
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards quantum corrosion chemistry: screening perfect Cr, Ni sites and stoichiometry on top of an Fe(110) surface using DFT
Zhihao Yanga,
Chi Zhangb,
Shuo Wanga,
Chengpeng Xuea,
Guangyuan Tiana,
Hui Sua,
Chengming Yana,
Zhifei Yanc,
Xiaoguang Liud and
Junsheng Wang *ae
*ae
aSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: junsheng.wang@bit.edu.cn; Tel: +86 10 68915043
bSchool of Materials Science and Engineering, Shenyang University of Technology, Shenyang 110870, China
cChina North Industries Group, No. 52 Research Institute, Baotou, Inner Mongolia 014030, China
dSchool of Materials Science and Engineering, University of Science & Technology Beijing, No. 30 Xueyuan Road, Beijing, 100083, China
eAdvanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
First published on 29th March 2023
Abstract
For decades, corrosion has been classified into many categories according to the microstructural morphology of the chemical reaction products. Until recently, the development of quantum chemistry has simplified the fundamental corrosion mechanism into only two processes: electrochemical dissolution and hydrogen evolution reaction (HER). Although Cr and Ni elements have been found to segregate towards the surface of stainless steel to form a protective layer and prevent Fe dissolution, the understanding of the exact chemistry on top of the Fe surface has not been reported in previous studies. In this study, we have identified suitable doping sites for simultaneous doping of several Cr and Ni atoms, and quantified the effects of different alloy compositions (Fe12Cr3Ni1, Fe11Cr4Ni1, Fe11Cr3Ni2, Fe10Cr4Ni2, Fe10Cr3Ni3) on the stability from two aspects: electron transfer and atomic dissolution. It was found that the doping atoms are more likely to be dispersed rather than aggregated in solid solution. When Cr atoms are symmetrically distributed and Ni atoms are located in the center, it is the site arrangement with the highest work function and stability. Fe10Cr4Ni2 has been found to possess a higher electron binding capacity and thus higher electrode potentials. This is determined by the change of dipole caused by both electronegativity difference between atoms and polarization between the doped layer and the substrate layer. By calculating the vacancy formation energy, it is shown that Fe11Cr4Ni2 is the perfect chemistry on top of the Fe(110) surface due to its high ability of preventing atomic dissolution.
1. Introduction
Iron-based alloys are widely used in modern industrial fields due to their abundant resources, low cost, good comprehensive performance, and wide range of performance.1,2 However, on a global scale, corrosion has always been a key issue that causes technical and economic losses to industries, and may even bring risks and hazards to personal safety.3,4 Therefore, the efforts on improving corrosion resistance have been a hot topic for the development of new iron-based alloys. By adding different kinds and proportions of alloying elements to iron, many new stainless steels have been created, which greatly improve the corrosion resistance of iron alloys. Ferrite is an α-Fe-based high alloyed steel with a Cr content of 15% to 30%. Good process ability and mechanical properties, excellent oxidation and corrosion resistance make ferrite widely used in proton exchange membrane fuel cells, automotive emission systems and many other fields.5,6 The corrosion resistance of stainless steel is mainly attributable to the passive film, which protects the material from continuous corrosive attack by the external environment. However, the passivated surface usually has local defects, and active ions and ligands are easily adsorbed at these sites to cause local damage of the passivation film. Or under certain conditions, the passivation film may also be damaged or even removed by the impact of solid particles.7 This leads to local metal exposure to corrosive media, resulting in localized corrosion, such as pitting corrosion, galvanic corrosion, etc., which has a serious negative impact on the properties of the alloy.8,9 Methods to resist this corrosion mainly include alloying element optimization and surface modification.7 It is therefore necessary to understand the effect of alloying elements on the corrosion of clean surfaces without natural passive films to minimize the corrosion hazards of stainless steel.10Researchers believe that Fe is easy to react with oxygen in humid environment and then corrosion occurs. The reaction formula is 4Fe + 3O2 + 6H2O = 4Fe(OH)3. Even at the high temperature of 600 °C, the presence of water vapor will still accelerate the corrosion.11 For Fe used in seawater equipment, such as ships and naval vessels, since seawater is an electrolyte solution, it will undergo electrochemical corrosion, and the corrosion mechanism can be explained by the depolarization process of oxygen.12 The rate of corrosion is directly related to the rate of oxygen diffusion in seawater. In addition, there are plenty of chloride ions in seawater, which will crack the oxide film on the metal surface, leading to stress corrosion cracking and pitting corrosion, and eventually the material properties degrade.12,13 By experimenting on different alloying additions, many studies have improved significantly the corrosion resistance of ferroalloys, inventing various types of stainless steels with good corrosion resistance. Wang et al.11 found that for Fe–Cr alloys in air, the oxidation rate decreases with the increase of Cr content. However, if the Cr content is increased to 15 wt% or higher, the mass of the alloy changes little due to the formation of protective Cr2O3 oxide film. Dong et al.14 found that by adding 0.3 wt% Cu and 0.3 wt% Cr to the steel bar, the corrosion resistance of the steel bar has been increased by 46.3% compared with adding 0.2 wt% Ni and 0.15 wt% Mo, and Cr is one of the main elements to improve the protection of the corrosion layer of the steel bar. Tian et al.15 believed that the surface oxide film of nickel-containing steel had a strong inhibition effect on corrosion attack. It can be found that the corrosion of ferroalloys by experimental methods is mostly concentrated on the passivation film and the corrosion layer, but it is difficult to study the corrosion resistance of the matrix itself.
Integrated Computational Materials Engineering (ICME) is a modern systems-based approach to designing materials that meet specific performance criteria by combining computational material models across multiple length scales.16,17 Many properties of alloys can be readily predicted based on tools such as density functional theory (DFT).18,19 It is generally believed that doping of alloying elements can change the atomic structure and electronic state of materials, thereby altering the physical and chemical properties of materials. The first-principles calculations based on DFT can analyze the corrosion mechanism from the perspective of the atomic scale, and obtain accurate results, which are difficult to achieve by traditional experimental techniques.20,21 The first-principles calculation of work function reflect the electrode potential on the surface of the material, which can be used to evaluate the corrosion behavior of the materials.22,23 Li et al.24 obtained the intrinsic potential difference between each phase by calculating the work function of α-Fe (110) plane, martensite (110) and (Fe,Cr)23C6 (111), and then evaluated the effect of annealing treatment on the corrosion resistance of X2CrNi12 steel by comparing the potential difference of each phase. Yin et al.13 calculated the surface energy and work function of the Fe(110), Cr(110) and Fe(110) surface doped with Cr to study the surface properties of the alloy, and found that the surface energy increases while the work function does not change much after doping with one or two Cr. However, the work function decreased significantly after adding the solvation model. Many works also often use first-principles calculations to study the adsorption, dissociation and other interactions between small molecules and metal surfaces, and then to analyze the corrosion mechanism of metals. Hu et al.25 studied the adsorption behavior of typical adsorbates H2O, H+, OH−, Cl− before and after doping Cr on Fe(110) surface based on DFT, and found that the Cr-doped Fe(110) surface is more stable than Fe(110) and Cr(110) surfaces. Cr doping greatly improves the electron-donating ability of adjacent Fe atoms, which in turn promotes the adsorption of positively charged H+.
However, most of the above studies focus on the effect of single atom or atoms of the same species on the surface alloying of Fe, and few studies have been done on the effects of multiple atoms of different species doping simultaneous. Actually, the properties of stainless steel are often related to the interaction and common influence of various elements. In the work reported in this paper, we focus on the corrosion-related properties of Fe–Cr–Ni base stainless steel systems under different alloy compositions. The formation enthalpy, work function, and vacancy formation energy were calculated by first-principles calculations. We identified suitable doping sites for Cr and Ni and discussed the effects of doping concentration on Fe surface stability. This computational approach can be successfully used to explain experimental phenomena, predict properties and even design materials.10 We hope that this study will help to reveal the mechanism of the effect of alloying elements on the corrosion of stainless steel at the atomic scale and provide a method that enables the rapid and large-scale search for new corrosion-resistant alloy systems, which is difficult for experimental research to achieve due to high cost and long period of time.
2. Computation methods
2.1 Models
All the simulation calculations in this study are conducted using spin-polarized density functional theory (DFT) as implemented in the Vienna ab initio simulation package (VASP).26,27 The projector augmented wave (PAW) method is used to characterize the interactions of core and valence electrons.28 The exchange–correlation effects are described using the Perdew–Burke–Ernzerhof (PBE) functions within the generalized gradient approximation (GGA).29 In the process of structure optimization for bulk metallic Fe of bcc cell, the plane wave kinetic energy cutoff is 450 eV, and the Γ-centered k-point mesh is 17 × 17 × 17. The optimized lattice constant of Fe is 2.83 Å, which is in good agreement with the experimental value (a1 = 2.86 Å)30 as well as the previous DFT calculated values (a2 = 2.841 Å and a3 = 2.834 Å).13,31Studies have shown that the (110) surface is the most stable surface and have the lowest energy among several low-index surfaces of bcc metals.32,33 Therefore, in this study, Cr and Ni are doped on Fe(110) surface. The model contains 80 atoms, arranged in 5 atomic layers, with 16 atoms in each layer, as shown in Fig. 1. A 10 Å vacuum layer is set in the c direction to ensure that the vacuum layer is large enough to calculate the work function and other related data. The electrochemical stability and the corrosion behavior of metals are very sensitive to the surface properties, and anodic dissolution always happens at surfaces and charge transfer also takes place at the surface or interface.34 Therefore, top surface layer doping is a reasonable and time-saving approach for studying the alloying effect on surface properties.13 As a result, this study used the atomic replacement method to dope Cr and Ni with different sites and contents only in the first layer. During the optimization process, the two bottom layers are fixed at their bulk coordinates, and the three top Fe layers are relaxed, so that the model has a surface-block-like structure.35 The Brillouin zone uses a 5 × 5 × 1 k-point mesh for the Fe(110) surface.
 | ||
| Fig. 1 The initial structure of Fe(110): (a) calculated slab model of Fe(110); (b) top view of the atomic structure of Fe(110). | ||
2.2 Formation enthalpy
Formation enthalpy (ΔH) is an important physical parameter that can be used to assess the stability of a molecule and estimate the energy released in a reaction.36 Formation enthalpy can be defined as the thermal effect of the most stable element in the standard state to generate the amount of a pure substance in the standard state at a certain temperature. Since the effect of pressure on the condensed phase is ignored in the calculation process, the energy calculated at 0 K does not have any entropy contribution, so the formation energy is exactly the formation enthalpy.37The formation enthalpy of each atom in the Fe bulk after doping with Cr and Ni can be expressed by the following formula:38,39
 | (1) |
2.3 Work function
Work function (Φ) is the minimum energy required for electrons to transfer from the Fermi level to the vacuum level on the metal surface, reflecting the ability of electrons to escape from the surface of the material and become free electrons.23,40 The work function is usually related to the distribution of electrons on the surface of the materials. The distribution of electrons on the surface can be changed by alloying, which in turn affects the physical and chemical properties such as bond strength and atomic stability.41 The work function can be calculated by the following formula:13,41| Φ = −eφ − EF | (2) |
2.4 Vacancy formation energy
Vacancy formation energy (Evac) refers to the energy required for atoms to break free from a material. The dissolution of atoms on the metal surface reflects the corrosion behavior of the metal to a certain extent. Therefore, we calculate the energy required for the dissolution of iron atoms to form vacancies by establishing a model for removing certain Fe atoms from the doped Fe surface. The vacancy formation energy can be calculated by the following formula:42,43| Evac = Eslab+vac − Eslab + Eatom | (3) |
3. Results and discussions
3.1 Selection of Cr and Ni doping sites on Fe surface
For most stainless steels, Cr and Ni elements account for the highest proportions besides Fe. Cr is the key alloying element to improve the corrosion resistance of stainless steel, due to its ability to inhibit water adsorption and increase surface electrode potential,10 while Ni has a higher corrosion potential than Cr, and can also promote the phase balance, element partitioning and corrosion resistance in acids.40 Therefore, this work attempts to study the corrosion behavior of the Fe–Cr–Ni basic system without considering the influence of Mn, Mo, Cu, Si, C and other trace elements or impurity elements.To determine the content of Cr and Ni doped in Fe, we have analyzed the compositions of the common 316L stainless steel and the QN1906 and QN2109 stainless steels of Qingshan Group with good corrosion resistance. The atomic fractions of the three grades after simplification are Fe11.64Cr2.84Ni1.51, Fe11.6–12.2Cr3.1–3.4Ni0.7–1.1, Fe10.8–11.4Cr3.4–3.7Ni1.2–1.5. Five different stainless steel compositions can be approximated after integration, namely Fe12Cr3Ni1, Fe11Cr4Ni1, Fe11Cr3Ni2, Fe10Cr4Ni2, Fe10Cr3Ni3. Therefore, in the first layer of Fe (110) surface, 16 atoms were replaced according to the above five compositions. However, since there are many arrangement options for the replacement sites in the 16 atoms, we first need to determine which arrangement Cr and Ni are more likely to be solvable. Therefore, the main content of this section is to find the most suitable doping sites for Cr and Ni on the surface of Fe (110) by calculating the formation enthalpy and work function.
We modeled various site schemes for the chemical compositions of Fe12Cr3Ni1 and Fe11Cr4Ni1 first and then calculated their formation enthalpies which are shown in Fig. 2 and 3. For the sake of clarity, only the top view of the arrangement of atoms in the first layer is shown. The model can be roughly divided into symmetric distribution of dopant atoms (A1, A2, B1, B2), distribution of dopant atoms with the farthest distance (A3, B3, B4), aggregated distribution of dopant atoms (A4, B5, B6), linear distribution of dopant atoms (A5).
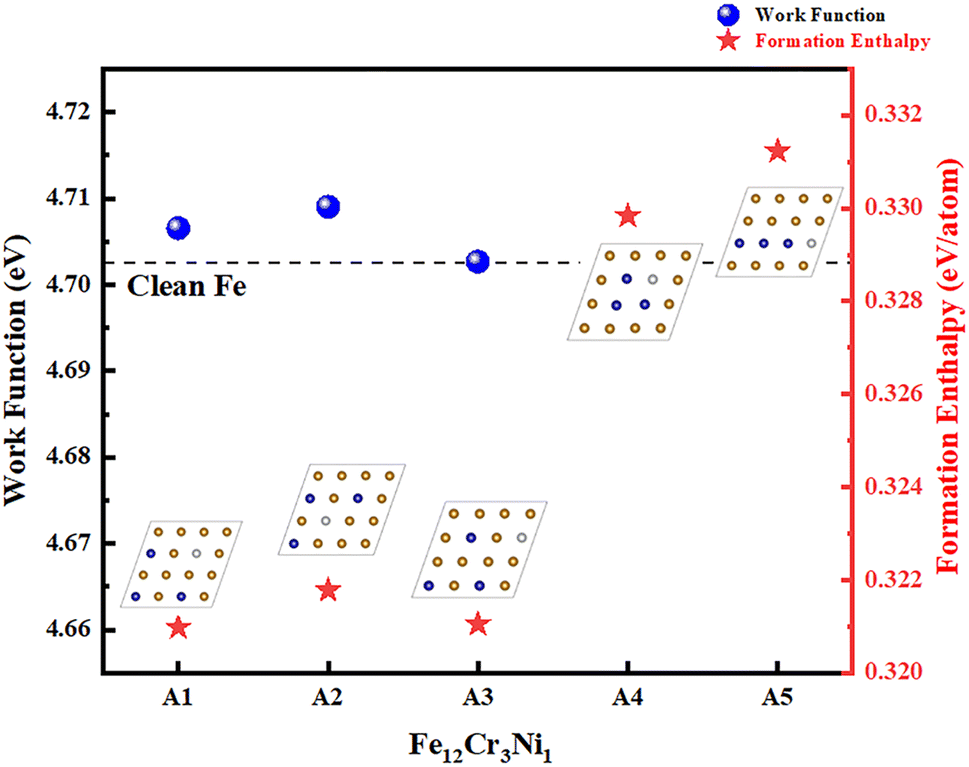 | ||
| Fig. 2 The work functions and the formation enthalpies of Fe(110) surface doped with three Cr atoms and one Ni atom. The illustrations show the corresponding atomic structures of the first layer of Fe(110) surface. The yellow atoms, blue atoms and white atoms represent Fe, Cr and Ni, respectively. The dashed line indicates the work function of the clean Fe surface. | ||
 | ||
| Fig. 3 The work functions and the formation enthalpies of Fe(110) surface doped with four Cr atoms and one Ni atom. The illustrations show the corresponding atomic structures of the first layer of Fe(110) surface. | ||
The level of formation enthalpy directly reflects the difficulty of solid solution formation. As shown in Fig. 2 and 3, we found that the formation enthalpies when the dopant atoms are aggregated or in a straight line (A4, A5, B5, B6) are generally 0.01–0.02 eV per atom higher than the formation enthalpies when the dopant atoms are dispersed (A1, A2, A3, B1, B2, B3, B4). This shows that the Fe(110) surface where the dopant atoms are aggregated is unstable, and Cr and Ni tend to be dissolved in a dispersed form. This is because the electronegativity of Cr is lower than that of Fe and Ni, so Cr doping on the Fe surface is more inclined to release electrons. When multiple Cr atoms aggregate, the electrons released by Cr will be unevenly distributed on the Fe surface, which will change the surface electron density distribution and improve the surface activity.
To further determine the substitution sites of the dopant atoms, we calculated the work functions of Fe12Cr3Ni1 and Fe11Cr4Ni1 when the atoms are dispersed (A1, A2, A3, B1, B2, B3, B4), as shown in Fig. 2 and 3. The work function can well reflect the overall ability of the surface to bind electrons. The surface with a lower work function has a weaker ability to bind electrons, and the electrons are usually more easily transferred, which in turn affects the occurrence of surface chemical reactions.41,44 The calculated work function value of clean Fe(110) surface is 4.702 eV, which is consistent with 4.74 eV and 4.79 eV in the previous studies.13,33 We found that A2 and B1 were the two structures with the highest work functions, both of which have a symmetrical distribution of Cr as Ni atoms are at the center of Cr atoms, indicating that the ferroalloys with this arrangement is less prone to lose electrons.
Furthermore, to determine the site arrangement when Ni is increased by one more than before, we continued to calculate the formation enthalpy and work function of several different models of Fe10Cr4Ni2. According to the above findings, four Cr atoms were fixed on the evenly distributed sites, and Ni atoms take a random distribution, as shown in Fig. 4. It can be found that the formation enthalpy is in good agreement with the work function, and the formation enthalpy of the structure with higher work function is correspondingly lower. This is because they are all affected by the electron density distribution on the solid surface,45 and a surface with a higher work function will have a higher electron binding capacity and a lower surface activity, resulting in a lower formation enthalpy and a more stable structure. It can be seen that the work functions of structures C3 and C4 are the highest. In fact, the Ni atoms after expanding these two structural units are exactly located at the central site of the Cr atoms, which is consistent with the previous conclusions on Fe12Cr3Ni1 and Fe11Cr4Ni1. This further demonstrates that Cr atoms are symmetrically distributed and Ni atoms are located in the center, which is the most stable site arrangement with the highest work function. In fact, the electronegativity of solid solution atoms can affect the number and direction of surface electron transfer, resulting in changes in the distribution of surface electrons, which in turn alters the work function.34 When the electronegativity of the dopant atoms is higher than that of the base atoms, the adjacent base atoms will transfer electrons to the dopant atoms and make the dopant atoms negatively charged, which will lead to a negative electric dipole moment on the metal surface, thereby increasing the work function.41,46 The relative electronegativities of Cr, Fe and Ni are 1.66, 1.83 and 1.91, respectively. It can be found that the difference in electronegativity between Cr and Ni is larger than that of Cr and Fe. When Ni is located in the center, the surrounding atoms, especially Cr, will transfer a large number of electrons to Ni, which thus produces a relatively negative electric dipole moment, resulting in a larger work function. It is worth mentioning that when we calculated the Fe10Cr4Ni2 composition, we also tried the structure of doped atoms with aggregated distribution, and found that it is difficult to relax, indicating that this is an unstable structure. This is consistent with the conclusions drawn above in terms of Fe12Cr3Ni1 and Fe11Cr4Ni1 compositions.
 | ||
| Fig. 4 The work functions and the formation enthalpies of Fe(110) surface doped with four Cr atoms and two Ni atoms. The illustrations show the corresponding atomic structures of the first layer of Fe(110) surface. | ||
3.2 Stability of the Fe surface with different doping compositions
The addition of alloying elements can significantly change the physical and chemical properties of the Fe surface. Different alloying elements would result in the changes of atomic structure and electron distributions on the surface, which would in turn vary the thermodynamic stability of the Fe surface. This effect on the stability changes the energy required for the electron transfer of the Fe surface and the separation of atoms from the Fe surface, both of which are closely related to the anodic dissolution behavior of Fe alloys, reflecting the corrosion resistance of ferroalloys to some extent.In Section 3.2, we have found the most suitable doping sites for Cr and Ni, and calculated the highest work function under the three chemical compositions including Fe12Cr3Ni1, Fe11Cr4Ni1 and Fe10Cr4Ni2. In this section, the work functions of Fe11Cr3Ni2 and Fe10Cr3Ni3 are presented, as shown in Fig. 5. The illustrations show the possible doping types with Fe11Cr3Ni2 composition and Fe10Cr3Ni3 composition in which Cr atoms are symmetrically distributed and Ni atoms are located in the center. For Fe11Cr3Ni2, it can be seen that the work functions of the three are higher than that of the clean Fe surface and the D3 arrangement has the highest work function. In addition, we also calculated their formation enthalpies, and found only a 10−4 order of magnitude difference, which shows the reliability of the conclusion in Section 3.1. Similarly, for Fe10Cr3Ni3, it can be seen that the work function is not much different, and the difference in their formation enthalpies has reached the order of 10−5 which is negligible.
 | ||
| Fig. 5 The work functions and the formation enthalpies of Fe(110) surface doped with three Cr atoms and two Ni atoms (D1, D2, D3), four Cr atoms and two Ni atoms (E1, E2, E3, E4). The illustrations show the corresponding atomic structures of the first layer of Fe(110) surface. | ||
In order to compare different compositions, we have listed the highest work functions of them in a graph, as shown in Fig. 6. It can be found that Fe10Cr4Ni2 composition has the highest work function, followed by the work function of Fe11Cr4Ni1. When the Cr content increases, the work function of the Fe alloy increases significantly; when the Cr content remains unchanged, a moderate increase in the Ni content also increases the work function of the Fe alloy. However, when the Ni content increases to the same level as Cr (Fe10Cr3Ni3), the contribution of Ni to the work function is not obvious or even tends to decrease. As we mentioned above, the effect of dopant atoms on the work function of the metal surface depends on the electronegativity of the dopant atoms.34 It is generally believed that if the electronegativity of the dopant atoms is greater than that of the substrate atoms, electrons will not only be transferred to the dopant atoms in the surface layer, but also transferred from the substrate layer to the dopant layer, resulting in excessive negative charges in the surface layer and excessive positive charges below the surface layer. This will create a negative dipole moment that points inwards, strengthening the dipoles of the original surface and increasing the work function.41,49 However, the electronegativity of Cr is lower than that of Fe and Ni, while Ni has the highest electronegativity. It seems that the results do not fit the traditional interpretation between work function change and charge transfer. The reasons why the simple electronegativity argument fails have been discussed previously.46,50 In fact, the change in work function depends not only on pure charge transfer, but also on the details of charge redistribution. For Fe–Cr–Ni alloys, there are basically metallic bonds, and the change in charge density is complicated, which has been explained with the general method proposed by Leung et al.46,50 They reported that the changes in work function were related to changes in dipole from both charge transfer and polarization. The theory explains the relationship among work function change, charge transfer, and polarization, which can be described by the following formula:46,49
 | (4) |
 | ||
| Fig. 6 The work functions of Fe(110) surface with five compositions. The illustrations show the corresponding atomic structures of the first layer of Fe(110) surface. The red line indicates the work function of the clean Fe surface. | ||
From above relationship, it is believed that the work function of a surface can be determined by the charge transfer caused by the difference in electronegativity of atoms, as given by ΔP1. Besides, the polarization between the layers can also result in the change in work function, as given by ΔP2. We believe that when the doping concentration of Cr reaches a certain level, the dipole change caused by the polarization between the doped layer and the substrate layer will play a dominant role. It will alter the sign of the dipole on the Fe surface, so the work function increases with increasing Cr content. When the Cr content remains unchanged, the charge transfer caused by an appropriate increase in Ni can generate a new negative dipole moment, thereby increasing the work function. However, when the amount of doping Ni atoms continues to increase, the polarization induced by Cr will be weakened, and the work function will tend to decrease. Therefore, the doping amount of Ni should be moderate. In previous reports by Sun et al.,49 it was found that doping Cr on the surface of Ni metal has a similar behavior as doping Cr on Fe surfaces. When the content of Cr on the surface of Ni (111) and (100) reaches 4Cr and 6Cr, respectively, the surface work function will increase with the Cr doping amount, which is a result of the dipole change due to the polarization between the doped layer and the underlying substrate.
Since there are several Fe atoms on the surface, Fe atoms at different sites will have different changes in atomic structure such as spatial displacement, and the bond length and bond strength formed with surrounding atoms will also be different. Therefore, the vacancy formation energy of Fe atoms at different sites will be bound to be different. In order to produce comprehensive and objective analysis, we differentiated Fe atoms at different sites with Ni as the center according to the principle of equivalence, namely the first, second nearest neighbor of doped atoms, and so on, as shown in Fig. 7(a). For the Fe11Cr4Ni1 composition, there are four difference sites: 1st nearest neighbor (yellow atoms), 2nd nearest neighbor (red atoms), 3rd nearest neighbor (green atoms) and 4th nearest neighbor (purple atoms). However, Fe10Cr4Ni2 only has Fe atoms at two different sites: 1st nearest neighbor (yellow atoms) and 2nd nearest neighbor (red atoms).
 | ||
| Fig. 7 (a)Vacancy formation energies of Fe atoms on Fe(110) surface doped with four Cr atoms and one Ni atom (Fe11Cr4Ni1, left), four Cr atoms and two Ni atoms (Fe10Cr4Ni2, right). The yellow atoms, red atoms, green atoms and purple atoms represent the first, second, third and fourth nearest neighboring Fe atom of the dopant, respectively; (b) vacancy formation energies of the first, second, third and fourth neighbors of doped atoms on Fe(110) surface with two compositions. | ||
In Fig. 7(b), we calculated the vacancy formation energies of the first, second, third and fourth nearest neighbor Fe atoms of doped atoms on the Fe(110) surface with two compositions. The vacancy formation energy is the energy required for atoms to escape from the metal surface, so the higher the vacancy formation energy, the more stable the iron atom at the site is. Conversely, the lower it is, the easier it is for atoms to escape from the surface, and the surface dissolution is more likely to occur.41 By comparing the vacancy formation energies at different sites, we can see that the second nearest neighbors of doped atoms require higher energy to escape the surface under both compositions, and the first nearest neighbor is the lowest. For Fe11Cr4Ni1 composition, the Fe atomic vacancy formation energy of the third and fourth neighbors is between the first and second neighbors. This is because the doping of Cr and Ni will change the distance between the first neighbor Fe atom and the doped atom, and in turn change the distance between the first neighbor and the second neighbor atom. This change has an important effect on the local atomic structure, thus changing the surface vacancy formation energy. More importantly, the vacancy formation energy of the first and second nearest neighbor Fe atoms of the chemistry Fe10Cr4Ni2 is higher than that of Fe11Cr4Ni1, which indicates that when 4 Cr atoms are doped, the addition of two Ni atoms is more conducive to the stability of Fe atoms. Combined with the conclusions in 3.2.1, Fe10Cr4Ni2 not only has a strong electron binding ability, but also has the potential to improve the stability of the first and second nearest neighbor Fe atoms, which reduces the anodic dissolution process of Fe to a certain level. However, it should be pointed out that the third nearest neighbor Fe atoms of Fe11Cr4Ni1 (the green atoms in Fig. 7(a)) becomes the first nearest neighbor Fe atoms of Fe10Cr4Ni2 when one more Ni is doped. For those four atoms in the model, the vacancy formation energy is reduced and there is a possibility of easier dissolution.
4. Conclusions
Based on first-principles calculations, the appropriate doping sites of alloying elements Cr and Ni on the Fe(110) surface were analyzed, and the effects of different alloy compositions (Fe12Cr3Ni1, Fe11Cr4Ni1, Fe11Cr3Ni2, Fe10Cr4Ni2, Fe10Cr3Ni3) on the surface stability of Fe(110) surface were also studied. We calculated the work function of Fe(110) surface and the vacancy formation energy of each Fe atomic site under different doping amounts, and studied the anodic dissolution process from the two aspects of electron transfer and atomic dissolution, which are then used as an indicator of the corrosion resistance of Fe alloys. The main conclusions are as follows:(1) By comparing the formation enthalpies and work functions of various doping schemes of Fe12Cr3Ni1, Fe11Cr4Ni1 and Fe10Cr4Ni2, it is found that the doping atoms are more likely to be dispersed rather than aggregated in solid solution. When the Cr atoms are symmetrically distributed and the Ni atoms are located in the center, the structure has the highest work function and the most stable site arrangement.
(2) Doping atoms have a great influence on the electron transfer on the Fe surface, and the work functions of the five calculated chemistries are ranked as follows: Fe10Cr4Ni2 > Fe11Cr4Ni1 > Fe11Cr3Ni2 > Fe10Cr3Ni3 > Fe12Cr3Ni1. This shows that Fe10Cr4Ni2 has the strongest electron binding ability among the five concentrations with the possibility of the highest corrosion potential. The increase of Cr content led to the increase of the work function of Fe(110) surface, while the addition of Ni is moderate, the excess of which on the surface will result in the decrease of the work function.
(3) By calculating the vacancy formation energies under Fe10Cr4Ni2 and Fe11Cr4Ni1 chemical compositions, it is found that the first neighbor of the dopant atoms is more unstable than the second neighbor, and the third and fourth neighbors of Fe11Cr4Ni1 are in between. Fe10Cr4Ni2 has higher vacancy formation energies of the first and second nearest neighbor Fe atoms than Fe11Cr4Ni1, indicating that most Fe atoms are more stable under the former composition, so it is more difficult to dissolve for surface atoms. However, since the first-neighbor sites under the two compositions are not exactly one-to-one correspondence, a small number of atoms in the model of Fe10Cr4Ni2 may be more likely to escape than Fe11Cr4Ni1.
Furthermore, this study provides a simple and efficient method of first-principles calculations for screening doping schemes and predict the corrosion resistance of alloying additions.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors appreciate tremendous helps from all members at the Integrated Computational Materials Engineering (ICME) lab, Beijing Institute of Technology, China. The research work is financially supported by National Natural Science Foundation of China (project number: 52073030) and National Natural Science Foundation of China-Guangxi Joint Fund (U20A20276).References
- Y. Zeng, M. Yu, Y. Meng, P. Fang, X. Lu and Y. Tong, Adv. Energy Mater., 2016, 6, 1601053 CrossRef.
- L. Martinelli, F. Balbaud-Célérier, A. Terlain, S. Delpech, G. Santarini, J. Favergeon, G. Moulin, M. Tabarant and G. Picard, Corrosion Sci., 2008, 50, 2523–2536 CrossRef CAS.
- J. Potgieter, P. Olubambi, L. Cornish, C. Machio and E.-S. M. Sherif, Corros. Sci., 2008, 50, 2572–2579 CrossRef CAS.
- T. Duong, Y. Wang, X. Yan, A. Couet and S. Chaudhuri, arXiv, preprint, arXiv:2104.10590, 2021 Search PubMed.
- K. Cashell and N. Baddoo, Thin-Walled Struct., 2014, 83, 169–181 CrossRef.
- Y. Inoue and M. Kikuchi, Nippon Steel Technical Report No. 88, July 2003 Search PubMed.
- T. Zhang and D. Li, J. Mater. Sci., 2001, 36, 3479–3486 CrossRef CAS.
- C. Punckt, M. Bolscher, H. H. Rotermund, A. S. Mikhailov, L. Organ, N. Budiansky, J. R. Scully and J. L. Hudson, Science, 2004, 305, 1133–1136 CrossRef CAS PubMed.
- M.-F. Ng, D. J. Blackwood, H. Jin and T. L. Tan, J. Phys. Chem. C, 2020, 124, 13799–13808 CrossRef CAS.
- C. Han, C. Zhang, X. Liu, H. Huang, S. Zhuang, P. Han and X. Wu, J. Mol. Model., 2015, 21, 1–9 CrossRef PubMed.
- F. Wang and Y. Shu, Oxid. Met., 2003, 59, 201–214 CrossRef CAS.
- L. Yang, C. Bo, W. Junwei, W. Zhiping and L. Wensheng, Rare Met. Mater. Eng., 2014, 43, 17–23 CrossRef.
- X. Yin, H. Wang, S. Sun and E.-H. Han, Mater. Today Commun., 2020, 24, 101122 CrossRef CAS.
- B. Dong, W. Liu, L. Chen, T. Zhang, Y. Fan, Y. Zhao, S. Li, W. Yang and W. Banthukul, Cem. Concr. Compos., 2022, 125, 104317 CrossRef CAS.
- Y. Tian, C. Dong, G. Wang, X. Cheng and X. Li, Cem. Concr. Compos., 2020, 246, 118462 CAS.
- C. Kuehmann and G. Olson, Mater. Sci. Technol., 2009, 25, 472–478 CrossRef CAS.
- P. Lu, J. E. Saal, G. B. Olson, T. Li, O. J. Swanson, G. Frankel, A. Y. Gerard, K. F. Quiambao and J. R. Scully, Scr. Mater., 2018, 153, 19–22 CrossRef CAS.
- S. Wang, C. Zhang, X. Li, H. Huang and J. Wang, J. Mater. Sci. Technol., 2020, 58, 205–214 CrossRef CAS.
- S. Wang, J. Wang, C. Zhang and C. Xue, J. Alloys Compd., 2022, 904, 163800 CrossRef CAS.
- X. Li, C. Zhang, J. Wang, H. Huang and S. Wang, Corros. Sci., 2020, 167, 108549 CrossRef CAS.
- S. Bhasker-Ranganath, C. Wick and B. Ramachandran, Mater. Today Chem., 2020, 17, 100298 CrossRef.
- J. Yao, N. Li, H. Grothe, Z. Qi and C. Dong, Appl. Surf. Sci., 2021, 554, 149597 CrossRef CAS.
- W. Li and D. Li, Acta Mater., 2006, 54, 445–452 CrossRef CAS.
- R. Li, B.-g. Fu, T.-s. Dong, G.-l. Li, J.-k. Li, X.-b. Zhao and J.-h. Liu, J. Mater. Res. Technol., 2022, 18, 448–460 CrossRef CAS.
- J. Hu, C. Wang, S. He, J. Zhu, L. Wei and S. Zheng, Coatings, 2018, 8, 51 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. Blöchl, O. Jepsen and O. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. Kittel, Introduction to Solid State Physics New York, 7th edn, 1976 Search PubMed.
- D. Jiang and E. A. Carter, Surf. Sci., 2003, 547, 85–98 CrossRef CAS.
- J. Wang and S.-Q. Wang, Surf. Sci., 2014, 630, 216–224 CrossRef CAS.
- R. Tran, X.-G. Li, J. H. Montoya, D. Winston, K. A. Persson and S. P. Ong, Surf. Sci., 2019, 687, 48–55 CrossRef CAS.
- Z. Luo, H. Zhu, T. Ying, D. Li and X. Zeng, Surf. Sci., 2018, 672, 68–74 CrossRef.
- J. A. Yuwono, N. Birbilis, R. Liu, Q. Ou, Q. Bao and N. V. Medhekar, J. Electrochem. Soc., 2017, 164, C918 CrossRef CAS.
- S. J. Mole, X. Zhou and R. Liu, J. Phys. Chem., 1996, 100, 14665–14671 CrossRef CAS.
- H. Zhang, S. Shang, J. E. Saal, A. Saengdeejing, Y. Wang, L.-Q. Chen and Z.-K. Liu, Intermetallics, 2009, 17, 878–885 CrossRef CAS.
- G. Ghosh and M. Asta, Acta Mater., 2005, 53, 3225–3252 CrossRef CAS.
- C. Wolverton, V. Ozoliņš and M. Asta, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144109 CrossRef.
- B. Jiang, T. Guo, Q. Peng, Z. Jiao, A. A. Volinsky, L. Gao, Y. Ma and L. Qiao, Corros. Sci., 2019, 157, 498–507 CrossRef CAS.
- C. Zhang, J. Wang, X. Li, S. Wang, S. Zhu and S. Guan, Phys. Chem. Chem. Phys., 2021, 23, 26887–26901 RSC.
- A. Sumer and S. Chaudhuri, Corrosion, 2017, 73, 596–604 CrossRef CAS PubMed.
- H. Ma, X.-Q. Chen, R. Li, S. Wang, J. Dong and W. Ke, Acta Mater., 2017, 130, 137–146 CrossRef CAS.
- H. L. Skriver and N. Rosengaard, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7157 CrossRef CAS PubMed.
- E. Zuo, X. Dou, Y. Chen, W. Zhu, G. Jiang, A. Mao and J. Du, Surf. Sci., 2021, 712, 121880 CrossRef CAS.
- T.-C. Leung, C. Kao, W. Su, Y. Feng and C. T. Chan, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195408 CrossRef.
- X. Huang, H. Lu and D. Li, Mater. Chem. Phys., 2016, 173, 238–245 CrossRef CAS.
- P. Bergveld, J. Hendrikse and W. Olthuis, Meas. Sci. Technol., 1998, 9, 1801 CrossRef CAS.
- X.-F. Sun, H.-T. Wang and E.-H. Han, Acta Metall. Sin., 2019, 32, 461–470 CrossRef CAS.
- G. Xu, Q. Wu, Z. Chen, Z. Huang and Y. P. Feng, J. Appl. Phys., 2009, 106, 043708 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |