
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the performance of Ni nanoparticle modified carbon felt towards glycerol electrooxidation: impact of organic additive
Mohamed E. Ghaith ,
Muhammad G. Abd El-Moghny*,
Hafsa H. Alalawy,
Mohamed E. El-Shakre and
Mohamed S. El-Deab*
,
Muhammad G. Abd El-Moghny*,
Hafsa H. Alalawy,
Mohamed E. El-Shakre and
Mohamed S. El-Deab*
Chemistry Department, Faculty of Science, Cairo University, Egypt. E-mail: mugamal@cu.edu.eg; msaada@sci.cu.edu.eg
First published on 6th April 2023
Abstract
Organic additives are widely used in the deposition baths of metals and alloys thanks to their special function which affects the growth and the building of the crystal. This study investigates the effect of glycerol on Ni deposition onto carbon felt (CF) and its effect on the catalytic activity towards glycerol electrooxidation. The impact of glycerol on the morphology, distribution, and particle size of the electrodeposited Ni is disclosed using a scanning electron microscope (SEM). X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and cyclic voltammetry (CV) techniques were used to probe the possible changes of the electrodeposited Ni oxide phases. Electrochemical measurements show that the as-synthesized Ni0.05@CF electrocatalyst prepared in the presence of 50 mM glycerol has a marked activity towards glycerol electrooxidation, as confirmed by the impressive increase of the oxidation current by about 1.6 times concurrently with a favorable negative shift of its onset potential. Moreover, the charge transfer resistance (Rct) is much reduced from 140 to 87 ohm. The addition of glycerol to the deposition bath is believed to retard the growth of the formed Ni deposits while enhancing the nucleation rate and thus increases the particle density and, consequently, the distribution of deposited Ni over the entire CF is improved along with increasing the surface concentration and surface-active sites. This assumption is supported by density functional theory (DFT) calculations.
1. Introduction
Direct alcohol fuel cells (DAFCs) are one of the recent technologies suggested for supplying sustainable energy because of the abundance of feeding fuels.1 The most common fuel used is methanol owing to its relatively cheap cost, being consistently available, and having high energy density (4820 W h L−1), good electrical activity, and biodegradability. However, it is toxic with poor stability and high volatility in addition to a crossover problem. Other fuels have been used, e.g. ethanol, due to its being nontoxic with high-energy density (6280 W h L−1), although it is difficult to undergo complete oxidation to CO2 at low temperatures due to its strong C–C bond. Considering this, glycerol seems to be promising to use as a fuel in DAFC in view of its nontoxicity, and cheap cost because it is considered a by-product of many processes especially biodiesel production, low flammability, low volatility, low crossover, and high energy density (6400 W h L−1).2,3 Additionally, the complete oxidation of glycerol generates 14 electrons producing many oxidized products. The overproduction of glycerol per year secures it as an alternative fuel of low cost. Moreover, the presence of the three hydroxyl groups in glycerol allows to produce of more valuable products in many ways and electrooxidation production is found to be the easiest way from which many valuable products can be obtained such as glyceraldehyde and dihydroxyacetone, glyceric acid, mesoxalic acid, tartronic acid, and glycolic acid. Remarkably, glycerol can be used to assist the anodic reaction in the water electrolysis process which is thermodynamically more favored than the oxygen evolution reaction to save the input energy.3–7Nickel-based catalysts have been reported previously as excellent catalysts for several applications, especially in electrocatalysis such as water splitting,8–15 fuel cells,16–19 oxidation of many organic molecules (methanol, ethanol, glucose, urea, and glycerol).20–37 This is attributed to its high stability in alkaline medium, low cost, high electro-catalytic activity, good electrical conductivity, and ease of active phase formation. Moreover, nickel-based electrocatalysts can be used as cheap and efficient electrocatalysts for DAFCs and become a substitute for expensive noble metals.28
The mechanism of the glycerol electrooxidation over the Ni catalyst according to Fleischmann et al. mechanism is illustrated in eqn (1)–(4):38
| Ni + 2OH → Ni(OH)2 + 2e− | (1) |
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (2) |
| NiOOH + RCH2OH + OH− → Ni(OH)2 + H2O + RCHO + e− | (3) |
| RCHO + 3OH− → RCOO− + 2H2O + 2e− | (4) |
From the previously mentioned mechanism, the active component in the Ni-based catalysts can be considered to be NiOOH.38
Various strategies are used to improve the catalytic activity of Ni electrocatalyst toward glycerol electro-oxidation. For instance, the addition of another metal such as Co, Cu, Fe, Bi, and Cr.39–42 Habibi et al.43 reported enhanced electrocatalytic activity of Ni by about 1.6 and 1.2 times via simple alloying of Ni with Co or Cu, respectively. El Nagar et al. observed that the insertion of Ni into Cu dendrites significantly enhances the activity and stability of the catalyst.44 Moreover, modification of the surface of the catalyst or substrate improves the morphology and distribution of Ni. Ghaith et al.45 showed that the catalytic activity of Ni was enhanced by 2.5 times via electrochemical treatment of the substrate in 1 M sulfuric acid. Houache et al.34 improved the catalytic performance of the Ni by 9 times after surface treatment of the catalyst by using a sinusoidal wave. Another way to improve the catalytic activity of Ni-based electrocatalysts could be achieved via controlling the rate of nucleation and growth of Ni during its electrodeposition. This can be verified by adjusting the electrodeposition bath chemistry by adding, e.g., organic molecules containing O, N, and/or S which form free radical adsorbed on the electrodeposited metal and at the same time adsorbed on the substrate thus addition of the organic molecules retards the rate of growth and enhances the nucleation rate consequently forming smaller particle size with the homogeneous distribution.46–48
Catalyst' supporting materials play a vital role in determining the performance of the catalyst where the main parameters affecting the efficiency of the supporting material are surface area, surface functional groups, porosity, electrical conductivity, and electrochemical stability.49–51 Carbon-based materials such as reticulated vitreous carbon (RVC), carbon sponge (CS), carbon felt (CF) or carbon paper (CP) are suitable candidates thanks to their high surface area, excellent porosity, chemical inertness, mechanical stability, good conductivity and favorable interaction between catalyst and its support. Compared to CF, these materials show some drawbacks as the high cost of preparation of RVC and rigid properties of CP (non-woven) due to the presence of the binder such as polytetrafluoroethylene.52 Therefore, CF is the most suitable candidate. On the other hand, CF suffers from some disadvantages such as low wettability (high hydrophobic nature) which prevent the uniform distribution of the catalyst, but this drawback can be overcome via surface treatment methods such as electrochemical and thermal techniques.53–59
Herein, a simple procedure is introduced to control the agglomeration of the (Ni nanoparticles) during its electrodeposition onto CF without any prior surface treatment by using glycerol as an additive. The growth and the building of the crystal are much affected as well as the Ni surface active sites depending on the concentration of glycerol in the deposition bath of Ni particle size of the catalyst, then the activity is investigated towards glycerol electrooxidation in an alkaline medium, and finally Density functional theory (DFT) calculation is used to rationalize the obtained results.
2. Experimental
2.1. Materials
All the used chemicals are of analytical grade and are used as received without any further purification. All the solutions are prepared using distilled water. Sodium hydroxide (NaOH), nickel sulfate hexahydrate (NiSO4·6H2O, 99.999%), sulfuric acid (99.95%) are purchased from Sigma-Aldrich, and carbon felt (CF, SGL, GFA 4 EA).2.2. Electrode's preparation
Commercial CF sheets are cut (4 mm nominal thickness, 2 mm width, and 3 mm length) connected with a GC rod with 1 mm diameter and are used as a substrate for further modification with Ni nanoparticles.The CF pieces are modified with Ni nanoparticles (Ni@CF) electrodeposited by employing the chronocoulometric technique from 0.1 M Na2SO4 containing 4 mM NiSO4 at −1.0 V vs. SCE. The metal loading for all the prepared electrodes is 30 μg. The electrodeposition in the presence of various glycerol concentrations (0.025, 0.05, 0.5, 0.75, 0.1, 0.2, 0.3, 0.4, and 0.5 M) is studied given a symbol Nix@CF where x is the concentration of glycerol added to the bath. After the electro-deposition of Ni on the CF, the obtained electrode is washed with distilled water to remove all the glycerol on the working electrode. The obtained metallic Ni is activated by cycling the potential in 0.1 M NaOH solution between 0.0 and 0.7 V vs. SCE at a potential scan rate of 200 mV s−1 for 40 cycles then the characteristic peaks were obtained by scanning the potential at 10 mV s−1 rate in the same solution. The mass of the electrodeposited Ni is estimated from the amount of passed charge during the electrodeposition using Faraday's law of electrolysis. As no significant hydrogen evolution development is observed, thus the deposition efficiency is considered as 100%.60
2.3. Electrochemical and material characterizations
3. Results and discussion
3.1. Material characterization
The effect of glycerol addition on the prepared catalyst morphology, composition, and structure is disclosed using SEM. SEM images of CF, Ni@CF, Ni0.05@CF, and Ni0.5@CF are shown in Fig. 1(A–D). There is a significant agglomeration of the electrodeposited Ni onto CF in the Ni@CF as clearly seen in Fig. 1(B). The addition of 0.05 M glycerol into the deposition bath improves the particle distribution of the catalyst as can be seen in mapping EDX and at the same time reduces the average particle size of the catalyst from 290 nm to 220 nm, calculated using ImageJ® software. Consequently, a significant increase in the surface-active sites of the catalyst is expected, c.f. Fig. 3(A). This enhances the catalytic activity of the deposited Ni. This enhancement can be attributed to the adsorption of glycerol on the Ni catalyst during the electrodeposition step which is confirmed by the negative adsorption energy as revealed from the DFT calculation see Fig. 2(A). So, the rate of nucleation is increased, and the rate of growth is retarded. Obviously as shown in Fig. 1(D and D′), the deposited Ni sheets are detached from CF and cracked as the concentration of the glycerol is increased by more than 0.05 M. This is due to covering the CF surface with glycerol which acts as an insulating barrier causing the detaching of the deposited catalyst. Thus, the number of active sites available on the substrate is reduced and Ni layer starts to detach away from the CF and form sheets as can be seen in Fig. 1(D and D′). Additionally, from DFT calculations based on the geometers shown in Fig. 2(A and B), the adsorption energy calculated using Monto Carlo simulation of the glycerol onto the CF is markedly higher than its adsorption energy on Ni. Thus, at lower concentration of glycerol, there are remaining active sites onto the CF available for further electrodeposition and at the same time the adsorption of the glycerol occurs onto the electrodeposited Ni, while at higher concentrations of glycerol, CF surface becomes enriched in glycerol thus hinders the adherence of Ni.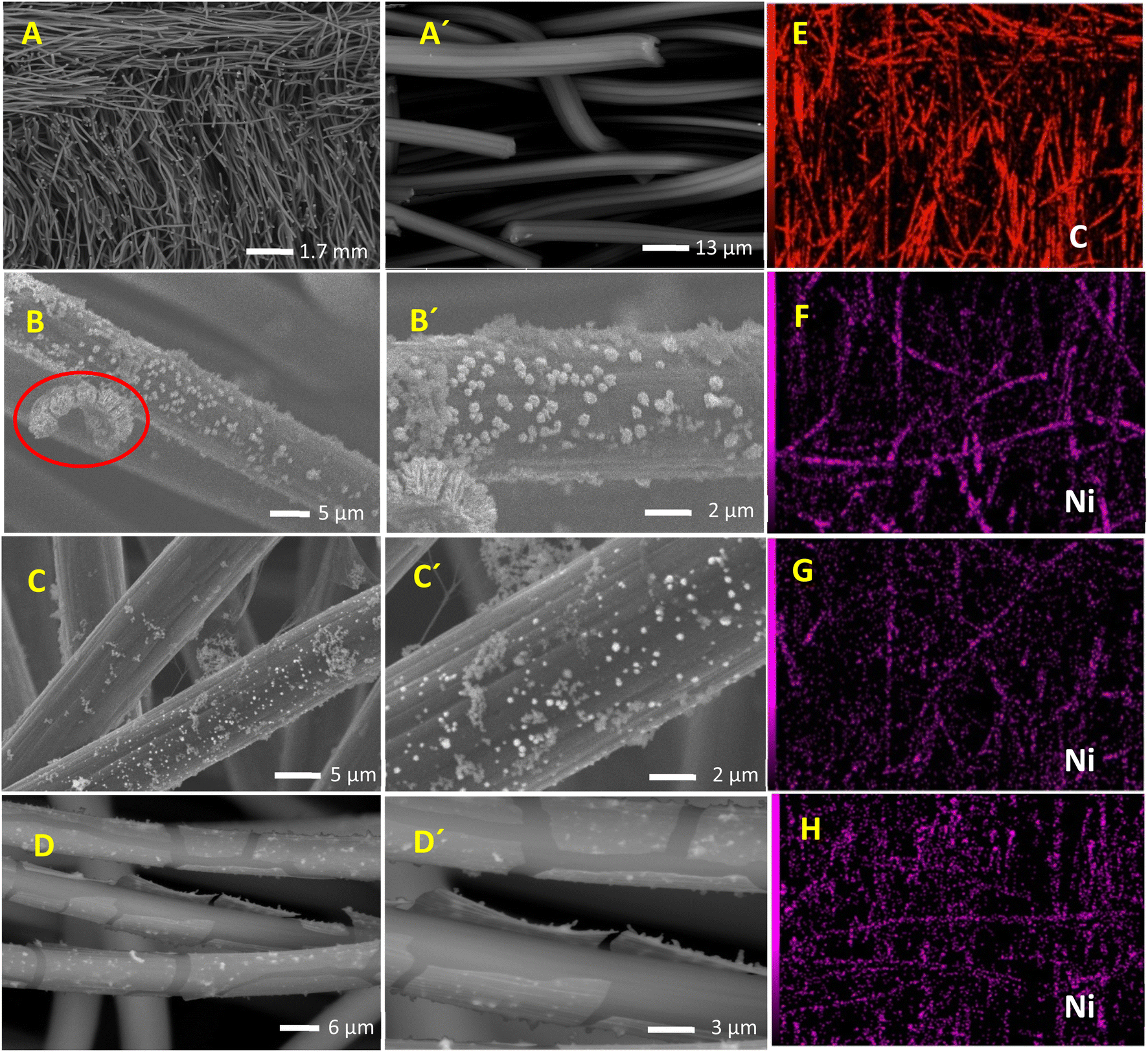 | ||
| Fig. 1 SEM images for: (A and A′) CF, (B and B′) Ni@CF, (C and C′) Ni0.05@CF, and (D and D′) Ni0.5@CF electrodes at different magnifications. EDX color mapping of Ni for: (E) CF, (F) Ni@CF, (G) Ni0.05@CF, and (H) Ni0.5@CF electrodes. | ||
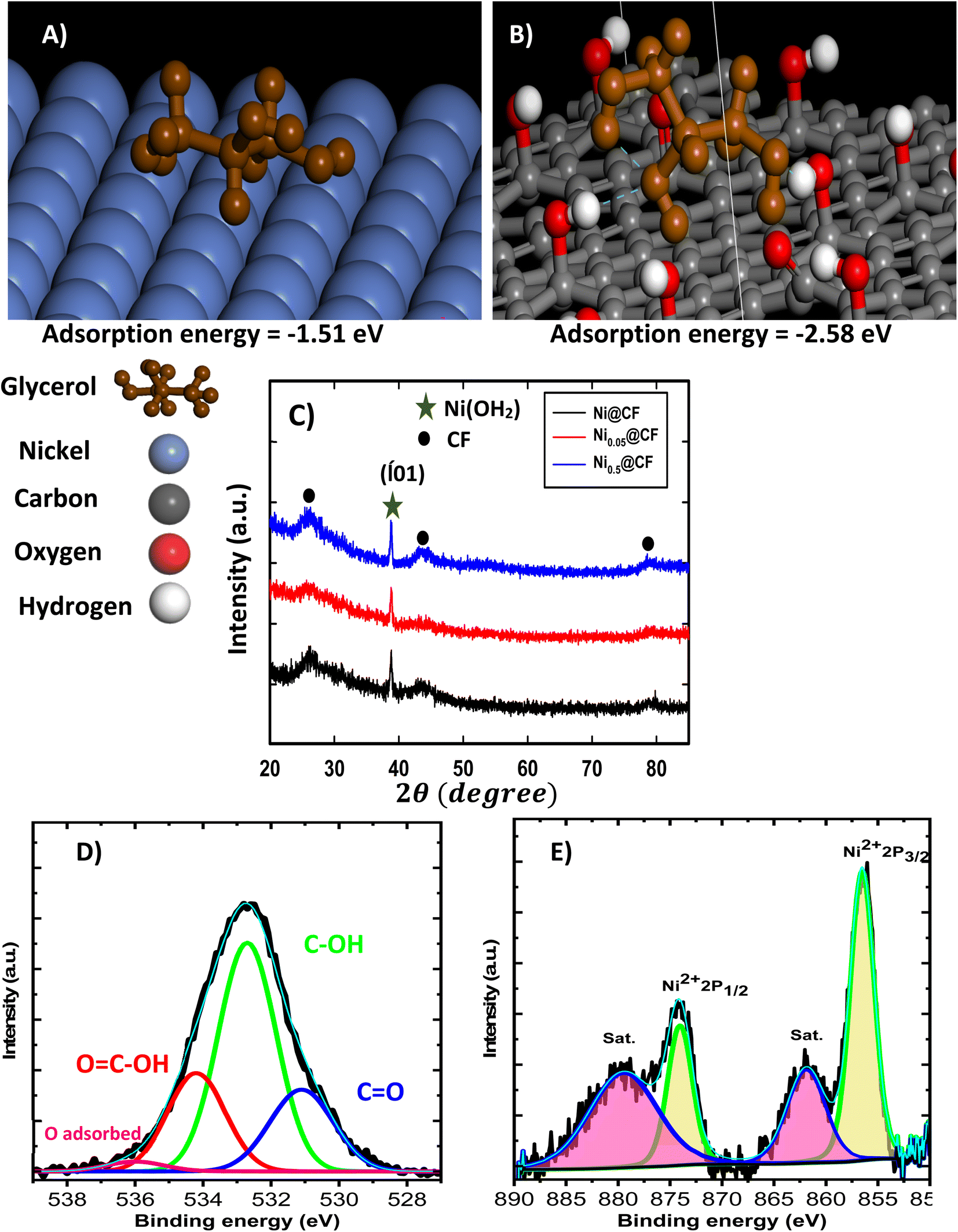 | ||
| Fig. 2 Monto Carlo simulation for glycerol on Ni and CF surface (A) and (B), respectively. (C) XRD patterns of Ni@CF, Ni0.05@CF, and Ni0.5@CF electrodes. High resolution XPS spectrum of O 1s and Ni 2p for CF and Ni0.05@CF electrode (D) and (E), respectively. | ||
XRD patterns of Ni@CF, Ni0.05@CF and Ni0.5@CF are displayed in Fig. 2(C) which are used to get information about their facets and crystallinity. As clearly seen, all the prepared electrodes show three diffraction peaks attributed to (002), (100), and (110) crystal planes of carbon which appear around 26°, 42°, and 77°, respectively, that agree with the (JCPDS ICDD card no. 00-001-0640). On the other hand, all the prepared electrodes show an additional peak at 38° attributed to the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01) crystal plane of Ni(OH)2 that is consistant with the reference code (COD 1011134). To figure out the effect of addition of glycerol on the crystal size of electrodeposited Ni, Scherrer's equation is used to estimate the average crystal size for the prepared electrodes, as described in eqn (5)
01) crystal plane of Ni(OH)2 that is consistant with the reference code (COD 1011134). To figure out the effect of addition of glycerol on the crystal size of electrodeposited Ni, Scherrer's equation is used to estimate the average crystal size for the prepared electrodes, as described in eqn (5)
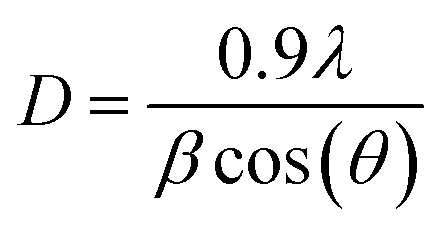 | (5) |
To figure out the surface state of the electrodeposited Ni, XPS analysis is performed, and the obtained O 1s and Ni 2p spectra are displayed in Fig. 2(D and E) for CF and Ni0.05@CF. The O 1s is deconvoluted into four peaks to confirm the presence of the carbonyl, hydroxyl, and carboxylic functional groups into the CF to can be used in the DFT calculation. Moreover, the Ni 2p spectrum for Ni0.05@CF typically shows Ni 2p3/2 and Ni 2p1/2 peaks with two satellites (“sat.”) at binding energies 856.3 and 873.9 eV, respectively. The spin energy separation between them is found to be 17.6 eV which is characteristic of Ni(OH)2.63,64
3.2. Glycerol electrooxidation
The performance of the prepared catalysts was investigated towards glycerol electrooxidation by using CV measurements. Fig. 3 shows the CVs of CF, Ni@CF, and Ni0.05@CF electrodes in 0.1 M NaOH solution in the absence (Fig. 3(A)) and in the presence (Fig. 3(B)) of 8 mM glycerol. According to many articles,15,34,41,60,65 the redox peak couple is observed at 0.45 V vs. SCE in Fig. 3(A) is attributed to NiOOH which is considered the active form toward glycerol electrooxidation where the larger the area under the peak, the more the activity obtained. As can be seen in Fig. 3(A) after adding 0.05 M glycerol in the deposition bath during the preparation of the catalyst, the NiOOH peak increases largely with the same metal loading and consequently, the activity of the catalyst is enhanced by about ≈1.6 times higher than Ni@CF and the onset potential is reduced by about 50 mV as can be concluded from Fig. 3(B). This is due to the adsorption of glycerol on the surface of the Ni catalyst during the electrodeposition on the CF which increases the rate of nucleation and retards the rate of growth. Therefore, better distribution and smaller particle size are obtained as shown in Fig. 1. Moreover, as notice peak (assigned by arrow d) in Fig. 3(B) represents the available sites from Ni catalyst and active for further glycerol electrooxidation which can be used if the concentration of the glycerol raised and can achieve enhancement higher than 1.6 times higher than Ni@CF thus, the electrooxidation of the glycerol for Ni@CF and Ni0.05@CF was performed at higher concentration of glycerol as clearly seen in Fig. 3(C). A systematic increase in the Ip is obtained by raising the concentration of the glycerol until 0.1 M glycerol is reached after that the steady state is obtained which means all of the active sites of Ni are participating in the electrooxidation. Moreover, the enhancement factor also increased by raising the concentration and reach 2 times higher than Ni@CF.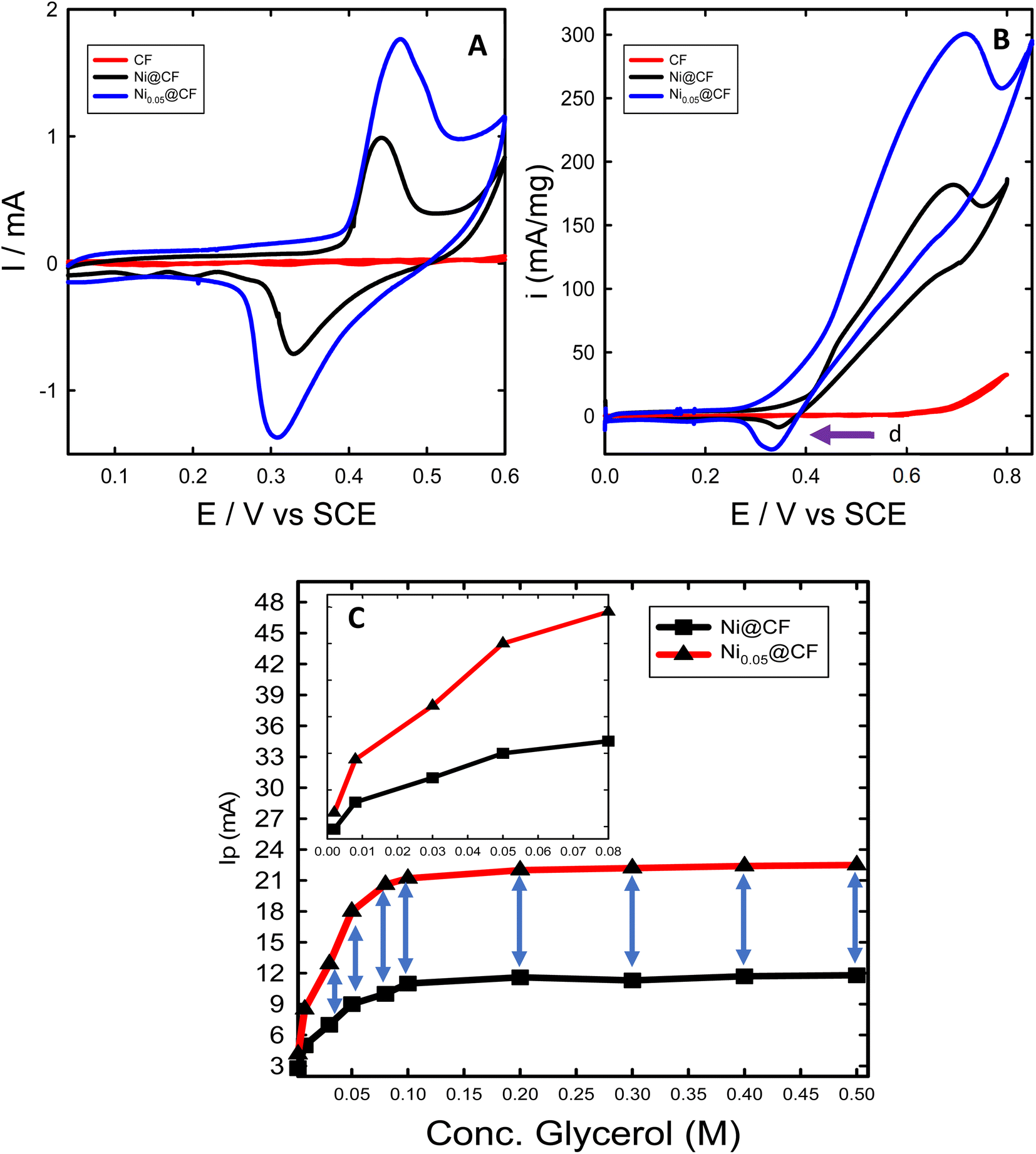 | ||
| Fig. 3 CVs of CF (red line), Ni@CF (black line), and Ni0.05@CF (blue line) measured in 0.1 M NaOH solution in absence (A) and in presence (B) of 8 mM glycerol with a same potential scan rate 10 mV s−1. Ip of the glycerol electrooxidation for Ni@CF and Ni0.05@CF as a function of glycerol concentration (0.002–0.5 M) (C) and inset of figure represent the zooming at the early stage of the two curves. | ||
To figure out the enhancement of the oxidation current of the transformation of Ni to NiOOH as can be seen in Fig. 3(A) doesn't attributed to the traces of glycerol remaining on the electrode from the glycerol added in the deposition bath, the reduction peak of NiOOH to Ni(OH)2 is also increased which means that the increase of current is due to better distribution of the catalyst' particles rather than the presence of glycerol. The surface concentration of the active phase is estimated from the amount of charge associated during the formation of the active phase using eqn (6):45
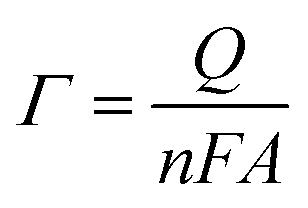 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 C mol−1) and A is the geometrical surface area of the substrate (0.52 cm2). The estimated Γ increases from ∼8 to ∼19 nmol cm−2 for Ni@CF and Ni0.05@CF, respectively. This means that the surface-active sites are increased 2.37 times by simple addition of 0.05 M glycerol to the deposition bath, which is adsorbed on the Ni catalyst during deposition, consequently better catalytic activity is obtained as can be seen in Fig. 3.
500 C mol−1) and A is the geometrical surface area of the substrate (0.52 cm2). The estimated Γ increases from ∼8 to ∼19 nmol cm−2 for Ni@CF and Ni0.05@CF, respectively. This means that the surface-active sites are increased 2.37 times by simple addition of 0.05 M glycerol to the deposition bath, which is adsorbed on the Ni catalyst during deposition, consequently better catalytic activity is obtained as can be seen in Fig. 3.
3.3. Optimization of glycerol concentration as additive
The effect of various glycerol concentrations added to the deposition bath was optimized for two deposition bath concentrations of NiSO4 (2 and 4 mM). The same amount of Ni was electrodeposited to single out the effect of glycerol addition on the obtained electrocatalytic activity for glycerol oxidation. The estimated Γ and Ip (oxidation peak current for glycerol electrooxidation) for both concentrations of NiSO4 for the prepared Ni@CF and Nix@CF electrodes as a function of glycerol concentration are shown in Fig. 4. It is clearly seen in Fig. 4 that 0.05 M is the optimum glycerol concentration to be added to the deposition bath where it exhibits the highest Ip and Γ associated with the lowest onset potential for the glycerol electrooxidation. This can be attributed to the adsorption of glycerol on the electrodeposited Ni and the active sites of the CF increasing the rate of nucleation and decreasing the rate of growth with the reduction of the crystal size and hence improving the distribution of the catalyst as can be evidenced from SEM and XRD analysis. The more the glycerol concentration is increased, the more the reduction in Ip and Γ is obtained because of the excessive adsorption of glycerol on CF acts as an insulating barrier for Ni deposition. So, the number of active sites available on the CF is reduced. This leads to Ni sheets detached far away from the CF and at the same time crystal size increases as can be seen in Fig. 1(D and D′) and XRD data. Raising the concentration of NiSO4 from 2 to 4 mM also enhances Ip and Γ due to increasing the deposition efficiency.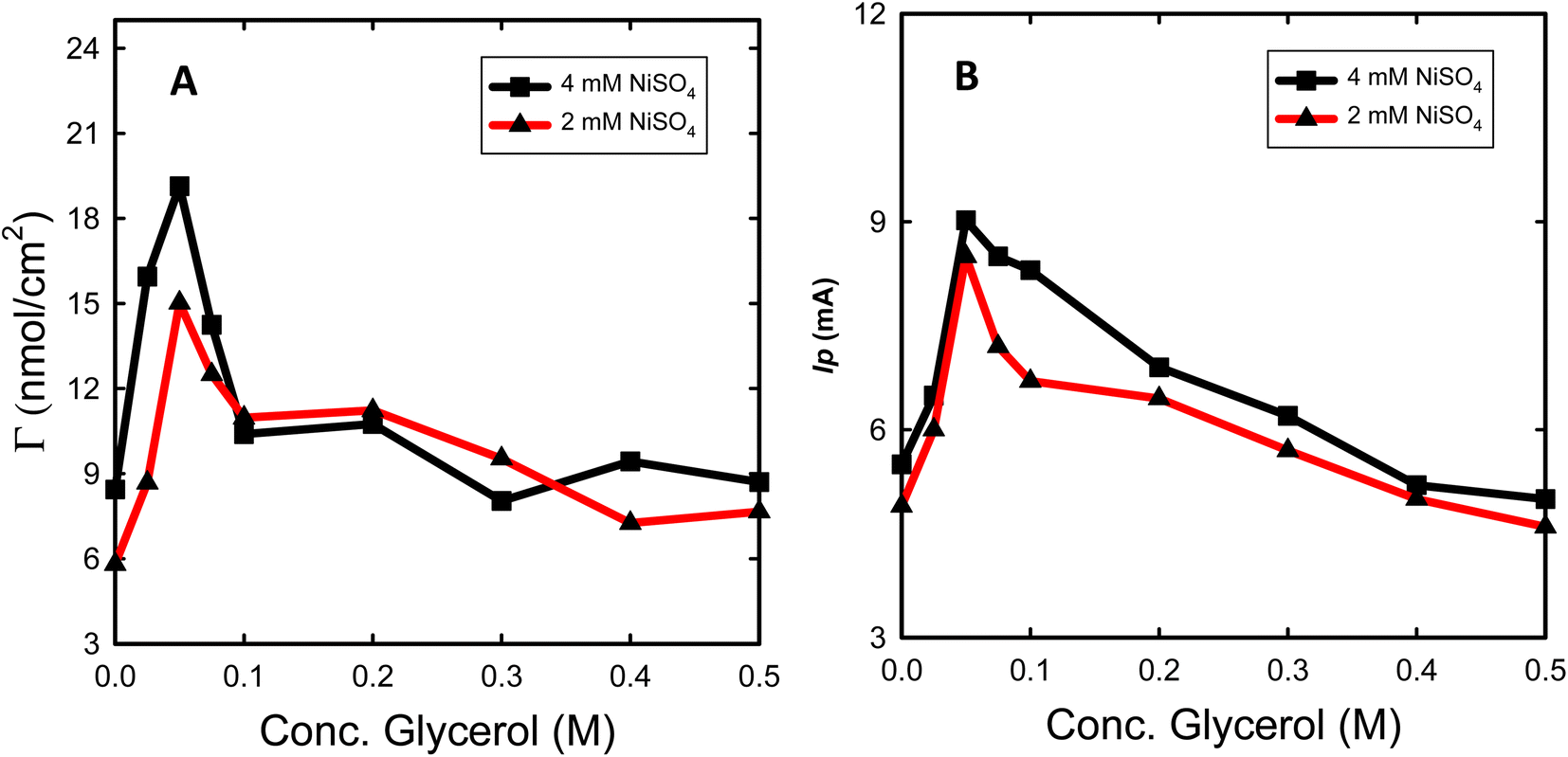 | ||
| Fig. 4 Γ (A) and Ip (B) for the prepared Nix@CF electrode as a function of glycerol concentration for (2 (red line) and 4 (black line) mM) NiSO4 with the same metal loading measured in 0.1 M NaOH without (A) and with (B) glycerol (8 mM) with a potential scan rate 10 mV s−1. | ||
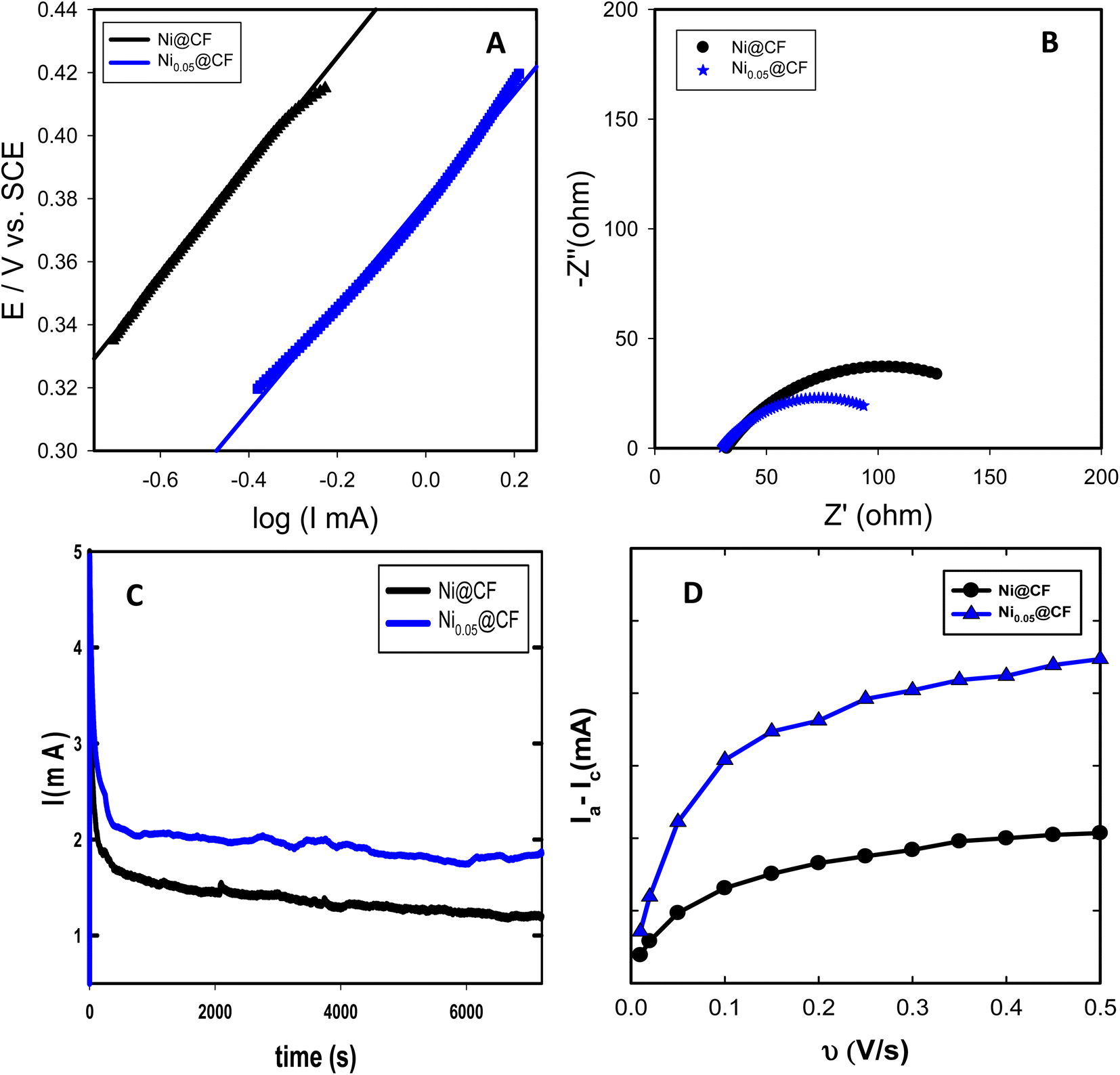 | ||
| Fig. 5 (A) Tafel plots measured at potential scan rate 0.01 V s−1. (B) Nyquist plots measured at 0.4 V. (C) Chronoamperometric curves measured at 0.6 V. All measurements are performed in 0.1 M NaOH solution containing 8 mM glycerol for Ni@CF and Ni0.05@CF electrodes. (D) The estimation of electrochemical double layer capacitance (Cdl) for Ni@CF and Ni0.05@CF by plotting (Ic − Ia) (capacitive current) vs. the potential scan rate in 0.1 M NaOH solution. | ||
Fig. 5(B) displays Nyquist plots of Ni@CF and Ni0.05@CF electrodes measured in 0.1 M NaOH solution containing 8 mM glycerol at 0.4 V vs. SCE and the electrodeposition of the catalyst performed using a deposition bath containing 4 mM NiSO4. The electrooxidation of glycerol on Ni@CF and Ni0.05@CF electrodes is found to be kinetically controlled due to the obtained semi-circle illustration for Nyquist plots.66,67 The experimentally measured Nyquist plots were fitted using Randle's equivalent circuit then calculating the charge transfer resistance (Rct). The Rct value is dropped from 140 ohm in the case of Ni@CF to 87 ohm using the Ni0.05@CF electrode which shows that the Ni0.05@CF electrode has a higher ability to oxidize glycerol which furtherly confirms that the addition of glycerol to the deposition bath enhances the catalytic activity of Ni catalyst towards glycerol electrooxidation.
The long-term stability of the prepared Ni@CF and Ni0.05@CF electrodes is addressed via chronoamperometric (I–t) measurements at 0.6 V vs. SCE for 2 h as displayed in Fig. 5(C). It is revealed from this data that the presence of 0.05 M glycerol in the deposition bath reduces the loss in the stability from 24 to 7% from their initial performance after only 750 s and at the same time increases the oxidation current by about 1.6 times after 2 h of continuous electrolysis as compared to Ni@CF electrode (in absence of glycerol in the Ni-deposition bath). Thus, Ni0.05@CF electrode has better stability performance than Ni@CF electrode. Furthermore, our prepared catalysts show better electrocatalytic activity towards glycerol electrooxidation than previously published work, as compared in Table 1.
| Catalyst | [NaOH] mol L−1 | [Glycerol] mol L−1 | Scan rate (mV s−1) | I (A g−1) at 1.6 V vs. RHE | Ref. |
|---|---|---|---|---|---|
| a CCE is carbon ceramic electrode. | |||||
| CoNi@C | 0.1 | 0.1 | 50 | 0.063 | 41 |
| FeCoNi@C | 0.1 | 0.1 | 50 | 0.065 | 41 |
| FeNi@C | 0.1 | 0.1 | 50 | 0.052 | 41 |
| NiCu@CCE | 0.1 | 1.0 | 50 | 43.871 | 40 |
| Ni@(CCE) | 0.1 | 1.0 | 50 | 24.372 | 40 |
| NiCo@CCE | 0.1 | 1.0 | 50 | 116.990 | 40 |
| Ni wire | 1 M KOH | 0.1 | 50 | 40.0 | 34 |
| Ni doped porous Cu/Cu2O | 0.1 | 0.1 | 10 | 6.423 | 70 |
| Ni@CF | 0.2 | 0.008 | 10 | 126.0 | This work |
| Ni0.05@CF | 0.2 | 0.008 | 10 | 223.0 | |
The reason behind the enhancement in the activity of the Ni0.05@CF towards glycerol electrooxidation is the increment in the number of the active sites which comes from the better nucleation by the action of the additive. That reason can be also evidenced from the relation between the non-faradic capacitive current and scan rate for Ni@CF and Ni0.05@CF to estimate the electrochemical surface area (ECSA) and the data are depicted in Fig. 5(D). As clearly seen in Fig. 5(D) the ECSA for Ni0.05@CF is much higher than Ni@CF.60,68 Moreover, the linear part obtained at low scan rates has a higher slope and consequently, indicating a larger fraction of the entire porous matrix is being contributing to the electrochemical surface area whereas at higher scan rates the top skin layer of the porous matrix contributes to the active area while the underlying porous matrix suffers from diffusion limitations.69
4. Conclusion
The presence of 0.05 M glycerol in the deposition bath of Ni electrodeposited on commercial CF resulted in boosting their performance towards glycerol electrooxidation. This enhancement was identified by about 2 times increase of the oxidation current as well as a ca. 50 mV negative shift of its onset potential. This enhancement can be attributed to the adsorption of glycerol on the Ni catalyst during the electrodeposition step which is confirmed by the negative adsorption energy in DFT calculation thus, the rate of nucleation increases and the rate of growth decreases. The distribution of deposited Ni over the entire CF was improved concurrently with increasing their surface concentration and surface-active sites. The average crystal size of the electrodeposited Ni nanoparticles is reduced from 54 nm to 36 nm by the simple addition of 0.05 M glycerol to the deposition bath. Various glycerol concentrations (as additives) were optimized for the two different concentrations of Ni. The optimum deposition bath was composed of 4 mM NiSO4 solution containing 0.05 M glycerol resulting in the electrocatalyst exhibiting the highest electrocatalytic activity for glycerol electrooxidation. The kinetics and stability of the Ni@CF towards glycerol electrooxidation are enhanced by adding 0.05 M glycerol, which is confirmed by EIS measurements, chronoamperometric curves, and Tafel plots. The charge transfer resistance is reduced from 140 to 87 ohm and loss in stability after 2 h of continuous electrolysis is also reduced from 24 to 7% by the simple addition of 0.05 M glycerol to the Ni deposition bath.Conflicts of interest
There are no conflicts to declare.References
- E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431–3450 CrossRef CAS.
- A. H. Abu-ghazala, H. H. Abdelhady, A. A. Mazhar and M. S. El-Deab, Renewable Energy, 2022, 200, 1120–1133 CrossRef CAS.
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrogen Energy, 2017, 42, 10142–10157 CrossRef CAS.
- H. Mou, Q. Chang, Z. Xie, S. Hwang, S. Kattel and J. G. Chen, Appl. Catal., B, 2022, 316, 121648 CrossRef CAS.
- S. P. Viviane, N. Júlio, R. Andrezza and O. N. Almir, J. Fuel Chem. Technol., 2022, 50, 474–483 CrossRef.
- M. R. Rizk, M. G. Abd El-Moghny, H. H. Abdelhady, W. M. Ragheb, A. H. Mohamed, H. F. Fouad, M. Mohsen, A. S. Kamel and M. S. El-Deab, Int. J. Hydrogen Energy, 2022, 47, 32145–32157 CrossRef CAS.
- M. S. E. Houache, K. Hughes and E. A. Baranova, Sustainable Energy Fuels, 2019, 3, 1892–1915 RSC.
- A. Shukla, S. C. Singh, C. S. Saraj, G. Verma and C. Guo, Mater. Today Chem., 2022, 23, 100691 CrossRef CAS.
- A. L. Wang, H. Xu and G. R. Li, ACS Energy Lett., 2016, 1, 445–453 CrossRef CAS.
- S. Li, C. Xi, Y. Z. Jin, D. Wu, J. Q. Wang, T. Liu, H. B. Wang, C. K. Dong, H. Liu, S. A. Kulinich and X. W. Du, ACS Energy Lett., 2019, 4, 1823–1829 CrossRef CAS.
- Q. Zhang, C. Zhang, J. Liang, P. Yin and Y. Tian, ACS Sustainable Chem. Eng., 2017, 5, 3808–3818 CrossRef CAS.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- D. Wang, Q. Li, C. Han, Z. Xing and X. Yang, ACS Cent. Sci., 2018, 4, 112–119 CrossRef CAS PubMed.
- S. Guan, X. Fu, Z. Lao, C. Jin and Z. Peng, Sustainable Energy Fuels, 2019, 3, 2056–2066 RSC.
- M. R. Rizk, M. G. Abd El-Moghny, A. Mazhar, M. S. El-Deab and B. E. El-Anadouli, Sustainable Energy Fuels, 2021, 5, 986–994 RSC.
- W. Ni, T. Wang, F. Héroguel, A. Krammer, S. Lee, L. Yao, A. Schüler, J. S. Luterbacher, Y. Yan and X. Hu, Nat. Mater., 2022, 21, 804–810 CrossRef CAS PubMed.
- L. Xie and D. W. Kirk, J. Electrochem. Soc., 2020, 167, 064519 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- R. M. Tesfaye, G. Das, B. J. Park, J. Kim and H. H. Yoon, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, J. Zang, L. Dong, H. Pan and Y. Yuan, Electrochim. Acta, 2013, 113, 583–590 CrossRef CAS.
- Z. Qi, H. Geng, X. Wang, C. Zhao, H. Ji, C. Zhang, J. Xu and Z. Zhang, J. Power Sources, 2011, 196, 5823–5828 CrossRef CAS.
- S. Lee, H. J. Kim, S. M. Choi, M. H. Seo and W. B. Kim, Appl. Catal., A, 2012, 429–430, 39–47 CrossRef CAS.
- M. Kapkowski, P. Bartczak, M. Korzec, R. Sitko, J. Szade, K. Balin, J. Lelątko and J. Polanski, J. Catal., 2014, 319, 110–118 CrossRef CAS.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef.
- Y. Z. Su, Q. Z. Xu, Q. S. Zhong, C. J. Zhang, S. T. Shi and C. W. Xu, Mater. Res. Bull., 2015, 64, 301–305 CrossRef CAS.
- A. Y. Tsivadze, M. R. Tarasevich, V. A. Bogdanovskaya and M. R. Ehrenburg, Dokl. Chem., 2008, 419, 54–56 CrossRef CAS.
- E. Lohrasbi and M. Asgari, Adv. Anal. Chem., 2015, 5, 9–18 Search PubMed.
- M. A. Goda, M. G. Abd El-Moghny and M. S. El-Deab, J. Electrochem. Soc., 2020, 167, 064522 CrossRef CAS.
- A. M. Ahmed, S. Y. Sayed, G. A. El-Nagar, W. M. Morsi, M. S. El-Deab and B. E. El-Anadouli, J. Electroanal. Chem., 2019, 835, 313–323 CrossRef CAS.
- Y. Zhao, L. Fan, J. Ren and B. Hong, Int. J. Hydrogen Energy, 2014, 39, 4544–4557 CrossRef CAS.
- A. Papaderakis, N. Pliatsikas, C. Prochaska, K. M. Papazisi, S. P. Balomenou, D. Tsiplakides, P. Patsalas and S. Sotiropoulos, Front. Chem., 2014, 2, 1–11 CAS.
- D. Soundararajan, J. H. Park, K. H. Kim and J. M. Ko, Curr. Appl. Phys., 2012, 12, 854–859 CrossRef.
- Y. Hong, H. J. Kim, H. J. Lee, J. Kim and S. Il Choi, Front. Chem., 2019, 7, 1–9 CrossRef.
- M. S. E. Houache, E. Cossar, S. Ntais and E. A. Baranova, J. Power Sources, 2018, 375, 310–319 CrossRef CAS.
- M. Rizk, M. Abd El-Moghny and M. El-Deab, J. Electrochem. Soc., 2020, 167, 114505 CrossRef CAS.
- Y. Kang, W. Wang, Y. Pu, J. Li, D. Chai and Z. Lei, Chem. Eng. J., 2017, 308, 419–427 CrossRef CAS.
- R. M. A. Tehrani and S. Ab Ghani, Electrochim. Acta, 2012, 70, 153–157 CrossRef CAS.
- D. P. M. Fleischmann and K. Korinek, J. Chem. Soc., Perkin trans. II, 1971, 1312–1315 Search PubMed.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, J. Electroanal. Chem., 2013, 703, 56–62 CrossRef CAS.
- B. Habibi and N. Delnavaz, RSC Adv., 2016, 6, 31797–31806 RSC.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, Electrochim. Acta, 2014, 117, 255–262 CrossRef CAS.
- M. S. E. Houache, K. Hughes, R. Safari, G. A. Botton and E. A. Baranova, ACS Appl. Mater. Interfaces, 2020, 12, 15095–15107 CrossRef CAS PubMed.
- B. Habibi and N. Delnavaz, RSC Adv., 2016, 6, 31797–31806 RSC.
- G. A. El-Nagar, I. Derr, T. Kottakkat and C. Roth, ECS Trans., 2017, 80, 1013–1022 CrossRef CAS.
- M. E. Ghaith, G. A. El-Nagar, M. G. Abd El-Moghny, H. H. Alalawy, M. E. El-Shakre and M. S. El-Deab, Int. J. Hydrogen Energy, 2020, 45, 9658–9668 CrossRef CAS.
- M. D. de Jesus Almeida, C. A. Della Rovere, L. R. P. de AndradeLima, D. V. Ribeiro and C. A. C. De Souza, Mater. Res., 2019, 22(4), e20180480 CrossRef.
- M. Mouanga, L. Ricq, J. Douglade and P. Berçot, J. Appl. Electrochem., 2007, 37, 283–289 CrossRef CAS.
- H. Nakano, T. Ura, S. Oue and S. Kobayashi, ISIJ Int., 2014, 54, 1653–1660 CrossRef CAS.
- K. Krishnadevi, S. RatnaKumari, D. Prasanna, H. B. N. Prasanna and V. Anuradha, J. Colloid Interface Sci., 2022, 607, 1776–1785 CrossRef CAS PubMed.
- B. Jaleh, M. Nasrollahzadeh, M. Eslamipanah, A. Nasri, E. Shabanlou, N. R. Manwar, R. Zboril, P. Fornasiero and M. B. Gawande, Carbon, 2022, 198, 301–352 CrossRef CAS.
- R. Zeng, K. Lian, B. Su, L. Lu, J. Lin, D. Tang, S. Lin and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 25055–25062 CrossRef CAS PubMed.
- F. C. Lee, M. S. Ismail, D. B. Ingham, K. J. Hughes, L. Ma, S. M. Lyth and M. Pourkashanian, Renewable Sustainable Energy Rev., 2022, 166, 112640 CrossRef CAS.
- T. X. H. Le, M. Bechelany and M. Cretin, Carbon, 2017, 122, 564–591 CrossRef CAS.
- R. E. G. Smith, T. J. Davies, N. D. B. Baynes and R. J. Nichols, J. Electroanal. Chem., 2015, 747, 29–38 CrossRef CAS.
- A. Di Blasi, O. Di Blasi, N. Briguglio, A. S. Aricò, D. Sebastián, M. J. Lázaro, G. Monforte and V. Antonucci, J. Power Sources, 2013, 227, 15–23 CrossRef CAS.
- Y. Wang and Y. Hasebe, Talanta, 2009, 79, 1135–1141 CrossRef CAS PubMed.
- L. Han, S. Tricard, J. Fang, J. Zhao and W. Shen, Biosens. Bioelectron., 2013, 43, 120–124 CrossRef CAS PubMed.
- Z. González, A. Sánchez, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Electrochem. Commun., 2011, 13, 1379–1382 CrossRef.
- K. J. Kim, Y. J. Kim, J. H. Kim and M. S. Park, Mater. Chem. Phys., 2011, 131, 547–553 CrossRef CAS.
- M. E. Ghaith, M. G. A. El-moghny, G. A. El-nagar, H. H. Alalawy, M. E. El-Shakre and M. S. El-Deab, RSC Adv., 2023, 13, 895–905 RSC.
- N. Benipal, J. Qi, J. C. Gentile and W. Li, Renewable Energy, 2017, 105, 647–655 CrossRef.
- I. H. Karahan, H. A. Çetinkara and H. S. Güder, Int. J. Surf. Eng. Coatings, 2013, 86, 157–161 Search PubMed.
- H. Jiang, Y. Guo, T. Wang, P. L. Zhu, S. Yu, Y. Yu, X. Z. Fu, R. Sun and C. P. Wong, RSC Adv., 2015, 5(17), 12931–12936 RSC.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- A. M. Abdelrahim, M. G. Abd El-Moghny, M. E. El-Shakre and M. S. El-Deab, RSC Adv., 2023, 13, 1811–1822 RSC.
- M. A. Goda, M. G. Abd El-Moghny and M. S. El-Deab, J. Electrochem. Soc., 2020, 167, 064522 CrossRef CAS.
- I.-M. Hsing, X. Wang and Y.-J. Leng, J. Electrochem. Soc., 2002, 149, A615 CrossRef CAS.
- I. O. Baibars, M. G. Abd El-Moghny and M. S. El-Deab, J. Environ. Chem. Eng., 2022, 10, 2 Search PubMed.
- C. Costentin and J. M. Savéant, ACS Appl. Energy Mater., 2019, 2, 4981–4986 CrossRef CAS.
- M. R. Rizk, M. G. Abd El-Moghny, G. A. El-Nagar, A. A. Mazhar and M. S. El-Deab, ChemElectroChem, 2020, 7, 951–958 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |