, H. Wang and Y. Luo, Small, 2020, 16, 1906985 CrossRef CAS PubMed.
- W. Jiang, X. Han, T. Zhang, D. Xie, H. Zhang and Y. Hu, Adv. Healthcare Mater., 2020, 9, 1901303 CrossRef CAS PubMed.
- K. Chen, X. Sun, Y. Liu, Y. Yang, M. Shi, J. Yu, S. Zhang and P. Shi, Inorg. Chem., 2022, 61, 16307–16316 CrossRef CAS PubMed.
- T. Wu, Y. Liu, Y. Cao and Z. Liu, Adv. Mater., 2022, 34, 2110364 CrossRef CAS PubMed.
- L. Kennedy, J. K. Sandhu, M. E. Harper and M. Cuperlovic-culf, Biomolecules, 2020, 10, 1–27 CrossRef PubMed.
- W. Cao, M. Jin, K. Yang, B. Chen, M. Xiong, X. Li and G. Cao, J. Nanobiotechnol., 2021, 19, 1–35 CrossRef PubMed.
- X. Xu, X. Gong, Y. Wang, J. Li, H. Wang, J. Wang, X. Sha, Y. Li, Z. Zhang, X. Xu, X. Gong, Y. Wang, J. Li, H. Wang, J. Wang, Y. Li, Z. Zhang and X. Sha, Adv. Thermoelectr., 2020, 3, 1900181 Search PubMed.
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- T. Wang, H. Zhang, W. Qiu, Y. Han, H. Liu and Z. Li, Bioact. Mater., 2022, 16, 418 CAS.
- Y. Zhang, K. Xi, X. Fu, H. Sun, H. Wang, D. Yu, Z. Li, Y. Ma, X. Liu, B. Huang, J. Wang, G. Li, J. Cui, X. Li and S. Ni, Biomaterials, 2021, 278, 121163 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- Z. Dong, L. Feng, Y. Chao, Y. Hao, M. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS PubMed.
- S. Xie, W. Sun, C. Zhang, B. Dong, J. Yang, M. Hou, L. Xiong, B. Cai, X. Liu and W. Xue, ACS Nano, 2021, 15, 7179–7194 CrossRef CAS PubMed.
- H. Fan, G. Yan, Z. Zhao, X. Hu, W. Zhang, H. Liu, X. Fu, T. Fu, X. B. Zhang and W. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477–5482 CrossRef CAS PubMed.
- Y. Yong, C. Zhang, Z. Gu, J. Du, Z. Guo, X. Dong, J. Xie, G. Zhang, X. Liu and Y. Zhao, ACS Nano, 2017, 11, 7164–7176 CrossRef CAS PubMed.
- G. N. d. A. Rego, A. da H. Alves, M. P. Nucci, J. B. Mamani, F. A. de Oliveira and L. F. Gamarra, Int. J. Mol. Sci., 2020, 21, 1–32 Search PubMed.
- Z. Lu, Y. Li, Y. Shi, Y. Li, Z. Xiao and X. Zhang, Adv. Funct. Mater., 2017, 27, 1703967 CrossRef.
- L. Vincent, P. Kermani, L. M. Young, J. Cheng, F. Zhang, K. Shido, G. Lam, H. Bompais-Vincent, Z. Zhu, D. J. Hicklin, P. Bohlen, D. J. Chaplin, C. May and S. Rafii, J. Clin. Invest., 2005, 115, 2992–3006 CrossRef CAS PubMed.
- Y. Kuang, X. Jiang, Y. Zhang, Y. Lu, H. Ma, Y. Guo, Y. Zhang, S. An, J. Li, L. Liu, Y. Wu, J. Liang and C. Jiang, Mol. Pharm., 2016, 13, 1599–1607 CrossRef CAS PubMed.
- D. Tseng, D. A. Vasquez-Medrano and J. M. Brown, Br. J. Cancer, 2011, 104, 1805 CrossRef CAS PubMed.
- M. Ehtesham, K. Y. Mapara, C. B. Stevenson and R. C. Thompson, Cancer Lett., 2009, 274, 305–312 CrossRef CAS PubMed.
- D. Séhédic, I. Chourpa, C. Tétaud, A. Griveau, C. Loussouarn, S. Avril, C. Legendre, N. Lepareur, D. Wion, F. Hindré, D. François and E. Garcion, Theranostics, 2017, 7, 4517–4536 CrossRef PubMed.
- L. H. Fu, C. Qi, Y. R. Hu, J. Lin and P. Huang, Adv. Mater., 2019, 31, 1808325 CrossRef PubMed.
- L. Zhang, Z. Wang, Y. Zhang, F. Cao, K. Dong, J. Ren and X. Qu, ACS Nano, 2018, 12, 10201–10211 CrossRef CAS PubMed.
- R. Ke, X. Zhen, H. S. Wang, L. Li, H. Wang, S. Wang and X. Xie, J. Colloid Interface Sci., 2022, 609, 307–319 CrossRef CAS PubMed.
- L. Qiao, X. Li, Y. Xiao, J. Yuan, D. Yu, M. Zuo, J. Chen, S. Han and D. Cheng, Biomater. Sci., 2022, 10, 4170–4183 RSC.
- C. Li, Y. Wan, Y. Zhang, L.-H. Fu, N. T. Blum, R. Cui, B. Wu, R. Zheng, J. Lin, Z. Li, P. Huang, C. Li, B. Wu, R. Zheng, Z. Li, P. Huang, Y. Wan, Y. Zhang, L.-H. Fu, N. T. Blum, R. Cui and J. Lin, Adv. Mater., 2022, 34, 2103980 CrossRef CAS PubMed.
- M. L. Macheda, S. Rogers and J. D. Best, J. Cell. Physiol., 2005, 202, 654–662 CrossRef CAS PubMed.
- G. Jego, A. Hazoumé, R. Seigneuric and C. Garrido, Cancer Lett., 2013, 332, 275–285 CrossRef CAS PubMed.
- Y. Zhou, F. Tozzi, J. Chen, F. Fan, L. Xia, J. Wang, G. Gao, A. Zhang, X. Xia, H. Brasher, W. Widger, L. M. Ellis and Z. Weihua, Cancer Res., 2012, 72, 304–314 CrossRef CAS PubMed.
- J. Zhu, A. Jiao, Q. Li, X. Lv, X. Wang, X. Song, B. Li, Y. Zhang and X. Dong, Acta Biomater., 2022, 137, 252–261 CrossRef CAS PubMed.
- L. Yang, S. Venneti and D. Nagrath, Annu. Rev. Biomed. Eng., 2017, 19, 163–194 CrossRef CAS PubMed.
- L. Xu, R. Xu, P. E. Saw, J. Wu, S. X. Cheng and X. Xu, Nano Lett., 2021, 21, 7569–7578 CrossRef CAS PubMed.
- A. Zinger, L. Koren, O. Adir, M. Poley, M. Alyan, Z. Yaari, N. Noor, N. Krinsky, A. Simon, H. Gibori, M. Krayem, Y. Mumblat, S. Kasten, S. Ofir, E. Fridman, N. Milman, M. M. Lübtow, L. Liba, J. Shklover, J. Shainsky-Roitman, Y. Binenbaum, D. Hershkovitz, Z. Gil, T. Dvir, R. Luxenhofer, R. Satchi-Fainaro and A. Schroeder, ACS Nano, 2019, 13, 11008 CrossRef CAS PubMed.
- W. C. Parks, C. L. Wilson and Y. S. López-Boado, Nat. Rev. Immunol., 2004, 4, 617–629 CrossRef CAS PubMed.
- V. P. Chauhan, J. D. Martin, H. Liu, D. A. Lacorre, S. R. Jain, S. v. Kozin, T. Stylianopoulos, A. S. Mousa, X. Han, P. Adstamongkonkul, Z. Popović, P. Huang, M. G. Bawendi, Y. Boucher and R. K. Jain, Nat. Commun., 2013, 4, 2516 CrossRef PubMed.
- Q. Zhao, X. He, X. Qin, Y. Liu, H. Jiang, J. Wang, S. Wu, R. Zhou, C. Yu, S. Liu, H. Zhang and M. Tian, Front. Immunol., 2022, 13, 938439 CrossRef CAS PubMed.
- M. Grabowska, B. F. Grześkowiak, K. Szutkowski, D. Wawrzyniak, P. Głodowicz, J. Barciszewski, S. Jurga, K. Rolle and R. Mrówczyński, PLoS One, 2019, 14(3), e0213852 CrossRef CAS PubMed.
- O. Cohen-Inbar and M. Zaaroor, J. Clin. Neurosci., 2016, 33, 52–58 CrossRef CAS PubMed.
- S. Agarwal, M. Sheikh Mohamed, T. Mizuki, T. Maekawa and D. Sakthi Kumar, J. Mater. Chem. B, 2019, 7, 5896–5919 RSC.
- W. L. Lin, D. Y. Lai, Y. J. Lee, N. F. Chen and T. H. Tseng, Toxicol. Lett., 2015, 232, 490–498 CrossRef CAS PubMed.
- L. Wang, H. Guo, L. Yang, L. Dong, C. Lin, J. Zhang, P. Lin and X. Wang, Mol. Cell. Biochem., 2013, 379, 7–18 CrossRef CAS PubMed.
- T. Fukai, K. Satoh, T. Nomura and H. Sakagami, Fitoterapia, 2003, 74, 720–724 CrossRef CAS PubMed.
- E. Mohiuddin and H. Wakimoto, Am. J. Cancer Res., 2021, 11, 3742 CAS.
- M. Piantino, A. Figarol and M. Matsusaki, Front. Toxicol., 2021, 3, 23 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |