
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent oxide microspheres with designed macroporosity (MICROSCAFS®): a customized platform for chemicals immobilization†
Mário Vale a,
Sofia Oriškováa,
António Mariquitoa,
Luís Reisb,
Moisés Pintoa and
Ana C. Marques*a
a,
Sofia Oriškováa,
António Mariquitoa,
Luís Reisb,
Moisés Pintoa and
Ana C. Marques*a
aCERENA, DEQ, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisboa, Portugal. E-mail: ana.marques@tecnico.ulisboa.pt
bIDMEC, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisboa, Portugal
First published on 25th April 2023
Abstract
A novel versatile, easily recoverable, and recyclable material platform is herein presented, consisting of multicomponent oxide microspheres, of silica–titania and silica–titania–hafnia composition, with tailored interconnected macroporosity (MICROSCAFS®). When functionalized or loaded with desired species, they are potential enablers of emerging applications in environmental remediation, among other fields. We combine emulsion templating for the spherical shape of the particles, with an adapted sol–gel technique involving polymerization-induced phase separation by spinodal decomposition. An advantage of our method regards the employed mix of precursors, which prevents the use of specific gelation additives and porogens and allows a high reproducibility of MICROSCAFS®. We present insight into their formation mechanism using cryo-scanning electron microscopy, and a systematic study of the effect of multiple synthesis parameters on the MICROSCAFS® size and porosity. The composition of the silicon precursors has the most significant effect on fine-tuning the pores size, ranging from the nanometer to the micron scale. Mechanical properties are correlated with morphological features. Larger macroporosity (68% open porosity, estimated by X-ray computed tomography) leads to less stiffness, higher elastic recovery, and compressibility values up to 42%. We believe this study creates a base for consistent custom MICROSCAFS® production, with a design for various future applications.
Introduction
MICROSCAFS®,1 a new class of support materials, are porous microspheres with controlled size, porosity and morphology (including pore interconnectivity), of hybrid or inorganic chemical nature, which display potential applications as support materials for chromatography, environmental remediation,2 photocatalysis2 and complex catalysis processes, such as those in polymers production, separation, drug delivery systems, energy storage and conversion and biomedical devices. They are prepared, without porogen agents, via an adapted sol–gel process involving polymerization-induced phase separation by spinodal decomposition to achieve the desired interconnected macroporosity, combined with emulsification techniques, which play the role of soft templating for achieving the spherical microparticles.3,4 Their tailoring is accomplished by modifying the synthesis parameters, as we already reported for the case of 100% silica (SiO2) porous microspheres.3The first reports on porosity control by polymerization-induced phase separation regard sol–gel derived SiO2 materials (monoliths),5–9 in the early 1990s. This was accomplished by using a phase separation inducer additive and porogen agent, and the effect of precursor and solvent composition,6 as well as the amount and molecular weight of the polymeric phase separation inducer additive,7 temperature,7 catalytic conditions,8 and organic solvents.9 Comprehensive studies on porous monoliths of varied compositions, achieved by this strategy, have been reported since then, but literature on equally porous microspheres is very scarce.4 As far as we are concerned, we are the first to report on multicomponent oxide silica–titania (ST) and silica–titania–hafnia (STH) microspheres with interconnected macroporosity. The biggest challenge in these systems is that the reactivities of the oxide precursors differ, leading to precipitation, material heterogeneity, and reproducibility issues. To overcome this disadvantage, we must strategically introduce chelating agents, fix the temperature and carefully control the reaction medium pH, and adequately combine the precursors' type and quantity. Such methodology has allowed us to develop reproducible multicomponent oxide MICROSCAFS® which are reported in the present paper.
A few studies have been published on tailored porous spheres by sol–gel synthesis combined with an emulsion process,3,10,11 dispersion process,12 self-assembly technology,13 nanocasting,14,15 or by using polystyrene beads as hard templates.16,17 The first attempt to synthesize silica microspheres with interconnected meso and macroporosity, following sol–gel synthesis and emulsification, was reported by Yang et al., where polyethylene oxide (PEO) and tri-block poly(ethylene oxide) poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO) Pluronic® F-127 copolymer were added to a pre-hydrolyzed silica sol.11 Colloid aggregation mechanism, in alkaline medium, was then promoted inside the water droplets of a water-in-oil (W/O) emulsion, forming the silica spheres. PEO was employed to accelerate gelation and act as auxiliary template to produce macropores, whereas Pluronic® F-127 copolymer acted as a surfactant to produce mesopores. Posteriorly, Shi et al. added PEO to the water phase of a W/O emulsion,10 which contained a pre-hydrolyzed silica sol (acidic catalyst). In this case, the gelation process, and the phase separation, were promoted by heating the emulsion at 60 °C for 20 h under magnetic stirring. For both cases, the solvent-rich phase and PEO had to be burnt off, to form the macropores, but in the latter case, treatment with an aqueous solution of NH4OH was necessary to provide mesoporosity within the spheres' skeleton through a dissolution–reprecipitation process.
Over the following years, similar silica microspheres have been obtained through the above-described emulsion method and applied to chromatography,10,18–23 drug delivery,24,25 adsorption,26 and the production of composites.27 Despite the successful development of macroporous spheres by combining acid or alkaline catalysts with porogen and phase separation agents, our group created a more straightforward approach, dispensing the need for such additives.2–4,28 Instead, we selected a specific silane combination (tetraethoxysilane, TEOS, and (3-glycidyloxypropyl) trimethoxysilane, GPTMS) to provide an inherent gelation/phase separation capability. Our method involves shorter production times and lower temperatures, since there is no need to calcination to get rid of polymeric additives to achieve the porous structure. A simple drying of the MICROSCAFS® is enough to eliminate the solvent remaining within the 3D interconnected co-continuous porous structure.
We have developed silica MICROSCAFS® with antifouling capability by contact.28 In this case, the amino groups of the biocide (ECONEA®) reacted with the epoxy groups remaining at the MICROSCAFS®. Posteriorly, we studied the effect of different synthesis parameters (hydrolysis time, hydrolysis catalyst, and surfactant quantities) on the size and pore size distribution of silica MICROSCAFS®,3 and the first experiments were conducted with these particles in a photocatalytic reactor by immobilizing TiO2 nanoparticles on their outer and inner surface. The supported photocatalyst degraded organic pollutants, such as methyl orange and paracetamol, in liquid media, through photocatalytic reactions driven by solar light.2 Also, the support material was reported to prevent nanoparticle aggregation and, most importantly, lead to a straightforward recovery of the photocatalytic nanoparticles. Its characteristic macroporosity favors the light harvesting and increases the capacity for mass transport, also very useful for efficient photocatalysis processes.
Aiming at further exploring the synergy between support materials (MICROSCAFS®) and TiO2 nanoparticles for photocatalytic applications, multicomponent oxide composition MICROSCAFS®, namely SiO2–TiO2 (ST) and SiO2–TiO2–HfO2 (STH) were developed and reported for the first time in the present paper. The present paper elucidates on the MICROSCAFS® formation mechanism and reports the effect of changing varied synthesis parameters, namely synthesis temperature, GPTMS/TEOS molar ratio, Ti precursor/chelating agent molar ratio and emulsion W/O mass ratio on the architecture, pore size and morphology, of the MICROSCAFS®, including a critical discussion around phase separation thermodynamics and sol–gel kinetics phenomena. Tuning the synthesis parameters and mastering this process allows to successfully control MICROSCAFS® morphology and final properties, to customize for specific applications, keeping up with the most recent technological trends.
Experimental
Synthesis of the ST and STH MICROSCAFS®
The samples reported in this paper are named using the following acronyms: composition of the MICROSCAFS® (ST or STH)/synthesis temperature/parameter change (AA+ = greater quantity of acetic acid, W+ = greater quantity of water in the W/O emulsion, GPTMS+ or − = higher or lower GPTMS/TEOS ratio). Table 1 lists the main samples prepared for this study, and corresponding synthesis parameters.| Sample acronym | Reaction temperature (°C) | GPTMS/TEOS molar ratio | TiPOT/CH3COOH molar ratio | W/O mass ratio | Volume of NH4OH 25% solution (ml) | Mass of HfOCl2·8H2O (g) |
|---|---|---|---|---|---|---|
| ST/85C | 85 | 0.87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
1:4 |
0.45:1 |
16.5 | 0 |
| ST/50C | 50 | 0.87:1 |
1:4 |
0.45:1 |
16.5 | 0 |
| ST/RT | RT (25) | 0.87:1 |
1:4 |
0.45:1 |
22.0 | 0 |
| ST/50C/AA+ | 50 | 0.87:1 |
1:6 |
0.45:1 |
16.5 | 0 |
| ST/50C/W+ | 50 | 0.87:1 |
1:4 |
0.68:1 |
16.5 | 0 |
| ST/50C/GPTMS+ | 50 | 1.09:1 |
1:4 |
0.45:1 |
16.5 | 0 |
| ST/50C/GPTMS− | 50 | 0.65:1 |
1:4 |
0.45:1 |
16.5 | 0 |
| STH/50C/GPTMS+ | 50 | 1.09:1 |
1:4 |
0.45:1 |
16.5 | 7.5 |
A typical ST and STH MICROSCAFS® synthesis involves the preparation of three solutions: 1 – hydrolyzate solution: silane or silane/hafnium precursor aqueous solution (sol); 2 – chelated solution: titanium(IV) isopropoxide (TiPOT) and acetic acid solution, and 3 – W/O emulsion.
For the reference synthesis ST/50C, the hydrolyzate solution (solution 1) was prepared as follows: 16.3 ml of TEOS (98%, Sigma-Aldrich), 13.8 ml of GPTMS (Xiameter OFS-6040, >98.5%, kindly supplied by Dow), and 11.6 ml of 0.28 M HCl solution were mixed at room temperature (RT) in a closed container, under magnetic stirring for 65 minutes. In the case of the STH/50C/GPTMS+ synthesis, an additional 7.49 g of hafnium dichloride oxide octahydrate (HfOCl2.8H2O, >98%, Alfa Aesar) and a total of 17.3 ml of GPTMS was added in this step. At the end of the hydrolysis, the pH of the hydrolyzate was measured using a Metria bench top meter model M21 with a Metria glass body dual pH electrode with an ATC probe.
In parallel, a complexation reaction of the TiPOT (solution 2) was carried out to decrease its reactivity and avoid the formation of titanium dioxide precipitates when in contact with water. 10 ml of TiPOT (98%, Acros Organics) and 8 ml of glacial acetic acid (≥ 99%, Fisher Chemical) were mixed separately and left under stirring at RT for 45 minutes.
Finally, for the W/O emulsion (solution 3), 100 g of decahydronaphthalene (decalin, mixture of cis and trans isomers, 98%, Merck), 45 g of distilled water, and 6 g of sorbitan monooleate surfactant (Span® 80, HLB: 4.3, Merck) were mixed at 13000 rpm for 10 minutes using a dispersing instrument (IKAT18 digital ULTRA-TURRAX®). Different concentrations of Span® 80, as well as a combination of Span® 80 with other surfactants, namely Tween® 80 and Pluronic® P-123, were also tested. Then we transferred the emulsion into the reactor and set the mechanical stirring speed to 600 rpm and the temperature to 50 °C. The reactor setup consisted of a three-neck round bottom flask reactor, a heating mantle with a thermocouple, and a mechanical stirrer.
Subsequently, the chelated TiPOT solution (solution 2) was added to the hydrolyzate solution (solution 1) drop-by-drop and stirred for 5 minutes. Then we immediately transferred it into the reactor, followed by the addition of 16.5 ml of aqueous ammonia solution (25%, Chem-Lab).
After 1 hour and 30 minutes, the MICROSCAFS® were collected by vacuum-assisted filtration, washed with acetone, and dried overnight at 45 °C.
Characterization
Cryo-scanning electron microscopy. The cryo-scanning electron microscopy (cryo-SEM) photos and energy-dispersive X-ray spectroscopy (EDS) data were obtained using a high-resolution scanning electron microscope with X-ray Microanalysis and Cryo-SEM experimental facilities: JSM 6301F microscope (JEOL, Ltd., Akishima, Tokyo, Japan)/INCA Energy 350 EDS spectrometer (Oxford Instruments plc, Abington, Oxfordshire, UK)/ALTO 2500 cryo transfer system (Gantan, Pleasanton, CA, USA).
Aliquots collected at different stages of the MICROSCAFS® synthesis were rapidly cooled (plunging them into sub-cooled nitrogen–slush nitrogen) and transferred under vacuum to the cold stage of the preparation chamber. They were then fractured and coated with Au/Pd by sputtering for 45 seconds before being transferred into the SEM chamber and studied at a temperature of −150 °C.
Particle size analysis
The particle size volume-weighted distribution data were obtained using a Mastersizer Hydro 2000S (Malvern Panalytical, Malvern, UK) particle size analyzer from the average of three measurements. Three different percentiles at 10%, 50%, and 90%, named D(0.1), D(0.5), and D(0.9), respectively, were analyzed to study the MICROSCAFS®' size distribution. Also, the relative span of the distribution was used to express the size dispersion, and it represents the curve's full width at half maximum (FWHM):
 | (1) |
Pore size analysis
Mercury intrusion porosimetry (MIP) was performed to quantify the pore size distribution, and cumulative pore volume, for a pore diameter interval from 0.01 to 10 μm using an Autopore IV 9500 (Micromeritics, Norcross, GA, USA). This procedure was employed to account for the interparticle spaces. Indeed, the porosity data achieved above 10 μm was not considered in this analysis, which does not mean that there are no interconnected macropores with a width or length above 10 μm. It should be stressed that these larger pores might not be considered in this work, leading to an underestimation of the macroporosity value. A constant contact angle of 140° was selected in the analysis. The samples were subjected to 120 °C for two hours before measurements.The specific surface areas and pore volumes of the microspheres were obtained by nitrogen (N2) adsorption/desorption BET method at 77 K using an ASAP 2010 adsorption analyzer (Micromeritics, Norcross, GA, USA). The pore size distributions were obtained through density functional theory (DFT), assuming slit-shaped pores. For these experiments, ∼300 mg of sample was sequentially outgassed at 200 °C for at least four hours, with a high vacuum pump, to remove moisture and other surface contaminants before the analyses.
Rheological characterization
The hydrolyzate apparent viscosity was determined using a rotary viscometer LV DV-II+Pro (Brookfield Engineering, Middleboro, MA, USA) using a CPE-52 spindle at 60 rpm and RT.The oscillatory time sweeps of the complex viscosity were measured using an MCR 92 modular compact rheometer (Anton Paar GmbH, Graz, Austria) with a CP50-1 cone-and-plate spindle with 50 mm diameter and 1° cone angle at a constant 0.101 mm gap. Approximately 1 ml of ST precursor sol was collected 5 min after the addition of chelated TiPOT solution (solution 2) to the hydrolyzate solution (Si precursors, solution 1). The setup was previously heated to the desired temperature (50 or 85 °C) before the start of the test. The complex viscosity was monitored under constant 1% shear strain and 2 Hz frequency over 30 minutes. These parameters were chosen to maintain the sol–gel samples in the viscoelastic regime.29 Rotational tests were also performed to obtain the viscosity curves and compare different emulsions in terms of viscosity evolution along a shear rate ranging from 1 to 100 s−1. Flow tests were also performed using the same system along a shear rate ranging from 1 to 100 s−1. The flow curves analysis allowed a better understanding of different emulsions used as templates for the spherical particles.
Fourier transform infrared (FTIR) spectroscopy
FTIR spectroscopy was used to assess the chemical structure of the MICROSCAFS® dried at 45 °C and heat treated for 1 hour at 150 and 700 °C. The used FTIR equipment was a Spectrum Two from PerkinElmer (Waltham, Massachusetts, USA) equipped with a UATR Two accessory (Attenuated Total Reflectance, ATR). The spectra were obtained at 4 cm−1 resolution, and eight scans of data accumulation.Thermogravimetric analysis (TGA)
Thermograms from the reference sample ST/50C dried at 45 °C and heat-treated for 1 hour at 700 °C were obtained to assess the water and organic content of the MICROSCAFS®. The equipment used was a HITACHI STA 7200 Thermal Analysis System (Ibaraki, Japan) under a controlled nitrogen atmosphere with a flow of 200 ml min−1, at a temperature increase rate of 10 °C min−1, in the range of 45–900 °C.X-ray computed tomography
X-ray computed tomography (CT) on the MICROSCAFS® ST/50C/GPTMS+ and ST/50C/GPTMS− was performed using a Skyscan 2214 (Bruker, Belgium) equipped with a flat panel and a CCD detector. Samples were placed inside a cylindrical plastic container without pressing, for analysis. For the acquisition of transmission images with the flat panel, the X-ray source was operated with a medium spot size at 50 kV and 120 μA of voltage and current, respectively, and the detector resolution was 1536 by 972 (2 × 2 pixel binning) at 7.15 μm pixel size, and a 180° sample rotation with a step of 0.2°. For the acquisition of transmission images with the CCD, the X-ray source was operated with a medium spot size at 90 kV and 89 μA of voltage and current, respectively. The detector resolution was 4032 by 2688 at 0.7 μm pixel size and a 360° sample rotation with a step of 0.15°. Reconstruction of the tomography images was carried out with the NRecon software (version 2.1.0.0, Bruker), based on the Feldkamp algorithm. The morphological (3D) analysis of the reconstructed images was done with the CTAn software (version 1.18.8.0, Bruker). A region of interest representative of the sample was selected and, the images were binarized. The objects of interest were represented in white color. The quantitative analyses of the sample were then performed through the 3D plug-in analysis. CTVox software (version 3.3.0, Bruker) was used for volume rendering.MICROSCAFS® mechanical properties analysis
MICROSCAFS®' powder compressibility, relaxation, stiffness, and elastic recovery were calculated using an Instron 5566 universal testing machine with a 500 N load cell (Instron, Norwood, MA, USA) and a designed compaction probe accessory, following a test method developed by Stable Micro Systems (application study ref. POW4/IPC0.5), in a compression force measurement mode, and powder relaxation sequence, originally developed to compare the compaction and stress relaxation properties of different powders. The test method consists of three phases: 1 – loading phase (compression), 2 – hold period (the stress relaxation phase), and 3 – unloading phase (decompression). 1 ml of powder was placed inside a cylindrical aluminum sample holder of 15.7 mm in height and 15.0 mm in diameter. In the loading phase, a crosshead was displaced at a constant rate of 0.5 mm s−1 and compressed the sample until it reached 50 N. Then, the crosshead stopped, and the strain (or probe displacement) was kept constant for over 120 seconds. Finally, the crosshead was unloaded at a constant rate of 0.25 mm s−1 until it reached the point where the displacement was zero. Every compression test was made in triplicate, and the parameters were calculated by computing graphical data using the following formulas:
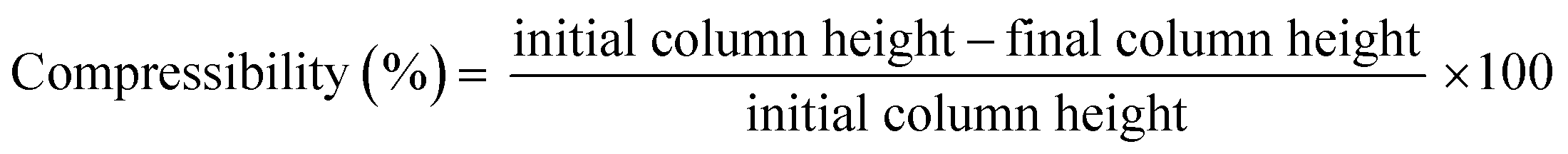 | (2) |
 | (3) |
 | (4) |
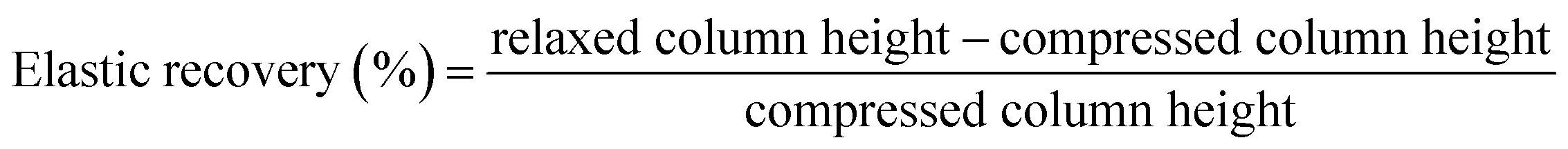 | (5) |
Results and discussions
MICROSCAFS® formation mechanism and other features
The scheme below (Fig. 1) shows the different main stages of the MICROSCAFS® formation process, with respective optical microscopy images of the reaction medium. In our methodology, a chelating agent (CH3COOH) is used to control the reactivity of TiPOT, while Si alkoxides, TEOS and GPTMS, are hydrolyzed before being mixed with chelated TiPOT (for ST MICROSCAFS®). Hf precursor in the form of salt may be added to the Si alkoxides solution at the beginning of the hydrolysis step (for STH MICROSCAFS®). Such mixture is added to a W/O emulsion, a soft template for the characteristic spherical shape of the MICROSCAFS®. The presence of Ti and Hf precursors increases the sol viscosity compared to 100% Si alkoxides. A proper combination of TEOS and GPTMS is selected to allow phase separation to occur via siloxane and Si–O–Ti formation by polycondensation of the silanol groups, and Ti–OH groups, but also from the epoxy ring opening reaction, being both an entropy and enthalpy-driven process. This phenomenon occurs inside the water droplets of the emulsion, resulting in droplets, containing an aqueous dispersion of polymerized species, dispersed in the oil phase. We believe that the relatively long alkyl chain and the oxirane group of GPTMS play an important role in the control of gelation and, consequently, the size and macroporosity of the MICROSCAFS®. However, ammonia can be added at this stage to increase the pH of the reaction medium and further catalyze the condensation reactions, favoring the chemical quenching, i.e. the freezing of the structure, at the moment spinodal decomposition occurs. Our approach eliminates the need for calcination to get rid of the polymeric additives typically employed to achieve macroporous materials. A simple drying of the spheres is enough to eliminate the solvent remaining within the wet gel MICROSCAFS®, resulting in a 3D interconnected co-continuous porous structure. In case purely inorganic MICROSCAFS® are desired, a heat treatment at temperatures above 500 °C shall be done. | ||
| Fig. 1 General scheme of ST MICROSCAFS® preparation, exhibiting the different main stages and associated optical microscopy images (scale bar = 400 μm). | ||
Synthesis temperatures as low as RT (25 °C), 50 °C, or 85 °C were used with success to obtain the wet gel MICROSCAFS®, followed by drying at 45 °C to get hybrid epoxy-ST MICROSCAFS®, and by further heat treatment at ca. 700 °C to achieve inorganic ST MICROSCAFS®. This is confirmed by the ATR-FTIR spectrum (ESI Fig. S1†) of a sample heat treated for one hour at 700 °C, typical of inorganic SiO2–TiO2 material, which reveals significant changes when compared to those from samples only dried or heat treated at 150 °C, namely: the OH stretching band (ca. 3400 cm−1, zone i) is drastically reduced, almost eliminated, as well those typical of phenol, from Span® 80, namely OH bending, 1410 cm−1, and C–O stretching at 1200 cm−1;30 the peaks at 2932 and 2872 cm−1 (zone i) due to C–H stretching (in CH2 units) and at 1440 cm−1 (zone ii) due to C–H bending disappear, suggesting the complete elimination of decalin and Span® 80 residues from the MICROSCAFS®, as well as of other organic residues, eventually from non-reacted alkoxides; also, the peak at 1735 cm−1, due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching from Span® 80, disappears; and, finally, contributions from the GPTMS oxirane ring, i.e. peaks assigned to C–O– stretching at ca. 1250, 915 and 890–800 cm−1 are not evident anymore (zone iii).
O stretching from Span® 80, disappears; and, finally, contributions from the GPTMS oxirane ring, i.e. peaks assigned to C–O– stretching at ca. 1250, 915 and 890–800 cm−1 are not evident anymore (zone iii).
The IR spectra of the MICROSCAFS® present a dominant band at about 1060 cm−1 (zone iii) due to the asymmetric stretching of the Si–O–Si bonding sequences (transverse optical, TO, component) and a weaker peak on its low-frequency side, at ∼940 cm−1, due to the overlap of Si–O− stretching vibrations in Si–O–Ti and a few Si–OH bonding environments.31 The Si–O–Ti stretching peak is characteristic of homogeneous gel/glassy network regions (achieved through heterocondensation), whereas the 1060 cm−1 peak is indicative of silica-rich regions in the network,32 achieved due to homocondensation. The inorganic MICROSCAFS® (heat-treated at 700 °C) exhibit robust scaffolding properties, including thermal and chemical stability. No significant weight loss is observed by TGA up to 900 °C, while the same MICROSCAFS® only dried at 45 °C displayed some weight loss at ca. 100 °C and 400 °C, due to water evaporation and organics elimination, respectively (ESI Fig. S2†).
Fig. 2a and b shows cryo-SEM images (backscattered electron detector mode) of MICROSCAFS® particles (cross-section of a purposedly broken particle) in an embryonic stage of development. They are located within the oil phase, given by the very large atomic (at)% of carbon (zone i, 87 at% C, 13 at% O), typical of the organic solvent decalin used for the oil phase. A thin layer that wraps the particle (at the water–oil interface) is already clearly formed (zone ii), while phase separation between Si and Ti oligomeric species and the water phase is developing, and the skeleton domains (zone iii) are growing inside the confined space of the emulsion droplet. Therefore, the mechanism of formation of the MICROSCAFS® is suggested to consist of the following different stages: 1 – migration of the hydrolyzate clusters to the water phase droplets of the emulsion; 2 – accumulation of the oligomers at the water–oil interface, either by kinetically trapping or migration, and formation of a thin layer; 3 – formation of Si and Ti-rich skeleton domains during early stages of phase separation; 4 – concomitant phase separation and gelation, with the formation of the MICROSCAFS® with interconnected macroporosity.
 | ||
| Fig. 2 Cryo-SEM images of ST/50C MICROSCAFS® located within the emulsion's oil phase (bar scale = 60 μm) (a, b) particle in an early development stage, one minute after the addition of the ammonia catalyst (backscattered electron detector mode) and (c) particle in a later development stage, wet gel MICROSCAFS® particle (secondary electron detector mode). | ||
The secondary electron detector micrograph (Fig. 2c) shows the topography of a MICROSCAFS® particle in a later stage of development.
Mesoporosity arises mainly from the oligomeric clusters that are bound together, forming the skeleton domains, while macroporosity is due to the phase separation process by spinodal decomposition. The thin layer that typically wraps the MICROSCAFS® is so thin (just some nanometers thickness) and porous that it will not compromise liquid flow inwards the MICROSCAFS® or species immobilization or grafting onto the inner pore surface of the MICROSCAFS®, according to previous experiments.2 However, by treating the wet particles right after the synthesis with a 0.06 M LiOH (0.01 g MICROSCAFS® per ml) at 50 °C, such thin outer layer dissolves, exposing the skeleton domains, wormlike structure typical of the MICROSCAFS®, as shown in ESI Fig. S3.†
ST MICROSCAFS® synthesis was revealed to be highly reproducible, despite the required control of the synthesis parameters to induce a simultaneous phase separation and gelation of the structure. Evidence for such reproducibility is shown in ESI Fig. S4† for the case of samples ST/85C and ST/50C/GPTMS+.
In general, greater quantities of surfactant Span® 80, in a range from 2 to 10 g, led to smaller particle diameters, narrower size distribution, and slightly smaller characteristic size (ESI Fig. S5†). In general, the same type of wormlike macropore structure is present in all samples, suggesting that the temporal space between the “freezing” of the structure (gel time, tg) and the onset of phase separation (tps) is very similar and, therefore, independent of the quantity of surfactant.
Different combinations of surfactants, Span® 80, Tween® 80, and Pluronic® P123, were also tested, keeping the mass of total surfactants fixed at 10 g, resulting in a drastic decrease of the MICROSCAFS® size, as shown in Fig. 3, revealing the impact that different surfactants and different values of HLB (4.3 versus 5.2) have on the W/O emulsion droplets' size. An HLB ≤ 6 is preferred for W/O emulsions. Indeed, when testing a further increase of the Tween® 80 amount to attain an HLB of 12, the obtained particles were not regular spheres anymore but an agglomerated thin powder, lacking the special features typical of the MICROSCAFS®.
 | ||
| Fig. 3 Optical microscopy images of the MICROSCAFS® ST/85C synthesized using different combinations of surfactants (from left to right): Span® 80 (HLB = 4.3), Span® 80+ Tween® 80 (HLB = 5.2) and Span® 80+ Pluronic® P123 (HLB = 5.2). Scale bar: 500 μm. | ||
It should be stressed that the quantity of surfactant Span® 80 was fixed at 6 g, and no combinations with other surfactants were carried out for the remaining studies in this work. This condition was chosen because preliminary tests with syntheses made at RT and 50 °C revealed that 2 g Span® 80 were not enough to attain a stable emulsion and regular size MICROSCAFS®, while those obtained with 6 and 10 g Span® 80 displayed already very similar features, such as homogeneous, regular spherical shape and size (ESI Fig. S6†).
Effect of synthesis temperature on the ST MICROSCAFS® morphology
Temperature plays a crucial role in phase separation thermodynamics and sol–gel condensation kinetics. On the one hand, it favors the solubility of the different phases and promotes homogenization in an upper critical solution temperature (UCST) type of sol–gel system.6 On the other hand, it accelerates gelation (silane condensation reactions).33,34The MICROSCAFS® size (diameter) distribution graphs (Fig. 4) shows a quite similar value for the mode of the three ST MICROSCAFS® samples prepared at RT (25 °C), 50 °C, and 85 °C, which reveals that temperatures within this range do not significantly affect the emulsion stability. However, sample ST/50C exhibits a few particles of larger size, shown by the increase in span and D (0.9) values (ESI Table S7†). Despite the similarity in MICROSCAFS® size, there is a significant change in pore morphology for different synthesis temperatures, as revealed by the SEM images (Fig. 4). MIP graphs show a strong increase in the pore size for higher reaction temperatures, as well as a larger cumulative intrusion pore volume (Fig. 4). For instance, the sample synthesized at 85 °C exhibits a broad and bimodal pore size distribution, peaked at ca. 135 and 1104 μm, mostly in the macropore range (>50 nm) and a cumulative pore volume of 1.54 ml g−1, more than three times larger than that of ST/RT. The fact that a higher synthesis temperature favors gelation (sol-to-gel transition) but not phase separation might lead to a delay in the onset of phase separation, explaining the presence of broader skeleton domains and larger macropores in the ST/85C MICROSCAFS®.
 | ||
| Fig. 4 Morphology of ST MICROSCAFS® synthesized at room temperature (ST/RT), 50 °C (ST/50C), and 85 °C (ST/85C). Top left: Optical microscopy photographs (scale bar = 600 μm); Top right: SEM photomicrographs (scale bar = 8 μm) of a particle cross-section; Bottom left: particle size distributions; Bottom right: MIP pore size distribution curves. | ||
On the other hand, we found that the average synthesis pH was lower than for ST/50C (ESI Table S8†), which is probably due to the evaporation of ammonia at higher synthesis temperatures. In this case, gelation is slower and might be delayed in relation to the onset of phase separation, resulting in larger pores and broader skeleton domains. Regarding mesoporosity (pores below 50 nm, in the MIP graphs, Fig. 4), it decreases with higher reaction temperatures, and ST/RT is the one that exhibits higher mesopore content. The best compromise in terms of larger macropore size and volume and higher amount of mesopores was achieved for ST/50C. Therefore, this sample served as a reference in subsequent studies.
It should also be noted that compared to a 100% SiO2 MICROSCAFS®, reported in a previous paper,3 ST/50C exhibits a larger average hydrolyzate viscosity (16.2 versus 8.7 cP), a monomodal size distribution peaked at 98 μm versus a bimodal distribution peaked at 70 and 130 μm, broader pore size distribution extended to the larger macropores side (pore size mode value at 184 nm versus 164 nm), larger cumulative pore volume (1.39 ml g−1 versus 0.55 ml g−1), larger total porosity (38% versus 28%, by MIP accounted for pores only below 10 μm) and a larger specific surface area (27.93 versus 14.50 m2 g−1).
The N2 adsorption–desorption isotherms (ESI Fig. S9†) also followed the previous observations regarding mesoporosity. At first analysis, all the isotherms seem to be of type III (IUPAC classification), which is typical of nonporous or macroporous materials. In the present case, they reveal macroporous materials, corroborated by the SEM images and MIP results, as well as some hysteresis, characteristic of mesoporosity, for ST/RT and ST/50C and no hysteresis for ST/85C, which is in line with the MIP results presented above.
Oscillatory time sweeps of the complex viscosity (η*) successfully monitor gelation and crosslinking phenomena over reaction time.35–37 The gelation reaction process of a polymer gel is divided into three stages: an initial induction, a fast crosslinking, and a final stable period.37 An abrupt increase in the complex viscosity is characteristic of the beginning of the gelation process.38 ESI Fig. S10† shows an oscillatory time sweep analysis, namely the evolution of η* along the reaction time for each of the three different reaction temperatures: RT, 50 and 85 °C, without the addition of the catalyst ammonia. As expected, the temperature strongly influenced the gelation of the precursors. At RT, the induction period was very long, and the viscosity began to increase only after ∼20 min, suggesting relatively slow gelation. At 50 °C, the reaction was much faster, having a sharp increase of complex viscosity starting at ∼2.5 min and reaching the stable period at ∼5 min. At 85 °C, the gelation was already in the final stable period, so no abrupt increase in viscosity is observed within the time frame of the analysis, which suggests that gelation quickly happens during the mixture of the different compounds and the launching of the testing. This analysis helped us to conclude the effect of the synthesis temperature on the kinetics of the gelation phenomenon, which is related to the chemical quenching, i.e. the freezing of the phase separated structure. However, the addition of ammonia to catalyze condensation might neutralize this effect, or even introduce another aspect, related to a stronger evaporation of ammonia at higher synthesis temperatures (as depicted by the lower pH measured for ST/85C).
Effect of the Si alkoxide type on the ST MICROSCAFS® morphology
Different sol–gel alkoxide precursors, and their combinations at different molar ratios, play a role in the final porosity of the material as they might influence the hydrolysis and condensation kinetics, as well as the phase separation between the siloxane-rich and the water-rich phases inside the water droplets of the emulsion.We studied three different molar ratios of the employed Si alkoxides (TEOS and GPTMS): GPTMS/TEOS at 0.65, 0.87, and 1.09. The MICROSCAFS® size distribution (Fig. 5) displays a slightly increased value of the mode for higher GPTMS/TEOS ratio but narrower size distribution, given by the span value (1.1) of the curve (ESI Table S7†). This effect might be linked to the slower condensation rate induced by higher GPTMS content since trialkoxysilanes are less prone to form 3D networks than tetraalkoxysilanes, and the presence of the glycidyloxy group might lead to some steric hindrance effect.
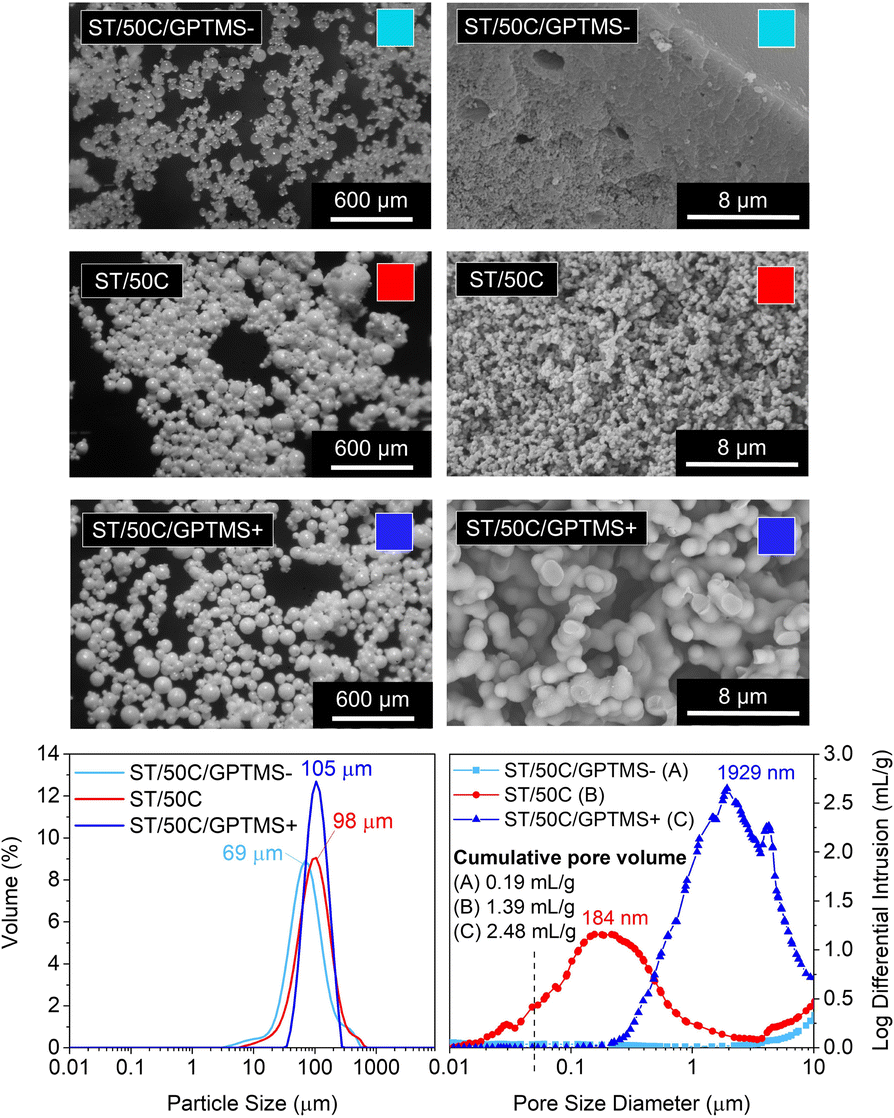 | ||
| Fig. 5 Morphology of ST MICROSCAFS® synthesized using 0.65 (ST/50C/GPTMS−), 0.86 (ST/50C), and 1.08 (ST/50C/GPTMS+) GPTMS/TEOS molar ratios. Top left: Optical microscopy photographs (scale bar = 600 μm); Top right: SEM photomicrographs (scale bar = 8 μm) of a particle cross-section; Bottom left: particle size distributions; Bottom right: MIP pore size distribution curves. | ||
Also, the alkyl chain and some remaining oxirane groups present in the hydrolyzed species of GPTMS, associated with the lower viscosity measured for ST/50C/GPTMS+ hydrolyzate (14.8 versus 25.0 cP for ST/50C/GPTMS−), might lead to a higher tendency for an even, controlled W/O emulsion droplets coalescence, which explains the larger diameter but lower span value of the MICROSCAFS® ST/50C/GPTMS+.
MIP data and SEM photomicrographs (Fig. 5) show a drastic change in pores morphology. A higher GPTMS/TEOS ratio strongly increases the pore size, exhibiting a larger macropore volume and size at the expense of mesopores. It should be noted that sample ST/50C/GPTMS+ displays a cumulative pore volume as high as 2.48 ml g−1 and a broad macropore size distribution peaked at ca. 1929 μm. These characteristics might be due to the delay in the chemical quench of the phase-separated system. On the contrary, the intrusion was almost zero for sample ST/50C/GPTMS− because of the fast overall gelation, which “freezes” the system much before the onset of phase separation by spinodal decomposition and, therefore, yields a denser structure. The SEM image in Fig. 5 shows that ST/50C/GPTMS− is denser close to the surface. The ammonia solution might have promoted this phenomenon, which further catalyzes the condensation reactions at the surface of the droplets. However, when more GPTMS is employed, such an effect is attenuated.
Fig. 6 shows a CT cross-section of MICROSCAFS® ST/50C/GPTMS− and ST/50C/GPTMS+ when placed, without pressing, inside a cylindrical container, together with pore and skeletons size distributions and calculated porosity obtained from the analysis of the image data set. ST/50C/GPTMS+ exhibited much more void space between the particles. CT corroborates the findings from SEM, MIP, and optical microscopy, in that ST/50C/GPTMS+ displays larger particle size and porosity. We segmented the images to consider only the areas/volumes corresponding to the particles as the region of interest for the morphological analysis, excluding the interparticle space. This strategy ensures that the obtained values represent the intraparticle region of interest. The obtained values of open and total porosity demonstrate that, for both samples, the pores detected by this technique are open to the external volume of the particles, with ST/50C/GPTMS+ displaying a significantly higher pore volume (ca. 67%) than ST/50C/GPTMS−. On the contrary, the volume of the silica walls (skeleton) is similar in both samples.
 | ||
| Fig. 6 Top: Cross-section images obtained by CT from MICROSCAFS® ST/50C/GPTMS− and ST/50C/GPTMS+ obtained with the CCD detector when placed, without pressing, inside a cylindrical container. Scale bar = 700 μm; Middle: Pore and skeleton size distributions, calculated from CT images; Bottom: open and total porosity calculated from CT images. For this calculation, a reconstructed 3D model of cross-sectional frames taken each 0.15° until 360° (2400 frames) was analyzed with the CTAn software. The images at the top are just one frame of a cross-section of the particles, being shown here as an example. | ||
The pore size distribution displays the presence of macropores of size above 10 microns, for ST/50C/GPTMS+, which reinforces the fact that macropore size and porosity estimation might be underestimated by MIP. This latter technique cannot distinguish internal from external (interparticle) volume and, therefore, we decided to consider a range only below 10 μm for MIP data treatment. On the other hand, the resolution of both techniques, CT and MIP is not the same. The former technique appears to be preferable in the larger size range (>10 μm) for these microparticles, while the latter one appears to be better in the smaller size range. These factors may explain the different values achieved for total porosity: e.g. 41% through MIP, and ca. 67% through CT, for ST/50C/GPTMS+.
The graphs resulting from oscillatory time sweep analyses (ESI Fig. S11†) show a longer induction period and a slower gelation process for a higher GPTMS/TEOS molar ratio (ST/50C/GPTMS+). Steric hindrance and the hydrophobic character from the relatively long glicidyloxypropyl chain of GPTMS may be the cause for slower condensation,34,39 but also for higher tendency for phase separation, which results in the coarsening of the skeleton domains and larger pore sizes and volumes, typical of ST/50C/GPTMS+. Corroborating this finding, the hydrolyzate for ST/50C/GPTMS+ exhibits a slightly lower viscosity than the reference ST/50C. This means the oligomers had lower molecular weight, due to a longer time required for gelation, explaining the obtained coarse macroporosity.
On the other hand, the relatively higher hydrolyzate viscosity measured for ST/50C/GPTMS− (ESI Table S8†) indicates a relevant condensation concomitant to the hydrolysis step. The presence of higher molecular weight oligomers places the system closer to the gel point so that the “freezing” of the structure occurs well before the onset of phase separation. Thus, the MICROSCAFS® exhibit a denser structure and a very different pore morphology from samples ST/50C and ST/50C/GPTMS+.
The N2 adsorption–desorption isotherms (ESI Fig. S12†) are of type III (IUPAC classification), suggesting a relatively weak physical adsorbent–adsorbate interaction.40 Both samples, ST/50C/GPTMS− and ST/50C/GPTMS+, show lower BET surface areas than the ST/50C reference, corroborating the MIP data, which shows no relevant intrusion below 50 nm of pore diameter. Sample ST/50C with an intermediate GPTMS/TEOS molar ratio, corresponds to the best compromise in terms of simultaneous meso and macroporosity. It should be emphasized that BET surface areas of ca. 5 m2 g−1 are very close to the limit value detected by this technique (e.g. the acceptable error of a standard aluminosilicate control sample with 214 m2 g−1 of surface area is ±6 m2 g−1). Also, the nanopore size distributions (insets of ESI Fig. S9 and ESI Fig. S12†) reveal peaks at 1.7 nm, which might be an effect from the model when applied to a low adsorption isotherm, with irregularities at low pressures.
EDS analysis (ESI Table S13†) showed that the higher the GPTMS/TEOS molar ratio, the closer the Si and Ti atomic percentage is to the theoretical (80% Si, 20% Ti). The slower condensation induced by a greater quantity of GPTMS improved the titanium incorporation into the network. Previous NMR studies for 100% SiO2 MICROSCAFS® have shown the presence of T and Q Si units, with T3 prevailing over T2 units.3 Greater amounts of trialkoxysilane GPTMS promote larger silica network disruption, which is expected to facilitate the incorporation of Ti species in the MICROSCAFS® 3D network.
The FTIR spectrum of these samples, when heat treated at 700 °C, reveal features that corroborate the above-mentioned findings. A slight shift of the TO peak to lower frequencies (from 1060 to 1050 cm−1), as seen in ESI Fig. S14,† is observed for higher ratios GPTMS/TEOS. This decrease in the frequency of the TO peak may be associated with the larger macroporosity observed for ST/50C/GPTMS+. This porosity, in turn, induces the strain in the Si–O–Si bonds at the surface of the pores, which leads to larger bridging angles and longer Si–O bonds, compared to ST/50C/GPTMS− and ST/50C, causing a decrease of the TO peak frequency.41 Also, the Si–O–Ti (Si–O−) stretching peak appears to be more intense for ST/50C/GPTMS+, corroborating the larger amount of Ti species in the network, detected by EDS.
Effect of the Ti precursor's chelating agent and emulsion characteristics on the ST MICROSCAFS®' morphology
The modification of TiPOT with glacial acetic acid (chelation) reduces the availability of groups that easily hydrolyze through the formation of a stable complex, typically Ti(OCOCH3)(Oi Pr)2.42 We studied the effect of increasing the amount of the Ti precursor's chelating agent, acetic acid (CH3COOH), up to the point of excess (TiPOT/CH3COOH molar ratio from 1/4 to 1/6), which resulted in a decrease of the average synthesis pH from 8.51 to 7.61. The MICROSCAFS® size distribution graph (Fig. 7) shows an apparent increase in the particle size mode and all the percentiles (ESI Table S7†) for a higher quantity of acetic acid (lower TiPOT/CH3COOH molar ratio, sample ST/50C/AA+).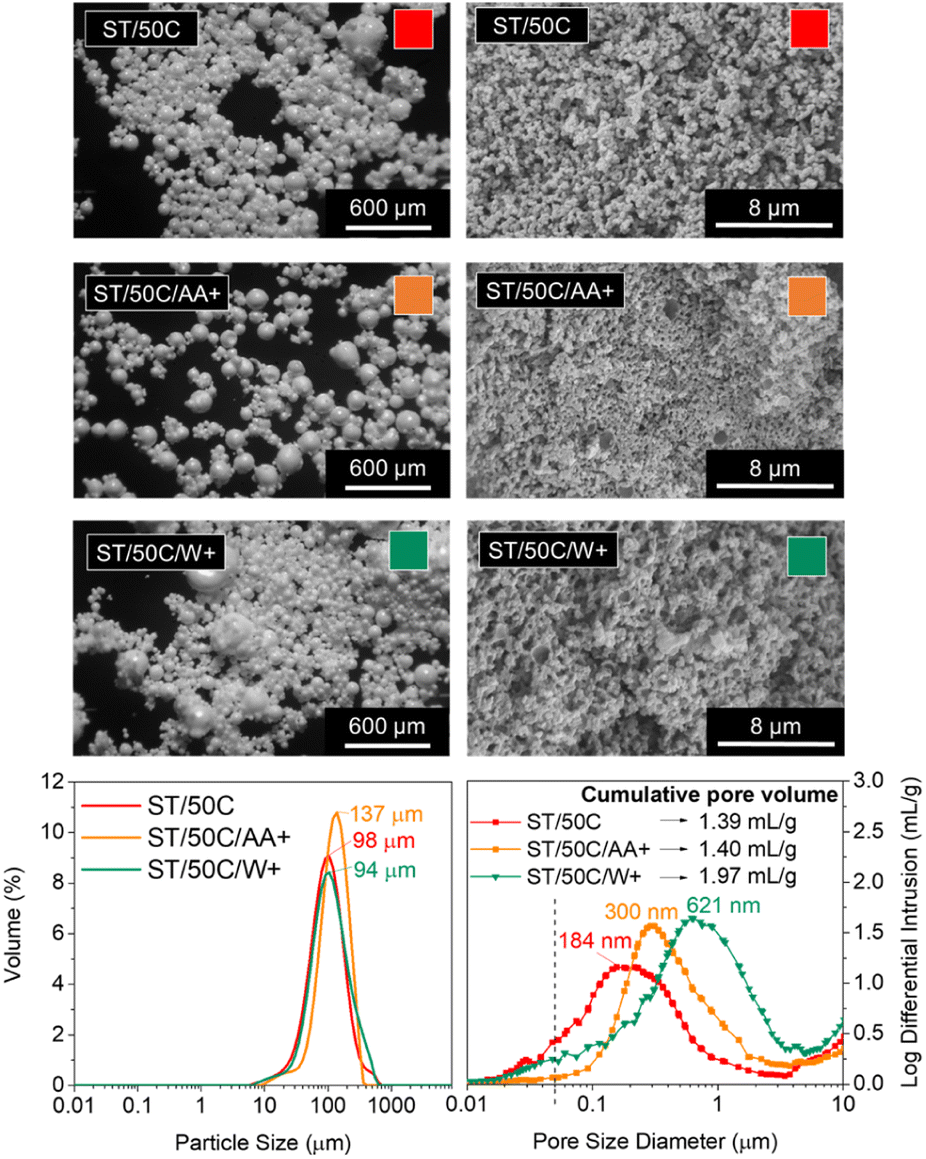 | ||
| Fig. 7 Morphology of ST MICROSCAFS® synthesized using 1:4 (ST/50C) and 1:6 (ST/50C/AA+) TiPOT/CH3COOH molar ratios, and 0.45 (ST/50C) and 0.68 (ST/50C/W+) W/O mass ratios. Top left: Optical microscopy photographs (scale bar = 600 μm); Top right: SEM photomicrographs (scale bar = 8 μm) of a particle cross-section; Bottom left: particle size distributions; Bottom right: MIP pore size distribution curves. | ||
On the contrary, a larger water phase volume in the emulsion and, therefore, a higher concentration of water droplets, acting as a soft template for the MICROSCAFS®, did not change the particle size mode significantly. Instead, there was an increase in the D(0.9) percentile and, consequently, the span value, indicating greater size dispersions (ESI Table S7†). This effect is also visible in the optical microscopy photograph of sample ST/50C/W+ (Fig. 7). Based on the theoretical models proposed by Pal et al., these emulsions are considered concentrated or highly concentrated.43 In these domains, the increase of dispersed phase concentration leads to more closely packed water droplets that interact much more with one another. Destabilizing phenomena such as coalescence and flocculation are more pronounced in these domains, leading to an increase in polydispersity of the template emulsion as the water phase increases,44,45 and to the presence of a fraction of larger MICROSCAFS®, with sizes ranging from 200 to 500 μm. The closely packed water droplet's structure will also be responsible for a pronounced increase in emulsion viscosity, found in the present work (ESI Fig. S15†), for increasing W/O mass ratios, in the range of 0.2 to 1.1. This behavior can be justified by increased friction between droplets imputable to movement restrictions for the overall 3D emulsion network.46
Regarding macroporosity, MIP data and SEM images (Fig. 7) showed that higher quantities of both the emulsion's water phase and TiPOT's chelating agent (acetic acid) lead to bigger macropore size, however, not so intensely as for samples ST/85C and ST/50C/GPTMS+.
An excess of acetic acid tends to slow down the alkaline condensation reaction, leading to a delay in the “freeze” of the phase-separated structure, which results in the coarser skeleton domains of ST/50C/AA+, and an increase of its macropores size, at the expense of mesopores.
However, when performing oscillatory time sweep analysis on the hydrolyzate without adding ammonia (ESI Fig. S16†), the acidic condensation reaction is kinetically more favorable. Indeed, an increase of η* along the reaction time occurs faster when more acetic acid is used. It has been reported that TiPOT only forms bidentate ligands with acetic acid at a 1:2 ratio. Therefore, all the excess acid remains in the solution and hinders the alkaline condensation of the precursors, increasing the pore size.42
By observing Fig. 7, a higher W/O mass ratio yielded MICROSCAFS® with larger pores. This might be caused by a dilution effect of the clusters, which delays the gelation as there is more water in the polymerized species-W/O dispersion. Also, protonic solvents (e.g., water) have been reported to decrease condensation reactions in alkaline media,47 which might also increase the time between the phase separation and gelation and yield greater pore sizes. Indeed, when we added an equivalent amount of the excess water to the ST/50C hydrolyzate solution and ran oscillatory time sweep measurements (ESI Fig. S16†), the gelation achieved approximately maximum η* one order of magnitude lower than the reference sample ST/50C. Regarding mesoporosity, a decrease in BET surface area was detected for this sample (8.73 m2 g−1 versus 27.93 m2 g−1 for ST/50C) (Supplementary Fig. S17†), in line with the lower amount of mesopores (Fig. 7). The isotherm of the ST/50C/W+ is also type III, characteristic of a relatively weak adsorbent–adsorbate interaction.
Development of multicomponent oxide SiO2–TiO2–HfO2 (STH) MICROSCAFS®
Hafnium dichloride oxide octahydrate salt was added during the silanes hydrolysis step of an ST/50C/GPTMS+ MICROSCAFS® synthesis to produce multicomponent oxide SiO2–TiO2–HfO2 (STH) MICROSCAFS®, aiming at enlarging the scope of applications for these particles. As a base, we chose ST/50C/GPTMS+ synthesis instead of the reference (ST/50C) due to its great macroporosity, so we can better evaluate any changes in the final porosity of the new MICROSCAFS®.Adding Hf salt led to a particle size distribution with larger dispersion (Fig. 8), indicated by a broader curve with a higher span value (ESI Table S7†). This might be related with the higher viscosity of the corresponding hydrolyzate (45.3 cP versus 16.2 cP for ST/50C, ESI Table S8†). However, all particles have a regular spherical shape. The SEM photomicrographs and MIP pore size distribution in Fig. 8 show a steep decrease in pore size and cumulative pore volume for STH/50C/GPTMS+ when compared to the ST analogue. However, they exhibited the same type of N2 adsorption/desorption isotherms, and a slightly larger surface area (ca. 10.3 m2 g−1) than that of ST/50C/GPTMS+. EDS data show that the initial relative molar concentration of 80% Si was kept in the final MICROSCAFS® network composition (ESI Table S13†). On the other hand, 7.6% of the Ti atoms was replaced by Hf atoms, suggesting their incorporation into the network. Doubling further the Hf precursor quantity generated irregular particles of non-spherical shapes and heterogeneity in terms of pore morphology and size (ESI Fig. S18†).
 | ||
| Fig. 8 Morphology of double (ST/50C/GPTMS+) and triple (STH/50C/GPTMS+) oxide MICROSCAFS®. Top left: Optical microscopy photographs (scale bar = 600 μm); Top right: SEM photomicrographs (scale bar = 8 μm) of a particle cross-section; Bottom left: particle size distributions; Bottom right: MIP pore size distribution curves. | ||
The reported high viscosity of the hydrolyzate in the presence of Hf precursor resulted in faster gelation. It is known that HfOCl2 provides HCl, which catalyzes the hydrolysis of the silanes present in this system. Also, high atomic number elements, such as Hf, when bonded to Cl increase reactivity and accelerate the gelation process. The large ionic strength of Hf–Cl bonds (large electronegativity difference) existing in the Hf precursor might easily destroy the protection of the repulsive double layer around primary particles, resulting in their aggregation and formation of a gel. The triple oxide system thus condensates and “freezes” the phase-separated structure faster than the double oxide system. This effect prevents coarsening and macropore enlargement expansion, originating smaller porosities than the double oxide system (Fig. 8). Indeed, when the Hf precursor is present, the delayed condensation, typical of greater quantities of GPTMS precursor, had no significant impact on the developed structure.
After heat treatment at 700 °C, the major difference when comparing STH MICROSCAFS® with ST MICROSCAFS®, consists of a much weaker peak at ∼936 cm−1, slightly shifted to lower frequencies (ESI Fig. S19†), assigned to Si–O− asymmetric stretching, which is characteristic of homogeneous gel/glass network regions, where Si–O–Ti and Si–O–Hf bonds are also included.48 Si–O–Hf peaks are typically much weaker than Si–O–Ti peaks, which might be due to a lower degree of homogeneity, in the former case, but it could also arise in part from a lower IR activity of the Si–O–Hf vibrational mode, due to structural differences, such as different distributions of the Si–O–Hf and Si–O–Ti angles.
MICROSCAFS®' mechanical properties
The mechanical properties of the MICROSCAFS® impact their performance, so their evaluation is relevant for future applications. If we consider a continuous flow setup for photocatalysis tests2 as a possible application in the framework of environmental remediation, it is beneficial to understand how the MICROSCAFS® behave when subject to compression in a confined space (reservoir). A more cohesive powder, i.e., a less free-flowing powder, exhibits higher compressibility and allows more MICROSCAFS® to fit inside the confined space, resulting in an increase of the bulk density under compressive stress. In the future, this characteristic may impact the photocatalytic activity and the flow of the solution that is being purified. The stiffness and mechanical strength of the materials give us an insight into how easily these catalytic supports will break, playing a significant role in the photocatalyst's activity, reusability, and recyclability.The different MICROSCAFS® were subject to a loading/hold/unloading process to evaluate various properties of the powders, namely compressibility, relaxation, elastic recovery, and stiffness. The loading period involves elastic and plastic deformation, while the hold period (stress relaxation) is generally purely plastic. It allows the study of the viscoelastic (time-dependent) behavior of the compacted MICROSCAFS® since the particles undergo plastic deformation and move into void spaces. The force usually drops off quickly, followed by a slighter decrease until reaching a plateau. The more viscoelastic the powder, the more the force drops during the hold period. The unloading period is generally purely elastic and (the initial slope) allows the measurement of the stiffness of the compact, while the elastic recovery upon unloading is a function of the loaded and relaxed column heights.
Fig. 9 compiles the impact of all the different reactional parameters on the MICROSCAFS® powder compression parameter (compressibility), force hold ratio (relaxation), unload strain (elastic recovery), and unload gradient (stiffness). The increase in reaction temperature, GPTMS/TEOS molar ratio, emulsion W/O mass ratio, and decrease in TiPOT/CH3COOH molar ratio yield MICROSCAFS® with higher compressibility and elastic recovery but lower stiffness than the corresponding reference ST/50C (marked with a red dashed line rectangle).
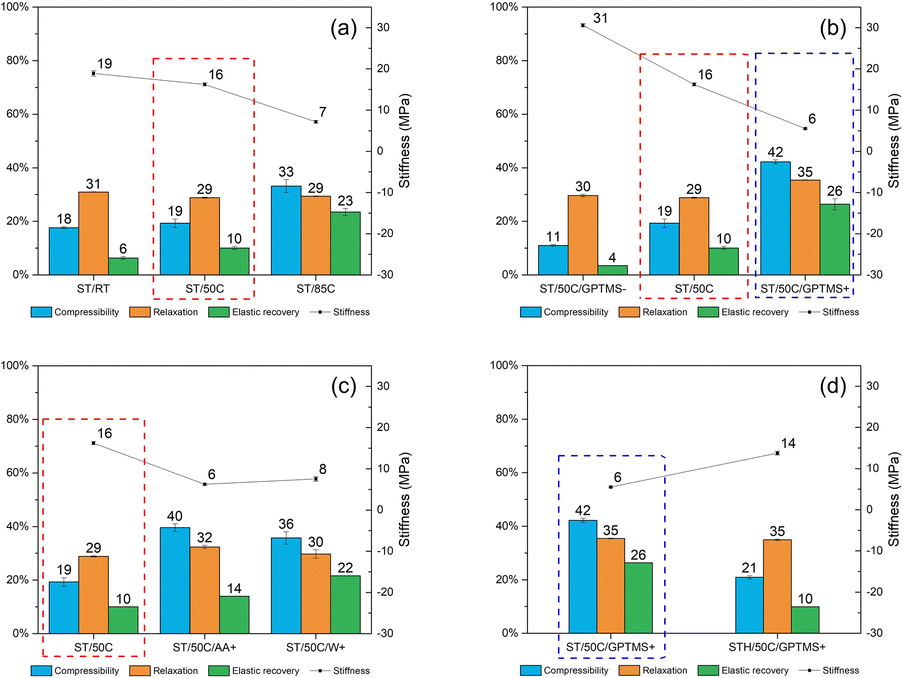 | ||
| Fig. 9 Effect of (a) reaction temperature, (b) GPTMS/TEOS molar ratio, (c) TiPOT/CH3COOH molar ratio and W/O mass ratio, and (d) addition of hafnium dichloride oxide salt (STH multicomponent oxides) in the MICROSCAFS®' mechanical properties. | ||
These parameters increase the temporal space between the “freezing” of the structure and the onset of phase separation, resulting in larger porosity and, therefore, less stiff MICROSCAFS® with larger elastic recovery upon unloading. The change in morphology (broader skeletons, larger pores) might also result in less free flowing, more cohesive, and, therefore, more compressible powders under compressive stress. There is clearly more free space between the particles, for MICROSCAFS® ST/50C/GPTMS+, before being pressed, as shown in Fig. 10. The CT scan results determined that the object (solid matter) volume was 53.2% for ST/50C/GPTMS− and 32.4% for ST/50C/GPTMS+. This fact results in higher compressibility when the sample ST/50C/GPTMS+ is subject to a compression force, explaining the recurrent high compressibility value obtained for higher contents of GPTMS. Inversely, adding Hf salt yielded stiffer MICROSCAFS®, exhibiting as well lower compressibility and lower elastic recovery. Finally, the stress relaxation (viscoelasticity) of the compacted MICROSCAFS® is only slightly affected (increased) for higher GPTMS/TEOS molar ratios, which suggests a similar level of plastic deformation of the MICROSCAFS® reported in this paper.
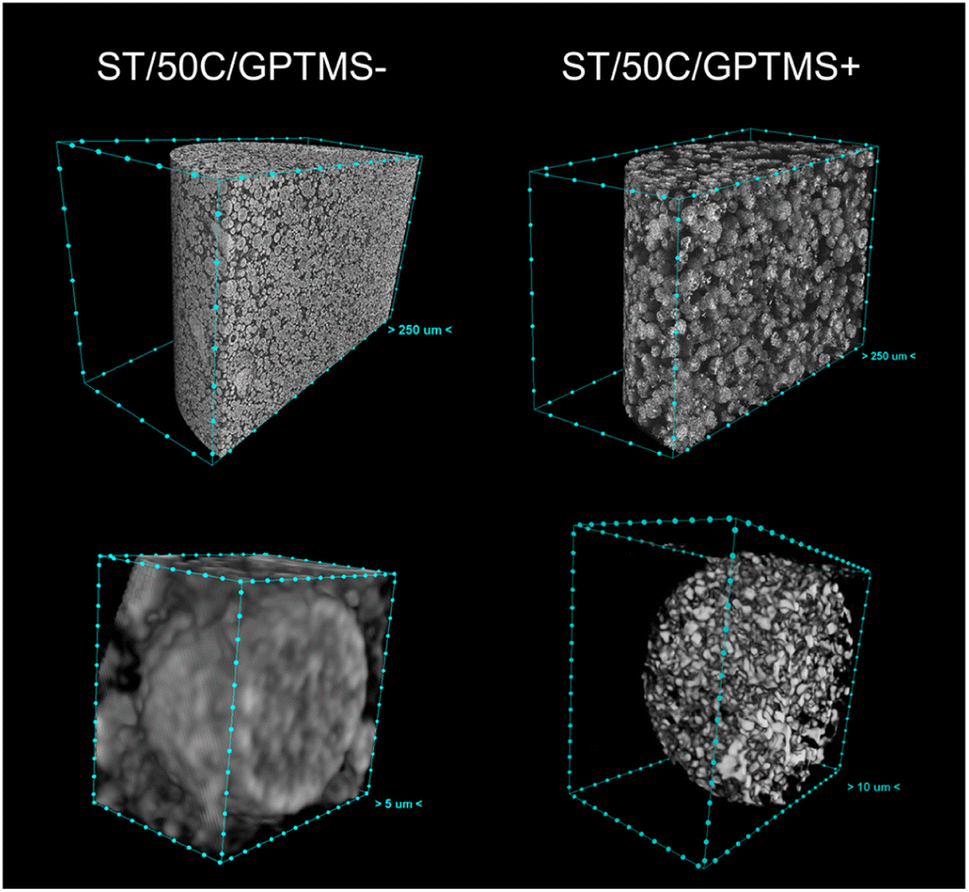 | ||
| Fig. 10 Cross-sectional 3D CT image of the powder (top line) and a single microsphere (bottom line) of the ST/50C/GPTMS− and ST/50C/GPTMS+ MICROSCAFS® (scale, distance between points: top left = 250 μm, top right = 250 μm, bottom left = 5 μm, bottom right = 10 μm). | ||
The effect of MICROSCAFS®' cumulative pore volume on the four evaluated parameters is represented in Fig. 11. Graphics containing the effect of pore size mode and particle size mode were also represented and can be found in ESI Fig. S20 and ESI Fig. S21.† Summarily, higher cumulative pore volumes and higher mode values of the pore size resulted in higher compressibility and elastic recovery, following an approximately linear trend. As for relaxation, no significant changes were observed, while stiffness decreases linearly for greater cumulative pore volumes, and exponentially for greater pore sizes. The effect of particle size, on the other hand, is not so evident in terms of MICROSCAFS® stiffness, because samples with similar particle size, but exhibiting different porosity, display different stiffness values.
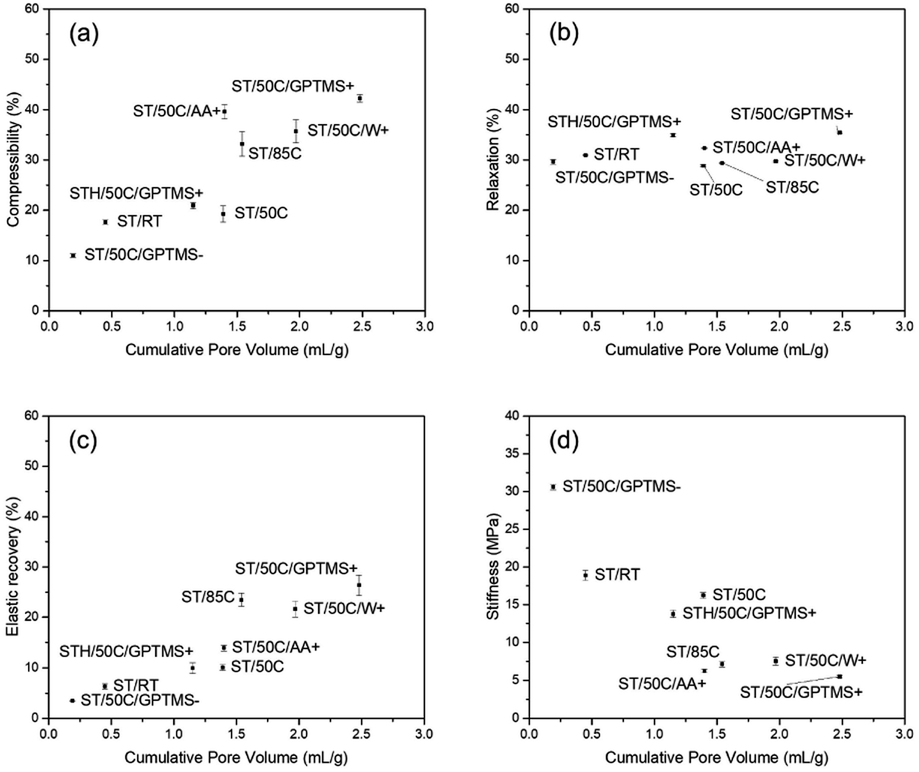 | ||
| Fig. 11 Effect of MICROSCAFS®' cumulative pore volume on (a) compressibility, (b) relaxation, (c) elastic recovery, and (d) stiffness. | ||
These results show that by changing synthesis parameters, such as GPTMS/TEOS molar ratio, we can finely tune the mechanical properties of the MICROSCAFS®, namely stiffness, compressibility, and elastic recovery. The recurrent lower stiffness observed in sample ST/50C/GPTMS+ is attributed to the higher quantity of GPTMS. Besides the larger macroporosity characteristic of this sample (Fig. 10), the presence of more glicidyloxypropyl groups, i.e., the higher number of organic fractions incorporated in the structure (more hybrid character) may also play a role in reducing the stiffness and increasing compressibility and elastic recovery. On the contrary, the sample with a lower GPTMS/TEOS molar ratio and, thus, a higher quantity of TEOS exhibits the highest stiffness due to its more inorganic character, less disrupted 3D network, and denser structure (less macroporosity). A higher amount of TEOS confers a stiffer glass-like structure to the MICROSCAFS®.
All the MICROSCAFS®' additional calculated and graphical data can be found in ESI Table S22 and ESI Fig. S23–S26.†
Conclusions
We developed a method to reproducibly obtain microspheres of tailored size, composition, interconnected macroporosity, and mechanical properties (named MICROSCAFS®). We achieved this despite the complexity of the simultaneous occurrence of phase separation and gelation in the confined space of emulsion droplets. The MICROSCAFS® display robust scaffolding properties, including thermal and chemical stability, making them an innovative multifunctional material for various applications.Polymerization-induced phase separation applied to the sol–gel synthesis of the microspheres, where an emulsion is used as a soft template, can effectively control the morphological features and mechanical properties of SiO2–TiO2 and SiO2–TiO2–HfO2 porous MICROSCAFS®. Such fine-tuning is achieved by a proper selection of the synthesis parameters and is expected to play a major role in terms of MICROSCAFS® performance. To sum up, larger macroporosity (in most of the cases at the expense of some of the mesoporosity) and particle sizes were achieved by using a higher reaction temperature (85 °C), higher GPTMS/TEOS molar ratio (1.09), or lower TiPOT/CH3COOH molar ratio (1:6). A higher W/O mass ratio (0.68:1) increased the pore size without effectively changing the particle size, while specific mixtures of surfactants (Span® 80 and Pluronic® P123, HLB = 5.2) resulted in a drastic decrease of particle size. The composition and content of the silane precursors, defined by the GPTMS/TEOS molar ratio, had the greatest effect on the final interconnected macroporosity and particle size, yielding pores with size mode ranging from a few nanometers to a few microns in diameter and particle sizes ranging from 69 to 105 μm. Greater GPTMS content also increases the material's hybrid inorganic/organic character, enabling further functionalization reactions, if desired. The presence of Hf precursor (STH MICROSCAFS®) leads to smaller interconnected macroporosity, explained by the higher gelation rate induced by the Hf precursor.
Finally, a correlation between the size and amount of macropores and the mechanical properties of the MICROSCAFS® was established. Greater macroporosity generally leads to MICROSCAFS® displaying less stiffness, higher elastic recovery, and compressibility values up to 42%, for the more porous samples (68% open porosity).
By precisely controlling the porosity and sizes of multicomponent silica-based spheres, we created a synthetic platform that yields a versatile product that can be adapted to multiple future applications. For instance, the potential for photocatalytic applications, and the effect of macroporosity, is being explored in current works, as well as the effect of mechanical properties on the MICROSCAFS® reusability and recyclability.
Author contributions
M. V.: conceptualization, investigation, methodology, writing – original draft, writing – review & editing. S. O: investigation, writing – review & editing, visualization. A. M.: rheological characterization, writing – review & editing; L. R.: MICROSCAFS® mechanical properties analysis, writing – review & editing. M. P.: X-ray computed tomography, writing – review & editing. A. C. M.: conceptualization, methodology, writing – original draft, writing – review & editing, funding acquisition, project administration, resources, supervision, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Fundação para a Ciência e a Tecnologia (FCT) through the support of CERENA (Strategic Project FCT-UIDB/04028/2020), and IDMEC, under LAETA (Strategic Project FCT-UIDB/50022/2020), the FCT grants SFRH/BD/138717/2018 (M. V.) and 2022.09822.BD (S. O.), and the FCT project PTDC/EQU/EQU/1056/2020 (SOLAR2CLEAN—Engineered solar light driven photocatalytic systems for wastewater purification). We would like to acknowledge Professor João Lopes and M.Sc. Nuno Costa from the Faculty of Pharmacy of the University of Lisbon for providing access to the particle size analyzer, Professor Frederico Ferreira for making available the rotary viscometer, and finally Professor Ana Paula Serro for providing access to the rheometer, both from Instituto Superior Técnico.Notes and references
- National Brand Registration no 684231 MICROSCAFS, by Instituto Superior Técnico. Granted in 2022.07.14. Published at the Intellectual Property Bulletin no 137/2022.
- A. C. Marques, M. Vale, D. Vicente, M. Schreck, E. Tervoort and M. Niederberger, Porous Silica Microspheres with Immobilized Titania Nanoparticles for In-Flow Solar-Driven Purification of Wastewater, Global Challenges, 2021, 5, 2000116 CrossRef.
- M. Vale, M. V. Loureiro, M. J. Ferreira and A. C. Marques, Silica-based microspheres with interconnected macroporosity by phase separation, J. Sol-Gel Sci. Technol., 2020, 95, 746–759 CrossRef CAS.
- A. C. Marques and M. Vale, Macroporosity Control by Phase Separation in Sol-Gel Derived Monoliths and Microspheres, Materials, 2021, 14, 4247 CrossRef CAS PubMed.
- K. Nakanishi and N. Soga, Phase Separation in Gelling Silica–Organic Polymer Solution: Systems Containing Poly(sodium styrenesulfonate), J. Am. Ceram. Soc., 1991, 74, 2518–2530 CrossRef CAS.
- K. Nakanishi and N. Soga, Phase separation in silica sol-gel system containing polyacrylic acid I. Gel formaation behavior and effect of solvent composition, J. Non-Cryst. Solids, 1992, 139, 1–13 CrossRef CAS.
- K. Nakanishi and N. Soga, Phase separation in silica sol-gel system containing polyacrylic acid II. Effects of molecular weight and temperature, J. Non-Cryst. Solids, 1992, 139, 14–24 CrossRef CAS.
- K. Nakanishi and N. Soga, Phase separation in silica sol-gel system containing polyacrylic acid. III. Effect of catalytic condition, J. Non-Cryst. Solids, 1992, 142, 36–44 CrossRef CAS.
- K. Nakanishi and N. Soga, Phase separation in silica sol-gel system containing polyacrylic acid. IV. Effect of chemical additives, J. Non-Cryst. Solids, 1992, 142, 45–54 CrossRef CAS.
- Z. G. Shi and Y. Q. Feng, Synthesis and characterization of hierarchically porous silica microspheres with penetrable macropores and tunable mesopores, Microporous Mesoporous Mater., 2008, 116, 701–704 CrossRef CAS.
- L. M. Yang, Y. J. Wang, Y. W. Sun, G. S. Luo and Y. Y. Dai, Synthesis of micrometer-sized hard silica spheres with uniform mesopore size and textural pores, J. Colloid Interface Sci., 2006, 299, 823–830 CrossRef CAS.
- T. Guo, C. Wang, L. Dong, J. Lü and T. Liang, Effect of Al2O3 on the Process Performance of ZrO2 Microspheres, J. Wuhan Univ. Technol.-Materials Sci. Ed., 2020, 35, 841–846 CrossRef CAS.
- X. Guo, Q. Zhang, X. Ding, Q. Shen, C. Wu, L. Zhang and H. Yang, Synthesis and application of several sol–gel-derived materials via sol–gel process combining with other technologies: a review, J. Sol-Gel Sci. Technol., 2016, 79, 328–358 CrossRef CAS.
- J. H. Smått, N. Schüwer, M. Järn, W. Lindner and M. Lindén, Synthesis of micrometer sized mesoporous metal oxide spheres by nanocasting, Microporous Mesoporous Mater., 2008, 112, 308–318 CrossRef.
- Y. Luan, Y. W. Xue and Z. G. Shi, Synthesis of hierarchically macro/meso/microporous carbon spheres and its application in fast rechargeable electric double layer capacitor, Mater. Lett., 2012, 88, 30–32 CrossRef CAS.
- D. Grosso, G. J. D. A. A. Soler Illia, E. L. Crepaldi, B. Charleux and C. Sanchez, Nanocrystalline transition-metal oxide spheres with controlled multi-scale porosity, Adv. Funct. Mater., 2003, 13, 37–42 CrossRef CAS.
- L. Lu, F. Teng, SenTapas, D. Qi, L. Wang and J. Zhang, Synthesis of visible-light driven CrxOy-TiO2 binary photocatalyst based on hierarchical macro-mesoporous silica, Appl. Catal. B Environ., 2015, 163, 9–15 CrossRef CAS.
- J. Li, L. Xu and Z. g. Shi, Waxberry-like hierarchically porous ethyl-bridged hybrid silica microsphere: a substrate for enzyme catalysis and high-performance liquid chromatography, J. Chromatogr., A, 2019, 1587, 79–87 CrossRef CAS PubMed.
- J. X. Wei, Z. G. Shi, F. Chen, Y. Q. Feng and Q. Z. Guo, Synthesis of penetrable macroporous silica spheres for high-performance liquid chromatography, J. Chromatogr., A, 2009, 1216, 7388–7393 CrossRef CAS PubMed.
- T. Long, Q. Z. Guo and L. Y. Xu, Fast separation of sulfanilamides using macroporous silica spheres as the separation media, J. Liq. Chromatogr. Relat. Technol., 2011, 34, 1391–1398 CrossRef CAS.
- C. r. Yin, L. y. Ma, J. g. Huang, L. Xu and Z. g. Shi, Fast profiling ecotoxicity and skin permeability of benzophenone ultraviolet filters using biopartitioning micellar chromatography based on penetrable silica spheres, Anal. Chim. Acta, 2013, 804, 321–327 CrossRef CAS.
- L. Ma, J. Li, J. Zhao, H. Liao, L. Xu and Z. G. Shi, Penetrable silica microspheres for immobilization of bovine serum albumin and their application to the study of the interaction between imatinib mesylate and protein by frontal affinity chromatography, Anal. Bioanal. Chem., 2016, 408, 805–814 CrossRef CAS PubMed.
- T. Qiao, L. y. Ma, X. Wang and Z. G. Shi, The flow-through silica as the matrix to immobilize gold nanoparticles for HPLC applications, J. Liq. Chromatogr. Relat. Technol., 2018, 41, 107–113 CrossRef CAS.
- Z. G. Shi, Q. Z. Guo, Y. T. Liu, Y. X. Xiao and L. Xu, Drug delivery devices based on macroporous silica spheres, Mater. Chem. Phys., 2011, 126, 826–831 CrossRef CAS.
- L. Y. Xu, Y. Y. Huang, T. Long and Z. G. Shi, Synthesis of macroporous silica spheres for drug delivery, Mater. Manuf. Processes, 2013, 28, 626–630 CAS.
- T. Ataei-Germi and A. Nematollahzadeh, Bimodal porous silica microspheres decorated with polydopamine nano-particles for the adsorption of methylene blue in fixed-bed columns, J. Colloid Interface Sci., 2016, 470, 172–182 CrossRef CAS PubMed.
- Q. Guo, L. Hu, C. Fan, Y. Zhang and Q. Zhang, Perforative silica microsphere-modified phenolphthalein-based poly(arylene ether sulfone) composites: tensile and thermal properties, Iran. Polym. J., 2018, 27, 611–619 CrossRef CAS.
- M. V. Loureiro, M. Vale, A. De Schrijver, J. C. Bordado, E. Silva and A. C. Marques, Hybrid custom-tailored sol-gel derived microscaffold for biocides immobilization, Microporous Mesoporous Mater., 2018, 261, 252–258 CrossRef CAS.
- X. Zhu, Y. Yang, Z. Zheng, B. Xiang and X. Cui, Multiwave rheology and dynamic light scattering characterizations for a two-step sol-gel transition of tetraethoxysilane hydrolysis and condensation, J. Sol-Gel Sci. Technol., 2018, 88, 255–262 CrossRef CAS.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons Ltd, West Sussex, England, 2004 Search PubMed.
- R. M. Almeida, Spectroscopy and Structure of Sol-Gel Systems, J. Sol-Gel Sci. Technol., 1998, 13, 51–59 CrossRef CAS.
- R. M. Almeida, P. J. Morais and A. C. Marques, Planar waveguides for integrated optics prepared by sol–gel methods, Philos. Mag., 2009, 82, 707–719 Search PubMed.
- S.-L. Chen, P. Dong, G.-H. Yang and J.-J. Yang, Kinetics of Formation of Monodisperse Colloidal Silica Particles through the Hydrolysis and Condensation of Tetraethylorthosilicate, Ind. Eng. Chem. Res., 1996, 35, 4487–4493 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol-gel science: the physics and chemistry of sol-gel processing, Academic Press, San Diego, US, 2013 Search PubMed.
- C. Wang, J. Wang, C. B. Park and Y. W. Kim, Cross-linking behavior of a polysiloxane in preceramic foam processing, J. Mater. Sci., 2004, 39, 4913–4915 CrossRef CAS.
- S. Y. Kim, D. G. Choi and S. M. Yang, Rheological analysis of the gelation behavior of tetraethylorthosilane/vinyltriethoxysilane hybrid solutions, Korean J. Chem. Eng., 2002, 19, 190–196 CrossRef CAS.
- Y. Sun, Y. Fang, A. Chen, Q. You, C. Dai, R. Cheng and Y. Liu, Gelation Behavior Study of a Resorcinol–Hexamethyleneteramine Crosslinked Polymer Gel for Water Shut-Off Treatment in Low Temperature and High Salinity Reservoirs, Energies, 2017, 10, 913 CrossRef.
- E. Katoueizadeh, M. Rasouli and S. M. Zebarjad, A comprehensive study on the gelation process of silica gels from sodium silicate, J. Mater. Res. Technol., 2020, 9, 10157–10165 CrossRef CAS.
- A. B. M. Giasuddin and D. W. Britt, Monitoring Silane Sol-Gel Kinetics with In-Situ Optical Turbidity Scanning and Dynamic Light Scattering, Molecules, 2019, 24, 2931 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- R. M. Almeida and A. C. Marques, Characterization of Sol-Gel Materials by Infrared Spectroscopy: Processing, Characterization and Applications, ed. L. Klein, M. Aparicio and A. Jitianu, Springer International Publishing, Cham, 2018, pp. 1121–1151 Search PubMed.
- R. Parra, M. S. Góes, M. S. Castro, E. Longo, P. R. Bueno and J. A. Varela, Reaction Pathway to the Synthesis of Anatase via the Chemical Modification of Titanium Isopropoxide with Acetic Acid, Chem. Mater., 2008, 20, 143–150 CrossRef CAS.
- R. Pal, Rheology of simple and multiple emulsions, Curr. Opin. Colloid Interface Sci., 2011, 16, 41–60 CrossRef CAS.
- R. Pal, Shear Viscosity Behavior of Emulsions of Two Immiscible Liquids, J. Colloid Interface Sci., 2000, 225, 359–366 CrossRef CAS.
- F. Goodarzi and S. Zendehboudi, A Comprehensive Review on Emulsions and Emulsion Stability in Chemical and Energy Industries, Can. J. Chem. Eng., 2019, 97, 281–309 CrossRef CAS.
- D. S. Kolotova, Y. A. Kuchina, L. A. Petrova, N. G. Voron'ko and S. R. Derkach, Rheology of Water-in-Crude Oil Emulsions: Influence of Concentration and Temperature, Colloids Interfaces Sci, 2018, 2, 64 CrossRef CAS.
- A. A. Issa and A. S. Luyt, Kinetics of Alkoxysilanes and Organoalkoxysilanes Polymerization: A Review, Polymers, 2019, 11, 537 CrossRef.
- D. A. Neumayer and E. Cartier, Materials characterization of ZrO2–SiO2 and HfO2–SiO2 binary oxides deposited by chemical solution deposition, J. Appl. Phys., 2001, 90, 1801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00895a |
| This journal is © The Royal Society of Chemistry 2023 |