
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution- and gas-phase study of binding of ammonium and bisammonium hydrocarbons to oxacalix[4]arene carboxylate†
Anna Cowart‡
ab,
Mari-Liis Brük‡ ab,
Nikita Žogloa,
Helena Roithmeyera,
Merle Uudsemaaa,
Aleksander Trummala,
Kaspar Selkea,
Riina Aavb,
Elina Kaleniusc and
Jasper Adamson*a
ab,
Nikita Žogloa,
Helena Roithmeyera,
Merle Uudsemaaa,
Aleksander Trummala,
Kaspar Selkea,
Riina Aavb,
Elina Kaleniusc and
Jasper Adamson*a
aLaboratory of Chemical Physics, National Institute of Chemical Physics and Biophysics, Akadeemia Tee 23, 12618 Tallinn, Estonia. E-mail: jasper.adamson@kbfi.ee
bDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia Tee 15, 12618 Tallinn, Estonia
cDepartment of Chemistry, NanoScience Center, University of Jyväskylä, Survontie 9B, FI-40014 JY, Finland
First published on 4th January 2023
Abstract
Oxacalixarenes represent a distinctive class of macrocyclic compounds, which are closely related to the parent calixarene family, offering binding motifs characteristic of calixarenes and crown ethers. Nevertheless, they still lack extensive characterization in terms of molecular recognition properties and the subsequent practical applicability. We present here the results of binding studies of an oxacalix[4]arene carboxylate macrocycle toward a variety of organic ammonium cationic species. Our results show that the substituents attached to the guest ammonium compound largely influence the binding strengths of the host. Furthermore, we show that the characteristic binding pattern changes upon transition from the gas phase to solution in terms of the governing intermolecular interactions. We identify the key factors affecting host–guest binding efficacy and suggest rules for the important molecular structural motifs of the interacting parts of ammonium guest species and the macrocycle to facilitate sensing of ammonium cations.
Introduction
Ammonium ions play a significant role in numerous biological processes and in many biochemical reactions that take place in the human body. For instance, the structures of biologically important amino acids and neurotransmitters, that include molecules such as dopamine, serotonin, or acetylcholine, all contain an amine as a functional group. These amine moieties in the structures are frequently protonated under physiological pH conditions, and therefore, the respective molecules circulate in human bodies as ammonium or indeed as zwitterionic species.1–3 Specific neurotransmitters and their concentrations in the neural system can be linked with various neurodegenerative disorders, such as Parkinson's disease or Alzheimer's disease.4–6 This correlation relates to the interest in designing biomimetic sensors and artificial receptors,7 that can be used to control and monitor the levels of the respective regulator molecules. It is similarly important to study the “host–guest” binding of drug molecules that contain the ammonium group. A lot of studies focus on obtaining the maximum binding strengths for these guest molecules. However, there might be advantages to regulating the binding strength and the respective rates of ingression and egression of the guest molecules. One such example is when the macrocycle–guest complexes are being delivered to the human body inside vesicles, similarly to mRNA,8–10 for targeted delivery of the drug molecule to the cell. Targeted delivery to the cell is still in its infancy but delivering macrocycle–drug complexes to the intracellular environment could be an advantage for precise localization for intended treatment.11–13 If the drug guest molecule is released from the macrocycle in the cell, it could have its local desired impact.We could consider the scenario where a vesicle, for example a dendrimersome nanoparticle, made of ionizable amphiphilic Janus dendrimers,8–10,14,15 carries the macrocycle–drug complex to the relevant cell and releases it there. The complex is subject to dilution in the cell and some of the drug molecule is being released. The complex release rate and extent depend on the concentration of the complex and the association strength (Ka). Lower association strength could be beneficial for releasing the maximum amount of the guest after the complex is subject to dilution. As an example, decreasing the association constant of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding between a host and a guest from 1000 to 100, would result in 91.6% of guest release compared to 61.8% guest release from a 1 mM solution, respectively. Similar considerations are applicable when the macrocycle–drug complex is delivered to any confined environment in the body, where the guest undergoes dissociation from the complex. In these instances, lower association strengths might be advantageous to obtain the maximized abundance of the guest species. Similarly, the reusability of sensors containing macrocycles could benefit from easy guest release. In this paper, we show that the carboxyl-substituted oxacalixarene 1 (Fig. 1) modulates the binding strength of different ammonium containing guest species compared to the noncyclic 2,6-dimethoxybenzoic acid 22 (Fig. 2). We suggest that this might arise from steric hindrance of the binding site in the macrocycle and that such modulated binding could be beneficial for applications that require controlled release of the active ingredient from the complex with the macrocycle.
:1 binding between a host and a guest from 1000 to 100, would result in 91.6% of guest release compared to 61.8% guest release from a 1 mM solution, respectively. Similar considerations are applicable when the macrocycle–drug complex is delivered to any confined environment in the body, where the guest undergoes dissociation from the complex. In these instances, lower association strengths might be advantageous to obtain the maximized abundance of the guest species. Similarly, the reusability of sensors containing macrocycles could benefit from easy guest release. In this paper, we show that the carboxyl-substituted oxacalixarene 1 (Fig. 1) modulates the binding strength of different ammonium containing guest species compared to the noncyclic 2,6-dimethoxybenzoic acid 22 (Fig. 2). We suggest that this might arise from steric hindrance of the binding site in the macrocycle and that such modulated binding could be beneficial for applications that require controlled release of the active ingredient from the complex with the macrocycle.
 | ||
| Fig. 1 Structure of monocarboxylic oxacalix[4]arene 1 with the optimized structure of the 1·12 complex in methanol illustrating the 1,3-alternate conformation of the macrocycle, that is characteristic of 4-membered heterocalixarenes. | ||
 | ||
| Fig. 2 Organic guest species 2–15 and 16–21 and 2,6-dimethoxybenzoic acid 22 screened in this work. Most of the guest compounds were used as their chloride salts (exceptions are mentioned in Experimental section). | ||
Examples of artificial amine receptors based on calixarenes with a carboxyl moiety have been investigated by a number of research groups.16–18 For example, in the case of calix[4]arenes, it has been shown that the cavity is too small for encapsulation of organic guest molecules and that complexation can be achieved by designing a binding site external to the macrocycle's cavity.19 In addition, chiral macrocycles with aromatic monomer repeat units have been utilized in the detection of chiral amines, such as in enantioselective complexation with amino acid derivatives.20–23 Such “host–guest” binding could also find use in environmental monitoring24 or in cavitands for label-free enzyme assays.25 These studies illustrate the potential of macrocycles with aromatic monomer repeat units in amine sensing. Oxacalixarenes have re-emerged recently as a prospective class of macrocycles,26,27 however, their characterization is still incomplete and molecular recognition properties of this class of host molecules are not yet well understood. Due to their structural features, oxacalixarenes have the potential to combine the intrinsic binding motifs of calixarenes and crown ethers and could thereby expand the applicability of these types of macrocycles. The scarce examples of oxacalixarene complexation studies include complexation with cationic guest species.28,29 For the purpose of ammonium sensing, Gattuso and co-workers synthesized a polycationic oxacalix[4]arene in 2015, which showed affinity towards paraquat and neutral aromatic compounds in aqueous media.30,31 Previously, our group has investigated complexation properties of “naked” oxacalixarenes32 and shown in 2020 that 1 can bind paraquat in methanol and water.33 To the best of our knowledge, there are only a few examples where calixarene complexation with amine molecules has been investigated in polar protic solvents.34,35 The present study focuses on how to best make use of these characteristics for ammonium sensing with oxacalixarenes, and to our surprise we find that the macrocycle modulates binding strengths in comparison to the noncyclic species 22. Furthermore, we study the complex stoichiometry in the gas phase via ESI-MS and determine the complexation site and structure using ion mobility mass spectrometry (IM-MS) studies paired with DFT simulations. We note that the environment of complexes, which affects the intramolecular interactions in solution and gas phase, is different. We show that in solution, electrostatic and π–π interactions, with possible contributions from cation–π and hydrogen bonding interactions, influence binding strengths. On the contrary, the ESI-MS spectra and DFT calculations in the gas phase show that complexes are formed between the ammonium guest and neutral host via a hydrogen bond only. This does not prevent the possibility of the formation of similar complexes with a neutral charge like seen in solution, but these complexes would not be visible in ESI-MS gas phase spectra due to the absence of net charge in the formed species.
Results and discussion
Oxacalix[4]arene 1 contains aromatic rings, where one of the monomer repeat units is substituted with a carboxylic acid moiety, making it feasible for the macrocycle to participate in various intermolecular interactions, such as electrostatic, π–π, cation–π, anion–π, or hydrogen bonding. The effect of these interactions for complex formation with host 1 was studied based on binding with 20 different ammonium and bis-ammonium guest species in methanol-d4. For the NMR studies, the macrocycle was used in the form of a tetrabutylammonium (TBA) salt to enhance its solubility in methanol and reduce the competition between the counterion and guest for the binding site. The structures of guest molecules investigated in this work are depicted in Fig. 2. The association constants were determined by NMR titration experiments and the data were fitted using an open-source online fitting tool Bindfit36,37 by simultaneously following four host 1 proton signals (H2, H5, H6, H9 in Fig. 1). For the monoammonium guest species 2–7 and 11–13 (Table 1), the best fits were obtained by fitting to a 1:2 complexation model, judged by residuals of the fit. In such complexes one host binds two ammonium guest species. Binding data showed very low binding constant and negative cooperativity (α = 0.4)36 for the second guest binding. Titration experiments for the bisammonium guest species 16–21 showed more complex changes, and therefore, quantitative analysis of binding was not possible based on the collected data. The NMR spectra and links to Bindfit binding isotherms are given in the ESI.†
| Entry | Guest | Kaa (M−1) with 1 | Kaa (M−1) with 22 |
|---|---|---|---|
| a Given errors of the association constant values are obtained from Bindfit and are based on single experiment's curve fitting calculation errors. Further details of titration data are given in Experimental section and in ESI. Association constants are expressed as K11 and K12 for the first and the second guest binding to carboxylates, respectively, since the complexation mode best fit points to the 1:2 complex formation. |
|||
| 1 | 2 | K11 167.1 ± 2 | K11 250 ± 20 |
| K12 15.9 ± 0.1 | K12 17 ± 1 | ||
| 2 | 3 | K11 97.1 ± 0.9 | — |
| K12 5.67 ± 0.05 | |||
| 3 | 4 | K11 105 ± 1 | — |
| K12 7.2 ± 0.1 | |||
| 4 | 6 | K11 103 ± 2 | — |
| K12 5.6 ± 0.1 | |||
| 5 | 7 | K11 46 ± 2 | — |
| K12 3.3 ± 0.1 | |||
| 6 | 11 | K11 124 ± 1 | K11 500 ± 100 |
| K12 8.73 ± 0.08 | K12 6.0 ± 0.4 | ||
| 7 | 12 | K11 818 ± 30 | K11 2000 ± 200 |
| K12 104 ± 5 | K12 100 ± 7 | ||
| 8 | 13 | K11 536 ± 8 | — |
| K12 31.4 ± 0.5 | |||
| 9 | 14 | K11 113 ± 2 | — |
| K12 3.13 ± 0.08 | |||
In the series of NMR titration experiments, the monoammonium guest molecules with aliphatic substituents (entries 1–5 in Table 1) were firstly investigated. The results show comparable binding strengths with the K11 values varying from 46 up to 167 M−1 for all the compounds.
We notice that the binding strength is dependent on how sterically hindered the ammonium group is in the guest structure. Guest 2 with the smallest alkyl chain has the largest association strength, which decreases with the increasing bulkiness of the side chain. Incorporation of longer alkyl (guest 3) or bulkier cycloalkyl groups (guests 4 and 6) leads to a decrease in the guest's binding ability. The sterically most crowded guest species, i.e., the secondary ammonium ion 7 has the lowest binding affinity for the macrocycle, further supporting the notion that steric availability of the ammonium group in the guest species is important for the binding event. It is also evident that complexation can take place if the ammonium group is the only interacting functional group in the guest species. The possible interactions leading to binding can be the formation of a hydrogen bond involving the N–H proton or electrostatic interactions between oppositely charged functional groups of the host and the guest. For a comparison, we undertook experiments with the TBA salt of 2,6-dimethoxybenzoic acid 22 (Fig. 2), which closely resembles the monomer repeat units that the macrocycle 1 is made of but does not provide the similar cyclic architecture. The NMR titration experiment with guest species 2 gave a higher binding constant (right hand column in Table 1), compared to host 1, while the second guest binding to host 1 and compound 22 had a similar strength (K12). We suggest that the difference for the first binding (K11) might arise from the steric hindrance of the carboxylate moiety in the host 1 binding pocket, as the subsequent second ammonium guest can freely approach the second O-atom of the carboxylate group, though with weaker electrostatic forces.
The next group of investigated guest molecules included the aniline derivatives 8, 9 and 10. The NMR spectra acquired upon addition of these guest molecules revealed that after the addition of the first equivalent of the guests to the host solution, significant changes in the chemical shifts in the spectra of 1 took place. Based on the 1H NMR spectrum, we identified host 1 to be in its carboxylic acid form after the preceding addition of the aniline derivatives (see Fig. S6–S8 in ESI†). We therefore suggest that proton transfer between anilinium 8, p-nitroanilium 9, p-anisidinium 10 and host 1 has taken place. This indicates that the ammonium groups of the protonated aniline derivatives are stronger Brønsted acids than the neutral carboxylic acid site in host 1 and they donate a proton attached to the nitrogen to the carboxyl group of the host compound. The structure of the complex between 1 and 8, optimized in implicit methanol solvent, supports the proton transfer towards the carboxylate moiety and is shown in Fig. 4a. The first of these experiments was conducted with aniline 8 and, later, guest 9 with electron-withdrawing (EW) and guest 10 with electron-donating (ED) substituents attached to the benzene core were investigated to see whether these could suppress or enhance the proton transfer due to conjugation with the ammonium cation. It can be concluded that proton transfer takes place similarly in all three aniline derivatives. Based on these results, however, we observed that upon continuing the titration experiments after the host has been protonated, no chemical shift changes were seen between this “neutral host” and the aniline guest species. This would indicate that the presence of the carboxylate moiety in the host is prerequisite for complex formation. Complexation through π–π interactions or hydrogen bonding alone with the guest molecules does not occur unless the complexation is supported by the electrostatic interaction between the carboxylate of host 1 and the ammonium group in the guest molecules. This hypothesis could further be rationalized by conducting a control experiment with nitrobenzene 15 (Fig. S13 in ESI†) to see whether complexation is possible solely as a result of π–π interactions. We did not detect complexation for this guest molecule and therefore, π–π interactions alone are suggested to not lead to complex formation with 1.
Equipped with the information that while the interactions between the carboxylate of the host and the ammonium group of the guest can lead to complexation, but π–π interactions alone do not, we set to explore the role of the combination of covalent/electrostatic and π–π bonding for binding. The studied guest molecules 11–14 show that the binding strength increases significantly upon the incorporation of the aromatic groups in the substituents of the ammonium guest molecules. An unsubstituted phenyl ring increased the binding ability of guest 13 5-fold relative to its aliphatic counterpart 6. We hypothesize that the interaction between the carboxylate group of host 1 and the ammonium group of the guest molecules triggers the π–π interactions between the same host and the guest. This notion is further corroborated by inspection of the structures of ion pairs formed between 1 and guests 11–12, optimized in methanol, where T-shape π–π interactions are visible in the optimized structures (Fig. 4b). The data suggest that two types of interactions appear in the complex, where π–π interactions require the concurrent presence of the covalent/electrostatic interaction between the carboxylate and the ammonium cation.
While comparing the results for guest 13 with that of guest 11, one can notice that the association strength decreases upon decreasing the size of the alkyl chain connecting the ammonium and aromatic groups. This observation suggests that the degree of freedom in the relative orientation of the two interacting sites to form bonds between the guest and 1 has likely an important influence for the formation of stable complexes. Additionally, we studied the effect of the nitro substituent in the aromatic rings on the guest molecules in solution. Comparing the association strength of guest 11 to that of guest 12 revealed that the introduction of the EW nitro group to the aromatic ring in the guest increased the binding ability of the guest. This is likely indicative of the favorable π–π interaction between the electron-deficient benzene ring of guest 12 and the electron-rich aromatic system of host 1 in solution. For comparison, we conducted experiments with guest 11 and 12 and the TBA salt of 22 to see how much the macrocyclic structure influences binding strengths (right hand column in Table 1). The binding strength was increased for both guest molecules with the noncyclic molecule 22 for the 1:1 complex (K11), while it remained similar to host 1 for the second guest binding (K12). We suggest that steric hindrance of the carboxylate binding site in the macrocyclic structure could lead again to the decrease of the binding constant. This suggests that the conformation of 1 modulates binding strengths between the host and the guest. If under certain conditions, for example the aforementioned targeted delivery of drugs or reusing sensor materials, the oxacalixarene would need to release its cargo, the affected modulated binding strength could prove to be beneficial for this release.
The addition of chiral naphthalene derivative 14 resulted in some unusual behavior upon complexation as distinction of diastereotopic protons of host 1 in the NMR spectra became evident in these experiments (Fig. 3c). These spectra indicate that upon complexation of host 1 with the chiral guest 14, the symmetry of host 1 is reduced. Therefore, there might be possibility for host 1 to act as a chirality sensor for ammonium guest species.38 We also tried to crystallize the formed complexes, but despite our attempts, with different crystallization techniques, the crystals obtained were too small to be analyzed by single crystal X-ray diffraction.
 | ||
| Fig. 3 Stacked titration plots showing 1H-NMR spectra of host 1 with host to guest ratio of 1:1 for (a) guests 2–4, 6, 7 and (b) 11–14, 19. (c) Distinction of diastereotopic protons during the titration with guest 14. | ||
 | ||
| Fig. 4 Optimized solution-phase structures of (a) [1 + 8], (b) [1 + 11] and [1 + 12] in methanol. Calculated at the B97D/6-311+G(2d,p) level of theory. | ||
As the following experiments, we decided to study the macrocycle in gas phase with ESI-MS and IM techniques, to understand if any other charged complexes between 1 and the guest species will form. The gas-phase properties of host 1 were studied using ESI-QTOF mass spectrometry and initially host 1 was analyzed in both positive and negative polarities. In the positive mode, singly-charged ions for 1 and its dimer 12, were detected through adduct ion formation with Na+ and K+. However, in the negative mode, the expected deprotonated ions were not detected, and instead, the decarboxylated ion of 1 at m/z 367 was seen (Fig. S23 in ESI†). Albeit decarboxylation being a common reaction for any compound containing a carboxylic acid group in ESI-MS, the detection of the decarboxylated compounds exclusively is relatively rare. Based on these results, MS analysis of the complexes was conducted in the positive mode due to the lack of stability of the macrocycle in the negative mode.
It was anticipated that the affinity of 1 towards ammonium cations would be driven by ion pair formation between macrocycle's carboxylate moiety and ammonium cation, similarly to the solution state studies. It should however be noted that in mass spectrometry, the ion pair complex is invisible due to its charge neutrality. During this investigation, the ammonium guests 2, 3, 5–7, 10–12 were analyzed and the respective host–guest pairs were mainly detected as 1:1 singly-charged positive ions (see ESI for (+)ESI-MS spectra and Table S2†). Notably, the ion m/z values revealed that the macrocycle was in its carboxylic acid form when interacting with the ammonium cations in these complexes, highlighting that the likely host–guest interactions involve hydrogen bonding in the gas phase. This observation does not rule out the possibility of ion pair formation as another contributing factor of complexation in ESI-MS experiments because the potential species would simply remain undetected due to zero net charge.
The detected complexes were further studied with drift tube IM-MS (DT-IMMS). IM-MS is a versatile gas-phase method that has been used for evaluating host–guest complex conformation, structures and geometries.39–43 In IM-MS, the collision cross section (CCS) value for analyte ions is measured. Additionally, in the presence of two or more binding modes, their respective structures can be separated. Identification and visualization of the structures is possible when IM-MS method is paired with theoretical modeling and the atomic coordinates from the modelled structures are used for calculation of theoretical CCS values. The DT-IMMS results of complexes of 1 with guests 2, 3, 5–7, 10–12 revealed that the drift times of the complexes are longer than the drift time for free host [1 + Na]+ (Fig. 5). This suggests that the size of the complex is increased compared to the free host and that the guest is located outside the oxacalixarene cavity. Furthermore, as the size of the ammonium guest increased (Table S2†), the drift times (and CCS values) increased as well, which evidence that binding takes place through a complex where the guest occupies space outside the macrocycle's cavity. It is noted here that the complexes observed with ESI-MS experiments are complexes between the neutral host and a cationic guest species, while by NMR titration experiments the complexes formed between the host with a carboxylate anion and cationic ammonium guest species.
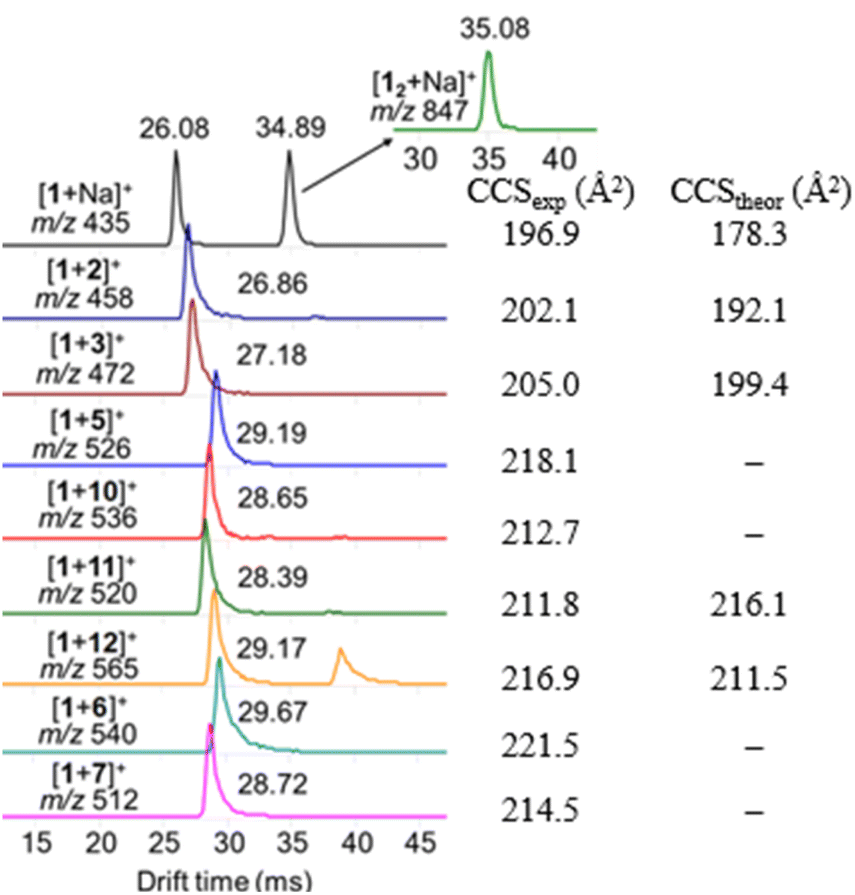 | ||
| Fig. 5 Arrival time distribution for (+)ESI-MS detected ions of [1 + guest]+ 1:1 complexes. Second peak occasionally observed ∼35–39 ms originates from dimer [12 + G]+ which fragmentates in drift tube to produce [1 + G]+. | ||
To rationalize the obtained experimental DTCCSN2 values, DFT calculations of [1 + 2]+, [1 + 3]+, [1 + 11]+ and [1 + 12]+ host–guest pairs were performed and the theoretical TMLJCCSN2 values were calculated based on the coordinates obtained from the optimized geometries of the hydrogen-bond-mediated complexes (Fig. 6). The theoretical simulations also suggest that binding occurs through an external binding site above the oxacalixarene cavity. The description of the theoretical simulations of the complexes in the gas phase as well as their counterparts obtained in methanol solution is brought in the ESI.† The calculated and theoretical CCS values of the four complexes are shown in Fig. 5 and Table S2.† The theoretical and experimental CCS values are in fair agreement, which demonstrates the similarity between the geometries of the measured gas-phase complexes and the calculated hydrogen-bonded structures. Therefore, complexation, albeit through different sets of intermolecular interactions, takes place both in solution and in gas phase.
 | ||
| Fig. 6 Optimized gas-phase structures of (a) [1 + 2]+, (b) [1 + 3]+, (c) [1 + 11]+ and (d) [1 + 12]+ host–guest complexes. Calculated at the B97D/6-311+G(2d,p) level of theory. | ||
Experimental section
NMR titration
The Ka values were determined in methanol-d4. All the spectra for 1H NMR titration experiment were collected on a Bruker AVANCE III 800 MHz spectrometer. Host 1 and compound 22 were used in its TBA salt forms and stock solutions (2–10 mM) were added to a vial containing a guest to keep the concentration of host 1 fixed throughout the titration experiment. All guest compounds were analyzed in their chloride salt form. All solutions were prepared using Hamilton® Gastight syringes and small aliquots of guest compounds were added to the solution using the same syringes. Compounds were weighed on a Sartorius microbalance (accuracy 15 μg), allowing preparation of stock solutions concentrations within 1% error. Increasing amounts of the guest stock solution were added to the NMR tube containing 600 μL of the host 1 stock solution. The concentrations of both the host and the guest stock solutions were based on the assumed association constants and all the details of total guest concentration are given in the ESI† below the titration spectra. All data points were measured at quantitative conditions and depending on the concentration of the host, data were collected using 8 or 14 scans with relaxation delay set to 10 s or 17 s, acquisition time set to 2.4 s and pulse width set to 3 μs (30° pulse). The chemical shifts were referenced based on methanol-d4 residual peak at 3.31 ppm. The Ka values were determined using Bindfit36,37 analysis tool (available at https://supramolecular.org) that is based on nonlinear regression analysis.Mass spectrometry
All the gas-phase studies were performed on an Agilent 6560 Ion Mobility Time-of-Flight mass spectrometer, equipped with a dual ESI ion source (Agilent Technologies, USA). The mass range for the ESI-MS and IM-MS experiments was set to m/z 300–1000.The 1 mM stock solution of host 1 and 5 mM stock solutions of ammonium guests 2, 3, 10, 12 as chloride salts were dissolved in methanol. Guests 5–7 and 11 were used in their amine form and dissolved in either methanol or water. Sample solution of host 1 was prepared in acetonitrile and diluted to the concentration of 10 μM. For the MS analysis of complexes, 5 μM sample of 1 was prepared in acetonitrile and an equimolar amount of guest was added to the sample. The sample flow rate was set to 2 μL min−1.
In ESI, N2 was used as the drying and nebulization gas. The source conditions for the IM-MS experiments were kept consistent throughout the screening of amines, with the following parameters: capillary voltage 4.5 kV, dry gas temperature 225 °C, drying gas flow 7 L min−1, nebulizer pressure 4 psi, fragmentor voltage 400 V, and Oct 1 RF Vpp 750 V.
The IM-MS spectra were measured with high purity N2 as the drift gas. The drift tube pressure was set to 3.95 torr and in the case of single-field IM-MS experiments the drift tube entrance and exit voltages were adjusted to 1700 V and 250 V, respectively. Trap filling time of 5000 μs and trap release time of 350 μs were used. The stepped-field IM-MS experiments for determination of DTCCSN2 were obtained by varying the drift tube entrance voltages from 1074 to 1674 V in 100 V steps. Reproducibility of the CCS values was checked using the ES tuning mix standard solution (Agilent Technologies) as a quality control.44,45 The data analysis was performed using Agilent MassHunter Qualitative Navigator (B.07.00, Agilent Technologies, USA) and MassHunter IM-MS Browser (B.08.00, Agilent Technologies, USA).
Theoretical CCS values were calculated with the ion mobility calculator IMoSSuite version 1.06.46,47 The trajectory method with Lennard-Jones potentials was used for calculation of theoretical TMLJCCSN2 values which appear to be in reasonable agreement with the measured DTCCSN2 values. The calculation was based on IM-MS experimental parameters that include gas, temperature and pressure. The number of rotations used was set to 3, applying 300000 gas molecules per rotation. Atomic coordinates of host 1 for the theoretical TMLJCCSN2 calculations were obtained from the already published crystal structure of the macrocycle.33 The respective coordinates of the ammonium cation complexes were obtained from the gas-phase DFT optimizations at the B97D/6-311+G(2d,p) level of theory. Further computational details are presented in the DFT calculations sections in ESI.†
Conclusions
The key structural aspects that contribute to binding between host 1 and ammonium guest molecules are presented and analysed in solution and in the gas phase. The results show that different ammonium guest structures bind to host 1 salt with various binding strengths in solution. We determine three important contributing factors that affect complexation. Firstly, the presence of a carboxylate moiety in the host is suggested to be prerequisite for complex formation even for guest molecules with aromatic substituents. Secondly, π–π interactions can support binding and increase binding strengths and thirdly, we hypothesize that the accessibility of the degrees of freedom for favourable mutual orientation of the host and the guest that participate in electrostatic and π–π interactions is important for stable complex formation. Furthermore, the binding is modulated and weaker compared to the noncyclic molecule 2,6-dimethoxybenzoic acid 22, which could have its advantages in the release of the desired guest molecules from the macrocycle for biomedical uses. We further show that a chiral guest molecule can change the symmetry of the oxacalixarene host macrocycle. The studies in the gas-phase and DFT calculations demonstrate that binding in the gas phase can occur solely through hydrogen bonding through the formation of a complex with a binding site above the oxacalixarene cavity. These results allowed to establish the geometries for the complexes in the gas phase by DFT methods.Author contributions
AC performed the ESI-MS and ion mobility experiments and preliminary synthesis, M-LB performed the synthesis of the macrocycle, NŽ and KS performed the NMR titration experiments, MU and AT performed the DFT calculations, HR helped with additional analysis for the revised manuscript, RA supervised the synthesis of the macrocycle and fitted the titration data, EK supervised the ESI-MS and ion mobility experiments, JA conceived the project, coordinated the project and supervised the synthesis and NMR experiments. NŽ and JA wrote the paper with contribution from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support by the Ministry of Education and Research, Republic of Estonia (grants PSG400, PRG661 and PRG399) and European Regional Development Fund (project TK134 “EQUITANT” and Center of Excellence in Molecular Cell Engineering TK143), and instrumentation of University of Jyväskylä.Notes and references
- B. Gorain, P. Sengupta, S. Dutta, M. Pandey and H. Choudhury, in Frontiers in Pharmacology of Neurotransmitters, Springer, 2020, pp. 213–240 Search PubMed.
- G. Rudnick, in Neurotransmitter Transporters: Structure, Function, and Regulation, ed. M. E. A. Reith, Springer, New York, 2nd edn, 2002, p. 31 Search PubMed.
- M. Lieberman and A. Peet, Marks' Essentials of Medical Biochemistry: A Clinical Approach, Wolters Kluwer, Philadelphia, 2nd edn, 2015 Search PubMed.
- L. Brichta, P. Greengard and M. Flajolet, Trends Neurosci., 2013, 36, 543–554 CrossRef CAS PubMed.
- S. A. Factor, W. M. McDonald and F. C. Goldstein, Eur. J. Neurol., 2017, 24, 1244–1254 CrossRef CAS PubMed.
- P. T. Francis, CNS Spectr., 2005, 10, 6–9 CrossRef PubMed.
- S. Moerkerke, V. Malytskyi, L. Marcélis, J. Wouters and I. Jabin, Org. Biomol. Chem., 2017, 15, 8967–8974 RSC.
- D. Zhang, E. N. Atochina-Vasserman, D. S. Maurya, N. Huang, Q. Xiao, N. Ona, M. Liu, H. Shahnawaz, H. Ni, K. Kim, M. M. Billingsley, D. J. Pochan, M. J. Mitchell, D. Weissman and V. Percec, J. Am. Chem. Soc., 2021, 143, 12315–12327 CrossRef CAS PubMed.
- D. Zhang, E. N. Atochina-Vasserman, D. S. Maurya, M. Liu, Q. Xiao, J. Lu, G. Lauri, N. Ona, E. K. Reagan, H. Ni, D. Weissman and V. Percec, J. Am. Chem. Soc., 2021, 143, 17975–17982 CrossRef CAS PubMed.
- D. Zhang, E. N. Atochina-Vasserman, J. Lu, D. S. Maurya, Q. Xiao, M. Liu, J. Adamson, N. Ona, E. K. Reagan, H. Ni, D. Weissman and V. Percec, J. Am. Chem. Soc., 2022, 144, 4746–4753 CrossRef CAS PubMed.
- P. T. Altenbuchner, P. D. L. Werz, P. Schöppner, F. Adams, A. Kronast, C. Schwarzenböck, A. Pöthig, C. Jandl, M. Haslbeck and B. Rieger, Chem.–Eur. J., 2016, 22, 14576–14584 CrossRef CAS PubMed.
- S. Tang, F. Zhang, H. Gong, F. Wei, J. Zhuang, E. Karshalev, B. Esteban-Fernández de Ávila, C. Huang, Z. Zhou and Z. Li, Sci. Robot., 2020, 5, eaba6137 CrossRef PubMed.
- Y. Zheng, L. Tang, L. Mabardi, S. Kumari and D. J. Irvine, ACS Nano, 2017, 11, 3089–3100 CrossRef CAS PubMed.
- S. Zhang, R. Moussodia, H. Sun, P. Leowanawat, A. Muncan, C. D. Nusbaum, K. M. Chelling, P. A. Heiney, M. L. Klein and S. André, Angew. Chem., Int. Ed., 2014, 126, 11079–11083 CrossRef.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim and F. S. Bates, Science, 2010, 328, 1009–1014 CrossRef CAS PubMed.
- J.-A. Richard, M. Pamart, N. Hucher and I. Jabin, Tetrahedron Lett., 2008, 49, 3848–3852 CrossRef CAS.
- C. Capici, G. Gattuso, A. Notti, M. F. Parisi, S. Pappalardo, G. Brancatelli and S. Geremia, J. Org. Chem., 2012, 77, 9668–9675 CrossRef CAS PubMed.
