
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlausible PEPPSI catalysts for direct C–H functionalization of five-membered heterocyclic bioactive motifs: synthesis, spectral, X-ray crystallographic characterizations and catalytic activity†
Donia Bensalaha,
Lamjed Mansourb,
Mathieu Sauthierc,
Nevin Gurbuzde,
Ismail Özdemirde,
Lotfi Bejif,
Rafik Gatrig and
Naceur Hamdi *a
*a
aResearch Laboratory of Environmental Sciences and Technologies (LR16ES09), Higher Institute of Environmental Sciences and Technology, University of Carthage, Hammam-Lif, Tunisia. E-mail: naceur.hamdi@isste.rnu.tn; Tel: +96 6556394839
bZoology Department, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
cEcole Nationale Superieure de Chimie de Lille, Unité de Catalyse et Chimie du Solide, UMR CNRS 8181, USTL, BP 90108, Villeneuve d’Ascq, 59652, France
dDepartment of Chemistry, Faculty of Science and Art, İnönü University, Malatya, 44280, Turkey
eİnönü University, Catalysis Research and Application Center, Malatya, 44280, Turkey
fDepartment of Physics, College of Sciences and Arts at Arras, Qassim University, Saudi Arabia
gLaboratoire de Synthèse Organique Sélective et Hétérocyclique Évaluation Biologique LR17ES01 Faculté des Sciences de Tunis Campus Universitaire, Université de Tunis El Manar, 1092, Tunis, Tunisia
First published on 7th November 2023
Abstract
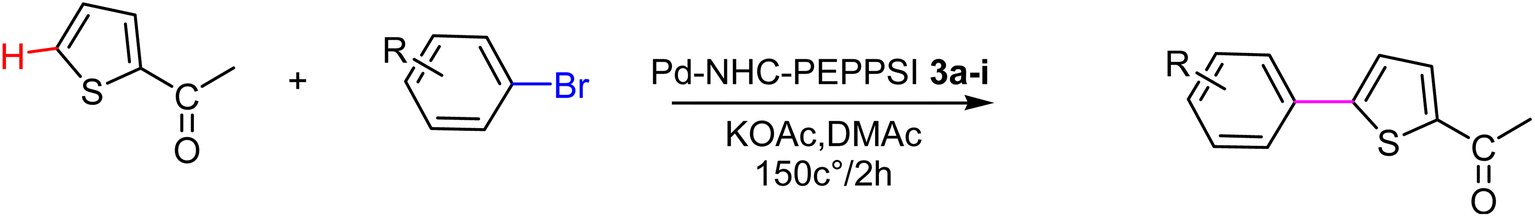
In this study, a series of benzimidazolium salts were synthesized as asymmetric N-heterocyclic carbene (NHC) precursors. Nine novel palladium complexes with the general formula [PdX2(NHC)(pyridine)] were synthesized using benzimidazolium salts in the PEPPSI (Pyridine Enhanced Precatalyst Preparation, Stabilization and Initiation) theme. All synthesized Pd(II) complexes are stable. The synthesized compounds were thoroughly characterized by respective spectroscopic techniques, such as 1HNMR, 13C NMR, FTIR spectroscopy, X-ray crystallography and elemental analysis. The geometric structure of the palladium N-heterocyclic carbene has been optimized in the framework of density functional theory (DFT) using the B3LYP-D3 dispersion functional with LANL2DZ as a basis set. The on/off mechanism of pyridine assisted Pd–NHC complexes made them the best C–H functionalized catalysts for regioselective C-5 arylated products. Five membered heterocyclic compounds such as 2-acetyl furan, furfuryl acetate 2-acetylthiophene and N-methylpyrrole-2-carboxaldehyde were treated with numerous aryl bromides and arylchlorides under optimal catalytic reaction conditions. Interestingly, all the prepared catalysts possessed essential structural features that facilitated the formation of desired coupling products in quantitative yield with excellent selectivity. The arylation reaction of bromoacetophenone was highly catalytically active with only 1 mol% catalyst loading at 150 °C for 2 hours. To check the efficiency of the synthesized complexes, three different five member heterocyclic substrates (2-acetylfuran, 2-acetylthiophen, 2-propylthaizole) were tested with a number of aryl bromides bearing both electron-donating and electron-withdrawing groups on para position. The data in Tables 2–4. Indicated that electron-donating groups on the para position of aryl halide decreased the catalytic conversion while electron-withdrawing groups increased the catalytic conversion this was due to the high nucleophilicity of the electron-donating substituents.
1 Introduction
The synthesis of arylated heterocyclic compounds has attracted extensive attention due to their biological and physical properties. Bi(hetero)aryl compounds can be prepared by Pd-catalyzed reactions such as Stille, Suzuki, Kumada or Negishi cross-coupling reactions.1–5 However, such reactions require organometallic nucleophiles and generate stoichiometric by-products. As early as 1985, Ohta et al. reported the direct arylation of heteroarenes with aryl halides via C–H bond activation using Pd(PPh3)4 catalysts.6–8 Owing to these exciting results, the Pd-catalyzed direct arylation of multiple heteroarenes has emerged as a very powerful approach to obtain a variety of arylated heterocycles in a simpler and more environmentally friendly manner.9–19 This approach is very attractive because it avoids the preparation of organometallic derivatives and the main by-product of the reaction is the base attached to HX rather than the metal salt (Scheme 1).20–36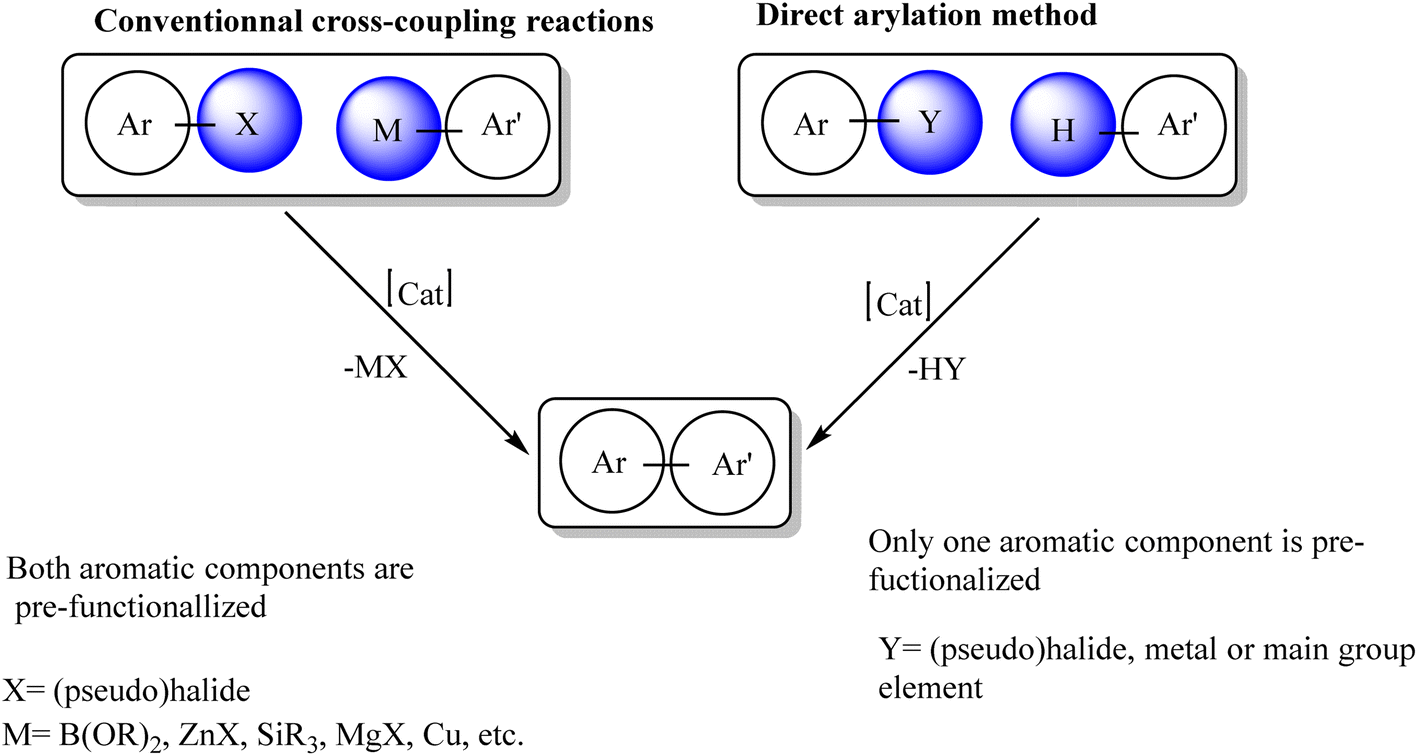 | ||
| Scheme 1 Comparison of conventional cross-coupling reactions and direct arylation method. | ||
Catalytic functionalization of C–H bonds present on arene and heteroarene backbones of commonly used organic building blocks is a powerful transformation in the modern era of stepeconomic chemical synthesis.37 Extensive research efforts, dedicated over several decades, blossomed a myriad of highly efficient homogeneous transition-metal catalysts for such an important transformation. Within the impressive library of metal complexes, the PdII–N-heterocyclic carbene (NHC) systems recently gained extraordinary attention for oxidative C–H transformation catalysis involving PdII/PdIV cycles, probably due to the strong σ-donation effect of NHC ligands in favoring efficient turn-over of high-valent Pd intermediates.38,39 Importantly, to exploit the diverse practical advantages primarily associated with the catalyst's reusability, separation, and compatibility to flow technology, a current focus intends to mould many Pd complexes including Pd–NHCs into single-site heterogeneous versions.40
Following these pioneering studies, the palladium-catalyzed direct arylation of heteroarenes with aryl halides has emerged as a practical and attractive approach for the synthesis of bi(hetero)aryls. To date, a large number of researchers have reported the palladium-catalyzed direct arylation of heteroarenes, especially five-membered heterocycles, such as thiophene41 and furan.42 N-heterocyclic carbene (NHC) is a neutral two-electron donor, considered a strong Lewis base and an excellent nucleophile, binding with metals better than phosphines.43 One of the main characteristics of NHCs is their stability compared to other classes of ligands. The strong σ-donor ability but poor π-accepting ability of NHC ligands leads to the formation of many stable metal–NHC complexes. Another feature of NHCs is that they can be tuned by adding various substituents to obtain complexes with desired electronic and steric properties.44
N-Heterocyclic carbenes (NHCs) are nitrogen-based heterocyclic compounds containing a divalent carbon atom. Previously, many researcher tried numerous synthetic methods to isolate the stable NHCs, but they were not successful until the first stable free-carbene was isolated in 1991 as a crystal solid by Arduengo and coworkers.45 Since then, the number of studies in carbene chemistry have increased considerably, and have become a stable in research laboratories throughout the world. Today, NHCs are one of the important classes of ligands for coordination chemistry. NHCs have strong σ-donating but, weak π-accepting properties, which show excellent support to stabilise various oxidation states of transition-metal. Also, they can provide steric and electronic properties for optimal design of transition-metal complexes.46–52 The modification at the nitrogen atoms of the NHCs significantly influence the reactivity and binding affinity of the ligand, thus NHCs make strong metal–carbon bond with different metals. Transition-metal complexes of NHCs are used as strong-, reactive-an selective-catalysts in many chemical reactions. Initially, the metal–NHC complexes were used extensively as a catalyst in organic transformations such as C–C, C–heteroatom cross-couplings, and C–H functionalization.53–64 In recent years, due to their strong σ-donors and easily tunable steric properties, low-toxic N-heterocyclic carbenes (NHCs) have been considered as effective ligands, which have the potential to overcome the shortcomings of phosphine-based catalytic instability and lead to achieve excellent catalytic performance.55–66 Doucet et al. for the first time, benzimidazolyl (NHCs)–Pd precatalysts have been shown to efficiently produce arylated thiophenes with a palladium loading of 1 mol% at 150 °C.67 In pyridine-enhanced precatalyst preparation stabilization and initiation (PEPPSI)-type palladium–NHC complexes, the methylene or phenylene linker of the NHC ligand allows additional degrees of rotation, which can guide the carbene ligand away from the metal center and avoid overloading. In addition, these ligands have flexible steric volumes that avoid steric crowding around the metal center caused by ligand chelation with N-substituents.
The design and synthesis of such PEPPSI-type palladium–NHC complexes has attracted great interest. In addition, substituents on NHC ring nitrogen atoms can change electronic and steric properties, which are important for their use as ligands in catalytic reactions. The first metal complexes containing NHC ligands were reported in 1968,68 but these ligands received little attention during those years. However, after the first isolation and characterization of stable free NHCs in 1991,69 interest in these ligands grew exponentially. Next, the first use of NHC in the Pd-catalyzed Heck reaction in 1995 (ref. 70) brought a new class of ligands into the field of catalysis. Today, NHCs are one of the most widely used ligand classes in organometallic chemistry and catalysis. After the discovery of Organ's PEPPSI (Pyridine Enhanced Precatalyst Preparation, Stabilization and Initiation) Pd complexes.71 Such complexes exhibit remarkable catalytic activity in various carbon–carbon and carbon–heteroatom coupling reactions. The PEPPSI-themed Pd–NHC complexes represent a new class of Pd catalysts that are quite different from other Pd–NHC complexes and are easier to synthesize and use.72 In recent years, Pd complexes containing PEPPSI have been developed for direct use as efficient arylation catalysts with successful results.73 In this regard, we have recently successfully reported the synthesis and structural characterization of PEPPSI-like Pd complexes with different NHC ligands and examined the catalytic activity of these complexes in direct arylation reactions.74
In our previous research,75–79 we reached the conclusion that the presence of electron-donating and bulky substituents on the nitrogen of the carbene ligand, along with substituents possessing lipophilic properties, enhances the antimicrobial activity of the Pd–NHCs. Due to this finding, we have chosen to prioritize sterically bulky benzyl substituents. Regarding the properties of the NHC ligand structure, the increase of electron-donating substituents can facilitate the oxidative addition process, while the bulky ligands can enhance the reductive elimination rate in the catalytic cycle.80 It has been hypothesized that sufficiently large NHC catalysts can protect the palladium center and inhibit catalyst degradation when exposed to air.81 According to ref. 82, positions C2 and C5 are favored due to the acidic nature of the H atom. The electronegativity of the oxygen atom results in more acidic protons at the C2 and C5 positions. C3 and C4 are unfavorable due to the higher electron density of the H atom. However, positions C3 and C4 can be arylated when C2 and C5 are blocked. In conclusion, controlling the regioselectivity of Pd-catalyzed direct arylation of furan derivatives remains a challenge (Scheme 2). In the case of unsubstituted furans, C2-arylated furans and in general, C2,C5-diarylated furans have been observed. The use of protecting groups such as esters at the C2 position enables the selective production of C5 monoarylated furans. Only some examples of C2 arylation of 3-substituted furans or C4 arylation with asymmetric 2,5-disubstituted furans have been reported. To be able to predict the regioselectivity of such arylations, it is necessary to investigate the influence of the nature of the coupling partner and the furanic substituent.
 | ||
| Scheme 2 Most favourable positions of furans for direct arylations. | ||
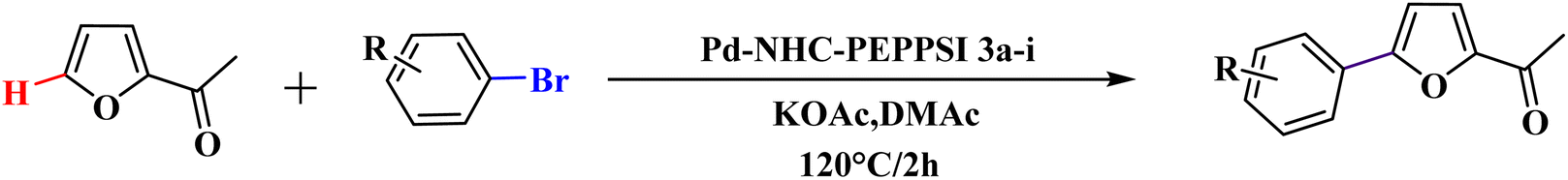
The significance of palladium(II) complexes lies in their ability to facilitate the creation of innovative metallodrugs. This is primarily due to their structural and electronic similarities to platinum(II) complexes. Numerous Pd(II) complexes have been documented in literature as potential agents in the fight against viruses, fungi, microbes, and tumors. Other studies have explored their interactions with DNA and bovine serum albumin (BSA), as well as their cytotoxic and antioxidant properties. Given the growing problem of bacterial resistance to existing antibiotics, there is a pressing need for the discovery of new complexes with antibacterial capabilities. As a result, recent efforts have been focused on the development of new palladium(II) drugs that specifically target multi-resistant bacterial strains.64–73 In this report, we present the successful creation of nine novel Pd–NHC complexes (3a–3i) following the PEPPSI theme. These complexes are represented by the general formula [PdX2(NHC)(pyridine)], where X can be either Cl or Br, and NHC refers to 1,3-disubstituted benzimidazole-2-ylidene. Various spectroscopic techniques were utilized to thoroughly characterize these complexes. Additionally, we conducted catalytic experiments using all Pd catalysts, specifically in the direct arylation of furan derivatives with aryl bromides. The experiments were conducted with a catalyst loading of 1 mol%, as depicted in Scheme 3. To optimize the yields of the monoarylated products, the reactive C2-position of heteroarenes was intentionally blocked.
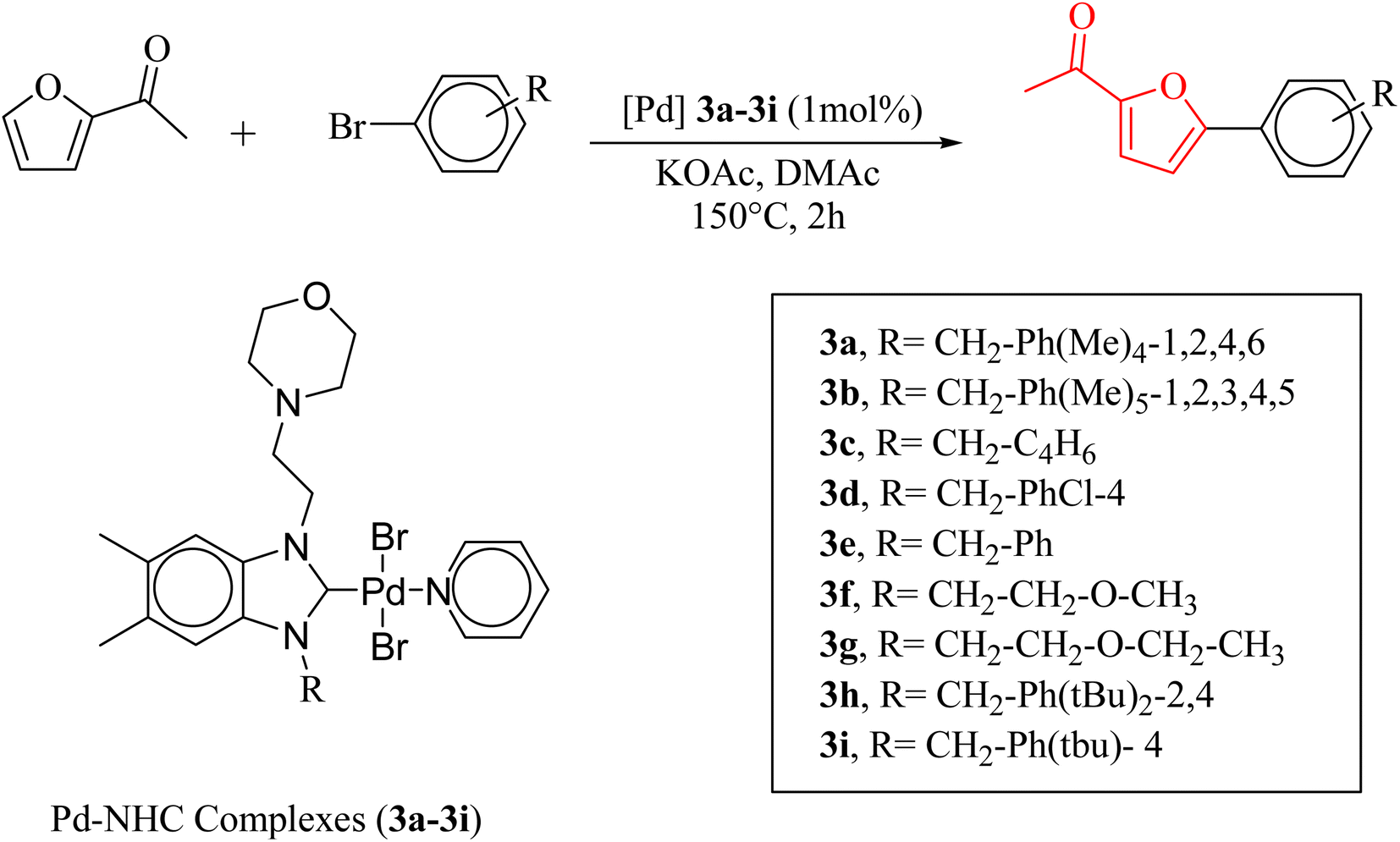 | ||
| Scheme 3 Pd-catalyzed direct arylation of C2-substituted furan derivatives with aryl bromides. | ||
Herein, we now report the successful synthesis of nine new PEPPSI-themed Pd–NHC complexes (3a–3i) of the general formula [PdX2 (NHC)(pyridine)], (X = Cl, Br; NHC = 1,3-disubstituted benzimidazole-2-ylidene), and their full characterization by various spectroscopic techniques. These complexes were found active against C–H bond activation of five membered heterocyclic compounds (Scheme 3). The reactive C2-position of heteroarenes was blocked in order to maximize the yields of the monoarylated products.
2 Results and discussion
2.1. Preparation of benzimidazolium salts 2
To initiate the N-alkylation reaction, 5,6-dimethylbenzimidazole (1 mmol) and 4-(2-chloroethyl)morpholine (1 mmol) were combined, and the reaction was conducted at room temperature for a duration of 2 hours in the presence of KOH. This procedure yielded the desired starting material, 1-(2-morpholinoethyl)-5,6-dimethylbenzimidazole (1). Subsequently, the precursor (2) was synthesized via the quaternization of intermediate (1) using the same alkyl halide in toluene. This reaction was carried out under reflux for 48 hours at 80 °C in degassed dimethylformamide. Scheme 4 illustrates this process. The resulting 5,6-dimethylbenzmidazolium salts were successfully obtained and purified as white solids that are resistant to both air and moisture. The yield of these salts exceeded 72%. The structures of the salts were conclusively determined through analysis of their characteristic spectroscopic data and elemental compositions. In the 13C NMR spectra of 2a–i, the characteristic signals of the imino carbon, (NCHN) were detected as typical singlets at δ 142.4, 142.1, 142.07, 143.2, 143.3, 142.8, 142.5, 152.2 and 152.5 ppm, respectively. The 1H NMR signals of the C(2)–H protons were observed as sharp singlets at chemical shifts of δ 10.16, 9.94, 11.03, 11.61, 11.57, 11.06, 11.08, 11.17 and 11.08 ppm, respectively for 2a–i, and further supported the assigned structures. These NMR values were in line with those found for other benzimidazolium salts of the literature.83 The formation of the benzimidazolium salts were also evidenced by their IR spectra, which showed (CN) bond absorption at ν 1556, 1557, 1562, 1563, 1566, 1566, 1563, 1564 and 1557 cm−1 for the respective CN bond vibration of 2a–i.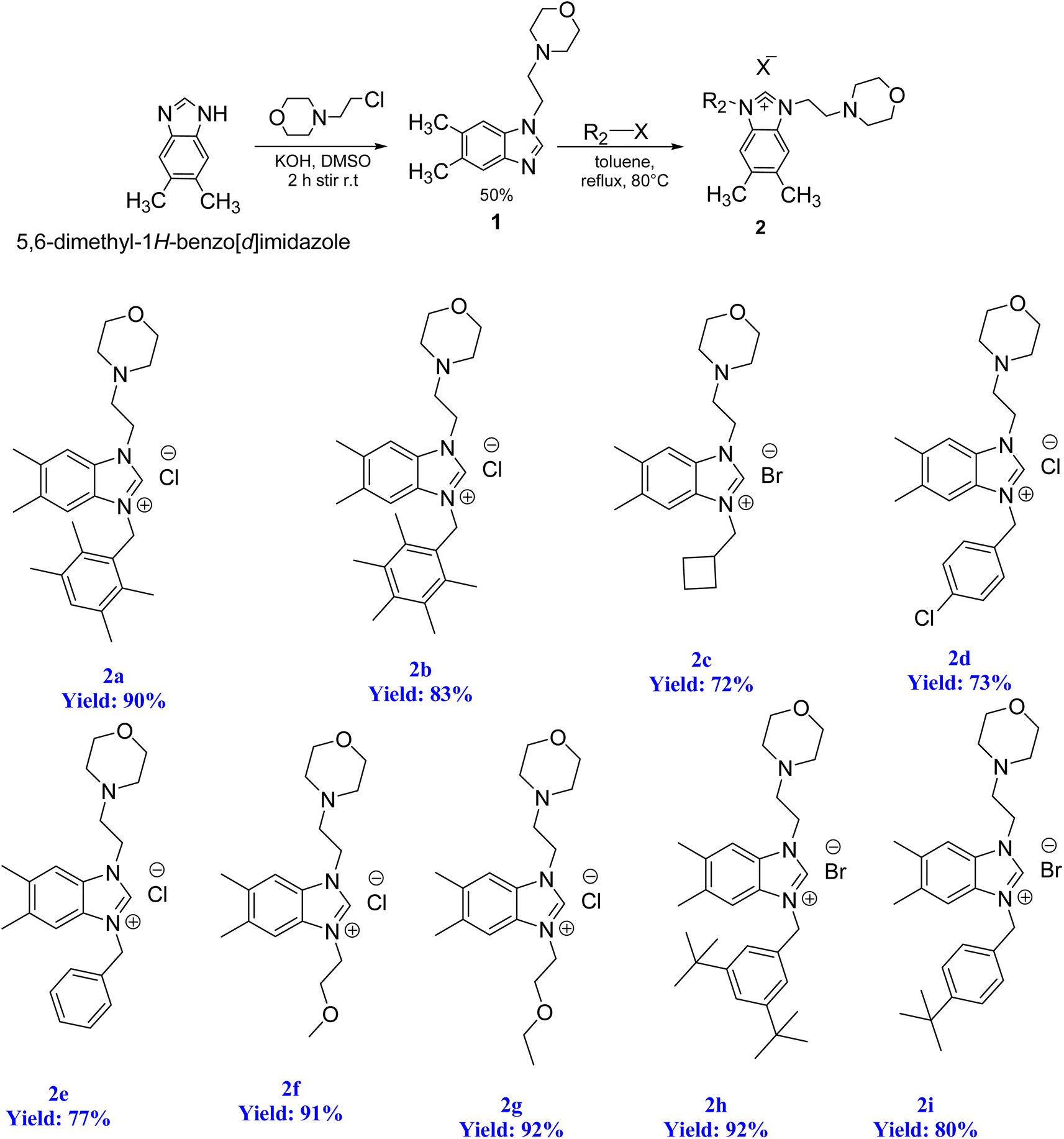 | ||
| Scheme 4 Synthesis of N-alkylated benzimidazole (1) and benzimidazolium salts (2). | ||
2.2. Preparation of the PEPPSI–Pd(II)–N-heterocyclic carbene (NHC) complexes 3a–i
Organ's methodology from 1975 was utilized in the preparation of the new complexes (3a–i). The process for creating the PEPPSI-type palladium–NHC complexes (3a–i) is outlined in Scheme 5. To accomplish this, the benzimidazolium salts were combined with PdCl2 in the presence of an excessive amount of K2CO3 and KBr in pyridine at a temperature of 80 °C for 16 h. Subsequently, the complexes were obtained through recrystallization, resulting in air and moisture resistant bright yellow crystals with a high yield (Scheme 5).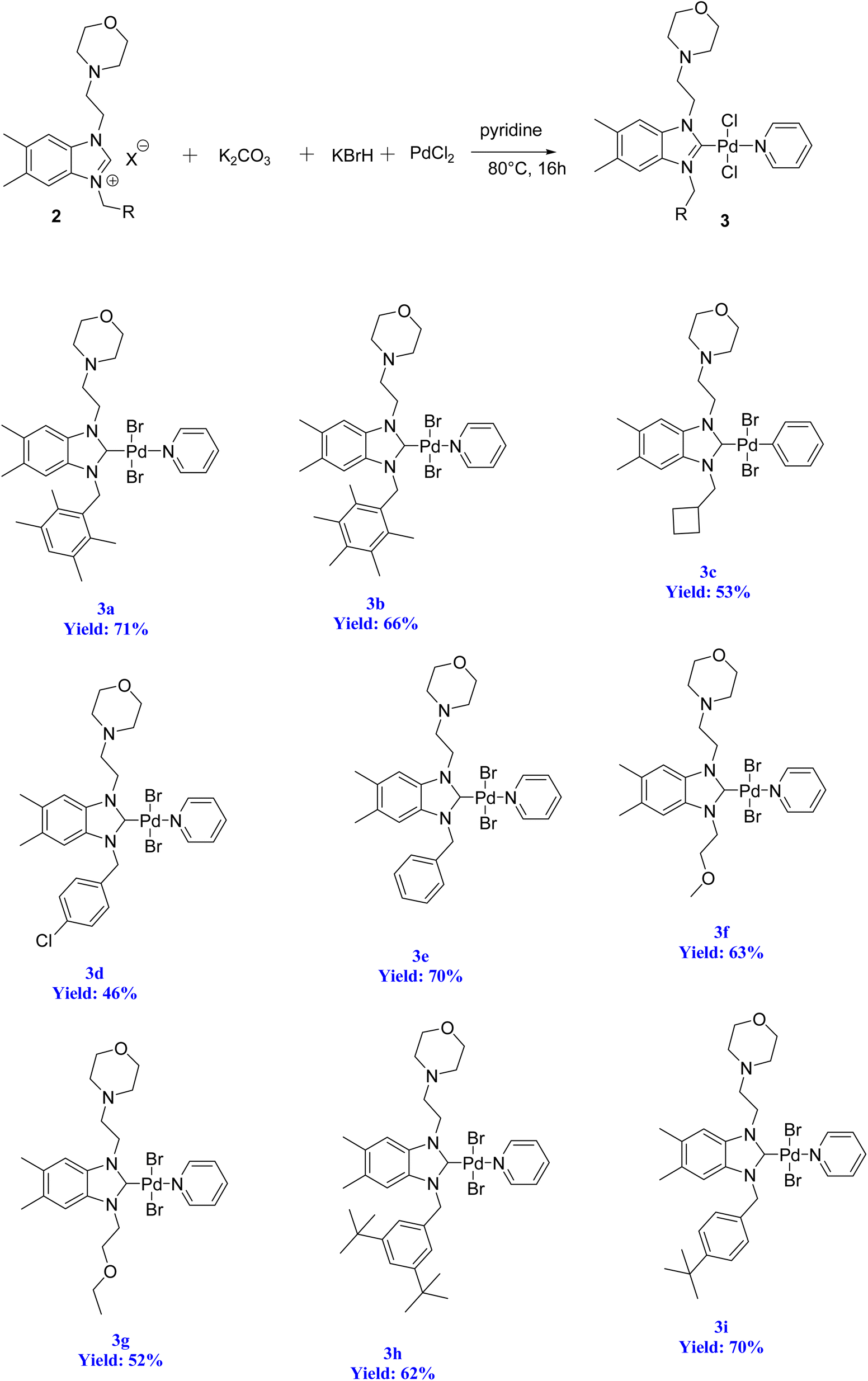 | ||
| Scheme 5 Synthesis of PEPPSI–Pd(II)–N-heterocyclic carbene (NHC) complexes 3a–i. | ||
The stability of these complexes was observed in both solution and solid state, demonstrating resistance to exposure from air, light, and moisture. Furthermore, these Pd–PEPPSI–NHC complexes 3 are soluble in most organic solvents, such as CH2Cl2, CHCl3, EtOAc and DMSO, with the exception of non-polar ones, such as pentane, hexane and Et2O.
The verification of the existence of the complexes was established by means of FT-IR, 1H NMR, and 13C{1H} NMR spectroscopic techniques as well as elementary analysis. These spectroscopic methods, known for their reliability, are in agreement with the suggested formula. The absence of the characteristic signals of the imino carbon (143–144 ppm) and the acidic imino proton (10–12 ppm) in the 13C NMR and 1H NMR spectra, which were present in the salts (2a–i), indicates the formation of the NHC–carbenes and their coordination to create the PEPPSI type palladium–NHC complexes. The signal of the benzylic proton (N–CH2–Ar) belonging to the Pd–NHC complexes 3a–i complexes was observed as a distinct singlet at approximately δ 6.03 ppm, 6.06 ppm, 4.97 ppm, 5.98 ppm, 6.08 ppm, 4.83 ppm, 4.96 ppm, and 6.05 ppm respectively. The proton signals for the pyridine ring exhibit a downfield shift, while the signals for the aromatic protons are observed between 7.11 and 7.77 ppm. In the 1H NMR spectra, distinct signals corresponding to the aromatic hydrogens of the pyridine ring are detected at δ = 8.98, 7.71, and 7.29 ppm. These signals indicate that the pyridine ring coordinates with the palladium center, resulting in the formation of PEPPSI-type palladium complexes 3. In the 13C NMR spectra, the aromatic carbon signals of the pyridine ring are observed at δ = 152.8, 152.2, and 128.1 ppm. The characteristic Pd–C2-carbene signals of the Pd-complex (3i) appear as a singlet at δ = 163.5 ppm in the 13C NMR spectra. The aromatic carbons of the benzene ring resonate between 112.03 and 152.6 ppm. These findings are consistent with the data obtained from similar complexes.84,85 The contents of C, H, and N in palladium PEPPSI-type complexes 3a–i were determined by elemental analysis. The results agreed well with the theoretical formula of the complexes. The obtained fragments are typical for each palladium PEPPSI-type complexes 3a–i and can provide further evidence for the characterization of the examined compounds. After the characterization of these complexes, their catalytic performances were evaluated as catalysts in direct C5-arylation of 2-acetylfuran derivatives.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Time (h) | Temp. [°C] | Conversionb [%] | Yieldc [%] |
| a Reaction conditions: 2-acetylfuran (2.0 mmol), p-bromobenzene (1.0 mmol), base (2.0 mmol), DMAc (2 mL), catalyst (1 mol%).b Conversion of the aryl bromide.c Yield of product were determined by GC with dodecane as internal standard.d Without any [Pd] catalyst. | |||||
| 1d | KOtBu | 2 | 100 | — | — |
| 2 | KOtBu | 0.5 | 100 | 76 | 74 |
| 3 | KOtBu | 1 | 100 | 92 | 88 |
| 4 | KOtBu | 1.5 | 100 | 95 | 90 |
| 5 | KOtBu | 2 | 100 | 95 | 91 |
| 6 | KOtBu | 0.5 | 130 | 93 | 90 |
| 7 | KOtBu | 1 | 130 | 94 | 88 |
| 8 | KOtBu | 1.5 | 130 | 93 | 91 |
| 9 | KOtBu | 2 | 130 | 92 | 90 |
| 10 | KOtBu | 2 | 150 | 98 | 95 |
| 11 | KOAc | 0.5 | 150 | 75 | 65 |
| 12 | KOAc | 1 | 150 | 89 | 78 |
| 13 | KOAc | 1.5 | 150 | 85 | 82 |
| 14 | KOAc | 2 | 150 | 96 | 85 |
| 15 | KOAc | 3 | 150 | 83 | 76 |
| 16 | KOAc | 4 | 150 | 66 | 83 |
| 17 | KOtBu | 2 | 90 | 68 | 75 |
| 18 | Na2CO3 | 2 | 150 | 56 | 62 |
| 19 | K2CO3 | 2 | 150 | 45 | 55 |
| 20 | Cs2CO3 | 2 | 150 | 69 | 72 |
| 21 | NaOAc | 2 | 150 | 62 | 67 |
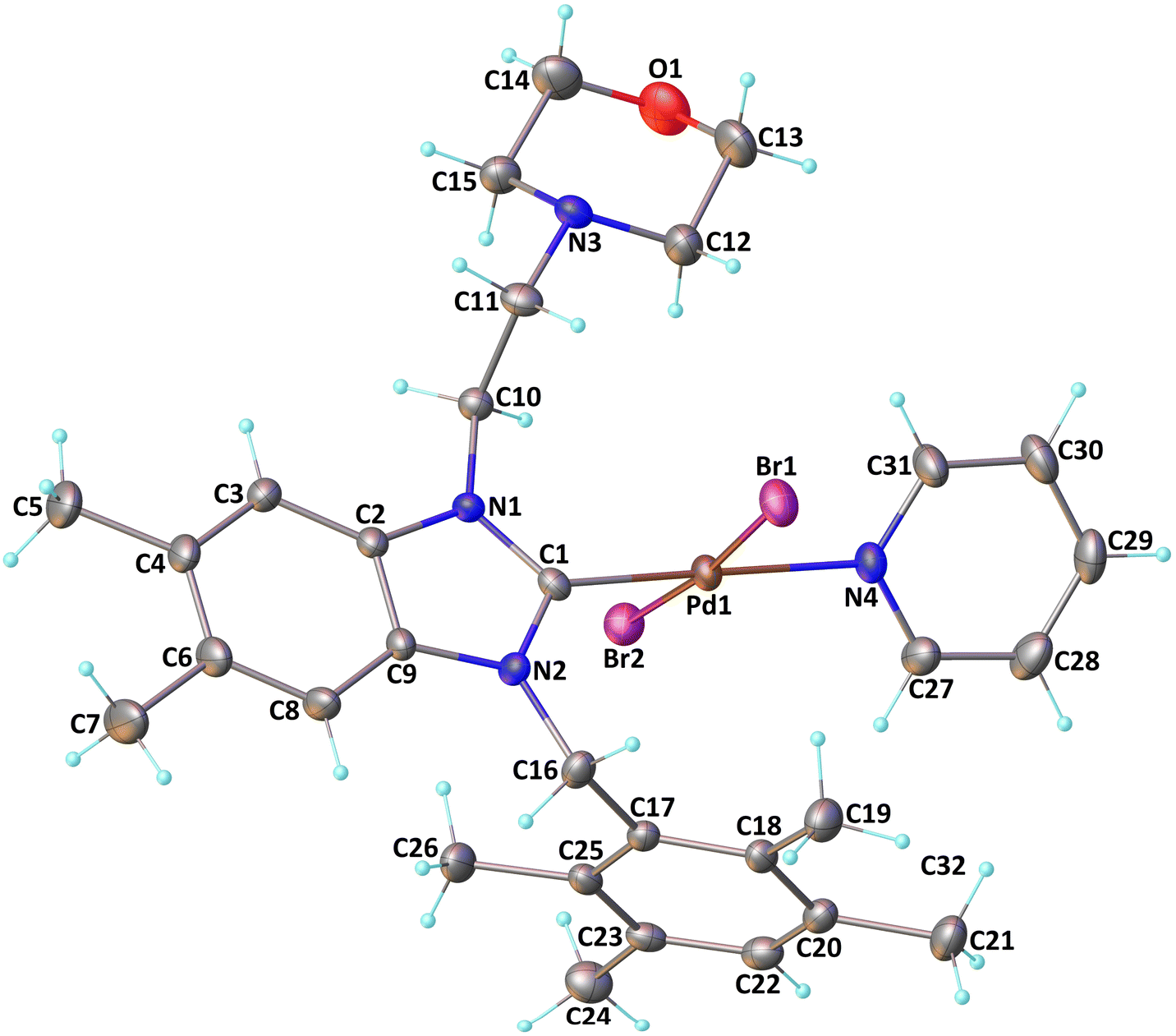 | ||
| Fig. 1 Molecular structure of complex 3 showing the atom-numbering scheme. | ||
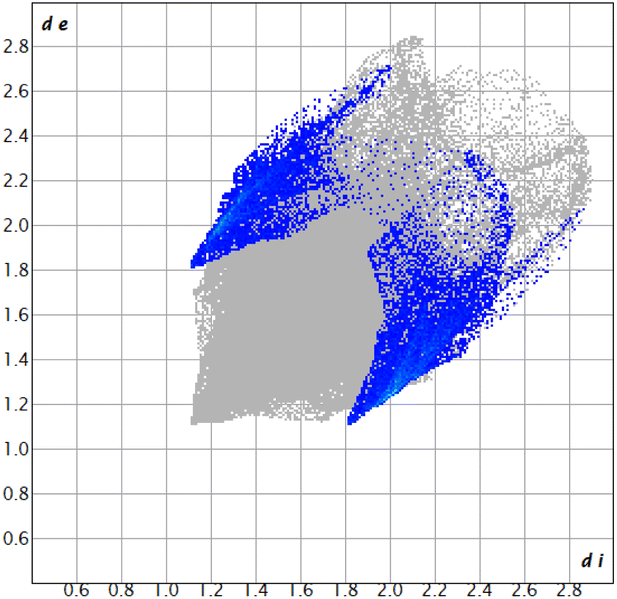 | ||
| Fig. 2 2D fingerprint plots of complex 3 (Br⋯H/H⋯Br 9.8%). | ||
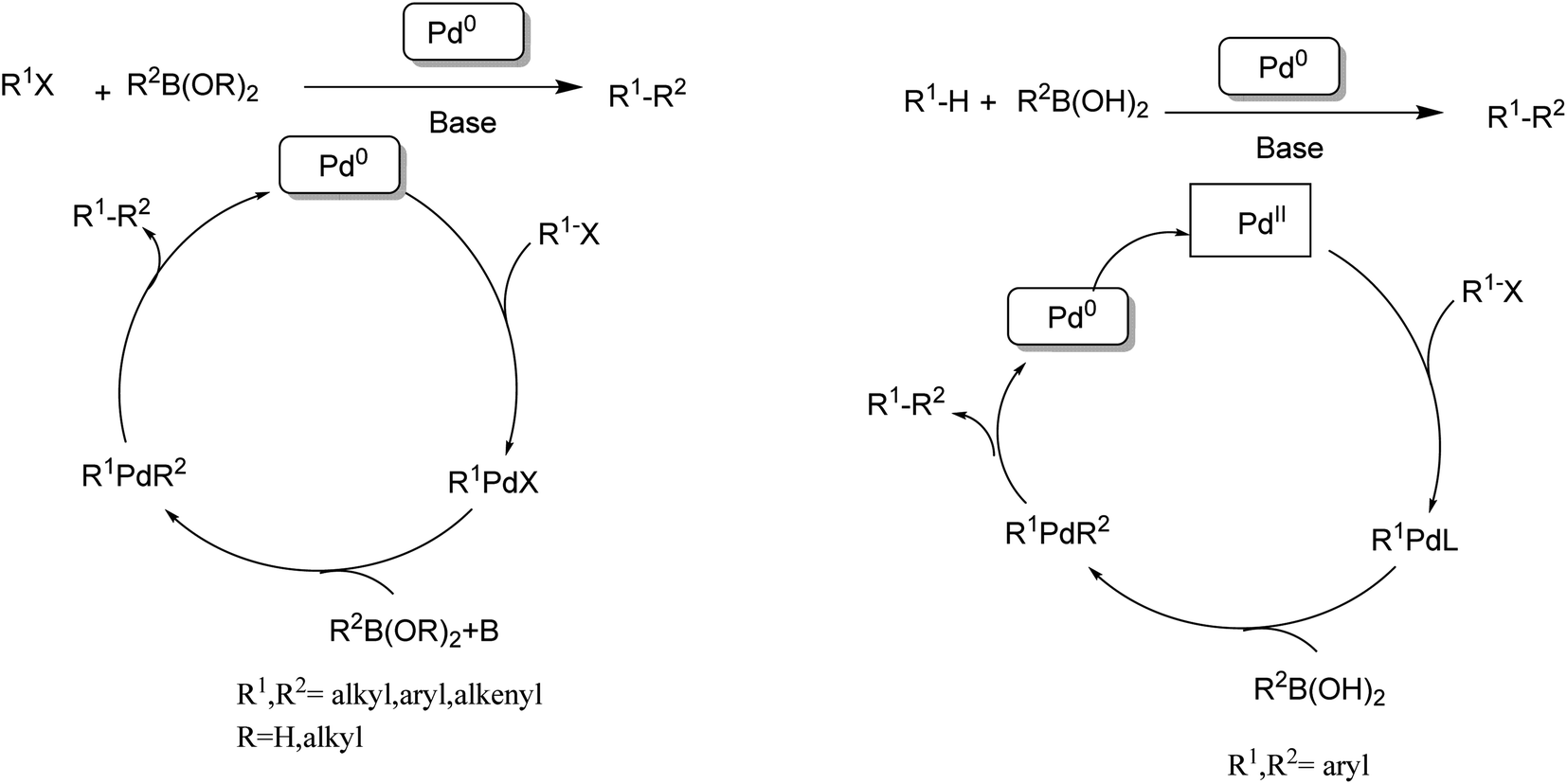 | ||
| Scheme 6 (Left) Traditional Suzuki–Miyaura coupling. (Right) Direct cross-coupling between CH bonds and CB bonds. | ||
In 1990, the first examples of Pd-catalyzed direct arylation of furans and thiophenes were reported by Ohta.103 In this pioneering work, the direct C(2)-arylation of furan and thiophene was carried out in medium to good yields with electron-rich or electron-poor aryl bromides using [Pd (PPh3)4] as the catalyst, potassium acetate (KOAc) as the base and dimethylacetamide (DMA) as the solvent. In the last two decades, Pd-catalyzed direct arylation was successfully performed using DMA/KOAc combination.104,105 Therefore, in this study, we selected DMA as the solvent, and KOAc as the base. The direct arylation of furan or thiophene itself to prepare C(2)-arylated products remains difficult as the formation of C(2)- and C(5)-diarylated products from C(2)-arylated products appears to be faster than the C(2)-arylation of furan or thiophene. Therefore, the use of a blocking group at the C(2)-position of furans and thiophenes in order to control the selectivity towards C(5)-arylation has also been described.106 Therefore, regioselective arylation on only the C(5)-position of C(2)-blocked furan or thiophene was observed. It is supposed that because the C(2)-position is blocked, the acidic C(5)-position is used for arylation and bonding of electron-deficient aryl groups is difficult compared with electron-rich aryl groups. For this reason, we selected C (2)-blocked 2-acetylfuran and 2-acetylthiophene as heteroaromatic substrates, and we focused on the direct arylation at the C(5)-position of these heteroarenes.
Also different bases such us: Na2CO3, K2CO3, NaOAc, Cs2CO3 led to different activity in the present protocol for C–H bond functionalization (Table 1, entries 17–20) but the most efficient base in this condition is KOtBu, which provides a 95% isolated yield in 2 h. Comparatively, lower yields were obtained with other bases (Table 1). While NaOAc and Cs2CO3 are inferior in reactivity (Table 1, entries 19, 20) and K2CO3 shut down the reaction completely (Table 1, entry 18). However further reduction to 90 °C decline the yield to 75% (Table 1, entry 16). After these preliminary trials, it was concluded that the best conditions for the direct arlyation were achieved at 150 °C, 2 h.
We tested the coupling of 2-acetylfuran and p-bromobenzene in the presence of, DMAc as a solvent because it is known to be a good solvent for direct arylations of heteroaromatics,107 and complex 3e (1 mol%) as a catalyst. The results of varying the other reaction conditions including used base, reaction time and reaction temperature are given in the Table 1. We initially examined the influence of the nature of the base on the conversion for this reaction and results showed good conversions of p-bromobenzene when we used KOBut as a base (Table 1, entries 1 to 9). The high temperature 150 °C leads to a slightly higher conversion and the conversion increases from 76 to 98% (Table 1, entries 1 and 9). When the base was changed from KOBut to KOAc at 150 °C with 0.5 h as time reaction the conversion raised to 75% with GC yield 65% (Table 1, entry 10). Increasing the time reaction from 0.5 h to 2 h had a noticeable effect on the conversion (Table 1, entries 11 to 13). Finally, the best conditions, leading to the 96% conversion of p-bromobenzene, with high selectivity in favor of the C5-arylated product, were obtained when the reaction was carried out in DMAc in the presence of KOAc (2 equiv.) at 150 °C for 2 h (Table 1, entry 13). According to the results, we can affirm that the Pd-complexes 3e is stable at high temperatures.108
With the optimized reaction conditions in hand, next, we evaluated the scope and limitations of the Pd-complexes 3a–3i for the direct arylation of 2-acetylfuran, 2-acetylthiophene and N-methylpyrrole-2-carboxaldehyde with different aryl bromides. The reaction worked well for a wide variety of aryl bromides such as bromobenzene, 4-bromotoluene, 4-bromoanisole, 4-bromobenzaldehyde, 1-bromo-4-fluorobenzene, 1-bromo-4-methyltrifluorobenzene, 3-bromoquinoline, 2-bromobenzonitrile, 1-bromo-4-methoxybenzene, 1-bromo-2-methylbenzene and 4-bromoacetophenone, and the results are summarized in Tables 2–4, respectively. The direct arylation reaction was carried out using electron-rich (4-bromotoluene, 4-bromoanisole), electron-poor (4-acetophenone) and electron-neutral (bromobenzene) substrates. The results are summarized in Tables 2–4 respectively. First, the 2-acetylfuran was coupled with several aryl bromides in the presence of Pd–PEPPSI complexes (3a–i). Complete conversion was obtained when 4-bromobenzaldehyde and 4-bromoacetophenone were used with 73–95% yields (Table 2, entries 28–40, 42–45). Good conversions were obtained with the rest of aryl bromides using all the complexes except the reaction with the 4-bromotoluene in the presence of 3a catalyst; it showed 89% of conversion (Table 2, entry 10). Second, we evaluated the reactivity of furfuryl acetate. The results are summarized in Table 3, high yield of C5-arylated products were obtained when we used bromobenzene, 4-bromotoluene, 4-bromoanisole, 4-bromobenzaldehyde and 4-bromoacetophenone with yields 50–92, 73–94, 57–95, 88–98 and 88–92%, respectively (Table 3, entries 1–45). Third, we evaluated the reactivity of furfural. The results are summarized in Table 4, complete conversion was obtained when 4-bromoacetophenone was used with 74–98% yields (Table 4, entries 37–45). Good conversions were obtained with the bromobenzene, 4-bromoanisole and 4-bromobenzaldehyde using all the complexes except the reaction with the 4-bromobenzaldehyde in the presence of 3f, 3g and 3h catalyst; it showed 100% of conversion (Table 4, entries 28, 30 and 32). Moderate conversion was obtained when we used 4-bromotoluene (79–98%) with yields 65–90% (Table 4, entries 10–18). However, with the 4-bromoanisole we obtained a moderate yield product (Table 4, entries 24, 25, 26). In addition, the use of a blocking group (acetyl group) in the C2 position for the furans increase the selectivity of the reaction towards C5 arylation. Under elevated temperature, the oxidative addition of aryl bromide to palladium is usually easy and does not necessitate the use of very selective ligands. Therefore, high conversions of aryl bromide were noted in all cases with all the catalysts. Therefore, the use of a blocking group at the C2 position of furans derivatives in order to control the selectivity towards C5 arylation has also been described. It is supposed that because the C2 position is blocked, the acidic C5 position is used for arylation and bonding of electron deficient aryl groups is difficult compared with electron rich aryl groups. For this reason, we selected C2 blocked 2-acetylfuran and we focused on the direct arylation at the C5 position of these heteroarenes. Consequently, aryl bromides were converted into the corresponding coupling products in high yields. The data in Tables 2–4 indicated that electron-donating groups on the para position of aryl halide (e.g. methoxy and methyl) decreased the catalytic conversion while electron-withdrawing groups (e.g. aldehyde and aceto) increased the catalytic conversion this was due to the high nucleophilicity of the electron-donating substituents. It should be noted that excellent yields are obtained by the coupling of 2-acetylfuran with aryl bromides bearing electron-withdrawing group while the increasing electron density on the aryl bromides lowered the catalyst activity. Deactivated aryl bromide such as 4-bromoanisol gave lower yields indicating that the reaction was sensitive to the electron density on the aryl bromides. The reaction of electronically neutral bromobenzene with phenylboronic acid also produced high yields of the product.
|
|||||
|---|---|---|---|---|---|
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] | Yieldc [%] |
| a Reaction conditions: 2-acetylfuran (1.3 mmol), aryl bromide (1.0 mmol), base (1.3 mmol), DMAc (2 mL), catalyst (1 mmol%).b Conversion of the aryl bromide determined by GC.c Isolated yield. | |||||
| 1 | 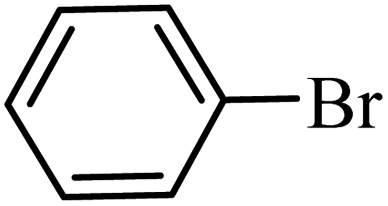 |
3a | 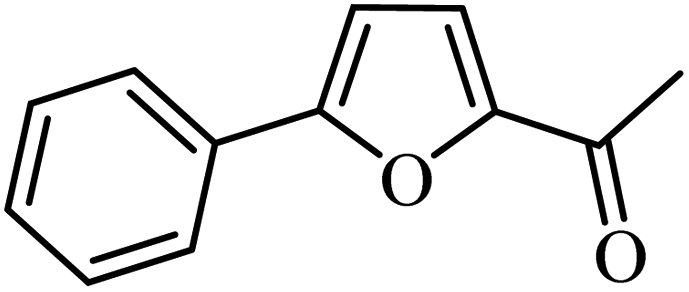 |
84 | 79 |
| 2 | 3b | 92 | 88 | ||
| 3 | 3c | 83 | 78 | ||
| 4 | 3d | 95 | 92 | ||
| 5 | 3e | 83 | 82 | ||
| 6 | 3f | 97 | 95 | ||
| 7 | 3g | 98 | 84 | ||
| 8 | 3h | 96 | 94 | ||
| 9 | 3i | 74 | 70 | ||
| 10 | 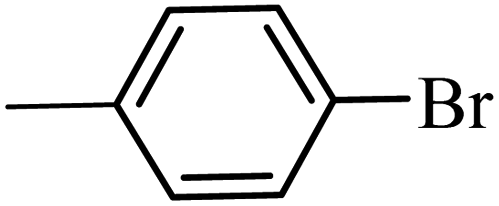 |
3a | 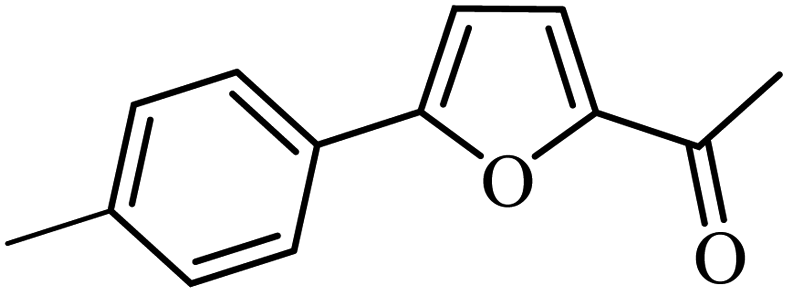 |
89 | 87 |
| 11 | 3b | 66 | 55 | ||
| 12 | 3c | 85 | 72 | ||
| 13 | 3d | 94 | 75 | ||
| 14 | 3e | 80 | 79 | ||
| 15 | 3f | 86 | 75 | ||
| 16 | 3g | 94 | 92 | ||
| 17 | 3h | 87 | 75 | ||
| 18 | 3i | 85 | 82 | ||
| 19 | 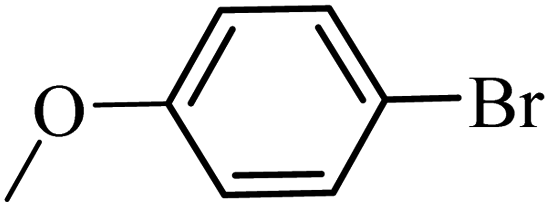 |
3a | 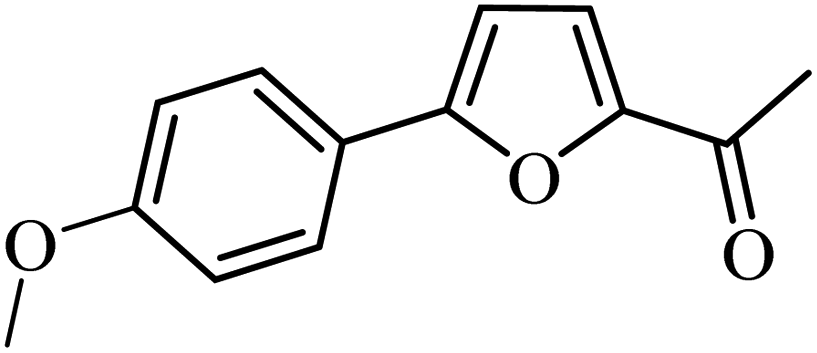 |
98 | 94 |
| 20 | 3b | 93 | 91 | ||
| 21 | 3c | 92 | 90 | ||
| 22 | 3d | 95 | 94 | ||
| 23 | 3e | 98 | 95 | ||
| 24 | 3f | 91 | 90 | ||
| 25 | 3g | 93 | 90 | ||
| 26 | 3h | 87 | 80 | ||
| 27 | 3i | 94 | 91 | ||
| 28 | 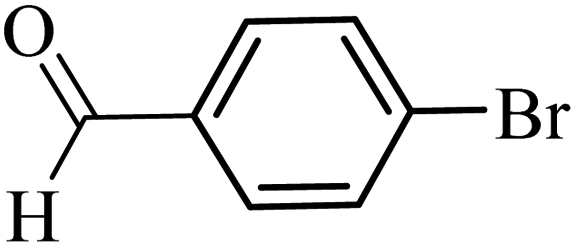 |
3a | 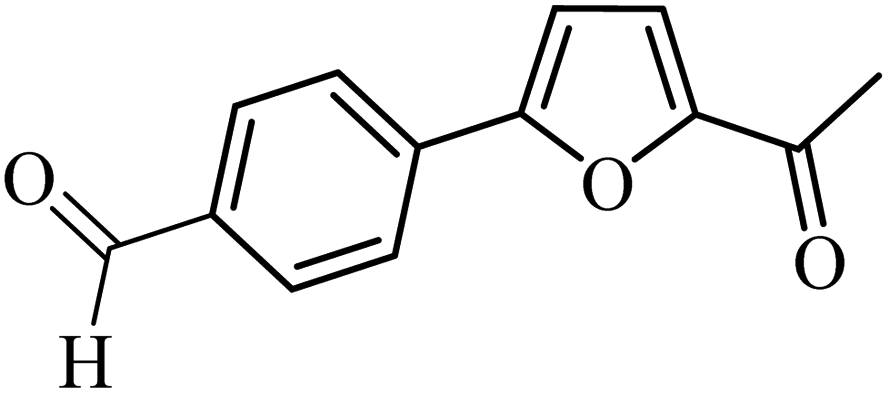 |
100 | 86 |
| 29 | 3b | 100 | 91 | ||
| 30 | 3c | 100 | 73 | ||
| 31 | 3d | 100 | 87 | ||
| 32 | 3e | 100 | 95 | ||
| 33 | 3f | 100 | 90 | ||
| 34 | 3g | 100 | 88 | ||
| 35 | 3h | 100 | 89 | ||
| 36 | 3i | 100 | 90 | ||
| 37 |  |
3a |  |
100 | 76 |
| 38 | 3b | 100 | 87 | ||
| 39 | 3c | 100 | 89 | ||
| 40 | 3d | 100 | 88 | ||
| 41 | 3e | 86 | 74 | ||
| 42 | 3f | 100 | 83 | ||
| 43 | 3g | 100 | 86 | ||
| 44 | 3h | 100 | 84 | ||
| 45 | 3i | 100 | 83 | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] | Yieldc [%] |
| a Reaction conditions: furfuryl acetate (1.3 mmol), aryl bromide (1.0 mmol), base (1.3 mmol), DMAc (2 mL), catalyst (1 mmol%).b Conversion of the aryl bromide determined by GC.c Isolated yield. | |||||
| 1 |  |
3a | 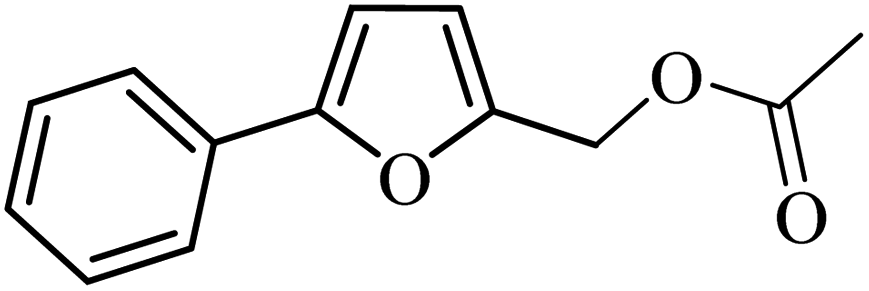 |
64 | 59 |
| 2 | 3b | 52 | 50 | ||
| 3 | 3c | 63 | 60 | ||
| 4 | 3d | 72 | 70 | ||
| 5 | 3e | 85 | 76 | ||
| 6 | 3f | 86 | 80 | ||
| 7 | 3g | 94 | 90 | ||
| 8 | 3h | 93 | 91 | ||
| 9 | 3i | 96 | 92 | ||
| 10 | 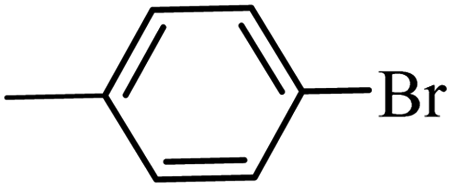 |
3a | 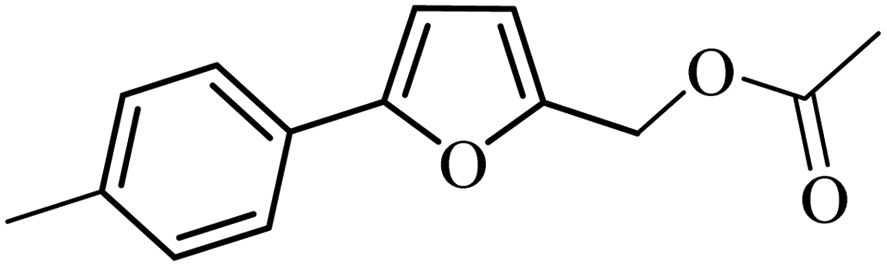 |
98 | 96 |
| 11 | 3b | 95 | 94 | ||
| 12 | 3c | 78 | 74 | ||
| 13 | 3d | 75 | 72 | ||
| 14 | 3e | 86 | 82 | ||
| 15 | 3f | 85 | 80 | ||
| 16 | 3g | 92 | 90 | ||
| 17 | 3h | 88 | 85 | ||
| 18 | 3i | 90 | 87 | ||
| 19 | 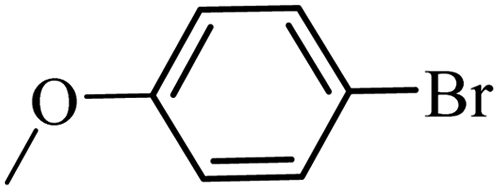 |
3a | 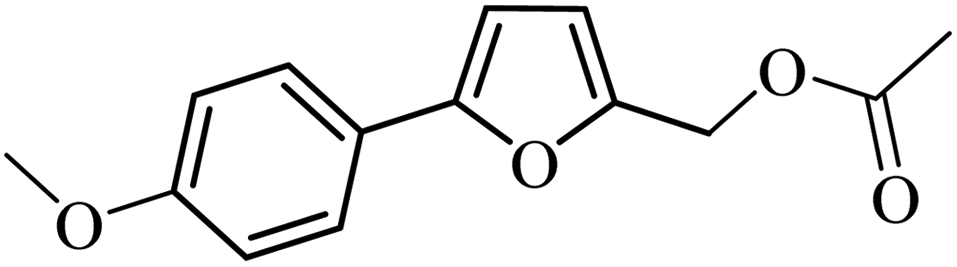 |
97 | 95 |
| 20 | 3b | 96 | 94 | ||
| 21 | 3c | 92 | 89 | ||
| 22 | 3d | 91 | 88 | ||
| 23 | 3e | 90 | 89 | ||
| 24 | 3f | 89 | 85 | ||
| 25 | 3g | 75 | 72 | ||
| 26 | 3h | 85 | 82 | ||
| 27 | 3i | 76 | 72 | ||
| 28 | 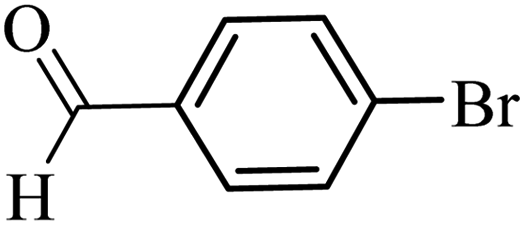 |
3a | 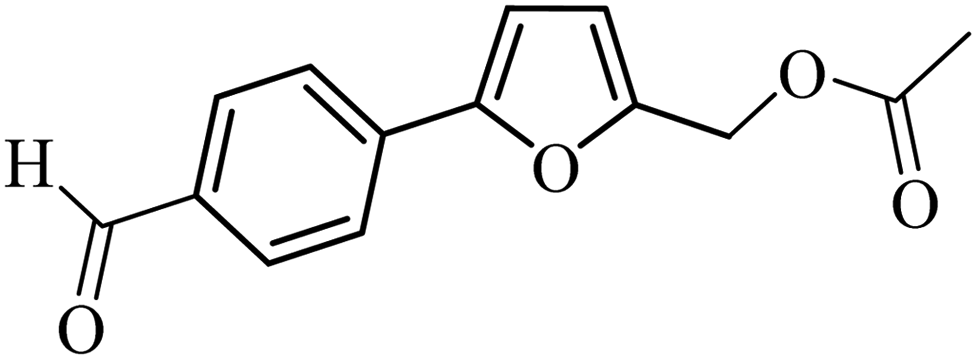 |
100 | 98 |
| 29 | 3b | 100 | 95 | ||
| 30 | 3c | 100 | 97 | ||
| 31 | 3d | 100 | 95 | ||
| 32 | 3e | 100 | 97 | ||
| 33 | 3f | 100 | 88 | ||
| 34 | 3g | 100 | 96 | ||
| 35 | 3h | 100 | 92 | ||
| 36 | 3i | 100 | 94 | ||
| 37 | 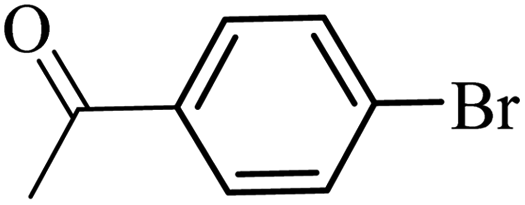 |
3a |  |
100 | 93 |
| 38 | 3b | 100 | 90 | ||
| 39 | 3c | 100 | 92 | ||
| 40 | 3d | 100 | 90 | ||
| 41 | 3e | 100 | 91 | ||
| 42 | 3f | 100 | 90 | ||
| 43 | 3g | 100 | 90 | ||
| 44 | 3h | 100 | 88 | ||
| 45 | 3i | 100 | 90 | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] | Yieldc [%] |
| a Reaction conditions: furfural (1.3 mmol), aryl bromide (1.0 mmol), base (1.3 mmol), DMAc (2 mL), catalyst (1 mmol%).b Conversion of the aryl bromide determined by GC.c Isolated yield. | |||||
| 1 |  |
3a |  |
75 | 68 |
| 2 | 3b | 75 | 72 | ||
| 3 | 3c | 76 | 71 | ||
| 4 | 3d | 78 | 68 | ||
| 5 | 3e | 84 | 68 | ||
| 6 | 3f | 75 | 72 | ||
| 7 | 3g | 71 | 65 | ||
| 8 | 3h | 81 | 71 | ||
| 9 | 3i | 72 | 68 | ||
| 10 | 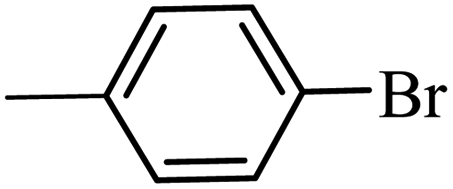 |
3a | 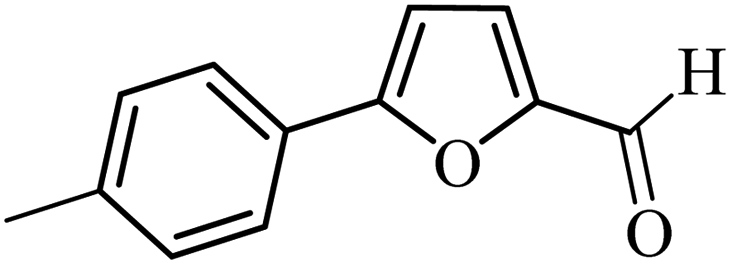 |
79 | 65 |
| 11 | 3b | 92 | 90 | ||
| 12 | 3c | 93 | 88 | ||
| 13 | 3d | 83 | 75 | ||
| 14 | 3e | 89 | 78 | ||
| 15 | 3f | 79 | 74 | ||
| 16 | 3g | 78 | 72 | ||
| 17 | 3h | 82 | 76 | ||
| 18 | 3i | 93 | 90 | ||
| 19 | 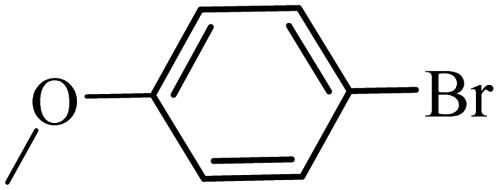 |
3a | 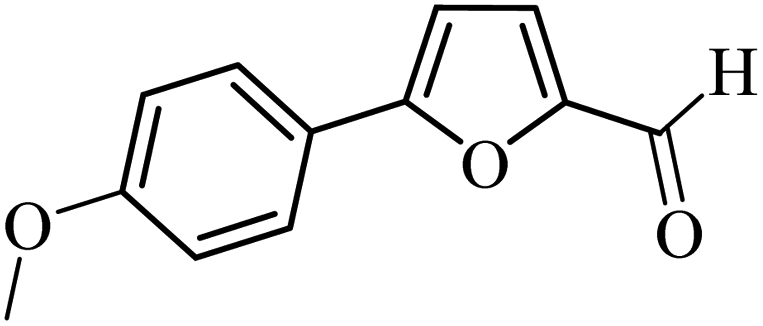 |
88 | 76 |
| 20 | 3b | 89 | 86 | ||
| 21 | 3c | 85 | 84 | ||
| 22 | 3d | 92 | 90 | ||
| 23 | 3e | 91 | 88 | ||
| 24 | 3f | 84 | 82 | ||
| 25 | 3g | 90 | 86 | ||
| 26 | 3h | 76 | 74 | ||
| 27 | 3i | 90 | 87 | ||
| 28 | 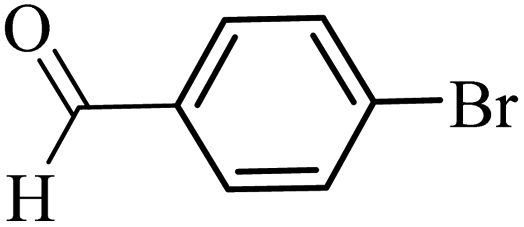 |
3a | 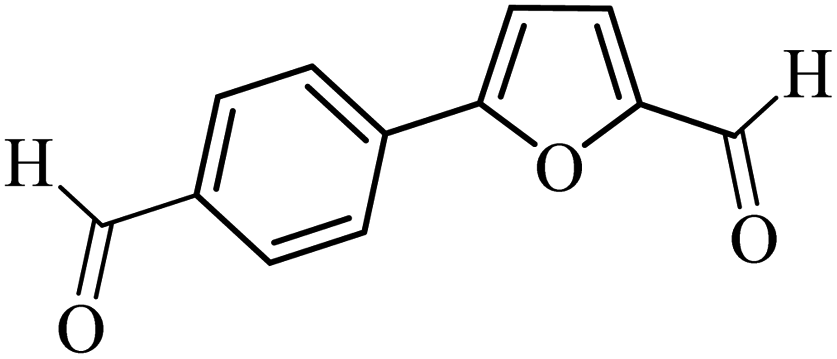 |
100 | 77 |
| 29 | 3b | 86 | 78 | ||
| 30 | 3c | 100 | 76 | ||
| 31 | 3d | 90 | 75 | ||
| 32 | 3e | 100 | 76 | ||
| 33 | 3f | 86 | 72 | ||
| 34 | 3g | 81 | 65 | ||
| 35 | 3h | 75 | 67 | ||
| 36 | 3i | 87 | 76 | ||
| 37 |  |
3a |  |
100 | 91 |
| 38 | 3b | 100 | 95 | ||
| 39 | 3c | 100 | 80 | ||
| 40 | 3d | 100 | 80 | ||
| 41 | 3e | 100 | 80 | ||
| 42 | 3f | 100 | 82 | ||
| 43 | 3g | 100 | 75 | ||
| 44 | 3h | 100 | 80 | ||
| 45 | 3i | 100 | 74 | ||

Second, we evaluated the reactivity of 2-acetylthiophene. The results are summarized in Table 5, high yield of C5-arylated products were obtained when we used bromobenzene, 4-bromotoluene, 4-bromobenzaldehyde and 4-bromoacetophenone with yields 70–95, 65–89, 87–98 and 87–95%, respectively (Table 5, entries 1–19). However, with the 4-bromoanisole we obtained a lower yield product (Table 5, entries 24, 25, 26). In addition the use of a blocking group (acetyl group) in the C2 position for the furans, thiophenes, and (n-propyl group) for the thiazole increase the selectivity of the reaction towards C5 arylation.109 Under elevated temperature, the oxidative addition of aryl bromide to palladium is usually easy and does not necessitate the use of very selective ligands. Therefore, high conversions of aryl bromide were noted in all cases with all the catalysts. Therefore, the use of a blocking group at the C2 position of furans, thiophenes and thiazole in order to control the selectivity towards C5 arylation has also been described.
|
||||
|---|---|---|---|---|
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] |
| a Reaction conditions: 2-acetylthiophene (2.0 mmol), aryl bromide (1.0 mmol), base (2.0 mmol), DMAc (2 mL), catalyst (1 mol%).b Conversion of the aryl bromide. | ||||
| 1 |  |
3a |  |
99 |
| 2 | 3b | 98 | ||
| 3 | 3c | 80 | ||
| 4 | 3d | 99 | ||
| 5 | 3e | 100 | ||
| 6 | 3f | 85 | ||
| 7 | 3g | 90 | ||
| 8 | 3h | 86 | ||
| 9 | 3i | 87 | ||
| 10 |  |
3a |  |
85 |
| 11 | 3b | 99 | ||
| 12 | 3c | 68 | ||
| 13 | 3d | 80 | ||
| 14 | 3e | 100 | ||
| 15 | 3f | 85 | ||
| 16 | 3g | 90 | ||
| 17 | 3h | 82 | ||
| 18 | 3i | 90 | ||
| 19 |  |
3a |  |
100 |
| 20 | 3b | 100 | ||
| 21 | 3c | 100 | ||
| 22 | 3d | 100 | ||
| 23 | 3e | 100 | ||
| 24 | 3f | 95 | ||
| 25 | 3g | 95 | ||
| 26 | 3h | 85 | ||
| 27 | 3i | 100 | ||
| 28 |  |
3a |  |
88 |
| 29 | 3b | 97 | ||
| 30 | 3c | 66 | ||
| 31 | 3d | 50 | ||
| 32 | 3e | 65 | ||
| 33 | 3f | 75 | ||
| 34 | 3g | 80 | ||
| 35 | 3h | 85 | ||
| 36 | 3i | 90 | ||
| 37 |  |
3a |  |
100 |
| 38 | 3b | 100 | ||
| 39 | 3c | 100 | ||
| 40 | 3d | 100 | ||
| 41 | 3e | 100 | ||
| 42 | 3f | 95 | ||
| 43 | 3g | 85 | ||
| 44 | 3h | 90 | ||
| 45 | 3i | 95 | ||
In presence of Pd-complexes 3c and 3d, we observed good conversion of the target product, 5-phenyl-1-methyl-2-formylpyrrole,110 (Table 6, entries 3 and 5). The reaction of N-methylpyrrole-2-carboxaldehyde with an electron-rich aryl bromide such as p-bromotoluene generated the 5-(4-methylphenyl)-1-methyl-2-formylpyrrole,111 with a conversion of 90% in the presence of 3d catalyst after 2 h. In the presence of para-substituted electron-deficient aryl bromide such as 4-bromobenzaldehyde, the expected compound, 5-(4-formylphenyl)-1-methyl-2-formylpyrrole,112 was obtained in moderate to high yields using only 1 mol% catalyst after 2 h (Table 6, entries 13, 14). This compound was obtained in a conversion of 92% using 3e catalyst (Table 6, entry 14). The coupling of 1-methylpyrrole-2-carboxaldehyde with electron-poor aryl bromide such as 4-bromoacetophenone proceeds nicely. 4-Bromoacetophenone gave the 5-(4-acetylphenyl)-1-methyl-2-formylpyrrole,97 with moderate to high conversion (Table 6, entries 16–20). When sterically hindered electrondonating 2-bromotoluene was used as aryl halide, 85% conversion of the 5-(2-methylphenyl)-1-methyl-2-formylpyrrole,113 was obtained in the presence of 3h catalyst after 2 h (Table 6, entry 35). 2-Bromobenzonitrile gave the 2-(5-formyl-1-methylpyrrol-2-yl)-benzonitrile, in 2 h,114 with moderate to high conversion after 2 h (Table 2, entries 42–46). This product was obtained with a conversion of 65% in the presence of 3i catalyst (Table 6, entry 45).
|
||||
|---|---|---|---|---|
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] |
| a Conditions: [Pd] 3a–3i (0.01 equiv., 1 mol%), N-methylpyrrole-2-carboxaldehyde (2 equiv.), (hetero)arylbromide (1 equiv.), KOAc (2 equiv.), DMAc (2 mL), 120 °C.b Conversion of the aryl bromide were determined by GC with dodecane as internal standard. | ||||
| 1 |  |
3a | 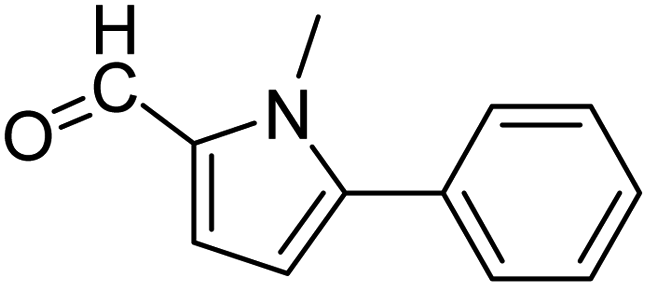 |
80 |
| 2 | 3b | 92 | ||
| 3 | 3c | 85 | ||
| 4 | 3d | 95 | ||
| 5 | 3e | 90 | ||
| 6 | 3f | 85 | ||
| 7 | 3g | 87 | ||
| 8 | 3h | 88 | ||
| 9 | 3i | 90 | ||
| 10 | 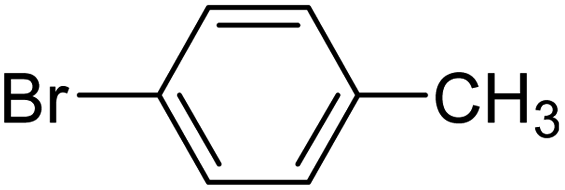 |
3a | 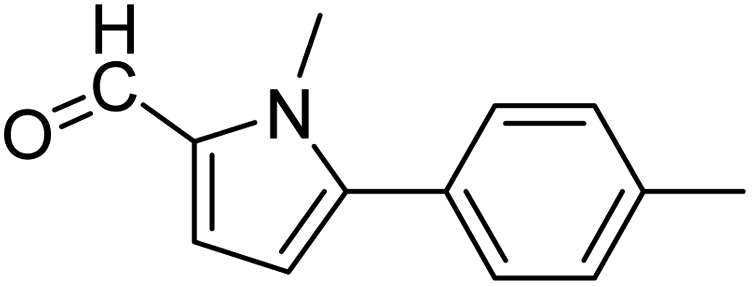 |
80 |
| 11 | 3b | 75 | ||
| 12 | 3c | 85 | ||
| 13 | 3d | 90 | ||
| 14 | 3e | 92 | ||
| 15 | 3f | 70 | ||
| 16 | 3g | 95 | ||
| 17 | 3h | 85 | ||
| 18 | 3i | 80 | ||
| 19 |  |
3a | 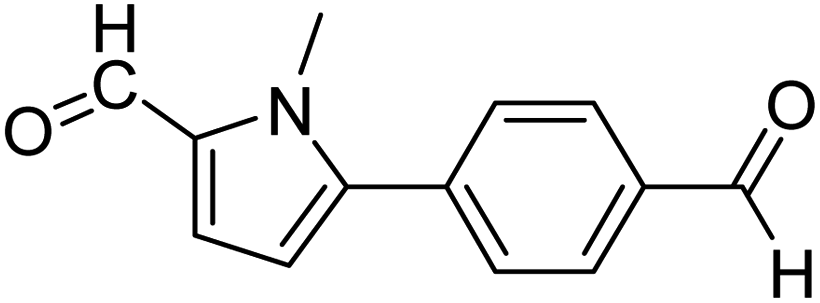 |
64 |
| 20 | 3b | 52 | ||
| 21 | 3c | 85 | ||
| 22 | 3d | 72 | ||
| 23 | 3e | 70 | ||
| 24 | 3f | 75 | ||
| 25 | 3g | 85 | ||
| 26 | 3h | 80 | ||
| 27 | 3i | 90 | ||
| 38 | 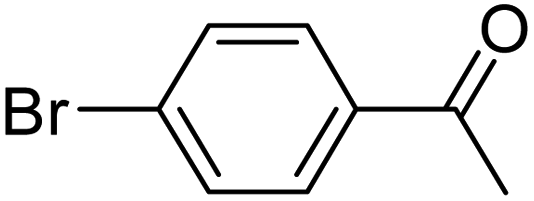 |
3a | 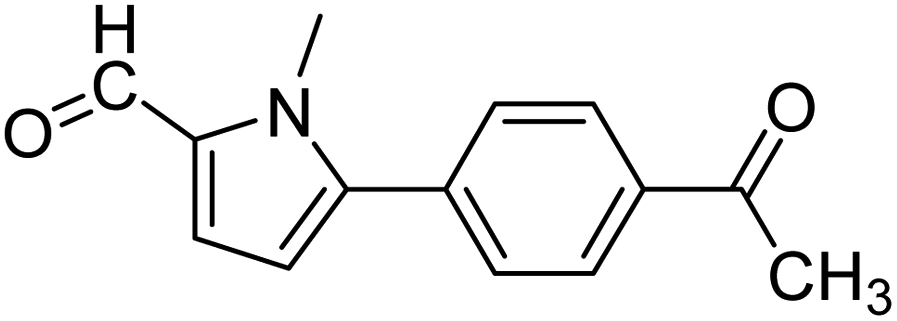 |
98 |
| 29 | 3b | 95 | ||
| 30 | 3c | 85 | ||
| 31 | 3d | 78 | ||
| 32 | 3e | 70 | ||
| 33 | 3f | 75 | ||
| 34 | 3g | 80 | ||
| 35 | 3h | 85 | ||
| 36 | 3i | 87 | ||
| 37 |  |
3a | 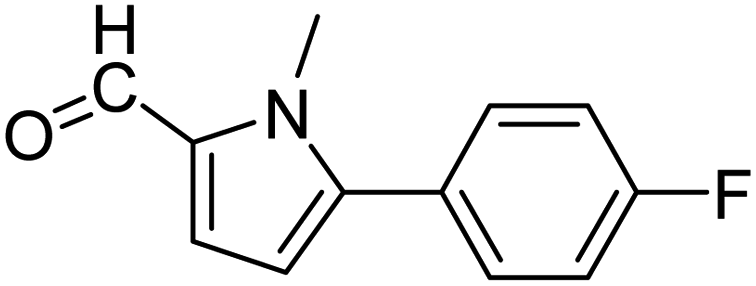 |
90 |
| 38 | 3b | 85 | ||
| 39 | 3c | 82 | ||
| 40 | 3d | 75 | ||
| 41 | 3e | 95 | ||
| 42 | 3f | 70 | ||
| 43 | 3g | 78 | ||
| 44 | 3h | 90 | ||
| 45 | 3i | 65 | ||
| 46 | 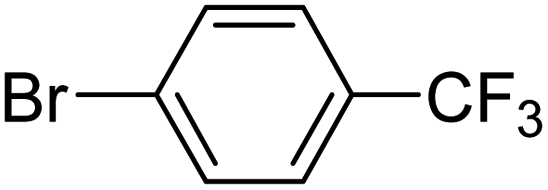 |
3a | 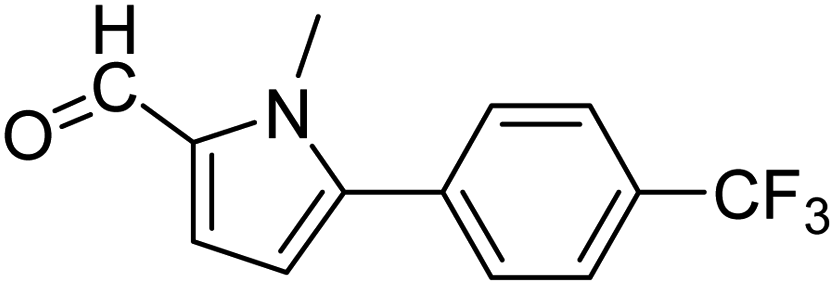 |
97 |
| 47 | 3b | 96 | ||
| 48 | 3c | 92 | ||
| 49 | 3d | 91 | ||
| 50 | 3e | 90 | ||
| 51 | 3f | 85 | ||
| 52 | 3g | 90 | ||
| 53 | 3h | 75 | ||
| 54 | 3i | 70 | ||
| 55 | 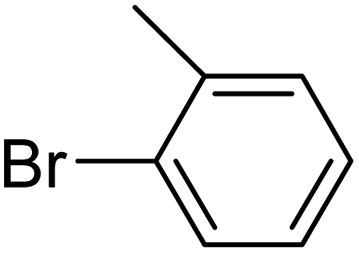 |
3a | 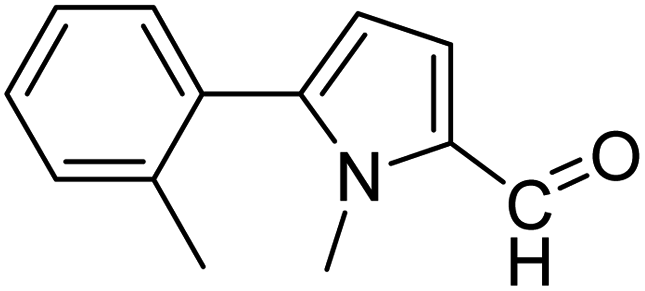 |
90 |
| 56 | 3b | 85 | ||
| 57 | 3c | 80 | ||
| 58 | 3d | 95 | ||
| 59 | 3e | 75 | ||
| 60 | 3f | 78 | ||
| 61 | 3g | 90 | ||
| 62 | 3h | 85 | ||
| 63 | 3i | 87 | ||
| 64 |  |
3a | 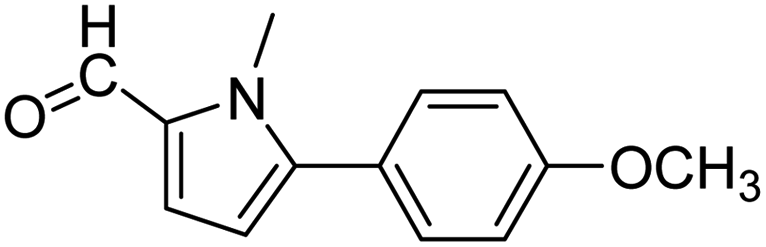 |
100 |
| 65 | 3b | 100 | ||
| 66 | 3c | 100 | ||
| 67 | 3d | 78 | ||
| 68 | 3e | 100 | ||
| 69 | 3f | 90 | ||
| 70 | 3g | 95 | ||
| 71 | 3h | 85 | ||
| 72 | 3i | 80 | ||
| 73 | 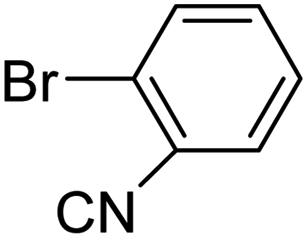 |
3a | 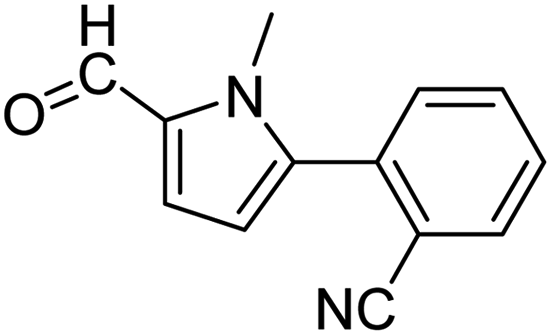 |
95 |
| 74 | 3b | 85 | ||
| 75 | 3c | 90 | ||
| 76 | 3d | 75 | ||
| 77 | 3e | 90 | ||
| 78 | 3f | 80 | ||
| 79 | 3g | 75 | ||
| 80 | 3h | 78 | ||
| 81 | 3i | 92 | ||
| 82 | 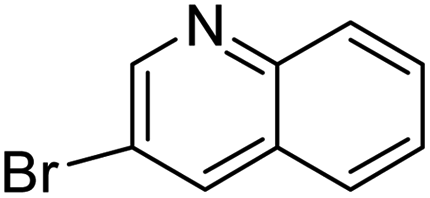 |
3a | 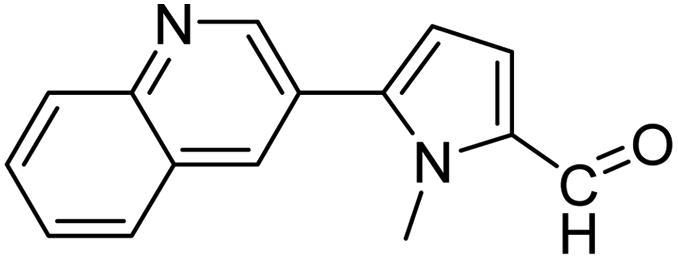 |
95 |
| 83 | 3b | 85 | ||
| 84 | 3c | 90 | ||
| 85 | 3d | 75 | ||
| 86 | 3e | 90 | ||
| 87 | 3g | 80 | ||
| 88 | 3g | 87 | ||
| 89 | 3h | 92 | ||
| 90 | 3i | 80 | ||

When chlorobenzene was used in the presence of 3d catalyst, which is the most active catalyst, 5a was obtained in 75% yield after 12 h (Table 7, entry 1). However, when p-chlorotoluene was used, 5b was obtained in 65% yield (Table 7, entry 2). When 4-chlorobenzaldehyde was used as the coupling partner, 5c was obtained in 87% yield after 15 h (Table 7, entry 3). High yields of expected C5-arylated product 5d was obtained for the coupling with 4-chloroacetophenone by using catalysts 3d (Table 7, entry 4).
| Entry | Aryl halide | Catalyst | Product | Conversionb [%] |
|---|---|---|---|---|
| a Conditions: [Pd] 3a–3i (0.01 equiv., 1 mol%), N-methylpyrrole-2-carboxaldehyde (2 equiv.), (hetero)arylbromide (1 equiv.), KOAc (2 equiv.), DMAc (2 mL), 120 °C.b Conversion of the aryl bromide were determined by GC with dodecane as internal standard. | ||||
| 1 | 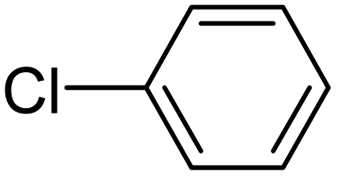 |
3a | 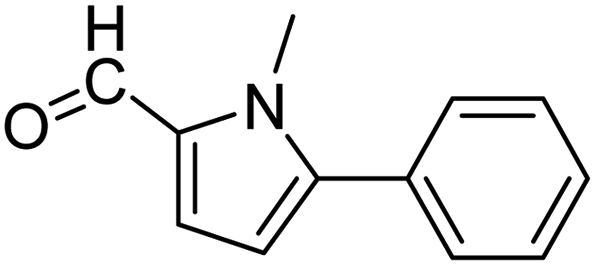 |
75 |
| 2 | 3b | 65 | ||
| 3 | 3c | 85 | ||
| 4 | 3d | 90 | ||
| 5 | 3e | 92 | ||
| 6 | 3f | 85 | ||
| 7 | 3g | 80 | ||
| 8 | 3h | 78 | ||
| 9 | 3i | 75 | ||
| 10 | 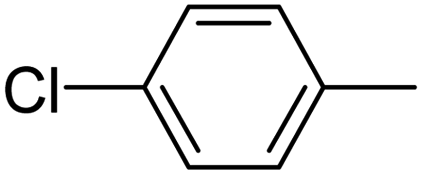 |
3a | 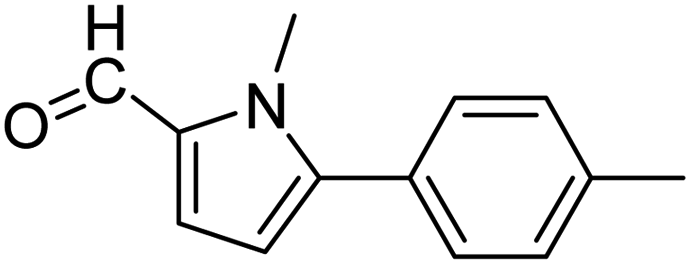 |
65 |
| 11 | 3b | 70 | ||
| 12 | 3c | 85 | ||
| 13 | 3d | 90 | ||
| 17 | 3e | 75 | ||
| 18 | 3f | 80 | ||
| 19 | 3g | 85 | ||
| 20 | 3h | 80 | ||
| 21 | 3i | 75 | ||
| 22 | 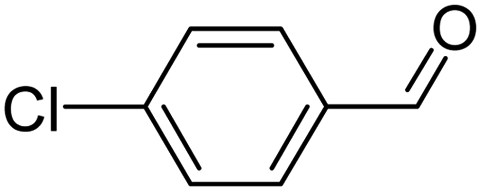 |
3a | 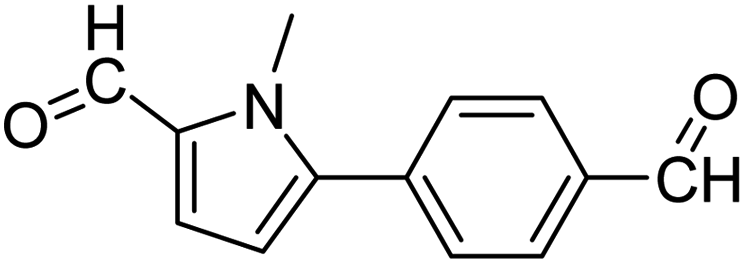 |
64 |
| 23 | 3b | 52 | ||
| 24 | 3c | 87 | ||
| 25 | 3d | 72 | ||
| 26 | 3e | 95 | ||
| 27 | 3f | 85 | ||
| 28 | 3g | 80 | ||
| 29 | 3h | 87 | ||
| 30 | 3i | 75 | ||
| 31 |  |
3a | 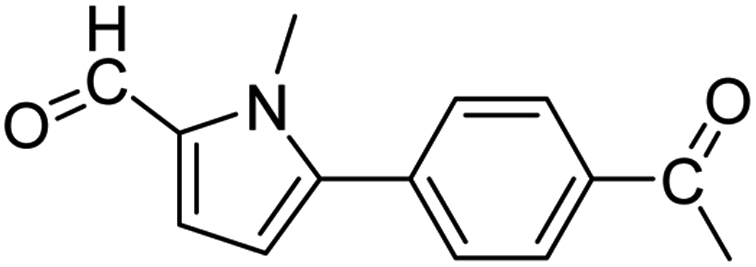 |
98 |
| 31 | 3b | 95 | ||
| 32 | 3c | 78 | ||
| 33 | 3d | 90 | ||
| 34 | 3e | 70 | ||
| 35 | 3f | 75 | ||
| 36 | 3g | 78 | ||
| 37 | 3h | 92 | ||
| 38 | 3i | 95 | ||
The Pd-catalyzed direct arylation of furan and thiophene with a variety of electrophilic reagents has been previously described.115,116 In previous studies, similar or close substrates have been employed with higher catalyst loading (1–20 mol%), and a higher reaction time (1–48 h) has been chosen for the direct arylation of furan and thiophene in the presence of Pd-catalysts. In the present work, 1 mol% catalyst loading was used, and the reaction time was shortened to 2 h. In most cases, high yields were observed with all complexes. Only a minor effect of the NHC ligand on the Pd-complex was observed for the coupling of aryl bromide with heteroaromatics. Surprisingly, similar conversions were obtained for the coupling of each aryl bromide. There is no significant difference between these complexes on the catalytic activity of direct arylation of heteroaromatics by aryl bromides. The only significant difference between 3a–3i complexes indicates that electronic and steric effects also play some role in these process. One of the main problems in studies of transition-metalcatalyzed reactions in solutions is the determination of the nature of active catalytic centers-if they are molecular complexes or metal clusters/nanoparticles.117–119 This task is of great importance both for catalysis with well-defined molecular metal complexes, for example, Pd/NHC complexes, which are typically considered homogeneous catalysts, and for supported metal catalysts, such as Pd/C.120–122 Thus, well-defined molecular metal complexes can undergo decomposition during preactivation or in the course of a catalytic process to give metal clusters and nanoparticles possessing substantially higher catalytic activities, allowing them to become the predominant active centers.123,124 On the other hand, for heterogeneous catalysts, the leaching of supported metals into the solution to give active molecular complexes is common.125,126 Reliably distinguishing the nature of the active centers and the type of catalyst (homogeneous or cluster/nanoparticle) is of great significance for tuning the reaction conditions and designing new generations of highly efficient, stable, and sustainable catalysts.127 Among the various methods, the mercury test (or “Hg test”, “mercury poisoning test”, “Hg poisoning test”, and “Hg drop test”) is one of the most frequently used rapid methods for distinguishing between truly homogeneous molecular catalysis and cluster/nanoparticle catalysis.128 The method is based on the assumption that metallic mercury will poison M0 clusters/nanoparticles that are acting as catalytically active centers and is inert toward molecular metal complexes. Inhibition of a catalytic reaction in the presence of metallic mercury is typically considered to be evidence of a cluster/nanoparticle in the catalytic mechanism. In previous mechanistic studies of several reactions catalyzed by complexes of Pd with N-heterocyclic carbenes (Pd/NHC), we performed control experiments to ascertain the inertness of the complexes toward metallic mercury. Surprisingly, solutions of Pd complexes, even highly stable PdII/NHC complexes, were decomposed by metallic mercury to give complex mixtures of products and dark metal precipitates on the surface of the mercury. This observation prompted us to thoroughly evaluate the applicability of the mercury test. The presence of Pd(0) species arising from Pd(0) nano particles cannot be excluded.129,130 However, as an attempt to destroy the nanoparticules,131,132 the reaction between 4-bromoacetophenone and furfural in the presence of Pd(II)–NHC complex 3b was carried out. Under the optimum reaction conditions, a typical catalytic coupling reaction was initiated by adding 300 equiv. Mercury into the reaction mixture and stirred vigorously. After a course of reaction time, the reaction did not stop and the corresponding coupling product was obtained in 89% yield. So this test not affect significantly the yield of the reaction. Thus the catalytic Pd(0) species seem to arise from the in situ reduction into Pd(0) species with base carbonate on heating. In our case the preactivation of catalyst involves the usual reduction of the Pd(II) spices into the Pd(0) species in the presence of base and heating.133 The oxidative addition of ArBr to the Pd(0) species leads to the Pd(II) intermediate and then to the substitution of bromide by acetate. The next step is the interaction of the heteroarene C5–H bond with the Pd(II)–OAc intermediate and intramolecular deprotonation of C5–H bond by acetate, the activation of C–H occurs at position 5.134 Finally, C5-arylated heteroarenes product was obtained by the result of reductive elimination. In the preactivation step Pd(II) is reduced to Pd(0) by the base and heating, as proposed. The Pd(II) catalysts are easily reduced into Pd(0) species in the presence of a base and heating. KOAc is basic enough to generate Pd(0) species. In addition NHC ligands are not strong pi-acceptor but they are electron-rich and they favour fast oxidative addition of R–X to the (NHC)Pd(0) species135–140 (Scheme 7).
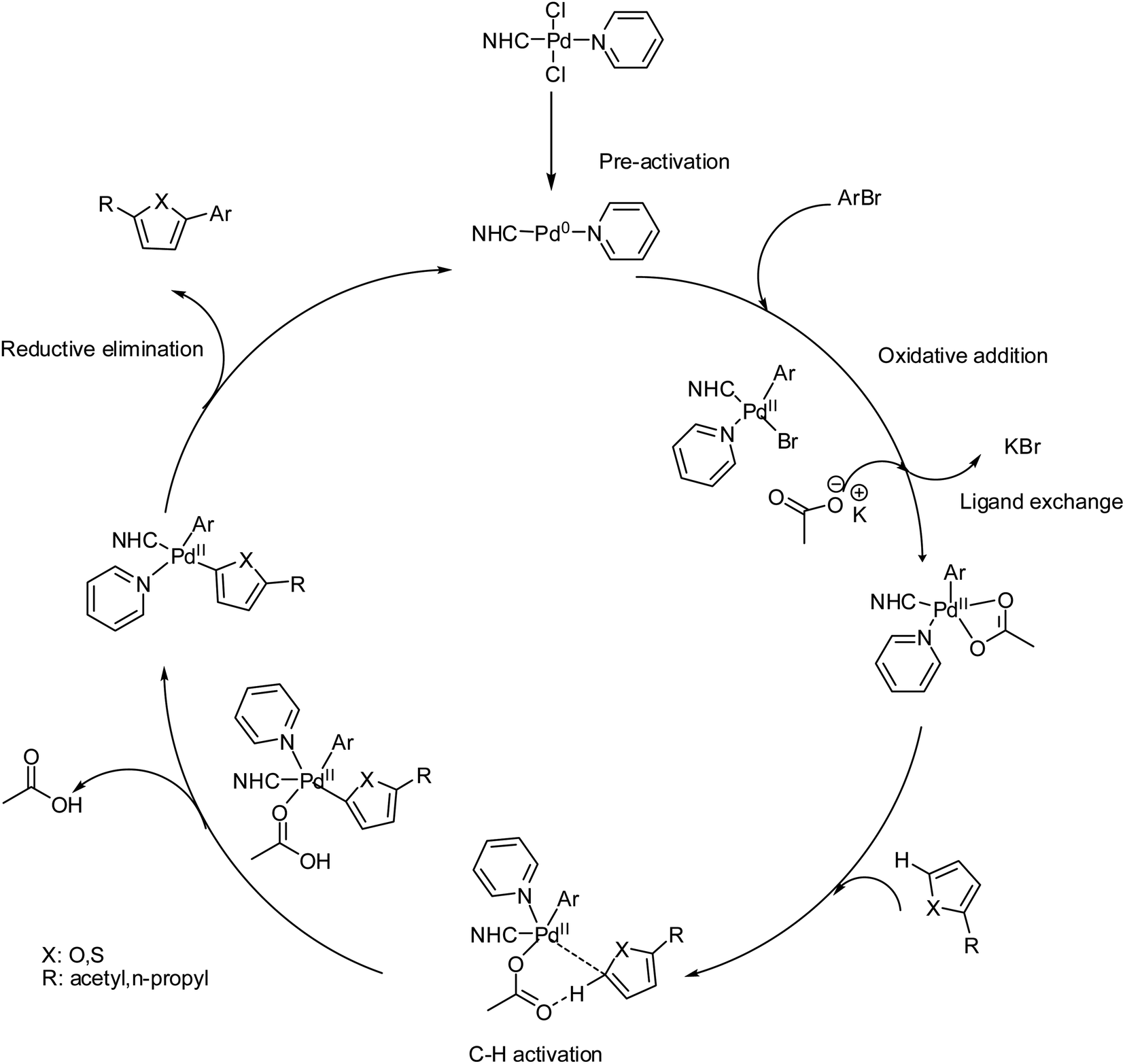 | ||
| Scheme 7 The catalytic mechanism for the direct C–H bond arylation for PEPPSI–Pd(II)–N-heterocyclic carbene (NHC) complexes 3a–i. | ||
3 Conclusion
This research work reported the synthesis and characterization of eighteen new compounds including benzimidazole and their asymmetric palladium PEPPSI complexes (3a–3e). Analytical techniques such as 1H, 13C NMR, IR, Mass, elemental analysis, melting points were used for thorough characterization of newly prepared compounds. Monoligated pyridine assisted PEPPSI complexes were crafted to achieve maximum output in direct arylation reaction of five membered heterocyclic compounds. Being tweaked ligands, NHCs with adjustable size and electronic properties were key to obtain desirable products in direct CH bond activation reactions of furfuryl acetate, 1-methyl-2-pyrrole carboxaldehyde, and acetyl furan with various hetero(aryl)bromides and chloride with excellent conversion rate and selectivity. These Pd(II) complexes feature high stability due to restricted rotation of bulky N-substitution on NHC under harsh catalytic conditions and producing pure product in excellent yield. This study is ecologically and economically significant due to the low catalyst loading and short reaction time. In this study, only AcOH and HBr were formed as by-products using the direct arylation method, thus minimizing the formation of by-products compared with multi-step conventional transition metal-catalyzed reactions. Furthermore, this study contributes to the literature on organometallic synthesis and preparation of bi(hetero)aryl derivatives. Further studies on the applicability of other reactions are currently underway in our laboratory. Poisoning experiments showed that homogeneous molecular palladium species containing NHC ligands were responsible for the catalytic activity. The catalytic system generated from these PEPPSI-type palladium–NHC complexes was very efficient at 1 mmol% catalyst loading from aryl halides in the presence of KOAc as base and DMAc as solvent at 150 °C selective formation of C–C bonds. Through evaluation of catalytic studies, all complexes were found to be suitable for the direct C5 arylation of 2-substituted furan derivatives with aryl bromides.4 Experimental
4.1. General methods
All manipulations were carried out under argon using standard Schlenk line techniques. Chemicals and solvents were purchased from Sigma-Aldrich Co. (Poole, Dorset, UK). The solvents used were purified by distillation over the drying agents indicated and were transferred under argon. DMAc (dimethylacetamide) analytical grade (99%) was not distilled before use. KOAc (99%) was employed. Melting points were measures in open capillary tubes with an Electrothermal-9200 melting points apparatus. IR spectra were recorded on ATR unit in the range of 400–4000 cm−1 with PerkinElmer Spectrum 100 Spectrophotometer. 1H NMR and 13C NMR spectra were recorded using Bruker Avance AMX and Bruker Avance III spectrometer operating at 400 MHz (1H NMR) and at 100 MHz (13C NMR) in CDCl3 with TMS added. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, hept = heptet, and m = multiplet signal. The NMR studies were carried out in high-quality 5 mm NMR tubes. The chemical shifts (d) are reported in ppm relative to CDCl3. Coupling constants (J values) are given in hertz. All catalytic reactions were monitored on an Agilent 6890 N GC and Shimadzu 2010 Plus GCMS system by GC-FID with an HP-5 column of 30 m length, 0.32 mm diameter, and 0.25 mm film thickness. Elemental analyses were performed by LECO CHNS-932 elementary chemical analyzer.4.2. Preparation of benzimidazolium salts 2
The reaction of 1-(2-morpholinoethyl)-5,6-dimethylbenzimidazole (1 mmol) with various alkyl chloride/alkyl bromide (1.04 mmol) in toluene (10 mL) at 80 °C for 72 hours afford benzimidazole salts. A white solid was obtained after adding diethyl ether (15 mL), which was subsequently filtered off. After washing with diethyl ether (3 × 15 mL) the solid was dried under vacuum.4.3 Synthesis of PEPPSI-type palladium NHC complexes (3a–i)
PEPPSI-type Pd(II)–NHC complexes were synthesized by using a modified protocol published elsewhere.20 In a typical procedure, under a continuous argon flow, a Schlenk tube was charged with PdCl2, (1.05 eq.), K2CO3 (5 eq.), KBr (10 eq.) and pyridine (1 eq.) in the presence of acetonitrile was heated at 80 °C for 16 h. The resulting mixture was stirred for 16 h at 80 °C. Pyridine was then removed by vacuum distillation. A short silica column was used for filtration. Then, the resulting complexes dried under vacuum. The yellow solid was crystallized from dichloromethane/n-pentane for further purification.4.4 General procedure for the arylation reaction
KOAc (2.0 mmol), arylbromide derivatives (1.0 mmol), heteroaryl derivatives 2-acetylfuran, PEPPSI-Pd(II)–N-heterocyclic carbene (NHC) complexes 3a–i (0.01 mmol) were dissolved in N,N-dimethylacetamide (DMAc) (2 mL) in a small Schlenk tube under argon as described in the literature. The reaction mixture was stirred in an oil bath at 150 °C for 2 h. After completion of the reaction, the solvent was evaporated under vacuum and the residue was solved with dichloromethane (2 mL). The chemical characterizations of the products were undertaken with GC. GC conversions and GC yields were calculated with respect to aryl bromide from the results of GC spectrometry with dodecane as internal standard. The GC result showed also the homo-coupling byproduct.4.5 Mercury poisoning test
To a small Schlenk tube filled with an excess of Hg (0.300 g, 1.50 mmol, Hg:Pd = 300:1), add 4-bromoacetophenone (1.0 mmol), furfural (1.3 mmol), KOAc (1.5 mmol), inner standard (50 μL) and 2 mL of DMAc, followed by the addition of 3b, Pd catalyst (0.01 mmol). The reaction mixture was stirred at 150 °C in an oil bath for 2 hours. After completion of the reaction, the solvent was evaporated in vacuo, the residue was dissolved in dichloromethane (2 mL), then filtered through a pad of silica gel and washed well, then concentrated and purified by silica gel flash chromatography. Reaction yields were determined by aryl halide based GC. The yield of the coupled product was 89%.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors extended their appreciation to the Researchers Supporting Project number (RSP2023R75), King Saud University, Riyadh, Saudi Arabia.References
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- C. Wolf and R. Lerebours, J. Org. Chem., 2003, 68, 7077–7084 CrossRef CAS PubMed.
- M. Allegretti, A. Arcadi, F. Marinelli and L. Nicolini, Synlett, 2001, 5, 609–612 CrossRef.
- M. G. Organ, M. Abdel-Hadi, S. Avola, N. Hadei, J. Nasielski, C. J. O'Brien and C. Valente, Chem.–Eur. J., 2006, 13, 150–157 CrossRef PubMed.
- J. M. L'Helgoual’ch, A. Seggio, F. Chevallier, M. Yonehara, E. Jeanneau, M. Uchiyama and F. Mongin, J. Org. Chem., 2008, 73, 177–183 CrossRef PubMed.
- Y. Akita, A. Inoue, K. Yamamoto, A. Ohta, T. Kurihara and M. Shimizu, Heterocycles, 1985, 23, 2327–2333 CrossRef CAS.
- A. Ohta, Y. Akita, T. Ohkuwa, M. Chiba, R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Heterocycles, 1990, 31, 1951–1958 CrossRef CAS.
- Y. Aoyagi, A. Inoue, I. Koizumi, R. Hashimoto, A. Miyafuji, J. Kunoh, R. Honma, Y. Akita and A. Ohta, Heterocycles, 1992, 33, 257–272 CrossRef CAS.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- B. J. Li, S. D. Yang and Z.-J. Shi, Synlett, 2008, 7, 949–957 Search PubMed.
- F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS.
- L. Ackermann, R. Vincente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS.
- X. F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 7316–7319 CrossRef CAS PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS.
- K. Yuan, J.-F. Soule and H. Doucet, ACS Catal., 2015, 5, 978–991 CrossRef CAS.
- L. Zhao, C. Bruneau and H. Doucet, Chem. Commun., 2013, 49, 5598–5600 RSC.
- S. Karthik and T. Gandhi, Org. Lett., 2017, 19, 5486–5489 CrossRef CAS PubMed.
- M. Schnurch, R. Flasik, A. F. Khan, M. Spina, M. D. Mihovilovic and P. Stanetty, Eur. J. Org. Chem., 2006, 15, 3283–3307 CrossRef.
- K. Yuan, J.-F. Souĺe and H. Doucet, ACS Catal., 2015, 5, 978–991 CrossRef CAS.
- K. Masui, H. Ikegami and A. Mori, J. Am. Chem. Soc., 2004, 126, 5074–5075 CrossRef CAS PubMed.
- E. David, S. Pellet-Rostaing and M. Lemaire, Tetrahedron, 2007, 63, 8999–9006 CrossRef CAS.
- M. Nakano, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1851–1854 CrossRef CAS PubMed.
- L. Chen, J. Roger, C. Bruneau, P. H. Dixneuf and H. Doucet, Chem. Commun., 2011, 6, 1872–1874 RSC.
- B. B. Toure, B. S. Lane and D. Sames, Org. Lett., 2006, 8, 1979–1982 CrossRef CAS PubMed.
- S. D. Yang, C. L. Sun, Z. Fang, B. J. Li, Y. Z. Li and Z. J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473–1476 CrossRef CAS PubMed.
- D. T. Gryko, O. Vakuliuk, D. Gryko and B. Koszarna, J. Org. Chem., 2009, 74, 9517–9520 CrossRef CAS PubMed.
- O. Vakuliuk, B. Koszarna and D. T. Gryko, Adv. Synth. Catal., 2011, 353, 925–930 CrossRef CAS.
- M. Wu, J. Luo, F. Xiao, S. Zhang, G.-J. Deng and H.-A. Luo, Adv. Synth. Catal., 2012, 354, 335–340 CrossRef CAS.
- L. Florentino, F. Aznar and C. Valdes, Chem. Eur., 2013, 19, 10506–10510 CrossRef CAS PubMed.
- R. Jin, K. Yuan, E. Chatelain, J.-F. Soule and H. Doucet, Adv. Synth. Catal., 2014, 356, 3831–3841 CrossRef CAS.
- A. Battace, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Orgamometallics, 2007, 26, 472–474 CrossRef CAS.
- R. Matsidik, J. Martin, S. Schmidt, J. Obermayer, F. Lombeck, F. Nübling, H. Komber, D. Fazzi and M. Sommer, J. Org. Chem., 2015, 80, 980–987 CrossRef CAS PubMed.
- P. Li, Z. Chai, G. Zhao and S. Z. Zhu, Tetrahedron, 2009, 65, 1673–1678 CrossRef CAS.
- C. Verrier, P. Lassalas, L. Théveau, G. Quéguiner, F. Trécourt, F. Marsais and C. Hoarau, Recent advances in direct C-H arylation: Methodology, selectivity and mechanism in oxazole series, Beilstein J. Org. Chem., 2011, 7, 1584–1601 CrossRef CAS PubMed.
- J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS.
- N. Nakamura, Y. Tajima and K. Sakai, Heterocycles, 1982, 17, 235–245 CrossRef CAS.
- Y. Akita, A. Inoue, K. Yamamoto, A. Ohta, T. Kurihara and M. Shimizu, Heterocycles, 1985, 23, 2327–2333 CrossRef CAS.
- E. David, S. Pellet Rostaing and E. Lemaire, Tetrahedron, 2007, 63, 8999–9006 CrossRef CAS.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- P. Kapitza, A. Scherfler, S. Salcher, S. Sopper, M. Cziferszky, K. Wurst and R. Gust, J. Med. Chem., 2023, 66(12), 8238–8250 CrossRef CAS PubMed.
- I. Petrov, S. L. Gorelsky and K. Fagnou, J. Org. Chem., 2010, 75, 1047–1060 CrossRef PubMed.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemine Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed.
- S. Díez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS.
- P. Małecki, K. Gajda, O. Ablialimov, M. Malinska, R. Gajda, K. Wozniak, A. Kajetanowicz and K. Grela, Organometallics, 2017, 36, 2153 CrossRef.
- G. Meng and M. Szostak, Org. Lett., 2018, 20, 6789 CrossRef CAS PubMed.
- X. Tian, J. Lin, S. Zou, J. Lv, Q. Huang, J. Zhu, S. Huang and Q. Wang, J. Organomet. Chem., 2018, 861, 125 CrossRef CAS.
- M. O. Karataş, S. Günal, A. Mansur, B. Alıcı and İ. Özdemir, Arch. Pharm., 2020, 353, e2000013 CrossRef PubMed.
- S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589 CrossRef CAS PubMed.
- D. Domyati, R. Latifi and L. Tahsini, J. Organomet. Chem., 2018, 860, 98 CrossRef CAS.
- M. O. Karataş, A. D. Giuseppe, V. Passarelli, B. Alıcı, J. J. Pérez-Torrente, L. A. Oro, İ. Özdemir and R. Castarlenas, Organometallics, 2018, 37, 191 CrossRef.
- M. O. Karataş, J. Organomet. Chem., 2019, 899, 120906 CrossRef.
- L. Boubakri, K. Dridi, A. S. Al-Ayed, İ. Özdemir, S. Yaşar and N. Hamdi, J. Coord. Chem., 2019, 72, 516 CrossRef CAS.
- N. Kaloğlu, İ. Özdemir, N. Gürbüz, H. Arslan and P. H. Dixneuf, Molecules, 2018, 23, 647 CrossRef PubMed.
- İ. Özdemir, S. Demir Düşünceli, N. Kaloğlu, M. Achard and C. Bruneau, J. Organomet. Chem., 2015, 799–800, 311 CrossRef.
- Z. Şahin, N. Gürbüz, İ. Özdemir, O. Şahin, O. Büyükgüngör, M. Achard and C. Bruneau, Organometallics, 2015, 34, 2296 CrossRef.
- N. Şahin, N. Özdemir, N. Gürbüz and İ. Özdemir, Appl. Organomet. Chem., 2019, 33, 4704 CrossRef.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- A. Battace, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Adv. Synth. Catal., 2007, 349, 2507–2516 CrossRef CAS.
- (a) H. W. Wanzlick and H. J. Schönherr, Angew. Chem., Int. Ed., 1968, 7, 141–142 CrossRef CAS; (b) K. Öfele, J. Organomet. Chem., 1968, 12, 42–43 CrossRef.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- W. A. Herrmann, N. W. Huber and O. Runte, Angew. Chem., Int. Ed., 1995, 34, 2187–2206 CrossRef CAS.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS.
- (a) N. Touj, N. Gürbüz, N. Hamdi, S. Yaşar and İ. Özdemir, Inorg. Chim. Acta, 2018, 78, 187–194 CrossRef; (b) L. Boubakri, K. Dridi, A. A. AlAyed, İ. Özdemir, S. Yaşar and N. Hamdi, J. Coord. Chem., 2019, 72, 516–527 CrossRef CAS.
- M. Kaloğlu, İ. Özdemir, V. Dorcet, C. Bruneau and H. Doucet, Eur. J. Inorg. Chem., 2017, 10, 1382–1391 CrossRef.
- I. Slimani, L. Boubakri, N. Özdemir, L. Mansour, I. Özdemir, N. Gürbüz, S. YAŞAR, M. Sauthier and N. Hamdi, Inorg. Chim. Acta, 2022, 532, 120747–120755 CrossRef CAS.
- (a) S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042–15053 CrossRef CAS PubMed; (b) W. de Almeida Bezerra, J. L. Sônego Milani, C. H. de Jesus Franco, F. T. Martins, Â. de Fátima, Á. F. A. da Mata and R. P. das Chagas, Mol. Catal., 2022, 530, 112632 CrossRef.
- (a) C. H. Wang, W. C. Shih, H. C. Chang, Y. Y. Kuo, W. C. Hung, T. G. Ong and W. S. Li, J. Med. Chem., 2011, 54, 5245–5249 CrossRef CAS PubMed; (b) Z. Nawaz, H. Ullah, N. Gürbüz, M. N. Zafar, F. Verpoort, M. N. Tahir, I. Özdemir and R. J. Trovitch, Mol. Catal., 2022, 526, 12369 Search PubMed.
- (a) R. A. Haque, A. W. Salman, S. Budagumpi, A. A. Abdullah and A. M. Majid, Metallomics, 2013, 5, 760–769 CrossRef CAS PubMed; (b) Y. Wu, L. Ma and Z. Song, et al., Carb. Neutrality, 2023, 2, 1, DOI:10.1007/s43979-022-00041-5.
- J. Y. Lee, J. Y. Lee, Y. Y. Chang, C. H. Hu, N. M. Wang and H. M. Lee, Organometallics, 2015, 34, 4359–4368 CrossRef CAS.
- J. Kuwabara, M. Sakai, Q. Zhang and T. Kanbara, Org. Chem. Front., 2015, 2, 520–525 RSC.
- (a) S. Fantasia and S. P. Nolan, Chem.–Eur. J, 2008, 14, 6987–6993 CrossRef CAS PubMed; (b) X.-C. Cai, S. Majumdar, G. C. Fortman, C. S. J. Cazin, A. M. Z. Slawin, C. Lhermitte, R. Prabhakar, M. E. Germain, T. Palluccio, S. P. Nolan, E. V. Rybak-Akimova, M. Temprado, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2011, 133, 1290–1293 CrossRef CAS PubMed.
- (a) M. G. Organ, G. A. Chass, D. C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2008, 2776–2797 CrossRef; (b) A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem.–Eur. J., 2012, 18, 4517–4521 CrossRef CAS PubMed.
- M. Fanelli, M. Formica, V. Fusi, L. Giorgi, M. Micheloni and P. Paoli, Coord. Chem. Rev., 2016, 310, 41–79 CrossRef CAS.
- A. Garoufis, S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 1384–1397 CrossRef CAS.
- G. Ayyannan, M. Mohanraj, M. Gopiraman, R. Uthayamalar, G. Raja, N. Bhuvanesh, R. Nandhakumar and C. Jayabalakrishnan, Inorg. Chim. Acta, 2020, 512, 119868–119875 CrossRef CAS.
- X. Wei, Y. Yang, J. Ge, X. Lin, D. Liu, S. Wang, J. Zhang, G. Zhou and S. Li, J. Inorg. Biochem., 2020, 202, 110857–110863 CrossRef CAS PubMed.
- M. Aminzadeh, M. Saeidifar and H. Mansouri-Torshizi, J. Mol. Struct., 2020, 1215, 128212–128219 CrossRef CAS.
- A. T. Fiori-Duarte, F. R. G. Bergamini, R. E. F. de Paiva, C. M. Manzano, W. R. Lustri and P. P. Corbi, J. Mol. Struct., 2019, 1186, 144–154 CrossRef CAS.
- N. Dheman, N. Mahoney, E. M. Cox, J. J. Farley, T. Amini and M. L. Lanthier, Infect. Dis., 2021, 73, 4444–4450 CrossRef PubMed.
- R. K. Dev, A. Bhattarai, N. K. Chaudhary and P. Mishra, Asian J. Chem., 2020, 32, 1473–1481 CAS.
- E. A. Nyawade, M. O. Onani, S. Meyer and P. Dube, Chem. Pap., 2020, 74, 3705–3715 CrossRef CAS.
- L. Boubakri, L. Mansour, A. H. Harrath, I. Ozdemir, S. Yasar and N. Hamdi, J. Coord. Chem., 2018, 71, 183–199 CrossRef CAS.
- H. Almallah, E. Brenner, D. Matt, J. Harrowfield, M. Jahajh and A. Hijazi, Palladium complexes of N-heterocyclic carbenes displaying an unsymmetrical N-alkylfluorenyl/N'-aryl substitution pattern and their behaviour in Suzuki–Miyaura cross coupling, Dalton Trans., 2019, 48, 14516–14529 RSC.
- N. Muniyappan and S. Sabiah, Synthesis, structure, and characterization of picolyl- and benzyl-linked biphenyl palladium N-heterocyclic carbene complexes and their catalytic activity in acylative cross-coupling reactions, Appl. Organomet. Chem., 2020, 34, e5421 CrossRef CAS.
- S. Guo and H. V. Huynh, Dinuclear triazole-derived, Janus-type N-heterocyclic carbene complexes of palladium: Syntheses, isomerizations, and catalytic studies toward direct C5-arylation of imidazoles, Organometallics, 2014, 33, 2004–2011 CrossRef CAS.
- N. Hamdi, J. Slimani, L. Mansour, F. Alresheedi, N. Gürbüz and I. Özdemir, N-heterocyclic carbene-palladium complexes and their catalytic activity in the direct C-H bond activation of heteroarene derivatives with aryl bromides: synthesis, and antimicrobial and antioxidant activities, New J. Chem., 2021, 45, 21248–21262 RSC.
- (a) C. F. McKenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems, IUCrJ, 2017, 4, 575–587 CrossRef PubMed; (b) P. R. Spackman; M. J. Turner; J. J. McKinnon; S. K. Wolff; D. J. Grimwood; D. Jayatilaka and M. A. Spackman, Crystal Explorer 21.5, University of Western Australia, Perth, Australia, 2021 Search PubMed.
- T. Ryhl, Thermodynamic properties of palladium(II) chloride and bromide complexes in aqueous solution, Acta Chem. Scand., 1972, 26, 2961–2962 CrossRef CAS.
- L. Boubakri, S. Yasar, V. Dorcet, T. Roisnel, C. Bruneau, N. Hamdi and I. Ozdemir, J. Chem., 2017, 41, 5105–5113 CAS.
- K. Yuan, J.-F. Soule and H. Doucet, ACS Catal., 2015, 5, 978–991 CrossRef CAS.
- E. Assen, B. Kantch and M. G. Organ, Angew. Chem., 2007, 46, 2768–2813 CrossRef PubMed.
- N. Touj, S. Yaşar, N. Özdemir and N. Hamdi, İ. Özdemir. J. Organomet. Chem., 2018, 860, 59–71 CrossRef CAS.
- (a) S. I. Gorelsky, D. Lapointe and K. Fagnou, Analysis of the palladium-catalyzed (aromatic) C–H bond metalation–deprotonation mechanism spanning the entire spectrum of arenes, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS PubMed; (b) S. I. Gorelsky, Tuning the regioselectivity of palladium-catalyzed direct arylation of azoles by metal coordination, Organometallics, 2012, 31, 794–797 CrossRef CAS.
- (a) M. Kaloğlu, N. Gürbüz, İ. Yıldırım, N. Özdemir and İ. Özdemir, Well-defined PEPPSI-themed palladium–NHC complexes: synthesis, and catalytic application in the direct arylation of heteroarenes, Appl. Organomet. Chem., 2020, 34, 5387–5394 CrossRef; (b) J. Roger, A. L. Gottumukkala and H. Doucet, Palladium-Catalyzed C3 or C4 Direct Arylation of Heteroaromatic Compounds with Aryl Halides by CH Bond Activation, ChemCatChem, 2010, 2, 20–40 CrossRef CAS; (c) S. Yanagisawa, K. Ueda, H. Sekizawa and K. Itami, Programmed synthesis of tetraarylthiophenes through sequential C−H arylation, J. Am. Chem. Soc., 2009, 131, 14622–14623 CrossRef CAS PubMed.
- (a) R. Giri, N. Maugel, J. Li, D. Wang, S. P. Breazzano, L. B. Saunders and J. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (b) L. Boubakri, A. Chakchouk-Mtibaa, A. Sulaiman Al-Ayed, L. Mansour, N. Abutaha, A. H. Harrath, L. Mellouli, I. Özdemir, S. Yasar and N. Hamdi, RSC Adv., 2019, 9, 34406–34420 RSC.
- (a) S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220–14221 CrossRef CAS PubMed; (b) F. Kakiuchi, M. Usui, S. Ueno, N. Chatani and S. Murai, J. Am. Chem. Soc., 2004, 126, 2706–2707 CrossRef CAS PubMed; (c) F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698–1699 CrossRef CAS PubMed.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed.
- C. Adamo, C. Amatore, I. Ciofini, A. Jutand and H. Lakmini, J. Am. Chem. Soc., 2006, 128, 6829–6836 CrossRef CAS PubMed.
- A. Ohta, Y. Akita, T. Ohkuwa, M. Chiba, R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Heterocycles, 1990, 31, 1951–1958 CrossRef CAS.
- E. David, S. PelletRostaing and E. Lemaire, Tetrahedron, 2007, 63, 8999–9006 CrossRef CAS.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS PubMed.
- C. B. Bheeter, L. Chen, J. F. Soulé and H. Doucet, Catal. Sci. Technol., 2016, 6, 2005–2049 RSC.
- C. R. Shahini, G. Achar, S. Budagumpi, M. Tacke and S. A. Patil, Appl. Organomet. Chem., 2017, 31, 3819–e3819 CrossRef.
- N. Sauermann, J. Loup, D. Kootz, V. R. Yatham, A. Berkessel and L. Ackermann, Synthesis, 2017, 49(49), 3476–3484 CAS.
- K. Xu, W. Li, R. Sun, L. Luo, X. Chen, C. Zhang, X. Zheng, M. Yuan, H. Fu, R. Li and H. Chen, Org. Lett., 2020, 22, 6107–6111 CrossRef CAS PubMed.
- J. Roger and H. Doucet, Adv. Synth. Catal., 2009, 351, 1977–1990 CrossRef CAS.
- M. C. R. Castro, M. Belsley and M. M. M. Raposo, Dyes Pigm., 2016, 131, 333–339 CrossRef CAS.
- E. David, C. Rangheard, S. PelletRostaing and M. Lemaire, Synlett, 2006, 13, 2016–2020 Search PubMed.
- (a) B. Glover, K. A. Harvey, B. Liu, M. J. Sharp and M. F. Tymoschenko, Org. Lett., 2003, 5, 301 CrossRef CAS PubMed; (b) L. N. Lewis, Chem. Rev., 1993, 93, 2693–2730 CrossRef CAS.
- Y. Lin and R. G. Finke, Nanocluster Catal., 1994, 33, 4891–4910 CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- A. Del Zotto and D. Zuccaccia, Catal. Sci. Technol., 2017, 7, 3934–3951 RSC.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- J. G. A de Vries, Dalton Trans., 2006, 421–429 RSC.
- L. A. Perego, L. Grimaud and F. Bellina, Adv. Synth. Catal., 2016, 358, 597–609 CrossRef CAS.
- S. Hübner, J. G. De Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- R. Cano, A. F. Schmidt and G. P. McGlacken, Chem. Sci., 2015, 6, 5338–5346 RSC.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. M. Hii, Catal. Today, 2014, 229, 95–103 CrossRef CAS.
- S. Chikhi, S. Djebbar, J. F. Soule and H. Doucet, Chem.–Asian J., 2016, 11, 2443–2452 CrossRef CAS PubMed.
- S. J. Meek, C. L. Pitman and A. J. M. Miller, J. Chem. Educ., 2016, 93, 275–286 CrossRef CAS.
- S. Karthik and T. Gandhi, Org. Lett., 2017, 19, 5486–5489 CrossRef CAS PubMed.
- R. N. Pease, J. Am. Chem. Soc., 1923, 45, 2296–2305 CrossRef CAS.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 14, 1559–1563 RSC.
- J. G. de Vries, Dalton Trans., 2006, 412, 421–429 RSC.
- M. M. Dell'Annaa, M. Malia, P. Mastrorilli, A. Rizzutia, C. Ponzonic and C. Leonellic, J. Mol. Catal. A: Chem., 2013, 366, 186–194 CrossRef.
- (a) A. Battace, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Organometallics, 2007, 26, 472–474 CrossRef CAS; (b) F. Pozgan, J. Roger and H. Doucet, ChemSusChem, 2008, 1, 404–407 CrossRef CAS PubMed.
- (a) J. Roger, F. Pozgan and H. Doucet, Green Chem., 2009, 11, 425–432 RSC; (b) M. Kaloğlu, İ. Özdemir, V. Dorcet, C. Bruneau and H. Doucet, Eur. J. Inorg. Chem., 2017, 10, 1382–1391 CrossRef.
- (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed; (b) Y. F. Yang, G. J. Cheng, P. Liu, D. Leow, T. Y. Sun, P. Chen, X. Zhang, J. Q. Yu, Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136(1), 344–355 CrossRef CAS PubMed.
- A. Wu, Q. Chen, W. Liu, L. You, Y. Fu and H. Zhang, Org. Chem. Front., 2018, 5, 1811–1814 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2294603. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06334h |
| This journal is © The Royal Society of Chemistry 2023 |