
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable, stretchable, toughened, bioadhesive composite hydrogel for bladder injury repair†
Zhouyang Fu ab,
Shuwei Xiaoad,
Pengchao Wangbc,
Jian Zhaoab,
Zhengyun Lingab,
Ziyan Anab,
Jinpeng Shaoab and
Weijun Fu*a
ab,
Shuwei Xiaoad,
Pengchao Wangbc,
Jian Zhaoab,
Zhengyun Lingab,
Ziyan Anab,
Jinpeng Shaoab and
Weijun Fu*a
aDepartment of Urology, The Third Medical Centre, Chinese PLA General Hospital, Beijing, 100853, China. E-mail: fuweijun@hotmail.com
bMedical School of Chinese PLA, Beijing, 100853, China
cDepartment of Urology, Hainan Hospital of PLA General Hospital, Hainan 572013, China
dDepartment of Urology, Air Force Medical Center, Beijing, 100142, China
First published on 6th April 2023
Abstract
The bladder is exposed to constant internal and external mechanical forces due to its deformation and the dynamic environment in which it is placed, which can hamper its repair after an injury. Traditional hydrogel materials have limitations regarding their use in the bladder owing to their poor mechanical and tissue adhesion properties. In this study, a composite hydrogel composed of methacrylate gelatine, methacrylated silk fibroin, and Pluronic F127 diacrylate was developed, which combines the characteristics of natural and synthetic polymers. The mechanical properties of the novel hydrogel, such as stretchability, viscoelasticity, and toughness, were improved by virtue of a particular molecular design strategy whereby covalent and non-covalent bond interactions create a cross-linking effect. In addition, the composite hydrogel has important usability properties; it can be injected in liquid format and rapidly transformed into a gel via photo-initiated crosslinking. This was demonstrated on an isolated porcine bladder where the hydrogel closed arbitrarily-shaped tissue defects within 90 s of its application, verifying its effective bioadhesive and sealing properties. This composite hydrogel has great potential for application in bladder injury repair as a tissue-engineering scaffold.
Introduction
The bladder is a hollow organ responsible for urine storage and voiding. Trauma, malignancy, neurogenic diseases, and congenital malformations cause anatomical or functional impairment of the bladder and require treatment involving bladder replacement. However, this surgical approach carries the risk of complications, such as metabolic disturbances, gall bladder stone formation, and urinary fistulae.1–3 Therefore, the search for new methods to achieve bladder repair and regeneration is necessary. Tissue engineering, which integrates seed cells, scaffold materials, and bioactive molecules, offers a possible solution for regenerative medicine,4 and scaffold materials have been extensively studied as an important component of this approach. Hydrogels are considered a preferred material for tissue engineering scaffolds, having a three-dimensional network structure incorporating a large number of hydrophilic groups, and their similarity to natural extracellular matrix.5 However, conventional hydrogels often exhibit poor mechanical properties, such as brittleness and a high swelling ratio. Moreover, because the bladder is exposed to physical activity and undergoes constant deformation, it is susceptible to mechanical forces, such as tensile and compressive forces. Failure of an injured bladder to adapt to dynamic mechanical alterations may increase the probability of local inflammatory reactions owing to leakage.6,7 Therefore, a hydrogel material suitable for bladder reconstruction should satisfy the following four criteria: (1) adaptability in terms of its mechanical properties (i.e. high levels of toughness, tensile strength, viscoelasticity, and fatigue resistance), (2) good biocompatibility, (3) reliable adhesion and sealing properties, and (4) biodegradability.8–10Hydrogels can be classified into natural and synthetic polymeric hydrogels. Natural polymeric hydrogels have strong biocompatibility and biological properties. However, they have disadvantages such as rapid degradation, extended gel formation times and poor mechanical capabilities.11 The mechanical and physicochemical properties of synthetic polymeric hydrogels can be better controlled. But they do not have natural sites of cell attachment and are less biocompatible, limiting their application in tissue engineering.12 The aim of this study is to create a mixture of natural and synthetic polymeric hydrogels suitable for the repair of bladder defects, which combines the benefits of both. From the perspective of bladder repair, gelatine and silk protein, which are important components of the extracellular matrix of bladder tissue, were selected as suitable natural polymers.13,14 Through molecular modification, methacrylate gelatine (GelMA) and methacrylated silk fibroin (SilMA) were synthesised, both having photo-crosslinking properties and good biocompatibility. However, GelMA is prone to mechanical fracture owing to its heterogeneous crosslinking density and extensive level of entanglement,15,16 whilst SilMA suffers from an absence of cell adhesion sites, a tendency to degrade rapidly, and its large swelling ratio.17 The poloxamer Pluronic F127 (PEO99–PPO65–PEO99) is an FDA-approved amphiphilic block copolymer, and F127DA synthetic polymer hydrogel is formed by self-assembly of F127 through hydrophobic association. The hydrogel incorporates a micelle structure with hydrophobic polypropylene glycol (PPO) as the core and hydrophilic polyethylene glycol (PEO) as the shell. It has a well-defined structure with small batch-to-batch variation and low risk of immune response by the human body.18,19 However, the F127 hydrogel network is characterised by its low strength, high level of random molecular entanglement, and hydrophobic interactions between the shell layers, resulting in poor stability and rapid disintegration of the network structure in an aqueous solution. Moreover, the lack of active sites for cellular interactions is not conducive to cell growth, which means its ability for integration with surrounding tissues is poor.20
It has been shown that the introduction of covalent and non-covalent interactions into a hydrogel network is an effective strategy for enhancing a hydrogel's mechanical properties. For example, Qu et al. prepared a hydrogel by blending quaternised chitosan with benzaldehyde-terminated poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide). The Schiff base bonding and PF127 micelle crosslinking in this system, as two dynamic crosslinking modes, resulted in a hydrogel that exhibited good ductility, compressibility, and self-healing ability under multi-cycle deformation conditions.21
In this study, composite hydrogels containing GelMA, SilMA, and F127DA were developed. GelMA and SilMA are covalently crosslinked by photoinitiated vinyl double bonds on their molecular chains to generate a covalent polymer network including two components. Photoinitiated F127DA molecules cause inter-micellar crosslinking, adjacent inter-micellar shell crosslinking and intra-micellar crosslinking, resulting in a dynamic non-covalent polymer network (Scheme 1). The chemical and physical properties of the composite hydrogels were characterised, including their specific chemical structure, equilibrium swelling ratio, and degradability. To investigate the challenges of repairing dynamic soft tissues in complex mechanical environments, the bioadhesive and sealing properties of the hydrogels were compared using a porcine bladder organ model. The biocompatibility of the hydrogels was also investigated to verify the feasibility of their application in soft tissue construction and regeneration.
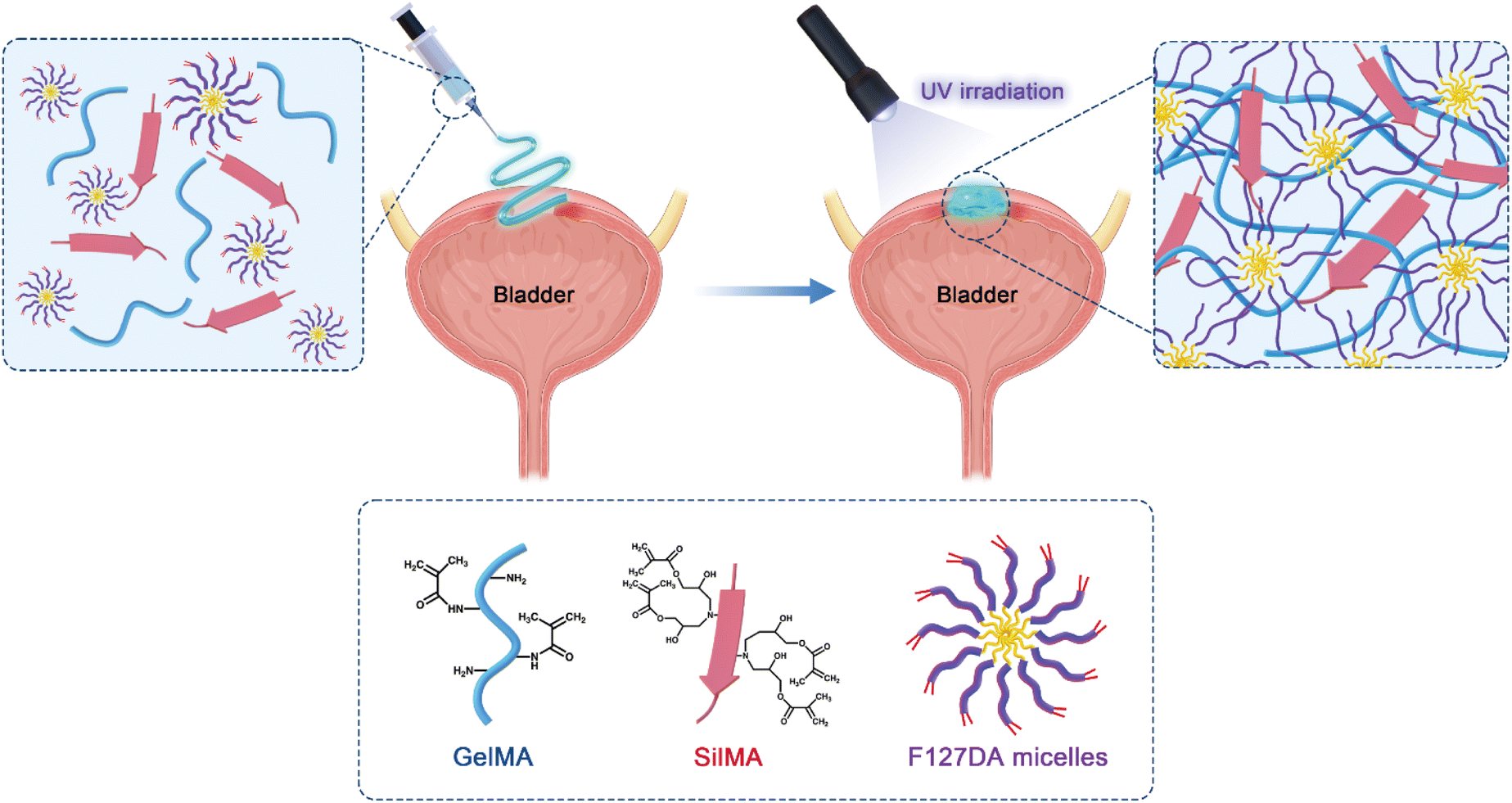 | ||
| Scheme 1 Components of composite hydrogels, formation of hydrogels based on covalent crosslinking and physical micellization initiated by UV irradiation, and a schematic diagram of their application in bladder tissue defects. | ||
Experimental section
Materials
Type A gelatine derived from pig skin was purchased from Beyotime Biotechnology (Shanghai, China). Bombyx mori cocoons were obtained from the Northwest Sericulture Base (Shanxi, China). Methacrylic anhydride (MA) (purity ≥ 94%) and glycidyl methacrylate (GMA) (purity ≥ 97%) were purchased from Aladdin Biochemical Technology (Shanghai, China). F127DA was purchased from Engineering for Life (Suzhou, China). Lithium bromide (LiBr; purity ≥ 99%) was purchased from Innochem Technology (Beijing, China). Type II collagenase was purchased from Worthington Biochemical (New Jersey, USA). Foetal bovine serum (FBS) was purchased from Bovogen Biologicals (Melbourne, Australia). The Cell Counting Kit-8 (CCK-8), Dulbecco's Modified Eagle's Medium (DMEM), trypsin–EDTA, penicillin–streptomycin, and live/dead cell viability/cytotoxicity kits were purchased from Thermo Fisher Scientific (Massachusetts, USA). All the reagents were used without further purification.Synthesis of methacrylate gelatine (GelMA)
GelMA was prepared according to the relevant literature.22–24 Briefly, 10 g of gelatine type A (derived from pig skin) was weighed and added to 100 mL of phosphate-buffered saline (PBS) at 50 °C to prepare a 10% (w/v) homogeneous solution. Subsequently, 1 mL of MA was added dropwise to the gelatine solution and fully reacted in an aluminium foil-covered reaction vessel at 50 °C for 3 h. Once the reaction was completed, the resultant solution was transferred to a dialysis bag (MWCO: 8–14 kDa) and dialysed with deionised water for approximately one week. Water changes were required three times during this period to remove unreacted MA and other by-products. Finally, the dialysis solution was frozen for 10 h and lyophilised with a freeze-dryer for approximately 5 days. The resulting samples were refrigerated at −20 °C until further use. The chemical structure of GelMA was determined by Fourier transform infrared spectroscopy (FTIR, Thermo Scientific Nicolet iS20) and nuclear magnetic resonance spectroscopy (1H NMR, Bruker Avance III HD 500 MHz).Synthesis of methacrylated silk fibroin (SilMA)
SilMA was prepared according to a protocol described in the literature.25 Briefly, cocoons were cut and added to boiling 0.05 M Na2CO3 solution. After 45 min, the degummed cocoons were removed, rinsed until clean with sericin and Na2CO3 prepared with deionised water, and dried in a blast drying oven at 60 °C. Subsequently, 5 g of degummed silk protein fibres were added to 25 mL of 9.3 M LiBr solution and dissolved at 60 °C, until the silk protein fibres could no longer be observed with the unaided eye. Subsequently, the fibres were transferred to a heated magnetic stirrer, and 2 mL of GMA was added dropwise with continuous stirring at 70 °C for 3 h, to fully react GMA with the silk protein. Finally, the resulting solution was dialysed (MWCO: 8–14 kDa) for one week and thereafter lyophilised for 5 days. The resulting samples were refrigerated at −20 °C until further use. The chemical structure of SilMA was determined by FTIR and 1H NMR.Hydrogel preparation
The hydrogels were divided into three groups: GS, GF, and GSF. For the GS hydrogel precursor solution, lyophilised GelMA and SilMA were dissolved in PBS solution at 40 °C, followed by the addition of the photo-initiator lithium trimethylbenzoylphosphinate (LAP). The final precursor hydrogel was composed of 5% (w/v) GelMA and 10% (w/v) SilMA. The GF hydrogel precursor solution, which was prepared in the same way as for GS, contained 5% (w/v) GelMA and 5% (w/v) F127DA. For the GSF hydrogel precursor solution, GelMA and SilMA were first premixed according to set proportions, whereafter F127DA was added until its dissolution, with final concentration being 5% (w/v) GelMA, 10% (w/v) SilMA, and 5% (w/v) F127DA. The concentration of the photo-initiators LAP in the hydrogel precursor solutions of all three groups was 0.25% (w/v). Finally, the hydrogel precursors were irradiated with UV light (405 nm, 100 mW cm−2, 90 s) to yield the composite hydrogel sample.Scanning electron microscopy analyses
Hydrogel samples from the three of the groups were freeze-fixed at −20 °C for 24 h, whereafter they were immediately transferred to a freeze-dryer and lyophilised for 48 h. The morphology and internal structure of the lyophilised samples were observed using scanning electron microscopy (SEM) (Zeiss Gemini 300, Germany) after spraying of cross-sections of the samples with gold particles.Swelling ratio
The swelling properties of the hydrogel samples were studied by equilibrium swelling experiments. The hydrogels were prepared as cylinders (diameter = 12 mm, height = 2 mm), using three samples from each group. The samples were lyophilised in a freeze-dryer and their dry weights (Wd) determined. The samples were then placed in PBS at 37 °C and removed from the buffer for weighing (Wi) at five different time points (0.5, 1, 2, 4, and 8 h). The water on the surfaces of the samples was gently wiped with absorbent paper each time. The swelling ratio was calculated as follows:| Swelling ratio = ((Wi − Wd)/Wd) × 100% | (1) |
Degradation test
The hydrogel samples were prepared according to the method above for the swelling ratio test, and the initial mass of each group of hydrogels (W0) was weighed and recorded. Subsequently, the hydrogel samples were degraded by immersing in PBS containing 2 U mL−1 type II collagenase at 37 °C. At different time points (2, 4, 8, 12, 24, 36, and 48 h), the degraded hydrogels were removed, rinsed with PBS, and weighed (Wt). The degradation ratio was calculated as follows:| Degradation ratio = ((W0 − Wt)/W0) × 100% | (2) |
Rheological properties
The rheological properties were measured using an MCR 302 rheometer (Anton Paar, Austria). First, the kinetic viscosity test was performed (rotational mode, 37 °C) and the shear rate measured in the range of 0.1–1000/s. The injectability of the hydrogel precursor solutions was characterised according to the logarithmic change in shear rate. Subsequently, the crosslinked hydrogels were cut into small discs (diameter = 20 mm, height = 2 mm), and the modulus-frequency relationship curves were measured by frequency scanning (oscillation mode, 37 °C, constant strain of 1%) in the range of 0.1–100 Hz.Mechanical properties
Tensile strength tests were performed using a universal mechanical testing machine equipped with a 1 kN tension sensor. Hydrogel samples from each group were prepared in a dumbbell shape (gauge length = 12 mm, gauge width = 2 mm, gauge thickness = 2.5 mm) using a polytetrafluoroethylene mould and stretched to fracture at a rate of 5 mm min−1. The tensile strength was the maximum stress at fracture, the Young's modulus was determined by the slope of the stress–strain curve, and the fracture toughness was calculated from the area enclosed by the stress–strain curve. Compression tests (using the same mechanical testing machine) were performed on cylindrical samples (diameter = 12 mm, height = 5 mm) of the hydrogel samples from each group with a speed setting of 3 mm min−1. The compression modulus was the slope of the stress–strain curve corresponding to a strain range of 20–30%. A loading-unloading cycle test was performed at a maximum strain of 70%, and the compression and load were recorded for five cycles. The energy loss was derived from the area below the curve for the 5th cycle as follows:| Energy loss ratio = ((area below loading curve − area below unloading curve)/area below loading curve) × 100% | (3) |
The tensile and compression tests were performed in triplicate for all of the hydrogel samples from each group, and the average value was used in the calculations.
Biocompatibility of the hydrogels and wound healing assay
To assess the biocompatibility of the hydrogels, hydrogel samples from each group were prepared as described above and after sterilisation with UV light, were immersed in DMEM (containing 10% FBS and 10 U mL−1 penicillin–streptomycin) for 24 h at 37 °C. The resultant hydrogel extracts were filtered (0.22 μm) to remove biological contaminants. Cell viability was then observed using a live/dead cytotoxicity kit, staining with calcein AM and ethidium homodimer-1 bromelain dimer on days 1, 3, and 5, and imaged with a fluorescence microscope. For further quantitative analyses, the proliferation of adipose-derived stem cells (ADSCs) was assessed using the CCK-8 method. Briefly, ADSCs were inoculated into 96-well plates and incubated (37 °C, 5% CO2, 24 h) to obtain a monolayer of cells. In a subsequent incubation step, the growth medium was replaced with the hydrogel extracts. The extracts were removed from the culture on days 1, 3 and 5, CCK-8 reagent was added, and cell proliferation was calculated by measuring the solution's absorbance at 450 nm using a microplate reader.For the cell scratch experiments, L929 fibroblasts were seeded into 6-well plates and cultured. Once the cell fusion was complete, the tip of a 200 μL pipette was used to scratch the cells perpendicular to the bottom of the wells. Subsequently, 100 μL of hydrogel precursor solution and serum-free medium were mixed and added to the wells, followed by incubation (37 °C, 5% CO2) for 12 and 24 h. Thereafter, the cells were photographed with use of a microscope. The migration area was calculated using ImageJ (version 1.52) software.
| Migration rate = ((W0 h − Wn h)/W0 h) × 100% | (4) |
In vitro tissue adhesion properties: lap-shear test
The lap shear test was performed based on the standard test method for determining the strength properties of tissue adhesives in lap-shear by tension loading (according to ASTM F2255-05). Briefly, fresh porcine bladders were cut into 12.5 mm × 35 mm rectangles and immersed in PBS. Hydrogel precursor solution (250 μL) was injected onto the moistened porcine bladder rectangles, and gelatine-coated slides were placed onto the hydrogel with an overlap area of 12.5 mm × 20 mm. The overlap area was exposed to UV light for 90 s and left at room temperature for 15 min before performing the lap-shear test at a stretch rate of 60 mm min−1. The adhesion properties of the hydrogels were evaluated based on their maximum shear strength at the separation point.In vitro tissue adhesion properties: wound closure test
Fresh porcine bladder tissue was used as the biological tissue to measure the wound closure adhesion strength of the hydrogels. Moistened porcine bladder tissue (20 mm × 50 mm) was sliced in the centre to simulate a damaged wound. Subsequently, hydrogel precursor solution (150 μL) was injected into the middle of the cut. The left and right adhesion areas were equivalent to one another, and the bonding area was 4 × 20 mm. After irradiating the gel with UV light and leaving it at 25 °C for 15 min, the two pieces of tissue were pulled apart in the shear direction at a rate of 60 mm min−1 until the hydrogel completely peeled off. The wound closure adhesion strength was calculated using the following formula:| Wound closure adhesion strength = 2F/S | (5) |
In vitro burst pressure test
A model burst device was designed based on the standard test method for wound closure strength of surgical tissue adhesives and sealants (ASTM F2458-05), and 3D printed from metal. Fresh bladder tissue was cut into circular slices, fixed to the measurement device, and a circular defect (diameter = 2 mm) was created in the centre of the tissue using surgical scissors. Hydrogel precursor solution (50 μL) was injected into the circular defect and exposed to UV light for crosslinking. Finally, the test gas was perfused into the bladder at a constant rate, and the burst pressure was recorded using a pressure transducer. Burst pressure was the peak pressure just before the pressure drop.In vitro organ sealing test
First, a rupture incision (diameter = 3 mm) in the isolated porcine bladder was made using surgical scissors, and then hydrogel precursor solution (100 μL) was injected into the incision and sealed with a UV light source used to induce crosslinking. Pure water was injected into the bladder to observe the hydrogel's sealing effect. Subsequently, test gas was instilled into the bladder at a constant rate using a syringe pump, and the rupture pressure values were recorded using a pressure transducer. The porcine tissues used in all of the above ex vivo experiments were collected from discarded animals in slaughterhouses.Statistical analyses
All numerical results were expressed as the mean ± standard deviation, and all experiments were repeated at least three times. One-way analysis of variance was performed using SPSS (version 26.0) software, followed by multiple comparison tests. P < 0.05 was considered at be statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).Results and discussion
Synthesis and characterisation of hydrogels
To prepare composite hydrogel samples with injectable and bioadhesive properties, GelMA was synthesised using type A gelatine derived from pig skin and MA. FTIR and 1H NMR were used to confirm the modification reaction of gelatine with MA.The FTIR spectrum of GelMA (Fig. S1A†) contained peaks at 1628 (amide I), 1538 (amide II), and 1480 cm−1 (amide III), attributable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching vibrations, N–H bond bending vibrations, and in-plane bending vibrations of C–N and N–H bonds in the chemical structure, respectively.26 The position of the amide peak remained unchanged after MA modification, but the peak intensity in the amide I region increased, attributable to the stress vibration of the methacrylate group's CC bond.27 The twin peaks of GelMA, which were caused by the alkenyl double bond (CC) of the methacrylate groups conjugated to gelatine, could be clearly observed at 5.43 and 5.67 ppm of the 1H-NMR spectrum (Fig. S1B†). The results indicated that GelMA had been successfully synthesised, and a double bond had been introduced.
O bond stretching vibrations, N–H bond bending vibrations, and in-plane bending vibrations of C–N and N–H bonds in the chemical structure, respectively.26 The position of the amide peak remained unchanged after MA modification, but the peak intensity in the amide I region increased, attributable to the stress vibration of the methacrylate group's CC bond.27 The twin peaks of GelMA, which were caused by the alkenyl double bond (CC) of the methacrylate groups conjugated to gelatine, could be clearly observed at 5.43 and 5.67 ppm of the 1H-NMR spectrum (Fig. S1B†). The results indicated that GelMA had been successfully synthesised, and a double bond had been introduced.
The synthesised SilMA was similarly verified using FTIR and 1H NMR (Fig. S2A and B,† respectively). The FTIR spectrum contained peaks at 1642 (amide I), 1511 (amide II), and 1232 cm−1 (amide III) for the silk fibroin and SilMA. The stretching of CHOH in the alcohol group, after opening of the epoxy group of GMA, resulted in a weaker peak at 1293 cm−1. In contrast, the small changes at 950 and 1112 cm−1 are attributable to CH2 vibration and stretching of the methacrylate vinyl groups, respectively.28 In comparison with the 1H NMR spectrum of the silk fibroin, the 1H NMR spectrum of SilMA showed the distinctive resonance of the methacrylate vinyl group at 6.13 and 5.69 ppm as well as the methyl group of GMA at 1.89 ppm were observed in. In summary, the outcomes demonstrate that GMA successfully modified the vinyl on silk fibroin molecules.
SEM analyses
The freeze-dried GF hydrogel was formed into a hard block, and SEM showed the microstructure to have dense stacking without much pore structure, which may be related to the hydrophobic region in the F127DA molecule that hinders the creation of ice crystals during the freeze-drying and is not conducive to the formation of pore structures (Fig. 1A). The internal pore diameters of GS and GSF hydrogels were in the range of 28.04–256.57 and 47.43–307.54 μm, respectively (Fig. S3†). The pore structure provides a channel for the exchange of oxygen, carbon dioxide, and nutrients and provides a suitable microenvironment for cell adhesion, proliferation, differentiation and tissue remodeling.29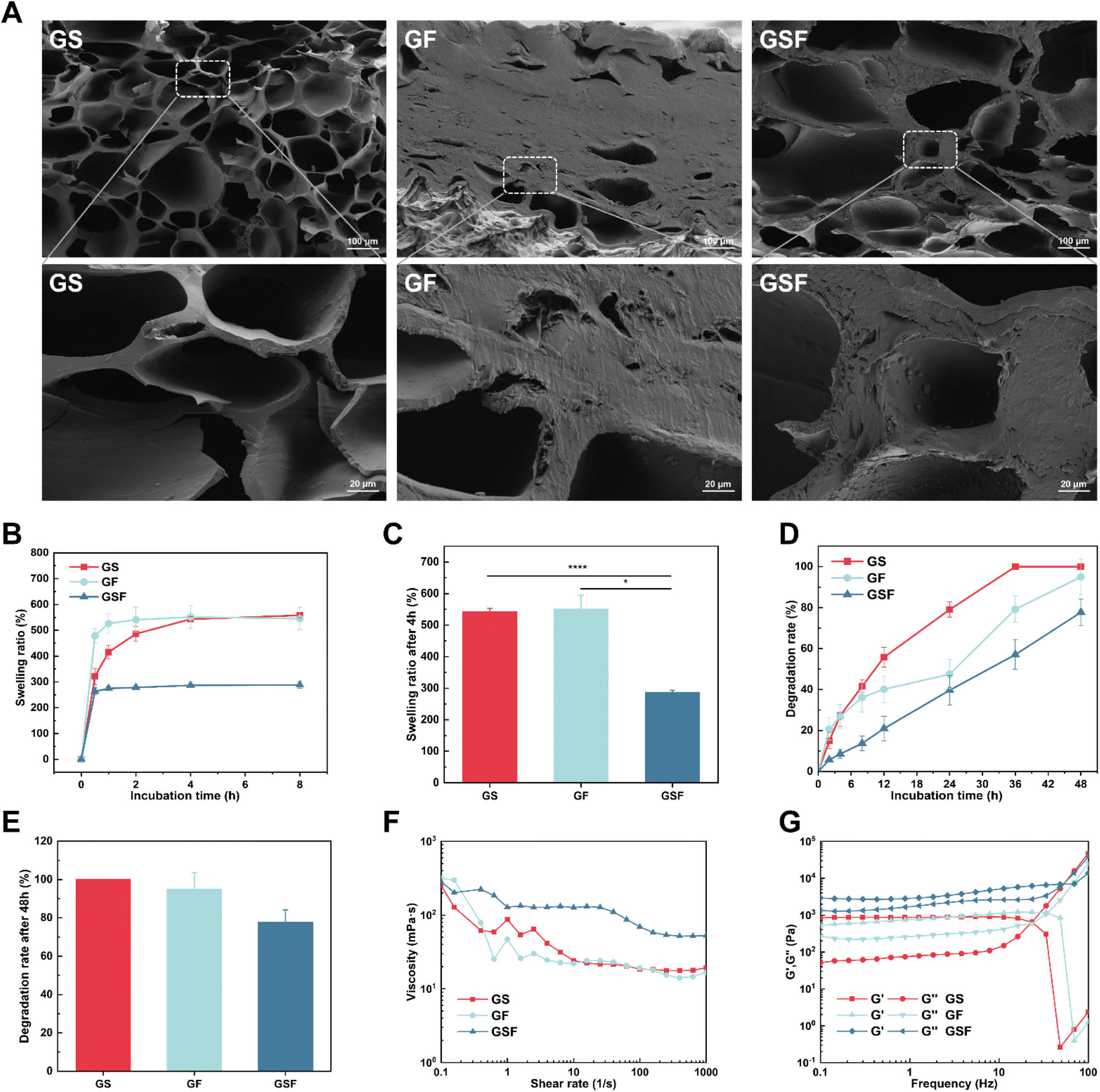 | ||
| Fig. 1 Correlation characterisation of the GS, GF, and GSF hydrogels: (A) SEM images and partially enlarged SEM images of hydrogel morphology, (B) time-dependent swelling curve of hydrogels in PBS (pH = 7.4) at 37 °C, (C) swelling rate of hydrogels after 4 h incubation, (D) time-dependent degradation curve of hydrogels in PBS containing type II collagenase at 37 °C, (E) degradation rate of hydrogels after 48 hours incubation, (F) variation curve of viscosity of hydrogel with shear rate at 37 °C, (G) variation curve of viscoelasticity of hydrogel with frequency at 37 °C and constant strain of%. | ||
Swelling ratio and degradation tests
The swelling and degradation of hydrogels can directly affect tissue repair and regeneration processes. A highly swollen hydrogel form (after water absorption) could cause exertion of stress at the tissue–hydrogel interface, which may result in delamination and peeling off of the adherent gel.30,31 The constant urine-induced swelling of the bladder creates a challenge in terms of an injured bladder (which has compromised mechanical properties) and also in terms of the optimal functioning of bioscaffolds that are in clinical use for bladder repair.32 The swelling ratio test results showed that the GS, GF, and GSF hydrogels reached basic swelling equilibrium after 4 h (Fig. 1B). The equilibrium swelling ratio of the GSF hydrogel (287%) was much lower than that of the other two hydrogels, which may be related to the higher crosslinking density of its hydrogel network structure (Fig. 1C). Therefore, the GSF hydrogel with its low swelling ratio exhibited better shape retention and was more suitable for repair and regeneration after bladder injury.Suitable degradation properties provide sufficient space for cell migration and proliferation and a sufficient area for new tissue growth.33 In the current study, we performed an in vitro enzymatic degradation assay using type II collagenase. All hydrogels exhibited biodegradation over time; however, the GS hydrogel had the fastest degradation rate and was completely degraded at 36 h. The degradation rate of the GSF hydrogel was the slowest, with 22.3% of the hydrogel remaining undegraded at 48 h (Fig. 1D and E). From the results, the fully naturally derived hydrogels degraded the fastest in the presence of collagenase, whilst the degradation rate of hydrogels introduced with nanomicelles was relatively slow. An appropriate degradation rate needs to match the rate of tissue regeneration in an organism. The composite hydrogels exhibited tuneable biodegradability, and the optimal degradation rate needs to be verified by subsequent animal studies.
Rheological properties
Rheological tests were performed on each of the three hydrogels groups to investigate their injectability and viscoelastic properties. Shear thinning is a characteristic parameter in determining injectability.34,35 Fig. 1F shows that the hydrogel precursor fluids demonstrate fluid-like flow and also shear-thinning characteristics under a shear force. This indicates that all three groups of hydrogel precursors can be injected into arbitrarily shaped defect sites to provide effective adhesion for non-flat complex-geometry wounds.Viscoelasticity is a universal characteristic of many tissues and extracellular matrices and affects the function and behaviour of cells during regeneration;35 the bladder itself has viscoelastic characteristics.36 As shown by the oscillatory frequency scan test (Fig. 1G), the energy storage modulus G′ was higher than the energy loss modulus G′′ for all of the hydrogels, in a certain frequency scan range, exhibiting solid-like characteristics and forming a stable network structure. Among them, the GSF hydrogel had the highest G′, indicating that it had relatively stronger mechanical properties.
Mechanical properties
The bladder has mechanical properties (i.e. basic stiffness and flexibility) and stability, allowing it to resist the mechanical forces and frequent deformations to which it is subjected.37 The composite hydrogel material therefore also needs to have good tensile properties to meet the cyclic urinary storage and voiding requirements of the bladder. In this study, tensile tests were performed on the hydrogels. The strain–stress curves in Fig. 2A show that the incorporation of both SilMA and F127DA improved the brittleness of GelMA. Tensile strains of 139.7 and 147.6% just prior to the breaking point were achieved for the GS and GF hydrogels, respectively (Fig. 2B). The inter-penetrating network and dissipative units significantly increased the tensile strength from 21.4 kPa to 78.7 kPa while imparting the corresponding tensile strain (126.3%) (Fig. 2D).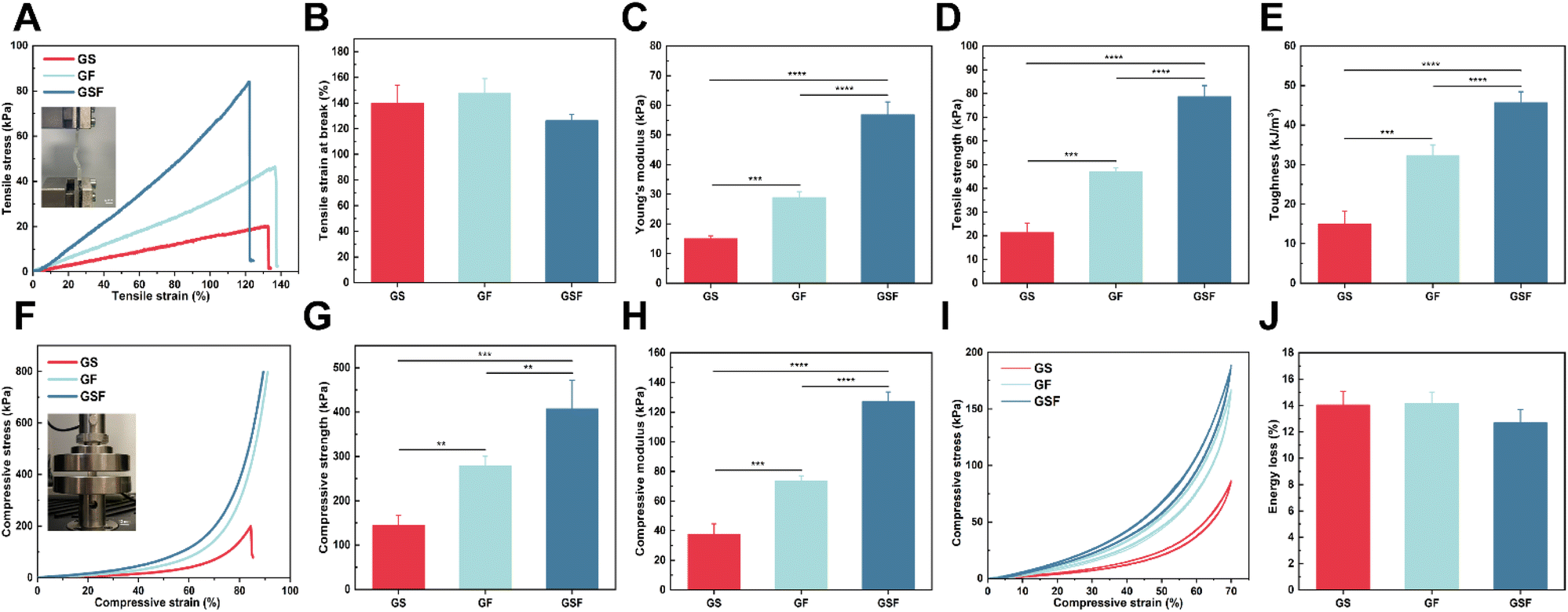 | ||
| Fig. 2 Mechanical properties of the GS, GF, and GSF hydrogels: (A) representative tensile stress–strain curves of hydrogels, (B) tensile strain at break, (C) Young's modulus, (D) tensile strength, (E) toughness, (F) representative compressive stress–strain curves of hydrogels, (G) compressive strength (the compression strain is set to 80%), (H) compressive modulus, (I) representative compressive cyclic loading-unloading curves, (J) energy loss of the 5th cycle of hydrogels. | ||
The Young's modulus of the GS and GF hydrogels were 15 and 29 kPa, respectively. The Young's modulus of the GSF hydrogel reached 56.9 kPa due to the increase in crosslinked network density (Fig. 2C). In addition, the toughness of the GS, GF, and GSF hydrogels was 15.01, 32.25, and 45.67 kJ m−3, respectively (Fig. 2E). These experimental results were superior to those of hydrogel materials previously used in bladder tissue engineering studies.38,39 It was also demonstrated that the addition of SilMA and F127DA micelles at appropriate concentrations imparted excellent tensile strength and toughness to the hydrogels.
It is well known that bladder position changes with body posture. When faced with unexpected situations, such as impact, the bladder, as a load-bearing soft tissue, needs to resist the pressure exerted by the surrounding tissues to ensure the organ does not rupture.40 From the stress–strain curves of compression test (Fig. 2F), it is evident that the introduced F127DA nanomicellar network is key to enhancing compressive resistance. During compression at the set maximum strain, both the GF and GSF hydrogels showed no rupture damage. In contrast, the GS hydrogel commonly ruptured when the compressive strain reached approximately 86%. At an 80% compressive strain, the GSF hydrogel exhibited the highest compressive strength (Fig. 2G). Compared with the other two hydrogels, the compressive modulus of GSF hydrogels increased from 37.6 to 127.1 kPa (Fig. 2H). The above results indicate that these crosslinking networks, including inter-micellar crosslinking, inter-micellar shell crosslinking, intra-micellar crosslinking, and covalent crosslinking, synergistically enhance the hydrogel's compressive properties. The hydrogels exhibited improved strength while maintaining their initial flexibility. Subsequently, cyclic compression loading-unloading tests were performed. Owing to the reversible nature of the hydrophobic association, the recovered hydrogel showed a stress–strain curve similar to that of the original hydrogel during five loading-unloading cycles with 70% strain applied (Fig. 2I). This proves that the structure can be recovered after conformational deformation. In other words, the composite hydrogel exhibited excellent fatigue resistance and self-recovery (Fig. 2J).
Biocompatibility of the hydrogels and wound healing assay
Cell survival and proliferation are essential for tissue repair and regeneration. To assess the biocompatibility of the hydrogels, live/dead cell staining and CCK-8 assays were used to investigate the effect of the hydrogels on cell viability and proliferation of ADSCs. Fig. 3A shows the fluorescence images of live and dead cells cultured in the hydrogel medium for 1, 3, and 5 days, for each of the three hydrogel groups. Compared with the control group, the addition of SilMA to GelMA had no effect on cell viability and indeed displayed cell growth-promoting effect. Although F127DA is an FDA-approved nanomaterial with good bio-descriptive properties, the results showed that it had a slight inhibitory effect on cell proliferation; therefore, cell proliferation in the GF and GSF hydrogels was slightly lower than that in the GS hydrogel. The subsequent CCK-8 test also showed that the hydrogels of all three groups promoted cell proliferation (Fig. 3B), and the cell viability was maintained above 80% within 5 days (Fig. 3C). According to the ISO 10993-5:2009 test for biological evaluation of medical devices, the relative value-added rate of biomedical materials to cells should be not less than 70%, which means that the hydrogels of all three groups have good biocompatibility and hence can provide a favourable growth microenvironment for cells.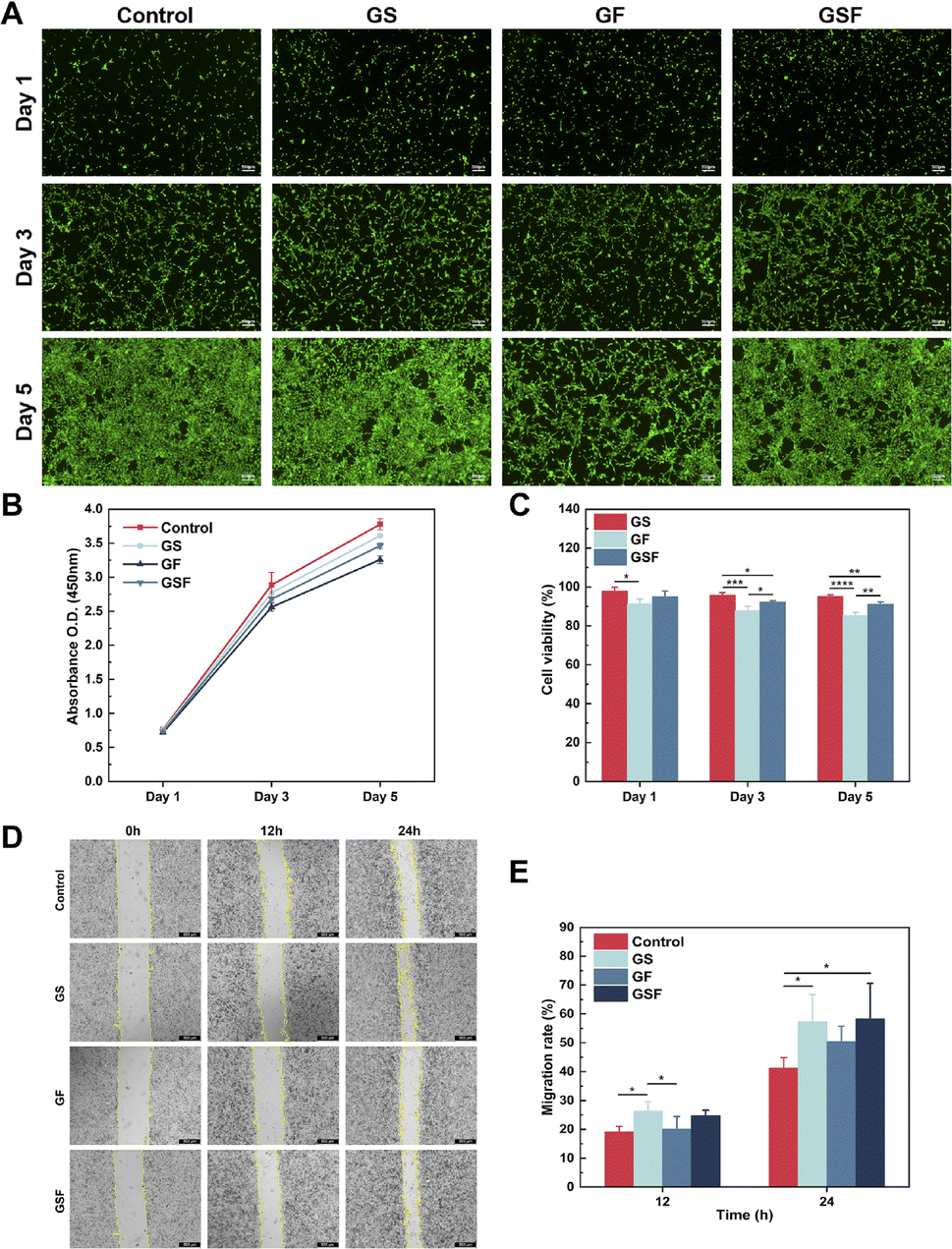 | ||
| Fig. 3 Biocompatibility evaluation of the GS, GF, and GSF hydrogels: (A) live/dead staining fluorescence images of ADSCs cells in hydrogel abstracts after 1, 3, and 5 days of incubation, (B) growth profile of ADSCs cells in hydrogel abstracts, (C) viability quantification of ADSCs cells in hydrogel extracts after 1, 3, and 5 days of incubation, (D) images of migration of L929 fibroblasts to the scratched area after hydrogel treatment, (E) migration rate of L929 fibroblasts after hydrogel treatment. | ||
In addition, cell scratching experiments were performed with a co-mix of hydrogel precursor solution and serum-free medium to assess the potential facilitation of cell migration by the hydrogels, using L929 fibroblasts. The results showed that L929 fibroblasts migrated from the edge of the scratch towards the blank area over time (Fig. 3D). Both the GS and GSF hydrogels induced higher migration rates than the blank control and the GF hydrogel, at both the 12 h and 24 h observation points, probably because the protein component of the hydrogel recruited more cells (Fig. 3E). This could enhance the integrated regeneration between scaffold materials and tissues in vivo.
In vitro tissue adhesion properties: lap-shear test
Adhesive properties of hydrogels are necessary for the repair of tissue breakage. As shown in Fig. S3,† hydrogels can bind to major organs, including the liver, lung, and kidney, without gravity-induced dislodgment. Lap-shear tests were performed on the hydrogels of all three groups using lengths of bladder tissue, in order to confirm their potential ability for use during repair of broken bladder tissue (Fig. 4A–D). The contact surface was a gelatine-coated slide, which may not sufficiently reproduce adhesion in real scenarios. However, the final results showed that the hydrogels nonetheless exhibited adhesion properties equal to or better than that of commercially available fibrin glue sealant.41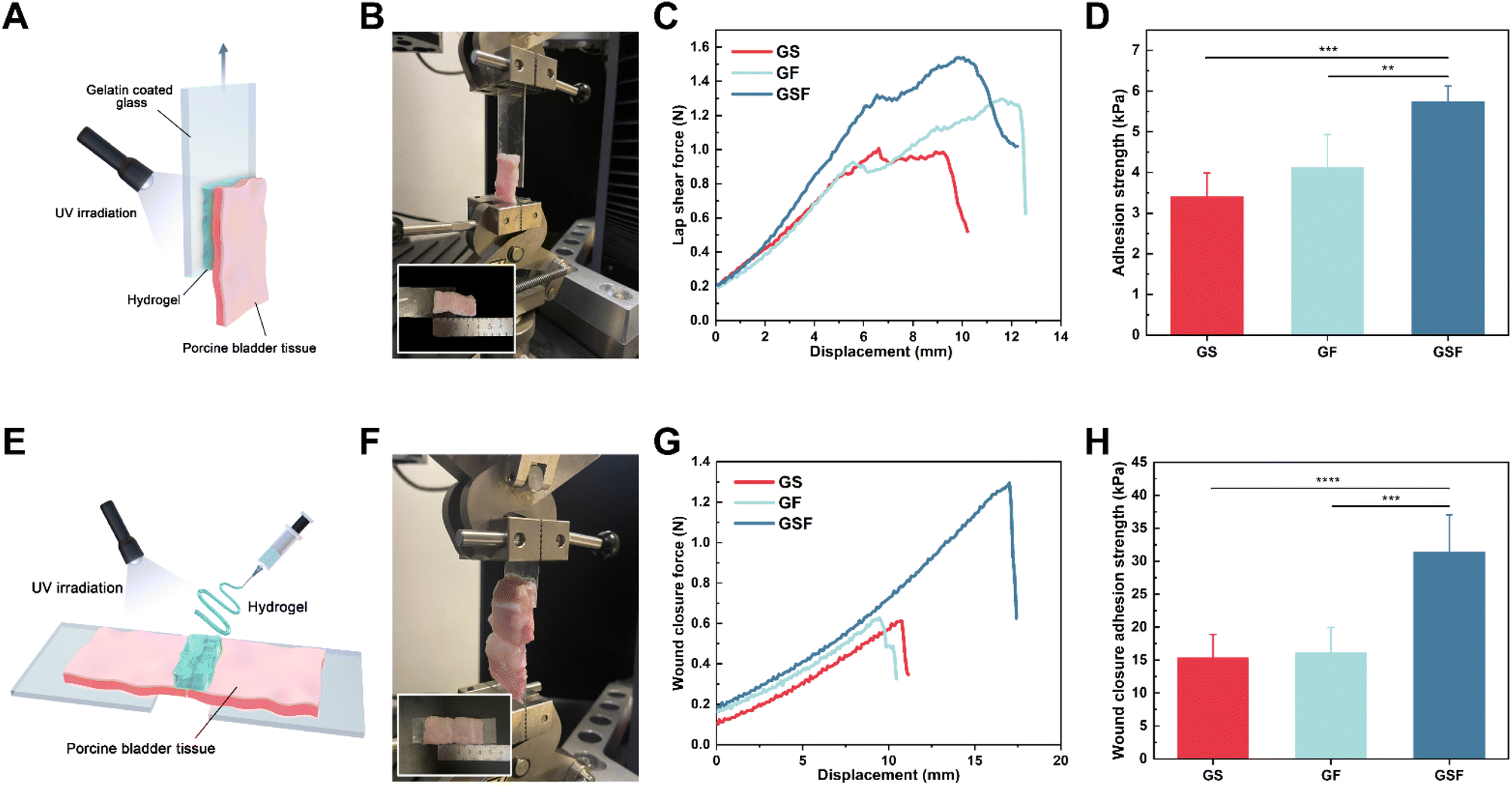 | ||
| Fig. 4 Evaluation of bioadhesive properties of the GS, GF and GSF hydrogels: (A) schematic diagram of in vitro lap-shear test, (B) image of in vitro lap shear evaluation, (C) variation curve of lap shear force–displacement of hydrogels, (D) adhesion strength in shear loading mode, (E) schematic diagram of wound closure test, (F) image of wound closure evaluation, (G) variation curve of force–displacement of wound closure test, (H) adhesion strength of wound closure. | ||
In vitro tissue adhesion properties: wound closure test
Fig. 4E shows a schematic of the in vitro wound closure test, which involves a tensile test that is performed after injection of hydrogel into the middle region of a long strip of torn bladder tissue. The wound-closure force versus displacement curve is shown in Fig. 4G. Adhesion failure occurs when the hydrogel detaches from bladder tissue. The closure strength was calculated based on maximum stress and the adhesive area of the hydrogel. The results in Fig. 4H show that the closure strengths of the GS and GF hydrogels were similar at 15.32 and 16.08 kPa, respectively. The closure strength of the GSF hydrogel increased to 31.42 kPa, which was approximately 95% higher than that of the other two groups. The strength of adhesion properties is determined by the surface adhesion and strong cohesion. The former ensures the stability of the binding interface and prevents peeling. The latter can maintain the structural stability of the hydrogel and resist fracture when large displacements are encountered.42 On the one hand, the strong adhesion properties of the GSF hydrogel are attributed to the covalent cross-linking network and the energy dissipation mechanism provided by F127DA. They both toughen the hydrogel in terms of its mechanical properties to achieve a higher cohesive strength. On the other hand, the hydrogen bonds generated between GelMA and the surface of amine-rich biological tissues, as well as the covalent bonding of amine–MA and thiol–MA groups.24 The unusual amino acid sequence, conformational transition from a random curl to a β-sheet and the intermolecular van der Waals forces in the SilMA structure also enhanced adhesion.43 Finally, the hydrogen bonds that the free PEO segment in F127DA can form with protein-rich substrates, and the hydrophobic interactions provided by the PPO segment with the cell membrane, promote the adhesion properties of the hydrogel.44In vitro burst pressure test
Hydrogels that bind tightly to tissues can complement or even replace sutures and staples. The burst pressure is a key metric for assessing the sealing properties of hydrogels for bladder repair. To quantify the bursting pressure, we designed and built a test device (Fig. 5A) using 3D printed metal, with reference to the standard test method for wound closure strength of tissue adhesives and sealants (ASTM F2458-05). A rupture notch was created in circular-shaped bladder tissue, whereafter hydrogel precursor solution was injected into the notch and photo-crosslinked. When the sealed hydrogel was damaged or peeled away the pressure dropped rapidly. With use of a pressure transducer, the burst pressure was recorded as 20.98, 21.24 and 32.23 kPa for the GS, GF, and GSF hydrogels, respectively (Fig. 5C). The GSF hydrogel, which can maintain a high interfacial bond strength, was not susceptible to overall fracture under peeling pressure. The results of this test are consistent with the results of the tensile stress, fracture toughness, and shear adhesion tests, which further verifies that the hydrogels' adhesion performance depends not only on the interfacial adhesion between the hydrogel and the tissue, but also on the cohesion of the hydrogel itself.45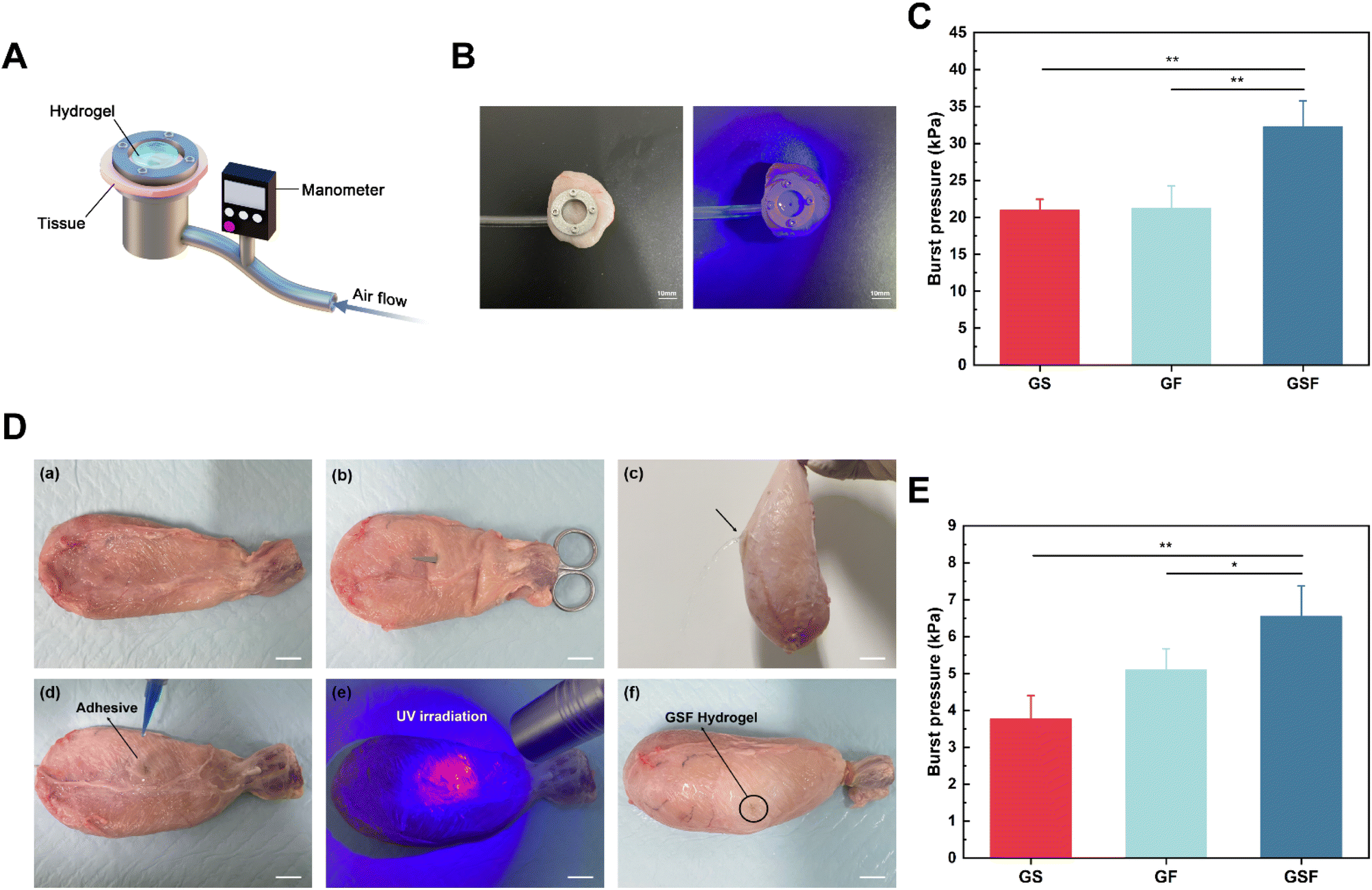 | ||
| Fig. 5 In vitro sealing performance evaluation of the GS, GF and GSF hydrogels: (A) schematic diagram of in vitro bursting pressure device, (B) burst pressure assessment image of fresh porcine bladder biotissue, (C) burst pressure values, (D) porcine bladder incision model: images showing (D, a) fresh porcine bladder, (D, b) superficial wound created with surgical scissors, (D, c) showing leakage, (D, d) hydrogel covering wound, (D, e) photo-crosslinking, (D, f) sealed bladder observed after filling with deionised water; scale bar = 10 mm, (E) burst pressure after filling with gas. | ||
In vitro organ sealing test
The sealing performance of the composite hydrogels was verified using isolated fresh porcine bladders to simulate the repair of a broken bladder in a realistic scenario. First, we created a breach notch using surgical scissors (Fig. 5Da–c), and confirmed the sealing properties of the applied hydrogels by instilling deionised water onto the test bladder (Fig. 5Dd–f). The composite hydrogels adhered to the bladder tissue under moist conditions as well as under various dynamic positions (vertical, inverted, and non-planar) where they resisted the effects of gravity. Subsequently, quantification of effective sealing was performed by filling the bladder with test gas. The results (Fig. 5E) show that the composite hydrogels influenced the maximum pressure that the sealed bladders could withstand. The burst pressure for the GS hydrogel was relatively low (∼3.77 kPa), whereas the addition of F127DA increased its burst pressure by ∼5.1 kPa, and the addition of SilMA and F127DA further enhanced the burst pressure value by ∼6.5 kPa. It has been reported in the literature that the bladder's location has a relatively stable, low-pressure range of 0–4 kPa.46 Therefore, the GF and GSF hydrogels have suitable structural stability and can fully withstand pressure variations within a clinically-acceptable range.Conclusion
In this study, we successfully prepared a composite hydrogel (GSF hydrogel), consisting of methacrylate gelatine (GelMA), methacrylated silk fibroin (SilMA), and Pluronic F127 diacrylate (PEO99–PPO65–PEO99) (F127DA). The hydrogel has injectability as well as mechanical and bioadhesive properties that are suitable for its clinical use, based on the dynamic organ of the bladder. The hydrogel has a network structure in which GelMA and SilMA are covalently crosslinked, and F127DA is physically connected between and within the micelles. This enhances the tensile-, compressive-, toughness-, and fatigue-resistant properties of the GSF hydrogel, providing effective structural support for the bladder organ during cyclic changes or large stress–strain encounters. During in vitro testing with a porcine bladder organ model, the composite hydrogel could rapidly adhere to and close slits or breaks in the bladder tissue. Chemical modification of the injector-applied hydrogel precursor via photo-initiation imparts crosslinking properties to the hydrogel, which combines with the hydrogel's covalent, hydrogen, and hydrophobic bonding interactions with biological tissues. In addition, the GSF hydrogel exhibited low swelling rate, good biodegradability, and excellent biocompatibility. Therefore, the prepared composite GSF hydrogel provides a new option for scaffold materials in the field of bladder tissue engineering and holds great potential for application in the sutureless repair and regeneration of an injured bladder.Author contributions
Z. Y. F.: experimental design, experimental operation, data collection and analysis, and manuscript writing; S. W. X., P. C. W., J. Z., and Z. Y. A.: manuscript revision; Z. Y. A., and J. P. S.: technical and material support; X. Z.: supervision; W. J. F.: experimental design, manuscript revision, and financial support.Conflicts of interest
All authors of this article declare no conflict of interest.Acknowledgements
This study was supported by funding from the National Natural Science Foundation of China (82270721, 81873600), Natural Science Foundation of Beijing Municipality (7182154), and Natural Science Foundation of Hainan Province (20158299).Notes and references
- M. Horst, D. Eberli, R. Gobet and S. Salemi, Front. Pediatr., 2019, 7, 91 CrossRef PubMed.
- S. T. Bazargani, H. Djaladat, H. Ahmadi, G. Miranda, J. Cai, A. K. Schuckman and S. Daneshmand, Eur. Urol. Focus, 2018, 4, 889–894 CrossRef PubMed.
- T. Drewa, J. Adamowicz and A. Sharma, Nat. Rev. Urol., 2012, 9, 561–572 CrossRef CAS PubMed.
- O. Lam Van Ba, S. Aharony, O. Loutochin and J. Corcos, Adv. Drug Delivery Rev., 2015, 82–83, 31–37 CrossRef CAS PubMed.
- F. Xu, C. Dawson, M. Lamb, E. Mueller, E. Stefanek, M. Akbari and T. Hoare, Front. Bioeng. Biotechnol., 2022, 10, 849831 CrossRef PubMed.
- S. Korossis, F. Bolland, E. Ingham, J. Fisher, J. Kearney and J. Southgate, Tissue Eng., 2006, 12, 635–644 CrossRef PubMed.
- D. Eberli, L. F. Filho, A. Atala and J. J. Yoo, Methods, 2009, 47, 109–115 CrossRef CAS PubMed.
- F. Ajalloueian, G. Lemon, J. Hilborn, I. S. Chronakis and M. Fossum, Nat. Rev. Urol., 2018, 15, 155–174 CrossRef PubMed.
- M. Casarin, A. Morlacco and F. Dal Moro, Int. J. Mol. Sci., 2022, 23, 6360 CrossRef CAS PubMed.
- M. Hanczar, M. Moazen and R. Day, Int. J. Mol. Sci., 2021, 22, 12657 CrossRef PubMed.
- Z. Bao, C. Xian, Q. Yuan, G. Liu and J. Wu, Adv. Healthcare Mater., 2019, 8, e1900670 CrossRef PubMed.
- S. Naahidi, M. Jafari, F. Edalat, K. Raymond, A. Khademhosseini and P. Chen, J. Controlled Release, 2013, 166, 182–194 CrossRef CAS PubMed.
- T. G. Tuttle, D. R. Morhardt, A. A. Poli, J. M. Park, E. M. Arruda and S. Roccabianca, J. Biomech. Eng., 2021, 143, 111007 CrossRef PubMed.
- W. Sun, D. A. Gregory, M. A. Tomeh and X. Zhao, Int. J. Mol. Sci., 2021, 22, 1499 CrossRef CAS PubMed.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- H. Montazerian, A. Baidya, R. Haghniaz, E. Davoodi, S. Ahadian, N. Annabi, A. Khademhosseini and P. S. Weiss, ACS Appl. Mater. Interfaces, 2021, 13, 40290–40301 CrossRef CAS PubMed.
- B. Kundu, R. Rajkhowa, S. C. Kundu and X. Wang, Adv. Drug Delivery Rev., 2013, 65, 457–470 CrossRef CAS PubMed.
- P. Ren, H. Zhang, Z. Dai, F. Ren, Y. Wu, R. Hou, Y. Zhu and J. Fu, J. Mater. Chem. B, 2019, 7, 5490–5501 RSC.
- Y. N. Sun, G. R. Gao, G. L. Du, Y. J. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS PubMed.
- E. Gioffredi, M. Boffito, S. Calzone, S. M. Giannitelli, A. Rainer, M. Trombetta, P. Mozetic and V. Chiono, Procedia CIRP, 2016, 49, 125–132 CrossRef.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed.
- S. R. U. Rehman, R. Augustine, A. A. Zahid, R. Ahmed, M. Tariq and A. Hasan, Int. J. Nanomed., 2019, 14, 9603–9617 CrossRef PubMed.
- W. Xiao, J. Li, X. Qu, L. Wang, Y. Tan, K. Li, H. Li, X. Yue, B. Li and X. Liao, Mater. Sci. Eng., C, 2019, 99, 57–67 CrossRef CAS PubMed.
- M. Tavafoghi, A. Sheikhi, R. Tutar, J. Jahangiry, A. Baidya, R. Haghniaz and A. Khademhosseini, Adv. Healthcare Mater., 2020, 9, e1901722 CrossRef PubMed.
- J. B. Costa, J. Park, A. M. Jorgensen, J. Silva-Correia, R. L. Reis, J. M. Oliveira, A. Atala, J. J. Yoo and S. J. Lee, Chem. Mater., 2020, 32, 8733–8746 CrossRef CAS PubMed.
- S. Sreekumaran, A. Radhakrishnan, A. A. Rauf and G. M. Kurup, Prog. Biomater., 2021, 10, 43–51 CrossRef CAS PubMed.
- K. Rahali, G. Ben Messaoud, C. J. F. Kahn, L. Sanchez-Gonzalez, M. Kaci, F. Cleymand, S. Fleutot, M. Linder, S. Desobry and E. Arab-Tehrany, Int. J. Mol. Sci., 2017, 18, 2675 CrossRef PubMed.
- S. H. Kim, Y. K. Yeon, J. M. Lee, J. R. Chao, Y. J. Lee, Y. B. Seo, M. T. Sultan, O. J. Lee, J. S. Lee, S. I. Yoon, I. S. Hong, G. Khang, S. J. Lee, J. J. Yoo and C. H. Park, Nat. Commun., 2018, 9, 1620 CrossRef PubMed.
- Y. Zhu, Q. Zhang, X. Shi and D. Han, Adv. Mater., 2019, 31, e1804950 CrossRef PubMed.
- D. G. Barrett, G. G. Bushnell and P. B. Messersmith, Adv. Healthcare Mater., 2013, 2, 745–755 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, F. Ma, X. Liu, Y. Liu, Y. Cao and R. Pei, J. Mater. Chem. B, 2022, 10, 915–926 RSC.
- M. Yang, Y. Zhang, C. Fang, L. Song, Y. Wang, L. Lu, R. Yang, Z. Bu, X. Liang, K. Zhang and Q. Fu, Adv. Mater., 2022, 34, e2109522 CrossRef PubMed.
- M. Sarem, F. Moztarzadeh and M. Mozafari, Carbohydr. Polym., 2013, 93, 635–643 CrossRef CAS PubMed.
- S. Bian, Z. Zheng, Y. Liu, C. Ruan, H. Pan and X. Zhao, J. Mater. Chem. B, 2019, 7, 6488–6499 RSC.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- S. C. Barnes, D. E. Shepherd, D. M. Espino and R. T. Bryan, J. Mech. Behav. Biomed. Mater., 2015, 42, 168–176 CrossRef CAS PubMed.
- M. Casarin, A. Morlacco and F. Dal Moro, Processes, 2021, 9, 1643 CrossRef CAS.
- S. Sivaraman, R. Ostendorff, B. Fleishman and J. Nagatomi, J. Biomater. Sci., Polym. Ed., 2015, 26, 196–210 CrossRef CAS PubMed.
- S. A. Bernal-Chavez, S. Alcala-Alcala, Y. S. Tapia-Guerrero, J. J. Magana, M. L. Del Prado-Audelo and G. Leyva-Gomez, RSC Adv., 2022, 12, 21713–21724 RSC.
- M. S. Jokandan, F. Ajalloueian, M. Edinger, P. R. Stubbe, S. Baldursdottir and I. S. Chronakis, J. Mech. Behav. Biomed. Mater., 2018, 79, 92–103 CrossRef PubMed.
- E. Lih, J. S. Lee, K. M. Park and K. D. Park, Acta Biomater., 2012, 8, 3261–3269 CrossRef CAS PubMed.
- H. Yuk, T. Zhang, S. Lin, G. A. Parada and X. Zhao, Nat. Mater., 2016, 15, 190–196 CrossRef CAS PubMed.
- R. Tutar, E. Yuce-Erarslan, B. Izbudak and A. Bal-Ozturk, J. Mater. Chem. B, 2022, 10, 2912–2925 RSC.
- H. Zhong, Z. Li, T. Zhao and Y. Chen, ACS Biomater. Sci. Eng., 2021, 7, 4828–4837 CrossRef CAS PubMed.
- H. Fan, J. Wang, Q. Zhang and Z. Jin, ACS Omega, 2017, 2, 6668–6676 CrossRef CAS PubMed.
- L. Chen, T. Q. Wei, Y. Wang, J. Zhang, H. Li and K. J. Wang, J. Urol., 2012, 188, 661–667 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00402c |
| This journal is © The Royal Society of Chemistry 2023 |