- A. Inthasot, M.-D. D. Thy, M. Lejeune, L. Fusaro, O. Reinaud, M. Luhmer, B. Colasson and I. Jabin, J. Org. Chem., 2014, 79, 1913–1919 CrossRef CAS PubMed.
- T. Pierro, C. Gaeta, F. Troisi and P. Neri, Tetrahedron Lett., 2009, 50, 350–353 CrossRef CAS.
- K. Jennings and D. Diamond, Analyst, 2001, 126, 1063–1067 RSC.
- A. Lledó, R. J. Hooley and J. Rebek Jr, Org. Lett., 2008, 10, 3669–3671 CrossRef PubMed.
- B. Setner and S. Agnieszka, Beilstein J. Org. Chem., 2019, 15, 1913–1924 CrossRef CAS PubMed.
- T. Panahi, H. L. Anderson, K. I. Castro, J. D. Lamb and R. G. Harrison, Supramol. Chem., 2020, 32, 71–80 CrossRef CAS.
- M. Chiesa, F. Rigoni, M. Paderno, P. Borghetti, G. Gagliotti, M. Bertoni, A. B. Denti, L. Schiavina, A. Goldoni and L. Sangaletti, J. Environ. Monit., 2012, 14, 1565–1575 RSC.
- D.-S. Guo, V. D. Uzunova, X. Su, Y. Liu and W. M. Nau, Chem. Sci., 2011, 2, 1722–1734 RSC.
- M.-X. Wang, Chem. Commun., 2008, 4541–4551 RSC.
- M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- D. Sobransingh, M. B. Dewal, J. Hiller, M. D. Smith and L. S. Shimizu, New J. Chem., 2008, 32, 24–27 RSC.
- M. Panchal, M. Athar, P. C. Jha, A. Kongor, V. Mehta and V. Jain, J. Lumin., 2017, 192, 256–262 CrossRef CAS.
- N. Manganaro, G. Lando, C. Gargiulli, I. Pisagatti, A. Notti, S. Pappalardo, M. F. Parisi and G. Gattuso, Chem. Commun., 2015, 51, 12657–12660 RSC.
- N. Manganaro, G. Lando, I. Pisagatti, A. Notti, S. Pappalardo, M. F. Parisi and G. Gattuso, Supramol. Chem., 2016, 28, 493–498 CrossRef CAS.
- A. Peterson, S. Kaabel, I. Kahn, T. Pehk, R. Aav and J. Adamson, ChemistrySelect, 2018, 3, 9091–9095 CrossRef CAS.
- A. Peterson, M.-L. Ludvig, J. Martõnova, S. Kaabel, P. Kerner, M. Uudsemaa, A. Trummal, M. Fomitšenko, T. Pehk and R. Aav, Supramol. Chem., 2020, 32, 313–319 CrossRef CAS.
- T. Oshima, K. Oishi, K. Ohto and K. Inoue, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 79–85 CrossRef CAS.
- N. K. Beyeh, F. Pan, A. Valkonen and K. Rissanen, CrystEngComm, 2015, 17, 1182–1188 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- L. Ustrnul, S. Kaabel, T. Burankova, J. Martõnova, J. Adamson, N. Konrad, P. Burk, V. Borovkov and R. Aav, Chem. Commun., 2019, 55, 14434–14437 RSC.
- D. V Dearden, T. A. Ferrell, M. C. Asplund, L. W. Zilch, R. R. Julian and M. F. Jarrold, J. Phys. Chem. A, 2009, 113, 989–997 CrossRef PubMed.
- M. Öeren, E. Shmatova, T. Tamm and R. Aav, Phys. Chem. Chem. Phys., 2014, 16, 19198–19205 RSC.
- A. Kiesilä, L. Kivijärvi, N. K. Beyeh, J. O. Moilanen, M. Groessl, T. Rothe, S. Götz, F. Topić, K. Rissanen and A. Lützen, Angew. Chem., Int. Ed., 2017, 56, 10942–10946 CrossRef PubMed.
- A. Kiesilä, J. O. Moilanen, A. Kruve, C. A. Schalley, P. Barran and E. Kalenius, Beilstein J. Org. Chem., 2019, 15, 2486–2492 CrossRef PubMed.
- E. Kalenius, M. Groessl and K. Rissanen, Nat. Rev. Chem., 2019, 3, 4–14 CrossRef CAS.
- S. M. Stow, T. J. Causon, X. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith and J. A. McLean, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS PubMed.
- V. Gabelica, A. A. Shvartsburg, C. Afonso, P. Barran, J. L. P. Benesch, C. Bleiholder, M. T. Bowers, A. Bilbao, M. F. Bush and J. L. Campbell, Mass Spectrom. Rev., 2019, 38, 291–320 CrossRef CAS PubMed.
- C. Larriba and C. J. Hogan Jr, J. Comput. Phys., 2013, 251, 344–363 CrossRef CAS.
- C. Larriba and C. J. Hogan Jr, J. Phys. Chem. A, 2013, 117, 3887–3901 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR titration data, ESI-MS data, DFT calculations details. See DOI: https://doi.org/10.1039/d2ra07614d |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |