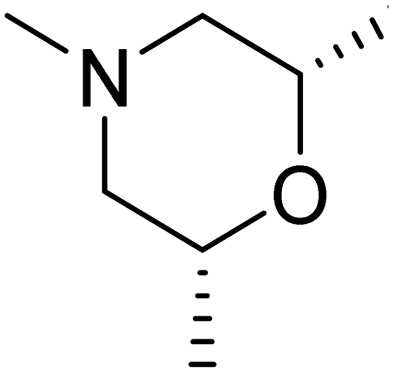
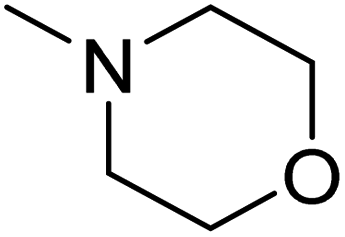
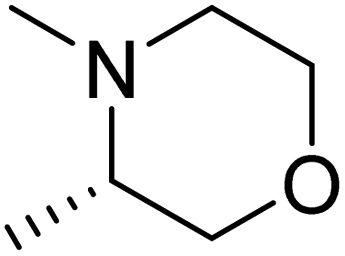
DOI:
10.1039/D3RA00056G
(Review Article)
RSC Adv., 2023,
13, 6872-6908
Medicinal chemistry perspective of pyrido[2,3-d]pyrimidines as anticancer agents
Received
4th January 2023
, Accepted 9th February 2023
First published on 28th February 2023
Abstract
Cancer is a major cause of deaths across the globe due to chemoresistance and lack of selective chemotherapy. Pyrido[2,3-d]pyrimidine is an emerging scaffold in medicinal chemistry having a broad spectrum of activities, including antitumor, antibacterial, CNS depressive, anticonvulsant, and antipyretic activities. In this study, we have covered different cancer targets, including tyrosine kinase, extracellular regulated protein kinases – ABL kinase, phosphatidylinositol-3 kinase, mammalian target of rapamycin, p38 mitogen-activated protein kinases, BCR-ABL, dihydrofolate reductase, cyclin-dependent kinase, phosphodiesterase, KRAS and fibroblast growth factor receptors, their signaling pathways, mechanism of action and structure–activity relationship of pyrido[2,3-d]pyrimidine derivatives as inhibitors of the above-mentioned targets. This review will represent the complete medicinal and pharmacological profile of pyrido[2,3-d]pyrimidines as anticancer agents, and will help scientists to design new selective, effective and safe anticancer agents.
1 Introduction
Cancer is the explosive growth of abnormal cells that typically grow beyond their original boundaries, invade surrounding areas, spread to other organs, and result in metastasis, which is one of the main causes of cancer-related death,and second most common cause of death across the globe. Around 10.0 million cancer-related fatalities (9.9 million excluding nonmelanoma skin cancer) and 19.3 million new cancer cases (18.1 million excluding nonmelanoma skin cancer) were estimated in 2020 across the globe. The most lethal malignancies include lung (1.8 million), colorectal (935![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), liver (830000), stomach (769000), and breast cancer. The most frequent cancers worldwide are lung (2.2 million), breast (2.09 million), colorectal (1.9 million), prostate (1.28 million), skin (1 million), and stomach (1 million) (627000).1 Cancer is the Latin term meaning crab. Because of the crab-like tenacity, a malignant tumor often appears to grab the tissues it invades, and the ancients used the term to indicate a malignancy.2 Cancer cells (defined by their uncontrollable growth and invasion of other tissues) do not show the same growth average as a normal heathly cell. Normal cells multiply and expand, controlled to produce additional cells as required to maintain a healthy body. Cells die as they grow old or are damaged and are replaced by new cells. Cancer cells, rather than dying, continue to proliferate and generate new, aberrant cells. Normal cells cannot invade (grow into) other tissues. The other cells can combine to form a mass of tissues known as a tumor. Not all tumors are cancerous; they might be benign (not cancerous) or malignant (cancerous).3 The worldwide burden of cancer is anticipated to rise significantly due to population growth, age, and increase in risk factors like smoking, inactivity, poor food, and infections that cause cancer. It is well established that cancer diagnosis and treatment have substantial and long-term consequences on both patient and caregiver physical, emotional, and spiritual well-being.3,4
000), liver (830000), stomach (769000), and breast cancer. The most frequent cancers worldwide are lung (2.2 million), breast (2.09 million), colorectal (1.9 million), prostate (1.28 million), skin (1 million), and stomach (1 million) (627000).1 Cancer is the Latin term meaning crab. Because of the crab-like tenacity, a malignant tumor often appears to grab the tissues it invades, and the ancients used the term to indicate a malignancy.2 Cancer cells (defined by their uncontrollable growth and invasion of other tissues) do not show the same growth average as a normal heathly cell. Normal cells multiply and expand, controlled to produce additional cells as required to maintain a healthy body. Cells die as they grow old or are damaged and are replaced by new cells. Cancer cells, rather than dying, continue to proliferate and generate new, aberrant cells. Normal cells cannot invade (grow into) other tissues. The other cells can combine to form a mass of tissues known as a tumor. Not all tumors are cancerous; they might be benign (not cancerous) or malignant (cancerous).3 The worldwide burden of cancer is anticipated to rise significantly due to population growth, age, and increase in risk factors like smoking, inactivity, poor food, and infections that cause cancer. It is well established that cancer diagnosis and treatment have substantial and long-term consequences on both patient and caregiver physical, emotional, and spiritual well-being.3,4
1.1 Pyrido[2,3-d]pyrimidine
Purines, quinazolines, pteridines, and pyrido-pyrimidines are examples of bicyclic nitrogen-containing heterocyclic compounds that are well-known pharmacophores in medicinal chemistry. Examples of commercial medications with a bicyclic main structure include the tyrosine kinase inhibitors gefitinib and erlotinib, and both are quinazoline derivatives. Both of them are used to manage non-small cell lung cancer. Pyrido[2,3-d]pyrimidines have been studied extensively as quinazoline analogs.5,6 Pyrido[2,3-d]pyrimidines have shown antitumor, antibacterial, CNS depressive, anticonvulsant, antipyretic, and analgesic effects, as shown in Fig. 1.6 Particularly, pyrido[2,3-d]pyrimidines have been shown to be effective against Toxoplasma gondii and Pneumocystis carinii (tg) culture of tumor cell lines, and the activity is attributed to dihydrofolate reductase inhibition.7–9 Due their diverse anticancer or antiproliferative properties, pyrido[2,3-d]pyrimidine derivatives were great interest of reseachers working in this field.10,11 Fusing a pyridine and a pyrimidine ring results in pyridopyrimidines, ortho-fused bicyclic heterocyclic structures. Pyridopyrimidines (1,3,8-triazanaphtalenes) are one of the four isomeric pyridopyrimidines.12,13 Because of their proximity to DNA bases, such structures are included in the favored heterocyclic scaffolds hypothesis for drug development, which was suggested by Evans in the late 1980s and recently refined by Altomare.14,15 Pyrido[2,3-d]pyrimidine is a favored heterocyclic scaffold that can act as ligands for various receptors in the body.16
 |
| | Fig. 1 Pharmacological activities of the pyrido[2,3-d]pyrimidine derivatives. | |
2 Synthetic strategies of pyrido[2,3-d]pyrimidines
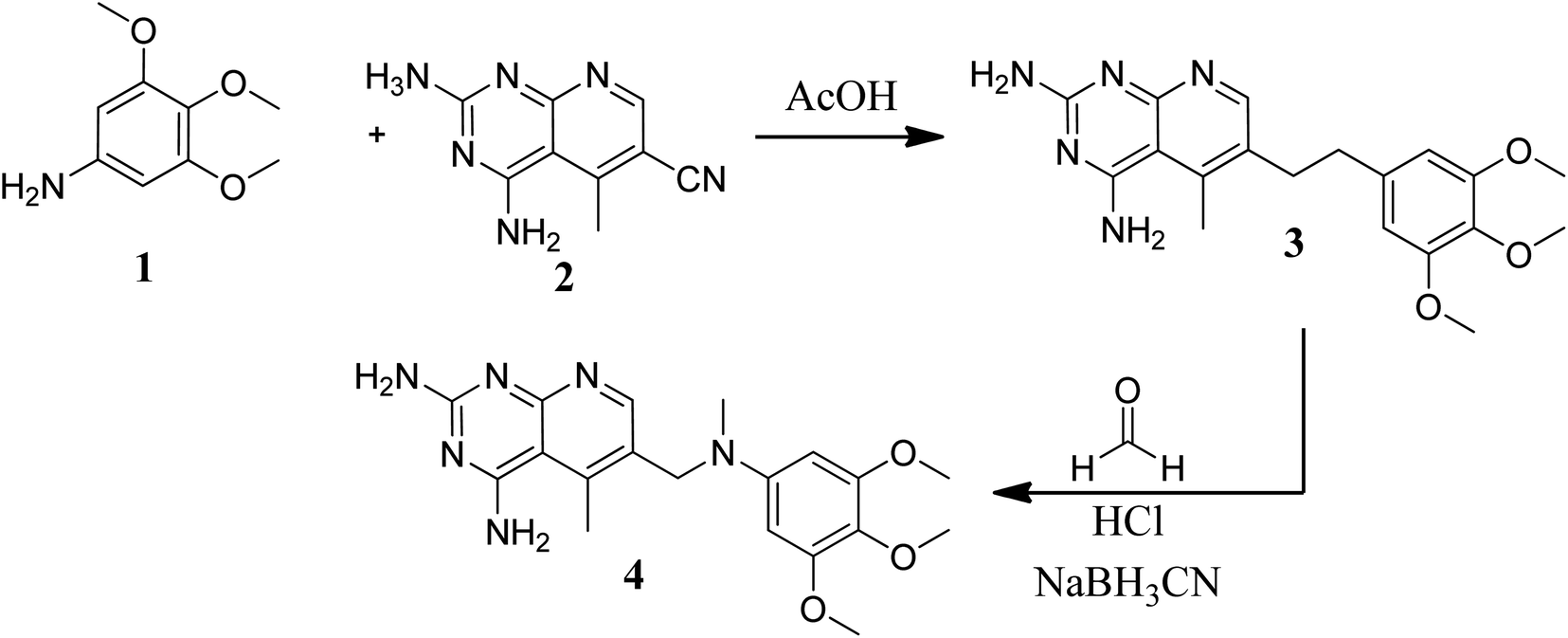
Various methods for developing pyrido[2,3-d]pyrimidines have been previously reported via condensation techniques and pyridine annelation reactions, in solid as well as solution phase. In 1993, Kisliuk et al. described the synthesis of pyrido[2,3-d]pyrimidine-2,4-diamines as shown in Fig. 2. They synthesized precursor (3) in the presence of RANEY® Ni (70%) in acetic acid by deductive condensation of 6-cyano-5-methyl-pyrido[2,3-d]pyrimidine-2,4-diamine (2) with 3,4,5-trimethoxyaniline (1), which was then methylated at the N10 position by reductive alkylation with formaldehyde and sodium cyanoborohydride.17,18
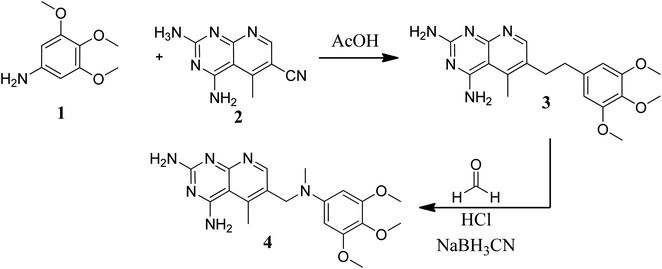 |
| | Fig. 2 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivative (4). | |
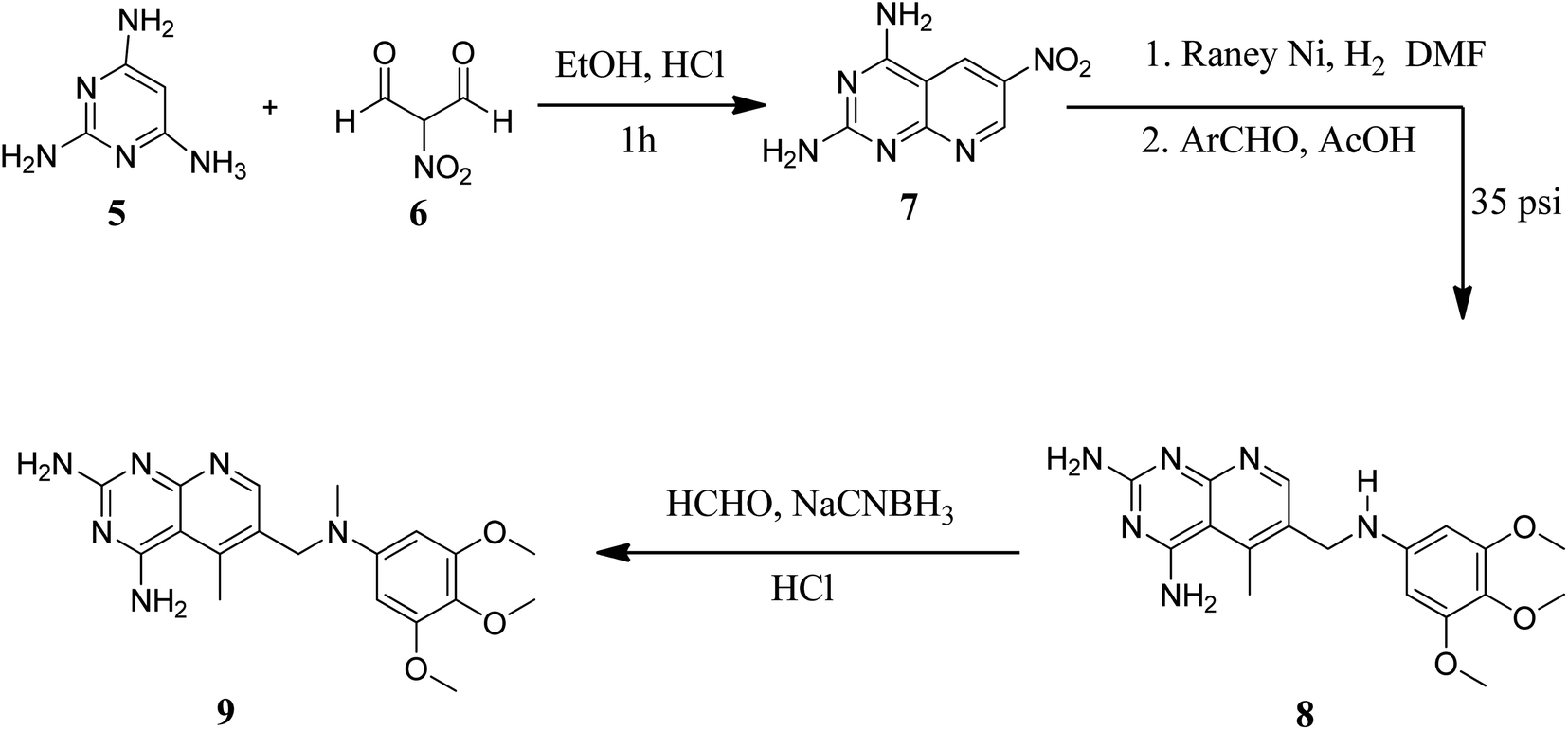
They also created a different method to synthesize pyrido[2,3-d]pyrimidine-2,4-diamines (9) by mixing 2,4,6-triaminopyrimidine (5) with the sodium salt of nitromalonaldehyde and obtained create the 2,4-diamino6-nitropyrido[2,3-d]pyrimidine (7) in a single step. This compound was subsequently reduced to its 6-amino homolog by RANEY® Ni using DMF. By using several aldehydes in reductive amination, the desired product 8 was produced (ArCHO = 3,4,5-trimethoxybenzaldehyde). In the final step, formaldehyde was used to N-methylate 8 in the presence of sodium cyanoborohydride (Fig. 3).18,19
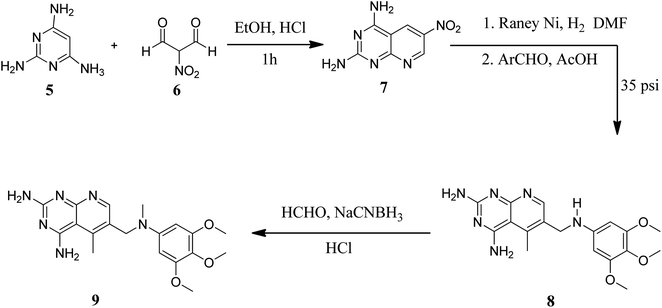 |
| | Fig. 3 Scheme for the synthesis of pyrido[2,3-d]pyrimidine diamine derivative (9). | |
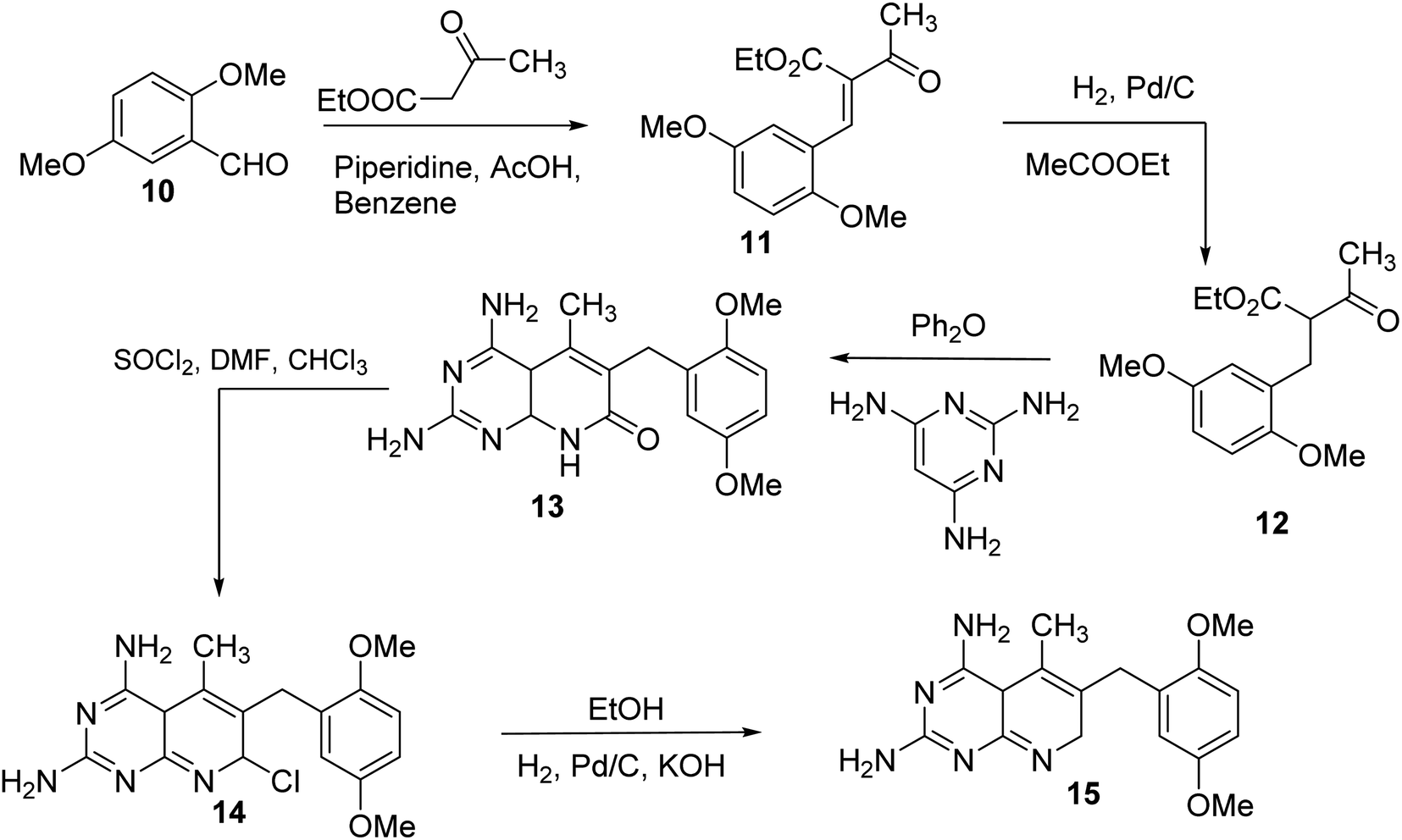
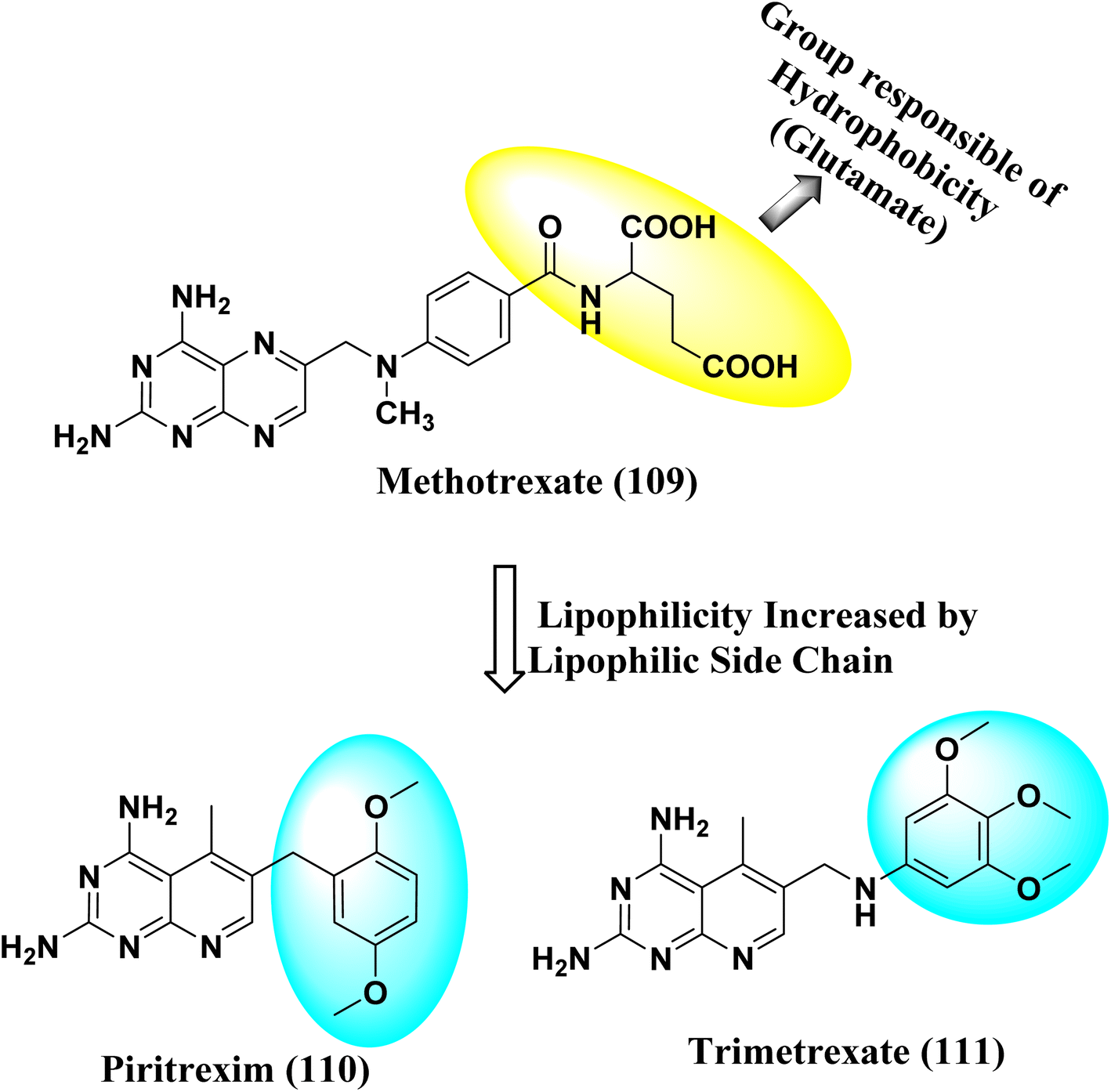
N6-[(2,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine was produced by Queener et al. In order to get the necessary 2,4,6-triaminoquinazoline, the 2,4-diamino-6-nitroquinazoline (7) was reduced with hydrogen and RANEY® nickel around 30–35 psi (10). Then, 2,5-dimethoxybenzaldehyde (ArCHO) was added to produce the N9–H precursor (11). Using sodium cyanoborohydride, the reductive N9-alkylation process was used to produce the end product. Queener et al. also investigated the biological effects of compound 12 as a lipophilic inhibitor of dihydrofolate reductase.20 Piritrexim was first synthesized by Grivsky et al. possessed anti-parasitic, anti-folate, anti-psoriatic, and anti-tumor characteristics. Piritrexim suppressed dihydrofolate reductase (DHFR) and had anticancer effects in rats with carcinosarcoma. Compared to certain analogs, Piritrexim has the advantage of not acting as a histamine metabolism inhibitor, reducing the danger of adverse metabolic effects.21 In the presence of a combination of piperidine and glacial acetic acid, 2,5-dimethoxybenzaldehyde (10) was reacted with ethyl acetoacetate in refluxing benzene to produce ethyl-acetyl-(2,5-dimethoxyphenyl) acrylate (11). The latter was subsequently hydrogenated with 5% Pd/C (catalyst) to produce the required ethyl-acetyl-(2,5-dimethoxyphenyl) propionate (12). Then, 12 was condensed using 2,4,6-triaminopyrimidine in diphenyl ether at 195–230 °C. 2,4-Diamino-7,8-dihydro-6-(2,5-dimethoxybenzyl)-5-methyl -7-oxopyrido[2,3-d]pyrimidine (13) was treated with a 1:1 combination of N,N-dimethylformamide thionyl chloride to produced -5-methylpyrido[2,3-d]pyrimidine2,4-diamine (14). The hydrogenolysis of 14 with Pd/C in the presence of potassium hydroxide produced the desired PTX (15) (Fig. 4).18,21
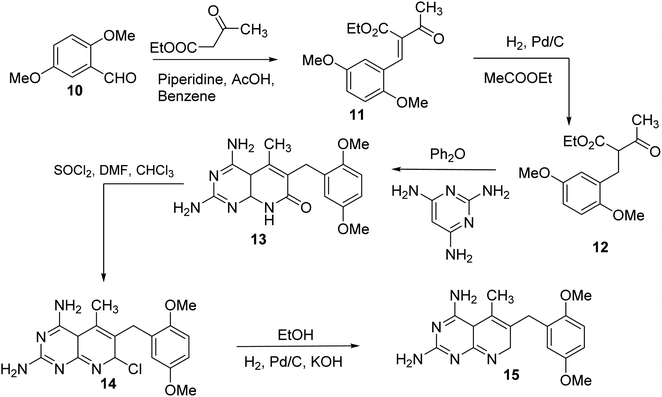 |
| | Fig. 4 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivative (15). | |
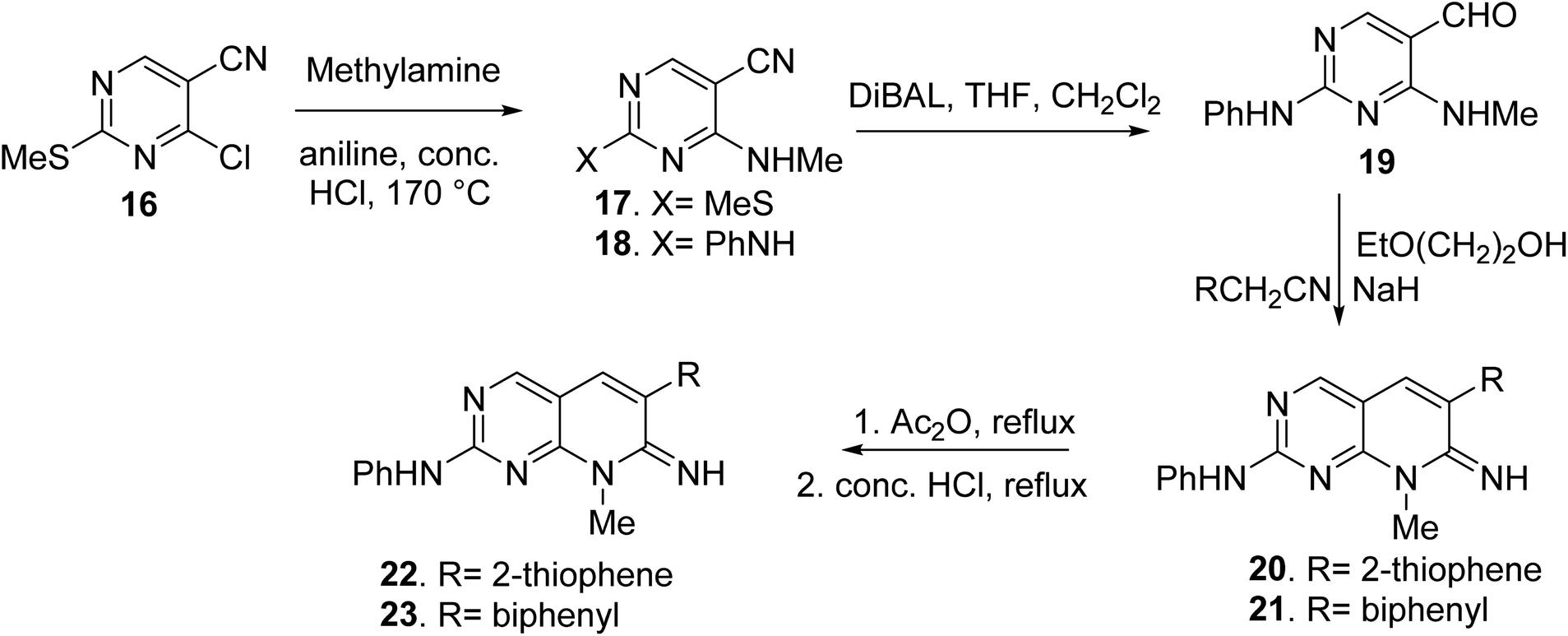
Blankley et al. synthesized 8-methyl-2-phenylamino-6-thiophene-2-yl-8H-pyrido-[2,3-d]pyrimidin-7-one (22) and 6-(biphenyl-4-yl)-8-methyl-2-phenylamino-8H-pyrido-[2,3-d]pyrimidin-7-one (23) by treating 4-chloro-5-cyano2-methylsulfanyl-pyrimidine (16) with methylamine (17). In the presence of a catalytic quantity of strong hydrochloric acid, the 2-methyl sulphide group of 17 was replaced with aniline, yielding compound 18. The aldehyde (19) was obtained by reducing the nitrile group of 18 with diisobutylaluminum hydride and then hydrolyzing it. The condensation of 19 with 2-thiopheneacetonitrile or 4-biphenylacetonitrile with 2-ethoxyethanol and sodium hydride, respectively, yielded the pyridopyrimidin-7-imines 20 and 21. Compounds 22 and 23 were obtained via acetylation of the imines followed by acid hydrolysis (Fig. 5).
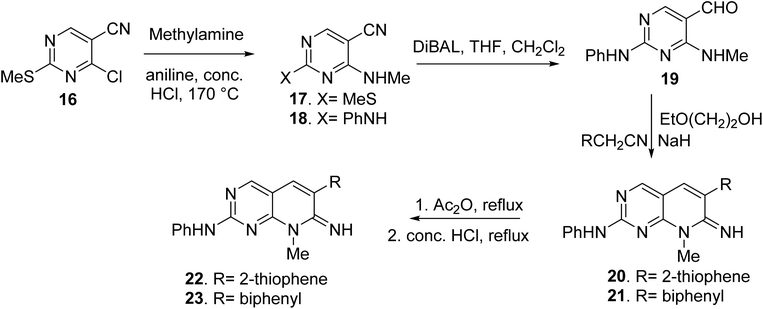 |
| | Fig. 5 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivatives. | |
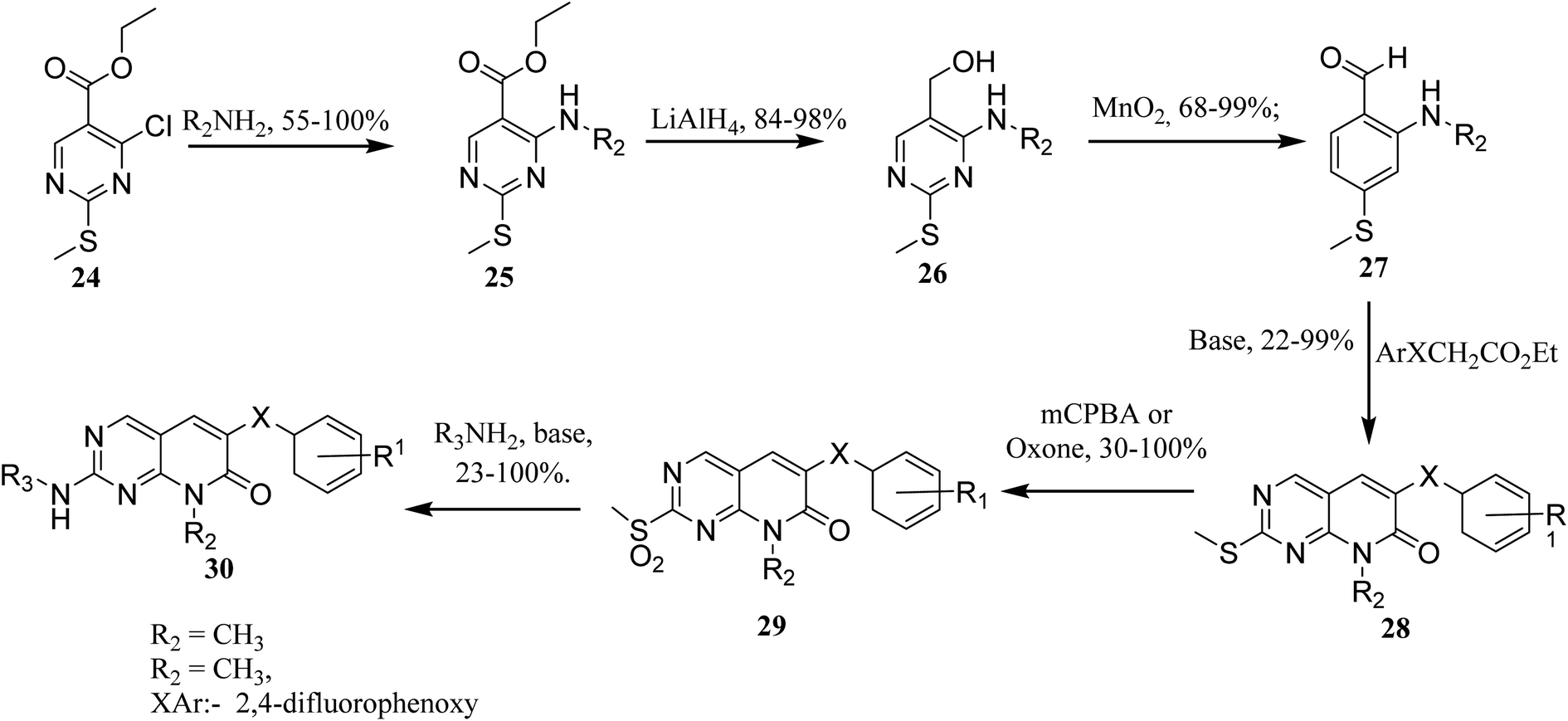
In order to synthesize intermediate esters (25), ethyl 4-chloro-2-methylthiopyrimidine-5-carboxylate (24) was combined with several amines. Alcohols (26) were then converted to 4-aminopyrimidine-5-carboxaldehydes through oxidation (27). To make the core pyrimidinopyridones, aldehydes (27) were condensed with appropriately substituted acetates. In order to synthesize the desired molecules (30), the methylthio groups of pyrimidinopyridones (28) were oxidized to produce the representative sulfones (29). The most frequent path was needed for 8-substituent chains since they were more intricate than straight chains (Fig. 6)22
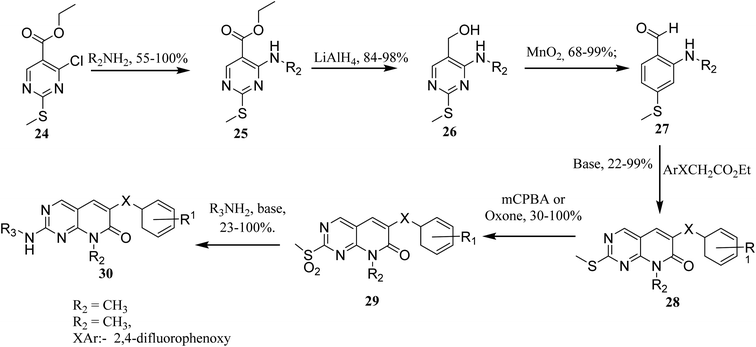 |
| | Fig. 6 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivatives. | |
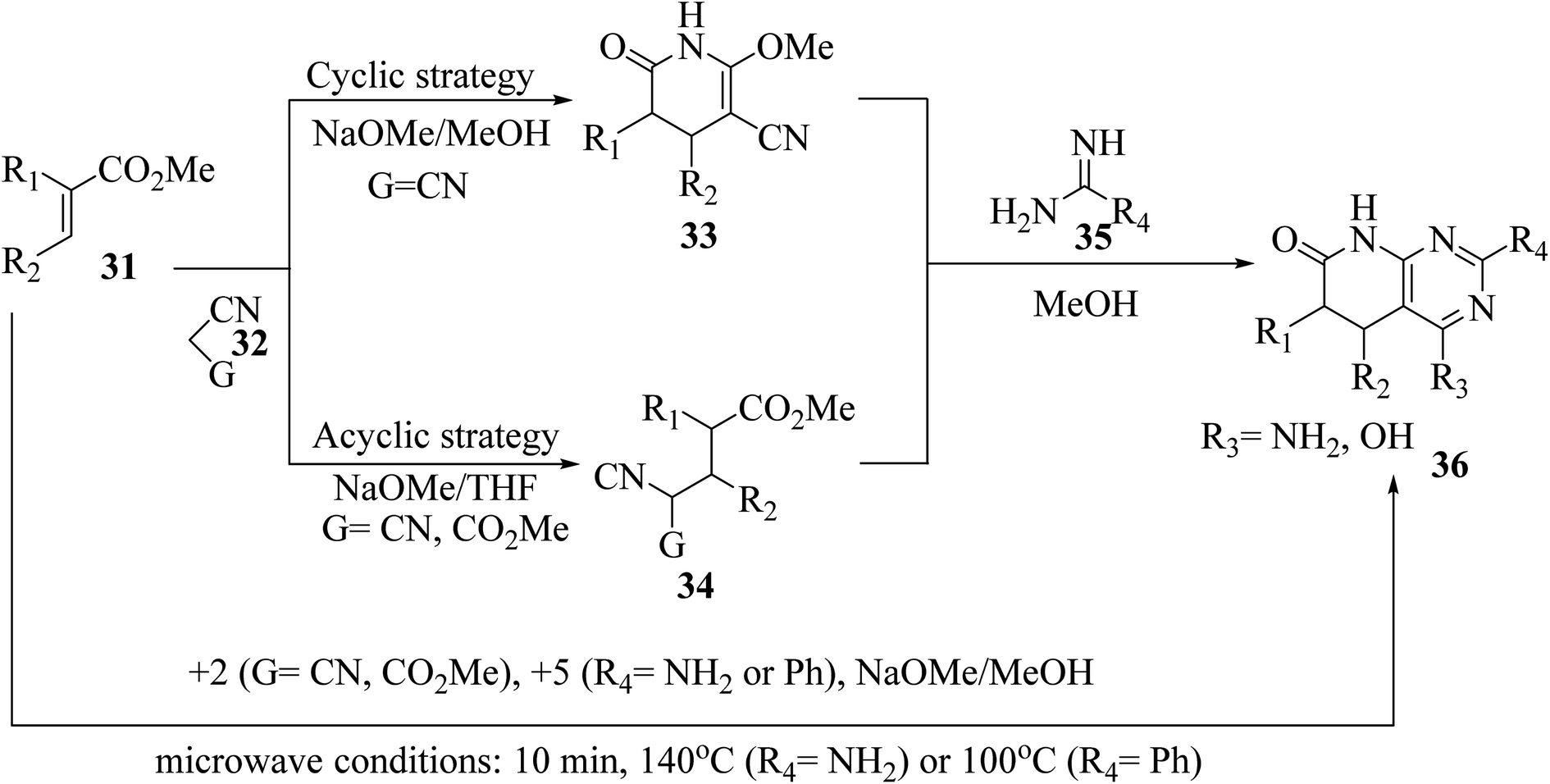
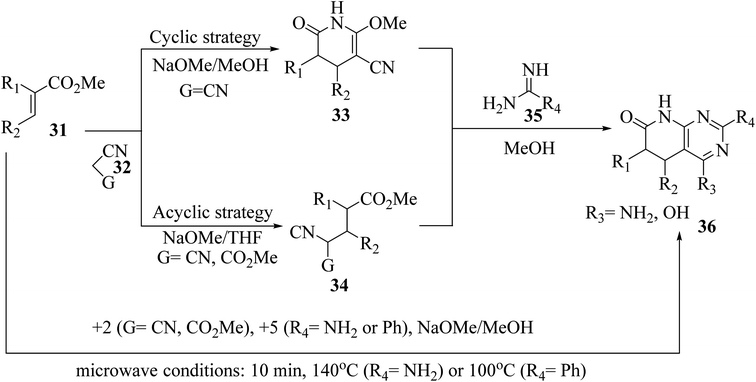
The synthesis of the reduced form of the target heterocycle in a highly regioselective manner has been reported using 6-aminouracil derivatives as the enamine component in a Michael or Hantzsch-type condensation. This strategy has several advantages over current methods, including using readily available alkynones as Michael acceptors, effectively eliminating the need for subsequent oxidation, and providing pyrido[2,3-d]pyrimidine derivatives directly without purification and with total regiochemical control.23 Victory and colleagues investigated a heterocyclic system that can be produced in a multistep sequence using 2-methoxy-6-oxo-1,4,5,6-tetra-hydropyridin-3-carbonitrile intermediates [created by reacting an α,β-unsaturated ester (31) with malononitrile (32, G = CN) in MeOH/NaOMe]. Because pyridones 33 include a highly reactive methoxy group, they are suitable substrates for a subsequent nucleophilic substitution or condensation processes. By treating pyridones 33 with amidine systems 35 (R4 = NH2, H, Me, Ph), they have created extensive procedures for the synthesis of bicyclic heterocycles such as pyrazolo[3,4-b]pyridines, 1,6-naphthyridines, and 4-amino-pyrido[2,3-d]pyrimidines 36 (R3 = NH2). In another study, they described an acyclic variation of the protocol for synthesizing pyridopyrimidines 36 (R3 = NH2), focusing on isolating the corresponding Michael adduct 34 (G = CN). This allowed them to also produce 4-oxopyrido[2,3-d]pyrimidines 36 (R3 = OH) by treating intermediates 34 (G = CO2Me), which were produced by the Michael addition of acrylate (31) and methyl cyanoacetate (32, G = CO2Me), with an amide analog (35). They showed that pyrido[2,3-d]pyrimidines 36 may be synthesized in a single step using a microwave-assisted cyclocondensation of α,β-unsaturated ester, amidine, with malononitrile/cyanoacetate (Fig. 7).24
 |
| | Fig. 7 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivatives. | |
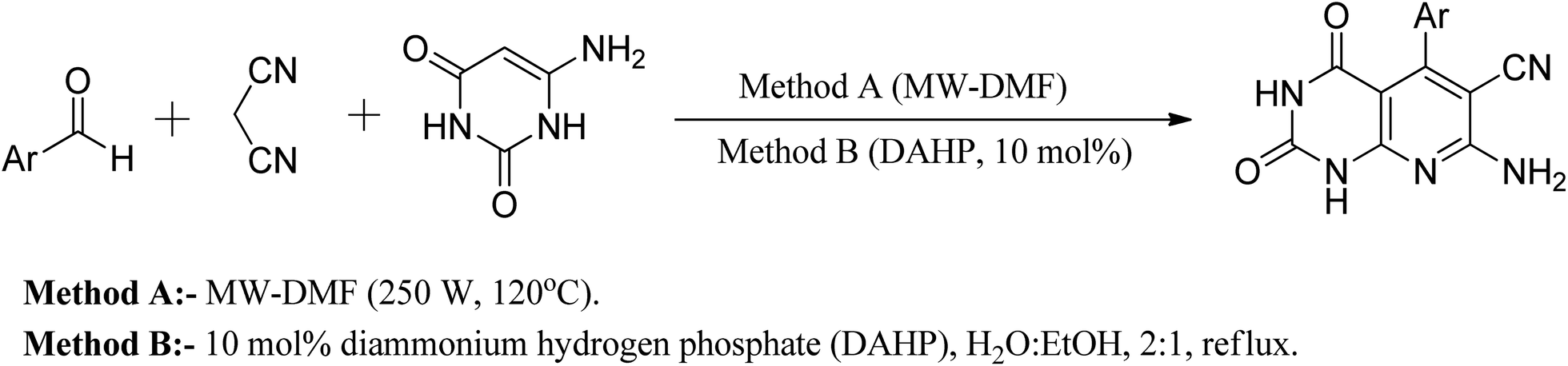
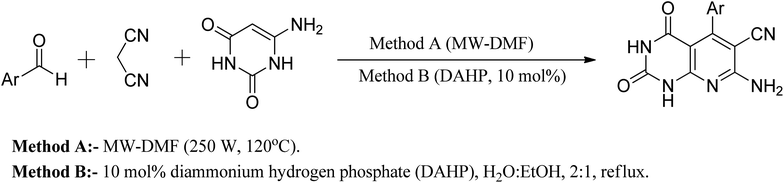
Another approach reported in the literature is using nanocrystalline MgO in the reaction of 6-aminouracil, 6-amino-2-thiouracil, or 6-amino-1,3-dimethyluracil with malononitrile and aldehydes in water at 80 °C to produce a series of pyrido[2,3-d]pyrimidine derivatives in high yields.25 In a different strategy, they demonstrated an efficient one-pot three-component reaction of aromatic aldehydes, malononitrile, and (6)-aminouracil and MWI for the synthesis of pyrido[2,3-d]pyrimidines using microwave irradiation (method A) and also in the presence of catalytic amounts of DAHP (10 mol percent) aqueous ethanol under reflux (method B) (Fig. 8).26
 |
| | Fig. 8 General method of synthesizing pyrido[2,3-d]pyrimidine derivatives. | |
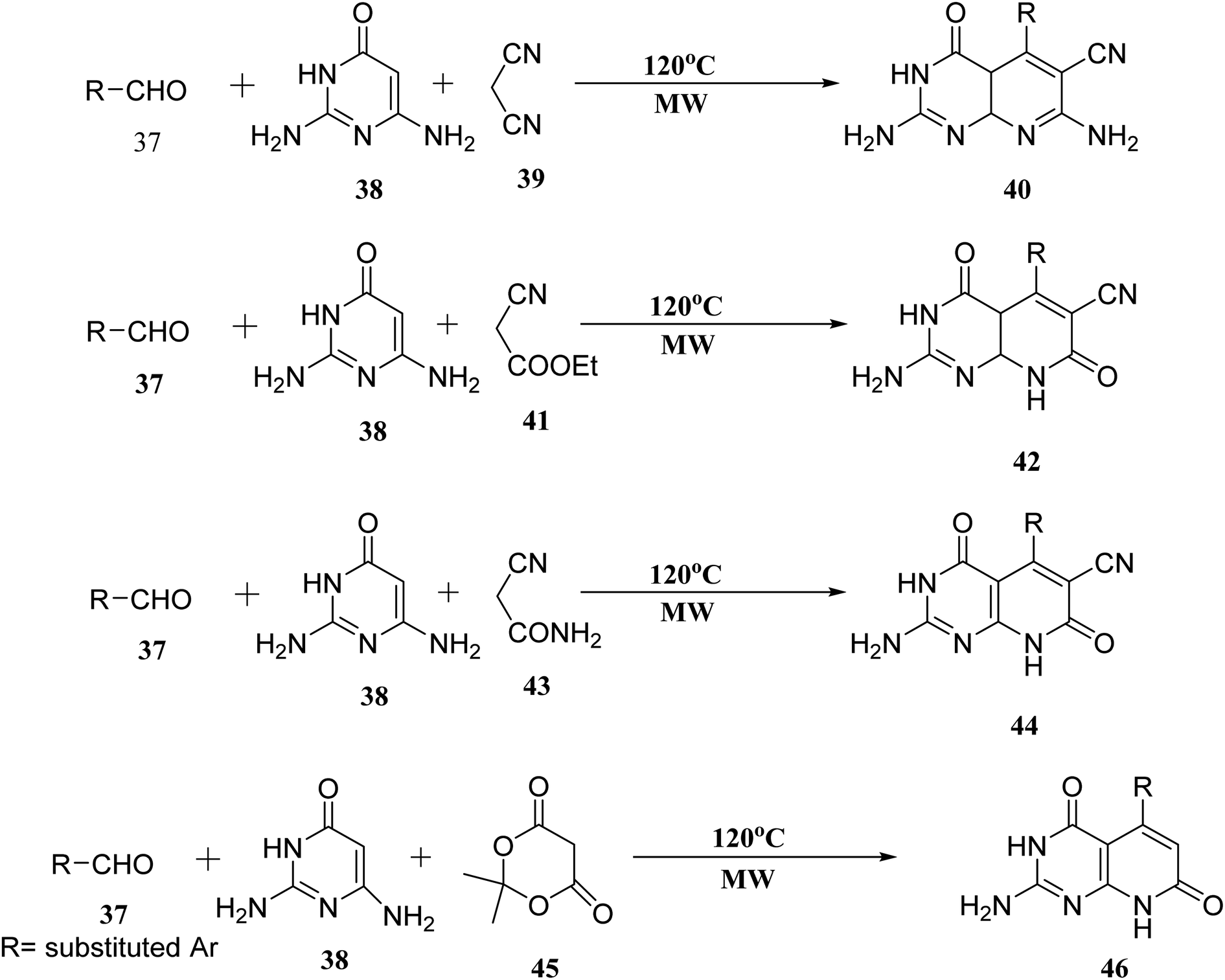
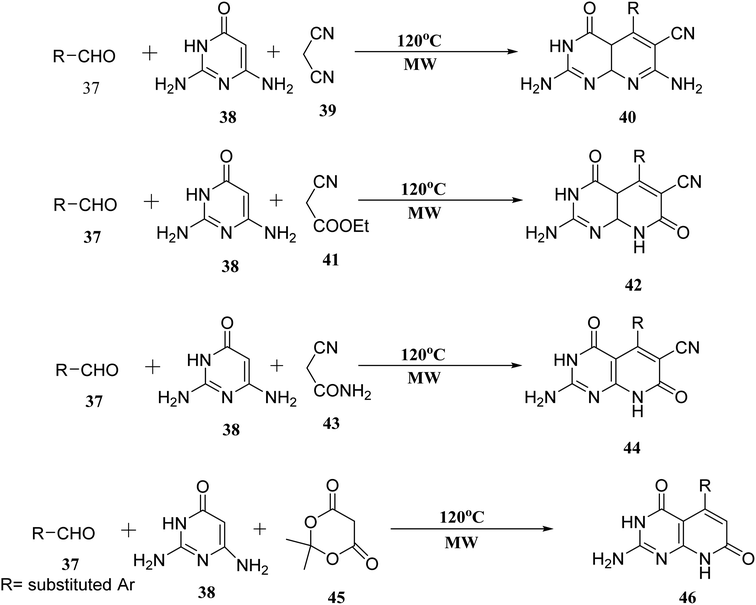
When the reaction of aldehyde, 2,6-diaminopyrimidin-4-one (38), and malononitrile (39) was performed in the presence of glycol under microwave irradiation, the derivative (40) was generated in good yields. Aldehyde (37), 2,6-diaminopyrimidin-4-one (38), and ethyl cyanoacetate (41) were carried out under the same conditions. When 2-cyanoacetamide was used in place of ethyl cyanoacetate (41), dehydrogenation of (42) was also accomplished (43). Meldrum's acid (45), (37), and (38), used as starting materials for microwave irradiation, produced the required compounds (46) in significant amounts (Fig. 9).27
 |
| | Fig. 9 Scheme for the synthesis of pyrido[2,3-d]pyrimidine derivatives. | |
3 Molecular targets of pyrido[2,3-d]pyrimidine derivatives
Many antitumor activities have been reported for pyrido[2,3-d]pyrimidines, which could be attributable to the inhibition of various enzymes involved in carcinogenesis.9
3.1 Tyrosine kinase
Tyrosine kinases are essential components of the signaling cascade that controls cell division, metabolism, migration, and the death of cells. Tyrosine kinases use ATP to catalyze the phosphorylation of particular tyrosine residues in their protein targets.28 Tyrosine kinases play an essential role in several stages of the emergence and spread of cancer. Tyrosine kinase signal transduction typically regulates excessive proliferation or increases apoptotic sensitivity. These signaling pathways are frequently genetically or epigenetically changed in cancer cells to provide specific benefits to cancer cells. As a result, it is no surprise that abnormally increased signaling from tyrosine kinases gives these enzymes a dominant oncoprotein status, resulting in signaling network failure.29
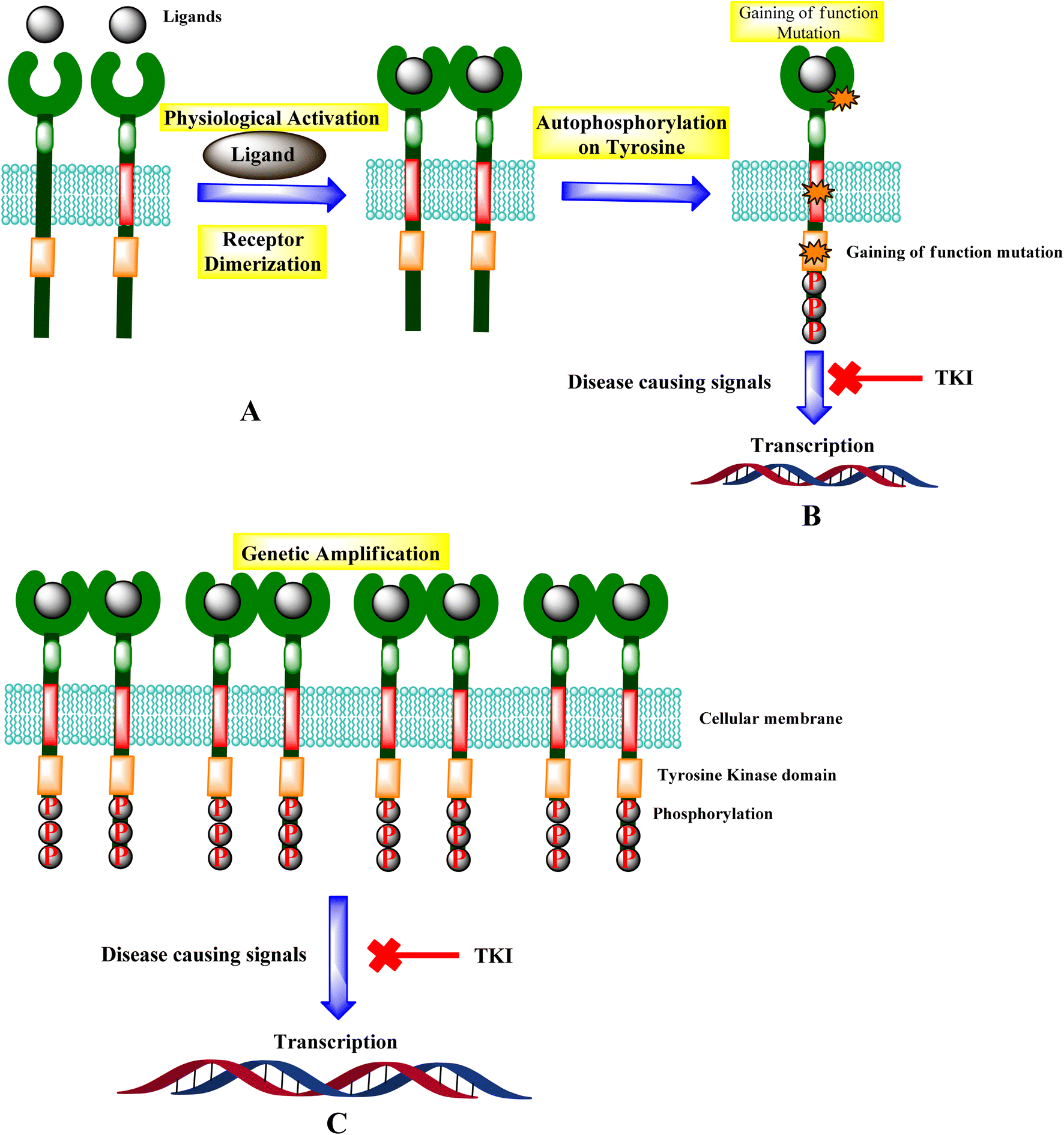
3.1.1 Biological pathway and inhibition of tyrosine kinase. Receptor tyrosine kinase (RTKs) are usually activated by ligands that target specific receptors. Growth factor ligands bind to RTK's extracellular regions, and the receptor activation is carried out by ligand-induced dimerization or oligomerization.30 Ligand attached to the extracellular domain stabilizes the generation of active dimers, which activates protein tyrosine kinases. The concentration of RTKs is increased by receptor oligomerization, enabling effective transphosphorylation of tyrosine residues inside the activation loop of the catalytic domain. For most RTKs, the resulting conformational alterations allow trans-autophosphorylation of each TKD and the release of cis-autoinhibition (Fig. 10).31,32
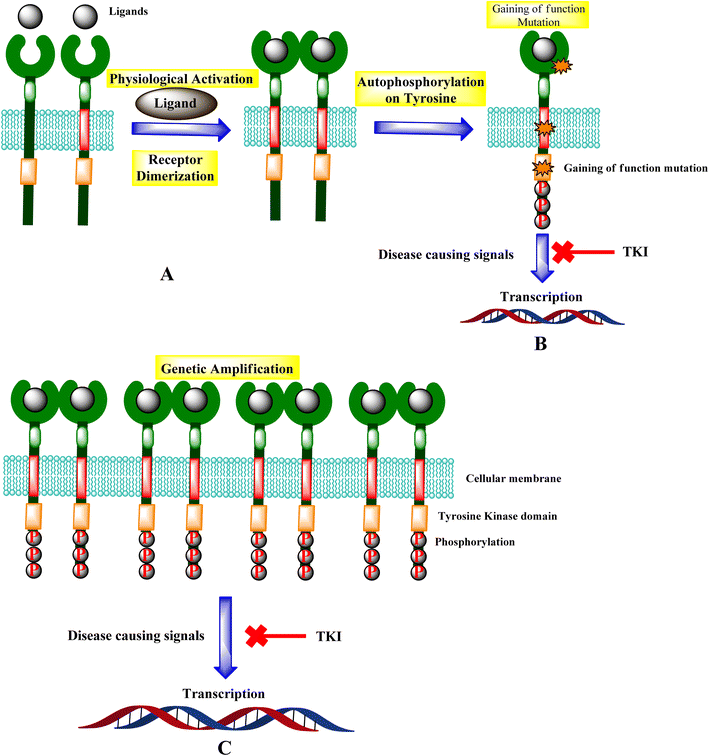 |
| | Fig. 10 Mechanisms of RTK activation. (A) RTK activation. (B) Potential gain-of-function mutations and the signal inhibition. (C) Genetic amplification leads to the transcription, and TKI leads to the inhibition of diseases causing signals. | |
Genetic amplification is the principal mechanism that leads to the overexpression of RTKs and transcription enhancement.33,34 SH2 domain proteins interact with phosphorylated receptors and become phosphorylated as well. The growth factor Ras/Raf/mitogen-activated protein map phosphorylated cascade then connects with the substance of the g-protein system, allowing the cell to migrate and express itself.30,35
3.1.2 Mechanism of action of tyrosine kinase inhibitors. Several amino acids of substrate enzymes are phosphorylated by tyrosine kinases, leading to different changes in the downstream cellular biology and signaling. TKs can alter cell growth, migration, differentiation, apoptosis, and death by triggering downstream signal transduction. Constitutive activation or inhibition, whether caused by mutations or other methods, can result in dysregulated signal cascades, which can lead to cancer and other diseases.35,36 The tyrosine kinase inhibitors inhibit this dysregulated signal cascade and block transcription in both genetic amplification and mutation, leading to the disease-causing signals.36 The compounds 47–51 showed excellent activity against various kinases, including TKs, PI3K, and CDK4/6. In molecule 47, the presence of dichloro and tertiary amine increased the activity. In molecule 48, piperazine with amine linkage and acetyl group at pyridine ring increased the activity, thiophene, phenyl with fluorine atom were responsible for activity in 49, presence of methoxy and morpholine were necessary for activity in molecule 50, also other triazole rings in 51 enhanced the activity (Fig. 11).37–40
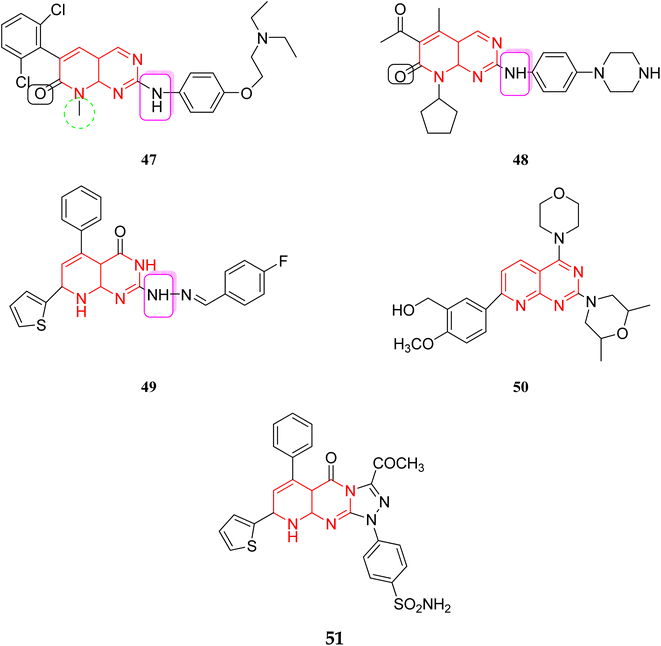 |
| | Fig. 11 Reported pyrido[2,3-d]pyrimidine as kinase inhibitors. | |
In 2018, Elzahabi et al. successfully synthesized substituted pyrido[2,3-d]pyrimidines and tested their ability to inhibit the growth of five cancer cell lines. Pyrido[2,3-d]pyrimidines, were found to have significant inhibitory effects against a variety of kinases, including TKs, PI3K, and CDK4/6.9
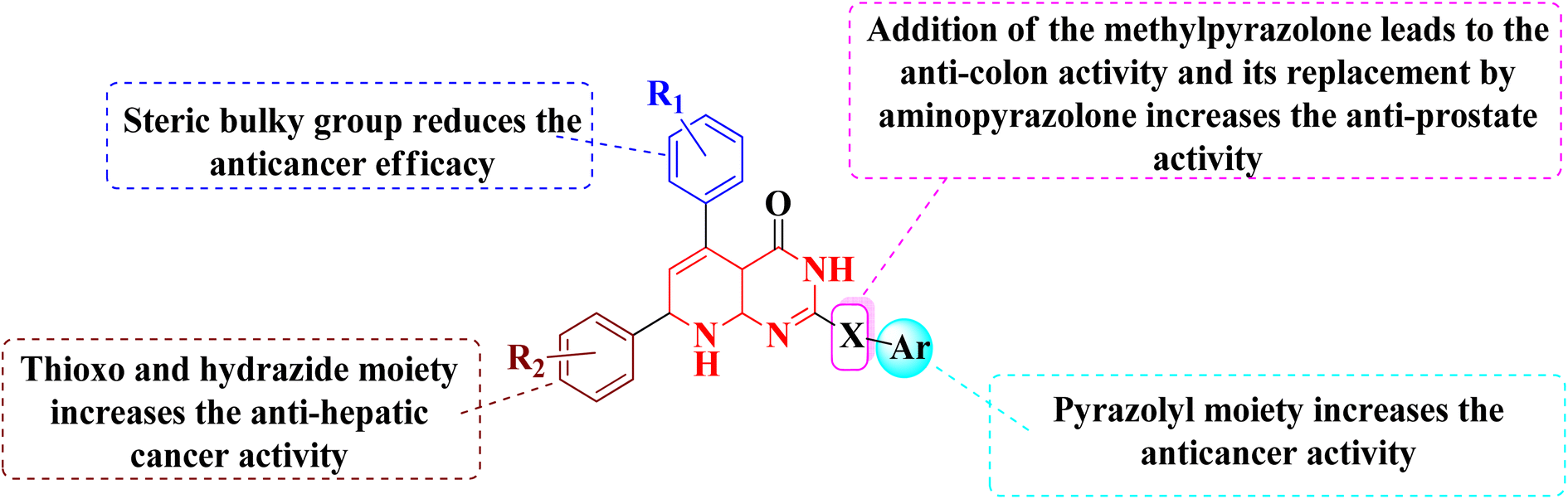
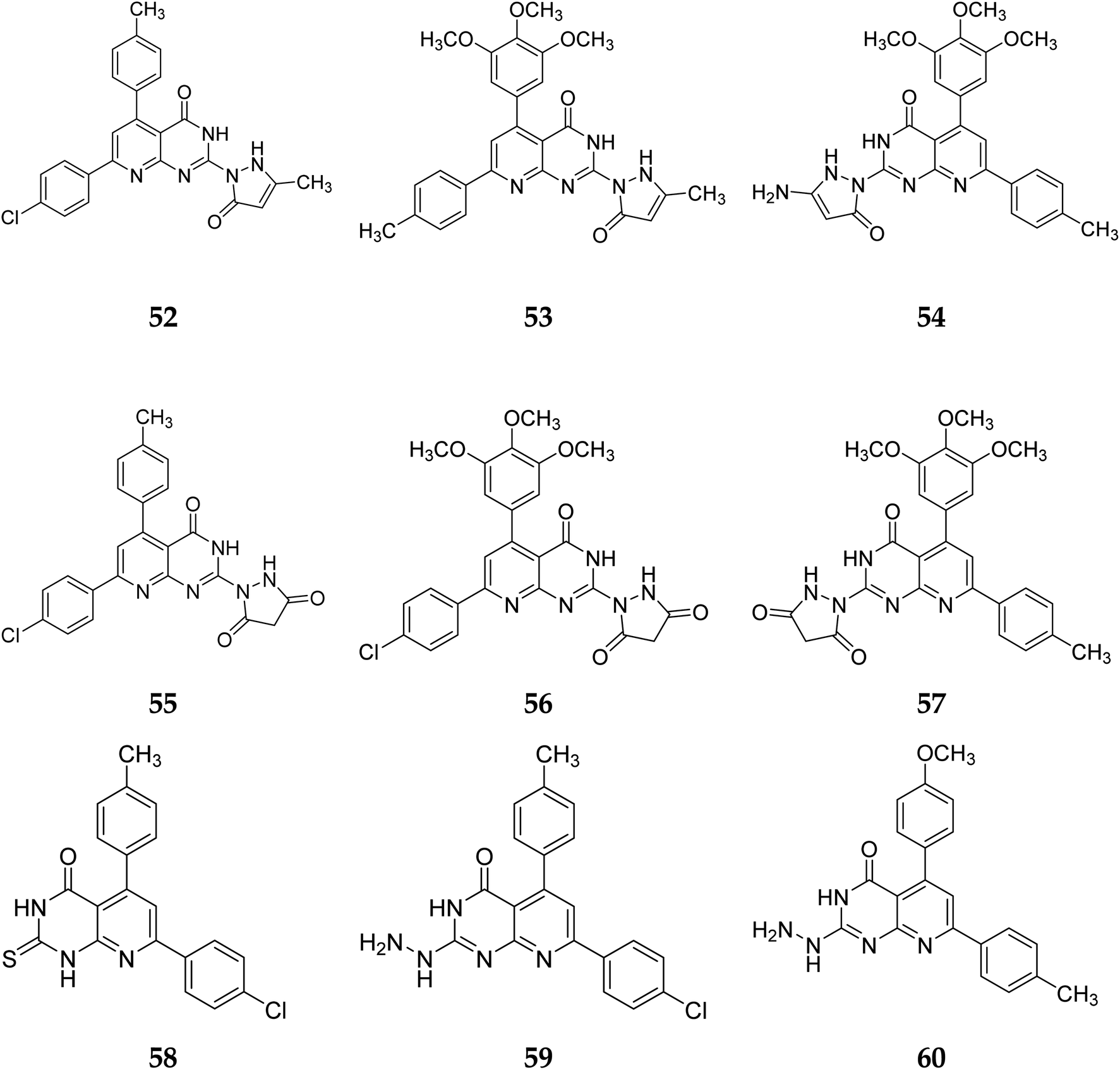
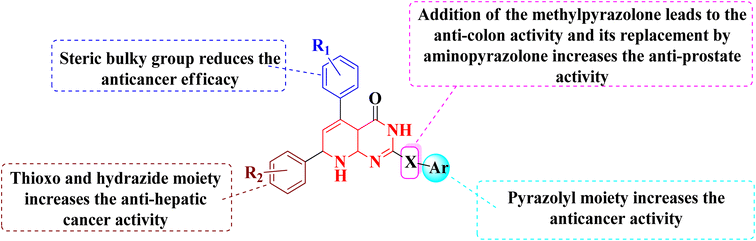
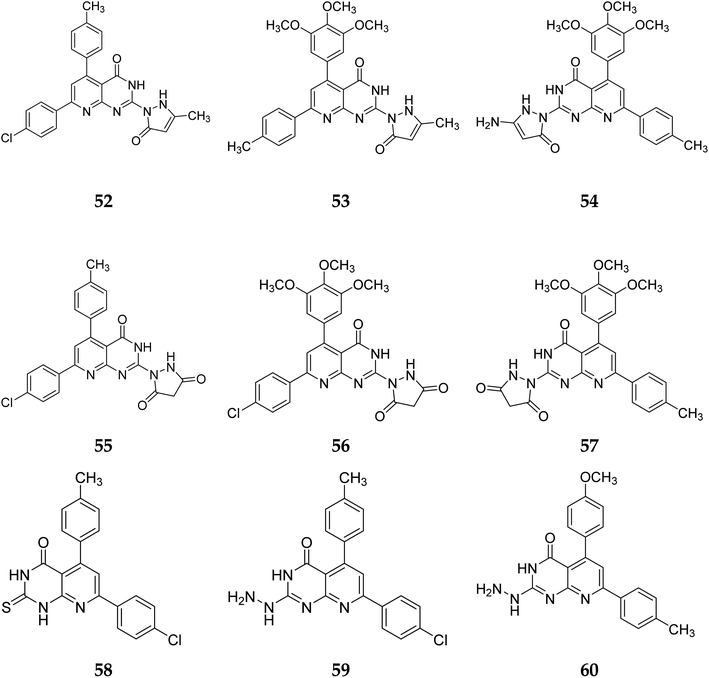
3.1.3 Structure activity relationship of tyrosine kinase inhibitors. According to the structure–activity relationships of the screened products, presence of carbonyl at C-2 of pyrido[2,3-d]pyrimidine derivatives (52–56) provided the maximum anticancer activity. Particularly 52 and 53, which had (4-CH3-phenyl) and (4-chlorophenyl) at C-5 and C-7, respectively, shared the same pyridopyrimidine scaffold. Compound 53 containing 3-methyl-5-oxopyrazolyl moiety of showed strong anticancer efficacy against prostate, colon, and liver malignancies. The anticancer action was switched from anti-colon to anti-prostate cancer when the methylpyrazolone moiety in 53 was replaced with aminopyrazolone in 54 (Fig. 12 and 13).
 |
| | Fig. 12 SAR of pyrido[2,3-d]pyrimidine derivatives. | |
 |
| | Fig. 13 Pyrido[2,3-d]pyrimidine derivatives reported as tyrosine kinase inhibitors. | |
In the same way, adding a steric bulky group to 56 and 57 reduced its anticancer efficacy. Converting the thioxo group in 58 to the hydrazide moiety in 59 significantly enhanced the anti-hepatic cancer activity. The electronic component was able to have a positive impact on anticancer activity due to the hydrophilic electron-rich nature of the hydrazide molecule.9
A novel class of substituted pyrido[2,3-d]pyrimidinones was investigated for their in vitro anticancer efficacy. Compounds 52 and 55 were more effective (IC50 = 0.3 μM) than doxorubicin in the HepG-2 cell line. In addition, compound 59 was approximately equivalent to doxorubicin (IC50 = 0.6 μM). Compounds 60 and 52 were having higher activity than doxorubicin (IC50 = 5.47, 6.6, and 6.8 μM, respectively) in a cellular screening on PC-3. The activity of 60, 52, and 53 was two-fold as that of doxorubicin in the HCT-116 cell line (IC50 of 6.9, 7, 5.9 μM, and 12.8 μM, respectively). Synthesized compounds showed significant growth inhibitory effects in HepG-2, HCT-116 as well as PC-3 cell lines with reference to doxorubicin, but on MCF-7 and A-549 cancer cells showed comparetively low activity. PDGFR β, EGFR, and CDK4/Cyclin D1 kinases were all inhibited by the potent anticancer molecule 52 (Fig. 13).9
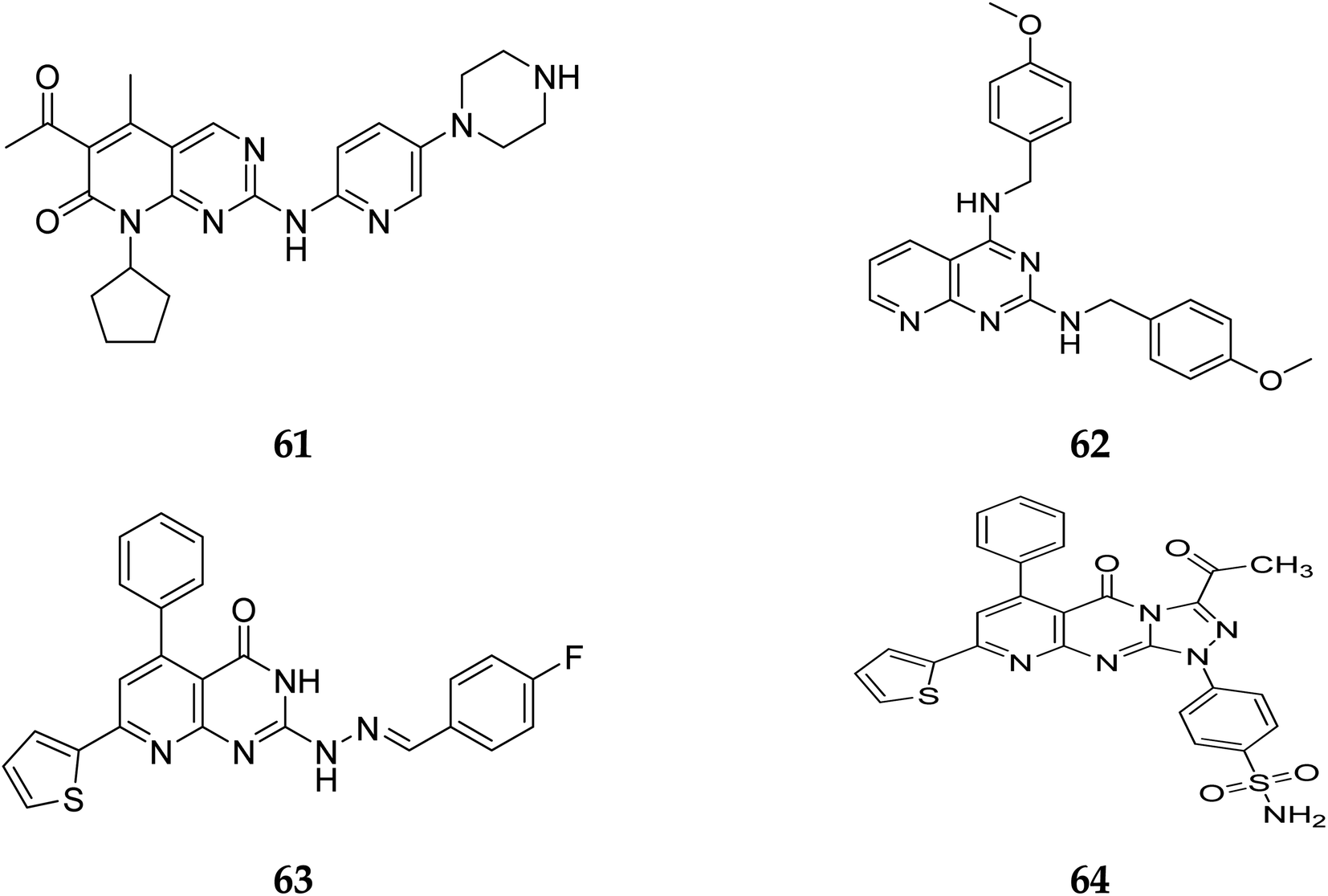
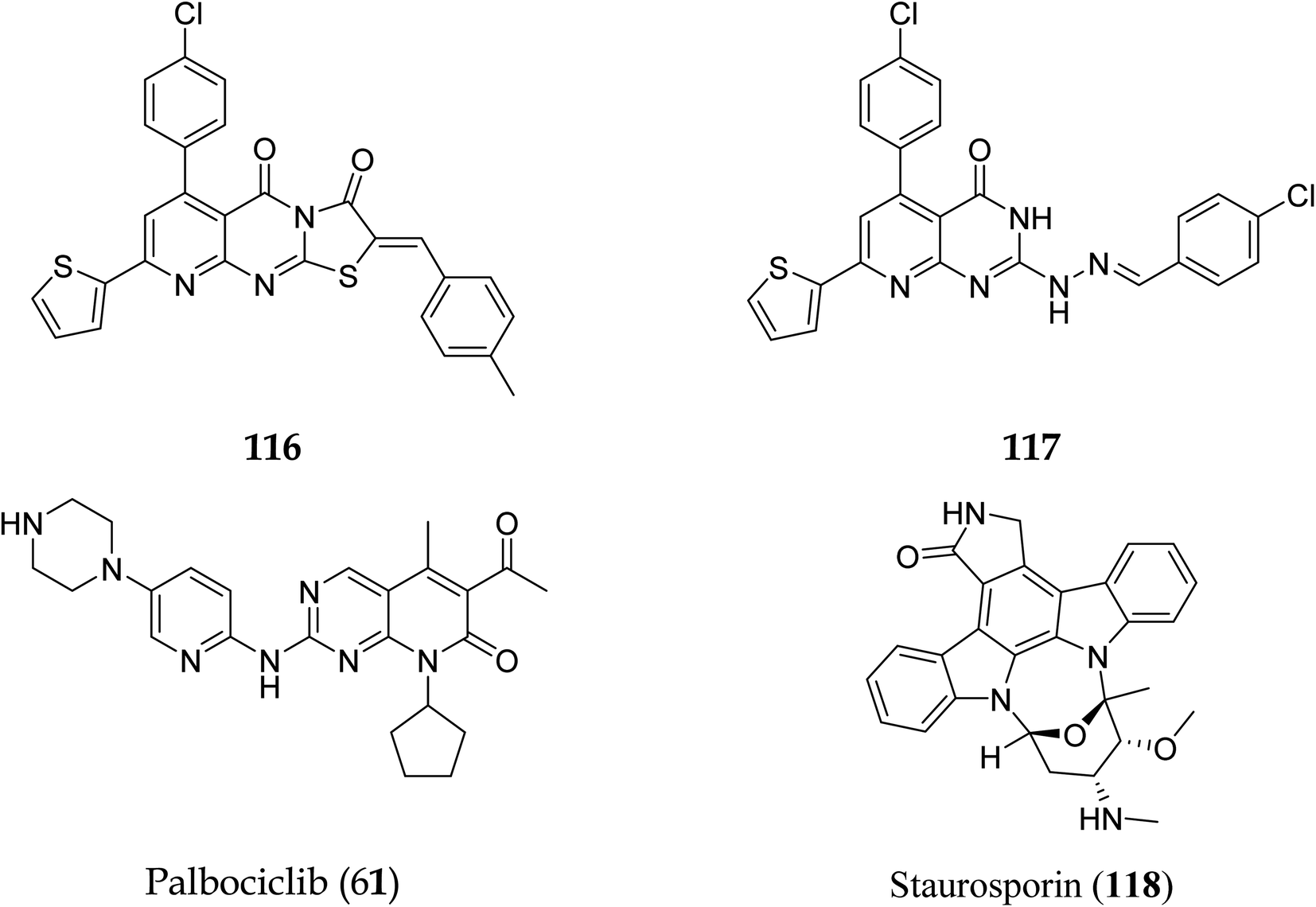
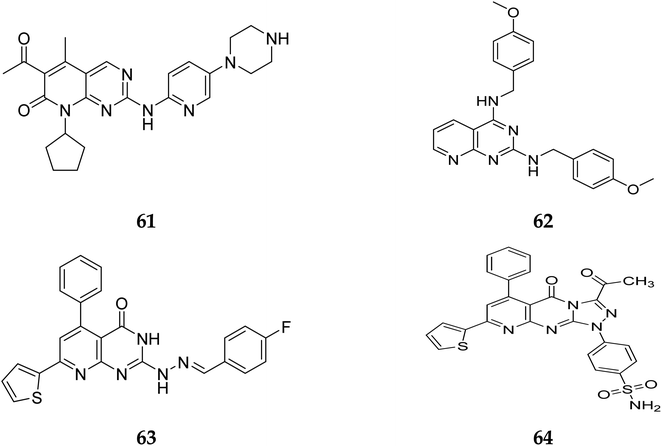
Pyrido[2,3-d]pyrimidine is a vital core known to play a role in many active compounds with diverse activities, particularly chemotherapeutic, which may be capable of inhibiting cyclin-dependent kinase (CDK). For example, The US Food and Drug Administration has authorized the CDK4/6 inhibitor palbociclib (61), which is used to treat breast cancer. In addition, 2,4-diaminopyrido[2,3-d]pyrimidine (62) is a promising anticancer drug with dose-dependent antiangiogenic and proapoptotic actions through induction of 2 separate signaling pathways, notably G2/M cell cycle arrest as well as induction of apoptosis by encouraging caspase-3 activation and DNA fragmentation. Furthermore, compound 63 showed remarkable anticancer activity against prostate (PC-3) and lung (A-549) cancer cells with IC50 values of 1.54 and 3.36 μM, respectively. Furthermore, at submicromolar concentrations (0.36, 0.41 μM, respectively), the fused triazolopyridopyrimidine (64) showed greater efficacy against both cell lines, causing cell cycle arrest by increasing the cell cycle inhibitor p21 and cancer cell death by activating caspase-3 (Fig. 14).41
 |
| | Fig. 14 Pyrido[2,3-d]pyrimidine derivatives reported as anticancer agents. | |
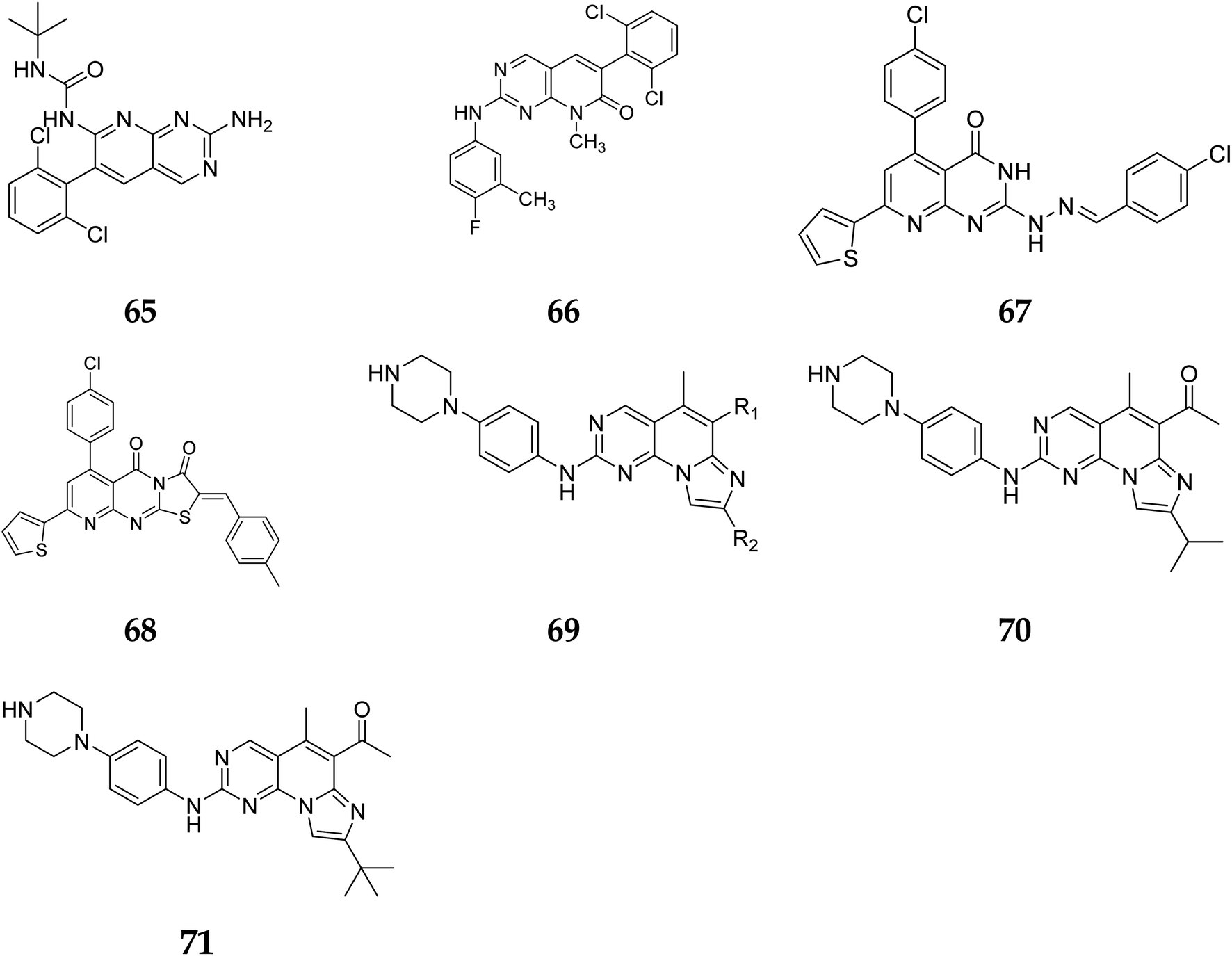
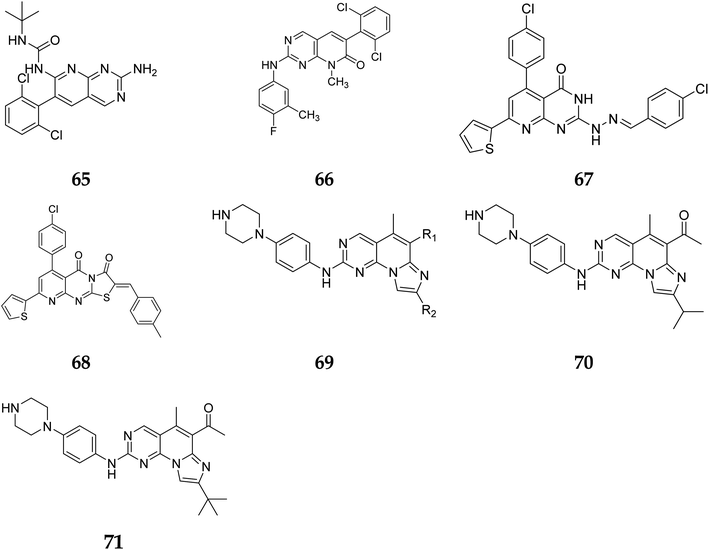
Another study discovered a highly potent tyrosine kinase inhibitor (65) having IC50 values of 1.11, 0.13, 0.45, and 0.22 μM against the tyrosine kinases, PDGFr, FGFr, EGFr, and c-src respectively.42 PD180970, the derivative of pyrido[2,3-d]pyrimidine, acted by inhibiting ATP-competitive protein tyrosine kinase. Dorsey et al. found that in human K562 chronic myelogenous leukemic cells, 66 reduced in vivo tyrosine phosphorylation of p210Bcr-Abl (IC50 = 170 nM), the p210Bcr-Abl substrates Gab2 and CrkL (IC50 = 80 nM).43 Compounds 65 and 66, with IC50 values of 115.38 nM and 726.25 nM, were the most effective direct CDK6 inhibitors in the series. Compound 65 caused apoptosis 1.9-fold in PC-3 cells and 1.8-fold in MCF-7 cells in the apoptotic assay. The molecule 65 triggered apoptosis primarily by activating caspase-3. Compound 66, on the other hand, promoted apoptosis primarily through the intrinsic route, with direct inhibition of CDK6.44 Shi et al. proposed a new class of CDK4/6 inhibitors: imidazo[1′,2′:1,6]pyrido(2,3-d)pyrimidine analogs.44,45 In the Colo-205 as well as U87MG cell lines, CDK4/6 was found to be effectively suppressed by compounds 70 and 71. The equipotent of palbociclib and abemaciclib, compounds 70 and 71 significantly inhibited CDK4/D3 (IC50 = 0.8 nM and 2.7 nM, respectively) and CDK6/D3 (IC50 = 2.0 nM and 4.8 nM, respectively). The acetyl and isopropyl moieties at C6 and C8 positions (compound 70) represented 2.5 more possible biological activities for CDK4/cyclinD3 than CDK6/cyclinD3.45 Changes to the functional group tert-butyl at the C8-substituent (compound 71) did not affect CDK4/6 targeting bioactivity. Compounds 70 and 71 showed favorable antiproliferative effects, remarkable metabolic properties, useful pharmacokinetic features, and significant tumour growth inhibitions with manageable toxicity in Colo-205 and U87MG xenograft models (Fig. 15).45
 |
| | Fig. 15 Pyrido[2,3-d]pyrimidine derivatives reported as anticancer agents. | |
3.2 Extracellular signal-regulated kinase and Phospatidylinositol-3 kinaseα
3.2.1 Extracellular regulated protein kinases (ERK). A serine/threonine protein kinase known as ERK1/2 (extracellular signal-regulated kinase) is frequently found in eukaryotic cells. It belongs to the family of MAPK (mitogen-activated protein kinases). The transfer of signals from surface receptors to the nucleus is significantly aided by ERK1/2. Apoptosis, cell proliferation, differentiation, and other activities are regulated by activated ERK1/2, which phosphorylates substrates in the cytoplasm or nucleus and causes the production or activation of particular proteins.46
3.2.2 Phospatidylinositol-3 kinase. Many cancers, including breast, gastric, ovarian, colorectal, prostate, glioma, and endometrial cancers, have altered or amplified the activity of the critical intracellular signalling system known as PI3K (phospatidylinositol-3 kinase, or PI3K). PI3K signalling is a viable therapeutic target since it is crucial for cancer cell survival, angiogenesis, and metastasis. Clinical trials employing PI3K inhibitors are continuing and have already been completed to identify effective PI3K inhibitors that could overcome resistance to existing treatments.47 Critical physiological and pathological processes like metabolism, cell development, proliferation, angiogenesis, and metastasis are influenced by PI3K/AKT signalling and contribute to the progression of tumours.48,49 PI3K proteins are categorized into three major classes based on the structural characteristics of the proteins as well as the peculiarities of the substrates (I, II, and III). Class, I PI3Ks are classified into subtypes A and B based on how they are regulated. Class IA PI3Ks combine their regulatory (p85, p85, p55, p55, and p50) and catalytic (p110, p110, p110) subunits to form dimers.50
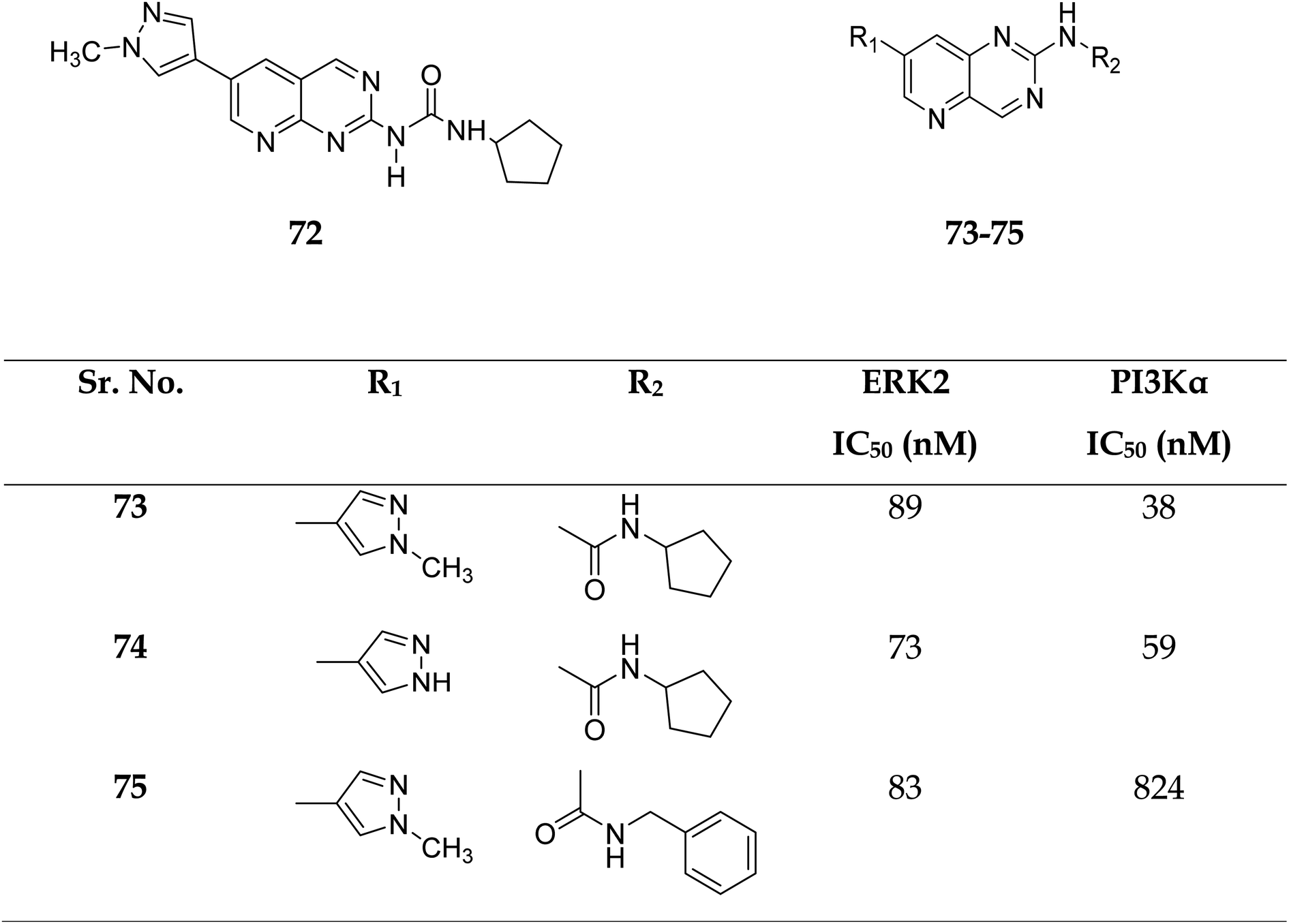
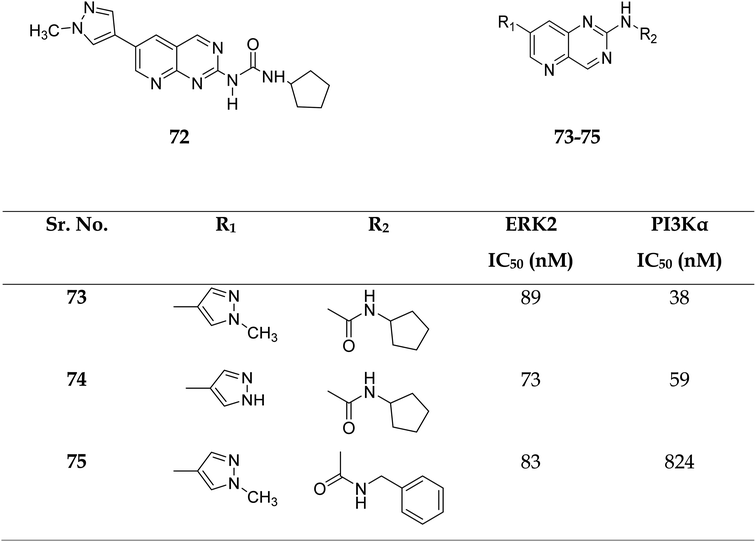
3.2.3 ERK2 and PI3Kα. The combined inhibition of the MAPK and PI3K signalling pathways has been identified as a viable cancer therapy that effectively overcomes the drug resistance of MAPK signalling pathway inhibitors.51Zhang et al. reported the scaffold-hopping synthesis of ERK/PI3K dual inhibitors by replacing 1H-pyrazolo[3,4-d]pyrimidine scaffold with pyrido[3,2-d]pyrimidine. Compound 73 with pyrido[3,2-d]pyrimidine scaffold exhibited suitable inhibitory activities on ERK2 and PI3Kα, while 72 with pyrido[2,3-d]pyrimidine scaffold had 1.8 and 15.1% inhibitory activity at the concentration of 1 μM on ERK2 and PI3Kα respectively (Fig. 16).51
 |
| | Fig. 16 Pyrido[2,3-d]pyrimidine and pyrido[3,2-d]pyrimidine derivatives as dual inhibitors of ERK2 and PI3Kα. | |
Compounds 73–75 were foundd to be dual inhibitors of ERK2 and PI3K-α. Compound 74, a potent and very effective ERK and PI3K dual inhibitor, was discovered during preliminary SAR study. Compound 74 had modest ERK and PI3K inhibitory and anti-proliferative properties. Although 74 had only acceptable pharmacokinetic characteristics in SD rats, with a moderate half-life (t1/2 = 2.32 h) after intravenous treatment, it displayed significant anticancer activity in vivo in an HCT-116 xenograft model without eliciting obvious adverse consequences.51
3.3 Antitumor activity
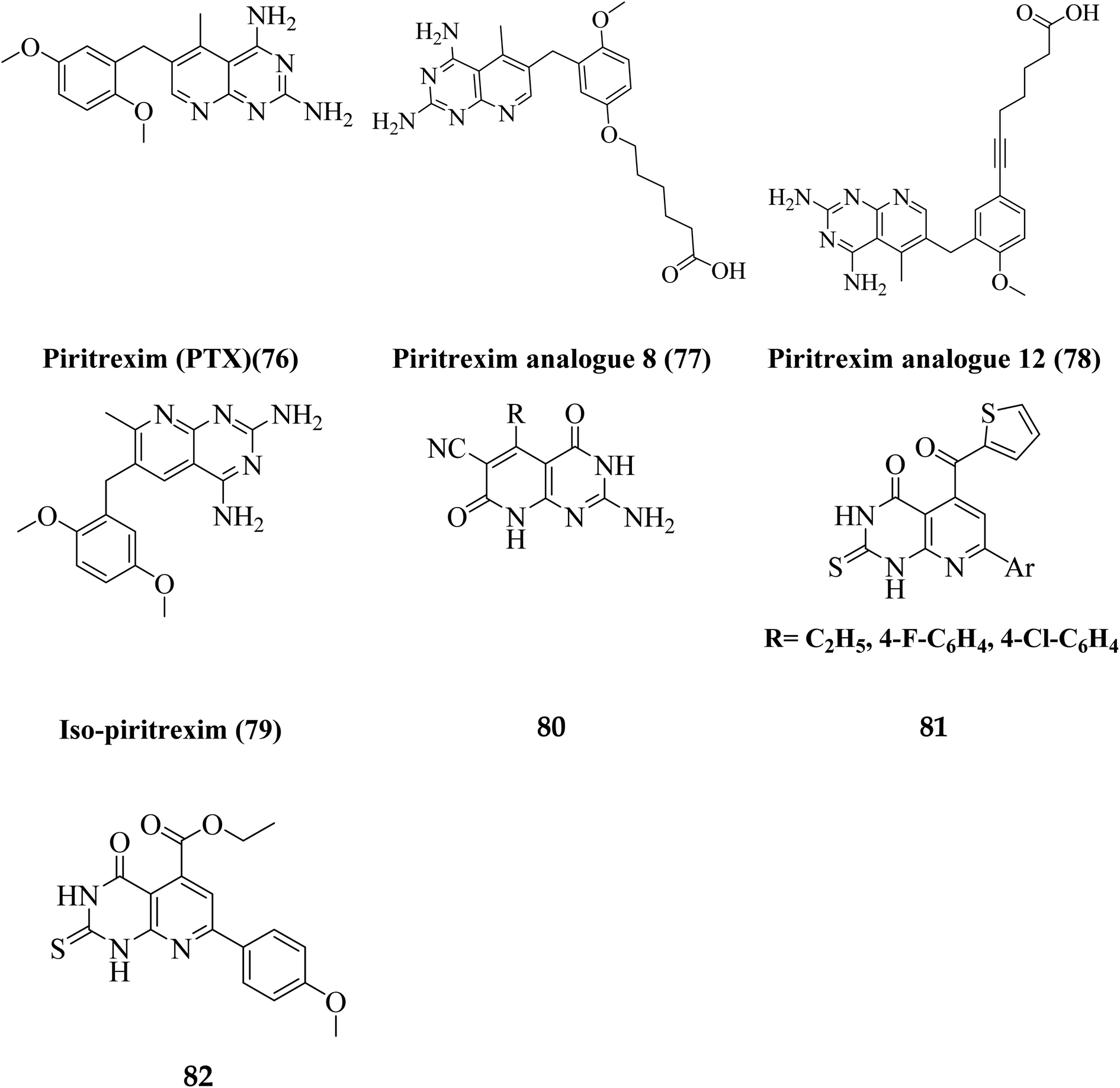
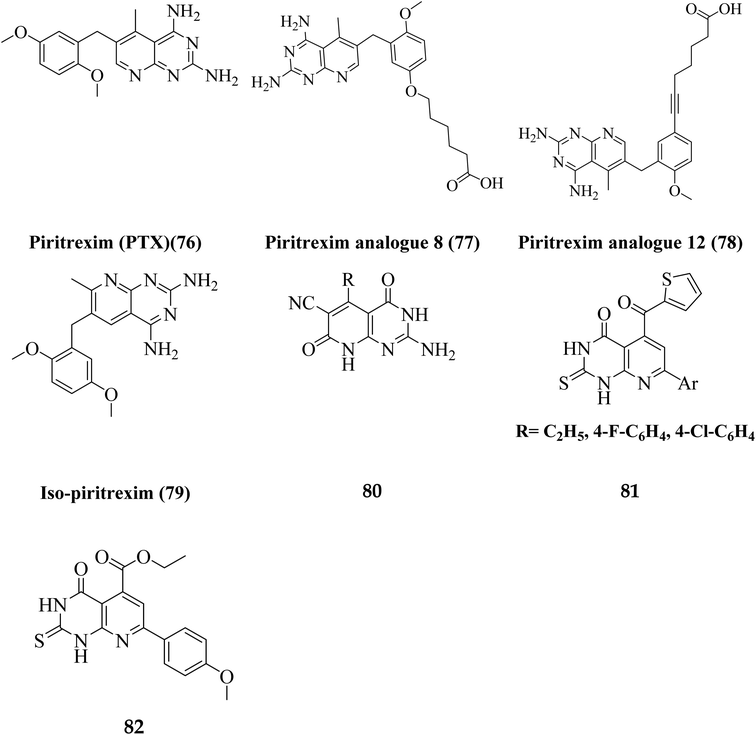
Literature review revealed, Piritrexim (76), and its analogues 77–82 (having pyrido[2,3-d]pyrimidine pharmacophore) showed excellent antitumor activity (Fig. 17).27,52–61
 |
| | Fig. 17 Pyrido[2,3-d]pyrimidine derivatives reported as antitumor agents. | |
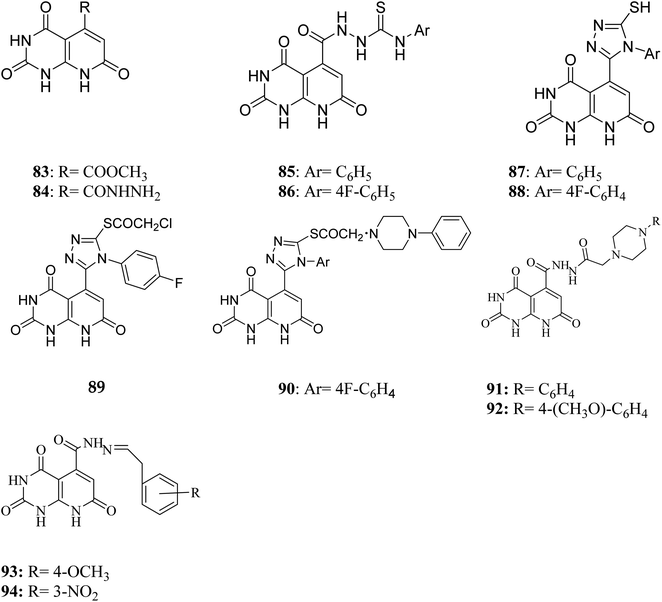
Gineinah et al. designed and synthesized pyrido[2,3-d]pyrimidine derivatives; this scaffold was then hybridized with one or more heterocyclic moieties (piperazines or triazoles).62 Despite structural alterations to the beginning molecule 83, the examined compounds' principal moiety (pyridopyrimidine) led to the conservation of biological activity. While adding the N-phenylthiosemicarbazide moiety, compound 85, at position five of the pyridopyrimidine, increased activity compared to carbohydrazide 84, adding the electron-withdrawing fluoro group to compound 86 at position four of the phenyl ring decreased activity, transferring the ester to acid hydrazide, compound 84, decreased activity. In general, compounds 87 and 88 were found more active than their non-cyclized counterparts, compounds 85 and 86, demonstrating that cyclization of thiosemicarbazide-containing compounds into [1,2,4]-triazoles resulted in more substantial and more robust antitumor activity. Furthermore, compound 89 had lower activity than compounds 87 and 88, indicating that the free sulfanyl group (–SH) had greater potency. In contrast to compound 89, compound 90 was very active as an anticancer drug, implying that the piperazine moiety enhanced antitumor activity. Similarly, including a piperazine moiety in compounds 91 and 92 resulted in high activity (Fig. 18).
 |
| | Fig. 18 Pyrido[2,3-d]pyrimidines reported as antitumor agents. | |
In contrast, the 1-phenylpiperazine, a methoxy group at the fourth position, decreased absorbance, rendering molecule 92 more efficient than molecule 91. This demonstrated that enhanced activity may occur when a piperazine moiety is modified at place four with a phenyl ring modified with an electron-donating compound, but not when an electron-withdrawing group or even with an unsubstituted group. It has been demonstrated that compound 93's methoxy group on fourth place of the Schiff base aromatic ring was more active than compound 94's nitro group. The presence of an electron-donating group on an aromatic nucleus is associated with increased anticancer action in part.62
3.4 BCR-ABL kinase
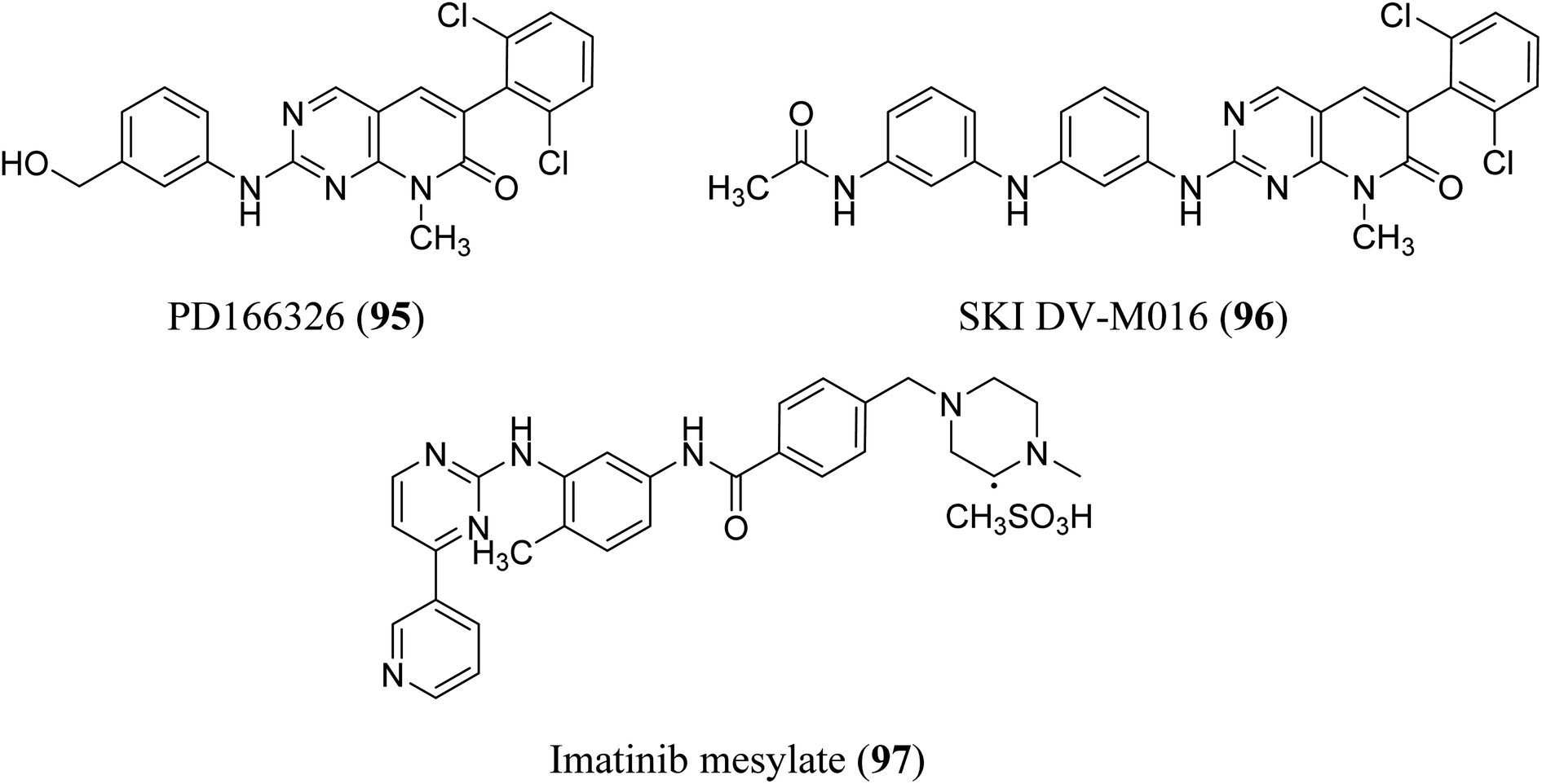
BCR-ABL is a mutation that results from the interaction of two genes, BCR and ABL. It is also known as a fusion gene. The BCR and ABL are present on chromosomes 22 and 9, respectively. When portions of the BCR and ABL genes break off and swap locations, the BCR-ABL mutation occurs. BCR-ABL genes were found in patients with certain types of leukemia and also detected in almost all patients with chronic myeloid leukemia (CML).63 Loss of auto-inhibition and constitutional activation of ABL1 may occur when the mutant BCR-ABL1 protein arises, with subsequent effects on signalling pathways related to cell cycle and apoptosis, such as the RAS/RAF/MEK/ERK pathway, the JAK2/STAT pathway, and the PI3K/AKT/mTOR pathway. Finally, it promotes the malignant transformation of hematopoietic cells.64 pyrido[2,3-d]pyrimidine are effective tyrosine kinase c-Abl and Bcr/Abl inhibitors. At an IC50 2.5 nM, PD180970 reduced Bcr/Abl tyrosine phosphorylation and triggered death in the human CML cell line K562 43. PD180970 was also effective against imatinib mesylate-resistant K562 cells and Ba/F3-P210 cells expressing BCR/ABL kinase domain mutations observed in imatinib mesylate-resistant CML patients.65 Compared to imatinib mesylate, PD173955 suppressed the growth of the Bcr/Abl cell line R10(−) with an IC50 of 2.5 nM.66 PD166326 (95) was the most potent inhibitor of CD34+ CML progenitor development among the six pyridopyrimidines studied, with an IC50 around 4-fold lower than PD173955. With an IC50 of 300 pM in K562 cells, PD166326 (95) reduced proliferation66 and demonstrated action against numerous imatinib mesylate-resistant Bcr/Abl mutants.66,67 western blot analysis of Bcr-abl autophosphorylation on K562 cells revealed that PD166326 (95) inhibited Bcr-abl activity in cells. In these cells, IC50 for the suppression of Bcr-abl autophosphorylation was 1 nm as compared with 100 nm for STI571. The biological activity and potency of PD166326 (95) was tested in K562 cells in cell growth assays. With an IC50 of 0.3 nm, compound 95 suppressed K562 cell growth. Additional hematopoetic and epithelial cell lines were only inhibited at two to three log higher doses with IC50s in the 0.8–2 nm range, indicating that PD166326's powerful biological activity was highly selective for Bcr-abl-driven cells.68 Bubnoff et al. tested 13 different pyridopyrimidine derivatives against wild-type as well as mutant BCR-ABL. All the compounds decreased the Bcr-Abl dependent phenotype more effectively than imatinib and inhibited Bcr-Abl kinase activity (Table 1). PD166326 (95) and SKI DV-M016 (96) were the most active compounds. Surprisingly, these drugs repressed Bcr-Abl H396P, a mutant with an activation loop, just as well as wild-type Bcr-Abl (Fig. 19).65
Table 1 Bcr-Abl kinase inhibitory activity of compounds 95 and 96
| Name |
IC50 value (nM) (mean ± SE) |
| SKI DV-M016(96) |
4.2 ± 0.6 |
| PD 166326(95) |
4.3 ± 0.8 |
 |
| | Fig. 19 Pyrido[2,3-d]pyrimidine derivatives as BCR-ABL kinase inhibitors. | |
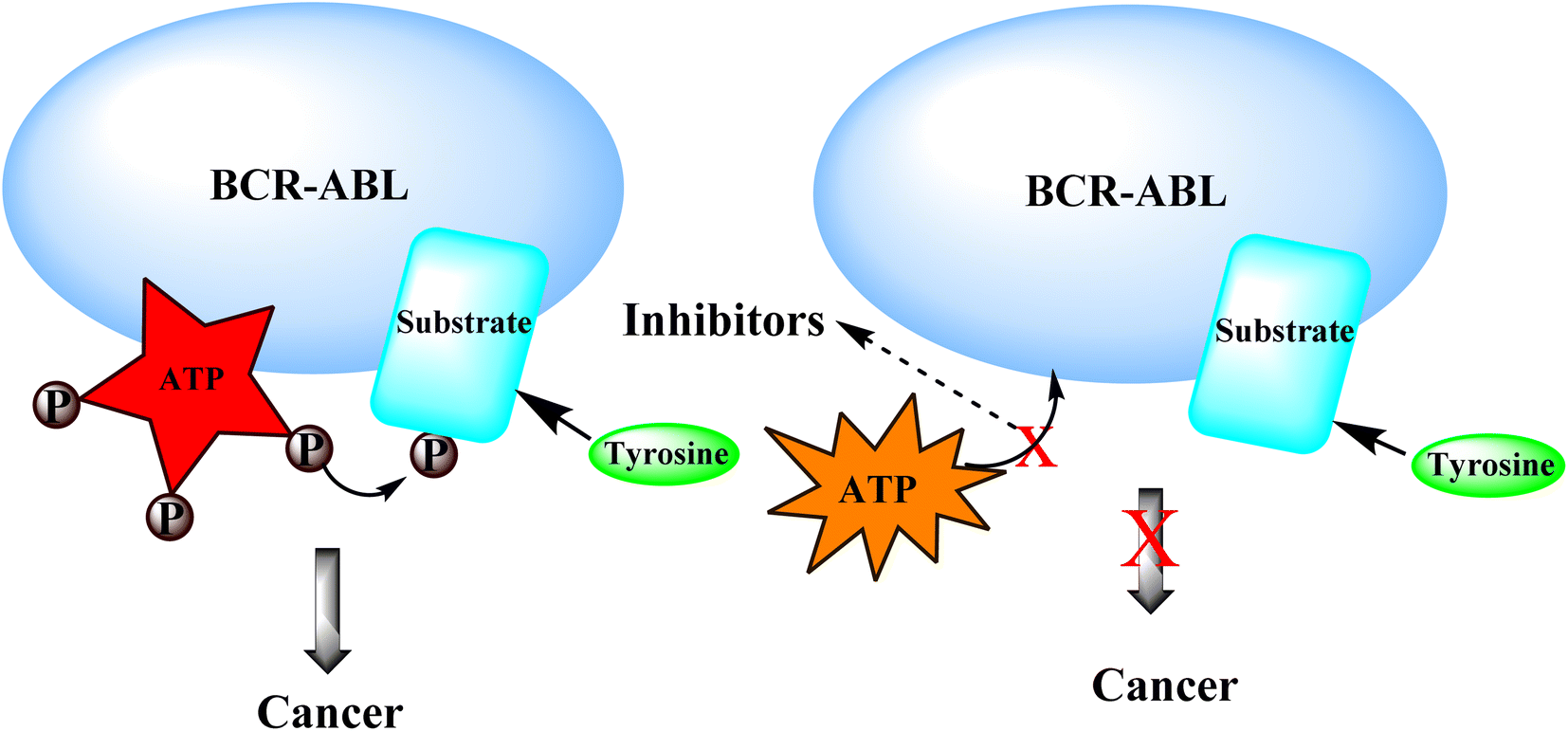
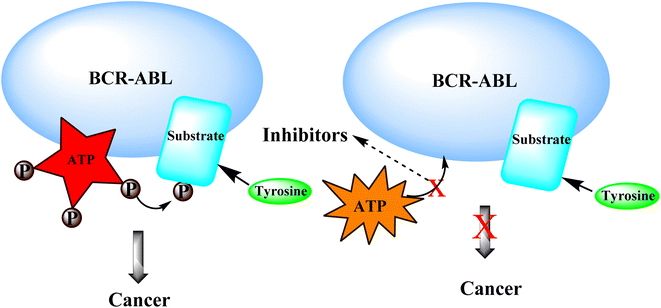
3.4.1 Mechanism of action of BCR-ABL inhibitors:-. BCR-ABL inhibitors compete with adenosine triphosphate (ATP) for the same binding site inside the BCR-ABL protein's kinase domain. When ATP is present in the binding site, BCR-ABL can catalyze the phosphorylation of tyrosine residues on substrate proteins. These BCR-ABL inhibitors (Imatinib mesylate) have a structure similar to ATP and can bind to the BCR-ATP-binding ABL's region. Because BCR-ABL inhibitors lack the necessary phosphate groups given by ATP, phosphorylation of tyrosine residues on substrate proteins is impossible. Unphosphorylated tyrosine residues in substrate proteins prevent them from adopting the required conformation for binding to effector molecules. As a result, downstream signal transduction pathways important for CML leukemogenesis are blocked (Fig. 20).69
 |
| | Fig. 20 Mechanism of action of BCR-ABL inhibitors. | |
3.5 p38 mitogen-activated protein kinases
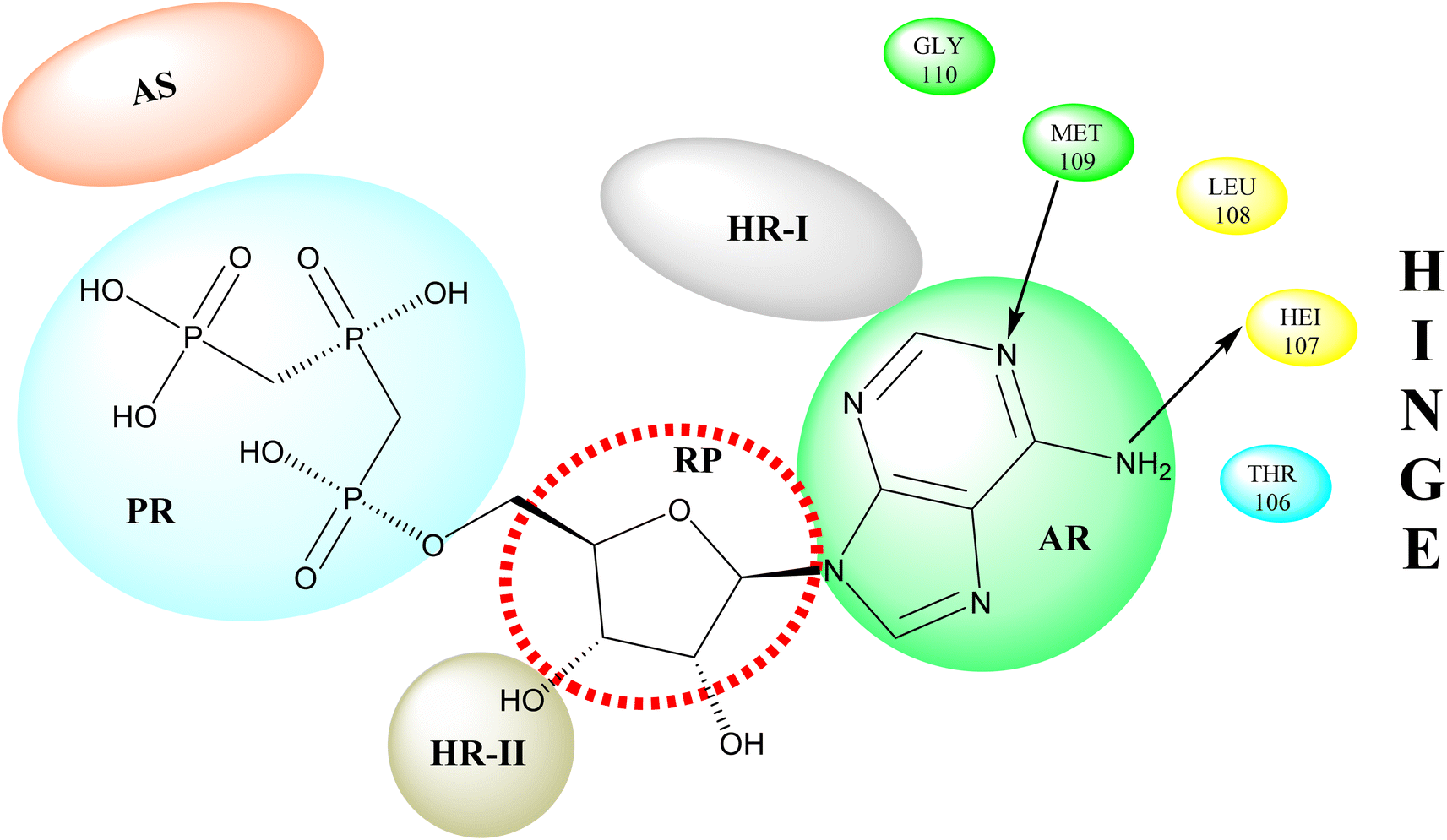
p38 Mitogen-activated protein kinases (p38 MAPKs) are proline-directed serine/threonine kinases that regulate pro-inflammatory signalling networks and are involved in the production of cytokines such as tumour necrosis factor-a (TNF-a) and interleukin-1b (IL-1b).70,71 Four isoforms of p38 MAPK have been found, namely α, β, γ, and δ, with p38a MAPK being the earliest identified, best described, and most significant isoform involved in the inflammatory response.72 The N- and C-terminal domains of p38a MAPK form the walls of a deep cleft in which the ATP cofactor is allocated in a pocket known as the ATP binding site.73 The adenine ring of ATP formed hydrogen bonds with His107 as well as Met109 residues of the p38α MAPK backbone. These two residues are found in the hinge region (residues 106–110), which connects the N and C-lobes, whereas the adenine binding region and ribose pocket, respectively, are located in the adenine binding region and ribose pocket, respectively.74 The dual interaction of ATP with the hinge region aligns the two protein lobes to produce a pocket large enough to hold the ATP triphosphate group; this pocket is known as the phosphate binding area and contains highly conserved kinase residues like Glu71 and Asp168.75 Three more hydrophobic areas have been discovered adjacent to the ATP binding site but are not directly involved in ATP binding (Fig. 6).76 The first section is known as hydrophobic region I (HR-I), and it is defined by Leu75, Lys53, Ala51, Leu104, Ile84, Leu167, and Thr106. It is placed at the back of the ATP binding site (Fig. 21).77 Because similar kinases have a bulkier residue at the same position, the tiny size of the gatekeeper residue Thr106 allows access to this pocket, which is essential for kinase inhibitor selectivity. The hydrophobic region II (HR-II), a solvent-exposed hydrophobic area defined mainly through residues Val30, Ile108, Gly110, Ala111, and Asp112, is the second section.78 The allosteric site is located next to HR-I and is only accessible when the kinase adopts the catalytically inactive conformation (DFG-out), in which the highly conserved Asp168-Phe169-Gly170 (DFG) motif is flipped “out” in comparison to its active state conformation (DFG-in).79
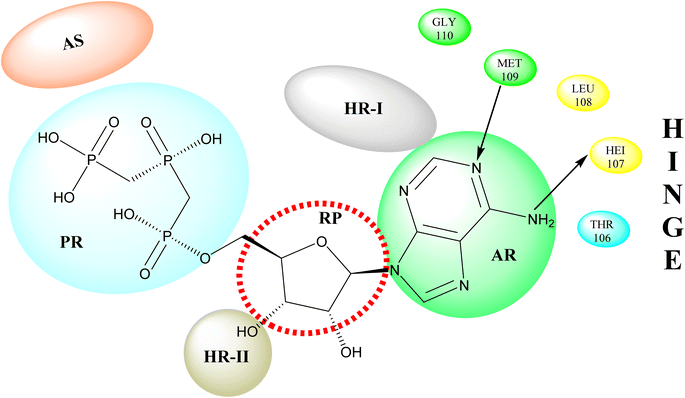 |
| | Fig. 21 Representation of ATP within p38a MAPK binding site of APT. AS: allosteric site; RP: ribose pocket; AR: adenine region; HR-I and HR-II: hydrophobic regions I and II; PR: phosphate region. | |
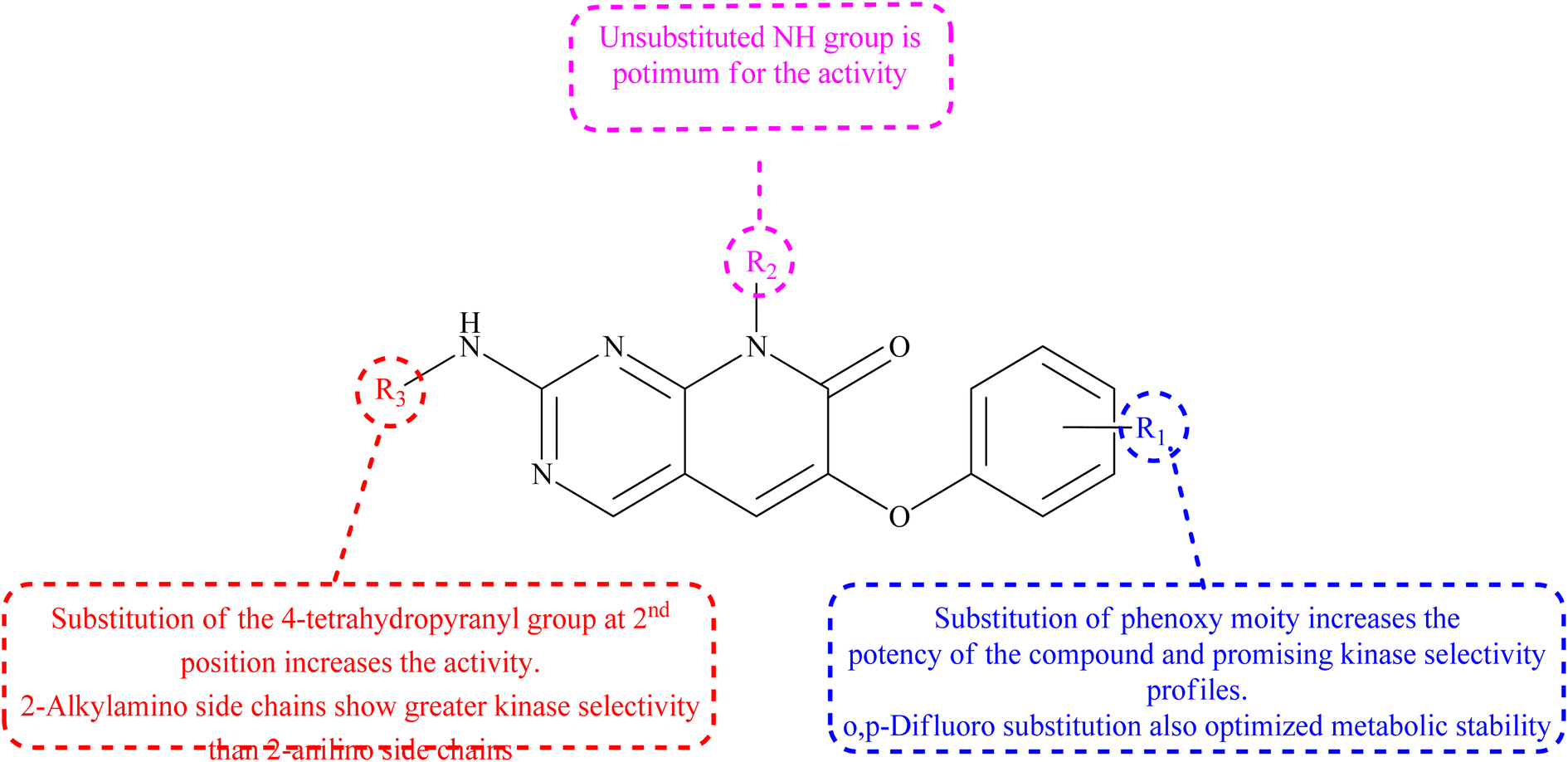
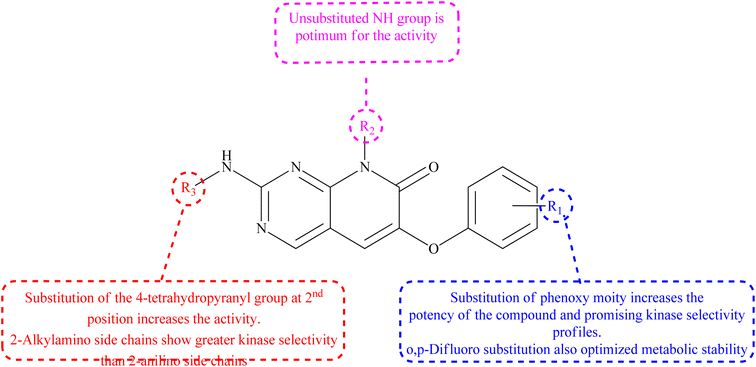
3.5.1 Structure–activity relationship of P38α inhibitors. The 6th-substituent substantially influences inhibitors' pan-kinase selectivity. In terms of potency, this position is also critical. 6-Aryl substituents, such as o-chlorophenyl in compound 98, are generally potent compared to 99 (Table 2). Phenoxy substituents were chosen because of their high activities and intriguing profile of kinase selectivity. SAR revealed that the phenoxy group found having tiny ortho and para substituents are preferred over no and meta substitutions. The ortho and para difluoro substitution improved the side chain's metabolic stability. At the 8th-position of the scaffold, many different side chains are allowed, reinforcing the idea that this variable points towards solvent. The kinase selectivity of 2-alkylamino side chains was higher than that of 2-anilino side chains. The molecule's potency was increased by substituting the 4-tetrahydropyranyl group in the second position (Fig. 22).22
Table 2 IC50 (nM) of compounds 98 and 99 as P38α inhibitors
| Compound |
X |
R1 |
R2 |
R3 |
Assay IC50 (nM) |
| P38α inhibition |
| 98 |
O |
2,4,-diF |
CH3 |
4-Tetrahydropyranyl |
10 ± 4 |
| 99 |
O |
2,4,-diF |
CH3 |
CH(CH2CH2OH)2 |
14 ± 2 |
 |
| | Fig. 22 Relationship between structure-modifications and an P38α inhibitors properties. | |
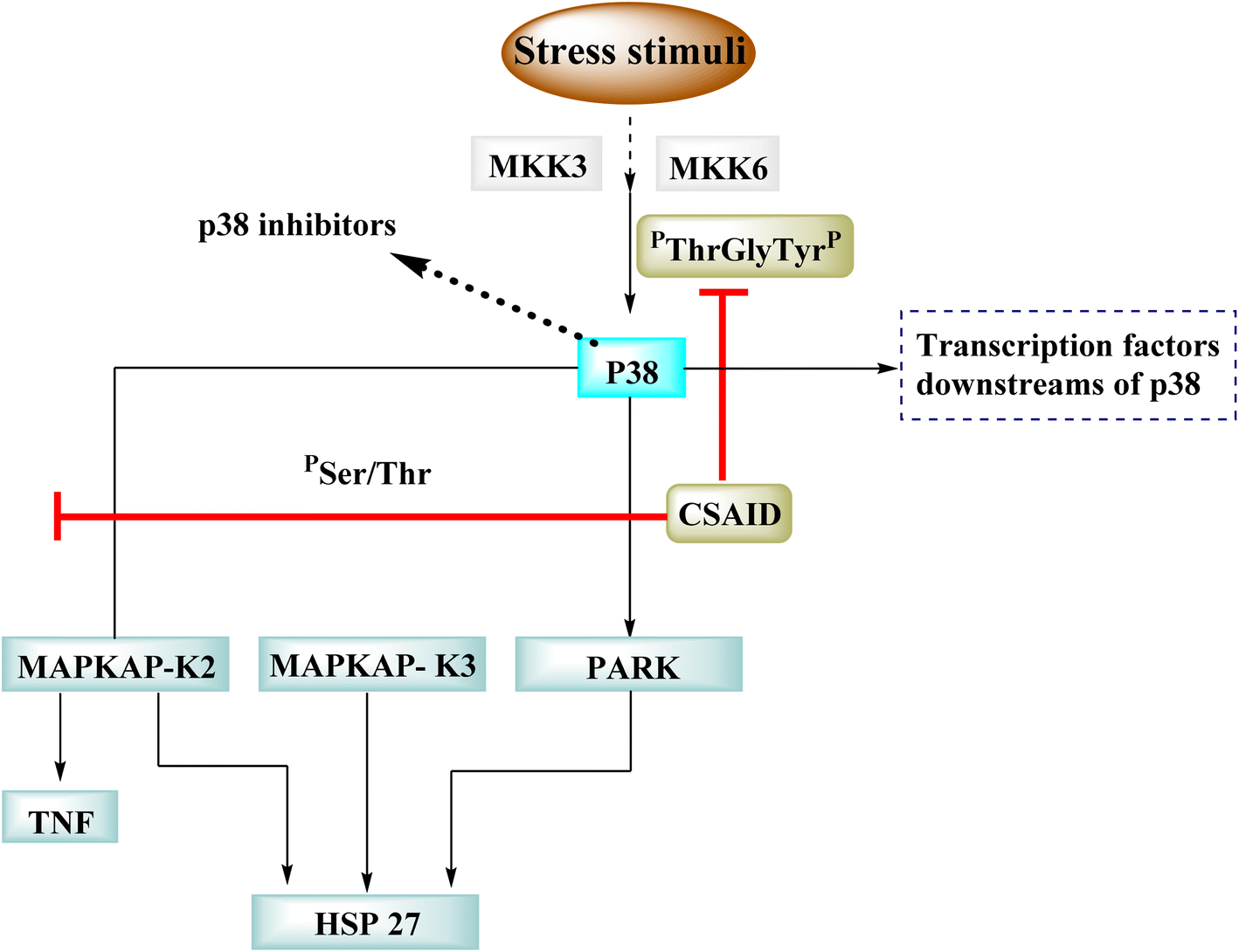
p38 is one of many kinases implicated in a stress-response signaling pathway similar to but not identical to the MAP kinase cascade. Phosphorylation of threonine 180 and tyrosine 182 by stress signals activates p38. MAPKAP kinase-2 and MAPKAP kinase-3 have been found as downstream targets of p38, which phosphorylates heat shock protein (Fig. 23).22
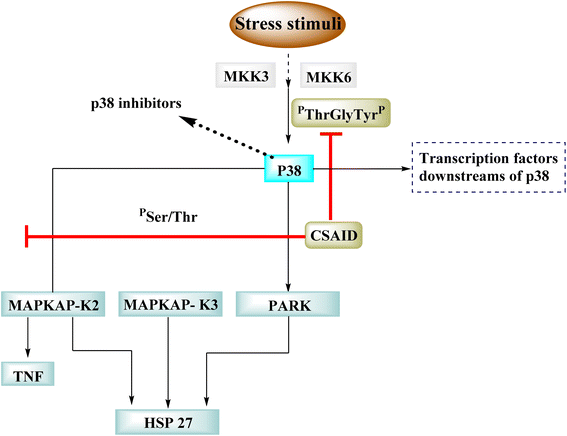 |
| | Fig. 23 Mechanism of p38 inhibitors. | |
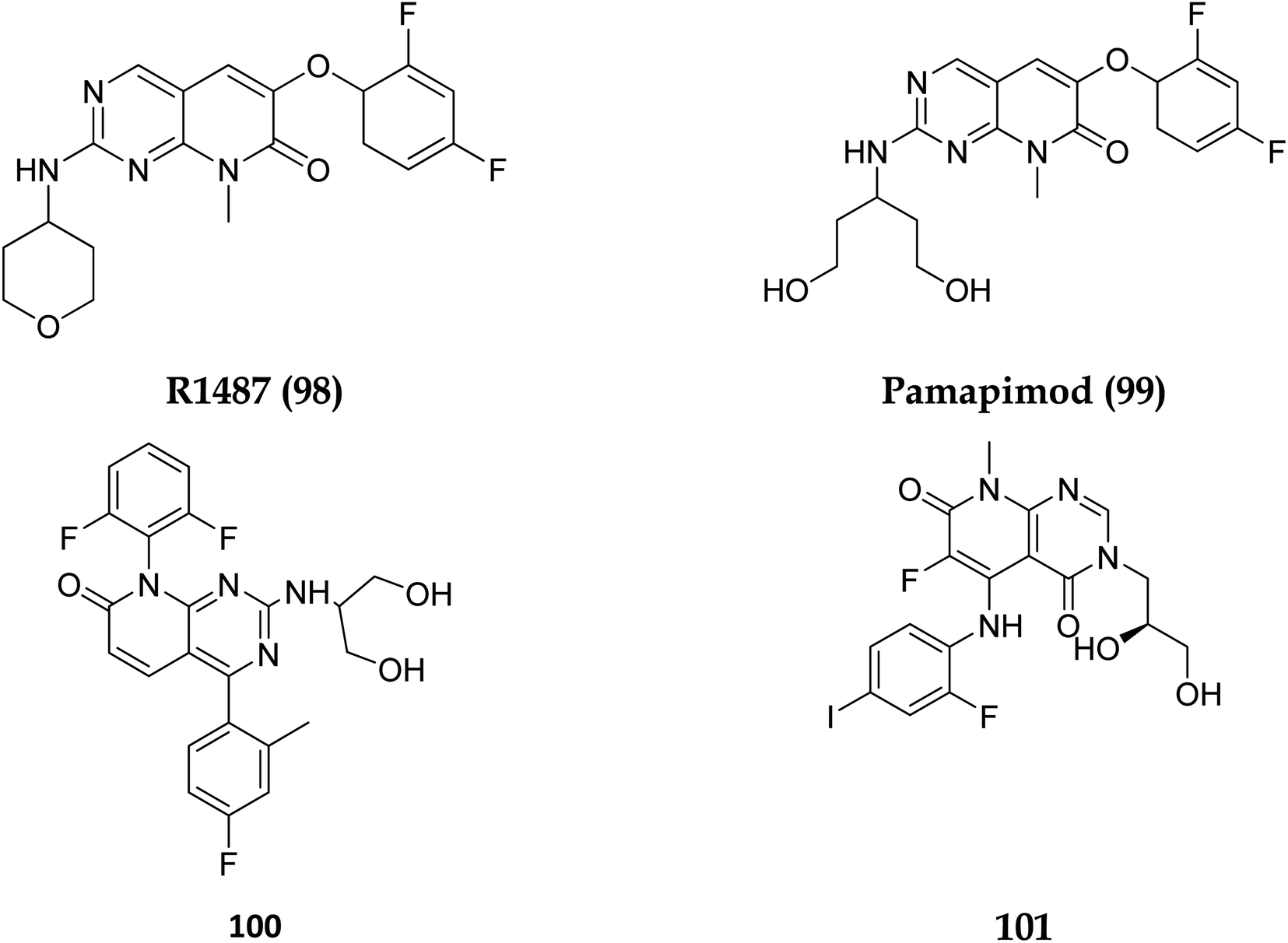
Dilmapimod (100, CAS 444606-18-2) is a p38 MAP-kinase inhibitor patented by SmithKline Beecham Corporation (GSK-681323) in 2001 and was used in clinical studies for inflammation, neuropathic pain, and cardiac problems but was eventually ceased due to liver damage.16 Clinical research examined TAK-733 (101, CAS 1035555-63-5), a MEK1 and MEK2 (MEK1/2) inhibitor, for advanced non-hematologic malignancies, including metastatic melanoma. It was created and patented by Takeda Pharmaceutical Company in 2007. TAK-733 was tested against non-small cell lung cancer and progressed to Phase I before being recalled off the market in 2015 (Fig. 24).16
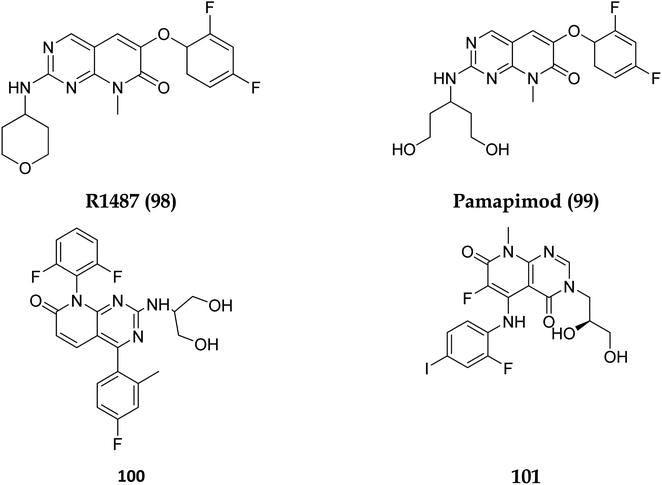 |
| | Fig. 24 Structures of compounds having p38 inhibitory activity. | |
3.6 Phosphatidylinositol 3 kinase and mTOR protein
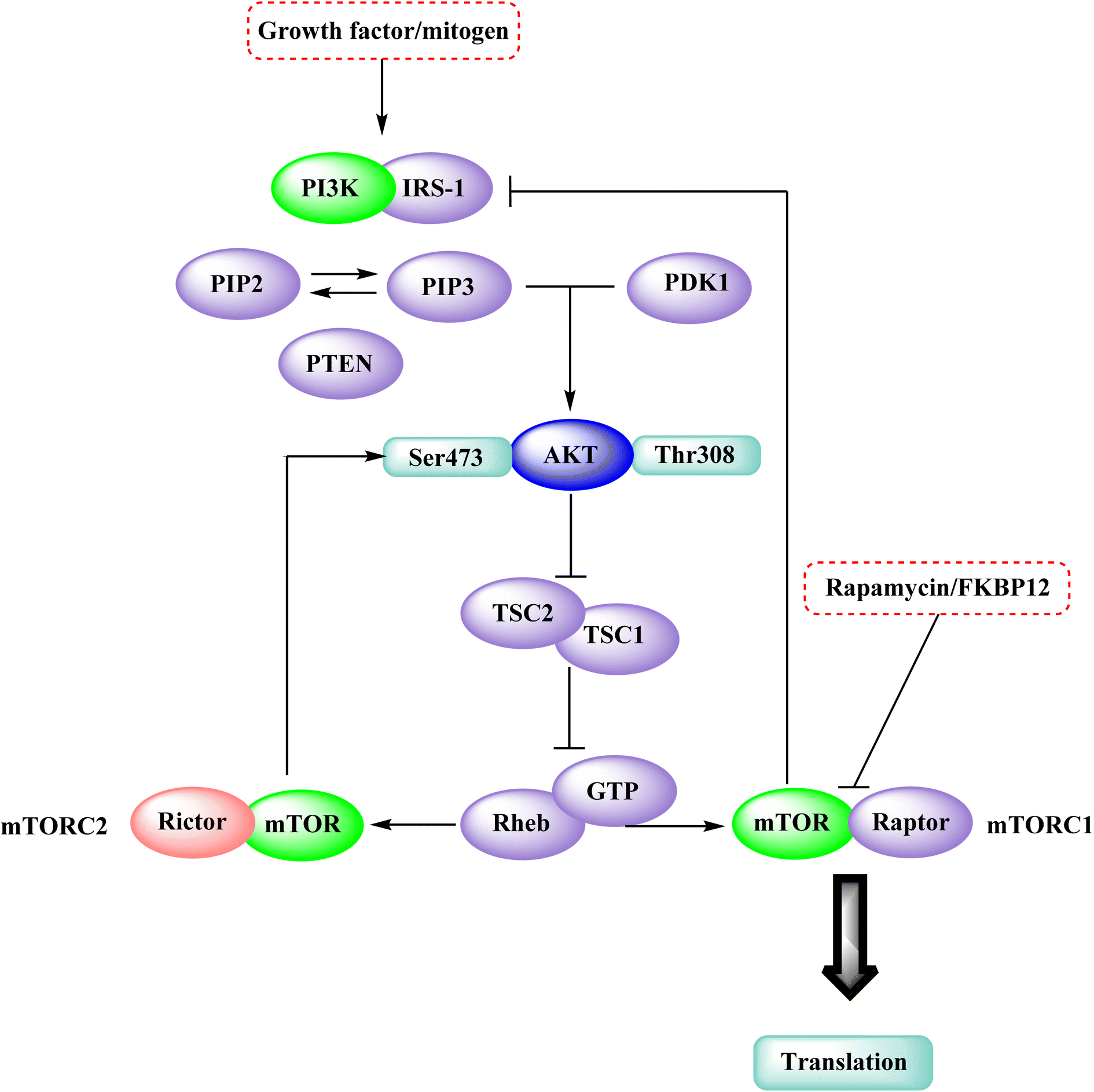
The phosphatidylinositol (PI) second messengers PI(3)P, PI(3,4)P2, and PI(3,4,5)P3 are produced by phosphoinositide 3-kinase (PI3-KS) enzyme (PIP3). The Class I PI3-KC isoform, which consists of catalytic (p110C) and adapter (p85) subunits is the most common PI3-kinase isoform. By producing 3-phosphorylated phospholipids (PIPs), PI3-KS activates kinases with lipid-binding domains, such as pleckstrin homology (PH) regions, which function as second messengers. For example, Akt as well as phosphoinositide-dependent kinase-1, Akt is moved to the cellular membranes when it links to membrane PIPs, which bind with PDK1 and becomes activated. A tumour suppressor protein, PTEN, dephosphorylates PIP, inhibiting the activation of Akt. The Akt, PDK1, and PI3-Ks are crucial players in controlling numerous cellular functions, including motility, proliferation, survival, and apoptosis. They are also crucial for disease-related molecular pathways, such as cancer, diabetes, and immune-mediated inflammation. Oncogenesis is linked to several components of the PI3K/Akt/PTEN pathway.80
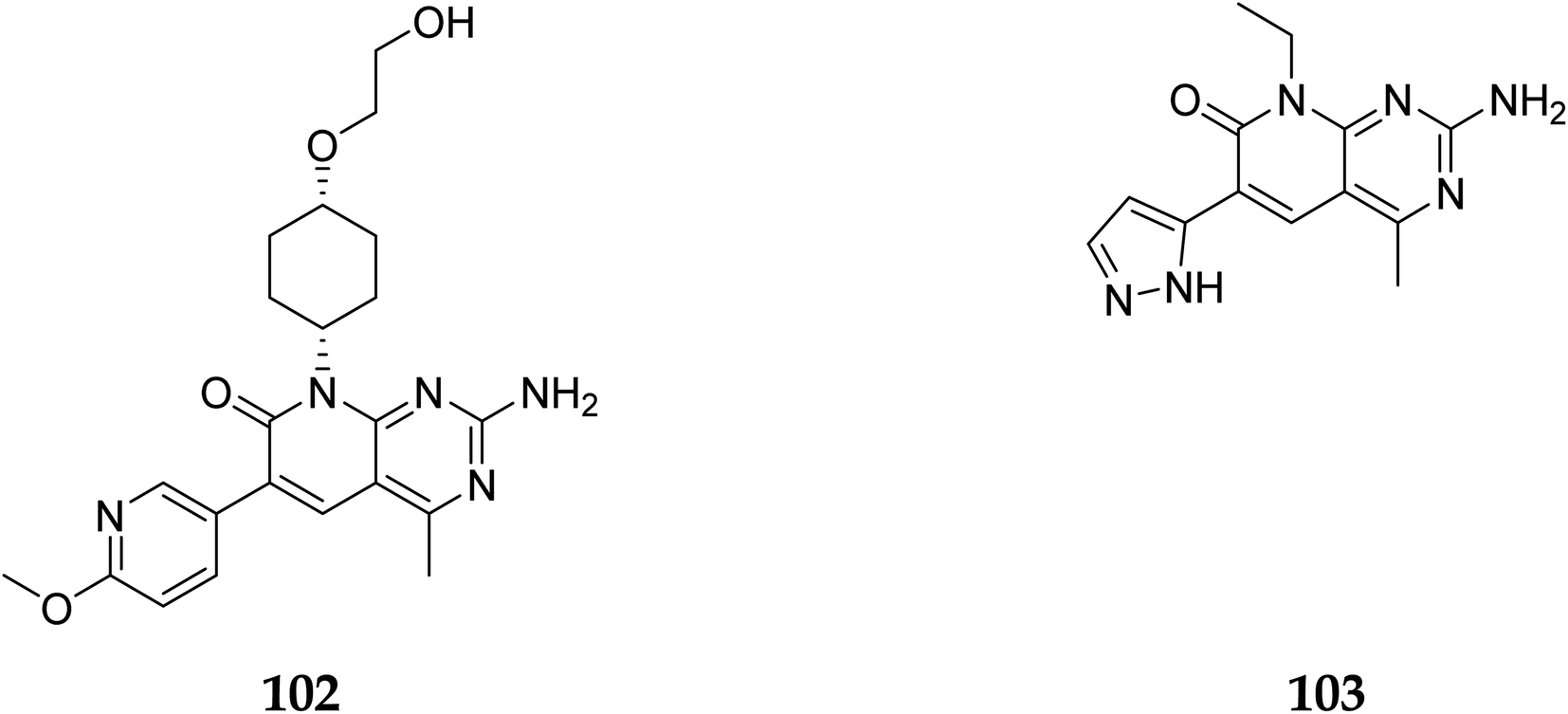
In 2007, Pfizer received a patent for compound 102, inhibiting the mTOR protein and phosphatidylinositol 3 kinases. Due to undesirable effects, it's clinical trial to treat endometrial cancer was stopped in 2012. N68 has also been tested in clinical trials for early (Phase 2) and late breast cancer (Phase 1b).16 Finally, Voxtalisib (CAS 934493-76-2) (103), a kinase inhibitor of PI3K and mammalian target of rapamycin in the PI3K/mTOR signaling cascade was patented by Exelis (XL-765) in 2006. In 2018, withdrawal of voxtalisib as a monotherapy and combo therapy for solid tumors (breast and ovarian cancer) took place due to serious side effects (Fig. 25).16
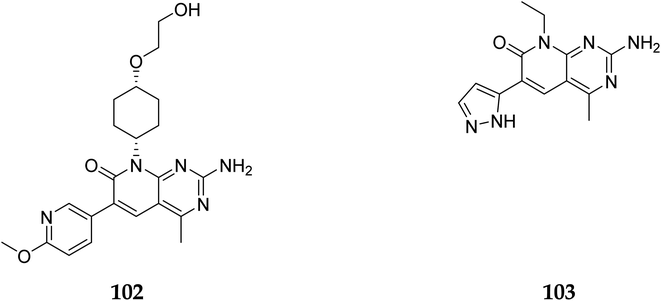 |
| | Fig. 25 Pyrido[2,3-d]pyrimidine derivatives (102 and 103) as PI3K/mTOR inhibitors. | |
In the discovery of anticancer therapies, the mammalian target of rapamycin (mTOR) is a critical target.81 The growth factor/mitogenic activation of the phosphatidylinositol 3-kinase (PI3K/Akt) signalling pathway activates this key regulator of cell growth and proliferation, frequently dysregulate route responsible for diseases. Serine and theonine kinases are the 2 mTOR found in the mammalian body. It's about 289 kDa in size and belongs to the evolutionary family. The eukaryotic PI3K like kinase (PIKK) family of proteins is highly conserved.82–84
3.6.1 Mammalian target of rapamycin pathway. The rapamycin and its analogues (RAD001, CCI-779, AP23573) bind to the FKBP12/rapamycin complex binding domain (FRB) and suppress signaling downstream substrates p70S6K and 4E-BP1.84–86 LY294002 and wortmannin, potent but non-specific PI3K inhibitors, have also been demonstrated to block the kinase activity of mTOR by targeting the protein's catalytic domain.87 According to the Brunn et al. and Edinger et al. mTOR is divided into two complexes: mTORC1, a rapamycin-sensitive complex that signals to p70S6K and 4E-BP1, and mTORC2, a rapamycin-insensitive complex that signals to Akt.87,88 Therefore, direct targeting of the mTOR kinase domain would suppress signalling via both mTORC1 and mTORC2 and such a drug would have a distinct pharmacological spectrum than rapamycin (Fig. 26).84
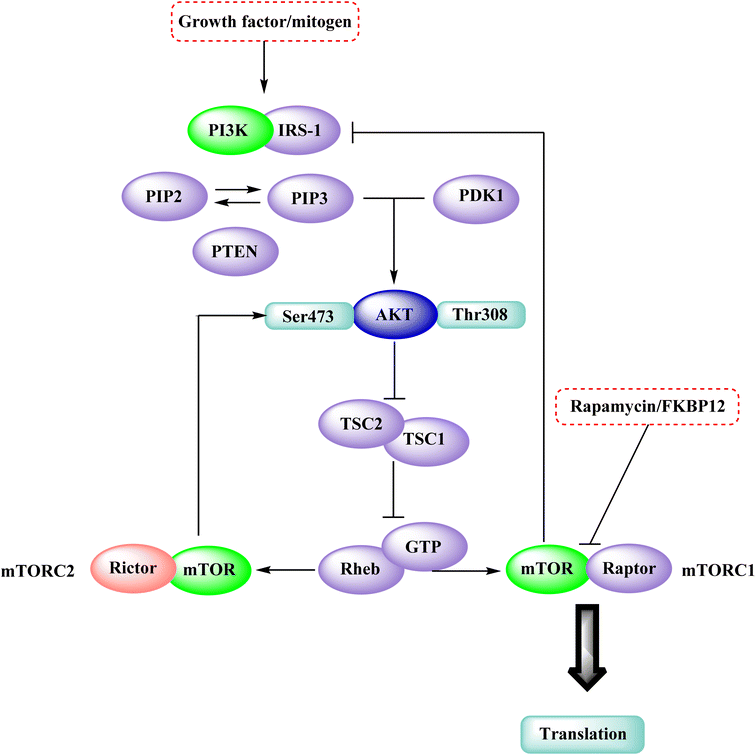 |
| | Fig. 26 The mTOR kinase inhibition in the mTOR-PI3K-AKT pathway. | |
3.7 Targeted inhibition of dihydrofolate reductase
NADPH (Nicotinamide adenine dinucleotide phosphate) serves as a cofactor for dihydrofolate reductase (DHFR), which transforms dihydrofolate (DHF) into tetrahydrofolate (THF). THF is necessary to form de novo purine and thymidylate (TMP) during cell growth.89 Inhibiting DHFR activity causes cell death,90 making DHFR an essential target enzyme in developing chemotherapy.91 Antifolates are categorized into two types: traditional/Classical: Due to the inhibitor's hydrophilic characteristic and glutamate moiety (which prevents the inhibitors from diffusing into cells), an active transport mechanism is needed. However, in the non-classical form, substituting the hydrophilic glutamate moiety with lipophilic groups improves its passive diffusion into cells.92,93 Dihydrofolate reductase (DHFR), specific kinases, such as the tyrosine-protein kinase transforming protein Abl or MAP kinases, and the biotin carboxylase are the most commonly described biological targets of pyrido[2,3-d]pyrimidine derivatives (Fig. 28).18
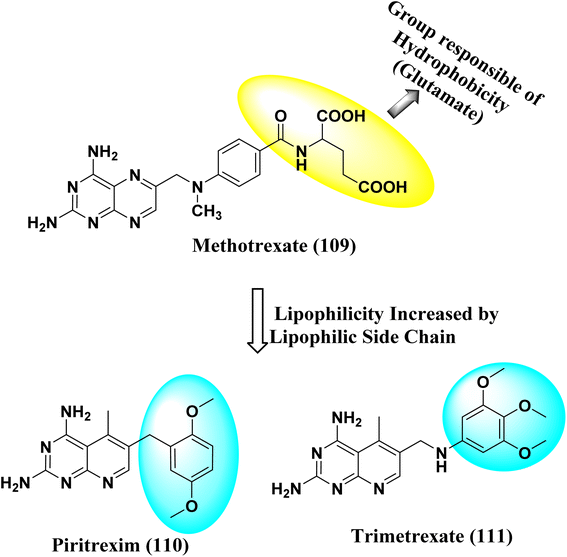 |
| | Fig. 28 Methotrexate (109) and pyrido[2,3-d]pyrimidine derivatives (110 and 111) DHFR inhibitors. | |
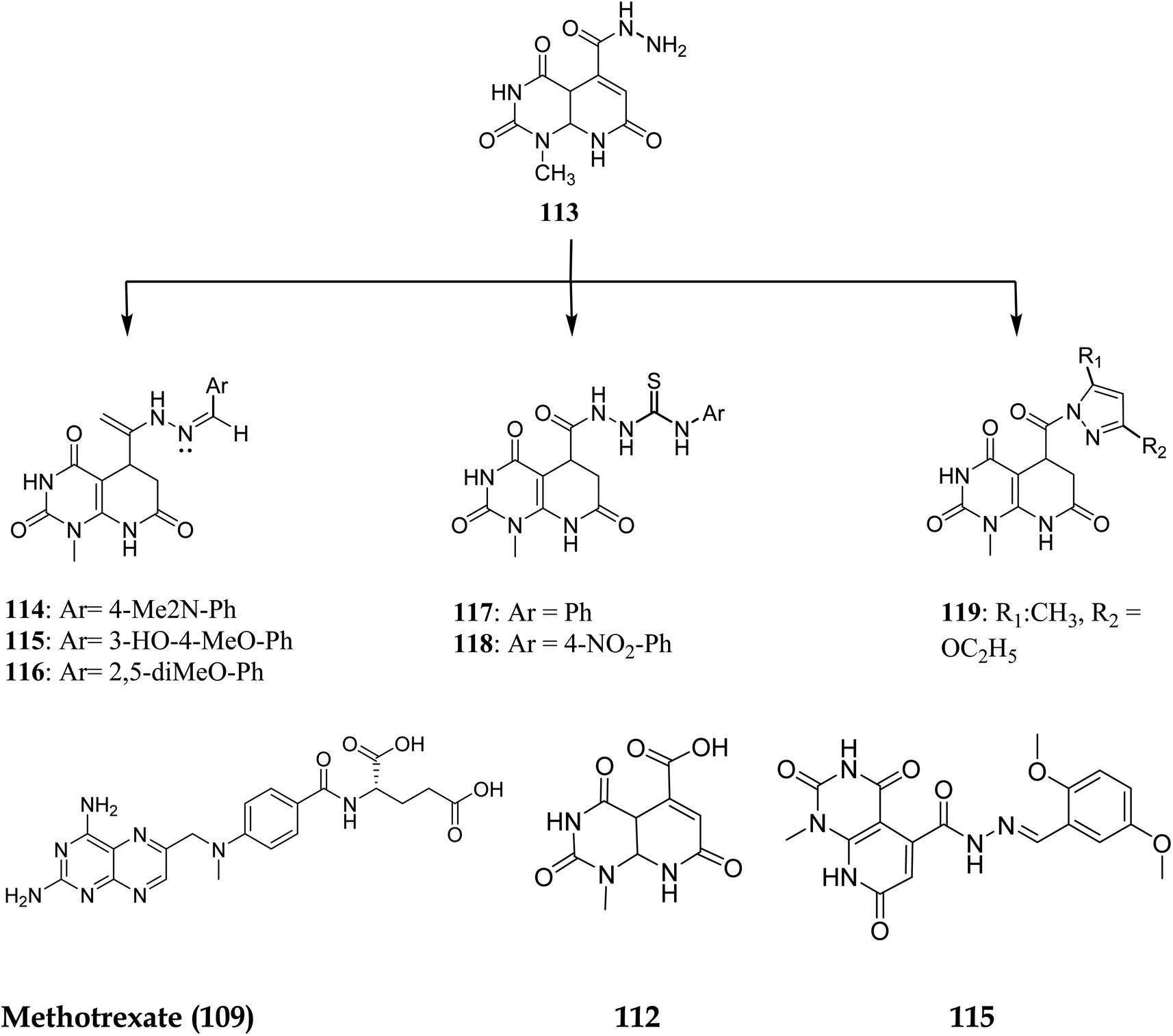
Walaa M. et al. (2019) synthesised pyrido[2,3-d]pyrimidine derivatives and evaluated their in vitro antitumor activity against five human cells lines: HePG2, MCF-7, PC3, HCT-116, and HeLa, using doxorubicin as a positive control. Compounds 112–119 (Fig. 29) showed the highest antitumor effects and were tested for enzymatic inhibition of the dihydrofolate reductase [compared to the reference medication methotrexate (MTX)] to explain the likely mechanism of action of the observed anticancer activity.94 Molecule 115 had the highest inhibitory activity at IC50 of 6.5 μM among all the tested molecules (IC50 of MTX = 5.57 μM, Table 4).
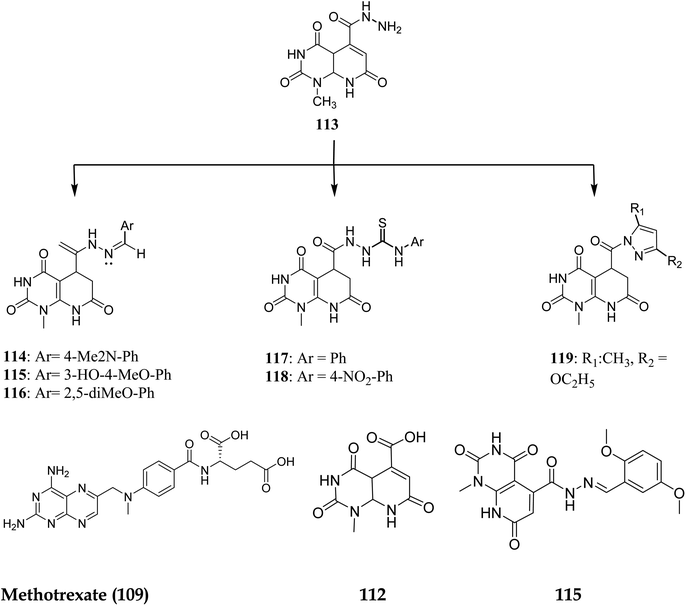 |
| | Fig. 29 Pyrido[2,3-d]pyrimidine derivatives as DHFR inhibitors. | |
Table 4 In vitro and in silico activity of compound 115 against DHFR
| Compound no. |
Kinase IC50 (μM) |
Docking score (kcal/mol) |
In vitro IC50 (μM) |
| MCF-7 |
| 115 |
6.5 |
−14.2 |
5.66 ± 0.4 |
| MTX(109) |
5.57 |
−14.7 |
|
3.7.1 Structure–activity relationship study of DHFR inhibitors. SAR study indicated that pyrido[2,3-d]pyrimidine scaffold was essential for activity. If the R was replaced with 2,5-dimethoxy substituents, it acted as PTX, and modification at NH position enhanced the activity. Both NH2 groups were responsible for hydrogen bonding (Fig. 30).
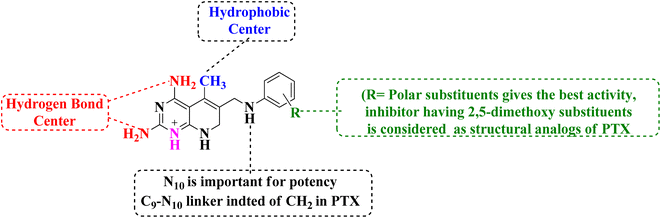 |
| | Fig. 30 SAR of DHFR inhibitors. | |
3.8 Apoptosis inducer
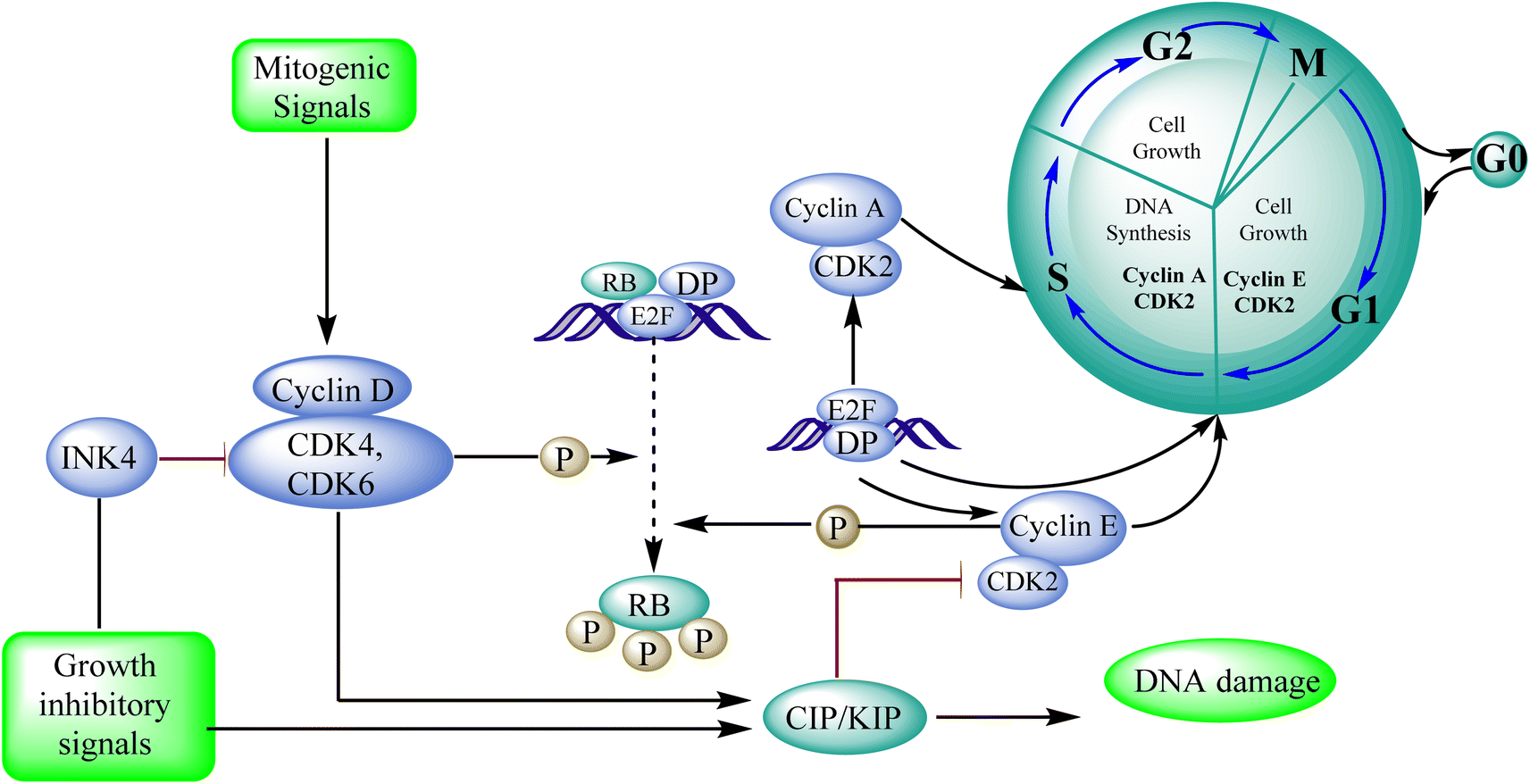
3.8.1 Cyclin-dependent kinase (CDK). In cancer, the cell-cycle regulatory mechanism, which is necessary for cellular division, is commonly disturbed, resulting in tumour growth. CDK4/6 inhibition inhibits cell proliferation and suppresses DNA replication in malignancies with functioning retinoblastoma. Many positive and negative regulators closely control cell division and the cell cycle. Mitogenic cues activate and upregulate cyclin D, getting complexed with CDK4/6, caused phospholylation leads to retinoblastoma, allowing bound E2F transcription factors to be released for the cell to divide. The cell cycle is positively regulated by CDKs and their cyclin partners, whereas the cell cycle is negatively regulated by retinoblastoma, other tumour suppressors (p16INK4b, p18INK4c, and p19INK4d), and the CDK-interacting protein/kinase inhibitory protein (Cip/Kip) family. Palbociclib is a CDK4/6 inhibitor effective in preclinical breast cancer models.95 After identifying cyclin-dependent kinases as essential regulators of cell proliferation, Pfizer Inc. developed palbociclib. Warner–Lambert received a patent for palbociclib in 2003. Palbociclib (pyrido[2,3-d]pyrimidin-7(8H)-one) interferes with the cell cycle. This inhibitor of CD4 kinase was picked out of the pyridopyrimidine class due to its intriguing structural and pharmacological characteristics. The FDA authorized it to treat HR-positive, advanced HER2-negative, or metastatic breast cancer in March 2015.95 Protein kinases, CDKs (cyclin-dependent kinases), are crucial for cellular processes. They participate in vital physiological processes like transcription and the cell cycle. Based on sequence similarities, the human genome contains 21 genes that encode CDKs, and five genes that encode a more distant group of proteins have been identified as CDK like (CDKL) kinases. The current CDK nomenclature includes 11 classical CDKs (CDK1-11), two newly proposed family members (CDK12-13), and additional proteins whose names are based on the presence of a cyclin-binding element (PFTAIRE and PCTAIRE proteins) or simply on sequence similarity to the original CDKs, such as CDC2-like kinases (CDC2L) or Cell cycle-related kinases (CCRK).96Apoptosis means allowing cells to die in a controlled manner. Apoptosis is a tightly controlled process in which cells self-destruct and eliminate undesired or diseased cells. The cell genome fractures, shrinks, and the cell portion disintegrate into smaller apoptotic entities during apoptosis.97 Cancer cells can potentially resist apoptosis, resulting in uncontrolled cancer development.98 The standard cellular mechanism called apoptosis keeps the ratio of healthy living cells to healthy dying cells in equilibrium. As a result, triggering apoptosis in cancer cells may be viewed as a cancer therapy strategy that does not harm healthy cells and prevents tumour resistance. As a result, finding novel drugs that target any phase of the cell cycle that leads to apoptosis might be a promising anticancer strategy.99
3.8.2 Cyclin-dependent kinase (CDK4/6). Pyrido[2,3-d]pyrimidine is a critical component in various bioactive compounds, including anticancer medicines, attributed to cyclin-dependent kinase inhibition.100,101 Palbociclib is a CDK4/6 inhibitor licensed by the US Food and Drug Administration to treat breast cancer.102 Safinaz et al., in 2019, prepared pyrido[2,3-d]pyrimidines derivatives and evaluated their cytotoxicity activity against various cell lines, including MCF-7, PC-3, and luA-549 and compared the results with doxorubicin. They synthesized different pyrido pyrimidine derivatives and checked their in vitro activity against human cancer lines (breast MCF-7, lungs A-549, and prostate PC-3),103 Cell cycle analysis,104 Apoptosis assay,105 Caspase activation assay,106 RT-PCR for (Bax, Bcl2,p53, and CDK4/6), and compound 116 showed excellent activity against Caspase-3 (Table 5). Furthermore, compounds 116 and 117 showed an excellent docking score and primary amino acid interaction with selected protein (Table 6 and Fig. 31). Compounds 116 and 117 were observed to promote apoptosis in PC-3 and MCF-7 cells via activating Bax, p53, and caspase-3, down-regulating Bcl2 and inhibiting CDK4/6. Furthermore, compound 116 directly inhibited CDK6 with an IC50 of 115.38 nM, whereas compound 117 inhibited it with an IC50 of 726.25 nM (Table 7).41
Table 5 Effect of compound 116 on Caspase-3
| Compound no |
Caspase-3 conc. (Pg ml−1) |
Optical density |
| Control |
43.45 |
0.103 |
| 116 |
603.4 |
0.741 |
Table 6 Docking scores of compounds 116, 117, and 118 with PDB ID 5L21
| Compound no |
Docking score (kcal mol−1) |
Interactions with amino acids |
| Palbociclib (61) |
−12.02 |
Asp163, Val101 |
| 116 |
−13.24 |
Val77, Phe164, Val101, Phe98, Asp163 |
| 117 |
−15.40 |
— |
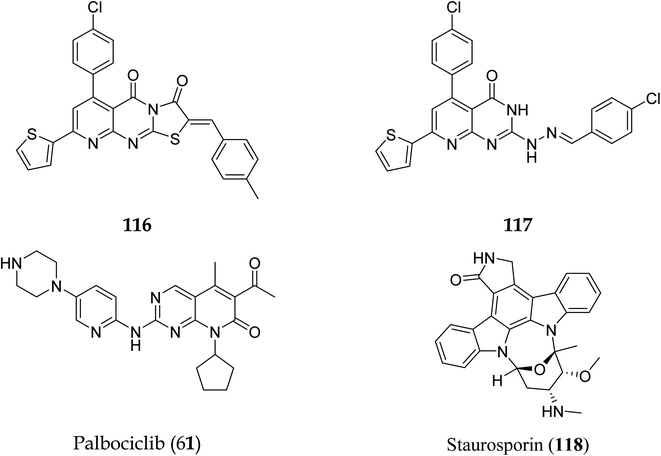 |
| | Fig. 31 Pyrido[2,3-d]pyrimidine derivatives with CDK 4/6 inhibitory activity. | |
Table 7 Activity of compounds 116 and 117 on cyclin-dependant kinase-6
| Compound no |
IC50 (nM) |
| 116 |
115.38 |
| 117 |
726.25 |
| Staurosporin (118) |
92.78 |
3.8.3 CDK2 roles in the cell cycle and cancer development. CDKs are key regulators of the cell cycle,107 and the proper regulation of CDK activity is crucial for the ordered execution of the cell cycle.108 CDK2/cyclin E expression abnormalities have been seen in colorectal, ovarian, breast, and prostate cancers.109 CDK2 is involved in many stages of the cell cycle, including DNA repair, gene transcription, the G1-S transition, and G2 progression modulation.110–112
3.8.4 The function of CDK2 in the proliferation of cells: -. CDK2 is involved in cell cycle control, transcription activity, and tumor epigenetic alterations. Certain malignancies, such as breast cancer,113 colo-rectal cancer,114 glioblastoma,115 and melanoma116 may benefit from CDK2 inhibition (Fig. 32).
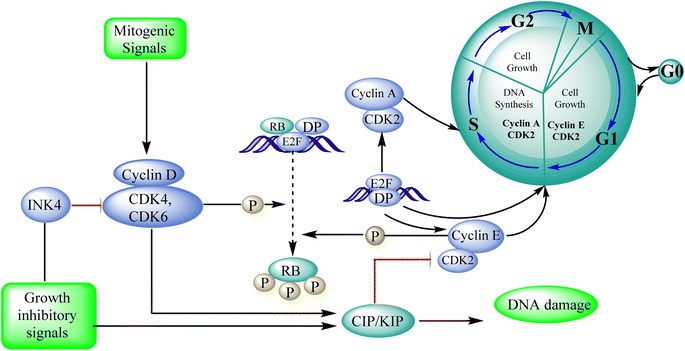 |
| | Fig. 32 Role of CDK2 in the cell cycle progression. | |
Cyclin-dependent kinase promotes the progression of the G1 phase into the S phase by phosphorylation of the retinoblastoma protein (RB). The E2F family of transcription factors is activated by the hyperphosphorylation of the RB, which reduces its capacity to regulate growth. Growth repressive signals promote CDK inhibitors from the INK4 and CIP/KIP families, which block G1-S progression. Cyclin and dependent kinase complexes are activated by mitogenic signals.117,118 Diaa et al., 2011 designed and synthesized 2,5,7-trisubstituted pyrido[2,3-d]pyrimidines by introducing a variety of substituents at C-2, C-5, and C-7 of the pyridopyridine ring for a potent and selective inhibitors of CDK2 (competitive inhibitors of ATP binding site) and tested them for CDK2, EGFR inhibitory activities, and cell growth inhibitory activities.10 Using the design of structural scaffolds addressing ATP interactions, they created pyrido[2,3-d]pyrimidine scaffolds (CDK2 inhibitors) using the X-ray crystalline structure of CDK2 (PDB ID: 1HCK). The CDK2 inhibitory and anti-proliferative activity is due to the side chains at the 2nd and 7th positions. The CDK2, CDK4, EGFR, and anti-proliferative activities of the Pyrido[2,3-d]pyrimidine derivatives were assessed against SNU638, A431, and HCT116, and the results showed that compounds 119 and 120 were more active than roscovitine with IC50 values of 0.3 and 0.09 μM, respectively, which shows that compound 120 has the good anti-proliferative activity. The compounds did not show any significant activity against the EGFR, which shows their selectivity against the CDK2 (Fig. 33).10
 |
| | Fig. 33 Pyrido[2,3-d]pyrimidine derivatives as CDK2 inhibitors. | |
3.8.5 Structure activity relationship of CDK2 inhibitors. SAR studies relealed that 7-amino group's lipophilic function (LF), such as the benzene, and sulfonamide group, significantly increased the CDK2 inhibitory effect. Further, small (LF) with an electron donating group on position 2 of the pyrido[2,3-d]pyrimidine moiety was required for CDK2 inhibitory activity.
3.9 Phosphodiesterase inhibitors
Phosphodiesterase 3 is also known as the cyclic GMP-inhibited PDE family. PDE3A and PDE3B are the two genes in the family, each has numerous splice variants. Harrison et al., Macphee et al., Maurice and Haslam et al. were the first to discover PDE3A as a significant PDE isoform in bovine heart and platelets.119 PDE3 enzymes play a role in modulation of cardiac and vascular smooth muscle contractility. PDE3 inhibitors were once examined for the treatment of heart failure. However, they are no longer being studied for that due to undesired arrhythmic side effects. The PDE3 inhibitor milrinone is licensed for intravenous usage in the treatment of heart failure.120 Abadi et al., 2013 synthesized 7-(3-bromophenyl)-5-(2-ethoxyphenyl) pyrido[2,3-d]- pyrimidin-4-amine and 7-(3-bromophenyl)-5-(2-methoxyphenyl)pyrido[2,3-d]- pyrimidin-4(3H)-one by cyclization of 2-amino-6-(3-bromophenyl)-4-(2-ethoxyphenyl) nicotineonitrile and 2-amino-6-(3-bromophenyl)-4-(2-methoxyphenyl)- nicotinonitrile by refluxing with formamide and formic acid respectively.121 PDEs could be promising targets for tumour cell growth control and apoptosis induction. These are; (I) modification of cyclic nucleotide signaling is known to contribute to one of the pathways regulating growth and operation of tumour cells. (II) Various PDE isozymes are discovered in various cancer tissues, (III) non-selective PDE inhibitors like theophylline or aminophylline affect the proliferation of cancer cell lines.122 HMG (Human malignant melanoma) has been found to have PDE3 activity, indicating that it might be a potential target for antineoplastic medications. However, the PDE3-specific inhibitors trequinsin and cilostamide did not affect HMG cell growth.123 The expansion of colon cancer cells was associated with the upregulation of PDE3B mRNA as well as protein in HT-29 cells. Through the dose-dependent activation of PDE3B, cyclic phosphatidic acid (cPA) reduced Akt phosphorylation, lowering HT-29 cell proliferation. As a result, the cPA-PDE3B-cAMP pathway plays an essential role in colon cancer growth, and cPA could be exploited for colon cancer targeted therapy.124
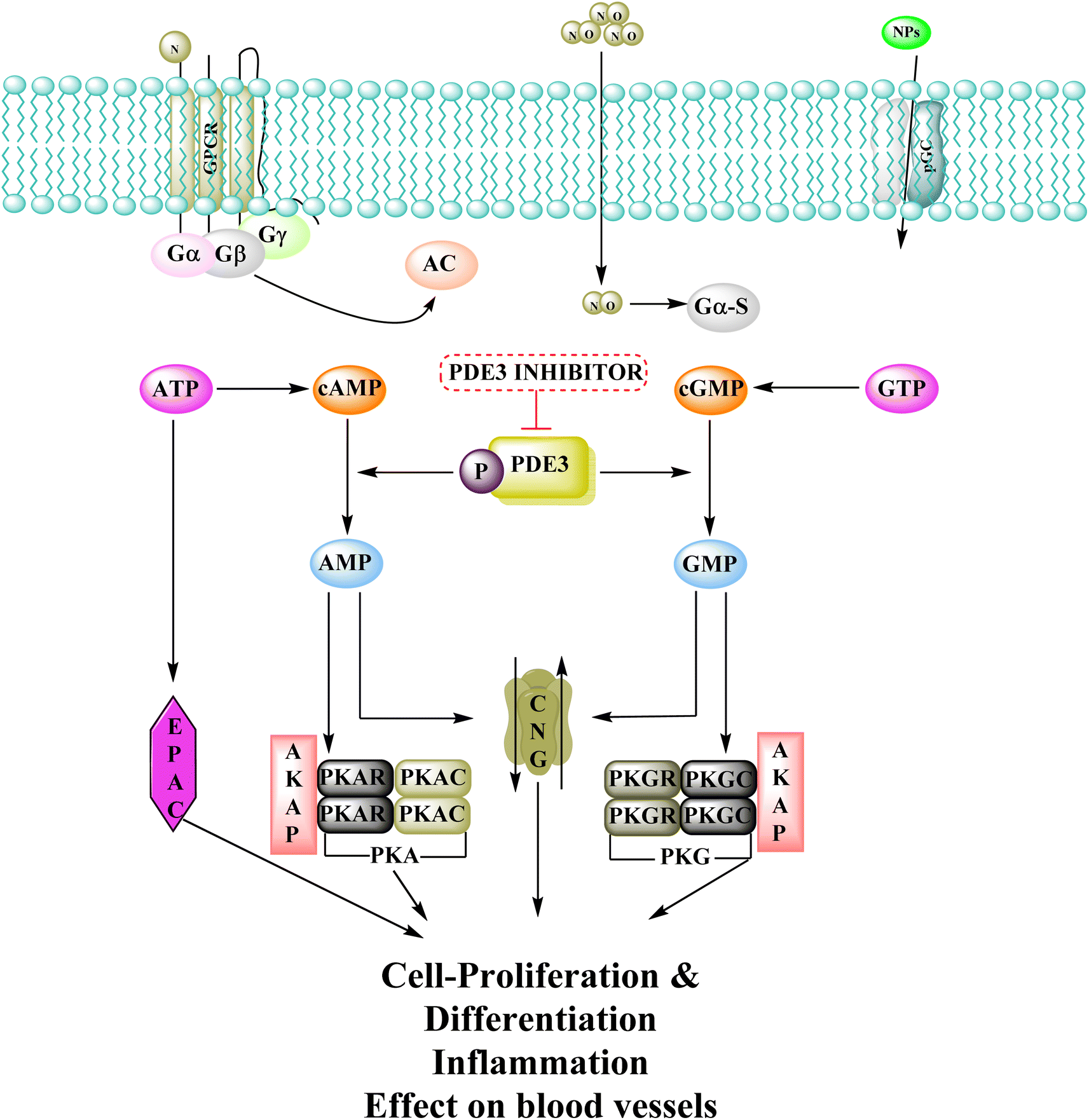
3.9.1 Mechanism of action of phosphodiesterase inhibitors. The cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) signalling systems are among the earliest signalling systems discovered, and are involved in a variety of physiological processes such as visual transduction, cell proliferation and differentiation, cell-cycle regulation, gene expression, inflammation, apoptosis, and metabolic function.125 Adenylyl-cyclase and guanylyl-cyclase produce cyclic nucleotides when G-protein-coupled receptors and molecules like natriuretic peptide or nitric oxide have been activated. cAMP then activates cAMP-dependent kinase (PKA) and other proteins, regulating biological processes like cell differentiation and proliferation. Proteins are phosphorylated when cGMP activates the cGMP dependent protein kinase (PKG), which is involved in physiological mechanisms like ion channel conductance as well as cell death. Phosphodiesterases 3 (PDEs) regulate cAMP and cGMP intracellular concentrations and their diverse biological consequences by catalyzing their hydrolysis.122PDEs can be expressed by every cell type, and their location is crucial in controlling cAMP or cGMP cellular functions.126 Interference with the cAMP/Cgmp signalling pathway has been linked to tumour growth. Increased intracellular cAMP/cGMP may suppress tumour growth and hence be a protective mechanism against tumour progression.3 PDE inhibitors that limit cAMP or cGMP hydrolysis have thus been identified as possible anticancer medicines (Fig. 34).125 However, some cancers, such as adenocarcinoma, can stimulate cell proliferation when cAMP levels are elevated.127,128
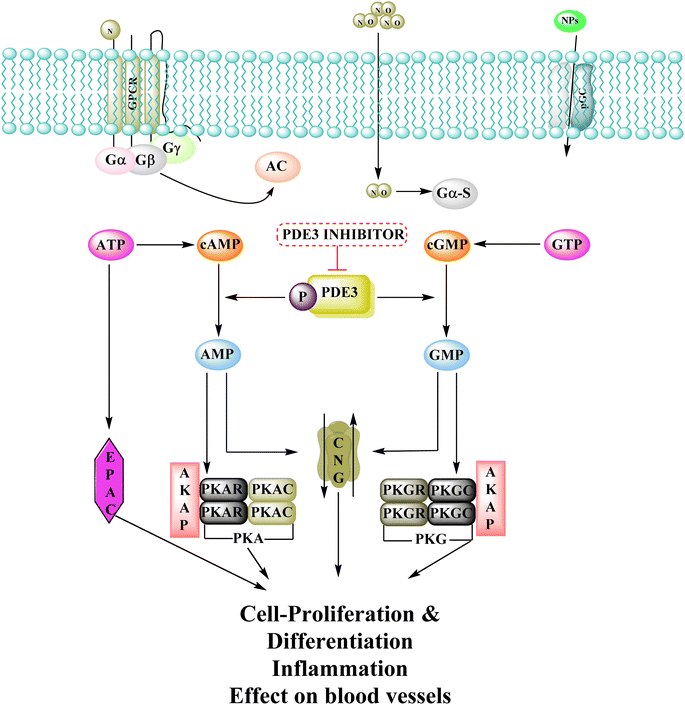 |
| | Fig. 34 Phosphodiesterase 3 signalling pathway and its inhibitors. NO: nitric oxide; PKA: protein kinase A; GPCR: G-protein-coupled receptors; NPs: atrial natriuretic peptide and B-type natriuretic peptide; AC: adenylate cyclase; EPAC: exchange protein activated by cAMP; PKG: protein kinase G; CNG: cyclic-nucleotide-gated ion channels; pGC: particulate guanylyl cyclase; sGC: soluble guanylyl cyclase. | |
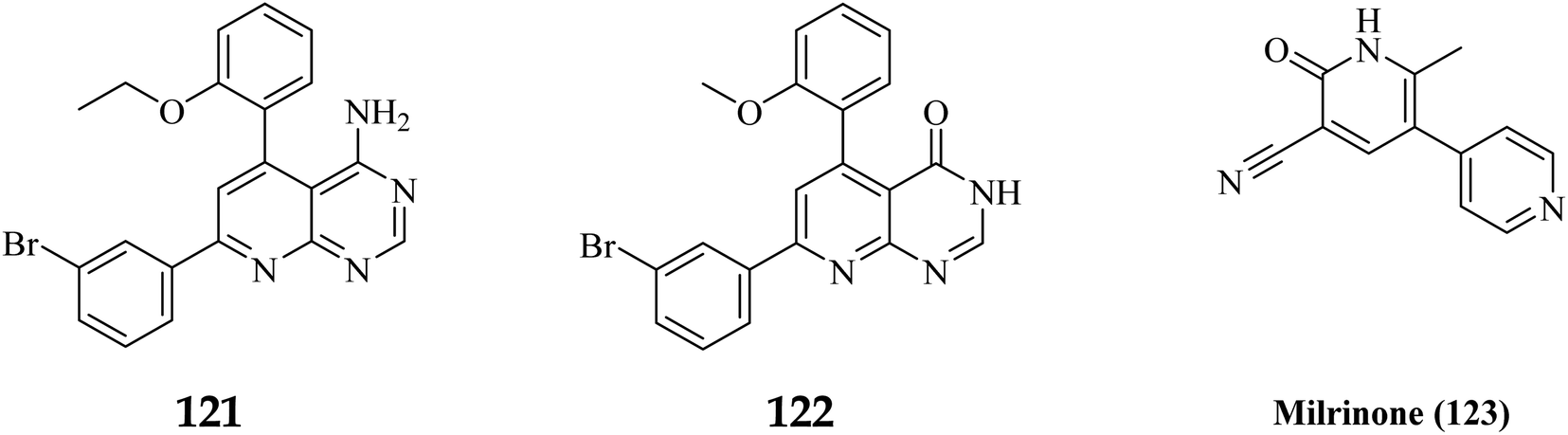
A cyclized pyridopyrimidone analog (122) has an IC50 as low as 1.34 μM. With cAMP and cGMP as substrates, these two (121, and 122) molecules efficiently inhibited PDE3. This indicates that for the best anticancer action, suppression of both cAMP, as well as cGMP hydrolysis to boost intracellular concentrations of both signalling molecules may be necessary (Fig. 35). Utilizing cGMP as the substrate, compound 121 had an IC50 of 10.9 μM and suppressed PDE3 (Table 8). Creating substrate-specific inhibitors for dual-functioning enzymes like PDE3 is a novel development that opens the way to modify the expression of one substrate instead of another, which may have implications for drug discovery in terms of safety and efficacy. An adaption of the IMAP (Fluorescence polarization phosphodiesterase assay) was used to detect PDE activity.
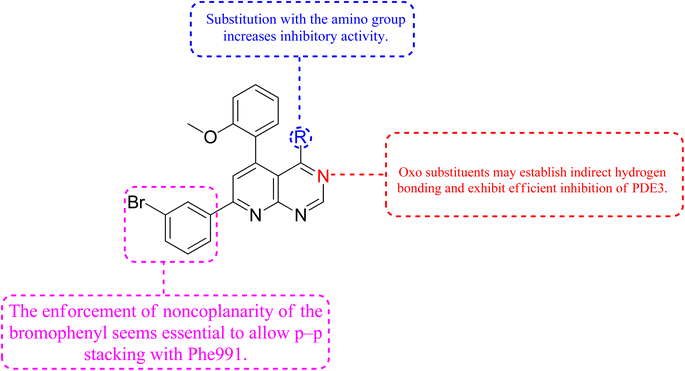 |
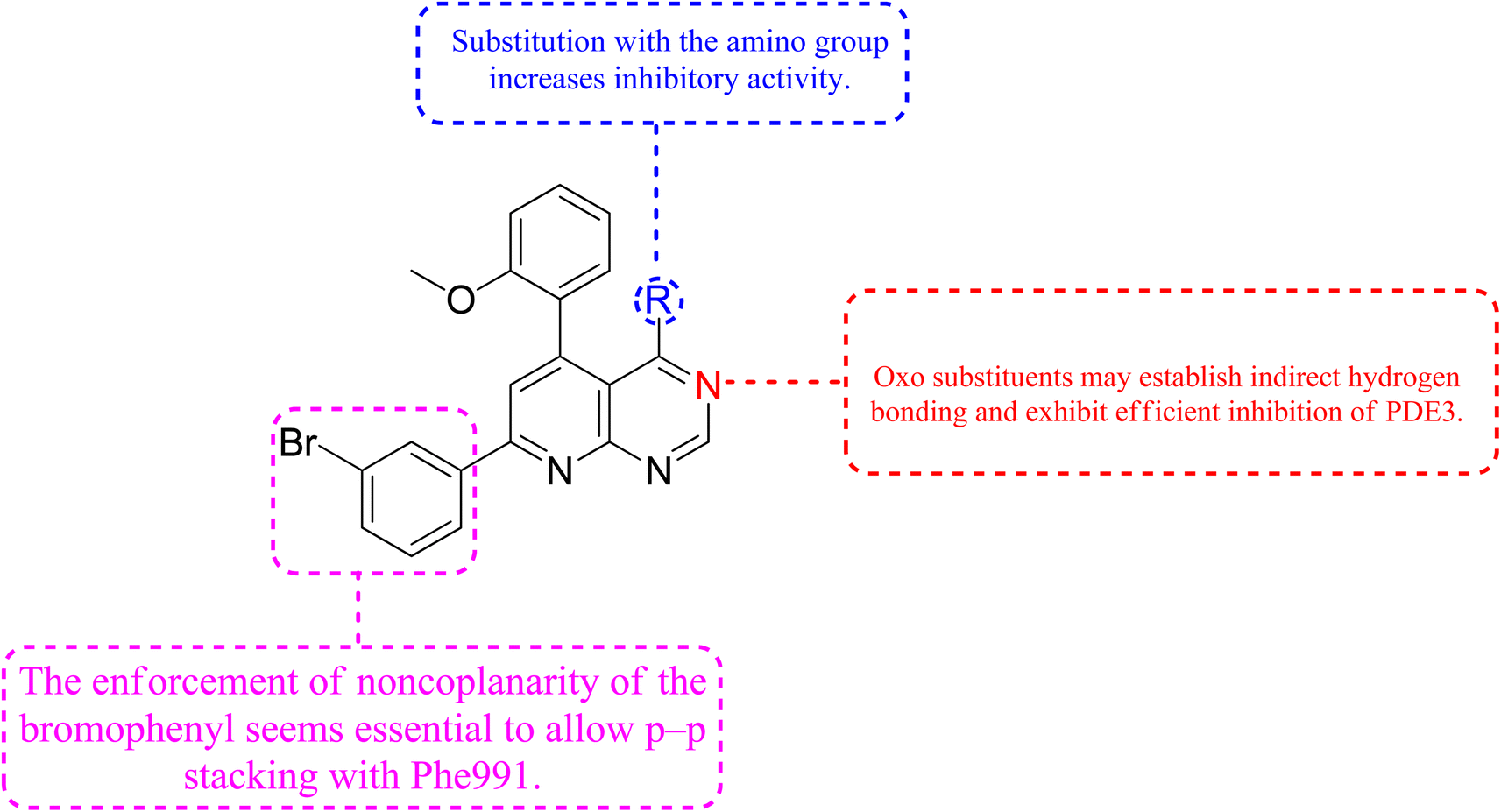
| | Fig. 35 SAR of phosphodiesterase inhibitors. | |
Table 8 Inhibitory activity of compounds 121 and 122 on Cell Line (HT-29) and phosphodiesterase 3B
| Compound no. |
HT-29 |
PDE3 inhibition |
| % Growth inhibition at 50 μm |
IC50 μm |
% PDE3 inhibition at 50 μm |
IC50 μm |
| cAMP |
cGMP |
Camp |
cGMP |
| 121 |
86.10 |
18.27 |
48 |
85 |
ND |
10.91 |
| 122 |
97.98 |
1.34 |
62 |
78.6 |
24.21 |
10.75 |
| Milrinone (123) |
10 |
>50 |
77 |
95 |
11.4 |
3.6 |
3.9.2 Relationship between structure-propeties and anticancer activities. The cyclized pyridopyrimidone derivative 121 reduced the tumour cell proliferation, with an IC50 of 1.34 μm (Fig. 36). With cAMP and cGMP as substrates, these three drugs efficiently inhibited PDE3. This indicated that the best strategy to treat cancer may be to limit both cAMP as well as cGMP hydrolysis to increase intracellular concentrations of both secondary messengers. The positive control milrinone's inability to cause apoptosis in the HT-29 cell line despite its dual inhibition of PDE3 and growth suggested that the other PDE and off proteins may contribute to the anticancer activity. Alkoxy and Bromo substituents at the ortho positions of the phenyl at positions 4 and 6, respectively, are believed to be responsible for the non-coplanarity between the two aryls and pyridone. The enforcement of bromophenyl non-coplanarity appears to be necessary for stacking with Phe991. Replacement of the oxygen atom of cyano-2-pyridones with an amino group may create the foundation for substrate-selective pharmacological regulation of this significant class of therapeutic targets (Fig. 35).121
 |
| | Fig. 36 Pyrido[2,3-d]pyrimidine derivatives, 122 and 123 with milrinone as phosphodiesterase inhibitors. | |
3.10 Gene Kirsten rat sarcoma viral oncogene homolog inhibitors
The Gene Kirsten rat sarcoma viral oncogene homolog is known as KRAS. It belongs to the group of epidermal growth factor receptor (EGFR) kinase. This multi-component signaling system helps to regulate cell growth, division, survival, and death by relaying signals from the outside to the inside of the cell. An essential signal for cellular differentiation in normal cells is the interaction of the epidermal growth factor to its target, mainly on the cell membrane. There are two more signals in the process including tyrosine kinase enzymes as well as a protein encoded by the KRAS gene, instead of separately promoting cell proliferation, the route's components typically cooperate to control cell division and growth. KRAS mutations are frequently linked to resistance to targeted medicines and poor outcomes in cancer patients, yet after more than three decades of research, no selective KRAS inhibitor has been authorized. However, in some tumors, EGFR becomes active even when EGF is not present, resulting in uncontrolled cell proliferation and division. EGFR or tyrosine kinase enzyme inhibitors are frequently used to treat these malignancies. However, an alteration in the KRAS gene found in a few of these malignancies leads to an abnormal K-Ras enzyme. The uncontrolled protein is constantly active and can promote cell growth despite the lack of signals from EGFR as well as other tyrosine kinase enzymes. Inhibitors of EGFR or tyrosine kinases will be ineffective in such tumours.129–131 KRas is the most often mutated oncogene among all types of human cancer. After the initial discovery of Ras oncogenes in 1982, In Ras-driven cancer, no approved medication specifically targets Ras.132 Ras proteins are chemical messengers that alternate between an active GTP-bound as well as an inactive GDP-bound state to control a number of cellular functions. Guanine nucleotide exchange factors (GEFs) enhance activation, and GTPase-activating proteins (GAPs) inactivates Ras protein by catalyzing GTP hydrolysis and are involved in these state changes.133,134 Ras protein mutations result in anomalies in the switch mechanism, a significant contributor to tumorigenesis. RAS mutations are present in three of the most lethal kinds of human malignancy, or around 25% of human tumors, including pancreatic cancer, and colon and lung cancer. KRas is the most frequently altered isoform of the Ras proteins (85%), followed by NRas (11%) and HRas (4%), with the most common mutations occurring at amino acid positions G12, G13, and Q61.132 Mutations at positions 12 and 13, which cause a steric clash with the catalytic arginine finger donated by the GAPs, are thought to be oncogenic. In contrast, mutations at position 61 disrupt the coordination of a nucleophilic water molecule, resulting in an accumulation of active GTP-bound Ras proteins in cells.135,136 The Shokat laboratory discovered the switch-II pocket in 2013, creating an opportunity for the creation of the KRasG12C inhibitors, which is investigated in the trials. A library of 480 tethering compounds was screened for covalent binding to KRasG12C using a disulfide-fragment-based screening method.137
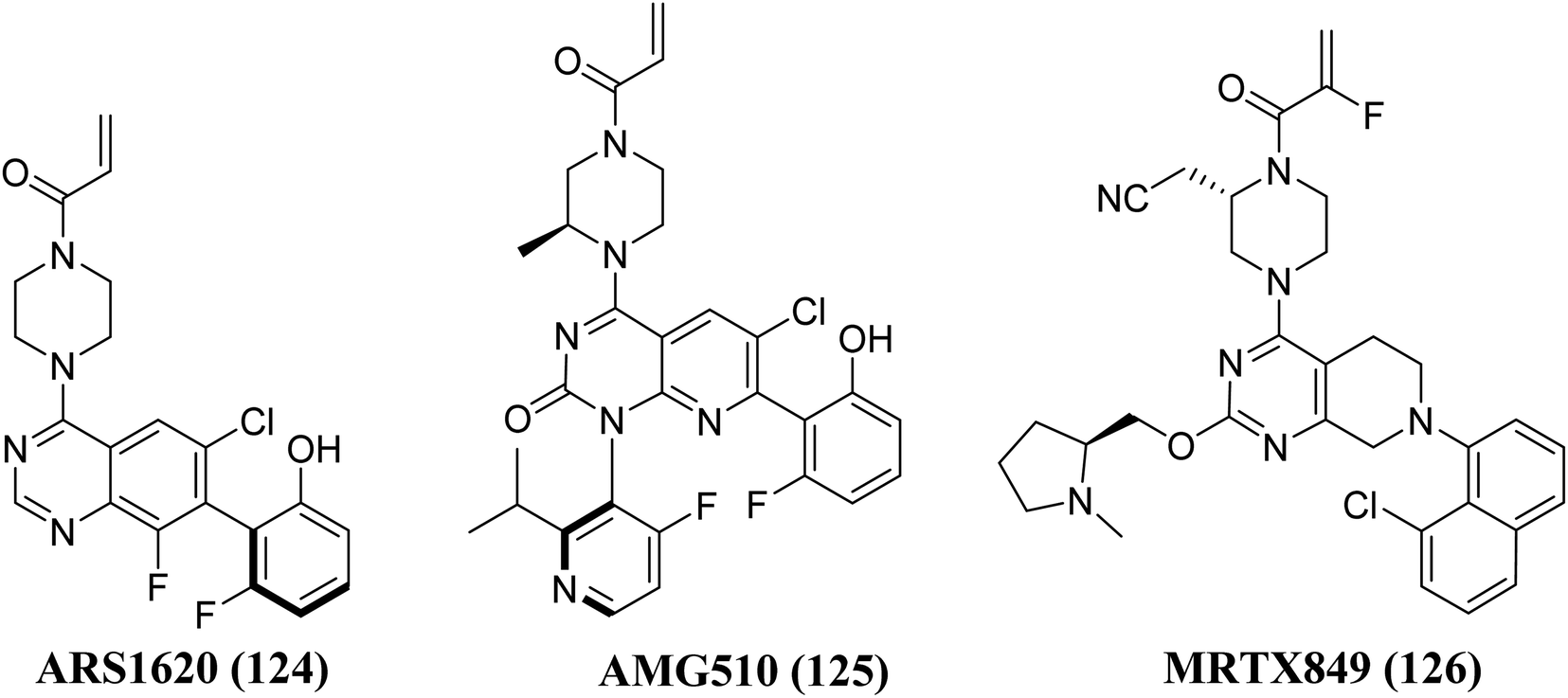
Structure–activity-relationship (SAR) investigations of 6H05, compound 6Ostrem, and KRASG12C in the GDP state resulted in a co-crystal structure with KRASG12C. Because crystallographic study revealed that the ligand does not attach to the nucleotide-binding pocket (S-IIP), it was given the switch-II pocket and placed near the activator binding switch-II. S-IIP is positioned between the core-sheet and switch-II, and when ligand binding occurs, it undergoes significant conformational changes to form a separate pocket, whereas switch-I conformation remains intact from the GDP-bound state.132 Many novel covalent allosteric KRASG12C inhibitors have been created as a result of this pioneering work, including ARS1620 (124),138 AMG510 (125),139,140 and MRTX849 (126),141,142 with AMG510 being the first KRASG12C inhibitor to reach clinical trials in August 2018 (Fig. 37). The findings of a phase I trial using this medication were recently released, revealing partial responses in 50% of non-small cell lung cancer (NSCLC) patients and stable disease in the majority of colorectal or appendix cancer patients, all of whom carried the KRASG12C mutation.143
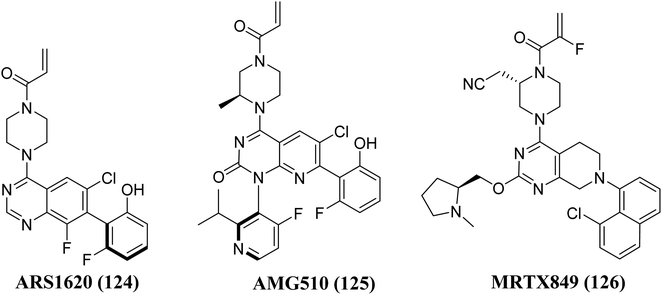 |
| | Fig. 37 Chemical structure of pyrido[2,3-d]pyrimidine derivatives 125, with 124, & MRTX849 (126) as KRAS inhibitors. | |
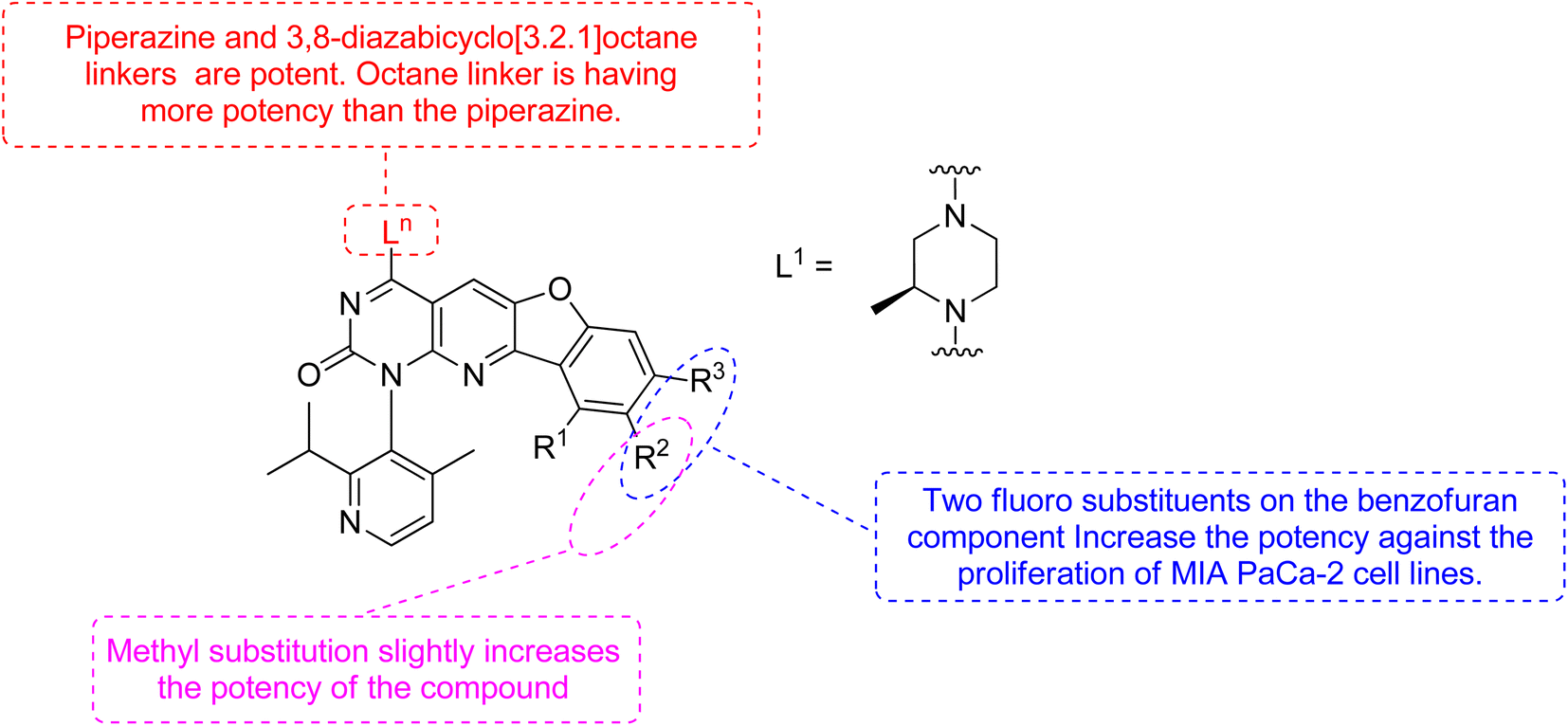
Xiao et al., 2021, designed new KRASG12C inhibitors using a novel approach. Two sets of pyrido[2,3-d]pyrimidine- and pyridinyl N-atom-shifted bicyclic pyrido[4,3-d]pyrimidine-containing derivatives of the clinical KRASG12C inhibitor AMG510 (125) were produced (Fig. 38). MIA PaCa-2 cells, clinical pancreatic cancerous cells with the KRASG12C mutations, were used to test the tetracyclic derivatives; investigating potential off-target effects, the inhibitory activity on A549 cells carrying other KRAS mutant G12S has also been investigated. Although compound 128 demonstrated considerable efficacy, including an IC50 value of 7.97 μM in MIA PaCa-2 cell line expressing KRASG12C, the tetracyclic molecules still exhibited selectivity toward the G12C mutation of KRAS (Table 9). According to the docking study, the shadow connection between 128 in the front entry and an open hydrophobic pocket may be the main reason for its less efficacy. The piperazine derivative was less effective than the 3,8-bicyclo[3.2.1]octane linker in the parent skeleton. However, it was found that the piperizinyl linker, which joins the bond formation warhead towards the tetracyclic nucleus, had some biological efficacy. Compounds 127 and 128 have the most effective R2-substituents, such as methyl or fluoro group.143
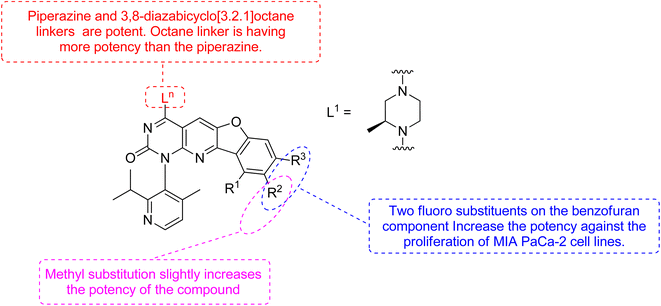 |
| | Fig. 38 SAR of KRAS inhibitors. | |
Table 9 Antiproliferative effects of 127 and 128 against MIA PaCa-2 cell lines
| Compound no |
Ln |
R1 |
R2 |
R3 |
IC50 |
| 127 |
L1 |
H |
CH3 |
H |
>10 μM |
| 128 |
L1 |
H |
F |
F |
7.97 μM |
| AMG510 (125) |
— |
— |
— |
— |
0.029 μM |
The more robust character of (pyrido[4,3-d]pyrimidin) in comparison to 128 (pyrido[2,3-d]pyrimidin), explained by fluorophenol group engaging a hydrophobic pocket and creating an H-bonds to Arg68. Additionally, the fluorophenol molecule appeared to occupy the space by the tiny fluoro group at the C-8 place (as in ARS1620). Only molecule 128 showed minor activity in the MIA PaCa-2 human cell expressing KRASG12C, with IC50 of 7.97 μM, whereas the tetracyclic molecules still showed specificity for the G12C form of KRAS. According to structural docking study, the shadow contact of 127 at the front entry and an unoccupied hydrophobic pocket may be the primary cause of its weak potency. In biochemical and cellular experiments, bicyclic compound 128 was found to be substantial inhibitor.143
3.11 Fibroblast growth factor receptors inhibitors
Several endothelial and tumour cells contain tyrosine kinases called FGFR (fibroblast growth factor receptors). They participate in tumour angiogenesis, tumour cell migration, survival, and proliferation. Many human malignancies have been connected to FGFR overexpression or aberrant activity control. As a result, reducing tumor cell proliferation, survival, and migration, as well as tumor angiogenesis by targeting FGFRs is an appealing technique for developing cancer therapeutic alternatives.144 The activation of downstream transduction pathways occurs when the intracellular kinase domain of the FGFRs undergoes transautophosphorylation due to the binding of FGFs, which also causes the dimerization of FGFRs.145,146 FGFRs contribute to crucial physiological activities, including cell migration, proliferation, differentiation, and survival, via activating downstream signalling pathways.147–149
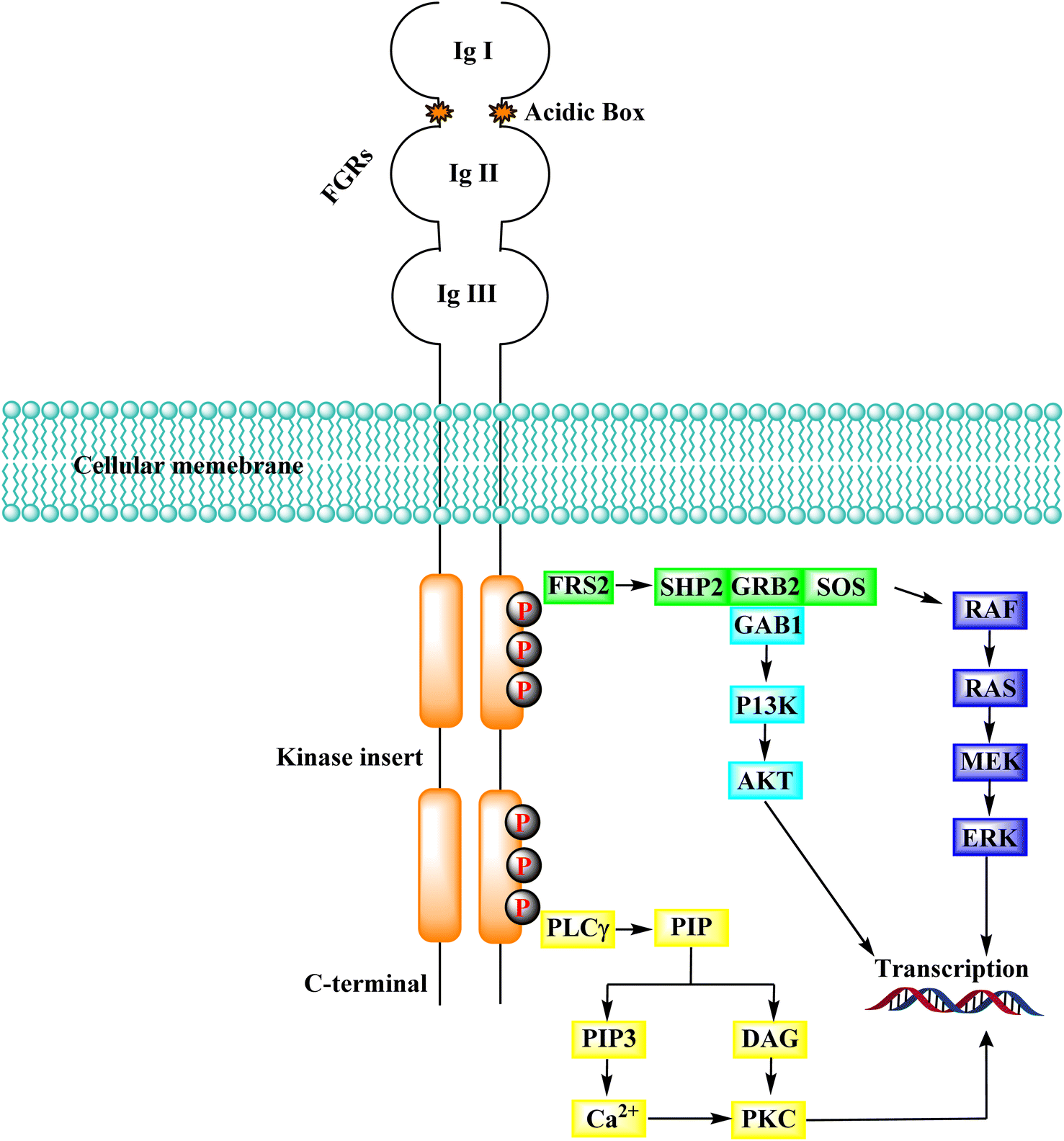
3.11.1 FGFR signaling pathways. FRS2, a prominent FGFR substrate, binds to the juxta membrane area of FGFR constitutively via its phospho-tyrosine binding domain (PTB), regardless of the kinase domain's activity or phosphorylation state. Several tyrosine residues in FRS2 are phosphorylated after FGFR activation and serve as docking sites for the following molecules.150 The RAS-MAPK-ERK signalling cascade is repeatedly activated by FRS2. RSA was chacterised by docking with different proteins, including SHP2 (SH2-containing tyrosine kinase phosphatase), GRB2 (growth factor receptor-bound protein 2), SOS (Son of Sevenless). The Src symmetry domain (SH2 domain) in SHP2 as well as GRB2 could bind directly phosphorylated tyrosine residues in GRB2 and FRS2. As a result, either directly or through the creation of the SHP2-GRB2-SOS complex, the GRB2-SOS complex is recruited to FRS2.151,152 In the RAS-MAPK-ERK signalling pathway, the complex then initiates a phosphorylation cascade. ERK1/2 is translocated from the cytoplasm to the nucleus after activation. It regulates the activity of various transcription factors to influence cell proliferation, differentiation, and signal transduction, making it one of the most persuasive signalling molecules in this pathway.153 When GRB2-related binding protein 1 (GAB1) binds to GRB2's SH3 domain, it can phosphorylate tyrosine itself and be dragged into the complex. Similarly, PI3K with an SH2 domain binds to the phosphorylated tyrosine residues of GRB2, activating the PI3K-AKT signalling cascade. AKT's downstream effector molecules include the well-known mTOR, which is involved in cell metabolism, transcription, and other processes (Fig. 39).154
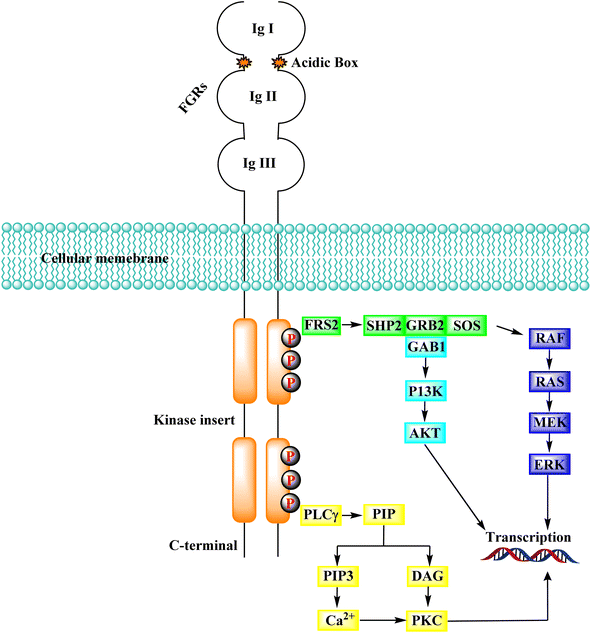 |
| | Fig. 39 FGFR signaling cascade. | |
In addition to FGFR, PIP2 (phosphatidylinositol 4,5-bisphosphate) is hydrolyzed by phospholipase C, which connects to a phosphorylated tyrosine in the kinase domain's C-terminus to produce the secondary mediators IP3 (inositol triphosphate) and DAG (diacyl glycerol). When IP3 binds to its receptor on the endoplasmic reticulum, Ca2+ is released from intracellular reserves, increasing Ca2+ concentration.155 DAG activates the PKC signalling pathway when it is coordinated with Ca2+, which produces crosstalk with the RAS-MAPK pathway (due to the rivalry between GRB2 and PLCγ) for FGFR binding.156
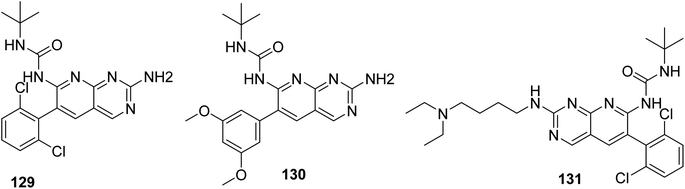
Connolly et al. from the Parke-Davis laboratory were the first to identify drugs tailored to target FGFR selectively. The authors used high-throughput screening approaches to find FGFR inhibitors. This led to the discovery of pyrido[2,3-d]pyrimidine (129), which inhibited FGFR potently (IC50 = 0.45 μM) in a biochemical assay; this indicates that the compound 129 binds at the ATP-binding site, and the molecule behaved in an ATP-competitive fashion.157
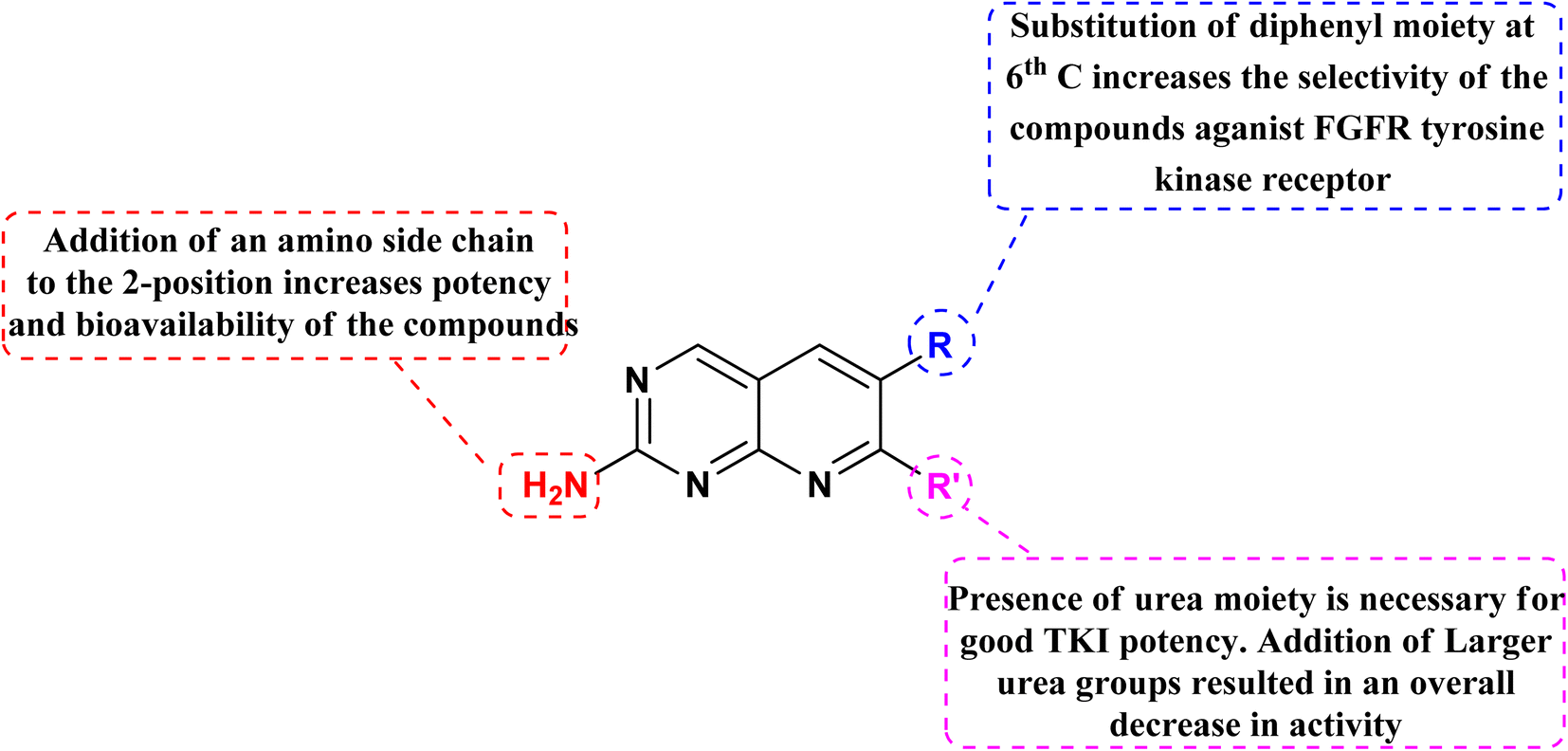
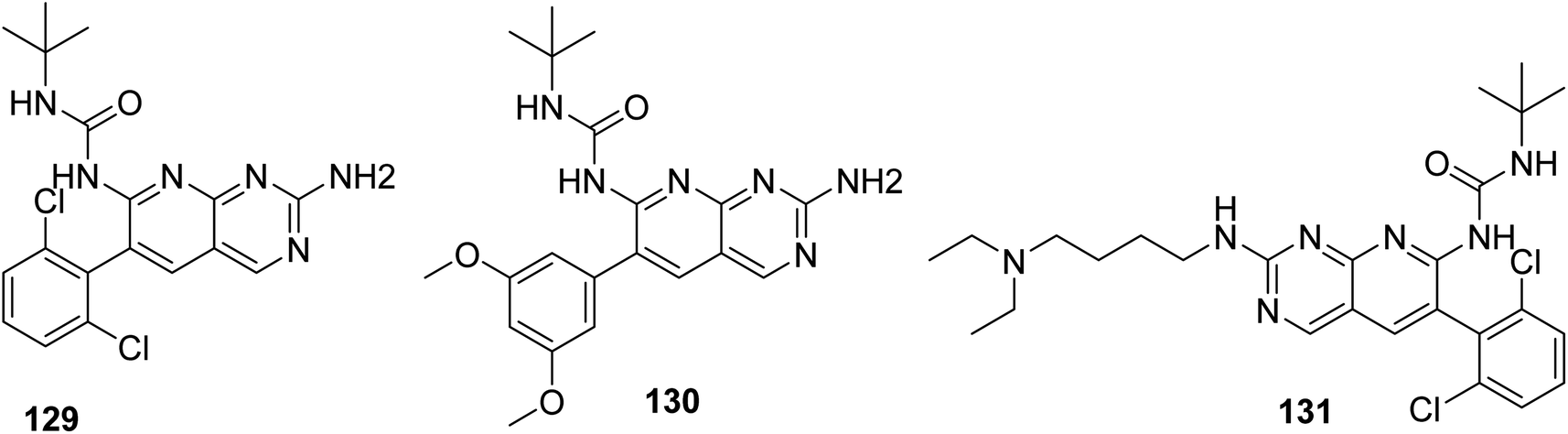
3.11.2 Structure–activity relationship of FGFR inhibitors. SAR analysis revealed the creation of new analogs that were more potent, soluble, and bioavailable than the parent lead. Compound 129 was transformed into compound 131 [by introducing a {4-(diethylamino) butyl} amine substituent], which had a higher potency as well as availability with an IC50 of 0.3 μM and reduced PDGF-stimulated vascular smooth muscle cell growth. A selective FGFr tyrosine kinase antagonist, 130, was also created by substituting 6-(2,6-dichlorophenyl) group of 129 with the 6-(3′,5′-dimethoxyphenyl). If, the pyrido[2,3-d]pyrimidine nucleus has a N′-substituted alkylurea group on the seventh position of parent skeleton, excellent tyrosine kinase antagonists can be synthesiszed.157 Compound 131 inhibited the FGFr tyrosine kinase with an IC50 of 0.060 M, whereas the IC50s for the PDGFr, FGFr, EGFr, c-src, and InsR tyrosine kinases were all greater than 50 μM (Fig. 40 and 41).157
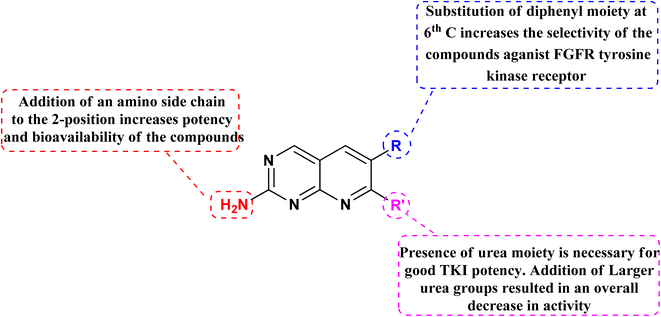 |
| | Fig. 40 SAR of FGFR inhibitors. | |
 |
| | Fig. 41 Structures of pyrido[2,3-d]pyrimidine derivatives as FGFR inhibitor. | |
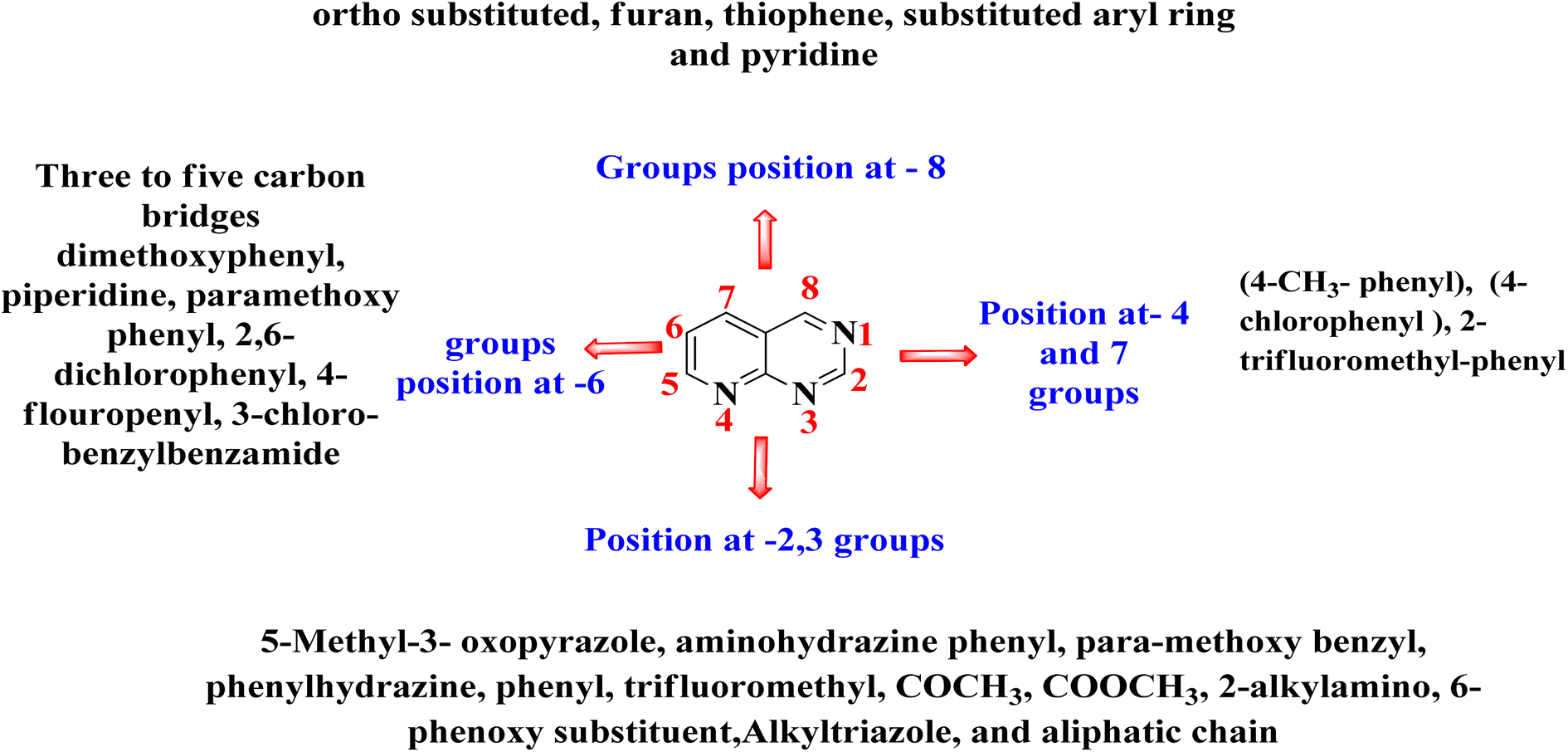
4 Summary of anticancer activity of pyrido[2,3-d]pyrimidine derivatives
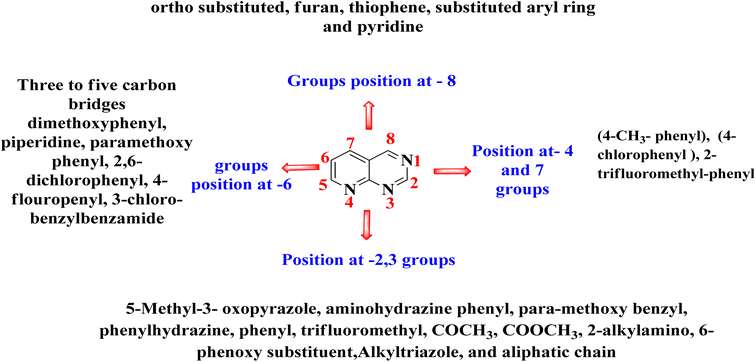
The overall SAR for the anticancer activity of pyrido[2,3-d]pyrimidine derivatives is shown below in Fig. 42.
 |
| | Fig. 42 Summary of SAR of pyrido[2,3-d]pyrimidine derivatives as anticancer agents. | |
5 Future perspective
Many FDA approved drugs like palbociclib, pamapimod, and piritrexim, having the pyrido[2,3-d]scaffold in their structure and are used to treat different form of cancer by via inhibiting CDK4/6, p38, and DHFR respectively. AMG510 and R1478 are under phase 1 clinical trails against KRAS and p38 respectively. Dilmapimod (100), TAK-733 (101), and compound 102 have been withdrawn from clinical trials due to undesirable side effects. Therefore, there is need to design selective molecules to combat resistance and problem of undesirable side effects. In this study, we have found many compounds showed excellent inhibition of cancer at the concentration of nM range. Researchers may design potent and selective molecules keeping in the consideration of stuructural requirements given in structure activity relationship study (Fig. 42).
6 Conclusion
Nitrogen containing heterocyclic compounds, especially pyrido[2,3-d]pyrimidines are a diverse class of chemicals having a number of biological properties. Tyrosine kinase, extracellular regulated protein kinases-ABL kinase, phosphatidylinositol-3 kinase, mammalian target of rapamycin, p38 mitogen-activated protein kinases, BCR-ABL, dihydrofolate reductase, Cyclin-dependent kinase, Phosphodiesterase, KRAS (the Gene Kirsten rat sarcoma viral oncogene homolog), and fibroblast growth factor receptors are the reported anticancer tagets of pyrido[2,3-d]pyrimidine. These derivatives have demonstrated significant anticancer action upon modifications at C-2, C-3, C-5, C-6, C-7, as well as C-8. Many researchers have explored its Structure activity relationship, as well as conformation and alignment parameters for ligand binding, employing computational modeling as well as docking investigations. The current study also emphasizes a variety of conventional, multi-component, and microwave-assisted approaches for synthesizing pyrido[2,3-d]pyrimidine derivatives and will also help scientists find highly effective, precise, and targeted pharmaceuticals.
Conflicts of interest
The authors declare no competing interests.
Author contributions
Conceptualization: P. K., and M. J.; data collection: A. K. S, and H. S.; writing the manuscript: A. K. and K. K. B; sketching of figures: A. K., T. A. and H. S.; data interpretation: A. H. E.; writing, review and final editing of the manuscript: A. V. and H. K.
Acknowledgements
Authors are highly thankful to Central University of Punjab, DST-FIST, India, to infrastructural support for completion of this study. The APC was funded by King Abdullah University of Science and Technology (KAUST), Thuwal, Jeddah, Saudi Arabia.
References
- T. Angre, A. Kumar, A. K. Singh, S. Thareja and P. Kumar, Role of collagen regulators in cancer treatment: A comprehensive review, Anti-Cancer Agents Med. Chem., 2022, 22, 1–7 Search PubMed.
- R. Weinberg and D. Hanahan, The hallmarks of cancer, Cell, 2000, 100(1), 57–70 CrossRef PubMed.
- L. Torre, R. Siegel and A. Jemal, Global cancer facts & figures, American Cancer Society, Atlanta, 2015, 2(3), pp. 1–64 Search PubMed.
- P. Vineis and C. P. Wild, Global cancer patterns: causes and prevention, Lancet, 2014, 383(9916), 549–557 CrossRef PubMed.
- K. M. Abu-Zied, T. K. Mohamed, O. K. Al-Duiaj and M. E. Zaki, A simple approach to fused pyrido[2,3-d]pyrimidines incorporating khellinone and trimethoxyphenyl moieties as new scaffolds for antibacterial and antifungal agents, Heterocycl. Commun., 2014, 20(2), 93–102 CrossRef CAS.
- R. K. Robins and G. H. Hitchings, Studies on condensed pyrimidine systems. XIX. A new synthesis of pyrido [2,3-d]pyrimidines. The condensation of 1, 3-diketones and 3-ketoaldehydes with 4-aminopyrimidines, J. Am. Chem. Soc., 1958, 80(13), 3449–3457 CrossRef CAS.
- Y. Yamamoto, Science of Synthesis, Houben-Weyl Methods of Molecular Transformations; Six-Membered Hetarenes with Two Identical Heteroatoms, Sci. Synth., 2004, 1–1568 Search PubMed.
- A. Gangjee, O. O. Adair and S. F. Queener, Synthesis and Biological Evaluation of 2, 4-Diamino-6-(arylaminomethyl) pyrido [2,3-d]pyrimidines as Inhibitors of Pneumocystis c arinii and Toxoplasma g ondii Dihydrofolate Reductase and as Antiopportunistic Infection and Antitumor Agents, J. Med. Chem., 2003, 46(23), 5074–5082 CrossRef CAS PubMed.
- H. S. Elzahabi, E. S. Nossier, N. M. Khalifa, R. A. Alasfoury and M. A. El-Manawaty, Anticancer evaluation and molecular modeling of multi-targeted kinase inhibitors based pyrido [2,3-d]pyrimidine scaffold, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 546–557 CrossRef CAS PubMed.
- D. A. Ibrahim and N. S. Ismail, Design, synthesis and biological study of novel pyrido [2,3-d]pyrimidine as anti-proliferative CDK2 inhibitors, Eur. J. Med. Chem., 2011, 46(12), 5825–5832 CrossRef CAS PubMed.
- T. Yang, H. He, W. Ang, Y.-H. Yang, J.-Z. Yang and Y.-N. Lin, et al., Syntheses and cell-based phenotypic screen of novel 7-amino pyrido [2,3-d]pyrimidine-6-carbonitrile derivatives as potential antiproliferative agents, Molecules, 2012, 17(3), 2351–2366 CrossRef CAS PubMed.
- M. Sako, Product Class 19: Pyridopyrimidines, Sci Synth, 2004, 16, 1155–1267 CAS.
- F. Buron, J. Mérour, M. Akssira, G. Guillaumet and S. Routier, Recent advances in the chemistry and biology of pyridopyrimidines, Eur. J. Med. Chem., 2015, 95(5), 76–95 CrossRef CAS PubMed.
- B. Evans, K. Rittle, M. Bock, R. DiPardo, R. Freidinger and W. Whitter, et al., Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists, J. Med. Chem., 1988, 31(12), 2235–2246 CrossRef CAS PubMed.
- C. D. Altomare, Privileged heterocyclic scaffolds in chemical biology and drug discovery: Synthesis and bioactivity, Chem. Heterocycl. Compd., 2017, 53(3), 239 CrossRef CAS.
- G. Jubete, R. Puig de la Bellacasa, R. Estrada-Tejedor, J. Teixidó and J. I. Borrell, Pyrido [2,3-d] pyrimidin-7 (8 H)-ones: synthesis and biomedical applications, Molecules, 2019, 24(22), 4161–4182 CrossRef PubMed.
- A. Gangjee, J. Shi, S. F. Queener, L. R. Barrows and R. L. Kisliuk, Synthesis of 5-methyl-5-deaza nonclassical antifolates as inhibitors of dihydrofolate reductases and as potential antipneumocystis, antitoxoplasma, and antitumor agents, J. Med. Chem., 1993, 36(22), 3437–3443 CrossRef CAS PubMed.
- J. F. Campos, T. Besson and S. Berteina-Raboin, Review on the Synthesis and Therapeutic Potential of Pyrido [2,3-d],[3, 2-d],[3,4-d] and [4, 3-d]pyrimidine Derivatives, Pharmaceuticals, 2022, 15(3), 352–379 CrossRef CAS PubMed.
- A. Gangjee, A. Vasudevan, S. F. Queener and R. L. Kisliuk, 2, 4-Diamino-5-deaza-6-substituted pyrido [2,3-d]pyrimidine antifolates as potent and selective nonclassical inhibitors of dihydrofolate reductases, J. Med. Chem., 1996, 39(7), 1438–1446 CrossRef CAS PubMed.
- A. Gangjee, O. O. Adair, M. Pagley and S. F. Queener, N9-substituted 2, 4-diaminoquinazolines: synthesis and biological evaluation of lipophilic inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase, J. Med. Chem., 2008, 51(19), 6195–6200 CrossRef CAS PubMed.
- E. M. Grivsky, S. Lee, C. W. Sigel, D. S. Duch and C. A. Nichol, Synthesis and antitumor activity of 2, 4-diamino-6-(2, 5-dimethoxybenzyl)-5-methylpyrido [2,3-d]pyrimidine, J. Med. Chem., 1980, 23(3), 327–329 CrossRef CAS PubMed.
- D. M. Goldstein, M. Soth, T. Gabriel, N. Dewdney, A. Kuglstatter and H. Arzeno, et al., Discovery of 6–(2,4-Difluorophenoxy)-2-[3-hydroxy-1-(2-hydroxyethyl) propylamino]-8-methyl-8 H-pyrido [2,3-d] pyrimidin-7-one (Pamapimod) and 6-(2, 4-Difluorophenoxy)-8-methyl-2-(tetrahydro-2 H-pyran-4-ylamino) pyrido [2,3-d] pyrimidin-7 (8 H)-one (R1487) as Orally Bioavailable and Highly Selective Inhibitors of p38α Mitogen-Activated Protein Kinase, J. Med. Chem., 2011, 54(7), 2255–2265 CrossRef CAS PubMed.
- M. C. Bagley, D. D. Hughes, R. Lloyd and V. E. Powers, A new and highly expedient synthesis of pyrido [2,3-d]pyrimidines, Tetrahedron Lett., 2001, 42(37), 6585–6588 CrossRef CAS.
- N. Mont, J. Teixidó, J. I. Borrell and C. O. Kappe, A three-component synthesis of pyrido [2,3-d]pyrimidines, Tetrahedron Lett., 2003, 44(29), 5385–5387 CrossRef CAS.
- A. M. Rad and M. Mokhtary, Efficient one-pot synthesis of pyrido [2,3-d]pyrimidines catalyzed by nanocrystalline MgO in water, Int. Nano Lett., 2015, 5(2), 109–123 CrossRef CAS.
- A. Shahrzad and B. Saeed, An efficient synthesis of pyrido [2,3-d]pyrimidine derivatives via one-pot three-component reaction in aqueous media, Int. J. Org. Chem., 2012, 2(1), 7–14 CrossRef.
- S. Tu, J. Zhang, X. Zhu, J. Xu, Y. Zhang and Q. Wang, et al., New potential inhibitors of cyclin-dependent kinase 4: design and synthesis of pyrido [2,3-d]pyrimidine derivatives under microwave irradiation, Bioorg. Med. Chem. Lett., 2006, 16(13), 3578–3581 CrossRef CAS PubMed.
- M. K. Paul and A. K. Mukhopadhyay, Tyrosine kinase–role and significance in cancer, Int. J. Med. Sci., 2004, 1(2), 101–115 CrossRef CAS PubMed.
- P. Blum-Jensen and T. Hunter, Oncogenic kinase signaling, Nature, 2001, 411(3), 355–365 CrossRef PubMed.
- J. Schlessinger, Cell signaling by receptor tyrosine kinases, Cell, 2000, 103(2), 211–225 CrossRef CAS PubMed.
- M. A. Lemmon and J. Schlessinger, Cell signaling by receptor tyrosine kinases, Cell, 2010, 141(7), 1117–1134 CrossRef CAS PubMed.
- Z. Du and C. M. Lovly, Mechanisms of receptor tyrosine kinase activation in cancer, Mol. Cancer, 2018, 17(1), 1–13 CrossRef PubMed.
- J. H. Ludes-Meyers, M. A. Subler, C. V. Shivakumar, R. M. Munoz, P. Jiang and J. E. Bigger, et al., Transcriptional activation of the human epidermal growth factor receptor promoter by human p53, Mol. Cell. Biol., 1996, 16(11), 6009–6019 CrossRef CAS PubMed.
- T. E. Reznik, Y. Sang, Y. Ma, R. Abounader, E. M. Rosen and S. Xia, et al., Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor, Mol. Cancer Res., 2008, 6(1), 139–150 CrossRef CAS PubMed.
- Q. Jiao, L. Bi, Y. Ren, S. Song, Q. Wang and Y.-s Wang, Advances in studies of tyrosine kinase inhibitors and their acquired resistance, Mol. Cancer, 2018, 17(1), 1–12 CrossRef PubMed.
- K. S. Kolibaba and B. J. Druker, Protein tyrosine kinases and cancer, Biochim. Biophys. Acta, Rev. Cancer, 1997, 1333(3), 217–248 CrossRef PubMed.
- M. Fares, S. M. Abou-Seri, H. A. Abdel-Aziz, S. E.-S. Abbas, M. M. Youssef and R. A. Eladwy, Synthesis and antitumor activity of pyrido [2,3-d]pyrimidine and pyrido [2,3-d][1, 2, 4] triazolo [4, 3-a]pyrimidine derivatives that induce apoptosis through G1 cell-cycle arrest, Eur. J. Med. Chem., 2014, 83, 155–166 CrossRef CAS PubMed.
- R. Rodriguez and M. Meuth, Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress, Mol. Biol. Cell, 2006, 17(1), 402–412 CrossRef CAS PubMed.
- L. Cordeu, E. Cubedo, E. Bandrés, A. Rebollo, X. Sáenz and H. Chozas, et al., Biological profile of new apoptotic agents based on 2, 4-pyrido [2,3-d]pyrimidine derivatives, Bioorg. Med. Chem., 2007, 15(4), 1659–1669 CrossRef CAS PubMed.
- M. Font, Á. González, J. A. Palop and C. Sanmartín, New insights into the structural requirements for pro-apoptotic agents based on 2, 4-diaminoquinazoline, 2, 4-diaminopyrido [2,3-d]pyrimidine and 2, 4-diaminopyrimidine derivatives, Eur. J. Med. Chem., 2011, 46(9), 3887–3899 CrossRef CAS PubMed.
- S. E. Abbas, R. F. George, E. M. Samir, M. M. Aref and H. A. Abdel-Aziz, Synthesis and anticancer activity of some pyrido [2,3-d]pyrimidine derivatives as apoptosis inducers and cyclin-dependent kinase inhibitors, Future Med. Chem., 2019, 11(18), 2395–2414 CrossRef CAS PubMed.
- J. M. Hamby, C. J. Connolly, M. C. Schroeder, R. T. Winters, H. H. Showalter and R. L. Panek, et al., Structure–activity relationships for a novel series of pyrido [2,3-d]pyrimidine tyrosine kinase inhibitors, J. Med. Chem., 1997, 40(15), 2296–2303 CrossRef CAS PubMed.
- J. F. Dorsey, R. Jove, A. J. Kraker and J. Wu, The pyrido [2,3-d]pyrimidine derivative PD180970 inhibits p210Bcr-Abl tyrosine kinase and induces apoptosis of K562 leukemic cells, Cancer Res., 2000, 60(12), 3127–3131 CAS.
- T. Adon, D. Shanmugarajan and H. Y. Kumar, CDK4/6 inhibitors: a brief overview and prospective research directions, RSC Adv., 2021, 11(47), 29227–29246 RSC.
- C. Shi, Q. Wang, X. Liao, H. Ge, G. Huo and L. Zhang, et al., Discovery of a novel series of imidazo [1′, 2’: 1, 6] pyrido [2,3-d] pyrimidin derivatives as potent cyclin-dependent kinase 4/6 inhibitors, Eur. J. Med. Chem., 2020, 193, 112239–112259 CrossRef CAS PubMed.
- T. Kong, M. Liu, B. Ji, B. Bai, B. Cheng and C. Wang, Role of the extracellular signal-regulated kinase 1/2 signaling pathway in ischemia-reperfusion injury, Front. Physiol., 2019, 10, 1038–1048 CrossRef PubMed.
- A. Kohen, Dihydrofolate reductase as a model for studies of enzyme dynamics and catalysis, F1000Research, 2015, 4, 1–8 Search PubMed.
- D. A. Fruman, H. Chiu, B. D. Hopkins, S. Bagrodia, L. C. Cantley and R. T. Abraham, The PI3K pathway in human disease, Cell, 2017, 170(4), 605–635 CrossRef CAS PubMed.
- F. Janku, T. A. Yap and F. Meric-Bernstam, Targeting the PI3K pathway in cancer: are we making headway?, Nat. Rev. Clin. Oncol., 2018, 15(5), 273–291 CrossRef CAS PubMed.
- L. M. Amzel, C.-H. Huang, D. Mandelker, C. Lengauer, S. B. Gabelli and B. Vogelstein, Structural comparisons of class I phosphoinositide 3-kinases, Nat. Rev. Cancer, 2008, 8(9), 665–669 CrossRef CAS PubMed.
- L. Zhang, Q. Ju, J. Sun, L. Huang, S. Wu and S. Wang, et al., Discovery of novel dual extracellular regulated protein kinases (erk) and phosphoinositide 3-kinase (pi3k) inhibitors as a promising strategy for cancer therapy, Molecules, 2020, 25(23), 5693 CrossRef CAS PubMed.
- H. B. El-Nassan, Synthesis and antitumor activity of novel pyrido [2,3-d][1, 2, 4] triazolo [4, 3-a] pyrimidin-5-one derivatives, Eur. J. Med. Chem., 2011, 46(6), 2031–2036 CrossRef CAS PubMed.
- C. Kurumurthy, P. S. Rao, P. S. Rao, B. Narsaiah, L. Velatooru and R. Pamanji, et al., Synthesis of novel alkyltriazole tagged pyrido [2,3-d]pyrimidine derivatives and their anticancer activity, Eur. J. Med. Chem., 2011, 46(8), 3462–3468 CrossRef CAS PubMed.
- F. Shi, J. Ding, S. Zhang, W.-J. Hao, C. Cheng and S. Tu, Substrate-controlled chemoselective synthesis and potent cytotoxic activity of novel 5, 6, 7-triarylpyrido [2,3-d] pyrimidin-4-one derivatives, Bioorg. Med. Chem. Lett., 2011, 21(5), 1554–1558 CrossRef CAS PubMed.
- N. R. Mohamed, M. M. T. El-Saidi, Y. M. Ali and M. H. Elnagdi, Utility of 6-amino-2-thiouracil as a precursor for the synthesis of bioactive pyrimidine derivatives, Bioorg. Med. Chem., 2007, 15(18), 6227–6235 CrossRef CAS PubMed.
- M. Zink, H. Lanig and R. Troschütz, Structural variations of piritrexim, a lipophilic inhibitor of human dihydrofolate reductase: synthesis, antitumor activity and molecular modeling investigations, Eur. J. Med. Chem., 2004, 39(12), 1079–1088 CrossRef CAS PubMed.
- A. Gangjee, O. Adair and S. F. Queener, Synthesis of 2, 4-diamino-6-(thioarylmethyl) pyrido [2,3-d]pyrimidines as dihydrofolate reductase inhibitors, Bioorg. Med. Chem., 2001, 9(11), 2929–2935 CrossRef CAS PubMed.
- V. K. Agrawal, R. Sohgaura and P. V. Khadikar, QSAR studies on biological activity of piritrexim analogues against pc DHFR, Bioorg. Med. Chem., 2002, 10(9), 2919–2926 CrossRef CAS PubMed.
- T. M. Joska and A. C. Anderson, Structure-activity relationships of Bacillus cereus and Bacillus anthracis dihydrofolate reductase: toward the identification of new potent drug leads, Antimicrob. Agents Chemother., 2006, 50(10), 3435–3443 CrossRef CAS PubMed.
- D. C. Chan, H. Fu, R. A. Forsch, S. F. Queener and A. Rosowsky, Design, synthesis, and antifolate activity of new analogues of piritrexim and other diaminopyrimidine dihydrofolate reductase inhibitors with ω-carboxyalkoxy or ω-carboxy-1-alkynyl substitution in the side chain, J. Med. Chem., 2005, 48(13), 4420–4431 CrossRef CAS PubMed.
- C. Sanmartín, M. Echeverría, B. Mendívil, L. Cordeu, E. Cubedo and J. García-Foncillas, et al., Synthesis and biological evaluation of new symmetrical derivatives as cytotoxic agents and apoptosis inducers, Bioorg. Med. Chem., 2005, 13(6), 2031–2044 CrossRef PubMed.
- M. M. Gineinah, M. N. Nasr, S. M. Badr and W. M. El-Husseiny, Synthesis and antitumor activity of new pyrido [2,3-d]pyrimidine derivatives, Med. Chem. Res., 2013, 22(8), 3943–3952 CrossRef CAS.
- A. L. Nashed, K. W. Rao and M. L. Gulley, Clinical applications of BCR-ABL molecular testing in acute leukemia, J. Mol. Diagn., 2003, 5(2), 63–72 CrossRef CAS PubMed.
- J. V. Melo and M. W. Deininger, Biology of chronic myelogenous leukemia—signaling pathways of initiation and transformation, Hematol. Oncol. Clin., 2004, 18(3), 545–568 CrossRef PubMed.
- M. Huang, J. F. Dorsey, P. Epling-Burnette, R. Nimmanapalli, T. H. Landowski and L. B. Mora, et al., Inhibition of Bcr–Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells, Oncogene, 2002, 21(57), 8804–8816 CrossRef CAS PubMed.
- D. Wisniewski, C. L. Lambek, C. Liu, A. Strife, D. R. Veach and B. Nagar, et al., Characterization of potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosine kinases, Cancer, Res2002, 62(15), 4244–4255 Search PubMed.
- N. Von Bubnoff, D. R. Veach, W. T. Miller, W. Li, J. Sänger and C. Peschel, et al., Inhibition of wild-type and mutant Bcr-Abl by pyrido-pyrimidine-type small molecule kinase inhibitors, Cancer Res., 2003, 63(19), 6395–6404 CAS.
- D. R. Huron, M. E. Gorre, A. J. Kraker, C. L. Sawyers, N. Rosen and M. M. Moasser, A novel pyridopyrimidine inhibitor of abl kinase is a picomolar inhibitor of Bcr-abl-driven K562 cells and is effective against STI571-resistant Bcr-abl mutants, Clin. Cancer Res., 2003, 9(4), 1267–1273 CAS.
- P. Vandenberghe, N. Boeckx, E. Ronsyn, R. Decorte, G. Verhoef and A. Hagemeijer, Imatinib mesylate induces durable complete remission of advanced CML persisting after allogeneic bone marrow transplantation, Leukemia, 2003, 17(2), 458–460 CrossRef CAS PubMed.
- A. Cuadrado and A. R. Nebreda, Mechanisms and functions of p38 MAPK signalling, Biochem. J., 2010, 429(3), 403–417 CrossRef CAS PubMed.
- L. R. Coulthard, D. E. White, D. L. Jones, M. F. McDermott and S. A. Burchill, p38MAPK: stress responses from molecular mechanisms to therapeutics, Trends Mol. Med., 2009, 15(8), 369–379 CrossRef CAS PubMed.
- A. Cuenda and S. Rousseau, p38 MAP-kinases pathway regulation, function and role in human diseases, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773(8), 1358–1375 CrossRef CAS PubMed.
- Z. Wang, P. C. Harkins, R. J. Ulevitch, J. Han, M. H. Cobb and E. J. Goldsmith, The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(6), 2327–2332 CrossRef CAS PubMed.
- P. Traxler and P. Furet, Strategies toward the design of novel and selective protein tyrosine kinase inhibitors, Pharmacol. Ther., 1999, 82(2–3), 195–206 CrossRef CAS PubMed.
- S. T. Wrobleski and A. M. Doweyko, Structural comparison of p38 inhibitor-protein complexes: a review of recent p38 inhibitors having unique binding interactions, Curr. Top. Med. Chem., 2005, 5(10), 1005–1016 CrossRef CAS PubMed.
- D. R. Hauser, T. Scior, D. M. Domeyer, B. Kammerer and S. A. Laufer, Synthesis, biological testing, and binding mode prediction of 6, 9-diarylpurin-8-ones as p38 MAP kinase inhibitors, J. Med. Chem., 2007, 50(9), 2060–2066 CrossRef CAS PubMed.
- E. L. Michelotti, K. K. Moffett, D. Nguyen, M. J. Kelly, R. Shetty and X. Chai, et al., Two classes of p38α MAP kinase inhibitors having a common diphenylether core but exhibiting divergent binding modes, Bioorg. Med. Chem. Lett., 2005, 15(23), 5274–5279 CrossRef CAS PubMed.
- S. A. Laufer, D. R. Hauser, D. M. Domeyer, K. Kinkel and A. J. Liedtke, Design, synthesis, and biological evaluation of novel Tri-and tetrasubstituted imidazoles as highly potent and specific ATP-mimetic inhibitors of p38 MAP kinase: Focus on optimized interactions with the enzyme's surface-exposed front region, J. Med. Chem., 2008, 51(14), 4122–4149 CrossRef CAS PubMed.
- Y. Liu and N. S. Gray, Rational design of inhibitors that bind to inactive kinase conformations, Nat. Chem. Biol., 2006, 2(7), 358–364 CrossRef CAS PubMed.
- H. Cheng, D. Bhumralkar, K. R. Dress, J. E. Hoffman, M. C. Johnson, R. S. Kania, et al., 4-Methylpyridopyrimidinone compounds, US Pat., US8273755B2, 2012 Search PubMed.
- S. Huang and P. J. Houghton, Targeting mTOR signaling for cancer therapy, Curr. Opin. Pharmacol., 2003, 3(4), 371–377 CrossRef CAS PubMed.
- T. A. Yap, M. D. Garrett, M. I. Walton, F. Raynaud, J. S. de Bono and P. Workman, Targeting the PI3K–AKT–mTOR pathway: progress, pitfalls, and promises, Curr. Opin. Pharmacol., 2008, 8(4), 393–412 CrossRef CAS PubMed.
- E. J. Brown, M. W. Albers, T. Bum Shin, C. T. Keith, W. S. Lane and S. L. Schreiber, A mammalian protein targeted by G1-arresting rapamycin–receptor complex, Nature, 1994, 369(6483), 756–758 CrossRef CAS PubMed.
- K. Malagu, H. Duggan, K. Menear, M. Hummersone, S. Gomez and C. Bailey, et al., The discovery and optimisation of pyrido [2,3-d]pyrimidine-2, 4-diamines as potent and selective inhibitors of mTOR kinase, Bioorg. Med. Chem. Lett., 2009, 19(20), 5950–5953 CrossRef CAS PubMed.
- D. C. Fingar, S. Salama, C. Tsou, E. Harlow and J. Blenis, Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E, Genes Dev., 2002, 16(12), 1472–1487 CrossRef CAS PubMed.
- D. C. Fingar, C. J. Richardson, A. R. Tee, L. Cheatham, C. Tsou and J. Blenis, mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E, Mol. Cell. Biol., 2004, 24(1), 200–216 CrossRef CAS PubMed.
- G. J. Brunn, J. Williams, C. Sabers, G. Wiederrecht, J. Lawrence Jr and R. T. Abraham, Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002, EMBO J., 1996, 15(19), 5256–5267 CrossRef CAS PubMed.
- D. J. VanderWeele and C. M. Rudin, Mammalian target of rapamycin promotes vincristine resistance through multiple mechanisms independent of maintained glycolytic rate, Mol. Cancer Res., 2005, 3(11), 635–644 CrossRef CAS PubMed.
- K. Misselbeck, L. Marchetti, M. S. Field, M. Scotti, C. Priami and P. J. Stover, A hybrid stochastic model of folate-mediated one-carbon metabolism: Effect of the common C677T MTHFR variant on de novo thymidylate biosynthesis, Sci. Rep., 2017, 7(1), 1–11 CrossRef CAS PubMed.
- T. C. Alexander, C. M. Simecka, F. Kiffer, T. Groves, J. Anderson and H. Carr, et al., Changes in cognition and dendritic complexity following intrathecal methotrexate and cytarabine treatment in a juvenile murine model, Behav. Brain Res., 2018, 346, 21–28 CrossRef CAS PubMed.
- L. H. Matherly, Molecular and cellular biology of the human reduced folate carrier, Prog. Nucleic Acid Res. Mol. Biol., 2001, 67(1), 131–162 CAS.
- H. Li, F. Fang, Y. Liu, L. Xue, M. Wang and Y. Guo, et al., Inhibitors of dihydrofolate reductase as antitumor agents: design, synthesis and biological evaluation of a series of novel nonclassical 6-substituted pyrido [3, 2-d]pyrimidines with a three-to five-carbon bridge, Bioorg. Med. Chem., 2018, 26(9), 2674–2685 CrossRef CAS PubMed.
- M. Wang, J. Yang, M. Yuan, L. Xue, H. Li and C. Tian, et al., Synthesis and antiproliferative activity of a series of novel 6-substituted pyrido [3, 2-d]pyrimidines as potential nonclassical lipophilic antifolates targeting dihydrofolate reductase, Eur. J. Med. Chem., 2017, 128, 88–97 CrossRef CAS PubMed.
- O. A. Abdelaziz, W. M. El Husseiny, K. B. Selim and H. M. Eisa, Dihydrofolate reductase inhibition effect of 5-substituted pyrido [2,3-d]pyrimidines: Synthesis, antitumor activity and molecular modeling study, Bioorg. Chem., 2019, 90(107), 2411–2502 Search PubMed.
- K. A. Cadoo, A. Gucalp and T. A. Traina, Palbociclib: an evidence-based review of its potential in the treatment of breast cancer, Breast Cancer: Targets Ther., 2014, 6(4), 123–133 CAS.
- M. Malumbres, E. Harlow, T. Hunt, T. Hunter, J. M. Lahti and G. Manning, et al., Cyclin-dependent kinases: a family portrait, Nat. Cell Biol., 2009, 11(11), 1275–1276 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri and E. Baehrecke, et al., Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009, Cell Death Differ., 2009, 16(1), 3–11 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, The hallmarks of cancer, Cell, 2000, 100(1), 57–70 CrossRef CAS PubMed.
- J. Plati, O. Bucur and R. Khosravi-Far, Dysregulation of apoptotic signaling in cancer: molecular mechanisms and therapeutic opportunities, J. Cell. Biochem., 2008, 104(4), 1124–1149 CrossRef CAS PubMed.
- P. L. Toogood, P. J. Harvey, J. T. Repine, D. J. Sheehan, S. N. VanderWel and H. Zhou, et al., Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6, J. Med. Chem., 2005, 48(7), 2388–2406 CrossRef CAS PubMed.
- M. Barvian, D. H. Boschelli, J. Cossrow, E. Dobrusin, A. Fattaey and A. Fritsch, et al., Pyrido [2,3-d] pyrimidin-7-one inhibitors of cyclin-dependent kinases, J. Med. Chem., 2000, 43(24), 4606–4616 CrossRef CAS PubMed.
- N. C. Turner, J. Ro, F. André, S. Loi, S. Verma and H. Iwata, et al., Palbociclib in hormone-receptor–positive advanced breast cancer, N. Engl. J. Med., 2015, 373(3), 209–219 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Sulforhodamine B colorimetric assay for cytotoxicity screening, Nat. Protoc., 2006, 1(3), 1112–1116 CrossRef CAS PubMed.
- A. Kamal, T. S. Reddy, S. Polepalli, N. Shalini, V. G. Reddy and A. S. Rao, et al., Synthesis and biological evaluation of podophyllotoxin congeners as tubulin polymerization inhibitors, Bioorg. Med. Chem., 2014, 22(19), 5466–5475 CrossRef CAS PubMed.
- C. P. Kumar, T. S. Reddy, P. S. Mainkar, V. Bansal, R. Shukla and S. Chandrasekhar, et al., Synthesis and biological evaluation of 5, 10-dihydro-11H-dibenzo [b, e][1, 4] diazepin-11-one structural derivatives as anti-cancer and apoptosis inducing agents, Eur. J. Med. Chem., 2016, 108, 674–686 CrossRef PubMed.
- V. Gurtu, S. R. Kain and G. Zhang, Fluorometric and colorimetric detection of caspase activity associated with apoptosis, Anal. Biochem., 1997, 251(1), 98–102 CrossRef CAS PubMed.
- A. W. Murray, Recycling the cell cycle: cyclins revisited, Cell, 2004, 116(2), 221–234 CrossRef CAS PubMed.
- A. Carnero, Targeting the cell cycle for cancer therapy, Br. J. Cancer, 2002, 87(2), 129–133 CrossRef CAS PubMed.
- K. R. Webster and S. D. Kimball, Novel drugs targeting the cell cycle, Emerging Drugs, 2000, 5(1), 45–59 CrossRef CAS.
- T. Ma, B. A. Van Tine, Y. Wei, M. D. Garrett, D. Nelson and P. D. Adams, et al., Cell cycle–regulated phosphorylation of p220NPAT by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription, Genes Dev., 2000, 14(18), 2298–2313 CrossRef CAS PubMed.
- L. De Boer, V. Oakes, H. Beamish, N. Giles, F. Stevens and M. Somodevilla-Torres, et al., Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events, Oncogene, 2008, 27(31), 4261–4268 CrossRef CAS PubMed.
- O. Flores, Z. Wang, K. E. Knudsen and K. L. Burnstein, Nuclear targeting of cyclin-dependent kinase 2 reveals essential roles of cyclin-dependent kinase 2 localization and cyclin E in vitamin D-mediated growth inhibition, Endocrinol, 2010, 151(3), 896–908 CrossRef CAS PubMed.
- S. Akli, C. S. Van Pelt, T. Bui, L. Meijer and K. Keyomarsi, Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin, E. Cancer Res., 2011, 71(9), 3377–3386 CrossRef CAS PubMed.
- H. Yamamoto, T. Monden, H. Miyoshi, H. Izawa, K. Ikeda and M. Tsujie, et al., Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells, Int. J. Oncol., 1998, 13(2), 233–242 CAS.
- V. Juric and B. Murphy, Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions, Cancer Drug Resist., 2020, 3(1), 48–62 CAS.
- B. M. Desai, J. Villanueva, T.-T. K. Nguyen, M. Lioni, M. Xiao and J. Kong, et al., The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling, PloS one, 2013, 8(3), 59588–59600 CrossRef PubMed.
- G. Angius, S. Tomao, V. Stati, P. Vici, V. Bianco and F. Tomao, Prexasertib, a checkpoint kinase inhibitor: from preclinical data to clinical development, Cancer Chemother. Pharmacol., 2020, 85(1), 9–20 CrossRef CAS PubMed.
- M. Shah, M. F. H. Qureshi, D. Mohammad, M. Lakhani, T. Urooj and S. Mushtaq, CDKs family-a glimpse into the past and present: from cell cycle control to current biological functions, Asian Pac. J. Cancer Biol., 2020, 5(1), 1–9 CrossRef CAS.
- J. Beavo, S. H. Francis and M. D. Houslay, Cyclic nucleotide phosphodiesterases in health and disease, CRC Press, 2007, p.728 Search PubMed.
- A. T. Bender and J. A. Beavo, Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use, Pharmacol. Rev., 2006, 58(3), 488–520 CrossRef CAS PubMed.
- A. H. Abadi, M. S. Hany, S. A. Elsharif, A. A. H. Eissa, B. D. Gary and H. N. Tinsley, et al., Modulating the cyclic guanosine monophosphate substrate selectivity of the phosphodiesterase 3 inhibitors by pyridine, pyrido [2,3-d]pyrimidine derivatives and their effects upon the growth of HT-29 cancer cell line, Chem. Pharm. Bull., 2013, 61(4), 405–410 CrossRef CAS PubMed.
- L. Hirsh, A. Dantes, B.-S. Suh, Y. Yoshida, K. Hosokawa and K. Tajima, et al., Phosphodiesterase inhibitors as anti-cancer drugs, Biochem. Pharmacol., 2004, 68(6), 981–988 CrossRef CAS PubMed.
- T. Murata, K. Shimizu, M. Narita, V. C. Manganiello and T. Tagawa, Characterization of phosphodiesterase 3 in human malignant melanoma cell line, Anticancer Res., 2002, 22(6A), 3171–3174 CAS.
- T. Tsukahara, Y. Matsuda and H. Haniu, Cyclic phosphatidic acid stimulates cAMP production and inhibits growth in human colon cancer cells, PloS one, 2013, 8(11), e81139 CrossRef PubMed.
- D. H. Maurice, H. Ke, F. Ahmad, Y. Wang, J. Chung and V. C. Manganiello, Advances in targeting cyclic nucleotide phosphodiesterases, Nat. Rev. Drug Discovery, 2014, 13(4), 290–314 CrossRef CAS PubMed.
- I. Levy, A. Horvath, M. Azevedo, R. B. de Alexandre and C. A. Stratakis, Phosphodiesterase function and endocrine cells: links to human disease and roles in tumor development and treatment, Curr. Opin. Pharmacol., 2011, 11(6), 689–697 CrossRef CAS PubMed.
- H. A. Al-Wadei and H. M. Schuller, Cyclic adenosine monophosphate-dependent cell type-specific modulation of mitogenic signaling by retinoids in normal and neoplastic lung cells, Cancer Detect. Prev., 2006, 30(5), 403–411 CrossRef CAS PubMed.
- T. Peng, J. Gong, Y. Jin, Y. Zhou, R. Tong and X. Wei, et al., Inhibitors of phosphodiesterase as cancer therapeutics, Eur. J. Med. Chem., 2018, 150, 742–756 CrossRef CAS PubMed.
- L. Huang, Z. Guo, F. Wang and L. Fu, KRAS mutation: from undruggable to druggable in cancer, Signal Transduction Targeted Ther., 2021, 6(1), 1–20 CrossRef PubMed.
- P. J. Roberts and T. E. Stinchcombe, KRAS mutation: should we test for it, and does it matter?, J. Clin. Oncol., 2013, 31(8), 1112–1121 CrossRef CAS PubMed.
- C. Tan and X. Du, KRAS mutation testing in metastatic colorectal cancer, World J. Gastroenterol., 2012, 18(37), 5171–5180 CAS.
- L. Goebel, M. P. Müller, R. S. Goody and D. Rauh, KRasG12C inhibitors in clinical trials: a short historical perspective, RSC Med. Chem., 2020, 11(7), 760–770 RSC.
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Drugging the undruggable RAS: Mission possible?, Nat. Rev. Drug Discovery, 2014, 13(11), 828–851 CrossRef CAS PubMed.
- I. R. Vetter and A. Wittinghofer, The guanine nucleotide-binding switch in three dimensions, Science, 2001, 294(5545), 1299–1304 CrossRef CAS PubMed.
- K. Scheffzek, M. R. Ahmadian, W. Kabsch, L. Wiesmuller, A. Lautwein and F. Schmitz, et al., The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants, Science, 1997, 277(5324), 333–339 CrossRef CAS PubMed.
- G. Buhrman, G. Holzapfel, S. Fetics and C. Mattos, Allosteric modulation of Ras positions Q61 for a direct role in catalysis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(11), 4931–4936 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions, Nature, 2013, 503(7477), 548–551 CrossRef CAS PubMed.
- M. R. Janes, J. Zhang, L.-S. Li, R. Hansen, U. Peters and X. Guo, et al., Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor, Cell, 2018, 172(3), 578–589 CrossRef CAS PubMed.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke and D. Bagal, et al., The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity, Nature, 2019, 575(7781), 217–223 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton and S. K. Booker, et al., Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors, J. Med. Chem., 2020, 52–65 CrossRef CAS PubMed.
- J. Hallin, L. D. Engstrom, L. Hargis, A. Calinisan, R. Aranda and D. M. Briere, et al., The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and PatientsTherapeutic Insight from the KRASG12C Inhibitor MRTX849, Cancer Discovery, 2020, 10(1), 54–71 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana and D. M. Briere, et al., Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer, J. Med. Chem., 2020, 63(13), 6679–6693 CrossRef CAS PubMed.
- X. Xiao, M. Lai, Z. Song, M. Geng, J. Ding and H. Xie, et al., Design, synthesis and pharmacological evaluation of bicyclic and tetracyclic pyridopyrimidinone analogues as new KRASG12C inhibitors, Eur. J. Med. Chem., 2021, 213, 1–59 Search PubMed.
- G. Zhao, W.-y Li, D. Chen, J. R. Henry, H.-Y. Li and Z. Chen, et al., A Novel, Selective Inhibitor of Fibroblast Growth Factor Receptors That Shows a Potent Broad Spectrum of Antitumor Activity in Several Tumor Xenograft ModelsSmall-Molecule Inhibitor of FGFR, Mol. Cancer Ther., 2011, 10(11), 2200–2210 CrossRef CAS PubMed.
- J. Schlessinger, Cell signaling by receptor tyrosine kinases: From basic concepts to clinical applications, Eur. J. Cancer Suppl., 2006, 4(6), 3–26 CrossRef.
- J. Schlessinger, Receptor tyrosine kinases: legacy of the first two decades, Cold Spring Harbor Perspect. Biol., 2014, 6(3), 1–13 Search PubMed.
- D. M. Ornitz and N. Itoh, The fibroblast growth factor signaling pathway, Wiley Interdiscip. Rev.: Dev. Biol., 2015, 4(3), 215–266 CrossRef CAS PubMed.
- A. Beenken and M. Mohammadi, The FGF family: biology, pathophysiology and therapy, Nat. Rev. Drug Discovery, 2009, 8(3), 235–253 CrossRef CAS PubMed.
- K. Dorey and E. Amaya, FGF signalling: diverse roles during early vertebrate embryogenesis, Development, 2010, 137(22), 3731–3742 CrossRef CAS PubMed.
- J. Zheng, W. Zhang, L. Li, Y. He, Y. Wei and Y. Dang, et al., Signaling Pathway and Small-Molecule Drug Discovery of FGFR: A Comprehensive Review, Front. Chem., 2022, 10, 1–24 Search PubMed.
- Y. Hadari, H. Kouhara, I. Lax and J. Schlessinger, Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation, Mol. Cell. Biol., 1998, 18(7), 3966–3973 CrossRef CAS PubMed.
- S. Ong, G. Guy, Y. Hadari, S. Laks, N. Gotoh and J. Schlessinger, et al., FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors, Mol. Cell. Biol., 2000, 20(3), 979–989 CrossRef CAS PubMed.
- Y. Guo, W. Pan, S. Liu, Z. Shen, Y. Xu and L. Hu, ERK/MAPK signalling pathway and tumorigenesis, Exp. Ther. Med., 2020, 19(3), 1997–2007 CAS.
- M.-Y. Quan, Q. Guo, J. Liu, R. Yang, J. Bai and W. Wang, et al., An FGFR/AKT/SOX2 signaling axis controls pancreatic cancer stemness, Front. Cell Dev. Biol., 2020, 8, 287–302 CrossRef PubMed.
- The IP3 receptor/Ca2+ channel and its cellular function, Biochemical Society Symposia, ed. M. J. Wakelam and K. Mikoshiba, Portland Press, 2007 Search PubMed.
- A. E. Fearon and R. P. Grose, Grb-ing receptor activation by the tail, Nat. Struct. Mol. Biol., 2014, 21(2), 113–114 CrossRef CAS PubMed.
- C. J. Connolly, J. M. Hamby, M. C. Schroeder, M. Barvian, G. H. Lu and R. L. Panek, et al., Discovery and structure-activity studies of a novel series of pyrido [2,3-d]pyrimidine tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett., 1997, 7(18), 2415–2420 CrossRef CAS.
Footnote |
| † Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Harshwardhan Singha,
Tanuja Angrea,
Amita Verma
a,
Harshwardhan Singha,
Tanuja Angrea,
Amita Verma